
Submitted:
22 February 2025
Posted:
24 February 2025
You are already at the latest version
Abstract
Hepatic stellate cells (HSC) are the major source of myofibroblasts (MFB) in fibrosis and cancer associated fibroblasts (CAF) in both primary and metastatic liver cancer. Over the past few decades, there has been significant progress in understanding the cellular and molecular mechanisms by which liver fibrosis and HCC occur and the key roles of HSC in the pathogenesis. HSC targeted approaches via specific surface markers and receptors may enable selective delivery of drugs, oligonucleotides, and therapeutic peptides that exert optimized anti-fibrotic and anti-HCC effects. Recent advances in omics, particularly single-cell sequencing and spatial transcriptomics, hold promise for identifying new HSC targets for diagnosing and treating liver fibrosis/cirrhosis and liver cancer.
Keywords:
Hepatic Stellate Cells
; Liver Cirrhosis
; Hepatocellular Carcinoma
; Targeting therapeutics
1. Introduction
Approximately 844 million people worldwide suffer from chronic liver diseases (CLD), resulting in approximately 2 million deaths annually, with a trend that continues to rise [1,2]. Liver fibrosis is a scar tissue repairing response following chronic liver injury from various etiologies. Persistent accumulation of scar tissue leads to irreversible changes in liver structure and function, resulting in decompensated cirrhosis and complications of portal hypertension, and a significantly increased risk of hepatocellular carcinoma (HCC). There is an urgent need for effective prevention and early diagnosis to manage and treat CLD, thus preventing cirrhosis-related morbidity and mortality. Hepatic stellate cells (HSC) are the major source of myofibroblasts (MFB) in fibrosis and cancer associated fibroblasts (CAF) in both primary and metastatic liver cancer [3,4]. Targeting the activation of HSC is not only important for fibrosis treatment (Figure 1), to alter their interaction with HCC cells is also one strategy for HCC treatment (Figure 2).
2. The Role of Hepatic Stellate Cells (HSC) in Liver Cirrhosis and HCC
2.1. HSC and Liver Fibrosis
HSC are non-parenchymal cells in the liver which play an essential role in fibrogenesis and the progression of liver cirrhosis. The activation of HSC, transitioning from quiescent, fat-storing (retinyl ester) pericytes residing in the liver’s peri-sinusoidal space to persistently activated myofibroblasts, is a critical event in the development of liver fibrosis [5]. Under stimuli such as cell death (apoptosis and necrosis), inflammation, oxidative stress, extracellular matrix degradation and remodeling, and pro-fibrotic cytokines, HSC are activated and transdifferentiated into MFB phenotypes characterized by high proliferative activity. They lose their characteristic lipid droplets containing retinoids, express α-smooth muscle actin (α-SMA) and extracellular matrix (ECM) components such as collagen, and acquire contractility, chemotaxis, and migratory capabilities. Activated HSC also produce large amounts of growth factors and pro-fibrotic cytokines, including transforming growth factor β (TGF-β) and platelet-derived growth factor (PDGF). Furthermore, activated HSC exhibit an inflammatory phenotype, releasing pro-inflammatory cytokines such as monocyte chemoattractant protein-1 (MCP-1), promoting the accumulation of inflammatory cells and amplifying the inflammatory response [6].
2.2. CAF and HCC
As a predominant complication of cirrhosis, liver cancer is characterized by crucial alterations of inflammation and fibrosis/cirrhosis in the microenvironment, with HSC as the main source of CAF in HCC stroma [7]. CAF can be located in the sinusoids, fibrous septa, and capsule of liver tumors, as well as within the tumor matrix. The cells increase matrix stiffness, enhance ECM remodeling, as well as secrete growth factors, chemokines, angiogenic factors, tumor-promoting microRNAs and exosomes that are taken up by tumor cells, support and facilitate HCC tumorigenesis, survival, growth and metastasis [8,9,10]. A combined single-cell RNA-sequencing (scRNA-seq) analysis with genetic tools to activate, deplete, inhibit, and manipulate HSC and their mediators during HCC development, it was revealed that there is a progressive imbalance of HSC subpopulations during disease progression. Quiescent HSC, which express protective factors, shift towards activated HSC, which express tumor-promoting mediators, secrete factors (e.g., VEGF and angiopoietin-1) that promote angiogenesis, thereby setting the stage for HCC development in CLD.
CAF may also play a key role in inducing immune tolerance and immune evasion of HCC cells [11,12]. HCC CAF modulates antigen presentation by dendritic cells (DC), recruits myeloid-derived suppressor cells (MDSC) and tumor-associated neutrophils, and recruit tumor-associated macrophages while inducing their polarization. HCC patients with high density of CAF around the tumor have poor long-term clinical prognosis and a high incidence of early recurrence and metastasis. The density of Foxp3+ regulatory T cells (Treg) and CD68+ macrophages around the tumor is positively correlated with the density of CAF. Activated HSC accelerate activated T cell apoptosis, increase Treg numbers, and inhibit T cell-mediated cytotoxicity. activated HSC inhibit T cell function by upregulating programmed death ligand PD-L1 (B7-H1) to bind to its receptor PD-1. PD-L1 expressed by HSC can induce T cell apoptosis, attenuate T cell infiltration, and suppress T cell mediated cytotoxicity. HSC activation causes massive production of Th2/Th3-like cytokines, downregulation of Th1-like cytokines and cytotoxic T cells (CTL) function, and further suppression of the host immune system. aHSC also induce expansion of two suppressive immune cell populations: myeloid-derived suppressor cells (MDSCs) and T helper 17 (Th17) cells, a subset of CD4+ effector T cells. Some cytokines secreted by HSC, such as IL-6, may also promote MDSC and inhibit T cell proliferation and function.
3. Targeting HSC for the Prevention and Treatment of Liver Cirrhosis
The activation of HSC is a determining factor in liver fibrosis progression and a critical target cell for anti-fibrotic treatments in various liver diseases [13,14]. Inhibiting stellate cell proliferation, differentiation and activation is an attractive strategy for ameliorating hepatic fibrosis. This may be achieved by blocking the synthesis of key molecules involved in fibrogenesis with nucleic acid drugs (e.g., siRNA, saRNA), blocking ATP binding to the receptors and transcriptional factors of fibrogenic signaling pathways by kinases inhibitors, and preventing key ligands from binding to its receptors by monoclonal antibodies and high-affinity ligand traps (Figure 1). Multiple signaling pathways and downstream transcription factors mediate the pro-fibrotic effects of HSC in liver fibrosis, including PDGF, integrin, TGF-β, Toll-like receptor 4 (TLR4) signaling pathways [15,16,17,18]. Various signaling molecules also contribute to fibrogenesis, such as platelet-derived growth factor (PDGF), TGFβ, and connective tissue growth factor (CTGF, or CCN2), damage-associated molecular patterns (DAMP) and pathogen associated molecular patterns (PAMP) [19,20]. Additional mediators promoting HSC activation include angiotensin, leptin, osteopontin and hedgehog ligands [21,22]. The represent clinical trials of potential antifibrotic therapies in chronic liver diseases targeting HSC activation are shown in Table 1.
3.1. Molecular Signaling Pathways Associated with HSC/MFB Activation
3.1.1. Reactive Oxygen Radicals (ROS)
Excessive production of intrahepatic ROS during liver injuries activates HSC and promotes the progression of liver fibrosis [23]. ROS activates signaling pathways and transcription factors such as JNK and NF-κB, and increases the expression of MCP-1, Col-1, and TIMP-1. ROS is produced by mitochondrial damage and activation of cytochrome P450 (especially cytochrome P450 2E1), xanthine oxidase, and NADPH oxidase (NOX). NOX is a group of enzymes with seven known homologs, and the activation of NOX1, NOX2, and NOX4 plays a key role in the activation of HSC and has been considered a therapeutic target [24,25].
3.1.2. TOLL-like Receptors (TLRs)
TLR4 is expressed by activated HSC and hepatic immune cells and is often a cofactor in the development and progression of liver fibrosis. TLR4 can be activated by exogenous PAMP ligands, such as lipopolysaccharides from intestinal bacteria, and endogenous DAMP such as high mobility group box-1 (HMGB1). The activation of TLR4 enhances TGF-β1 signaling and promotes the fibrogenic effects of inflammatory chemokines (such as CCL2, IL-6) by downregulating the expression of osteoblastin and TGF-β1 transmembrane inhibitor (BAMBI) [26]. Therefore, TLR4 inhibition has been considered in clinical trials for the treatment of liver fibrosis in NASH (ClinicalTrials.gov ID: NCT02442687, NCT04255069). In addition, TLR3 promotes liver fibrosis activity by stimulating IL-10 production.
3.1.3. Hedgehog Signaling Pathway
The Hedgehog (Hh) pathway is a highly conserved signaling pathway that has been implicated in cell proliferation, adhesion, migration, differentiation, and embryogenesis. In the liver, Hh signaling in HSC is pro-fibrotic, mediated through multiple downstream transcriptional targets including Smo, Ptc, Gli1 and Gli2. Hh signaling promotes the activation of HSC and liver repair [27]. The interaction of Hh and Notch signaling pathways regulates key cells in the liver repair process by regulating epithelial-mesenchymal transition (EMT). The Hh pathway also regulates metabolic pathways (e.g., glycolysis, glutaminolysis) [28], as well as DNA methylation and DNA methyl-binding protein (MeCP2). Forskolin, a Hh signaling inhibitor, can alleviate the progression of CCl4-induced murine liver fibrosis [29]. Ligustrazine plays an anti-fibrotic role by blocking Hh signaling to arrest the cell cycle and trigger apoptosis of HSCs. Curcumin induces apoptosis of HSC, as well as regulates glycolysis and other metabolic pathways of HSCs by inhibiting the Hh signaling pathway [30]. Therefore, specific Hh signal inhibitors can be used as promising therapeutic agents for anti-liver fibrosis treatment.
3.1.4. Wnt Signaling Pathway
The Wnt signaling pathway can be classified into classical (i.e., Wnt/β-catenin) and non-canonical Wnt signaling pathways, involving proteins such as Wnt5a and Wnt11. The Wnt signaling pathway is involved in cell proliferation, differentiation, and apoptosis, promoting HSC activation and inhibiting apoptosis [31]. The classical Wnt signaling pathway inhibits the activation of HSCs by increasing MeCP2 protein expression, which subsequently inhibits peroxisome proliferator-activated receptor γ (PPAR-γ) [32]. MiR-17-5p has been implicated in liver fibrosis progression by inhibiting Wnt inhibitory factor 1 (WIF1) expression and activating the Wnt/β-catenin signaling pathway [33]. Hesperetin derivative-7 (HDND-7) reverses liver fibrosis by modulating the Wnt/β-catenin signaling pathway to inhibit HSC activation and proliferation [34]. Wnt5a may be involved in the regulation of inflammatory cytokine production in HSCs and cell proliferation, making it a potential therapeutic target for liver fibrosis [35].
3.1.5. Chemokine and Cytokine
In addition to TGF-β, a number of other growth factors and chemokine targets are being pursued, including interleukin (IL)-11, connective tissue growth factor (CCN2), and CCL24. Interleukin 11 (IL-11) has remarkably pleiotropic activity on epithelial cells and mesenchymal cells across a number of tissues, including liver. Antagonism or knockout of IL-11 attenuates HSC activation while reducing steatosis and metabolic derangements within hepatocytes in MASH [36]. A neutralizing antibody to IL-11 has been evaluated in a clinical trial (NCT05658107) for its efficacy and safety in NASH. CCL24 is a circulating chemokine produced by epithelial cells and fibroblasts, which binds to its cognate receptor CCR3 to promote inflammation, cell trafficking and fibrosis [37]. Serum levels of CCL24 correlate with severity of fibrosisand and are particularly elevated in patients with primary sclerosing cholangitis. CCL24 levels also correlate with disease stage in systemic sclerosis [38]. A monoclonal antibody targeting CCL24 has demonstrated efficacy in several animal models of liver disease, leading to its evaluation in early clinical trials [39]. A completed Phase 1B trial in patients with MASLD but not MASH demonstrated good tolerability and improvement in several serum markers of collagen turnover and inflammation, and a Phase 2A randomized, placebo-controlled trial is currently underway (NCT05824156).
3.2. Molecular Signaling Pathways Associated with HSC/MFB Proliferation
PDGF is the most potent proliferative stimulus for HSC. PDGF belongs to the family of growth factors consisting of four secreted extracellular ligands encoded by four different genes. They assemble into disulfide-bonded dimers via homo- or heterodimerization. All members share the highly conserved and specific PDGF/VEGF homology domain necessary for receptor binding and activation. The PDGF ligands exert their biological effects through the two structurally related tyrosine kinase receptors PDGFR-α and PDGF-β. PDGF-β is upregulated during HSC activation both in vitro and in vivo. PDGF-B and -D bind to PDGF-β, resulting in downstream phosphorylation of extracellular signal-regulated protein kinase/mitogen-activated protein kinase (Erk/MAPK) and protein kinase B (Akt/PKB) in the phosphoinositide-3-kinase (PI3K) pathways, leading to significant HSC proliferation.
Blocking the biological effects of PDGF inhibits HSC proliferation and reduces the degree of liver fibrosis. Imatinib mesylate (Gleevec), a clinically used PDGF receptor (PDGFR) tyrosine kinase inhibitor could be a promising molecular targeted approach to limit the liver fibrosis development. PDGF antagonists, PDGF-specific neutralizing antibodies (e.g., AbyD3263, MOR8457), soluble dominant-negative PDGF receptor inhibitors can serve as targeted therapies to block PDGF. In addition, several multi-kinase inhibitors targeting PDGF receptors (e.g., imatinib, nilotinib, sorafenib) are currently under clinical trials [40]. Although extensive effort has been made to study PDGF systems in liver fibrosis, most PDGF antagonists that block fibrogenic activation and ECM production of MFB work well in culture and in some rodent models of liver fibrosis, but carry a high risk of unwanted side effects in patients due to their lack of specificity. Inhibition of PDGFR-β signaling with tyrosine kinase inhibitors has proven effective in early phases but not in advanced cirrhotic models in rodents. Nevertheless, they remain among the more promising developments due to longstanding use in cancer and established safety and tolerability.
3.3. Molecular Signaling Pathways Associated with Pro-Liver Fibrosis
Antifibrotic targets may be conserved across multiple organs. The concept of core and regulatory pathways has been proposed to identify the optimal targets for antifibrotic drug discovery/design. Core pathways are conserved in different organs and species, whereas regulatory pathways may be confined to specific cell types or organs. Potential core pathway components include lysyl oxidase 2 (LOXL2), αv-integrin, TGF-β and their downstream signaling effector molecule CTGF, and drugs targeting these molecules are being evaluated in clinical trials.
3.3.1. Transforming Growth Factor β (TGF-β)
The TGF-β pathway plays a crucial role in maintaining physiological homeostasis, including immunomodulation and tumor suppression. It has long been recognized as the most potent driver of fibrosis in all tissues and remains the primary antifibrotic target in HSCs and other fibrogenic cell populations [41]. Blocking the TGF-β1 pathway inhibits liver fibrosis development, but systemic inhibition of TGF-β1 can promote inflammation and adversely affect liver parenchyma and precursor cells, Integrins αvβ6 and αvβ8 play a key role in activating latent TGF-β1 [42,43]. Therefore, small-molecule integrin inhibitors and function-blocking antibodies may serve as therapeutic strategies for liver fibrosis treatment.
However, the pleiotropic effects, multiple activation modes, and diverse signaling pathways of TGF-β, which are cell-type- and cell-state-specific, have made this a challenging target. Moreover, systemic antagonism of TGF-β is unsafe, as inhibiting its developmental, antiproliferative, antiapoptotic, and anti-inflammatory activities can disrupt tissue homeostasis and promote cancer [44,45,46]. Therefore, TGF-β antagonists are sought that antagonize only its fibrogenic activity while preserving other functions. One strategy seeks to inhibit cell surface integrins that contribute to TGF-β activation at the cell membrane, which underlies the promise of using a small molecule (Bexotegrast, PLN-74809) that blocks activity of αvβ1 and αvβ6 in pulmonary fibrosis [47] and primary sclerosing cholangitis (NCT04480840). An exciting new approach has leveraged the discovery that latent TGF-β is complexed with different proteins, each of which mediates different activities of the cytokine. Specifically, whereas release of latent TGF-β from either GARP or LRRC33 largely regulates its immunogenic activity [48,49], its binding to latent TGF-β binding protein (LTBP) controls its fibrogenic activity [50]. With this knowledge, investigators have developed an antibody that only prevents the release of LTBP-bound TGF-β but does not block the release of TGF-β from GARP or LRRC33, thereby inhibiting fibrosis while preserving TGF-β’s immunoregulatory and other activities [51]. As proof-of-principle, this antibody attenuates progression of renal fibrosis in two mechanistically distinct mouse models, but no studies in MASH models have been reported yet. Another approach to antagonizing TGF-β activity distinguishes between the differential fibrogenic activities of the three major TGF-β isoforms, TGF-β1, TGF-β2 and TGF-β3. A recent study indicates that most of TGF-β’s fibrogenic activity can be to TGF-β2 and TGF-β3 [52], whose antagonism avoids the liabilities of inhibiting TGF-β1; this strategy shows promise in systemic sclerosis but has not yet been explored in liver fibrosis [53].
Connective tissue growth factor (CTGF) is a molecular downstream of TGF-β1 signaling that amplifies its signaling effect, and a monoclonal antibody to CTGF (FG-3019) completed clinical trials II but failed to show efficacy in Phase 3 trials against pulmonary fibrosis (NCT01890265, NCT04419558) [54,55].
3.3.2. FAP
Activated hepatic stellate cells adopt a more myofibroblast-like phenotype and express α-smooth muscle actin (αSMA), glial fibrillary acidic protein (GFAP), and fibroblast activation protein (FAP) [56]. Intrahepatic expression of FAP, but not GFAP or αSMA, correlated with the degree of liver fibrosis in patients with hepatitis C virus (HCV) infection [57]. FAP activity was 14- to 18-fold higher in cirrhotic livers compared to healthy livers, and circulating FAP levels nearly doubled in patients with alcoholic cirrhosis [58]. The concentration and activity of circulating FAP were significantly increased in patients with liver cirrhosis compared to healthy livers, and these elevated levels correlated with increased cleavage of α-2 antiplasmin. N-terminally cleaved α-2 antiplasmin is a more potent inhibitor of fibrinolysis than its uncleaved form. Therefore, it has been proposed that elevated circulating FAP may contribute to hemostasis-related bleeding and thrombotic events in liver cirrhosis. Low levels of circulating FAP can be used clinically to rule out clinically significant liver fibrosis in patients with non-alcoholic fatty liver disease [59].
3.3.3. Cannabinoid Receptors (CB)
The cannabinoid receptors CB1 and CB2 are components of the G-protein-coupled receptor family and the endocannabinoid system, both of which play key roles in the progression of liver fibrosis in chronic liver disease. Activated HSC express CB1 receptors, which promote fibrosis. The first-generation CB1 antagonist rimonabant was withdrawn from clinical use due to its association with depression. Currently, peripherally restricted CB1 antagonists that do not cross the blood-brain barrier are under development. CB2 receptors are currently regarded as promising targets for anti-inflammatory and antifibrotic therapy, and treatment with CB1 antagonists and CB2 analogues may represent an ideal multi-target antifibrotic approach [60,61].
3.4. Molecular Signaling Pathways Associated with HSC/MFB Contraction Responses
Endothelin-1 (ET-1) is a potent hepatic vasoconstrictor [62]. In a healthy liver, ET-1 is primarily produced by endothelial cells, whereas in liver injury, it is predominantly produced by hepatic stellate cells (HSC). Activated HSC exhibit high expression of ET-1, angiotensin-II (Ang-II), and their receptors. The renin-angiotensin system (RAS) regulates the formation of liver fibrosis. Blocking RAS through angiotensin receptor 1 (AT1) blockers and angiotensin-converting enzyme inhibitors (ACE inhibitors) may be an effective strategy for the treatment of liver fibrosis. There is a crosstalk between Ang-II/AT1 and ET-1 systems, with Ang-II inducing the expression of ET-1 in HSC via the PI3K/Akt signaling pathway, and ET-1 promotes the role of Ang-II in the transdifferentiation of HSC into MFB-like cells [63].
3.5. Molecular Signaling Pathways Associated with Reversal of Liver Fibrosis
3.5.1. Activated HSC Return to Resting Molecular Signaling Pathways
PPAR-γ is a nuclear transcription factor that can be activated by peroxisome proliferators and plays an important role in maintaining HSC in a quiescent state. Reduced PPAR-γ expression promotes HSC activation, while PPAR-γ agonists or ligands can inhibit HSC activation and reduce ECM deposition, making them potential targets for antifibrotic therapy [64]. The combination of PPAR-α/δ stimulant (GFT505) has been shown to have significant hepatoprotective effects in animal models of nonalcoholic fatty liver disease, making it a highly promising direction for NAFLD/NASH-targeted drug therapy [65].
3.5.2. Molecular Signaling Pathways That Induce Apoptosis and Senescence of Activated-HSC/MFB
Inducing apoptosis in activated HSC is an important strategy for liver fibrosis regression, and HSC contain multiple molecular families that mediate apoptosis, such as Fas/Fas-L, NF-κB, nerve growth factor receptor, Bcl-2/Bax. These pathways can be deeply studied and applied to the treatment of anti-liver fibrosis. NK cells are generally considered to have antifibrotic therapeutic potential because they promote activated HSC apoptosis through TRAIL/DR5 and NKG2D-RAE1 [66].
Senescent HSC are a proposed target for antifibrotic therapies, based on their reported roles in hepatic fibrosis, MASH, and HCC development. The core features of senescence are irreversible growth arrest and increased expression of β-galactosidase (SA-Bgal) associated with cell morphology and senescence. Their senescence mechanisms of HSC are mainly governed by the p16-Rb and Arf-p53-p21 pathways. The adenosine A2A receptor can promote the progression of liver fibrosis by reducing p53 and Rb through the PKA/Rac1/p38 MAPK pathway, thereby reducing HSC senescence and promoting HSC proliferation, which provides strong evidence for adenosine to regulate the progression of liver fibrosis [67].
Integrated high-resolution single-nucleus RNA sequencing, combined with immunostaining and flow cytometry, has identified urokinase plasminogen activator receptor (uPAR) as a marker of senescent HSC. uPAR is expressed by activated HSC during early injury and by immune cells as liver injury progresses [68]. Senescent HSC can be selectively eliminated using senolytic CAR T cells targeting uPAR or other senescence-associated proteins, such as mannose receptor CD206, to reduce liver fibrosis. HSC-derived CAF might be depleted by targeting fibroblast activation protein (FAP) via DNA vaccines, CAR T cell therapy or oncolytic viruses, to potentially reduce hepatic fibrosis and tumor burden.
Activated HSC exhibit elevated expression of αV integrin and TGF-β receptor, both of which are potential therapeutic targets for fibrosis reduction. A small molecule CWHM12 pharmacologically blocks αV integrin to attenuate fibrosis [69], and TGFβ receptor signaling can also be locally inhibited by targeting caveolin, hyaluronic acid synthase 2, CD147, hydrogen peroxide inducible clone 5, or galectin-1. After prolonged activation, HSC can senesce and secrete the SASP component IL-33 via the gasdermin D pore, which promotes tumor development [70]. Disulfiram reduces tumor burden in mice by inhibiting the gasdermin D pore [71].
3.6. ECM and Liver Fibrosis
Liver fibrosis is a dynamic process resulting from an imbalance between ECM synthesis and degradation. The regulation of ECM is primarily controlled by the balance between matrix metalloproteinases (MMPs) and matrix metalloproteinase inhibitors (TIMPs). Enhancing MMP activity while reducing TIMP activity promotes ECM degradation and suppresses ECM synthesis, contributing to antifibrotic effects [72]. HSCs are the main source of MMP-2, MMP-9 and MMP13. MMP-2 inhibits collagen I production, and promotes HSC apoptosis via cadherin signaling. HSCs are also the main source of TIMPs, and TIMP1 is an anti-apoptotic factor in activated HSCs, so TIMP1 can be used as one of the key targets for the treatment of liver fibrosis.
In addition, osteopontin, an ECM cytokine expressed by HSCs, promotes collagen I expression through integrin αvβ3 and activation of the PI3K/pAkT/NFκB signaling pathway [73], thereby promoting ECM synthesis and increasing hepatic fibrosis.
Lysyl oxidase like 2 (LOXL2) plays a role in matrix stiffness and collagen cross-linking [74]. Moreover, LOXL2 has been shown to inhibit ECM degradation in animal models of CCl4-induced liver fibrosis, and humanized antibody inhibitors of LOXL2 were tested in clinical trials against liver fibrosis [75,76]. Notably, phase 2 clinical trials targeting LOXL2 or the collagen chaperone heat shock protein 47 (HSP47) in HSCs (NCT04267393) have not been successful in patients with end-stage MASH [77].
3.7. Epigenetic Regulation Associated with Liver Fibrosis
3.7.1. DNA Methylation and Related Histone Modifications
DNA methylation in HSCs may contribute to the maintenance of their quiescent phenotype. Upon activation, HSCs express MeCP2, which promotes the silencing of antifibrotic genes (e.g., IκB, PPARγ) and increases the expression of histone methyltransferases. This leads to enhanced transcription of COL-1, TIMP-1, and TGF-β. In mammals, five DNA methyltransferases (DNMTs) have been identified: DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L. In the CCl4-induced liver fibrosis model, DNMT1 was upregulated following HSC activation, while RNAi-mediated silencing of DNMT1 inhibited both HSC activation and proliferation [78]. Guadecitabine (SGI-110), a DNMT inhibitor and a novel hypomethylated dinucleotide of decitabine, is in Phase II clinical trials for hepatocellular carcinoma but has not yet been tested for liver fibrosis [79]. In addition, histone deacetylase (HDAC) inhibitors have the potential to serve as therapeutic targets for liver fibrosis [80].
3.7.2. MicroRNAs
microRNAs (miR) regulate HSC proliferation and fibrosis formation by modulating the expression of proliferative proteins and profibrotic signaling pathways [81]. miR-27a, miR-27b, and miR-29b are significantly upregulated upon HSC activation. The physiological role of miR-29b is to inhibit the production of various extracellular matrix (ECM) proteins. Both TGF-β and lipopolysaccharide (LPS) regulate its expression levels, while hepatocyte growth factor (HGF) upregulates miR-29b expression. Additionally, overexpression of miR-29b suppresses TGF-β-induced collagen production through the Smad signaling pathway [82]. The target molecules of miR-15b and miR-16 include Bcl-2. These miRNAs promote the expression of apoptosis-related proteins by downregulating Bcl-2 and accelerating the apoptosis of activated HSC. Recent studies have confirmed that miR-30 inhibits fibrosis by suppressing the expression of Kruppel-like factor 11 (KLF11) and attenuating TGF-β signaling in CCl4-induced animal models [83]. These findings provide new insights into the mechanisms underlying liver fibrosis development.
3.8. Cell Therapies to Treat Fibrosis
CART cells are now being developed for a growing number of indications including solid tumors, autoimmunity and, most recently, senescence and fibrosis. To generate CART, DNA constructs encoding transmembrane chimeric receptors are transduced into T cells. Their general structure includes an antigen binding domain on the cell surface linked to an intracellular domain that activates T cells upon ligand engagement [84]. Studies have implicated HSC as drivers of liver injury and inflammation, leading to an effort to identify cell surface markers of senescence in this cell type to target them for CAR T mediated clearance. A pioneering study combined the knowledge of CART production with senescence biology to seek markers of senescence in HSC [85]. Using an informatics-based approach, the cell surface protein urokinase plasminogen activated receptor (uPAR) was identified as one such marker, and administration of a uPAR-directed CAR T in a murine model of MASH attenuated fibrosis, cleared senescent cells and improved serum albumin levels. Complementary to the CAR T approach to clear senescent HSC, studies by Epstein and colleagues engineered CAR T to target only cells expressing FAP-1, which is a cell surface receptor that marks fibrogenic cells in several tissues, including heart and joints, among others [86]. Administration of CAR T cells that were transduced ex vivo reduced fibroblast number and fibrosis, and improved cardiac function in a model of chronic cardiac injury [87]. These findings have established a target that does not rely on senescence and is more specific than uPAR. A remarkably elegant strategy by the same group built upon the conventional CAR T approach, instead developing a method for in vivo programming of CAR T cells [88,89,90,91]. To do so, mRNA designed to program T cells into CAR T is delivered by lipid nanoparticles that target T cells in vivo, which instruct T cells to express CAR T directed to FAP-1 on the fibroblast cell surface, yielding similar therapeutic benefit in the heart as conventional CAR T. This in vivo methodology has at least two distinct advantages. First, therapeutic CAR T-generating nanoparticles can be produced in advance and therefore be available immediately as an “off-the-shelf” technology, greatly expanding their availability beyond only facilities that can generate ex vivo CAR T onsite. Second, the use of RNA-expressing lipid nanoparticles avoids integration of genetic material into the cell genome, thereby enabling titration of CAR T activity and allowing for discontinuation or repeat administration, while avoiding unrestrained HSC clearance. Studies attempting to use in vivo CAR T targeting to FAP1 expressing cells in liver are underway, complemented by studies using FAP-1 imaging to quantify fibrosis [92]. While these reports underscore the potential benefit of selectively depleting HSC populations to reduce fibrosis, their complete elimination is potentially risky. A study in mice has demonstrated that when > 99% HSCs are depleted using a cell therapy similar to CAR T, the livers fail to regenerate due to loss of paracrine signals from HSCs that support hepatocyte replication [93], highlighting the importance of HSC in maintaining liver homeostasis, as discussed above. Selective clearance of only those HSC populations that promote fibrosis may be a more rational approach than total HSC clearance.
4. Targeting Hepatic Stellate Cells for the Prevention and Treatment of Hepatocellular Carcinoma
4.1. Angiogenesis Provide Basic Survival and Metastasis Condition of Tumor Cells
Angiogenesis is the main characteristic of HCC. Activated HSCs promote the growth of the vascular epithelium by upregulating angiopoietin-I (Ang-I). aHSCs secrete vascular endothelial growth factor (VEGF) to promote angiogenesis. Additionally, the LPS/TLR4 pathway can induce HSCs to secrete pro-angiogenic factors, promoting endothelial cell migration and tubulogenesis, enhancing angiogenesis in HCC and providing nutrients for tumor cell growth [10]. The TLR4 signaling pathway in HSCs promotes tumor progression and metastasis via the production of laminin-4 [9], and contributes to the formation of immune-tolerant microenvironment in tumors.
In patients with cirrhosis, portal hypertension causes the translocation of the gut microbiota to the liver. PAMP such as lipopolysaccharides (LPS) derived from the outer surface membrane of gram-negative bacteria, activate TLR4 in Kupffer cells and activated HSC. As a result, increased VEGF, Ang-I, and platelet-derived growth factor (PDGF) levels facilitate tumor angiogenesis. Additionally, the hypoxic environment resulting from liver inflammation, fibrosis, and rapid tumor growth promotes the expression of hypoxia-inducible factor 1 (HIF-1), upregulating VEGF and promoting the formation of new vessels.
4.2. Matrix Stiffness Promotes Tumor Growth, Invasion and Metastasis
Tumor cells have a favorable environment because of ECM deposits, collagen cross-linking, and increased matrix stiffness, which positively regulate tumor growth and metastasis. Cancer stem cells (CSC) originate from human stem cells and tumor cells. CSC, characterized by its ability to transform into different types of tumor cells, sustains the overall number of tumor cells through continuous proliferation and transformation. A higher matrix stiffness upregulates the stemness of HCC. Integrin β1 promotes the stemness of HCC cells by activating the serine-threonine protein kinase/animal rapamycin/sex-determining region Y-box 2 (AKT/mTOR/SOX2) pathway.
When comparing the effects of different mesenchymal stiffnesses on the epithelial-mesenchymal transition (EMT) and the invasive ability of tumor cells, higher matrix stiffness was found to promote tumor cell invasion and metastasis by upregulating EMT via integrins β1 and α5. Tumor progression may be related to the activation of TGF-β/Smad. Lysyl oxidase (LOX) increases matrix stiffness through collagen cross-linking. LOX is mainly involved in the EMT process and improves tumor invasion, which is reflected in patients with high LOX expression, who are more likely to relapse. HIF-1 can promote the expression of LOX. Researchers have found that LOX accelerates angiogenesis by upregulating angiogenic factors such as VEGF. Chemotherapy-resistant tumor cells can become tumor-initiating cells. These cells exhibit traits similar to CSC and often lead to tumor recurrence. These tumor-initiating cells secrete LOX, suggesting that LOX plays a crucial role in tumor progression and lesion formation.
4.3. Matrix Remodeling via MMP/TIMPs Are Crucial for Tumor Invasion and Metastasis
MMPs decompose components of the basement membrane and spread to other parts of the body, forming new lesions through blood circulation. Multiple studies have revealed that patients with HCC exhibit increased levels of MMP-2, MMP-4, MMP-9, and MMP-14. The expression level of MMP-9 correlates with HCC tumor cell invasion, potentially through MMP-mediated degradation of the basement membrane, which enhances the invasion of nearby blood vessels. Patients with high MMP-9 expression are more likely to experience relapse after radical resection.
4.4. Reprogramming of Cancer-Associated Fibroblasts
CAF, activated fibroblasts, regulate the biology of both stromal and tumor cells via cell–cell contact, release large amounts of cytokines and chemokines, and ultimately remodel the ECM. CAF secrete immunosuppressive ligands such as TGFβ and CXCL12. Reprogramming CAF with a TGFβ-R inhibitor, a CXCR4 blocker, or other methods increases T cell activation and infiltration, while also decreasing CAF recruitment. This, in combination with immune checkpoint inhibitors (ICIs), may further provide clinical benefits and improve prognosis. It has shown better outcomes compared with monotherapy in basic and preclinical research, but more clinical trial data are needed to confirm these findings [94]. A combination of the PD-1 inhibitor Nivolumab and the TGF-β inhibitor Galunisertib (LY2157299) has been proposed in a clinical study (NCT02433343) involving HCC patients to determine the maximum tolerated dose (MTD) and progression-free survival (PFS).
5. Translational Barriers in Targeting HSC for Anti-Fibrosis and Anti-Tumor Therapy
5.1. Heterogeneity of HSC
The progression of liver fibrosis is a highly dynamic process with the potential for both progression and regression. HSC exhibit heterogeneity at different stages of liver fibrosis and cirrhosis progression and regression, as well as in the tumor microenvironment, where they function as CAF. This heterogeneity is reflected in varying activation phenotypes, differential expression of surface molecules, activation of signaling molecules, transcription factors, and the secretion of active molecules. Single-cell RNA sequencing (scRNA-Seq) studies have revealed that quiescent HSC form a highly homogeneous population, characterized by high expression of platelet-derived growth factor receptor (PDGFR). In contrast, in mouse livers with carbon tetrachloride-induced fibrosis, PDGFR-positive myofibroblasts can be divided into four distinct subpopulations, distinguished by the most significantly expressed marker genes [95]. Subpopulations MFB I, III, and IV express genes associated with collagen fiber formation, while MFB II expresses fewer matrix-related genes but more genes involved in the positive regulation of leukocyte activity and immune modulation. scRNA-Seq analysis also identified S100 calcium-binding protein A6 (S100A6) as a new marker specifically expressed in activated MFB but not in quiescent HSC. Furthermore, activated MFB secrete chemokines, such as CXCL1, which attract neutrophils from the circulation, whereas quiescent HSC and MFB express CCL2, a chemokine that recruits monocytes. The scRNA-Seq data confirm that activation of HSC into MFB is a key feature of collagen expression, while quiescent HSC and selective MFB subpopulations secrete chemokines that regulate the inflammatory environment around them. With recent discoveries indicating that functionally and genetically distinct subtypes exist among activated HSC [96,97,98,99,100,101], efforts to target antifibrotic molecules to all activated stellate cells may not be as effective as selectively targeting only those subsets that clearly promote fibrosis. However, current strategies assume that most activated HSC share sufficient common features to make them viable therapeutic targets, a conclusion supported by a recent study documenting strong similarities in the activated HSC transcriptome across different etiologies of liver disease [102].
scRNA-Seq analyses have demonstrated the transition from quiescent HSCs to several activated HSC and CAF phenotypes. In silico analyses describe the transcriptional differentiation of activated HSCs into distinct subtypes, including proliferative (pHSC), inflammatory (iHSC), contractile/migratory (cmHSC), and fibrogenic myofibroblast (myHSC) phenotypes. Other subtypes expressing microvasculature genes have been identified within the vascular HSC (vHSC) subpopulation, yet their origins remain unclear. A deactivated HSC (dHSC) subpopulation emerges during liver fibrosis regression, characterized by an intermediate gene expression profile between quiescent and activated HSCs. However, full reversion to the quiescent phenotype has not been observed. However, full transdifferentiation of this subtype back to the quiescent phenotype has not been described.
Similarly, CAF undergo a transcriptional transition from an inflammatory (iCAF) to a fibrogenic (myCAF) phenotype, with intermediary subpopulations, including vascular CAFs (vCAFs) and antigen-presenting CAFs (apCAFs). Type 1 collagen-expressing myHSC increase liver stiffness, thereby promoting tumor cell proliferation. Conversely, HSC expressing cytokines and growth factors (iHSC) suppress HCC growth via hepatocyte growth factor (HGF) and its receptor MET. In iCCA, myCAF produce hyaluran synthase 2, the enzyme responsible for hyaluronic acid (HA) production, promoting tumor growth and progression. On the other hand, type 1 collagen produced by the myCAF subpopulation contributes to liver stiffness, but without effects on tumor growth. The vCAF subpopulation promotes tumor growth via the interleukin (IL)-6/IL-6R axis. In liver metastasis, type 1 collagen derived from myCAFs suppresses tumor growth by mechanically restraining the tumor, whereas HA promotes tumor growth. In both iCCA and metastases, iCAF promote tumor growth via the HGF-MET axis.
Due to the remarkable plasticity of HSC and CAF during liver fibrogenesis and carcinogenesis, their distinct subpopulations show complementary or ambiguous functions in response to specific chronic inflammatory and tumor microenvironments.Further identification of specific surface markers, signaling molecules, and therapeutic targets associated with liver fibrosis progression and regression, particularly in activated HSC, is needed. These could serve as both therapeutic targets and functional imaging biomarkers.
5.2. Lack of Specific Targeted Methods for HSC
Despite the identification of numerous anti-fibrotic candidate targets and the demonstration of potent anti-fibrotic effects in experimental animal models, their clinical efficacy remains limited or suboptimal. To date, no antifibrotic drugs have been approved for clinical use. Current treatments for liver fibrosis primarily focus on addressing the underlying causes of liver disease, such as inhibiting hepatitis B virus (HBV) and hepatitis C virus (HCV) replication or suppressing immune responses in autoimmune hepatitis. The liver, characterized by its extensive vasculature and unique metabolic capacity, exhibits high overall drug uptake. However, most therapeutic agents are either metabolized by hepatic stem cells or cleared by the reticuloendothelial system, resulting in minimal drug delivery to hepatic stellate cells (HSC) and consequently limiting therapeutic efficacy. Furthermore, the failure of many anti-fibrotic drugs in clinical trials is largely attributed to their low efficacy, resulting from off-target uptake and rapid clearance. Consequently, the development of targeted drug delivery strategies that specifically activate HSC represents a critical approach to enhancing therapeutic responses across different stages of liver disease.
5.3. Barriers to the Translation of Basic Research into Clinical Practice
Significant progress has been made in elucidating the cellular and molecular mechanisms of liver fibrosis; however, only a few findings have been successfully translated into clinical applications. Firstly, the high cost of drug development and target validation necessitates prolonged timelines and substantial financial investment. Secondly, as regulatory requirements become more stringent, there is an increasing demand for drugs with well-defined clinical efficacy and safety profiles. Moreover, the efficacy observed in animal models often fails to fully translate to clinical settings due to differences in pharmacokinetics, extracellular matrix (ECM) cross-linking, and disease pathophysiology. Despite advancements in anti-fibrotic drug development, accurately identifying ideal noninvasive biomarkers for fibrotic activity and establishing consensus on optimal clinical endpoints remain significant challenges [103,104].
Currently, addressing the underlying cause remains the only proven strategy to halt or reverse liver fibrosis progression, while the development of effective anti-fibrotic therapies continues to pose a major challenge in liver disease management. Over the past few decades, substantial progress has been made in elucidating the cellular and molecular mechanisms underlying liver fibrosis. Liver fibrosis is a complex pathological change involving multiple cells, multiple factors, and multiple pathways, and the study of the cellular and molecular mechanism of its occurrence and development provides an important theoretical basis and therapeutic target for clinical drug development. It is anticipated that improved animal models and well-designed clinical trials will facilitate the successful translation of anti-fibrotic research into effective clinical treatments in the near future.
6. Promoting Targeted Hepatic Stellate Cell-Based Strategies for the Prevention and Treatment of Liver Fibrosis and Hepatocellular Carcinoma
6.1. Utilizing New Omics Technologies to Identify Markers and Therapeutic Targets for Activated HSCs
scRNA-Seq technology enables genome amplification of isolated single cells, followed by high-throughput sequencing after exonic capture, allowing for the identification of cell population heterogeneity and cellular evolutionary relationships. A key advantage of scRNA-Seq is its ability to generate multi-dimensional data, including differences in cell composition and abundance across tissues, gene expression variability and functional alterations within seemingly homogeneous cell populations, as well as insights into cellular interactions and microenvironmental regulatory networks [105,106]. Integrating scRNA-Seq with spatial mapping has provided critical insights into the pathological and physiological mechanisms underlying liver fibrosis. This approach has been utilized to delineate metabolic zoning and molecular patterning in hepatocytes, endothelial cells, and HSC within human and murine liver lobules [107,108,109]. Single-cell analysis of human liver macrophage subpopulations has identified a novel scar-associated TREM2+CD9+ macrophage subset, which originates from circulating monocytes and expands during liver fibrosis. These cells reside in the fibrotic microenvironment and promote mesenchymal cell activation and scar deposition [110]. Additionally, ACKR+ and PLVAP+ endothelial cells have been shown to expand during fibrosis, contributing to the establishment of the fibrotic microenvironment. Crosstalk among scar-associated macrophages, endothelial cells, and PDGFR+ collagen-producing mesenchymal cells unveils multiple fibrotic pathways, including the activation of TNFRSF12A, PDGFR, and NOTCH signaling. The interactions among these cells and subpopulations in liver fibrosis provide a molecular framework for targeting key pathogenic cell populations [111,112]. Moreover, fibrosis progression fingerprints derived from liver tissue and serum [113], along with dynamic network biomarkers (DNB) identified via systems biology approaches, offer promising targets for anti-fibrotic drug development [114].
6.2. Receptor-Mediated Targeted Treatment Strategies for HSC and Clinical Translation
Hepatic stellate cells (HSC) express surface markers, including PDGF receptors, which facilitate HSC-targeted delivery of therapeutic cargo to mitigate liver fibrosis and its associated complications, such as portal hypertension and HCC [115,116,117,118,119,120,121,122]. HSC targeted approaches may enable selective delivery of drugs and oligonucleotides, and therapeutic peptides. The delivered drugs may include chemical compounds or antibodies with antifibrotic, anti-proliferative, anti-inflammatory, and pro-apoptotic properties, as well as collagen synthesis inhibitors, tyrosine kinase inhibitors, and angiotensin inhibitors. Oligonucleotide-based therapies include small interfering RNA (siRNA), self-amplifying RNA (saRNA), and CRISPR-based approaches for gene silencing or activation [123].
PDGFR plays a critical role in liver fibrosis and is highly expressed in activated HSCs. PDGFR-recognizing peptides (PPB) can facilitate the targeted delivery of anti-fibrotic biologics, such as interferon (IFN)-mimicking peptides containing IFN signaling domains, thereby enhancing HSC-specific uptake and therapeutic efficacy while minimizing systemic side effects. This approach has shown significant inhibition of CCl4-induced liver fibrosis in murine models [124]. These strategies present novel therapeutic opportunities for the treatment of liver fibrosis, other fibrotic diseases, and liver cancer. Additionally, these peptides could also serve as diagnostic tools for monitoring the progression of fibrosis.
In addition to PDGFR, other receptors implicated in HSC-targeting include integrins, mannose-6-phosphate/insulin-like growth factor II receptor (M6P/IGF-IIR), fibroblast growth factor receptor 1 (FGFR1), elastin receptor RXFP1, retinol (vitamin A)-binding protein (RBP) receptors, type VI collagen receptors, low-density lipoprotein receptors (LDLR), synaptophysin (SYN), cluster of differentiation 44 (CD44), and tumor necrosis factor-related apoptosis-inducing ligand receptor (TRAILR) [125]. SYN is a membrane glycoprotein expressed on differentiate portal fibroblasts. SYN is advantageous over other markers for targeted delivery of antifibrotic agents because it forms part of endocytosing vesicles, leading to increased chance of endocytosis of its ligand and associated cargos. Studies have shown that a single-chain antibody (scAb) C1-3 that binds to SYN on activated HSCs but not hepatocytes, supporting the feasibility of using C1-3 scAb as a targeting ligand for HSC-specific delivery [126]. One potential limitation of this strategy is the nonspecific delivery to neuroendocrine and neural cells, although there are no evidence showing that scAb can cross the blood-brain barrier. Several specific receptor-recognizing peptides or siRNAs have been designed targeting these receptors for anti-fibrosis treatments.
The insulin-like growth factor 2 receptor (IGF2R) is a multifunctional receptor that is overexpressed on activated HSCs and is a specific molecular marker of activated HSCs in the fibrotic liver. IGF2R-specific peptide significantly increases the binding affinity and uptake of a protein-based siRNA nanocomplex to activated HSCs, and the nanocomplex significantly improved the serum stability and silencing activity of siRNA, which has the potential to be a useful delivery system to specifically deliver therapeutic siRNAs to activated HSCs in the fibrotic liver [127,128,129].
Hsp47 is a collagen chaperone and its inhibition not only alters collagen expression and alignment but also promotes HSC death due to intracellular collagen misfolding. HSC-selective delivery of Hsp47 siRNA via Vitamin A-coupled liposomes has shown promising data in a rat model of cirrhosis [115], phase 2 clinical trials were initiated in patients with advanced fibrosis or compensated liver cirrhosis who had eradicated HCV infection or MASH and compensated liver cirrhosis. In patients with eradicated HCV infection and F3-F4 fibrosis (NCT03420768), BMS-986263 achieved an improvement in fibrosis by ≥ 1 stage in 17-21% compared to 13% in the placebo group and a reduction of HSP47 mRNA in most patients in the higher dose group. However, the effects of BMS-986263 on target gene expression were disappointing, showing only a reduction of 5.9% in HSP47 mRNA and 10.1% in HSP47 protein levels [130]. Notably, the phase 2 trial of BMS-986263 in patients with compensated MASH cirrhosis was terminated due to a lack of efficacy (NCT04267393). It is possible that the low target gene reduction, possibly due to suboptimal delivery to HSCs, contributed to the low efficacy.
Targeted delivery of antifibrotic agents to activated HSCs is critical for the successful treatment of liver fibrosis. A number of clinical trials have been conducted for the treatment of liver fibrosis using small molecules, proteins, monoclonal antibodies, and nucleic acids. Among them, one clinical trial (NCT02227459) used vitamin A as a targeting ligand for the antifibrotic siRNA [129].
Currently, most investigators focus on natural ligands, which are very limited and generally have low affinity for these receptors. In addition, some of the natural ligands may activate downstream signaling pathways after binding to their receptors on HSC. A potential solution for this challenge is to use affinity selection technologies such as phage display biopanning to discover peptide-or antibody-based ligands, or SELEX to discover aptamer-based ligands. Compared with natural ligands, artificial ligands—including peptides, antibody fragments, antibodies, and aptamers—exhibit higher affinity and greater flexibility for chemical modification and coupling to antifibrotic agents or delivery systems. In particular, peptides and aptamers are attractive ligands because of their small size, ease of production, and lack of immunogenicity.
6.3. Design and Progress of Peptide Drugs
Peptides targeting activated hepatic stellate cells, which are compounds formed by linking multiple amino acids through peptide bonds, are common bioactive substances found in living organisms. The advent of the 21st century ushered in a new era in peptide drug development, with advancements in structural biology, recombinant biotechnology, and new synthesis and analytical technologies playing a significant role in accelerating this process. This has led to the establishment of a comprehensive peptide drug development framework, encompassing peptide drug discovery, design, synthesis, structural modifications, and activity evaluation. Peptide drugs offer advantages such as high specificity, selectivity, and potent efficacy at low concentrations. With the continuous development of structural biology and proteomics, many protein-protein interactions (PPIs) have been studied, involving cellular pathways and biological functions [131,132], as well as regulation of signaling pathways related to physiological and pathological processes [133]. Therefore, peptides act as natural agonists or antagonists with inherent advantages over small molecule drugs. Compared to small molecule drugs, peptides exhibit stronger target affinity, higher specificity, and fewer side effects. In contrast to large molecule drugs, peptides demonstrate lower immunogenicity and reduced production costs. Thus, the rational design of peptides based on known crystal structures is regarded as a promising strategy for discovering and developing new peptide drugs.
Rational design of peptides involves computationally analyzing the crystal structure of the target protein using bioinformatics techniques. For example, crystal structure analysis can determine the secondary and tertiary structures of peptides, followed by alanine scanning, structure-activity relationship (SAR) analysis, and the identification of essential amino acids and potential substitutable sites on protein surfaces involved in PPIs. These essential amino acids, known as “hot spots,” serve as the basis for deriving initial peptide sequences from continuous fragments or residues [134]. Further modifications, such as peptide cyclization and scaffold modifications, can enhance peptide activity, improve physicochemical properties, extend half-life, and increase stability [135,136,137]. Multiple formulations are also explored in later stages of development. To date, more than 80 peptide drugs have received FDA approval for clinical use. [138]. Liraglutide, a synthetic analog of human glucagon-like peptide 1 (GLP-1), is a successful example of peptide drug development and is widely used in the treatment of obesity and type 2 diabetes [139,140]. This success in peptide drug development provides valuable insights for targeted HSC therapies in the treatment of fibrosis, cirrhosis, and liver cancer.
7. Conclusions
In summary, there is currently a lack of direct anti-fibrotic or anti-cirrhotic therapeutic agents that have demonstrated clinical success. Identifying reversible tipping points in liver fibrosis/cirrhosis, establishing ideal diagnostic biomarkers or methods for non-invasively assessing the staging, progression, and reversal of liver fibrosis/cirrhosis, developing effective anti-fibrotic/cirrhotic therapies, and devising methods to prevent or treat tumors by improving the tumor microenvironment represent key challenges and frontiers in current research. Recent advances in omics, particularly single-cell sequencing and spatial transcriptomics, hold promise in identifying new HSC targets for the diagnose and treatment of liver fibrosis/cirrhosis and liver cancer. Establishing specific HSC targeted delivery methods is a critical step in translating these discoveries into effective clinical diagnostic and therapeutic strategies.
Author Contributions
Conceptual design of the manuscript (JG), drafting and writing (HX, JG). All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Fund of Nature Science of P.R. China (Nos. 91129705, 81070340) and the Shanghai Pujiang Talent Program (09PJ1402600).
Data Availability Statement
All the citations included in this manuscript are available upon request by contact with the corresponding author.
Conflicts of Interest
The authors have no conflict of interests related to this publication.
References
- Ginès, P., A. Krag, J. G. Abraldes, E. Solà, N. Fabrellas, and P. S. Kamath. "Liver Cirrhosis." Lancet 398, no. 10308 (2021): 1359-76.
- Marcellin, P. , and B. K. Kutala. "Liver Diseases: A Major, Neglected Global Public Health Problem Requiring Urgent Actions and Large-Scale Screening." Liver Int 38 Suppl 1 (2018): 2-6.
- Wang, S. S., X. T. Tang, M. Lin, J. Yuan, Y. J. Peng, X. Yin, G. Shang, G. Ge, Z. Ren, and B. O. Zhou. "Perivenous Stellate Cells Are the Main Source of Myofibroblasts and Cancer-Associated Fibroblasts Formed after Chronic Liver Injuries." Hepatology 74, no. 3 (2021): 1578-94.
- Mederacke, I., C. C. Hsu, J. S. Troeger, P. Huebener, X. Mu, D. H. Dapito, J. P. Pradere, and R. F. Schwabe. "Fate Tracing Reveals Hepatic Stellate Cells as Dominant Contributors to Liver Fibrosis Independent of Its Aetiology." Nat Commun 4 (2013): 2823.
- Friedman, S. L. "Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver." Physiol Rev 88, no. 1 (2008): 125-72.
- Guo, J. , and S. L. Friedman. "Hepatic Fibrogenesis." Semin Liver Dis 27, no. 4 (2007): 413-26.
- Barry, A. E., R. Baldeosingh, R. Lamm, K. Patel, K. Zhang, D. A. Dominguez, K. J. Kirton, A. P. Shah, and H. Dang. "Hepatic Stellate Cells and Hepatocarcinogenesis." Front Cell Dev Biol 8 (2020): 709.
- Bárcena, C., M. Stefanovic, A. Tutusaus, G. A. Martinez-Nieto, L. Martinez, C. García-Ruiz, A. de Mingo, J. Caballeria, J. C. Fernandez-Checa, M. Marí, and A. Morales. "Angiogenin Secretion from Hepatoma Cells Activates Hepatic Stellate Cells to Amplify a Self-Sustained Cycle Promoting Liver Cancer." Sci Rep 5 (2015): 7916.
- Santamato, A., E. Fransvea, F. Dituri, A. Caligiuri, M. Quaranta, T. Niimi, M. Pinzani, S. Antonaci, and G. Giannelli. "Hepatic Stellate Cells Stimulate Hcc Cell Migration Via Laminin-5 Production." Clin Sci (Lond) 121, no. 4 (2011): 159-68.
- Lu, Y., J. Xu, S. Chen, Z. Zhou, and N. Lin. "Lipopolysaccharide Promotes Angiogenesis in Mice Model of Hcc by Stimulating Hepatic Stellate Cell Activation Via Tlr4 Pathway." Acta Biochim Biophys Sin (Shanghai) 49, no. 11 (2017): 1029-34.
- Zhao, W., L. Zhang, Y. Xu, Z. Zhang, G. Ren, K. Tang, P. Kuang, B. Zhao, Z. Yin, and X. Wang. "Hepatic Stellate Cells Promote Tumor Progression by Enhancement of Immunosuppressive Cells in an Orthotopic Liver Tumor Mouse Model." Lab Invest 94, no. 2 (2014): 182-91.
- Zhou, Z., Y. Hu, Y. Wu, Q. Qi, J. Wang, L. Chen, and F. Wang. "The Immunosuppressive Tumor Microenvironment in Hepatocellular Carcinoma-Current Situation and Outlook." Mol Immunol 151 (2022): 218-30.
- Higashi, T., S. L. Friedman, and Y. Hoshida. "Hepatic Stellate Cells as Key Target in Liver Fibrosis." Adv Drug Deliv Rev 121 (2017): 27-42.
- Wiering, L., P. Subramanian, and L. Hammerich. "Hepatic Stellate Cells: Dictating Outcome in Nonalcoholic Fatty Liver Disease." Cell Mol Gastroenterol Hepatol 15, no. 6 (2023): 1277-92.
- Guo, J., J. Loke, F. Zheng, F. Hong, S. Yea, M. Fukata, M. Tarocchi, O. T. Abar, H. Huang, J. J. Sninsky, and S. L. Friedman. "Functional Linkage of Cirrhosis-Predictive Single Nucleotide Polymorphisms of Toll-Like Receptor 4 to Hepatic Stellate Cell Responses." Hepatology 49, no. 3 (2009): 960-8.
- Zeng, Z., Y. Wu, Y. Cao, Z. Yuan, Y. Zhang, D. Y. Zhang, D. Hasegawa, S. L. Friedman, and J. Guo. "Slit2-Robo2 Signaling Modulates the Fibrogenic Activity and Migration of Hepatic Stellate Cells." Life Sci 203 (2018): 39-47.
- Seki, E. , and R. F. Schwabe. "Hepatic Inflammation and Fibrosis: Functional Links and Key Pathways." Hepatology 61, no. 3 (2015): 1066-79.
- Rockey, D. C. "Translating an Understanding of the Pathogenesis of Hepatic Fibrosis to Novel Therapies." Clin Gastroenterol Hepatol 11, no. 3 (2013): 224-31.e1-5.
- Zhang, Z., C. Lin, L. Peng, Y. Ouyang, Y. Cao, J. Wang, S. L. Friedman, and J. Guo. "High Mobility Group Box 1 Activates Toll Like Receptor 4 Signaling in Hepatic Stellate Cells." Life Sci 91, no. 5-6 (2012): 207-12.
- Guo, J. , and S. L. Friedman. "Toll-Like Receptor 4 Signaling in Liver Injury and Hepatic Fibrogenesis." Fibrogenesis Tissue Repair 3 (2010): 21.
- Bansal, M. B. , and N. Chamroonkul. "Antifibrotics in Liver Disease: Are We Getting Closer to Clinical Use?" Hepatol Int 13, no. 1 (2019): 25-39.
- Cannito, S., E. Novo, and M. Parola. "Therapeutic Pro-Fibrogenic Signaling Pathways in Fibroblasts." Adv Drug Deliv Rev 121 (2017): 57-84.
- Richter, K. , and T. Kietzmann. "Reactive Oxygen Species and Fibrosis: Further Evidence of a Significant Liaison." Cell Tissue Res 365, no. 3 (2016): 591-605.
- Liang, S., T. Kisseleva, and D. A. Brenner. "The Role of Nadph Oxidases (Noxs) in Liver Fibrosis and the Activation of Myofibroblasts." Front Physiol 7 (2016): 17.
- Crosas-Molist, E., E. Bertran, and I. Fabregat. "Cross-Talk between Tgf-Β and Nadph Oxidases during Liver Fibrosis and Hepatocarcinogenesis." Curr Pharm Des 21, no. 41 (2015): 5964-76.
- Liu, C., X. Chen, L. Yang, T. Kisseleva, D. A. Brenner, and E. Seki. "Transcriptional Repression of the Transforming Growth Factor Β (Tgf-Β) Pseudoreceptor Bmp and Activin Membrane-Bound Inhibitor (Bambi) by Nuclear Factor Κb (Nf-Κb) P50 Enhances Tgf-Β Signaling in Hepatic Stellate Cells." J Biol Chem 289, no. 10 (2014): 7082-91.
- Yang, J. J., H. Tao, and J. Li. "Hedgehog Signaling Pathway as Key Player in Liver Fibrosis: New Insights and Perspectives." Expert Opin Ther Targets 18, no. 9 (2014): 1011-21.
- Du, K., J. Hyun, R. T. Premont, S. S. Choi, G. A. Michelotti, M. Swiderska-Syn, G. D. Dalton, E. Thelen, B. S. Rizi, Y. Jung, and A. M. Diehl. "Hedgehog-Yap Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells." Gastroenterology 154, no. 5 (2018): 1465-79.e13.
- El-Agroudy, N. N., R. N. El-Naga, R. A. El-Razeq, and E. El-Demerdash. "Forskolin, a Hedgehog Signalling Inhibitor, Attenuates Carbon Tetrachloride-Induced Liver Fibrosis in Rats." Br J Pharmacol 173, no. 22 (2016): 3248-60.
- Lian, N., Y. Jiang, F. Zhang, H. Jin, C. Lu, X. Wu, Y. Lu, and S. Zheng. "Curcumin Regulates Cell Fate and Metabolism by Inhibiting Hedgehog Signaling in Hepatic Stellate Cells." Lab Invest 95, no. 7 (2015): 790-803.
- Moon, R. T., B. Bowerman, M. Boutros, and N. Perrimon. "The Promise and Perils of Wnt Signaling through Beta-Catenin." Science 296, no. 5573 (2002): 1644-6.
- Kweon, S. M., F. Chi, R. Higashiyama, K. Lai, and H. Tsukamoto. "Wnt Pathway Stabilizes Mecp2 Protein to Repress Ppar-Γ in Activation of Hepatic Stellate Cells." PLoS One 11, no. 5 (2016): e0156111.
- Yu, F., Z. Lu, K. Huang, X. Wang, Z. Xu, B. Chen, P. Dong, and J. Zheng. "Microrna-17-5p-Activated Wnt/Β-Catenin Pathway Contributes to the Progression of Liver Fibrosis." Oncotarget 7, no. 1 (2016): 81-93.
- Lin, X., L. N. Kong, C. Huang, T. T. Ma, X. M. Meng, Y. He, Q. Q. Wang, and J. Li. "Hesperetin Derivative-7 Inhibits Pdgf-Bb-Induced Hepatic Stellate Cell Activation and Proliferation by Targeting Wnt/Β-Catenin Pathway." Int Immunopharmacol 25, no. 2 (2015): 311-20.
- Dong, S., C. Wu, J. Hu, Q. Wang, S. Chen, Zhirong Wang, and W. Xiong. "Wnt5a Promotes Cytokines Production and Cell Proliferation in Human Hepatic Stellate Cells Independent of Canonical Wnt Pathway." Clin Lab 61, no. 5-6 (2015): 537-47.
- Widjaja, A. A., B. K. Singh, E. Adami, S. Viswanathan, J. Dong, G. A. D'Agostino, B. Ng, W. W. Lim, J. Tan, B. S. Paleja, M. Tripathi, S. Y. Lim, S. G. Shekeran, S. P. Chothani, A. Rabes, M. Sombetzki, E. Bruinstroop, L. P. Min, R. A. Sinha, S. Albani, P. M. Yen, S. Schafer, and S. A. Cook. "Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis." Gastroenterology 157, no. 3 (2019): 777-92.e14.
- Mor, A., S. Friedman, S. Hashmueli, A. Peled, M. Pinzani, M. Frankel, and R. Safadi. "Targeting Ccl24 in Inflammatory and Fibrotic Diseases: Rationale and Results from Three Cm-101 Phase 1 Studies." Drug Saf 47, no. 9 (2024): 869-81.
- De Lorenzis, E., A. Mor, R. L. Ross, S. Di Donato, R. Aricha, I. Vaknin, and F. Del Galdo. "Serum Ccl24 as a Biomarker of Fibrotic and Vascular Disease Severity in Systemic Sclerosis." Arthritis Care Res (Hoboken) 76, no. 9 (2024): 1269-77.
- Segal-Salto, M., N. Barashi, A. Katav, V. Edelshtein, A. Aharon, S. Hashmueli, J. George, Y. Maor, M. Pinzani, D. Haberman, A. Hall, S. Friedman, and A. Mor. "A Blocking Monoclonal Antibody to Ccl24 Alleviates Liver Fibrosis and Inflammation in Experimental Models of Liver Damage." JHEP Rep 2, no. 1 (2020): 100064.
- Borkham-Kamphorst, E. , and R. Weiskirchen. "The Pdgf System and Its Antagonists in Liver Fibrosis." Cytokine Growth Factor Rev 28 (2016): 53-61.
- Massagué, J. , and D. Sheppard. "Tgf-Β Signaling in Health and Disease." Cell 186, no. 19 (2023): 4007-37.
- Munger, J. S., X. Huang, H. Kawakatsu, M. J. Griffiths, S. L. Dalton, J. Wu, J. F. Pittet, N. Kaminski, C. Garat, M. A. Matthay, D. B. Rifkin, and D. Sheppard. "The Integrin Alpha V Beta 6 Binds and Activates Latent Tgf Beta 1: A Mechanism for Regulating Pulmonary Inflammation and Fibrosis." Cell 96, no. 3 (1999): 319-28.
- Stockis, J., S. Liénart, D. Colau, A. Collignon, S. L. Nishimura, D. Sheppard, P. G. Coulie, and S. Lucas. "Blocking Immunosuppression by Human Tregs in Vivo with Antibodies Targeting Integrin Avβ8." Proc Natl Acad Sci U S A 114, no. 47 (2017): E10161-e68.
- Guo, W., H. Liu, Y. Yan, D. Wu, H. Yao, K. Lin, and X. Li. "Targeting the Tgf-Β Signaling Pathway: An. Updated Patent Review (2021-Present)." Expert Opin Ther Pat 34, no.
- Dickson, M. C., J. S. Martin, F. M. Cousins, A. B. Kulkarni, S. Karlsson, and R. J. Akhurst. "Defective Haematopoiesis and Vasculogenesis in Transforming Growth Factor-Beta 1 Knock out Mice." Development 121, no. 6 (1995): 1845-54.
- Danielpour, D. "Advances and Challenges in Targeting Tgf-Β Isoforms for Therapeutic Intervention of Cancer: A Mechanism-Based Perspective." Pharmaceuticals (Basel) 17, no. 4 (2024).
- Lancaster, L., V. Cottin, M. Ramaswamy, W. A. Wuyts, R. G. Jenkins, M. B. Scholand, M. Kreuter, C. Valenzuela, C. J. Ryerson, J. Goldin, G. H. J. Kim, M. Jurek, M. Decaris, A. Clark, S. Turner, C. N. Barnes, H. E. Achneck, G. P. Cosgrove, A. Lefebvre É, and K. R. Flaherty. "Bexotegrast in Patients with Idiopathic Pulmonary Fibrosis: The Integris-Ipf Clinical Trial." Am J Respir Crit Care Med 210, no. 4 (2024): 424-34.
- Duan, Z., X. Lin, L. Wang, Q. Zhen, Y. Jiang, C. Chen, J. Yang, C. H. Lee, Y. Qin, Y. Li, B. Zhao, J. Wang, and Z. Zhang. "Specificity of Tgf-Β1 Signal Designated by Lrrc33 and Integrin A(V)Β(8)." Nat Commun 13, no. 1 (2022): 4988.
- Liénart, S., R. Merceron, C. Vanderaa, F. Lambert, D. Colau, J. Stockis, B. van der Woning, H. De Haard, M. Saunders, P. G. Coulie, S. N. Savvides, and S. Lucas. "Structural Basis of Latent Tgf-Β1 Presentation and Activation by Garp on Human Regulatory T Cells." Science 362, no. 6417 (2018): 952-56.
- Lack, J., J. M. O'Leary, V. Knott, X. Yuan, D. B. Rifkin, P. A. Handford, and A. K. Downing. "Solution Structure of the Third Tb Domain from Ltbp1 Provides Insight into Assembly of the Large Latent Complex That Sequesters Latent Tgf-Beta." J Mol Biol 334, no. 2 (2003): 281-91.
- Jackson, J. W., C. Streich Frederick, Jr., A. Pal, G. Coricor, C. Boston, C. T. Brueckner, K. Canonico, C. Chapron, S. Cote, K. B. Dagbay, F. T. Danehy, Jr., M. Kavosi, S. Kumar, S. Lin, C. Littlefield, K. Looby, R. Manohar, C. J. Martin, M. Wood, A. Zawadzka, S. Wawersik, S. B. Nicholls, A. Datta, A. Buckler, T. Schürpf, G. J. Carven, M. Qatanani, and A. I. Fogel. "An Antibody That Inhibits Tgf-Β1 Release from Latent Extracellular Matrix Complexes Attenuates the Progression of Renal Fibrosis." Sci Signal 17, no. 844 (2024): eadn6052.
- Sun, T., Z. Huang, W. C. Liang, J. Yin, W. Y. Lin, J. Wu, J. M. Vernes, J. Lutman, P. Caplazi, S. Jeet, T. Wong, M. Wong, D. J. DePianto, K. B. Morshead, K. H. Sun, Z. Modrusan, J. A. Vander Heiden, A. R. Abbas, H. Zhang, M. Xu, E. N. N'Diaye, M. Roose-Girma, P. J. Wolters, R. Yadav, S. Sukumaran, N. Ghilardi, R. Corpuz, C. Emson, Y. G. Meng, T. R. Ramalingam, P. Lupardus, H. D. Brightbill, D. Seshasayee, Y. Wu, and J. R. Arron. "Tgfβ2 and Tgfβ3 Isoforms Drive Fibrotic Disease Pathogenesis." Sci Transl Med 13, no. 605 (2021).
- Sun, T., J. A. Vander Heiden, X. Gao, J. Yin, S. Uttarwar, W. C. Liang, G. Jia, R. Yadav, Z. Huang, M. Mitra, W. Halpern, H. S. Bender, H. D. Brightbill, Y. Wu, P. Lupardus, T. Ramalingam, and J. R. Arron. "Isoform-Selective Tgf-Β3 Inhibition for Systemic Sclerosis." Med 5, no. 2 (2024): 132-47.e7.
- Spagnolo, P. , and T. M. Maher. "The Future of Clinical Trials in Idiopathic Pulmonary Fibrosis." Curr Opin Pulm Med 30, no. 5 (2024): 494-99.
- Richeldi, L., E. R. Fernández Pérez, U. Costabel, C. Albera, D. J. Lederer, K. R. Flaherty, N. Ettinger, R. Perez, M. B. Scholand, J. Goldin, K. H. Peony Yu, T. Neff, S. Porter, M. Zhong, E. Gorina, E. Kouchakji, and G. Raghu. "Pamrevlumab, an Anti-Connective Tissue Growth Factor Therapy, for Idiopathic Pulmonary Fibrosis (Praise): A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial." Lancet Respir Med 8, no. 1 (2020): 25-33.
- Levy, M. T., G. W. McCaughan, C. A. Abbott, J. E. Park, A. M. Cunningham, E. Müller, W. J. Rettig, and M. D. Gorrell. "Fibroblast Activation Protein: A Cell Surface Dipeptidyl Peptidase and Gelatinase Expressed by Stellate Cells at the Tissue Remodelling Interface in Human Cirrhosis." Hepatology 29, no. 6 (1999): 1768-78.
- Levy, M. T., G. W. McCaughan, G. Marinos, and M. D. Gorrell. "Intrahepatic Expression of the Hepatic Stellate Cell Marker Fibroblast Activation Protein Correlates with the Degree of Fibrosis in Hepatitis C Virus Infection." Liver 22, no. 2 (2002): 93-101.
- Yang, A. T., Y. O. Kim, X. Z. Yan, H. Abe, M. Aslam, K. S. Park, X. Y. Zhao, J. D. Jia, T. Klein, H. You, and D. Schuppan. "Fibroblast Activation Protein Activates Macrophages and Promotes Parenchymal Liver Inflammation and Fibrosis." Cell Mol Gastroenterol Hepatol 15, no. 4 (2023): 841-67.
- Williams, K. H., A. J. Viera de Ribeiro, E. Prakoso, A. S. Veillard, N. A. Shackel, Y. Bu, B. Brooks, E. Cavanagh, J. Raleigh, S. V. McLennan, G. W. McCaughan, W. W. Bachovchin, F. M. Keane, A. Zekry, S. M. Twigg, and M. D. Gorrell. "Lower Serum Fibroblast Activation Protein Shows Promise in the Exclusion of Clinically Significant Liver Fibrosis Due to Non-Alcoholic Fatty Liver Disease in Diabetes and Obesity." Diabetes Res Clin Pract 108, no. 3 (2015): 466-72.
- Teixeira-Clerc, F., B. Julien, P. Grenard, J. Tran Van Nhieu, V. Deveaux, L. Li, V. Serriere-Lanneau, C. Ledent, A. Mallat, and S. Lotersztajn. "Cb1 Cannabinoid Receptor Antagonism: A New Strategy for the Treatment of Liver Fibrosis." Nat Med 12, no. 6 (2006): 671-6.
- Mallat, A., F. Teixeira-Clerc, and S. Lotersztajn. "Cannabinoid Signaling and Liver Therapeutics." J Hepatol 59, no. 4 (2013): 891-6.
- Khimji, A. K. , and D. C. Rockey. "Endothelin and Hepatic Wound Healing." Pharmacol Res 63, no. 6 (2011): 512-8.
- He, C., X. Miao, J. Li, and H. Qi. "Angiotensin Ii Induces Endothelin-1 Expression in Human Hepatic Stellate Cells." Dig Dis Sci 58, no. 9 (2013): 2542-9.
- Lee, Y. A., M. C. Wallace, and S. L. Friedman. "Pathobiology of Liver Fibrosis: A Translational Success Story." Gut 64, no. 5 (2015): 830-41.
- Staels, B., A. Rubenstrunk, B. Noel, G. Rigou, P. Delataille, L. J. Millatt, M. Baron, A. Lucas, A. Tailleux, D. W. Hum, V. Ratziu, B. Cariou, and R. Hanf. "Hepatoprotective Effects of the Dual Peroxisome Proliferator-Activated Receptor Alpha/Delta Agonist, Gft505, in Rodent Models of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis." Hepatology 58, no. 6 (2013): 1941-52.
- Fernández-Álvarez, S., V. Gutiérrez-de Juan, I. Zubiete-Franco, L. Barbier-Torres, A. Lahoz, A. Parés, Z. Luka, C. Wagner, S. C. Lu, J. M. Mato, M. L. Martínez-Chantar, and N. Beraza. "Trail-Producing Nk Cells Contribute to Liver Injury and Related Fibrogenesis in the Context of Gnmt Deficiency." Lab Invest 95, no. 2 (2015): 223-36.
- Ahsan, M. K. , and W. Z. Mehal. "Activation of Adenosine Receptor A2a Increases Hsc Proliferation and Inhibits Death and Senescence by down-Regulation of P53 and Rb." Front Pharmacol 5 (2014): 69.
- Yashaswini, C. N., T. Qin, D. Bhattacharya, C. Amor, S. Lowe, A. Lujambio, S. Wang, and S. L. Friedman. "Phenotypes and Ontogeny of Senescent Hepatic Stellate Cells in Metabolic Dysfunction-Associated Steatohepatitis." J Hepatol 81, no. 2 (2024): 207-17.
- Huang, X., H. Cai, R. Ammar, Y. Zhang, Y. Wang, K. Ravi, J. Thompson, and G. Jarai. "Molecular Characterization of a Precision-Cut Rat Liver Slice Model for the Evaluation of Antifibrotic Compounds." Am J Physiol Gastrointest Liver Physiol 316, no. 1 (2019): G15-g24.
- Yamagishi, R., F. Kamachi, M. Nakamura, S. Yamazaki, T. Kamiya, M. Takasugi, Y. Cheng, Y. Nonaka, Y. Yukawa-Muto, L. T. T. Thuy, Y. Harada, T. Arai, T. M. Loo, S. Yoshimoto, T. Ando, M. Nakajima, H. Taguchi, T. Ishikawa, H. Akiba, S. Miyake, M. Kubo, Y. Iwakura, S. Fukuda, W. Y. Chen, N. Kawada, A. Rudensky, S. Nakae, E. Hara, and N. Ohtani. "Gasdermin D-Mediated Release of Il-33 from Senescent Hepatic Stellate Cells Promotes Obesity-Associated Hepatocellular Carcinoma." Sci Immunol 7, no. 72 (2022): eabl7209.
- Hu, J. J., X. Liu, S. Xia, Z. Zhang, Y. Zhang, J. Zhao, J. Ruan, X. Luo, X. Lou, Y. Bai, J. Wang, L. R. Hollingsworth, V. G. Magupalli, L. Zhao, H. R. Luo, J. Kim, J. Lieberman, and H. Wu. "Fda-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation." Nat Immunol 21, no. 7 (2020): 736-45.
- Robert, S., T. Gicquel, T. Victoni, S. Valença, E. Barreto, B. Bailly-Maître, E. Boichot, and V. Lagente. "Involvement of Matrix Metalloproteinases (Mmps) and Inflammasome Pathway in Molecular Mechanisms of Fibrosis." Biosci Rep 36, no. 4 (2016).
- Urtasun, R., A. Lopategi, J. George, T. M. Leung, Y. Lu, X. Wang, X. Ge, M. I. Fiel, and N. Nieto. "Osteopontin, an Oxidant Stress Sensitive Cytokine, up-Regulates Collagen-I Via Integrin A(V)Β(3) Engagement and Pi3k/Pakt/Nfκb Signaling." Hepatology 55, no. 2 (2012): 594-608.
- Chen, W., A. Yang, J. Jia, Y. V. Popov, D. Schuppan, and H. You. "Lysyl Oxidase (Lox) Family Members: Rationale and Their Potential as Therapeutic Targets for Liver Fibrosis." Hepatology 72, no. 2 (2020): 729-41.
- Meissner, E. G., M. McLaughlin, L. Matthews, A. M. Gharib, B. J. Wood, E. Levy, R. Sinkus, K. Virtaneva, D. Sturdevant, C. Martens, S. F. Porcella, Z. D. Goodman, B. Kanwar, R. P. Myers, M. Subramanian, C. Hadigan, H. Masur, D. E. Kleiner, T. Heller, S. Kottilil, J. A. Kovacs, and C. G. Morse. "Simtuzumab Treatment of Advanced Liver Fibrosis in Hiv and Hcv-Infected Adults: Results of a 6-Month Open-Label Safety Trial." Liver Int 36, no. 12 (2016): 1783-92.
- Muir, A. J., C. Levy, H. L. A. Janssen, A. J. Montano-Loza, M. L. Shiffman, S. Caldwell, V. Luketic, D. Ding, C. Jia, B. J. McColgan, J. G. McHutchison, G. Mani Subramanian, R. P. Myers, M. Manns, R. Chapman, N. H. Afdhal, Z. Goodman, B. Eksteen, and C. L. Bowlus. "Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results with Insights on the Natural History of the Disease." Hepatology 69, no. 2 (2019): 684-98.
- Harrison, S. A., M. F. Abdelmalek, S. Caldwell, M. L. Shiffman, A. M. Diehl, R. Ghalib, E. J. Lawitz, D. C. Rockey, R. A. Schall, C. Jia, B. J. McColgan, J. G. McHutchison, G. M. Subramanian, R. P. Myers, Z. Younossi, V. Ratziu, A. J. Muir, N. H. Afdhal, Z. Goodman, J. Bosch, and A. J. Sanyal. "Simtuzumab Is Ineffective for Patients with Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis." Gastroenterology 155, no. 4 (2018): 1140-53.
- Bian, E. B., B. Zhao, C. Huang, H. Wang, X. M. Meng, B. M. Wu, T. T. Ma, L. Zhang, X. W. Lv, and J. Li. "New Advances of DNA Methylation in Liver Fibrosis, with Special Emphasis on the Crosstalk between Micrornas and DNA Methylation Machinery." Cell Signal 25, no. 9 (2013): 1837-44.
- Kuang, Y., A. El-Khoueiry, P. Taverna, M. Ljungman, and N. Neamati. "Guadecitabine (Sgi-110) Priming Sensitizes Hepatocellular Carcinoma Cells to Oxaliplatin." Mol Oncol 9, no. 9 (2015): 1799-814.
- Hardy, T. , and D. A. Mann. "Epigenetics in Liver Disease: From Biology to Therapeutics." Gut 65, no. 11 (2016): 1895-905.
- Hyun, J., J. Park, S. Wang, J. Kim, H. H. Lee, Y. S. Seo, and Y. Jung. "Microrna Expression Profiling in Ccl₄-Induced Liver Fibrosis of Mus Musculus." Int J Mol Sci 17, no. 6 (2016).
- Roderburg, C., G. W. Urban, K. Bettermann, M. Vucur, H. Zimmermann, S. Schmidt, J. Janssen, C. Koppe, P. Knolle, M. Castoldi, F. Tacke, C. Trautwein, and T. Luedde. "Micro-Rna Profiling Reveals a Role for Mir-29 in Human and Murine Liver Fibrosis." Hepatology 53, no. 1 (2011): 209-18.
- Tu, X., X. Zheng, H. Li, Z. Cao, H. Chang, S. Luan, J. Zhu, J. Chen, Y. Zang, and J. Zhang. "Microrna-30 Protects against Carbon Tetrachloride-Induced Liver Fibrosis by Attenuating Transforming Growth Factor Beta Signaling in Hepatic Stellate Cells." Toxicol Sci 146, no. 1 (2015): 157-69.
- Høydahl, L. S., G. Berntzen, and GÅ Løset. "Engineering t-Cell Receptor-Like Antibodies for Biologics and Cell Therapy." Curr Opin Biotechnol 90 (2024): 103224.
- Amor, C., J. Feucht, J. Leibold, Y. J. Ho, C. Zhu, D. Alonso-Curbelo, J. Mansilla-Soto, J. A. Boyer, X. Li, T. Giavridis, A. Kulick, S. Houlihan, E. Peerschke, S. L. Friedman, V. Ponomarev, A. Piersigilli, M. Sadelain, and S. W. Lowe. "Senolytic Car T Cells Reverse Senescence-Associated Pathologies." Nature 583, no. 7814 (2020): 127-32.
- Aghajanian, H., T. Kimura, J. G. Rurik, A. S. Hancock, M. S. Leibowitz, L. Li, J. Scholler, J. Monslow, A. Lo, W. Han, T. Wang, K. Bedi, M. P. Morley, R. A. Linares Saldana, N. A. Bolar, K. McDaid, C. A. Assenmacher, C. L. Smith, D. Wirth, C. H. June, K. B. Margulies, R. Jain, E. Puré, S. M. Albelda, and J. A. Epstein. "Targeting Cardiac Fibrosis with Engineered T Cells." Nature 573, no. 7774 (2019): 430-33.
- Amrute, J. M., X. Luo, V. Penna, S. Yang, T. Yamawaki, S. Hayat, A. Bredemeyer, I. H. Jung, F. F. Kadyrov, G. S. Heo, R. Venkatesan, S. Y. Shi, A. Parvathaneni, A. L. Koenig, C. Kuppe, C. Baker, H. Luehmann, C. Jones, B. Kopecky, X. Zeng, T. Bleckwehl, P. Ma, P. Lee, Y. Terada, A. Fu, M. Furtado, D. Kreisel, A. Kovacs, N. O. Stitziel, S. Jackson, C. M. Li, Y. Liu, N. A. Rosenthal, R. Kramann, B. Ason, and K. J. Lavine. "Targeting Immune-Fibroblast Cell Communication in Heart Failure." Nature 635, no. 8038 (2024): 423-33.
- Friedman, S. L. "Fighting Cardiac Fibrosis with Car T Cells." N Engl J Med 386, no. 16 (2022): 1576-78.
- Rurik, J. G., I. Tombácz, A. Yadegari, P. O. Méndez Fernández, S. V. Shewale, L. Li, T. Kimura, O. Y. Soliman, T. E. Papp, Y. K. Tam, B. L. Mui, S. M. Albelda, E. Puré, C. H. June, H. Aghajanian, D. Weissman, H. Parhiz, and J. A. Epstein. "Car T Cells Produced in Vivo to Treat Cardiac Injury." Science 375, no. 6576 (2022): 91-96.
- Basalova, N., N. Alexandrushkina, O. Grigorieva, M. Kulebyakina, and A. Efimenko. "Fibroblast Activation Protein Alpha (Fapα) in Fibrosis: Beyond a Perspective Marker for Activated Stromal Cells?" Biomolecules 13, no. 12 (2023).
- Zhao, X., J. Lin, M. Liu, D. Jiang, Y. Zhang, X. Li, B. Shi, J. Jiang, C. Ma, H. Shao, Q. Xu, H. Ping, J. Li, and Y. Gao. "Targeting Fap-Positive Chondrocytes in Osteoarthritis: A Novel Lipid Nanoparticle Sirna Approach to Mitigate Cartilage Degeneration." J Nanobiotechnology 22, no. 1 (2024): 659.
- Eriksson, O. , and I. Velikyan. "Radiotracers for Imaging of Fibrosis: Advances during the Last Two Decades and Future Directions." Pharmaceuticals (Basel) 16, no. 11 (2023).
- Trinh, V. Q., T. F. Lee, S. Lemoinne, K. C. Ray, M. D. Ybanez, T. Tsuchida, J. K. Carter, J. Agudo, B. D. Brown, K. M. Akat, S. L. Friedman, and Y. A. Lee. "Hepatic Stellate Cells Maintain Liver Homeostasis through Paracrine Neurotrophin-3 Signaling That Induces Hepatocyte Proliferation." Sci Signal 16, no. 787 (2023): eadf6696.
- Li, M., B. Wu, L. Li, C. Lv, and Y. Tian. "Reprogramming of Cancer-Associated Fibroblasts Combined with Immune Checkpoint Inhibitors: A Potential Therapeutic Strategy for Cancers." Biochim Biophys Acta Rev Cancer 1878, no. 5 (2023): 188945.
- Krenkel, O., J. Hundertmark, T. P. Ritz, R. Weiskirchen, and F. Tacke. "Single Cell Rna Sequencing Identifies Subsets of Hepatic Stellate Cells and Myofibroblasts in Liver Fibrosis." Cells 8, no. 5 (2019).
- Khan, M. A., J. Fischer, L. Harrer, F. Schwiering, D. Groneberg, and A. Friebe. "Hepatic Stellate Cells in Zone 1 Engage in Capillarization Rather Than Myofibroblast Formation in Murine Liver Fibrosis." Sci Rep 14, no. 1 (2024): 18840.
- Yamagata, K., S. Takasuga, M. Tatematsu, A. Fuchimukai, T. Yamada, M. Mizuno, M. Morii, and T. Ebihara. "Foxd1 Expression Identifies a Distinct Subset of Hepatic Stellate Cells Involved in Liver Fibrosis." Biochem Biophys Res Commun 734 (2024): 150632.
- Rosenthal, S. B., X. Liu, S. Ganguly, D. Dhar, M. P. Pasillas, E. Ricciardelli, R. Z. Li, T. D. Troutman, T. Kisseleva, C. K. Glass, and D. A. Brenner. "Heterogeneity of Hscs in a Mouse Model of Nash." Hepatology 74, no. 2 (2021): 667-85.
- Cogliati, B., C. N. Yashaswini, S. Wang, D. Sia, and S. L. Friedman. "Friend or Foe? The Elusive Role of Hepatic Stellate Cells in Liver Cancer." Nat Rev Gastroenterol Hepatol 20, no. 10 (2023): 647-61.
- Wang, S., K. Li, E. Pickholz, R. Dobie, K. P. Matchett, N. C. Henderson, C. Carrico, I. Driver, M. Borch Jensen, L. Chen, M. Petitjean, D. Bhattacharya, M. I. Fiel, X. Liu, T. Kisseleva, U. Alon, M. Adler, R. Medzhitov, and S. L. Friedman. "An Autocrine Signaling Circuit in Hepatic Stellate Cells Underlies Advanced Fibrosis in Nonalcoholic Steatohepatitis." Sci Transl Med 15, no. 677 (2023): eadd3949.
- Filliol, A., Y. Saito, A. Nair, D. H. Dapito, L. X. Yu, A. Ravichandra, S. Bhattacharjee, S. Affo, N. Fujiwara, H. Su, Q. Sun, T. M. Savage, J. R. Wilson-Kanamori, J. M. Caviglia, L. Chin, D. Chen, X. Wang, S. Caruso, J. K. Kang, A. D. Amin, S. Wallace, R. Dobie, D. Yin, O. M. Rodriguez-Fiallos, C. Yin, A. Mehal, B. Izar, R. A. Friedman, R. G. Wells, U. B. Pajvani, Y. Hoshida, H. E. Remotti, N. Arpaia, J. Zucman-Rossi, M. Karin, N. C. Henderson, I. Tabas, and R. F. Schwabe. "Opposing Roles of Hepatic Stellate Cell Subpopulations in Hepatocarcinogenesis." Nature 610, no. 7931 (2022): 356-65.
- Merens, V., E. Knetemann, E. Gürbüz, V. De Smet, N. Messaoudi, H. Reynaert, S. Verhulst, and L. A. van Grunsven. "Hepatic Stellate Cell Single Cell Atlas Reveals a Highly Similar Activation Process across Liver Disease Aetiologies." JHEP Rep 7, no. 1 (2025): 101223.
- Fallowfield, J. A. "Therapeutic Targets in Liver Fibrosis." Am J Physiol Gastrointest Liver Physiol 300, no. 5 (2011): G709-15.
- Friedman, S. L., D. Sheppard, J. S. Duffield, and S. Violette. "Therapy for Fibrotic Diseases: Nearing the Starting Line." Sci Transl Med 5, no. 167 (2013): 167sr1.
- Ramachandran, P., K. P. Matchett, R. Dobie, J. R. Wilson-Kanamori, and N. C. Henderson. "Single-Cell Technologies in Hepatology: New Insights into Liver Biology and Disease Pathogenesis." Nat Rev Gastroenterol Hepatol 17, no. 8 (2020): 457-72.
- Roehlen, N., E. Crouchet, and T. F. Baumert. "Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives." Cells 9, no. 4 (2020).
- Dobie, R., J. R. Wilson-Kanamori, B. E. P. Henderson, J. R. Smith, K. P. Matchett, J. R. Portman, K. Wallenborg, S. Picelli, A. Zagorska, S. V. Pendem, T. E. Hudson, M. M. Wu, G. R. Budas, D. G. Breckenridge, E. M. Harrison, D. J. Mole, S. J. Wigmore, P. Ramachandran, C. P. Ponting, S. A. Teichmann, J. C. Marioni, and N. C. Henderson. "Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis." Cell Rep 29, no. 7 (2019): 1832-47.e8.
- Su, T., Y. Yang, S. Lai, J. Jeong, Y. Jung, M. McConnell, T. Utsumi, and Y. Iwakiri. "Single-Cell Transcriptomics Reveals Zone-Specific Alterations of Liver Sinusoidal Endothelial Cells in Cirrhosis." Cell Mol Gastroenterol Hepatol 11, no. 4 (2021): 1139-61.
- Saviano, A., N. C. Henderson, and T. F. Baumert. "Single-Cell Genomics and Spatial Transcriptomics: Discovery of Novel Cell States and Cellular Interactions in Liver Physiology and Disease biology." J Hepatol 73, no. 5 (2020): 1219-30.
- Ramachandran, P., R. Dobie, J. R. Wilson-Kanamori, E. F. Dora, B. E. P. Henderson, N. T. Luu, J. R. Portman, K. P. Matchett, M. Brice, J. A. Marwick, R. S. Taylor, M. Efremova, R. Vento-Tormo, N. O. Carragher, T. J. Kendall, J. A. Fallowfield, E. M. Harrison, D. J. Mole, S. J. Wigmore, P. N. Newsome, C. J. Weston, J. P. Iredale, F. Tacke, J. W. Pollard, C. P. Ponting, J. C. Marioni, S. A. Teichmann, and N. C. Henderson. "Resolving the Fibrotic Niche of Human Liver Cirrhosis at Single-Cell Level." Nature 575, no. 7783 (2019): 512-18.
- Chen, Z., A. Jain, H. Liu, Z. Zhao, and K. Cheng. "Targeted Drug Delivery to Hepatic Stellate Cells for the Treatment of Liver Fibrosis." J Pharmacol Exp Ther 370, no. 3 (2019): 695-702.
- Bansal, R. , and K. Poelstra. "Hepatic Stellate Cell Targeting Using Peptide-Modified Biologicals." Methods Mol Biol 2669 (2023): 269-84.
- Qian, T., N. Fujiwara, B. Koneru, A. Ono, N. Kubota, A. K. Jajoriya, M. G. Tung, E. Crouchet, W. M. Song, C. A. Marquez, G. Panda, A. Hoshida, I. Raman, Q. Z. Li, C. Lewis, A. Yopp, N. E. Rich, A. G. Singal, S. Nakagawa, N. Goossens, T. Higashi, A. P. Koh, C. B. Bian, H. Hoshida, P. Tabrizian, G. Gunasekaran, S. Florman, M. E. Schwarz, S. P. Hiotis, T. Nakahara, H. Aikata, E. Murakami, T. Beppu, H. Baba, Warren Rew, S. Bhatia, M. Kobayashi, H. Kumada, A. J. Fobar, N. D. Parikh, J. A. Marrero, S. H. Rwema, V. Nair, M. Patel, S. Kim-Schulze, K. Corey, J. G. O'Leary, G. B. Klintmalm, D. L. Thomas, M. Dibas, G. Rodriguez, B. Zhang, S. L. Friedman, T. F. Baumert, B. C. Fuchs, K. Chayama, S. Zhu, R. T. Chung, and Y. Hoshida. "Molecular Signature Predictive of Long-Term Liver Fibrosis Progression to Inform Antifibrotic Drug Development." Gastroenterology 162, no. 4 (2022): 1210-25.
- Guo, J., W. Liu, Z. Zeng, J. Lin, X. Zhang, and L. Chen. "Tgfb3 and Mmp13 Regulated the Initiation of Liver Fibrosis Progression as Dynamic Network Biomarkers." J Cell Mol Med 25, no. 2 (2021): 867-79.
- Schnittert, J., R. Bansal, G. Storm, and J. Prakash. "Integrins in Wound Healing, Fibrosis and Tumor Stroma: High Potential Targets for Therapeutics and Drug Delivery." Adv Drug Deliv Rev 129 (2018): 37-53.
- Kurniawan, D. W., R. Booijink, L. Pater, I. Wols, A. Vrynas, G. Storm, J. Prakash, and R. Bansal. "Fibroblast Growth Factor 2 Conjugated Superparamagnetic Iron Oxide Nanoparticles (Fgf2-Spions) Ameliorate Hepatic Stellate Cells Activation in Vitro and Acute Liver Injury in Vivo." J Control Release 328 (2020): 640-52.
- Huang, L., J. Xie, Q. Bi, Z. Li, S. Liu, Q. Shen, and C. Li. "Highly Selective Targeting of Hepatic Stellate Cells for Liver Fibrosis Treatment Using a D-Enantiomeric Peptide Ligand of Fn14 Identified by Mirror-Image Mrna Display." Mol Pharm 14, no. 5 (2017): 1742-53.
- Moreno, M., T. Gonzalo, R. J. Kok, P. Sancho-Bru, M. van Beuge, J. Swart, J. Prakash, K. Temming, C. Fondevila, L. Beljaars, M. Lacombe, P. van der Hoeven, V. Arroyo, K. Poelstra, D. A. Brenner, P. Ginès, and R. Bataller. "Reduction of Advanced Liver Fibrosis by Short-Term Targeted Delivery of an Angiotensin Receptor Blocker to Hepatic Stellate Cells in Rats." Hepatology 51, no. 3 (2010): 942-52.
- Lee, J., J. Byun, G. Shim, and Y. K. Oh. "Fibroblast Activation Protein Activated Antifibrotic Peptide Delivery Attenuates Fibrosis in Mouse Models of Liver Fibrosis." Nat Commun 13, no. 1 (2022): 1516.
- Klein, S., M. M. Van Beuge, M. Granzow, L. Beljaars, R. Schierwagen, S. Kilic, I. Heidari, S. Huss, T. Sauerbruch, K. Poelstra, and J. Trebicka. "Hsc-Specific Inhibition of Rho-Kinase Reduces Portal Pressure in Cirrhotic Rats without Major Systemic Effects." J Hepatol 57, no. 6 (2012): 1220-7.
- Bansal, R., J. Prakash, E. Post, L. Beljaars, D. Schuppan, and K. Poelstra. "Novel Engineered Targeted Interferon-Gamma Blocks Hepatic Fibrogenesis in Mice." Hepatology 54, no. 2 (2011): 586-96.
- Sato, Y., K. Murase, J. Kato, M. Kobune, T. Sato, Y. Kawano, R. Takimoto, K. Takada, K. Miyanishi, T. Matsunaga, T. Takayama, and Y. Niitsu. "Resolution of Liver Cirrhosis Using Vitamin a-Coupled Liposomes to Deliver Sirna against a Collagen-Specific Chaperone." Nat Biotechnol 26, no. 4 (2008): 431-42.
- Salvati, A. , and K. Poelstra. "Drug Targeting and Nanomedicine: Lessons Learned from Liver Targeting and Opportunities for Drug Innovation." Pharmaceutics 14, no. 1 (2022).
- Bansal, R., J. Prakash, M. De Ruiter, and K. Poelstra. "Targeted Recombinant Fusion Proteins of Ifnγ and Mimetic Ifnγ with Pdgfβr Bicyclic Peptide Inhibits Liver Fibrogenesis in Vivo." PLoS One 9, no. 2 (2014): e89878.
- Yazdani, S., R. Bansal, and J. Prakash. "Drug Targeting to Myofibroblasts: Implications for Fibrosis and Cancer." Adv Drug Deliv Rev 121 (2017): 101-16.
- Pringle, T. A., E. Ramon-Gil, J. Leslie, F. Oakley, M. C. Wright, J. C. Knight, and S. Luli. "Synthesis and Preclinical Evaluation of a (89)Zr-Labelled Human Single Chain Antibody for Non-Invasive Detection of Hepatic Myofibroblasts in Acute Liver Injury." Sci Rep 14, no. 1 (2024): 633.
- Lin, C. Y., U. F. Mamani, Y. Guo, Y. Liu, and K. Cheng. "Peptide-Based Sirna Nanocomplexes Targeting Hepatic Stellate Cells." Biomolecules 13, no. 3 (2023).
- Jain, A., A. Barve, Z. Zhao, J. P. Fetse, H. Liu, Y. Li, and K. Cheng. "Targeted Delivery of an Sirna/Pna Hybrid Nanocomplex Reverses Carbon Tetrachloride-Induced Liver Fibrosis." Adv Ther (Weinh) 2, no. 8 (2019).
- Zhao, Z., Y. Li, A. Jain, Z. Chen, H. Liu, W. Jin, and K. Cheng. "Development of a Peptide-Modified Sirna Nanocomplex for Hepatic Stellate Cells." Nanomedicine 14, no. 1 (2018): 51-61.
- Lawitz, E. J., D. E. Shevell, G. S. Tirucherai, S. Du, W. Chen, U. Kavita, A. Coste, F. Poordad, M. Karsdal, M. Nielsen, Z. Goodman, and E. D. Charles. "Bms-986263 in Patients with Advanced Hepatic Fibrosis: 36-Week Results from a Randomized, Placebo-Controlled Phase 2 Trial." Hepatology 75, no. 4 (2022): 912-23.
- Hill, T. A., N. E. Shepherd, F. Diness, and D. P. Fairlie. "Constraining Cyclic Peptides to Mimic Protein Structure Motifs." Angew Chem Int Ed Engl 53, no. 48 (2014): 13020-41.
- Petta, I., S. Lievens, C. Libert, J. Tavernier, and K. De Bosscher. "Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics." Mol Ther 24, no. 4 (2016): 707-18.
- Lee, A. C., J. L. Harris, K. K. Khanna, and J. H. Hong. "A Comprehensive Review on Current Advances in Peptide Drug Development and Design." Int J Mol Sci 20, no. 10 (2019).
- Geppert, T., B. Hoy, S. Wessler, and G. Schneider. "Context-Based Identification of Protein-Protein Interfaces and "Hot-Spot" Residues." Chem Biol 18, no. 3 (2011): 344-53.
- Bhat, A., L. R. Roberts, and J. J. Dwyer. "Lead Discovery and Optimization Strategies for Peptide Macrocycles." Eur J Med Chem 94 (2015): 471-9.
- Villar, E. A., D. Beglov, S. Chennamadhavuni, J. A. Porco, Jr., D. Kozakov, S. Vajda, and A. Whitty. "How Proteins Bind Macrocycles." Nat Chem Biol 10, no. 9 (2014): 723-31.
- Liu, D., D. Xu, M. Liu, W. E. Knabe, C. Yuan, D. Zhou, M. Huang, and S. O. Meroueh. "Small Molecules Engage Hot Spots through Cooperative Binding to Inhibit a Tight Protein-Protein Interaction." Biochemistry 56, no. 12 (2017): 1768-84.
- Wang, L., N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang, and C. Fu. "Therapeutic Peptides: Current Applications and Future Directions." Signal Transduct Target Ther 7, no. 1 (2022): 48.
- Mantovani, A., C. D. Byrne, and G. Targher. "Efficacy of Peroxisome Proliferator-Activated Receptor Agonists, Glucagon-Like Peptide-1 Receptor Agonists, or Sodium-Glucose Cotransporter-2 Inhibitors for Treatment of Non-Alcoholic Fatty Liver Disease: A Systematic Review." Lancet Gastroenterol Hepatol 7, no. 4 (2022): 367-78.
- Malm-Erjefält, M., I. Bjørnsdottir, J. Vanggaard, H. Helleberg, U. Larsen, B. Oosterhuis, J. J. van Lier, M. Zdravkovic, and A. K. Olsen. "Metabolism and Excretion of the Once-Daily Human Glucagon-Like Peptide-1 Analog Liraglutide in Healthy Male Subjects and Its in Vitro Degradation by Dipeptidyl Peptidase Iv and Neutral Endopeptidase." Drug Metab Dispos 38, no. 11 (2010): 1944-53.
Figure 1.
Targeting hepatic stellate cells activation for the treatment of liver fibrosis and cirrhosis.
Figure 1.
Targeting hepatic stellate cells activation for the treatment of liver fibrosis and cirrhosis.
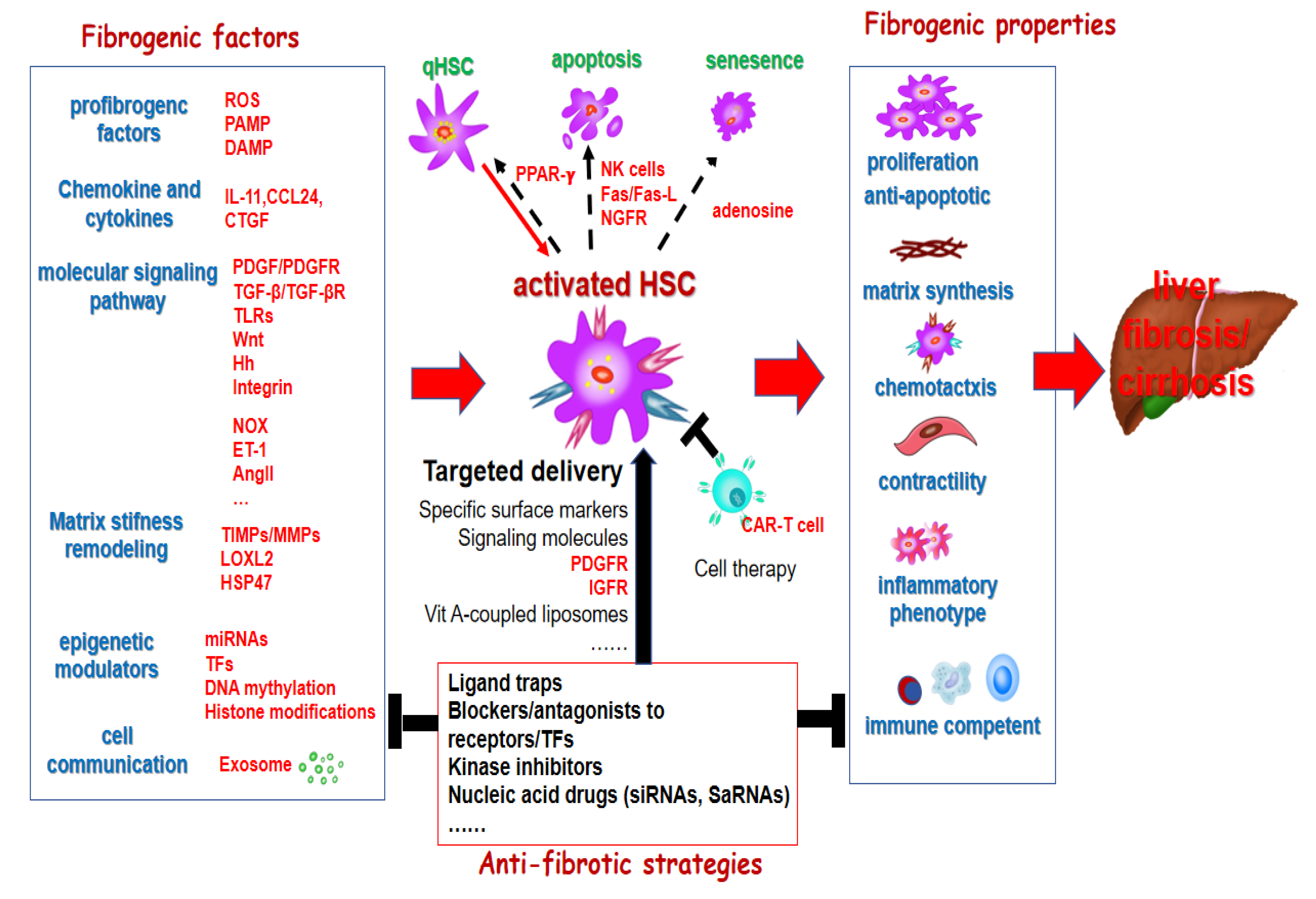
Figure 2.
Targeting cancer associated fibroblasts for the treatment of hepatocellular carcinoma.
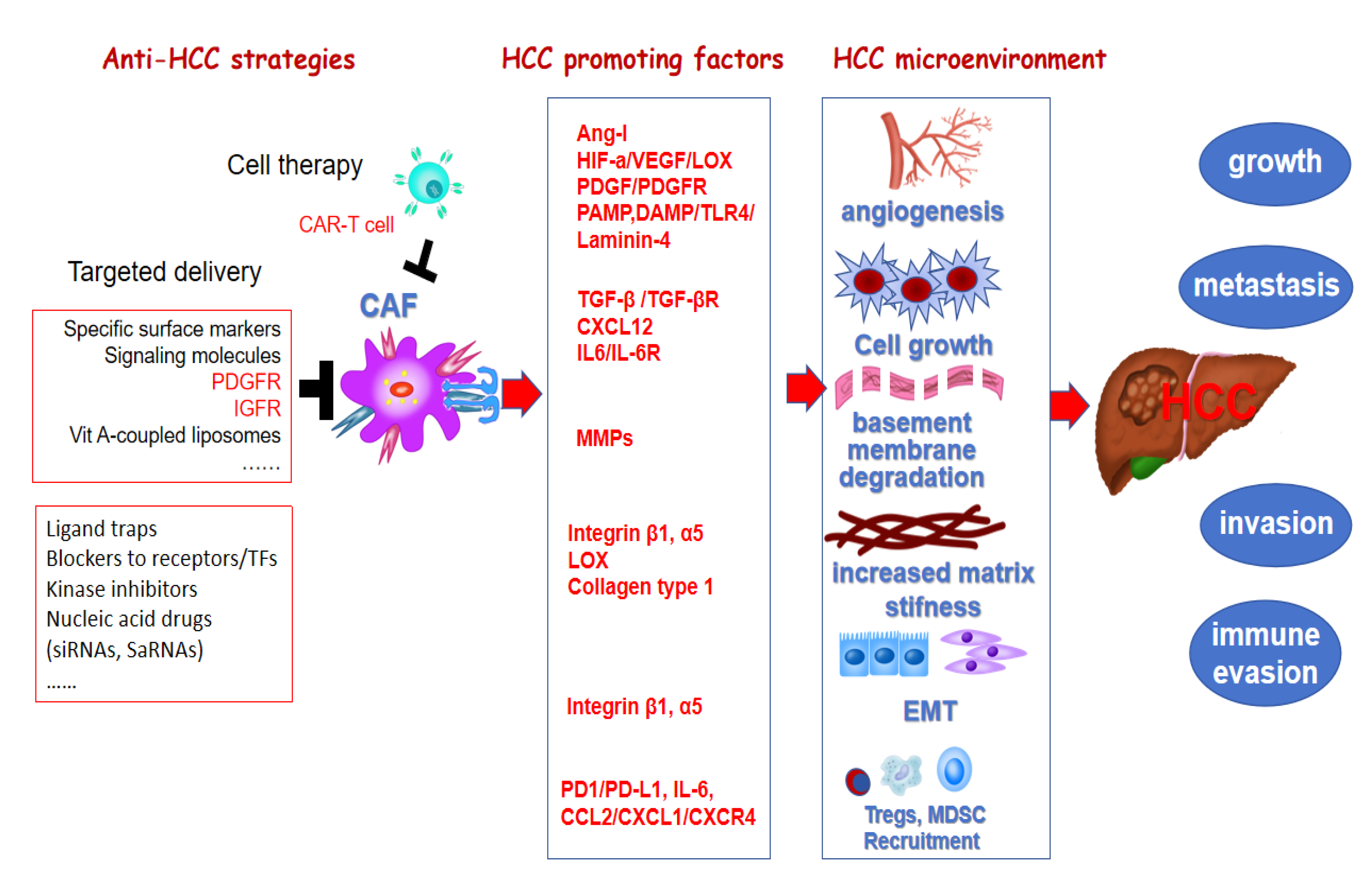

Table 1.
The represent clinical trials of potential antifibrotic therapies in chronic liver diseases.
Table 1.
The represent clinical trials of potential antifibrotic therapies in chronic liver diseases.
| targeting HSC activation Target | Drug name | Drug category | Population(n) | Highest status (phase) | NCT | Status |
| TGF-β/TGFβR | Hydronidone | Small molecule | Liver fibrosis (248) | III | NCT05115942 | Completed |
| FGF21 | BIO89-100 | Fusion proteins | MASH (101) | II | NCT04048135 | Completed |
| Efruxifermin | Fusion proteins | MASH (110) | II | NCT03976401 | Completed | |
| Pergbelfermin | Fusion proteins | Liver cirrhosis andMASH (155) | II | NCT03486912 | Completed | |
| FGF19 | Aldafermin | Fusion proteins | MASH (171) | II | NCT03912532 | Completed |
| WNT/β-catenin | PRI-724 | Small molecule | Liver cirrhosis (27) | II | NCT03620474 | Completed |
| LOXL2 | PXS-5382A | Small molecule | MASH (18) | I | NCT04183517 | Completed |
| PPARα/γ | Saroglitazar | Small molecule | MASH (20) | II | NCT03639623 | Completed |
| PPARα/δ/γ | Lanifibranor | Small molecule | MASH (1000) | III | NCT04849728 | Recruiting |
| PPARα | Pemafibrate | Small molecule | MASH (118) | II | NCT03350165 | Completed |
| PPARα/δ | ZSP0678 | Small molecule | MASH (104) | I | NCT04137055 | Completed |
| TLR4 | JKB-121 | Small molecule | MASH (65) | II | NCT02442687 | Completed |
| JKB-122 | Small molecule | MASH (300) | II | NCT04255069 | Unknown | |
| GLP-1 receptor | Semaglutide | Small molecule | MASH (1200) | III | NCT04822181 | Active, not recruiting |
| GLP-1/GIP receptor | Trizepatide | Small molecule | MASH (190) | II | NCT04166773 | Completed |
| GLP-1/Glucagon receptor | Cotadutide | Small molecule | MASH (54) | II | NCT05364931 | Completed |
| GLP-1/GIP/Glucagon | HM-15211 | Small molecule | MASH (240) | II | NCT04505436 | Recruiting |
| THRβ | Resmetirom | Small molecule | MASH (1759) | III | NCT03900429 | Active, not recruiting |
| VK2809 | Small molecule | MASH (248) | II | NCT04173065 | Completed | |
| MPC | Azemiglitazone | Small molecule | NASH (1800) | III | NCT03970031 | Unknown |
| Deuterium-stabilized (R)-Piglitazone | Small molecule | MASH (117) | II | NCT04321343 | Completed | |
| PDEs (mainly PED2) | NZSP1601 | Small molecule | MASH (37) | II | NCT04140123 | Completed |
| LOXL2, PDE3/4 | Epeleuton | Small molecule | MAFLD (96) | II | NCT02941549 | Completed |
| AMPK | PXL-770 | Small molecule | MAFLD (211) | II | NCT03763877 | Completed |
| MMP(MMP2,MMP9. VEGF-A) | ALS-L1023 | Small molecule | MASH (60) | II | NCT04342793 | Completed |
| A3AR | Namodenoson | Small molecule | MASH (60) | II | NCT02927314 | Completed |
| FASN | TVB-2640 | Small molecule | MASH and MAFLD (2000) | III | NCT06692283 | Not yet recruiting |
| Biodentical testosterone | LPCN1144 | Small molecule | MASH (56) | II | NCT04134091 | Completed |
| Stem cell | HepaStem | Cell transplant therapies | MASH (23) | II | NCT03963921 | Completed |
| HSP47 | BMS-986263 | siRNA | MASH (124) | II | NCT04267393 | Terminated |
Note: data source: https://clinicaltrials.gov.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



