
Submitted:
25 March 2025
Posted:
26 March 2025
You are already at the latest version
Abstract
Systemic sclerosis (SSc) is a multifaceted autoimmune disease in which the complex interplay of genetic predisposition and environmental factors triggers aberrant immune responses, ultimately leading to vasculopathy and fibrosis. This review offers a com-prehensive overview of current perspectives on SSc pathogenesis, integrating classical concepts with recent breakthroughs enabled by advanced analytical techniques. We delve into the foundational trans-organ pathophysiology of SSc, encompassing epige-netic dysregulation, chronic inflammation, vascular injury, vasculopathy, and fibrosis. Furthermore, we explore the organ-specific modifiers that contribute to the heteroge-neity of SSc manifestations across different organ systems, including the skin, gastro-intestinal tract, lungs, and heart. Recent studies employing single-cell transcriptomics, spatial proteomics, and epigenomic profiling are highlighted, demonstrating how these technologies are revolutionizing our understanding of SSc cellular and molecular pa-thology. This evolving landscape of SSc pathogenesis research is critical for identifying novel therapeutic targets and advancing personalized medicine approaches for SSc pa-tients.
Keywords:
systemic sclerosis
; pathogenesis
; fibrosis
; vasculopathy
; immune dysfunction
; skin
; single-cell analysis
; transcriptomics
; proteomics
; epigenetics
1. Introduction
Systemic sclerosis (SSc) is a multisystem connective tissue disease of unknown etiology, characterized by aberrant immune activation, vascular injury leading to defective neovascularization and impaired vascular remodeling, and subsequent tissue fibrosis affecting the skin and various internal organs [1]. The etiology of SSc is still unknown, and there is currently no single hypothesis that uniquely explains the variety of pathophysiologic manifestations of the disease. However, our insight into its pathogenesis is rapidly growing, driven by clinical investigations of patient-derived samples, basic science research with animal models, and progress in targeted molecular treatments. Recent breakthroughs in sophisticated analytical methods, particularly single-cell analysis, have revolutionized the field. These advancements are vital for confirming disease mechanisms in patient tissues and for facilitating the discovery of new therapeutic approaches. This manuscript provides a summary of current perspectives on SSc pathogenesis, with a focus on the latest advancements.
2. Systemic Sclerosis Pathogenesis: Foundational Trans-Organ Pathophysiology
2.1. “. Genetics” in SSc
Genetic and etiological investigations have established that SSc arises from a complex interplay between genetic predisposition and environmental factors. The strongest identified risk factor for SSc is a positive family history [2]; however, twin concordance rates for SSc are low (4.7%) and comparable between monozygotic and dizygotic twins (4.2% versus 5.6%). Notably, concordance for autoantibody presence against nuclear or cytoplasmic antigens is significantly elevated in healthy monozygotic twin siblings compared to healthy dizygotic twin siblings of SSc patients (95% versus 60%, p < 0.05) [3]. This observation suggests that genetic factors are associated with autoimmunity, thereby increasing SSc susceptibility, but are insufficient for the development of clinically definite SSc. Consistent with this notion, the majority of SSc susceptibility genes are Human Leukocyte Antigen (HLA) haplotypes and non-HLA genes implicated in immunity and inflammation, which are also implicated in other connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus (SLE) [4,5]. Conversely, multiple case-control and genome-wide association studies demonstrate that single nucleotide polymorphisms (SNPs) within certain disease-susceptibility genes correlate with SSc disease severity [6,7,8,9]. For example, a specific SNP associated with the downregulation of interferon regulatory factor 5 (IRF5) is more frequently observed in SSc patients exhibiting milder clinical manifestations [10]. Thus, genetic factors influence both the susceptibility to and the severity of SSc.
2.2. “. Epigenetics” in SSc
Epigenetics refers to the mechanisms by which cells regulate and transmit gene expression without alterations to the DNA base sequence. It also encompasses the field of study dedicated to these mechanisms. The pathogenesis of SSc is investigated from three perspectives: immune dysregulation, vasculopathy, and fibrosis. However, the ultimate goal of these investigations is to elucidate the mechanisms through which fibroblasts maintain a persistently activated state across various organs. While fibroblast activation is generally considered a response to the cumulative stimuli from the extracellular microenvironment in vivo, fibroblasts isolated from the skin and lungs of SSc patients retain their activated phenotype even after in vitro serial passage. This observation suggests the existence of a mechanism for maintaining a "pathological memory" intrinsic to these cells. Epigenetics is considered to be one such mechanism. DNA methylation, histone acetylation and methylation, microRNAs (miRNAs), and long non-coding RNAs (lncRNAs) have been identified as epigenetic modifications implicated in sustaining the activated state of fibroblasts derived from SSc patients.
Focusing on genetic factors, genome-wide association studies, as noted earlier, have identified that most susceptibility genes for SSc are located within the HLA region and in non-HLA immune-related genes. This suggests that genetically determined immune abnormalities play a central role in the development of SSc [5]. Indeed, antinuclear antibodies are detected in over 90% of SSc patients, and numerous other autoantibodies with pathogenic functions have been identified [11,12]. In the lesional skin of patients with early-stage SSc, perivascular lymphocytic infiltration is observed. This inflammation is thought to induce structural abnormalities in blood vessels characteristic of SSc, such as the loss of capillaries and thickening of small artery walls. Furthermore, it activates fibroblasts, leading to fibrosis. Although the precise mechanisms remain to be fully elucidated, it is proposed that SSc-specific phenotypic changes arise in various cells through epigenetic regulation influenced by environmental factors and potentially shaped by genetic predisposition. These changes result in the breakdown of immune tolerance, excessive inflammatory responses, impaired vascular remodeling, and persistent and aberrant activation of vascular endothelial cells and fibroblasts. Consequently, these processes contribute to the complex pathogenesis of this disease [13]. Moreover, epigenetic modifications are deeply implicated in the mechanisms by which various cells maintain their pathological phenotypes within the highly fibrotic environment. In summary, epigenetics is considered to be involved in cellular phenotypic changes from two perspectives: responses to external environmental factors that trigger SSc onset and the mechanisms maintaining pathological homeostasis against the internal microenvironment.
In SSc, the transcription factor FLI1 was the first gene identified as being under epigenetic control of its expression. Wang et al. [14] reported that, in both cultured SSc dermal fibroblasts and lesional SSc skin, CpG methylation is markedly increased in the FLI1 promoter region. Furthermore, in cultured SSc dermal fibroblasts, a significant decrease in histone H3 and H4 acetylation has been observed. Immunohistochemical analysis of skin tissue revealed that FLII expression in fibroblasts and vascular endothelial cells is mildly reduced in non-lesional SSc skin compared to healthy skin. A more pronounced reduction was observed in lesional SSc skin [15]. In the skin of Fli1+/- mice, fibroblasts are constitutively activated and exhibit increased type I collagen production; however, dermal thickening is not histologically apparent. Furthermore, mild structural abnormalities are present in skin microvessels, and phenotypic alterations resembling those in SSc skin vascular endothelial cells are evident at the molecular level [16]. Conversely, when bleomycin-induced SSc model mice are generated using Fli1+/- mice, an exacerbation of SSc-like phenotypes is observed across all aspects: inflammation, vasculopathy, and fibrosis [17]. These findings indicate that reduced FLI1 expression can induce activation of immune cells, vascular endothelial cells, and fibroblasts, molecularly resembling SSc, but this anomaly alone is insufficient to trigger SSc onset.
KLF5 is a crucial transcription factor regulating diverse fibrotic responses, acting as a pro-fibrotic factor in cardiac fibroblasts and a fibrosis-suppressing factor in renal tubular cells [18,19]. DNA microarray analysis has revealed that the expression of this transcription factor is downregulated in lesional SSc skin [20]. A study using cultured SSc dermal fibroblasts has revealed a marked downregulation of KLF5 expression at both the mRNA and protein levels. It was demonstrated that the application of an epigenetic inhibitor could recover KLF5 expression to levels comparable to those in healthy cells. Moreover, increased CpG methylation in the KLF5 promoter region was also detected [16]. These findings elucidate that KLF5 expression is robustly suppressed by epigenetic mechanisms in SSc dermal fibroblasts.
Building upon these findings, Klf5+/-; Fli1+/- mice were generated to serve as a model for skin fibrosis. Notably, these mice not only faithfully recapitulated skin and lung fibrosis pathologies of SSc but also mirrored SSc vasculopathy, inflammation, and autoimmunity. Importantly, inflammation and autoimmunity emerged at 4 weeks of age, vasculopathy at 4-8 weeks, and skin fibrosis at 8-12 weeks. The temporal sequence of these major pathologies mirrored that observed in SSc [16]. Thus, Klf5+/-; Fli1+/- mice can be considered a novel SSc model that spontaneously develops the three major SSc pathologies in a temporally similar manner to human SSc. This research highlights a significant finding: by focusing on factors governing pathological memory in SSc patient-derived cells, we can potentially identify key players in the pathogenesis of this disease.
Analyses of DNA methylation, histone modifications, miRNAs, and lncRNAs have been conducted in various cell types derived from SSc patients, including fibroblasts, vascular endothelial cells, CD4+ T cells, B cells, and plasmacytoid dendritic cells (pDCs). Consequently, a substantial number of epigenetic aberrations have been reported. Detailed descriptions of individual findings are omitted in this paper, as they are summarized in several review articles; readers are referred to these for further details [21,22,23,24,25,26,27].
2.3. Genetic Polymorphisms of FLI1 and SSc Susceptibility
Yamashita et al. [4] conducted a case-control study in a Japanese population, directly genotyping this FLI1 (GA)n microsatellite. Their findings revealed a significant association between extended repeat alleles of the FLI1 (GA)n microsatellite and increased susceptibility to SSc. Specifically, alleles with 22 or more GA repeats (L alleles) were more frequent in SSc patients compared to healthy controls. Furthermore, these L alleles were associated with reduced FLI1 mRNA levels in healthy individuals, suggesting a functional consequence of this genetic polymorphism on gene expression. This genetic association with FLI1 further strengthens the evidence implicating FLI1 as a key player in SSc pathogenesis, acting not only through epigenetic modifications but also through inherent genetic predispositions that influence its expression and potentially its function. These findings suggest that FLI1 genetic variants, particularly microsatellite polymorphisms, may contribute to the "missing heritability" in SSc.
2.4. Inflammation and Immunological Dysfunction in SSc
As previously described, the interaction between endothelial cells and circulating immune cells, mediated by cell adhesion molecules and chemokines, facilitates the activation of inflammatory cells and their infiltration into SSc-affected organs. Typically, infiltration by T cells, macrophages, and mast cells predominates in SSc-involved skin, whereas B cell infiltration is comparatively limited [28,29,30,31]. In contrast, numerous lymphoid aggregates, characterized by a substantial accumulation of B cells and relatively fewer T cells and macrophages, are commonly observed in the lung tissue of patients with SSc-associated interstitial lung disease (ILD) [32]. Notably, genes associated with activated B cells are upregulated in SSc-involved skin [20], and the B cell count in the skin correlates with the progression of skin fibrosis [31]. Consequently, while the composition of infiltrating cell types may vary across different affected organs, increased infiltration of B cells, T cells, and innate immune cells is a shared feature in the organs involved in SSc.
Alterations in T cell subsets are a well-documented feature of SSc. The balance of Th1/Th2 and Th17/Treg immune responses is skewed toward Th2 and Th17 dominance [33,34,35,36], and regulatory T cell (Treg) function is impaired during the active phase of SSc [37]. In the early stage of diffuse cutaneous SSc (dcSSc), serum levels of interleukin-6 (IL-6) and IL-10 are significantly elevated, whereas they decrease to normal levels in the late stage of dcSSc, which is characterized by the regression of skin sclerosis [38]. IL-4 levels remain within the normal range in the early stage of dcSSc but decrease concurrently with the resolution of skin sclerosis. Conversely, serum IL-12 levels are reduced in the early dcSSc stage, subsequently increase gradually with disease duration, and ultimately reach significantly higher levels than normal controls in the late dcSSc stage [33]. Regarding Th17-associated cytokines, the expression levels of IL-17A, IL-21, and IL-22, but not IL-17F, are elevated in the lesional skin of early dcSSc [36,39]. Moreover, the proportions of circulating Th17 cells and IL-17 production are increased in peripheral blood mononuclear cells from SSc patients, and the Th17 cell count correlates with disease activity [34]. With respect to Tregs, the proportion of Th2-like Tregs is increased in the affected skin of SSc patients [30].
Currently, the direct role of SSc-related antinuclear antibodies (ANAs), including antibodies against topoisomerase I (topo I), centromere, and RNA polymerase III (RNAP III) antigens, is not fully understood, although a potential role for anti-topoisomerase I antibodies has been suggested (described below). Nevertheless, the strong correlation of these ANAs with clinical manifestations implies that altered B cell phenotypes may be associated with the core abnormality driving disease progression. This could occur through genetic and epigenetic mechanisms shared across cell types and/or through complex interactions with other immune and non-immune cells. Beyond antibody production, B cells exert diverse functions within the immune system, encompassing cytokine production, antigen presentation, macrophage differentiation and activation, and lymphoid tissue development [40]. SSc B cells exhibit constitutive activation, as evidenced by the upregulated expression of CD19, a B cell co-receptor [41], and the upregulated expression of CD80 and CD86 on memory B cells [42]. Further underscoring the significance of B cells in the SSc-specific disease cascade, B cell depletion therapy with rituximab, an anti-CD20 antibody, has demonstrated improvements in skin fibrosis and ILD in multiple case series and open-label studies [43,44,45,46]. Furthermore, several case reports and case series have documented the amelioration of calcinosis, digital ulcers, and arterial stiffness following rituximab therapy [47,48,49]]. Thus, B cells contribute to the activation of vascular and fibrotic processes, in addition to immune system activation in SSc, reinforcing the concept that immune cells are upstream mediators in the SSc-specific disease cascade.
Beyond adaptive immune cells, innate immune cells are also abundantly present in SSc-involved organs. In SSc lesional skin, mast cells secrete excessive levels of transforming growth factor-β (TGF-β) [50]. Moreover, M2 macrophages appear to be critical regulators of tissue fibrosis, as the M2 macrophage-associated gene program, which is upregulated in the skin of early SSc patients, is suppressed in conjunction with the resolution of skin fibrosis following treatment with tocilizumab (an anti-IL-6 receptor antibody) [51].
pDCs are a major source of interferon-α (IFN-α), produced through the activation of Toll-like receptors (TLR) 7 and 9. In SSc-involved skin, the expression of chemerin, a potent chemoattractant for pDCs, is increased in dermal small vessels, and pDCs are localized around these vessels [52,53]. Additionally, the expression of LL-37, which interacts with self-DNA and facilitates its conversion into a stimulatory ligand for TLR7 and 9, is elevated in dermal small vessels [54]. Given that endothelial cell death resulting from autoimmune attacks releases self-DNA, it is hypothesized that pDCs produce excessive IFN-α in the vicinity of dermal small vessels through TLR7 and 9 activation, mediated by self-DNA and LL-37 complexes derived from endothelial cells. Furthermore, disease-associated autoantibodies, particularly anti-topoisomerase I antibodies, may contribute to this process. Anti-topoisomerase I antibodies react with nuclear antigens from endothelial cells, and immune complexes formed with nucleic acids, especially RNA, induce IFN-α production from pDCs [55]. Indeed, prior clinical observations and experimental studies support the concept that IFN-α promotes SSc development and progression. For example, in a clinical trial of recombinant IFN-α in SSc patients, protocol withdrawal was more frequent in patients treated with IFN-α compared to placebo, and most patients discontinuing IFN-α treatment experienced exacerbation of ILD [56]. Furthermore, IFN-α therapy for other conditions, such as multiple sclerosis and chronic hepatitis C virus infection, can induce the onset of typical SSc or SSc-like symptoms [57,58,59,60,61,62]. Considering that sustained exposure to IFN-α induces endothelial cell senescence [63], potentially amplifying IFN-α production from pDCs by providing self-DNA, endothelial cell death and pDCs may establish a feedforward loop that promotes SSc progression via vascular injury and immune system activation mediated by IFN-α. Intriguingly, a recent study has demonstrated ectopic TLR8 expression in SSc pDCs and a critical role for TLR8-expressing pDCs in experimental skin fibrosis, suggesting that TLR8 is the primary RNA-sensing TLR and that IFN-α overproduction is implicated in the establishment of SSc-associated tissue fibrosis [64]. In addition to their role in IFN-α production, recent findings indicate that pDCs can also contribute to fibrosis through endoplasmic reticulum (ER) stress-mediated mechanisms, as demonstrated by Ferreira et al. [65] who revealed that ER stress induction in pDCs promotes fibroblast activation via direct cell-cell contact, suggesting a novel pathway contributing to fibrosis development in SSc. Collectively, the continuous release of autoantigens from damaged and senescent endothelial cells serves as a fundamental driver of SSc pathology, acting through the induction of chronic inflammation.
2.5. Vascular Injury in SSc
As previously discussed, the pathogenesis of SSc begins with immune dysregulation, while histopathologically detectable structural abnormalities first manifest as vascular damage [66,67,68]. Indeed, vasculopathy is a critical element in the early clinical picture of SSc, manifesting in patient-reported symptoms such as Raynaud's phenomenon and digital edema [69]. Crucially, the presence of disease-specific autoantibodies, hallmarks of SSc's autoimmune nature, can be detected even before these initial clinical manifestations emerge, highlighting the early involvement of autoimmunity in vascular injury [69,70,71]. Following this initial vascular insult, the vasculature in SSc undergoes significant structural abnormalities [72]. These changes arise from a combination of dysfunctional vascular remodeling processes and the development of various vascular functional impairments [72].
In the early stages of SSc, capillary fragility leads to capillary dilation, which in turn results in the extravasation of erythrocytes [69]. The observation of dilated and hemorrhaging nailfold capillaries is diagnostically significant, serving as an important early indicator of the disease [70]. As SSc progresses, a progressive loss of capillaries occurs, with capillary numbers gradually diminishing and eventually being replaced by fibrotic tissue [70]. In parallel with this capillary rarefaction and fibrotic replacement, vascular endothelial cells and pericytes, which are crucial components of blood vessels, undergo differentiation into myofibroblasts through processes known as endothelial-to-mesenchymal transition (EndoMT) and pericyte-to-mesenchymal transition (PMT), respectively [72,73]. These transformed cells acquire resistance to apoptosis, or programmed cell death, and exhibit cellular senescence phenotypes. This altered cellular behavior significantly contributes to the establishment and perpetuation of the extensive fibrosis characteristic of SSc [72]. The ensuing loss of capillaries leads to tissue hypoxia, a state of oxygen deficiency, which in turn acts as a potent stimulus for further myofibroblast activation and promotes the fibrotic process across a range of organs, including the skin, lungs, heart, and intestines [72]. In contrast, within arterioles and small to medium-sized arteries, the vascular endothelial cells, when injured, also undergo differentiation into myofibroblasts [72]. Furthermore, the proliferative capacity of vascular smooth muscle cells, another key cell type in blood vessels, and their ability to produce extracellular matrix (ECM) components are enhanced, culminating in fibrotic stenosis, or narrowing of the blood vessels [72]. Arterial lesions in SSc are marked by the abnormal accumulation of myofibroblasts within the vessel wall, leading to the development of fibroproliferative vascular lesions that impede blood flow [72].
The vascular functional derangements in SSc are multifaceted and encompass a range of abnormalities. These include diminished vascular endothelial function, which refers to the impaired ability of the endothelium to regulate vascular tone and permeability; a reduced capacity for thrombus inhibition, increasing the risk of blood clot formation; impaired physiological anticoagulation mechanisms, further exacerbating the pro-thrombotic state; aberrant expression of cell adhesion molecules and chemokines, which contribute to chronic inflammation and immune cell recruitment to the vessel wall; and an augmented generation of reactive oxygen species (ROS), leading to oxidative stress and cellular damage [17,74,75]. These functional aberrations play a crucial role in activating fibroblasts, the primary effector cells in fibrosis, mainly by promoting tissue hypoxia and chronic inflammation [17,74,75,76,77]. Vasospasm affecting arterioles and arteries, clinically manifested as Raynaud's phenomenon, further exacerbates fibroblast activation through ischemia-reperfusion injury, a process involving tissue damage caused by alternating periods of insufficient blood supply and reperfusion [78]. Fibroblasts themselves undergo phenotypic modulation, acquiring a profibrotic phenotype, and exhibit dysregulated responses to inflammatory signals. Notably, they demonstrate excessive ECM synthesis, contributing to the tissue fibrosis [72]. The cellular origins of myofibroblasts in SSc are diverse, encompassing not only resident tissue fibroblasts but also vascular wall-resident endothelial cells and pericytes, epithelial cells, adipocytes, and bone marrow-derived fibrocytes [72]. Myofibroblasts originating from this heterogeneous array of cellular sources collectively orchestrate the pathogenesis of the extensive fibrosis observed in SSc [72]. The preceding discussion elucidates the fundamental pathophysiology of SSc, highlighting a shared mechanistic basis that operates across various organs affected by fibrosis, and thus can be conceptualized as a foundational trans-organ pathophysiology [66,67].
2.6. Fibrosis in SSc
Fibroblast activation represents the ultimate consequence of the SSc-specific disease cascade. In affected organs, numerous α-smooth muscle actin (α-SMA)-positive myofibroblasts are present, which produce excessive quantities of ECM. These myofibroblasts originate from various sources, including resident fibroblasts, bone marrow-derived fibrocytes [79], epithelial-mesenchymal transition [80], endothelial-mesenchymal transition (EndoMT) [81,82], and adipocyte-myofibroblast transdifferentiation [83]. These cells employ both intrinsic and extrinsic mechanisms to sustain their constitutively activated state throughout disease progression.
A key growth factor implicated in the activation of SSc dermal fibroblasts is TGF-β, a potent inducer of ECM components, including fibrillar collagens that constitute the dermis (types I, III, and V). TGF-β expression in affected skin is elevated in patients with early active disease but is diminished or undetectable in those with established fibrosis. The expression profile of the three TGF-β isoforms in SSc is characterized by: (i) detection of all three isoforms within the ECM, and (ii) prominent expression of TGF-β1 and TGF-β2 around dermal vessels, associated with perivascular infiltrating mononuclear cells [84,85,86]. Consequently, in the early stages of SSc, TGF-β appears to promote inflammation by recruiting leukocytes through the modulation of cell adhesion molecules and the establishment of chemokine gradients, by activating leukocytes, and by inducing various proinflammatory cytokines and mediators. Conversely, in the sclerotic phase, SSc dermal fibroblasts exhibit constitutive activation with a profibrotic phenotype, resembling that of normal fibroblasts treated with TGF-β1, even when TGF-β expression is diminished or undetectable in the skin [87]. This suggests that SSc fibroblasts possess a self-activation system, at least partially mediated via autocrine TGF-β signaling. The increased expression of latent TGF-β receptors, including integrin αVβ3, integrin αVβ5, and thrombospondin-1, contributes to this process in SSc fibroblasts [88,89,90,91,92]. These receptors recruit and activate latent TGF-β on the cell surface, efficiently increasing the concentration of active TGF-β in the cellular microenvironment. Further expanding on the mechanisms of TGF-β-driven fibrosis, Meng et al. [93] identified ADAM19 as a significantly upregulated metalloproteinase in SSc skin fibrosis, demonstrating its role in promoting TGF-β-induced ECM deposition and fibroblast activation through the shedding of pro-fibrotic neuregulin-1 (NRG1), thereby contributing to the development of skin fibrosis in SSc. Thus, dermal fibroblasts are constitutively activated, at least through autocrine TGF-β signaling, in SSc lesional skin.
SSc dermal fibroblasts exhibit selective responsiveness to profibrotic stimuli from T cells. In normal dermal fibroblasts, collagen production is suppressed by Th1 cells via membrane-associated IFN-γ [94], and by Th2 cells via membrane-associated tumor necrosis factor-α (TNF-α) [95], which counter the profibrotic effect of IL-4. In contrast, in SSc dermal fibroblasts, increased collagen synthesis is resistant to Th1 and Th2 cell-mediated suppression, particularly to the latter [94,95]. A potential mechanism underlying this characteristic is the secretion of excessive amounts of progranulin, an intrinsic antagonist of TNF receptors, which renders SSc dermal fibroblasts resistant to the antifibrotic effect of TNF-α in an autocrine and paracrine manner [96]. Given the Th2-skewed immune polarization observed early in the course of dcSSc, unresponsiveness to Th2 cell-mediated suppression may contribute to fibroblast activation in the early stages of dcSSc. Adding to the complexity of fibroblast activation, Bergmann et al. [97] discovered a mutual amplification loop between GLI2/Hedgehog and JUN/AP-1 signaling pathways within SSc fibroblasts, where these pathways synergistically enhance each other's activity, resulting in sustained fibroblast activation and collagen production, highlighting a potential target for combined therapeutic interventions.
Conversely, SSc dermal fibroblasts influence the transdifferentiation of inflammatory cells. For instance, SSc dermal fibroblasts regulate the tissue-localized transdifferentiation of Tregs into Th2-like cells via IL-33 in affected skin [30,98]. Furthermore, SSc dermal fibroblasts suppress IFN-γ expression in skin-infiltrating CD4+ T cells through galectin-9 overproduction, promoting skin fibrosis development within the Th2/Th17-predominant microenvironment [99]. Thus, SSc dermal fibroblasts may influence the immune response in the skin more broadly than previously recognized.
Overall, SSc fibroblasts maintain their activated state through a self-activation system and feedforward loops involving interactions with other cell types, ultimately leading to irreversible fibrotic remodeling of multiple organs.
3. Verification of Pathogenesis Hypotheses: Insights from Recent Analytical Innovations
Pathogenesis hypotheses of SSc, as previously outlined, have been largely formulated based on conventional clinical and basic research methodologies, utilizing clinical samples from SSc patients (primarily lesional skin and lung biopsy tissues, and peripheral blood) and animal models. However, recent years have witnessed a transformative shift with the advent of cutting-edge analytical techniques, including single-cell RNA sequencing (scRNA-seq), spatial transcriptomics, imaging mass cytometry, and single-nucleus Assay for Transposase-Accessible Chromatin with sequencing (snATAC-seq), particularly in the context of lesional skin analysis. These advanced technologies have provided unprecedented opportunities for successive validation and refinement of the aforementioned pathogenesis hypotheses. Representative studies employing these innovative approaches are introduced below.
3.1. Scleroderma-Associated Fibroblast (ScAF) Identification via scRNA-seq Analysis
In a landmark study in 2022, Gur et al. [100] leveraged scRNA-seq to analyze skin tissues from a large cohort comprising 97 patients with SSc and 56 healthy individuals, subsequently publishing transformative findings. Their comprehensive analysis led to the refined classification of skin fibroblasts into 10 distinct cellular subpopulations. Notably, myofibroblasts, which have historically been considered central effector cells in the pathogenesis of SSc, were found to represent only a minor fraction, constituting approximately 1% of the total cellular population. Conversely, the most abundant fibroblast subpopulation was identified as LGR5 (Leucine-rich repeat-containing G-protein coupled Receptor 5)-positive fibroblasts, accounting for approximately 30% of cells in healthy individuals. Gene expression pattern analysis indicated that this LGR5+ fibroblast population plays a critical homeostatic role in maintaining normal skin architecture. However, a significant reduction in the abundance of this cell population was observed in SSc patients. Furthermore, these Scleroderma-Associated Fibroblasts (ScAFs) exhibited a constellation of molecular characteristics consistent with previously described SSc skin fibroblasts, including: 1, pathologically excessive ECM production coupled with suppressed degradation, 2, aberrant activation of Type I interferon signaling, TGF-β pathway, and IL-1 pathway, 3, dysregulation of Wnt signaling and IGF1 signaling, abnormal expression of CCN1 family proteins, 4, pathologically activated angiogenesis, increased vascular fragility, and enhanced coagulation and platelet aggregation, 5, diminished antioxidation and adipogenesis capacity, and 6, overexpression of CDKN2A (p16) and CDKN1A (p21), indicative of enhanced cellular senescence. Notably, the study also demonstrated a significant inverse correlation between the cellular density of this ScAF population and skin score. These compelling findings suggest that ScAFs, identified as the principal fibroblast subpopulation orchestrating the fibrotic pathology of SSc, may represent promising novel therapeutic targets. Conversely, the investigation also revealed a concomitant increase in vascular endothelial cells and pericytes alongside the progression of skin sclerosis. In particular, RGS5 (Regulator of G-protein Signaling 5)-positive vascular pericytes demonstrated a positive correlation with skin score. These observations may be interpreted as providing support for conventional pathogenesis hypotheses that posit vascular endothelial cells and pericytes as primary cellular origins of pathogenic fibroblasts in SSc.
3.2. Spatial Transcriptomics Analysis
Ma et al. [101] performed an in-depth analysis of SSc pathogenesis using single-cell and spatial transcriptomics. They analyzed scRNA-seq data from skin biopsies of 22 SSc patients and 18 healthy controls, alongside spatial RNA-seq data from 4 SSc patients, to map disease-associated cells and their interactions within SSc lesions. The study reported four key observations. First, fibroblasts, vascular endothelial cells, and pericytes were diffusely present in fibrotic areas of SSc skin. Second, fibroblasts were classified into seven distinct populations; SFRP2+ fibroblasts activated and differentiated into COL8A1+ fibroblasts (with myofibroblast features) during fibrosis progression. Third, vascular endothelial cells demonstrated heterogeneity, with categorization into seven distinct subpopulations, including arteriolar endothelial cells (EC2) and activated endothelial cells (EC5). EndoMT maturity served as a differentiating factor among these subpopulations, with EC2 identified as the dominant subpopulation within SSc lesions. Fourth, ligand-receptor network analysis indicated that fibroblast-vascular endothelial cell interactions were most pronounced, with EC2 and COL8A1+ myofibroblast-like fibroblasts as key communicators. These results reinforce existing pathogenesis models implicating vascular wall cells as myofibroblast origins in SSc.
3.3. Integrated Analysis of scRNA-seq and snATAC-seq Focusing on Vascular Endothelial Cells
In their 2024 study, Huang et al. [102] performed a comprehensive analysis using scRNA-seq and snATAC-seq data to investigate SSc vasculopathy. Analyzing skin biopsies from 27 SSc patients and 10 healthy controls via scRNA-seq, and snATAC-seq data from 8 SSc patients and 6 controls, they explored the role of transcription factors in SSc-associated vascular pathology. The scRNA-seq analysis of lesional SSc skin revealed two key observations: first, increased apoptosis and decreased cell numbers in arteriolar endothelial cells; and second, an elevation in tip and stalk cell populations, indicative of constitutively enhanced angiogenesis in dermal microvascular endothelial cells. These findings align with established pathogenesis hypotheses of SSc, particularly regarding destructive vasculopathy and angiogenic abnormalities. Furthermore, snATAC-seq analysis indicated increased chromatin accessibility at the ETS motif in SSc vascular endothelial cells, supporting the involvement of ETS transcription factors, especially FLI1, in SSc vasculopathy, consistent with prior research. These integrated analyses using scRNA-seq and snATAC-seq reinforce the conventional understanding of SSc pathogenesis by highlighting the roles of vascular endothelial cell apoptosis, dysregulated angiogenesis, and ETS transcription factors in the development of SSc vasculopathy.
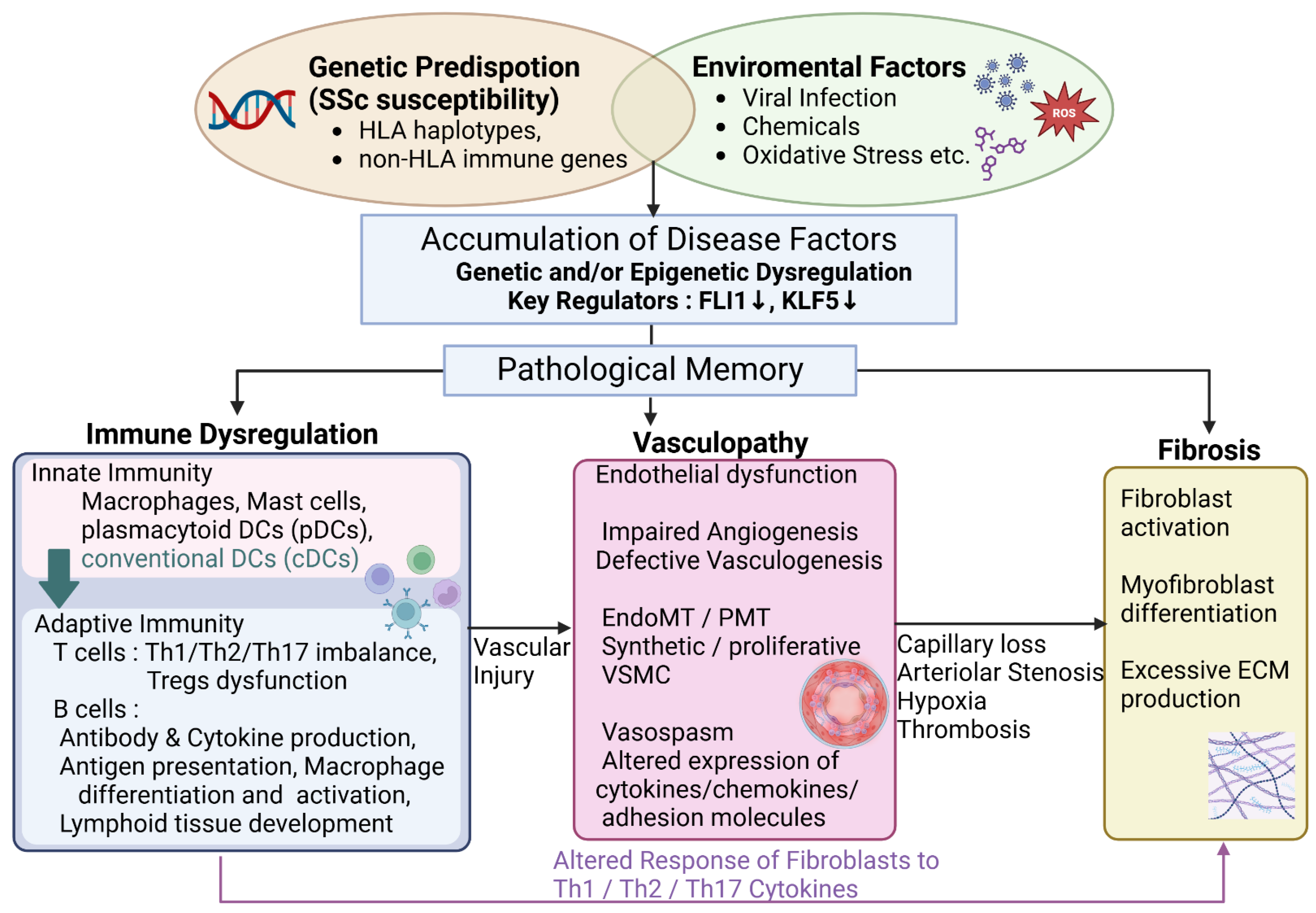
Figure 1.
Systemic sclerosis pathogenesis: An integrated pathway. This flowchart illustrates the multi-factorial pathogenesis of systemic sclerosis (SSc), highlighting the interplay between genetic predisposition, epigenetic dysregulation (particularly reduced FLI1 and KLF5 expression), immune dysregulation, vasculopathy, and fibrosis. Vascular dysfunction is depicted as a critical and early event in disease progression, ultimately leading to the diverse clinical manifestations of SScThis is a figure. Schemes follow the same formatting.
Figure 1.
Systemic sclerosis pathogenesis: An integrated pathway. This flowchart illustrates the multi-factorial pathogenesis of systemic sclerosis (SSc), highlighting the interplay between genetic predisposition, epigenetic dysregulation (particularly reduced FLI1 and KLF5 expression), immune dysregulation, vasculopathy, and fibrosis. Vascular dysfunction is depicted as a critical and early event in disease progression, ultimately leading to the diverse clinical manifestations of SScThis is a figure. Schemes follow the same formatting.
3.4. Vascular Niche Analysis by Spatial Proteomics Using Imaging Mass Cytometry
Rius Rigau et al. [103] employed imaging mass cytometry to conduct a vascular niche analysis in skin samples from 19 SSc patients and 14 healthy individuals. Their spatial proteomics approach identified seven subpopulations of vascular endothelial cells based on their unique protein expression profiles. In SSc patients, the researchers reported an increased population of CD34+;αSMA+;CD31+ cells alongside a reduction in vascular endothelial progenitor cells. The perivascular microenvironment of CD34+;αSMA+;CD31+ cells was characterized by a significant presence of immune cells, predominantly CD4+ T cells and myeloid cells, as well as myofibroblasts. Moreover, CD34+;αSMA+;CD31+ cells exhibited markers of EndoMT, such as SNAI1, SNAI2, TWIST, and ZEB1. The density of CD34+;αSMA+;CD31+ cells was found to correlate with the clinical progression of skin sclerosis. These observations reinforce the established pathogenesis model wherein vascular endothelial cells contribute to the myofibroblast population through EndoMT in SSc-related fibrosis.
3.5. Novel Pathogenesis Mechanism of SSc Skin Fibrosis Suggested by Epigenetic Analysis Using ATAC-seq
Liu et al. [104] conducted an epigenetic analysis to explore novel pathogenesis mechanisms in SSc skin fibrosis, employing ATAC-seq. Using flow cytometry, they isolated eight skin-resident cell types—fibroblasts, vascular endothelial cells, epidermal cells, CD4+ T cells, CD8+ T cells, dendritic cells, Langerhans cells, and macrophages—from healthy, lesional, and non-lesional SSc skin of 7 SSc patients and 6 healthy controls, totaling 19 samples. Leveraging the known enrichment of disease-susceptibility SNPs in non-coding regulatory DNA, they hypothesized that cell-type-specific chromatin accessibility analysis at SSc-associated SNP loci could pinpoint pathogenic cell types. Their analysis revealed significantly increased chromatin accessibility in SSc-associated SNP regions specifically within dendritic cells (DCs), compared to other skin cell types. Re-analysis of time-series RNA-seq data from SSc lesions further supported this, showing a strong positive correlation between the DC gene signature and skin fibrosis score. Specifically, conventional DCs (cDCs) were identified as a key cellular population. Immunohistochemical validation using ZBTB46, a cDC marker, confirmed significant infiltration of ZBTB46+ cells into SSc lesions. These epigenetic findings suggest a previously unappreciated role for cDCs in SSc skin fibrosis, offering a novel perspective on SSc pathogenesis beyond conventional hypotheses. The importance of dendritic cells in the pathogenesis of SSc is also demonstrated in the following articles [105,106]
3.6. Novel Therapeutic Targets Identified by Gene Expression Meta-Analysis of Lung Tissue
Yang et al.[107] published a gene expression meta-analysis of lung tissue, examining 38 patients with SSc-ILD and 18 healthy controls. Their analysis, utilizing three public datasets (GSE48149, GSE81292, GSE76808), identified the activation of epithelial-mesenchymal transition, cellular senescence, coagulation, and DNA repair pathways as characteristic changes in SSc-ILD lung tissue. Consistent with an aging phenotype, telomere length in type II alveolar epithelial cells from SSc-ILD lungs was found to be reduced, indicating enhanced cellular senescence. Current therapeutic development for SSc-ILD primarily targets myofibroblasts and inflammation/autoimmunity. However, this study suggests that cellular senescence and coagulation pathways could offer novel therapeutic avenues for SSc-ILD.
Plasminogen Activator Inhibitor-1 (PAI-1) is a serpin that inhibits tissue-type Plasminogen Activator (tPA) and urokinase-type Plasminogen Activator (uPA), and regulates the plasmin activation and the fibrinolytic system [108]. Emerging research underscores the significance of PAI-1 in regulating cellular senescence, with findings demonstrating that PAI-1 not only serves as a senescence marker but also actively mediates senescence pathways, impacting lifespan and age-related pathologies [109,110,111]. tPA, uPA, and PAI-1 are considered to play an important role in the maintenance of endothelial homeostasis, and are associated with the endothelial dysfunction of SSc [112,113]. Therefore, PAI-1 inhibition may be therapeutic for SSc-ILD.
4. Organ-Specific Pathophysiology Modifiers: Refining the Landscape of SSc Organ Involvement
To fully elucidate the organ-specific manifestations of SSc, it is essential to consider organ-specific pathophysiology modifiers, in addition to the broadly acting trans-organ basic pathophysiology. The following sections detail the principal organ-specific modifiers across major organ systems affected by SSc [13].
4.1. Cutaneous Pathology
Epidermal cells and adipocytes function as key pathophysiology modifiers in the skin in SSc (Figure 2).
4.1.1. Keratinocytes
Over the past three decades, numerous studies have demonstrated the upregulation of disease-associated molecules in the epidermis of skin involved by SSc. These molecules include endothelin-1, TGF-β, chemokine ligand 2 (CCL2), vascular endothelial growth factor (VEGF), IL-21 receptor, wound healing-associated cytokeratins (keratin 6 and keratin 16), IL-1α, connective tissue growth factor (CTGF), IL-6, TNF-α, CCL5, psoriasin, and galectin-7 [114,115,116,117,118,119,120,121,122,123]. Given the potent profibrotic effects of IL-1α, CTGF, and IL-6, SSc keratinocytes likely contribute to the activation of dermal fibroblasts.
A potential role for epithelial cells, including keratinocytes, stratified squamous epithelia of the esophagus, and medullary thymic epithelial cells, in SSc development has been proposed based on animal experiments using a novel SSc murine model [124]. Deficiency of the transcription factor Fli1, a potential SSc susceptibility factor [14], induces SSc-like properties in various cell types, including fibroblasts, endothelial cells, keratinocytes, T cells, B cells, cDC, and macrophages [17,124,125,126,127,128]. Notably, epithelial cell-specific Fli1 knockout mice (Fli1flox/flox; K14-Cre+/- mice), which exhibit SSc-like phenotypic features in epithelial cells, spontaneously develop dermal and esophageal fibrosis due to epithelial cell activation in the skin and esophagus. Furthermore, these mice develop ILD, mediated at least in part by T cells autoreactive to lung antigens, resulting from impaired negative selection and Treg development in the thymus. A component of this impaired central tolerance is attributed to the downregulation of autoimmune regulator (Aire), which modulates the processing and presentation of self-antigens in medullary thymic epithelial cells [129,130]. Importantly, epithelial cell-specific Fli1 knockout mice lacking acquired immune systems (Rag1-/-; Fli1flox/flox; K14-Cre+/- mice) spontaneously develop dermal and esophageal fibrosis, along with mast cell infiltration in the skin [131]. This suggests that epithelial cell activation alone can induce tissue fibrosis through the activation of innate immunity. This novel murine model indicates that abnormally activated epithelial cells underlie selective organ fibrosis and autoimmunity in SSc.
Another potential aspect of keratinocyte-dependent regulation of dermal fibrosis is the interplay between the immune system and the skin microbiota. This area of research has recently garnered significant attention regarding inflammatory skin diseases, such as atopic dermatitis [132] and SLE [133]. This interaction is initiated by keratinocytes sensing pathogen-associated molecular patterns derived from microorganisms via pattern recognition receptors. This is followed by the secretion of antimicrobial peptides (AMPs) from keratinocytes, ultimately leading to the death or inactivation of a diverse array of microorganisms and the activation of various immune and non-immune cells, including dermal fibroblasts and dermal microvascular endothelial cells [134]. While some AMPs are constitutively expressed, the expression of others is transient and regulated by components of the skin microbiota. To date, the contribution of skin microbiota to the altered phenotype of keratinocytes and skin immunity in SSc remains unclear, although microbiome dysbiosis has been reported in SSc-involved skin [135].
4.1.2. Adipocytes
The presence of adjacent and abundant adipose tissue is a distinctive histological characteristic of the skin. In recent decades, the potential contribution of subcutaneous adipose tissue to skin fibrosis in SSc has been increasingly recognized. As previously mentioned, myofibroblasts can originate from non-fibroblast lineage cells within the profibrotic microenvironment [136]. According to lineage-tracing studies [137], subcutaneous adipocytes are highly plastic cells capable of transdifferentiating into myofibroblasts [138]. Indeed, a significant proportion of activated myofibroblasts in SSc-involved skin appear to derive from adipocytes located adjacent to the deep dermis [83,139]. Furthermore, adipocytes produce a diverse array of adipokines, which exert pleiotropic effects on various cell types through autocrine, paracrine, and endocrine mechanisms [140]. An altered adipokine balance resulting from adipocyte loss or dysfunction may contribute to the inflammation, vasculopathy, and fibrosis that are characteristic of SSc [141,142,143,144,145,146,147,148,149,150,151]. Among the adipokine family, the role of adiponectin has been extensively investigated. Serum levels and tissue expression of adiponectin exhibit an inverse correlation with skin score in SSc patients [141,150,152]. AdipoP-/- mice develop attenuated dermal fibrosis in response to bleomycin administration [153], and AdipoRon, a pharmacological inhibitor of adiponectin signaling, mitigates the bleomycin-induced development of SSc-like skin fibrosis, vasculopathy, and immune abnormalities [154]. Collectively, subcutaneous adipose tissue appears to function as a critical driver of skin fibrosis in SSc.
4.2. Gastrointestinal Pathology
Gastrointestinal (GI) symptoms are prevalent in approximately 90% of patients with SSc and are a leading cause of morbidity. These symptoms are characterized by hypomotility, dysmotility, and impaired secretion of digestive enzymes throughout the GI tract, from the oral cavity to the anus [155,156,157]. The prevalence of upper and lower GI symptoms is estimated at 70–90% and 20–70%, respectively. The esophagus is the most frequently affected organ, followed by the anorectal region, small bowel, stomach, and colon. Esophageal dysfunction, including gastroesophageal reflux disease (GERD) and dysphagia, is the most common GI manifestation. A spectrum of lower GI symptoms can also occur, such as small intestinal bacterial overgrowth, malabsorption, malnutrition, diarrhea, pseudo-obstruction, constipation, pneumatosis intestinalis, and fecal incontinence [158].
Consistent with observations in the skin and other internal organs, the common SSc-specific pathological cascade broadly impacts the GI system, ultimately leading to extensive atrophy and fibrosis of the gastrointestinal smooth muscle [159]. Additionally, SSc exhibits a GI organ-specific pathology relevant to the complex and highly organized enteric nervous system. Vascular structural changes, such as capillary rarefaction and arteriolar stenosis, induce tissue hypoxia throughout the GI tract, resulting in autonomic axonal degeneration [160]. Thus, SSc-associated GI involvement is attributed to hypomotility and dysmotility stemming from extensive atrophy and fibrosis of the enteric smooth muscle, as well as disturbances in the enteric nervous system. Indeed, SSc-associated esophageal dysfunction comprises three pathological components: (i) reduced lower esophageal sphincter pressure, (ii) ineffective esophageal body peristalsis, particularly in the lower esophagus, and (iii) discoordination of peristaltic and lower esophageal sphincter function [155,159,161,162,163]. Importantly, esophageal dysfunction often manifests early in the SSc disease course, even in very early cases without skin sclerosis, reflecting enteric nervous system impairment secondary to vasculopathy. Conversely, esophageal dysmotility progresses rapidly (within years) in dcSSc, whereas it progresses more slowly (within decades) in limited cutaneous SSc (lcSSc) [164,165]. This temporal difference reflects the atrophic and fibrotic changes in the gastrointestinal smooth muscle as a late consequence of SSc-associated GI involvement.
A subset of SSc patients harbor antibodies targeting myenteric neurons [166], including antibodies against the muscarinic acetylcholine receptor M3 [167,168]. These antibodies interact with myenteric neurons and disrupt intestinal peristalsis in animal models [166,167]. Furthermore, SSc patients with higher titers of anti-muscarinic acetylcholine receptor M3 antibodies exhibit more severe GI phenotypes [169], suggesting a pathogenic role for these antibodies in humans. Consistent with this, the motility of the pharynx and proximal esophagus, which is regulated by the somatic nervous system, remains normal in SSc [169,170]. Overall, current evidence supports the concept that a combination of autoimmunity, vasculopathy, and fibrosis underlies GI involvement in SSc.
As discussed previously regarding the skin, stratified squamous epithelia can act as a potential driver of esophageal fibrosis. The esophagus of Fli1flox/flox; K14-Cre+/- mice exhibits molecular features characteristic of the SSc esophagus, such as increased IL-8 expression, and prominently expresses IL-1β in its stratified squamous epithelia. Importantly, it is suggested that esophageal epithelium-derived inflammatory cytokines contribute to GERD-related and functional heartburn in patients who continue to experience heartburn despite receiving adequate-dose proton pump inhibitor therapy [171]. Indeed, several studies have failed to demonstrate any correlation between GI symptoms and the severity of GI physiological changes in SSc patients [155,172]. Although further research is warranted, the altered phenotype of esophageal epithelium may be implicated in the development of esophageal fibrosis and heartburn in SSc patients.
Another significant component of GI-specific pathology is the gut microbiota, which influences the development and function of the immune system and appears to play a role in autoimmune diseases through microbiota-related immune dysfunction [173,174,175]. In SSc, multiple cohorts have demonstrated that intestinal microbiota composition differs from that of healthy individuals, notably showing decreased levels of commensal bacteria, such as Faecalibacterium, Clostridium, and Bacteroides, and increased levels of pathobiont bacteria, such as Fusobacterium and γ-Proteobacteria [176,177]. Currently, it remains unclear whether microbiota alterations precipitate and perpetuate the SSc-associated immune system dysregulation or are a consequence of SSc itself and/or related therapies. Further basic and clinical investigations are needed to elucidate the mechanisms by which gut microbiota interact with key inflammatory and fibrotic mediators underlying the development of SSc-specific clinical symptoms.
In conclusion, the abnormally activated stratified squamous epithelia and enteric nervous system dysfunction constitute organ-specific pathological processes within the GI tract in SSc (Figure 3).
4.3. Pulmonary Pathology
Pulmonary involvement, encompassing ILD and pulmonary hypertension (PH), represents the primary cause of SSc-related mortality [178,179]. ILD in SSc arises from the common SSc-specific pathological cascade and can be further influenced by microaspiration of gastric contents due to GERD. Pulmonary arterial hypertension (PAH) in SSc is attributed to pulmonary arteriolar stenosis resulting from occlusive vascular fibrosis. In contrast, other forms of SSc-PH are linked to cardiac involvement and ILD. These distinct pathologies can indeed coexist to varying extents, collectively contributing to elevated pulmonary arterial pressure in SSc-PH patients.
The World Health Organization (WHO) classification categorizes PH into five groups: group 1, PAH, characterized by pulmonary arteriole abnormalities; group 2, PH associated with left heart disease; group 3, PH associated with lung disease or hypoxia; group 4, PH resulting from pulmonary embolism or thrombosis, also known as chronic thromboembolic pulmonary hypertension (CTEPH); and group 5, PH due to miscellaneous causes not classified in the other groups. SSc-PH can fall into groups 1, 2, or 3, and SSc patients with antiphospholipid antibody syndrome may develop group 4 PH. SSc-PAH is more prevalent in patients with lcSSc and those with anticentromere antibodies (ACA) [180]. Histologically, SSc-PAH is defined by fibrotic occlusion of pulmonary arterioles, a consequence of the shared SSc-specific pathological cascade. SSc-PAH often coexists with histological changes in pulmonary venules reminiscent of pulmonary veno-occlusive disease (PVOD) [181,182].
ILD is detectable in 50–60% of SSc patients via high-resolution computed tomography (HRCT) [183,184]. Risk factors for the development of SSc-ILD encompass dcSSc [185], African American ethnicity [186], shorter disease duration [187], older age at disease onset [185], and the presence of anti-topoisomerase I antibody and/or absence of ACA [185]. ILD typically manifests early in the course of dcSSc, particularly within the first 3 years of disease onset [185,187,188], while in lcSSc patients, ILD can arise at any point during the disease course [189]. The clinical trajectory of SSc-ILD is heterogeneous; some patients maintain stable forced vital capacity (FVC), whereas others experience a progressive decline in pulmonary function [190]. ILD progression is generally most pronounced within the initial 4 years following SSc onset, subsequently slowing or ceasing entirely, even without therapeutic intervention [191]. Severe ILD, defined by an FVC decline below 50%, is reported to affect approximately 15% of the total SSc population [191,192]. The predominant histological pattern in SSc-ILD is nonspecific interstitial pneumonia (NSIP), observed in roughly two-thirds of patients [193]. Usual interstitial pneumonia (UIP) is present in a smaller proportion of SSc-ILD cases [193,194,195] and may correlate with less favorable prognoses [196].
Histologically, nonspecific interstitial pneumonia (NSIP) in SSc-ILD is categorized into four stages [197]:
* Stage 1 (Initial): Characterized by microvessel overdevelopment with structural abnormalities and alveolar septal thickening with numerous α-SMA-positive myofibroblasts. The overdeveloped microvessels contain blood cells within their lumina, indicating maintained functional circulation.
* Stage 2 (Progressive ECM Deposition): Marked by substantial and progressive ECM deposition, irregular and indistinct alveolar septal borders, further structural disorganization of microvessels, and obliteration of larger blood vessels. Disarray or partial loss of the alveolar epithelium is also evident.
* Stage 3 (Extensive Fibrosis): Progression of fibrosis extensively damages vital lung structures, including alveoli and vasculature.
* Stage 4 (Final): The lung transforms into a contracted fibrous organ devoid of alveoli and vasculature.
The early microvascular alterations and subsequent progressive fibrotic changes reinforce the concept that SSc-ILD is driven by the common SSc-specific pathological cascade, similar to other organ involvements.
Above pathological findings suggest the presence of foundational trans-organ pathophysiology in the lung tissue.
Microaspiration of gastric contents due to GERD is a potential factor driving the progression of SSc-ILD. Clinical data, histological analyses, and animal studies support this hypothesis. Several clinical studies have shown a positive correlation between increased lung fibrosis severity and more frequent reflux episodes, as well as greater proximal extension of refluxate [198]. In a rat GERD model, pulmonary parenchymal fibrosis was induced by introducing gastric content into the lungs [199]. Analysis of lung biopsy specimens identified a distinct histological pattern of lung disease, centrilobular fibrosis (CLF), particularly prevalent in SSc patients with severe GERD [200]. CLF is characterized by a predominantly bronchocentric distribution of lesions and the presence of intraluminal basophilic material and foreign bodies within the bronchi, sometimes accompanied by multinucleated giant cell reactions. In a prior study examining open lung biopsies from 22 SSc-ILD patients [201], isolated CLF was observed in 21% of cases, and a CLF pattern was present in 84% of patients with a predominant NSIP pattern, suggesting that GERD may exacerbate underlying NSIP in SSc-ILD. Although clinical trial data have not yet demonstrated pulmonary function improvement in SSc-ILD following aggressive GERD management, aggressive GERD treatment may still benefit the majority of SSc patients. Overall, GERD acts as an organ-specific disease modifier in SSc-ILD. (Figure 4)
4.4. Cardiovascular Pathology
Histological examination of autopsy tissues from SSc patients without prior clinical cardiac symptoms reveals evidence of myocardial disease in all cases [202]. Thus, cardiac involvement is nearly ubiquitous in SSc patients, although often clinically silent [203,204]. Once clinically manifest, however, cardiac involvement carries a poor prognosis [203,205,206,207]. Primary cardiac involvement in SSc encompasses a wide spectrum of clinical manifestations, including arrhythmias, conduction system defects, myocarditis, pericarditis, systolic and diastolic ventricular dysfunction, and heart failure [208,209]. Primary myocardial involvement is estimated to account for approximately 30% of deaths in SSc patients [179,207,210].
In a study employing cardiovascular magnetic resonance (CMR) parametric mapping in SSc patients, Purevsuren et al. [211] demonstrated that native T1 mapping effectively detects early myocardial changes and correlates with left ventricular diastolic dysfunction, with more pronounced myocardial involvement observed in dcSSc compared to lcSSc. This finding of early diffuse myocardial edema-like lesions on contrast-enhanced MRI mirrors the edematous induration seen in early skin lesions of SSc, suggesting a parallel pathological process affecting both skin and internal organs in the initial stages of the disease.
Although the precise molecular mechanisms of SSc-related cardiomyopathy remain incompletely understood [208,212,213,214], prevailing consensus attributes a central role to microvascular disease. Specifically, vascular structural abnormalities, such as capillary rarefaction and arteriolar stenosis, are thought to induce tissue hypoxia, subsequently promoting inflammation and excessive ECM production by cardiac fibroblasts [203]. Indeed, histological evaluation of myocardial specimens from SSc autopsy cases has demonstrated inflammation, vascular alterations, and ECM deposition to a greater extent than in specimens from age- and sex-matched control autopsy subjects [202]. Supporting the crucial role of the microvasculature, rather than coronary arteries, fibrotic changes in the myocardium are frequently patchy and distributed across both ventricles, independent of coronary artery perfusion territories [212,215].
Abnormal vasoreactivity of small arteries and arterioles, termed “myocardial Raynaud’s phenomenon,” contributes to SSc-associated cardiac involvement. This concept is supported by evidence indicating that the absence of prior calcium channel blocker therapy is an independent factor associated with left ventricular dysfunction. Furthermore, vasodilators, such as nifedipine, nicardipine, and captopril, acutely mitigate both myocardial perfusion and function [216,217,218,219], possibly through the reduction of myocardial vascular resistance. Importantly, approximately 30% of SSc patients with a history of Raynaud’s phenomenon exhibit myocardial Raynaud’s phenomenon upon exposure to cold air, an effect that is inhibited by calcium channel blocker administration [220].
Overall, from a management perspective, it is critical to recognize that two distinct categories of vascular changes—structural (capillary rarefaction and arteriolar stenosis) and functional (myocardial Raynaud’s phenomenon)—contribute to cardiac involvement in SSc.
5. Conclusions
This review has described current models of SSc pathogenesis and their corroboration using recent analytical innovations. The skin, being the most frequently involved organ in SSc, allows for less invasive tissue sampling, rendering it a primary target for pathogenesis studies. The future application of advanced analytical methods to internal organs, including the lungs, promises to significantly refine our comprehension of both the foundational trans-organ pathophysiology and organ-specific modifiers in SSc. This deeper understanding is anticipated to promote the creation of more effective and individualized treatment strategies, ultimately improving disease management and prognosis for patients with SSc and revealing novel therapeutic perspectives.
Author Contributions
Conceptualization, T.T. and Y.A.; Writing – Original Draft Preparation, T.T.; Writing – Review & Editing, T.T. and Y.A.; Supervision, Y.A. All authors have read and agreed to the published version of the manuscript.
Funding
None.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
None.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Denton, C.P.; Khanna, D. Systemic Sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Arnett, F.C.; Cho, M.; Chatterjee, S.; Aguilar, M.B.; Reveille, J.D.; Mayes, M.D. Familial Occurrence Frequencies and Relative Risks for Systemic Sclerosis (Scleroderma) in Three United States Cohorts. Arthritis Rheum. 2001, 44, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Feghali-Bostwick, C.; Medsger, T.A., Jr; Wright, T.M. Analysis of Systemic Sclerosis in Twins Reveals Low Concordance for Disease and High Concordance for the Presence of Antinuclear Antibodies. Arthritis Rheum. 2003, 48, 1956–1963. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Kawasaki, A.; Matsushita, T.; Furukawa, H.; Kondo, Y.; Okiyama, N.; Nagaoka, S.; Shimada, K.; Sugii, S.; Katayama, M.; et al. Association of Functional (GA)n Microsatellite Polymorphism in the FLI1 Gene with Susceptibility to Human Systemic Sclerosis. Rheumatology (Oxford) 2020, 59, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K. The Genetics of Systemic Sclerosis. Discov. Med. 2010, 10, 134–143. [Google Scholar]
- Dieudé, P.; Guedj, M.; Wipff, J.; Avouac, J.; Fajardy, I.; Diot, E.; Granel, B.; Sibilia, J.; Cabane, J.; Mouthon, L.; et al. Association between the IRF5 Rs2004640 Functional Polymorphism and Systemic Sclerosis: A New Perspective for Pulmonary Fibrosis. Arthritis Rheum. 2009, 60, 225–233. [Google Scholar] [CrossRef]
- Ito, I.; Kawaguchi, Y.; Kawasaki, A.; Hasegawa, M.; Ohashi, J.; Hikami, K.; Kawamoto, M.; Fujimoto, M.; Takehara, K.; Sato, S.; et al. Association of a Functional Polymorphism in the IRF5 Region with Systemic Sclerosis in a Japanese Population. Arthritis Rheum. 2009, 60, 1845–1850. [Google Scholar] [CrossRef]
- Radstake, T.R.D.J.; Gorlova, O.; Rueda, B.; Martin, J.-E.; Alizadeh, B.Z.; Palomino-Morales, R.; Coenen, M.J.; Vonk, M.C.; Voskuyl, A.E.; Schuerwegh, A.J.; et al. Genome-Wide Association Study of Systemic Sclerosis Identifies CD247 as a New Susceptibility Locus. Nat. Genet. 2010, 42, 426–429. [Google Scholar] [CrossRef]
- Tang, L.; Chen, B.; Ma, B.; Nie, S. Association between IRF5 Polymorphisms and Autoimmune Diseases: A Meta-Analysis. Genet Mol Res 2014, 13, 4473–4485. [Google Scholar] [CrossRef]
- Sharif, R.; Mayes, M.D.; Tan, F.K.; Gorlova, O.Y.; Hummers, L.K.; Shah, A.A.; Furst, D.E.; Khanna, D.; Martin, J.; Bossini-Castillo, L.; et al. IRF5 Polymorphism Predicts Prognosis in Patients with Systemic Sclerosis. Ann. Rheum. Dis. 2012, 71, 1197–1202. [Google Scholar] [CrossRef]
- Benfaremo, D.; Svegliati Baroni, S.; Manfredi, L.; Moroncini, G.; Gabrielli, A. Putative Functional Pathogenic Autoantibodies in Systemic Sclerosis. Eur. J. Rheumatol. 2020, 7, S181–S186. [Google Scholar] [CrossRef] [PubMed]
- Senécal, J.-L.; Hoa, S.; Yang, R.; Koenig, M. Pathogenic Roles of Autoantibodies in Systemic Sclerosis: Current Understandings in Pathogenesis. J. Scleroderma Relat. Disord. 2020, 5, 103–129. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y. The Pathogenesis of Systemic Sclerosis: An Understanding Based on a Common Pathologic Cascade across Multiple Organs and Additional Organ-Specific Pathologies. J. Clin. Med. 2020, 9, 2687. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, P.-S.; Kahaleh, B. Association between Enhanced Type I Collagen Expression and Epigenetic Repression of the FLI1 Gene in Scleroderma Fibroblasts. Arthritis Rheum. 2006, 54, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Czuwara-Ladykowska, J.; Moussa, O.; Markiewicz, M.; Smith, E.; Silver, R.M.; Jablonska, S.; Blaszczyk, M.; Watson, D.K.; Trojanowska, M. Persistent Down-Regulation of Fli1, a Suppressor of Collagen Transcription, in Fibrotic Scleroderma Skin. Am. J. Pathol. 2003, 163, 571–581. [Google Scholar] [CrossRef]
- Noda, S.; Asano, Y.; Nishimura, S.; Taniguchi, T.; Fujiu, K.; Manabe, I.; Nakamura, K.; Yamashita, T.; Saigusa, R.; Akamata, K.; et al. Simultaneous Downregulation of KLF5 and Fli1 Is a Key Feature Underlying Systemic Sclerosis. Nat. Commun. 2014, 5, 5797. [Google Scholar] [CrossRef]
- Taniguchi, T.; Asano, Y.; Akamata, K.; Noda, S.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Trojanowska, M.; Sato, S. Fibrosis, Vascular Activation, and Immune Abnormalities Resembling Systemic Sclerosis in Bleomycin-Treated Fli-1-Haploinsufficient Mice: SSc Phenotype Induction by Bleomycin in Fli-1+/−Mice. Arthritis Rheumatol. 2015, 67, 517–526. [Google Scholar] [CrossRef]
- Takeda, N.; Manabe, I.; Uchino, Y.; Eguchi, K.; Matsumoto, S.; Nishimura, S.; Shindo, T.; Sano, M.; Otsu, K.; Snider, P.; et al. Cardiac Fibroblasts Are Essential for the Adaptive Response of the Murine Heart to Pressure Overload. J Clin Invest. 1 2010, 120, 254–265. [Google Scholar] [CrossRef]
- Fujiu, K.; Manabe, I.; Nagai, R. Renal Collecting Duct Epithelial Cells Regulate Inflammation in Tubulointerstitial Damage in Mice. J. Clin. Invest. 2011, 121, 3425–3441. [Google Scholar] [CrossRef]
- Whitfield, M.L.; Finlay, D.R.; Murray, J.I.; Troyanskaya, O.G.; Chi, J.-T.; Pergamenschikov, A.; McCalmont, T.H.; Brown, P.O.; Botstein, D.; Connolly, M.K. Systemic and Cell Type-Specific Gene Expression Patterns in Scleroderma Skin. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12319–12324. [Google Scholar] [CrossRef]
- Yu, J.; Tang, R.; Ding, K. Epigenetic Modifications in the Pathogenesis of Systemic Sclerosis. Int. J. Gen. Med. 2022, 15, 3155–3166. [Google Scholar] [CrossRef] [PubMed]
- Riemekasten, G.; Distler, J.H.W. A Broad Look into the Future of Systemic Sclerosis. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X221109404. [Google Scholar] [CrossRef]
- Angiolilli, C.; Marut, W.; van der Kroef, M.; Chouri, E.; Reedquist, K.A.; Radstake, T.R.D.J. New Insights into the Genetics and Epigenetics of Systemic Sclerosis. Nat. Rev. Rheumatol. 2018, 14, 657–673. [Google Scholar] [CrossRef]
- Altorok, N.; Kahaleh, B. Epigenetics and Systemic Sclerosis. Semin. Immunopathol. 2015, 37, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Lu, Q. The Critical Importance of Epigenetics in Autoimmune-Related Skin Diseases. Front. Med. 2023, 17, 43–57. [Google Scholar] [CrossRef]
- Truchetet, M.E.; Brembilla, N.C.; Chizzolini, C. Current Concepts on the Pathogenesis of Systemic Sclerosis. Clin. Rev. Allergy Immunol. 2023, 64, 262–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Maskan Bermudez, N.; Sa, B.; Maderal, A.D.; Jimenez, J.J. Epigenetic Mechanisms Driving the Pathogenesis of Systemic Lupus Erythematosus, Systemic Sclerosis and Dermatomyositis. Exp. Dermatol. 2024, 33, e14986. [Google Scholar] [CrossRef]
- Higashi-Kuwata, N.; Jinnin, M.; Makino, T.; Fukushima, S.; Inoue, Y.; Muchemwa, F.C.; Yonemura, Y.; Komohara, Y.; Takeya, M.; Mitsuya, H.; et al. Characterization of Monocyte/Macrophage Subsets in the Skin and Peripheral Blood Derived from Patients with Systemic Sclerosis. Arthritis Res. Ther. 2010, 12, R128. [Google Scholar] [CrossRef]
- Yukawa, S.; Yamaoka, K.; Sawamukai, N.; Shimajiri, S.; Kubo, S.; Miyagawa, I.; Sonomoto, K.; Saito, K.; Tanaka, Y. Dermal Mast Cell Density in Fingers Reflects Severity of Skin Sclerosis in Systemic Sclerosis. Mod. Rheumatol. 2013, 23, 1151–1157. [Google Scholar] [CrossRef]
- MacDonald, K.G.; Dawson, N.A.J.; Huang, Q.; Dunne, J.V.; Levings, M.K.; Broady, R. Regulatory T Cells Produce Profibrotic Cytokines in the Skin of Patients with Systemic Sclerosis. J. Allergy Clin. Immunol. 2015, 135, 946–955.e9. [Google Scholar] [CrossRef]
- Bosello, S.; Angelucci, C.; Lama, G.; Alivernini, S.; Proietti, G.; Tolusso, B.; Sica, G.; Gremese, E.; Ferraccioli, G. Characterization of Inflammatory Cell Infiltrate of Scleroderma Skin: B Cells and Skin Score Progression. Arthritis Res. Ther. 2018, 20, 75. [Google Scholar] [CrossRef] [PubMed]
- Lafyatis, R.; O’Hara, C.; Feghali-Bostwick, C.A.; Matteson, E. B Cell Infiltration in Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheum. 2007, 56, 3167–3168. [Google Scholar] [CrossRef]
- Matsushita, T.; Hasegawa, M.; Hamaguchi, Y.; Takehara, K.; Sato, S. Longitudinal Analysis of Serum Cytokine Concentrations in Systemic Sclerosis: Association of Interleukin 12 Elevation with Spontaneous Regression of Skin Sclerosis. The Journal of rheumatology 2006, 33, 275–284. [Google Scholar]
- Yang, X.; Yang, J.; Xing, X.; Wan, L.; Li, M. Increased Frequency of Th17 Cells in Systemic Sclerosis Is Related to Disease Activity and Collagen Overproduction. Arthritis Res. Ther. 2014, 16, R4. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Fujimoto, M.; Matsushita, T.; Hamaguchi, Y.; Hasegawa, M.; Takehara, K.; Komura, K.; Sato, S. Clinical Association of Serum Interleukin-17 Levels in Systemic Sclerosis: Is Systemic Sclerosis a Th17 Disease? J. Dermatol. Sci. 2008, 50, 240–242. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, W.; Xu, K.; Han, D.; Jiang, C.; Mou, K.; Li, Y.; Meng, L.; Lu, S. The Elevated Expression of Th17-Related Cytokines and Receptors Is Associated with Skin Lesion Severity in Early Systemic Sclerosis. Hum. Immunol. 2015, 76, 22–29. [Google Scholar] [CrossRef]
- Frantz, C.; Auffray, C.; Avouac, J.; Allanore, Y. Regulatory T Cells in Systemic Sclerosis. Front. Immunol. 2018, 9, 2356. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hasegawa, M.; Takehara, K. Serum Levels of Interleukin-6 and Interleukin-10 Correlate with Total Skin Thickness Score in Patients with Systemic Sclerosis. J. Dermatol. Sci. 2001, 27, 140–146. [Google Scholar] [CrossRef]
- Nakashima, T.; Jinnin, M.; Yamane, K.; Honda, N.; Kajihara, I.; Makino, T.; Masuguchi, S.; Fukushima, S.; Okamoto, Y.; Hasegawa, M.; et al. Impaired IL-17 Signaling Pathway Contributes to the Increased Collagen Expression in Scleroderma Fibroblasts. J. Immunol. 2012, 188, 3573–3583. [Google Scholar] [CrossRef]
- Yoshizaki, A. B Lymphocytes in Systemic Sclerosis: Abnormalities and Therapeutic Targets. J. Dermatol. 2016, 43, 39–45. [Google Scholar] [CrossRef]
- Sato, S.; Hasegawa, M.; Fujimoto, M.; Tedder, T.F.; Takehara, K. Quantitative Genetic Variation in CD19 Expression Correlates with Autoimmunity. J. Immunol. 2000, 165, 6635–6643. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Fujimoto, M.; Hasegawa, M.; Takehara, K. Altered Blood B Lymphocyte Homeostasis in Systemic Sclerosis: Expanded Naive B Cells and Diminished but Activated Memory B Cells. Arthritis Rheum. 2004, 50, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Distler, J.H.W.; Maurer, B.; Huscher, D.; van Laar, J.M.; Allanore, Y.; Distler, O. ; EUSTAR Rituximab study group Effects and Safety of Rituximab in Systemic Sclerosis: An Analysis from the European Scleroderma Trial and Research (EUSTAR) Group. Ann. Rheum. Dis. 2015, 74, 1188–1194. [Google Scholar] [CrossRef]
- Daoussis, D.; Melissaropoulos, K.; Sakellaropoulos, G.; Antonopoulos, I.; Markatseli, T.E.; Simopoulou, T.; Georgiou, P.; Andonopoulos, A.P.; Drosos, A.A.; Sakkas, L.; et al. A Multicenter, Open-Label, Comparative Study of B-Cell Depletion Therapy with Rituximab for Systemic Sclerosis-Associated Interstitial Lung Disease. Semin. Arthritis Rheum. 2017, 46, 625–631. [Google Scholar] [CrossRef]
- Bosello, S.L.; De Luca, G.; Rucco, M.; Berardi, G.; Falcione, M.; Danza, F.M.; Pirronti, T.; Ferraccioli, G. Long-Term Efficacy of B Cell Depletion Therapy on Lung and Skin Involvement in Diffuse Systemic Sclerosis. Semin. Arthritis Rheum. 2015, 44, 428–436. [Google Scholar] [CrossRef]
- Ebata, S.; Yoshizaki, A.; Fukasawa, T.; Miura, S.; Takahashi, T.; Sumida, H.; Asano, Y.; Sato, S. Rituximab Therapy Is More Effective than Cyclophosphamide Therapy for Japanese Patients with Anti-Topoisomerase I-Positive Systemic Sclerosis-Associated Interstitial Lung Disease. J. Dermatol. 2019, 46, 1006–1013. [Google Scholar] [CrossRef]
- Daoussis, D.; Antonopoulos, I.; Liossis, S.-N.C.; Yiannopoulos, G.; Andonopoulos, A.P. Treatment of Systemic Sclerosis-Associated Calcinosis: A Case Report of Rituximab-Induced Regression of CREST-Related Calcinosis and Review of the Literature. Semin. Arthritis Rheum. 2012, 41, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Khor, C.-G.; Chen, X.L.-F.; Lin, T.-S.; Lu, C.-H.; Hsieh, S.-C. Rituximab for Refractory Digital Infarcts and Ulcers in Systemic Sclerosis. Clin. Rheumatol. 2014, 33, 1019–1020. [Google Scholar] [CrossRef]
- Maslyanskiy, A.; Lapin, S.; Kolesova, E.; Peñín, I.; Cheshuina, M.D.; Feist, E.; Konradi, A. Effects of Rituximab Therapy on Elastic Properties of Vascular Wall in Patients with Progressive Systemic Sclerosis. Clin. Exp. Rheumatol. 2014, 32, S–228. [Google Scholar]
- Hügle, T.; Hogan, V.; White, K.E.; van Laar, J.M. Mast Cells Are a Source of Transforming Growth Factor β in Systemic Sclerosis. Arthritis Rheum. 2011, 63, 795–799. [Google Scholar] [CrossRef]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and Efficacy of Subcutaneous Tocilizumab in Adults with Systemic Sclerosis (FaSScinate): A Phase 2, Randomised, Controlled Trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Akamata, K.; Asano, Y.; Taniguchi, T.; Yamashita, T.; Saigusa, R.; Nakamura, K.; Noda, S.; Aozasa, N.; Toyama, T.; Takahashi, T.; et al. Increased Expression of Chemerin in Endothelial Cells Due to Fli1 Deficiency May Contribute to the Development of Digital Ulcers in Systemic Sclerosis. Rheumatology (Oxford) 2015, 54, 1308–1316. [Google Scholar] [CrossRef]
- Duan, H.; Fleming, J.; Pritchard, D.K.; Amon, L.M.; Xue, J.; Arnett, H.A.; Chen, G.; Breen, P.; Buckner, J.H.; Molitor, J.A.; et al. Combined Analysis of Monocyte and Lymphocyte Messenger RNA Expression with Serum Protein Profiles in Patients with Scleroderma. Arthritis Rheum. 2008, 58, 1465–1474. [Google Scholar] [CrossRef]
- Takahashi, T.; Asano, Y.; Nakamura, K.; Yamashita, T.; Saigusa, R.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Yoshizaki, A.; Tamaki, Z.; et al. A Potential Contribution of Antimicrobial Peptide LL-37 to Tissue Fibrosis and Vasculopathy in Systemic Sclerosis. Br. J. Dermatol. 2016, 175, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Peck, A.; Santer, D.; Patole, P.; Schwartz, S.M.; Molitor, J.A.; Arnett, F.C.; Elkon, K.B. Induction of Interferon-Alpha by Scleroderma Sera Containing Autoantibodies to Topoisomerase I: Association of Higher Interferon-Alpha Activity with Lung Fibrosis. Arthritis Rheum. 2008, 58, 2163–2173. [Google Scholar] [CrossRef]
- Black, C.M.; Silman, A.J.; Herrick, A.I.; Denton, C.P.; Wilson, H.; Newman, J.; Pompon, L.; Shi-Wen, X. Interferon-Alpha Does Not Improve Outcome at One Year in Patients with Diffuse Cutaneous Scleroderma: Results of a Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheum. 1999, 42, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Hügle, T.; Gratzl, S.; Daikeler, T.; Frey, D.; Tyndall, A.; Walker, U.A. Sclerosing Skin Disorders in Association with Multiple Sclerosis. Coincidence, Underlying Autoimmune Pathology or Interferon Induced? Ann. Rheum. Dis. 2009, 68, 47–50. [Google Scholar] [CrossRef]
- Pelidou, S.-H.; Tsifetaki, N.; Giannopoulos, S.; Deretzi, G.; Voulgari, P.; Kyritsis, A. Multiple Sclerosis Associated with Systemic Sclerosis. Rheumatol. Int. 2007, 27, 771–773. [Google Scholar] [CrossRef]
- Spadaro, A.; Sensi, F.; Barrella, M.; Francia, A. Systemic Sclerosis and Multiple Sclerosis. J. Neurol. 1999, 246, 497–499. [Google Scholar] [CrossRef]
- Tahara, H.; Kojima, A.; Hirokawa, T.; Oyama, T.; Naganuma, A.; Maruta, S.; Okada, K.; Ban, S.; Yoshida, K.; Takagi, H.; et al. Systemic Sclerosis after Interferon Alphacon-1 Therapy for Hepatitis C. Intern. Med. 2007, 46, 473–476. [Google Scholar] [CrossRef]
- Esteban, I.; Vilardell, M. R. Solans, J.A. Bosch. Clin. Exp. Rheumatol. 2004, 22, 625–628. [Google Scholar]
- Beretta, L.; Caronni, M.; Vanoli, M.; Scorza, R. Systemic Sclerosis after Interferon-Alfa Therapy for Myeloproliferative Disorders. Br. J. Dermatol. 2002, 147, 385–386. [Google Scholar] [CrossRef]
- Pammer, J.; Reinisch, C.; Birner, P.; Pogoda, K.; Sturzl, M.; Tschachler, E. Interferon-Alpha Prevents Apoptosis of Endothelial Cells after Short-Term Exposure but Induces Replicative Senescence after Continuous Stimulation. Lab. Invest. 2006, 86, 997–1007. [Google Scholar] [CrossRef]
- Ah Kioon, M.D.; Tripodo, C.; Fernandez, D.; Kirou, K.A.; Spiera, R.F.; Crow, M.K.; Gordon, J.K.; Barrat, F.J. Plasmacytoid Dendritic Cells Promote Systemic Sclerosis with a Key Role for TLR8. Sci. Transl. Med. 2018, 10, eaam8458. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, B.H.; Silva, I.S.; Mendes, A.; Leite-Pinheiro, F.; Paton, A.W.; Paton, J.C.; Duarte, I.F.; Pierre, P.; Almeida, C.R. Promoting ER Stress in a Plasmacytoid Dendritic Cell Line Drives Fibroblast Activation. Cell Commun. Signal. 2025, 23, 66. [Google Scholar] [CrossRef]
- Abraham, D.J.; Krieg, T.; Distler, J.; Distler, O. Overview of Pathogenesis of Systemic Sclerosis. Rheumatology (Oxford) 2009, 48 Suppl 3, iii3–7. [Google Scholar] [CrossRef]
- Asano, Y. Future Treatments in Systemic Sclerosis. J. Dermatol. 2010, 37, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Sato, S. Vasculopathy in Scleroderma. Semin. Immunopathol. 2015, 37, 489–500. [Google Scholar] [CrossRef]
- Prescott, R.J.; Freemont, A.J.; Jones, C.J.; Hoyland, J.; Fielding, P. Sequential Dermal Microvascular and Perivascular Changes in the Development of Scleroderma. J. Pathol. 1992, 166, 255–263. [Google Scholar] [CrossRef]
- Sgonc, R.; Gruschwitz, M.S.; Dietrich, H.; Recheis, H.; Gershwin, M.E.; Wick, G. Endothelial Cell Apoptosis Is a Primary Pathogenetic Event Underlying Skin Lesions in Avian and Human Scleroderma. J. Clin. Invest. 1996, 98, 785–792. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Gordon, S.M.; Waldman, M.; Edison, J.D.; Little, D.J.; Stitt, R.S.; Bailey, W.T.; Hughes, J.B.; Olson, S.W. Autoantibodies Are Present before the Clinical Diagnosis of Systemic Sclerosis. PLoS One 2019, 14, e0214202. [Google Scholar] [CrossRef] [PubMed]
- Ihn, H. Scleroderma, Fibroblasts, Signaling, and Excessive Extracellular Matrix. Curr. Rheumatol. Rep. 2005, 7, 156–162. [Google Scholar] [CrossRef]
- Romano, E.; Rosa, I.; Fioretto, B.S.; Matucci-Cerinic, M.; Manetti, M. New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy. Life (Basel) 2021, 11, 610. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Asano, Y.; Amiya, E.; Hatano, M.; Tamaki, Z.; Takata, M.; Ozeki, A.; Watanabe, A.; Kawarasaki, S.; Taniguchi, T.; et al. Clinical Correlation of Brachial Artery Flow-Mediated Dilation in Patients with Systemic Sclerosis. Mod. Rheumatol. 2014, 24, 106–111. [Google Scholar] [CrossRef]
- Cerinic, M.M.; Valentini, G.; Sorano, G.G.; D’Angelo, S.; Cuomo, G.; Fenu, L.; Generini, S.; Cinotti, S.; Morfini, M.; Pignone, A.; et al. Blood Coagulation, Fibrinolysis, and Markers of Endothelial Dysfunction in Systemic Sclerosis. Semin. Arthritis Rheum. 2003, 32, 285–295. [Google Scholar] [CrossRef]
- Ihn, H. Autocrine TGF-Beta Signaling in the Pathogenesis of Systemic Sclerosis. J. Dermatol. Sci. 2008, 49, 103–113. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Kelley, K.; Melichian, D.S.; Tamaki, Z.; Fang, F.; Su, Y.; Feng, G.; Pope, R.M.; Budinger, G.R.S.; Mutlu, G.M.; et al. Toll-like Receptor 4 Signaling Augments Transforming Growth Factor-β Responses: A Novel Mechanism for Maintaining and Amplifying Fibrosis in Scleroderma. Am. J. Pathol. 2013, 182, 192–205. [Google Scholar] [CrossRef]
- Kahaleh, M. Raynaud’s Phenomenon and Vascular Disease in Scleroderma. Curr. Opin. Rheumatol. 1994, 6, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Tourkina, E.; Bonner, M.; Oates, J.; Hofbauer, A.; Richard, M.; Znoyko, S.; Visconti, R.P.; Zhang, J.; Hatfield, C.M.; Silver, R.M.; et al. Altered Monocyte and Fibrocyte Phenotype and Function in Scleroderma Interstitial Lung Disease: Reversal by Caveolin-1 Scaffolding Domain Peptide. Fibrogenesis Tissue Repair 2011, 4, 15. [Google Scholar] [CrossRef]
- Nikitorowicz-Buniak, J.; Denton, C.P.; Abraham, D.; Stratton, R. Partially Evoked Epithelial-Mesenchymal Transition (EMT) Is Associated with Increased TGFβ Signaling within Lesional Scleroderma Skin. PLoS One 2015, 10, e0134092. [Google Scholar] [CrossRef]
- Mendoza, F.A.; Piera-Velazquez, S.; Farber, J.L.; Feghali-Bostwick, C.; Jiménez, S.A. Endothelial Cells Expressing Endothelial and Mesenchymal Cell Gene Products in Lung Tissue from Patients with Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2016, 68, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Romano, E.; Rosa, I.; Guiducci, S.; Bellando-Randone, S.; De Paulis, A.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Endothelial-to-Mesenchymal Transition Contributes to Endothelial Dysfunction and Dermal Fibrosis in Systemic Sclerosis. Ann. Rheum. Dis. 2017, 76, 924–934. [Google Scholar] [CrossRef]
- Marangoni, R.G.; Korman, B.D.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in Murine Cutaneous Fibrosis Originate from Adiponectin-Positive Intradermal Progenitors: Adipocyte-Myofibroblast Transition in Skin Fibrosis. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef]
- Gruschwitz, M.; Müller, P.U.; Sepp, N.; Hofer, E.; Fontana, A.; Wick, G. Transcription and Expression of Transforming Growth Factor Type Beta in the Skin of Progressive Systemic Sclerosis: A Mediator of Fibrosis? J. Invest. Dermatol. 1990, 94, 197–203. [Google Scholar] [CrossRef]
- Querfeld, C.; Eckes, B.; Huerkamp, C.; Krieg, T.; Sollberg, S. Expression of TGF-Beta 1, -Beta 2 and -Beta 3 in Localized and Systemic Scleroderma. J. Dermatol. Sci. 1999, 21, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Kulozik, M.; Hogg, A.; Lankat-Buttgereit, B.; Krieg, T. Co-Localization of Transforming Growth Factor Beta 2 with Alpha 1(I) Procollagen MRNA in Tissue Sections of Patients with Systemic Sclerosis. J. Clin. Invest. 1990, 86, 917–922. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Kubo, M.; Tamaki, K. Impaired Smad7-Smurf-Mediated Negative Regulation of TGF-Beta Signaling in Scleroderma Fibroblasts. J. Clin. Invest. 2004, 113, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Increased Expression of Integrin Alpha(v)Beta3 Contributes to the Establishment of Autocrine TGF-Beta Signaling in Scleroderma Fibroblasts. J. Immunol. 2005, 175, 7708–7718. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Kubo, M.; Tamaki, K. Increased Expression Levels of Integrin Alphavbeta5 on Scleroderma Fibroblasts. Am. J. Pathol. 2004, 164, 1275–1292. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Involvement of Alphavbeta5 Integrin-Mediated Activation of Latent Transforming Growth Factor Beta1 in Autocrine Transforming Growth Factor Beta Signaling in Systemic Sclerosis Fibroblasts. Arthritis Rheum. 2005, 52, 2897–2905. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Tamaki, K. Increased Expression of Integrin Alphavbeta5 Induces the Myofibroblastic Differentiation of Dermal Fibroblasts. Am. J. Pathol. 2006, 168, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Mimura, Y.; Ihn, H.; Jinnin, M.; Asano, Y.; Yamane, K.; Tamaki, K. Constitutive Thrombospondin-1 Overexpression Contributes to Autocrine Transforming Growth Factor-Beta Signaling in Cultured Scleroderma Fibroblasts. Am. J. Pathol. 2005, 166, 1451–1463. [Google Scholar] [CrossRef]
- Meng, Q.; Bao, D.; Liu, S.; Huang, J.; Guo, M.; Dai, B.; Ding, L.; Xie, S.; Meng, M.; Lv, C.; et al. ADAM Metallopeptidase Domain 19 Promotes Skin Fibrosis in Systemic Sclerosis via Neuregulin-1. Mol. Med. 2024, 30, 269. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, C.; Rezzonico, R.; Ribbens, C.; Burger, D.; Wollheim, F.A.; Dayer, J.M. Inhibition of Type I Collagen Production by Dermal Fibroblasts upon Contact with Activated T Cells: Different Sensitivity to Inhibition between Systemic Sclerosis and Control Fibroblasts. Arthritis Rheum. 1998, 41, 2039–2047. [Google Scholar] [CrossRef]
- Chizzolini, C.; Parel, Y.; De Luca, C.; Tyndall, A.; Akesson, A.; Scheja, A.; Dayer, J.-M. Systemic Sclerosis Th2 Cells Inhibit Collagen Production by Dermal Fibroblasts via Membrane-Associated Tumor Necrosis Factor Alpha: Th2 Cells Inhibit Collagen Synthesis by Dermal Fibroblasts. Arthritis Rheum. 2003, 48, 2593–2604. [Google Scholar] [CrossRef]
- Ichimura, Y.; Asano, Y.; Akamata, K.; Noda, S.; Taniguchi, T.; Takahashi, T.; Toyama, T.; Tada, Y.; Sugaya, M.; Sato, S.; et al. Progranulin Overproduction Due to Fli-1 Deficiency Contributes to the Resistance of Dermal Fibroblasts to Tumor Necrosis Factor in Systemic Sclerosis: THE ROLE OF PROGRANULIN IN SSc. Arthritis Rheumatol. 2015, 67, 3245–3255. [Google Scholar] [CrossRef]
- Bergmann, C.; Chenguiti Fakhouri, S.; Trinh-Minh, T.; Filla, T.; Rius Rigau, A.; Ekici, A.B.; Merlevede, B.; Hallenberger, L.; Zhu, H.; Dees, C.; et al. Mutual Amplification of GLI2/Hedgehog and Transcription Factor JUN/AP-1 Signaling in Fibroblasts in Systemic Sclerosis: Potential Implications for Combined Therapies. Arthritis Rheumatol. 2025, 77, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Asano, Y.; Taniguchi, T.; Hirabayashi, M.; Nakamura, K.; Miura, S.; Yamashita, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; et al. Fli1-Haploinsufficient Dermal Fibroblasts Promote Skin-Localized Transdifferentiation of Th2-like Regulatory T Cells. Arthritis Res. Ther. 2018, 20, 23. [Google Scholar] [CrossRef]
- Saigusa, R.; Asano, Y.; Nakamura, K.; Hirabayashi, M.; Miura, S.; Yamashita, T.; Taniguchi, T.; Ichimura, Y.; Takahashi, T.; Yoshizaki, A.; et al. Systemic Sclerosis Dermal Fibroblasts Suppress Th1 Cytokine Production via Galectin-9 Overproduction Due to Fli1 Deficiency. J. Invest. Dermatol. 2017, 137, 1850–1859. [Google Scholar] [CrossRef]
- Gur, C.; Wang, S.-Y.; Sheban, F.; Zada, M.; Li, B.; Kharouf, F.; Peleg, H.; Aamar, S.; Yalin, A.; Kirschenbaum, D.; et al. LGR5 Expressing Skin Fibroblasts Define a Major Cellular Hub Perturbed in Scleroderma. Cell 2022, 185, 1373–1388.e20. [Google Scholar] [CrossRef]
- Ma, F.; Tsou, P.-S.; Gharaee-Kermani, M.; Plazyo, O.; Xing, X.; Kirma, J.; Wasikowski, R.; Hile, G.A.; Harms, P.W.; Jiang, Y.; et al. Systems-Based Identification of the Hippo Pathway for Promoting Fibrotic Mesenchymal Differentiation in Systemic Sclerosis. Nat. Commun. 2024, 15, 210. [Google Scholar] [CrossRef]
- Huang, M.; Tabib, T.; Khanna, D.; Assassi, S.; Domsic, R.; Lafyatis, R. Single-Cell Transcriptomes and Chromatin Accessibility of Endothelial Cells Unravel Transcription Factors Associated with Dysregulated Angiogenesis in Systemic Sclerosis. Ann. Rheum. Dis. 2024, 83, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Rius Rigau, A.; Li, Y.-N.; Matei, A.-E.; Györfi, A.-H.; Bruch, P.-M.; Koziel, S.; Devakumar, V.; Gabrielli, A.; Kreuter, A.; Wang, J.; et al. Characterization of Vascular Niche in Systemic Sclerosis by Spatial Proteomics. Circ. Res. 2024, 134, 875–891. [Google Scholar] [CrossRef]
- Liu, Q.; Zaba, L.C.; Satpathy, A.T.; Longmire, M.; Zhang, W.; Li, K.; Granja, J.; Guo, C.; Lin, J.; Li, R.; et al. Chromatin Accessibility Landscapes of Skin Cells in Systemic Sclerosis Nominate Dendritic Cells in Disease Pathogenesis. Nat. Commun. 2020, 11, 5843. [Google Scholar] [CrossRef]
- Miura, S.; Asano, Y.; Saigusa, R.; Yamashita, T.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Yoshizaki, A.; Sato, S.; et al. Regulation of Skin Fibrosis by RALDH1-Producing Dermal Dendritic Cells via Retinoic Acid-Mediated Regulatory T Cell Induction: A Role in Scleroderma. J. Dermatol. Sci. 2020, 97, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Watanabe, Y.; Saigusa, R.; Yamashita, T.; Nakamura, K.; Hirabayashi, M.; Miyagawa, T.; Yoshizaki, A.; Trojanowska, M.; Sato, S.; et al. Fli1 Deficiency Suppresses RALDH1 Activity of Dermal Dendritic Cells and Related Induction of Regulatory T Cells: A Possible Role in Scleroderma. Arthritis Res. Ther. 2021, 23, 137. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.M.; Lee, S.; Neely, J.; Hinchcliff, M.; Wolters, P.J.; Sirota, M. Gene Expression Meta-Analysis Reveals Aging and Cellular Senescence Signatures in Scleroderma-Associated Interstitial Lung Disease. Front. Immunol. 2024, 15, 1326922. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Rana, T.; Jiang, C.; Liu, G.; Miyata, T.; Antony, V.; Thannickal, V.J.; Liu, R.-M. PAI-1 Regulation of TGF-Β1-Induced Alveolar Type II Cell Senescence, SASP Secretion, and SASP-Mediated Activation of Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2020, 62, 319–330. [Google Scholar] [CrossRef]
- Eren, M.; Boe, A.E.; Murphy, S.B.; Place, A.T.; Nagpal, V.; Morales-Nebreda, L.; Urich, D.; Quaggin, S.E.; Budinger, G.R.S.; Mutlu, G.M.; et al. PAI-1-Regulated Extracellular Proteolysis Governs Senescence and Survival in Klotho Mice. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 7090–7095. [Google Scholar] [CrossRef]
- Khan, S.S.; Shah, S.J.; Klyachko, E.; Baldridge, A.S.; Eren, M.; Place, A.T.; Aviv, A.; Puterman, E.; Lloyd-Jones, D.M.; Heiman, M.; et al. A Null Mutation in SERPINE1 Protects against Biological Aging in Humans. Sci. Adv. 2017, 3, eaao1617. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y. The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis. Int. J. Mol. Sci. 2019, 20, 619. [Google Scholar] [CrossRef]
- Kanno, Y. The UPA/UPAR System Orchestrates the Inflammatory Response, Vascular Homeostasis, and Immune System in Fibrosis Progression. Int. J. Mol. Sci. 2023, 24, 1796. [Google Scholar] [CrossRef]
- Vancheeswaran, R.; Azam, A.; Black, C.; Dashwood, M.R. Localization of Endothelin-1 and Its Binding Sites in Scleroderma Skin. J. Rheumatol. 1994, 21, 1268–1276. [Google Scholar]
- Rudnicka, L.; Varga, J.; Christiano, A.M.; Iozzo, R.V.; Jimenez, S.A.; Uitto, J. Elevated Expression of Type VII Collagen in the Skin of Patients with Systemic Sclerosis. Regulation by Transforming Growth Factor-Beta. J. Clin. Invest. 1994, 93, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Pap, T.; Kowal-Bielecka, O.; Meyringer, R.; Guiducci, S.; Landthaler, M.; Schölmerich, J.; Michel, B.A.; Gay, R.E.; Matucci-Cerinic, M.; et al. Overexpression of Monocyte Chemoattractant Protein 1 in Systemic Sclerosis: Role of Platelet-Derived Growth Factor and Effects on Monocyte Chemotaxis and Collagen Synthesis. Arthritis Rheum. 2001, 44, 2665–2678. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.A.; Jeziorska, M.; Freemont, A.J.; Herrick, A.L. The Differential Expression of VEGF, VEGFR-2, and GLUT-1 Proteins in Disease Subtypes of Systemic Sclerosis. Hum. Pathol. 2006, 37, 190–197. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Jüngel, A.; Kowal-Bielecka, O.; Michel, B.A.; Gay, R.E.; Sprott, H.; Matucci-Cerinic, M.; Chilla, M.; Reich, K.; Kalden, J.R.; et al. Expression of Interleukin-21 Receptor in Epidermis from Patients with Systemic Sclerosis: Altered Expression Pattern of Keratinocytes in SSc. Arthritis Rheum. 2005, 52, 856–864. [Google Scholar] [CrossRef]
- Aden, N.; Nuttall, A.; Shiwen, X.; de Winter, P.; Leask, A.; Black, C.M.; Denton, C.P.; Abraham, D.J.; Stratton, R.J. Epithelial Cells Promote Fibroblast Activation via IL-1alpha in Systemic Sclerosis. J. Invest. Dermatol. 2010, 130, 2191–2200. [Google Scholar] [CrossRef]
- Nikitorowicz-Buniak, J.; Shiwen, X.; Denton, C.P.; Abraham, D.; Stratton, R. Abnormally Differentiating Keratinocytes in the Epidermis of Systemic Sclerosis Patients Show Enhanced Secretion of CCN2 and S100A9. J. Invest. Dermatol. 2014, 134, 2693–2702. [Google Scholar] [CrossRef]
- McCoy, S.S.; Reed, T.J.; Berthier, C.C.; Tsou, P.-S.; Liu, J.; Gudjonsson, J.E.; Khanna, D.; Kahlenberg, J.M. Scleroderma Keratinocytes Promote Fibroblast Activation Independent of Transforming Growth Factor Beta. Rheumatology (Oxford) 2017, 56, 1970–1981. [Google Scholar] [CrossRef]
- Takahashi, T.; Asano, Y.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Miura, S.; Ichimura, Y.; Toyama, T.; Hirabayashi, M.; Taniguchi, T.; et al. A Potential Contribution of Psoriasin to Vascular and Epithelial Abnormalities and Inflammation in Systemic Sclerosis. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 291–297. [Google Scholar] [CrossRef]
- Saigusa, R.; Yamashita, T.; Miura, S.; Hirabayashi, M.; Nakamura, K.; Miyagawa, T.; Fukui, Y.; Yoshizaki, A.; Sato, S.; Asano, Y. A Potential Contribution of Decreased Galectin-7 Expression in Stratified Epithelia to the Development of Cutaneous and Oesophageal Manifestations in Systemic Sclerosis. Exp. Dermatol. 2019, 28, 536–542. [Google Scholar] [CrossRef]
- Takahashi, T.; Asano, Y.; Sugawara, K.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Akamata, K.; et al. Epithelial Fli1 Deficiency Drives Systemic Autoimmunity and Fibrosis: Possible Roles in Scleroderma. J. Exp. Med. 2017, 214, 1129–1151. [Google Scholar] [CrossRef]
- Asano, Y.; Stawski, L.; Hant, F.; Highland, K.; Silver, R.; Szalai, G.; Watson, D.K.; Trojanowska, M. Endothelial Fli1 Deficiency Impairs Vascular Homeostasis: A Role in Scleroderma Vasculopathy. Am. J. Pathol. 2010, 176, 1983–1998. [Google Scholar] [CrossRef]
- Asano, Y.; Markiewicz, M.; Kubo, M.; Szalai, G.; Watson, D.K.; Trojanowska, M. Transcription Factor Fli1 Regulates Collagen Fibrillogenesis in Mouse Skin. Mol. Cell. Biol. 2009, 29, 425–434. [Google Scholar] [CrossRef]
- Bujor, A.M.; El Adili, F.; Parvez, A.; Marden, G.; Trojanowska, M. Fli1 Downregulation in Scleroderma Myeloid Cells Has Profibrotic and Proinflammatory Effects. Front. Immunol. 2020, 11, 800. [Google Scholar] [CrossRef]
- Taniguchi, T.; Miyagawa, T.; Toyama, S.; Yamashita, T.; Nakamura, K.; Saigusa, R.; Ichimura, Y.; Takahashi, T.; Toyama, T.; Yoshizaki, A.; et al. CXCL13 Produced by Macrophages Due to Fli1 Deficiency May Contribute to the Development of Tissue Fibrosis, Vasculopathy and Immune Activation in Systemic Sclerosis. Exp. Dermatol. 2018, 27, 1030–1037. [Google Scholar] [CrossRef]
- Anderson, M.S.; Venanzi, E.S.; Klein, L.; Chen, Z.; Berzins, S.P.; Turley, S.J.; von Boehmer, H.; Bronson, R.; Dierich, A.; Benoist, C.; et al. Projection of an Immunological Self Shadow within the Thymus by the Aire Protein. Science 2002, 298, 1395–1401. [Google Scholar] [CrossRef]
- Mathis, D.; Benoist, C. Aire. Annu. Rev. Immunol. 2009, 27, 287–312. [Google Scholar] [CrossRef]
- Saigusa, R.; Asano, Y.; Yamashita, T.; Takahashi, T.; Nakamura, K.; Miura, S.; Ichimura, Y.; Toyama, T.; Taniguchi, T.; Sumida, H.; et al. Systemic Sclerosis Complicated with Localized Scleroderma-like Lesions Induced by Köbner Phenomenon. J. Dermatol. Sci. 2018, 89, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The Human Skin Microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Terui, H.; Yamasaki, K.; Wada-Irimada, M.; Onodera-Amagai, M.; Hatchome, N.; Mizuashi, M.; Yamashita, R.; Kawabe, T.; Ishii, N.; Abe, T.; et al. Staphylococcus Aureus Skin Colonization Promotes SLE-like Autoimmune Inflammation via Neutrophil Activation and the IL-23/IL-17 Axis. Sci. Immunol. 2022, 7, eabm9811. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The Skin Microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Franks, J.M.; Cai, G.; Mehta, B.K.; Wood, T.A.; Archambault, K.; Pioli, P.A.; Simms, R.W.; Orzechowski, N.; Arron, S.; et al. Microbiome Dysbiosis Is Associated with Disease Duration and Increased Inflammatory Gene Expression in Systemic Sclerosis Skin. Arthritis Res. Ther. 2019, 21, 49. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wei, J.; Varga, J. Understanding Fibrosis in Systemic Sclerosis: Shifting Paradigms, Emerging Opportunities. Nat. Rev. Rheumatol. 2011, 8, 42–54. [Google Scholar] [CrossRef]
- Lynch, M.D.; Watt, F.M. Fibroblast Heterogeneity: Implications for Human Disease. J. Clin. Invest. 2018, 128, 26–35. [Google Scholar] [CrossRef]
- Zhang, Z.; Shao, M.; Hepler, C.; Zi, Z.; Zhao, S.; An, Y.A.; Zhu, Y.; Ghaben, A.L.; Wang, M.-Y.; Li, N.; et al. Dermal Adipose Tissue Has High Plasticity and Undergoes Reversible Dedifferentiation in Mice. J. Clin. Invest. 2019, 129, 5327–5342. [Google Scholar] [CrossRef]
- Varga, J.; Marangoni, R.G. Systemic Sclerosis in 2016: Dermal White Adipose Tissue Implicated in SSc Pathogenesis: Systemic Sclerosis in 2016. Nat. Rev. Rheumatol. 2017, 13, 71–72. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Shimomura, I.; Kihara, S.; Funahashi, T. Importance of Adipocytokines in Obesity-Related Diseases. Horm. Res. 2003, 60 Suppl 3, 56–59. [Google Scholar] [CrossRef]
- Masui, Y.; Asano, Y.; Shibata, S.; Noda, S.; Aozasa, N.; Akamata, K.; Yamada, D.; Tamaki, Z.; Tada, Y.; Sugaya, M.; et al. Serum Adiponectin Levels Inversely Correlate with the Activity of Progressive Skin Sclerosis in Patients with Diffuse Cutaneous Systemic Sclerosis: Significance of Serum Adiponectin Levels in SSc. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Masui, Y.; Asano, Y.; Takahashi, T.; Shibata, S.; Akamata, K.; Aozasa, N.; Noda, S.; Taniguchi, T.; Ichimura, Y.; Toyama, T.; et al. Clinical Significance of Monitoring Serum Adiponectin Levels during Intravenous Pulse Cyclophosphamide Therapy in Interstitial Lung Disease Associated with Systemic Sclerosis. Mod. Rheumatol. 2013, 23, 323–329. [Google Scholar] [CrossRef]
- Masui, Y.; Asano, Y.; Shibata, S.; Noda, S.; Akamata, K.; Aozasa, N.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; et al. A Possible Contribution of Visfatin to the Resolution of Skin Sclerosis in Patients with Diffuse Cutaneous Systemic Sclerosis via a Direct Anti-Fibrotic Effect on Dermal Fibroblasts and Th1 Polarization of the Immune Response. Rheumatology (Oxford) 2013, 52, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Aozasa, N.; Asano, Y.; Akamata, K.; Noda, S.; Masui, Y.; Yamada, D.; Tamaki, Z.; Tada, Y.; Sugaya, M.; Kadono, T.; et al. Serum Apelin Levels: Clinical Association with Vascular Involvements in Patients with Systemic Sclerosis: Significance of Serum Apelin Levels in SSc. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 37–42. [Google Scholar] [CrossRef]
- Masui, Y.; Asano, Y.; Akamata, K.; Aozasa, N.; Noda, S.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Sumida, H.; et al. Serum Resistin Levels: A Possible Correlation with Pulmonary Vascular Involvement in Patients with Systemic Sclerosis. Rheumatol. Int. 2014, 34, 1165–1170. [Google Scholar] [CrossRef]
- Toyama, T.; Asano, Y.; Takahashi, T.; Aozasa, N.; Akamata, K.; Noda, S.; Taniguchi, T.; Ichimura, Y.; Sumida, H.; Tamaki, Z.; et al. Clinical Significance of Serum Retinol Binding Protein-4 Levels in Patients with Systemic Sclerosis: Significance of Serum RBP4 Levels in SSc. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Asano, Y.; Noda, S.; Aozasa, N.; Akamata, K.; Taniguchi, T.; Ichimura, Y.; Toyama, T.; Sumida, H.; Kuwano, Y.; et al. A Possible Contribution of Lipocalin-2 to the Development of Dermal Fibrosis, Pulmonary Vascular Involvement and Renal Dysfunction in Systemic Sclerosis. Br. J. Dermatol. 2015, 173, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Asano, Y.; Saigusa, R.; Yamashita, T.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Tamaki, Z.; Tada, Y.; et al. Serum Omentin Levels: A Possible Contribution to Vascular Involvement in Patients with Systemic Sclerosis. J. Dermatol. 2015, 42, 461–466. [Google Scholar] [CrossRef]
- Miura, S.; Asano, Y.; Saigusa, R.; Yamashita, T.; Taniguchi, T.; Takahashi, T.; Ichimura, Y.; Toyama, T.; Tamaki, Z.; Tada, Y.; et al. Serum Vaspin Levels: A Possible Correlation with Digital Ulcers in Patients with Systemic Sclerosis. J. Dermatol. 2015, 42, 528–531. [Google Scholar] [CrossRef]
- Lakota, K.; Wei, J.; Carns, M.; Hinchcliff, M.; Lee, J.; Whitfield, M.L.; Sodin-Semrl, S.; Varga, J. Levels of Adiponectin, a Marker for PPAR-Gamma Activity, Correlate with Skin Fibrosis in Systemic Sclerosis: Potential Utility as Biomarker? Arthritis Res. Ther. 2012, 14, R102. [Google Scholar] [CrossRef]
- Tomčík, M.; Arima, K.; Hulejová, H.; Kuklová, M.; Filková, M.; Braun, M.; Beláček, J.; Novák, M.; Bečvář, R.; Vencovský, J.; et al. Adiponectin Relation to Skin Changes and Dyslipidemia in Systemic Sclerosis. Cytokine 2012, 58, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Jinnin, M.; Muchemwa, F.C.; Makino, T.; Kajihara, I.; Makino, K.; Honda, N.; Sakai, K.; Fukushima, S.; Ihn, H. Adiponectin Expression Is Decreased in the Involved Skin and Sera of Diffuse Cutaneous Scleroderma Patients: Letter to the Editor. Exp. Dermatol. 2011, 20, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, R.G.; Masui, Y.; Fang, F.; Korman, B.; Lord, G.; Lee, J.; Lakota, K.; Wei, J.; Scherer, P.E.; Otvos, L.; et al. Adiponectin Is an Endogenous Anti-Fibrotic Mediator and Therapeutic Target. Sci. Rep. 2017, 7, 4397. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Lakota, K.; Taniguchi, T.; Yoshizaki, A.; Sato, S.; Hong, W.; Zhou, X.; Sodin-Semrl, S.; Fang, F.; Asano, Y.; et al. An Orally-Active Adiponectin Receptor Agonist Mitigates Cutaneous Fibrosis, Inflammation and Microvascular Pathology in a Murine Model of Systemic Sclerosis. Sci. Rep. 2018, 8, 11843. [Google Scholar] [CrossRef]
- Sjogren, R. Gastrointestinal Features of Scleroderma. Curr. Opin. Rheumatol. 1996, 8, 569–575. [Google Scholar] [CrossRef]
- Poirier, T.; Rankin, G. Gastrointestinal Manifestations of Progressive Systemic Scleroderma Based on a Review of 364 Cases. Am. J. Gastroenterol. 1972, 58, 30–44. [Google Scholar]
- Young, M.A.; Rose, S.; Reynolds, J.C. Gastrointestinal Manifestations of Scleroderma. Rheum. Dis. Clin. North Am. 1996, 22, 797–823. [Google Scholar] [CrossRef]
- Emmanuel, A. Current Management of the Gastrointestinal Complications of Systemic Sclerosis. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 461–472. [Google Scholar] [CrossRef]
- Lock, G.; Holstege, A.; Lang, B.; Schölmerich, J. Gastrointestinal Manifestations of Progressive Systemic Sclerosis. Am. J. Gastroenterol. 1997, 92, 763–771. [Google Scholar]
- Malandrini, A.; Selvi, E.; Villanova, M.; Berti, G.; Sabadini, L.; Salvadori, C.; Gambelli, S.; De Stefano, R.; Vernillo, R.; Marcolongo, R.; et al. Autonomic Nervous System and Smooth Muscle Cell Involvement in Systemic Sclerosis: Ultrastructural Study of 3 Cases. J. Rheumatol. 2000, 27, 1203–1206. [Google Scholar]
- Lepri, G.; Guiducci, S.; Bellando-Randone, S.; Giani, I.; Bruni, C.; Blagojevic, J.; Carnesecchi, G.; Radicati, A.; Pucciani, F.; Marco, M.-C. Evidence for Oesophageal and Anorectal Involvement in Very Early Systemic Sclerosis (VEDOSS): Report from a Single VEDOSS/EUSTAR Centre. Ann. Rheum. Dis. 2015, 74, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Weston, S.; Thumshirn, M.; Wiste, J.; Camilleri, M. Clinical and Upper Gastrointestinal Motility Features in Systemic Sclerosis and Related Disorders. Am. J. Gastroenterol. 1998, 93, 1085–1089. [Google Scholar] [CrossRef]
- Roman, S.; Hot, A.; Fabien, N.; Cordier, J.-F.; Miossec, P.; Ninet, J.; Mion, F. ; Réseau Sclérodermie des Hospices Civils de Lyon Esophageal Dysmotility Associated with Systemic Sclerosis: A High-Resolution Manometry Study: Systemic Sclerosis and Esophageal HRM. Dis. Esophagus 2011, 24, 299–304. [Google Scholar] [CrossRef]
- Clements, P.J.; Becvar, R.; Drosos, A.A.; Ghattas, L.; Gabrielli, A. Assessment of Gastrointestinal Involvement. Clin. Exp. Rheumatol. 2003, 21, S15–8. [Google Scholar]
- Weber, P.; Ganser, G.; Frosch, M.; Roth, J.; Hülskamp, G.; Zimmer, K.P. Twenty-Four Hour Intraesophageal PH Monitoring in Children and Adolescents with Scleroderma and Mixed Connective Tissue Disease. J. Rheumatol. 2000, 27, 2692–2695. [Google Scholar] [PubMed]
- Eaker, E.Y.; Kuldau, J.G.; Verne, G.N.; Ross, S.O.; Sallustio, J.E. Myenteric Neuronal Antibodies in Scleroderma: Passive Transfer Evokes Alterations in Intestinal Myoelectric Activity in a Rat Model. J. Lab. Clin. Med. 1999, 133, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Nakamura, Y.; Matsumoto, I.; Nishimagi, E.; Satoh, T.; Kuwana, M.; Sumida, T.; Hara, M. Muscarinic-3 Acetylcholine Receptor Autoantibody in Patients with Systemic Sclerosis: Contribution to Severe Gastrointestinal Tract Dysmotility. Ann. Rheum. Dis. 2009, 68, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Mehendiratta, V.; Del Galdo, F.; Jimenez, S.A.; Cohen, S.; DiMarino, A.J.; Rattan, S. Immunoglobulins from Scleroderma Patients Inhibit the Muscarinic Receptor Activation in Internal Anal Sphincter Smooth Muscle Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1206–13. [Google Scholar] [CrossRef]
- Kafaja, S.; Valera, I.; Divekar, A.A.; Saggar, R.; Abtin, F.; Furst, D.E.; Khanna, D.; Singh, R.R. PDCs in Lung and Skin Fibrosis in a Bleomycin-Induced Model and Patients with Systemic Sclerosis. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Dantas, R.O.; Aprile, L.R.O. Esophageal Striated Muscle Contractions in Patients with Gastroesophageal Reflux Symptoms. Dig. Dis. Sci. 2002, 47, 2586–2590. [Google Scholar] [CrossRef]
- Miwa, H.; Kondo, T.; Oshima, T. Gastroesophageal Reflux Disease-Related and Functional Heartburn: Pathophysiology and Treatment. Curr. Opin. Gastroenterol. 2016, 32, 344–352. [Google Scholar] [CrossRef]
- Dessein, P.H.; Joffe, B.I.; Metz, R.M.; Millar, D.L.; Lawson, M.; Stanwix, A.E. Autonomic Dysfunction in Systemic Sclerosis: Sympathetic Overactivity and Instability. Am. J. Med. 1992, 93, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Wu, C.; Zeng, X.; Wang, Q. The Role of Gut Microbiota in the Pathogenesis of Rheumatic Diseases. Clin. Rheumatol. 2018, 37, 25–34. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Shoenfeld, Y. The Microbiome in Autoimmune Diseases. Clin. Exp. Immunol. 2019, 195, 74–85. [Google Scholar] [CrossRef]
- Rosser, E.C.; Mauri, C. A Clinical Update on the Significance of the Gut Microbiota in Systemic Autoimmunity. J. Autoimmun. 2016, 74, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Bellocchi, C.; Volkmann, E.R. Update on the Gastrointestinal Microbiome in Systemic Sclerosis. Curr. Rheumatol. Rep. 2018, 20, 49. [Google Scholar] [CrossRef]
- Volkmann, E.R. Intestinal Microbiome in Scleroderma: Recent Progress. Curr. Opin. Rheumatol. 2017, 29, 553–560. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A. Changes in Causes of Death in Systemic Sclerosis, 1972-2002. Ann. Rheum. Dis. 2007, 66, 940–944. [Google Scholar] [CrossRef]
- Elhai, M.; Meune, C.; Boubaya, M.; Avouac, J.; Hachulla, E.; Balbir-Gurman, A. ; Allanore Mapping and Predicting Mortality from Systemic Sclerosis. Annals of the rheumatic diseases 2017, 76, 1897–1905. [Google Scholar] [CrossRef]
- Steen, V.; Medsger, T.A. , Jr Predictors of Isolated Pulmonary Hypertension in Patients with Systemic Sclerosis and Limited Cutaneous Involvement. Arthritis Rheum. 2003, 48, 516–522. [Google Scholar] [CrossRef]
- Daraban, A.M.; Enache, R.; Predescu, L.; Platon, P.; Constantinescu, T.; Mihai, C.; Coman, I.M.; Ginghina, C.; Jurcuţ, R. Pulmonary Veno-Occlusive Disease: A Rare Cause of Pulmonary Hypertension in Systemic Sclerosis. Case Presentation and Review of the Literature. Rom. J. Intern. Med. 2015, 53, 175–183. [Google Scholar] [CrossRef]
- Duarte, A.C.; Cordeiro, A.; Loureiro, M.J.; Ferreira, F. Pulmonary Veno-Occlusive Disease: A Probably Underdiagnosed Cause of Pulmonary Hypertension in Systemic Sclerosis. Clin. Rheumatol. 2020, 39, 1687–1691. [Google Scholar] [CrossRef]
- Steele, R.; Hudson, M.; Lo, E.; Baron, M. ; Canadian Scleroderma Research Group Clinical Decision Rule to Predict the Presence of Interstitial Lung Disease in Systemic Sclerosis. Arthritis Care Res. (Hoboken) 2012, 64, 519–524. [Google Scholar] [CrossRef] [PubMed]
- White, B. Interstitial Lung Disease in Scleroderma. Rheum. Dis. Clin. North Am. 2003, 29, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Nihtyanova, S.I.; Schreiber, B.E.; Ong, V.H.; Rosenberg, D.; Moinzadeh, P.; Coghlan, J.G.; Wells, A.U.; Denton, C.P. Prediction of Pulmonary Complications and Long-Term Survival in Systemic Sclerosis: Pulmonary Complications and Survival in SSc. Arthritis Rheumatol. 2014, 66, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.; Domsic, R.T.; Lucas, M.; Fertig, N.; Medsger, T.A. , Jr A Clinical and Serologic Comparison of African American and Caucasian Patients with Systemic Sclerosis. Arthritis Rheum. 2012, 64, 2986–2994. [Google Scholar] [CrossRef]
- Jaeger, V.K.; Wirz, E.G.; Allanore, Y.; Rossbach, P.; Riemekasten, G.; Hachulla, E.; Distler, O.; Airò, P.; Carreira, P.E.; Balbir Gurman, A.; et al. Incidences and Risk Factors of Organ Manifestations in the Early Course of Systemic Sclerosis: A Longitudinal EUSTAR Study. PLoS One 2016, 11, e0163894. [Google Scholar] [CrossRef]
- Ostojić, P.; Damjanov, N. Different Clinical Features in Patients with Limited and Diffuse Cutaneous Systemic Sclerosis. Clin. Rheumatol. 2006, 25, 453–457. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Kubo, M.; Tamaki, K. The Prevalence and Clinical Significance of Anti-U1 RNA Antibodies in Patients with Systemic Sclerosis. J. Invest. Dermatol. 2003, 120, 204–210. [Google Scholar] [CrossRef]
- Man, A.; Davidyock, T.; Ferguson, L.T.; Ieong, M.; Zhang, Y.; Simms, R.W. Changes in Forced Vital Capacity over Time in Systemic Sclerosis: Application of Group-Based Trajectory Modelling. Rheumatology (Oxford) 2015, 54, 1464–1471. [Google Scholar] [CrossRef]
- Steen, V.D.; Conte, C.; Owens, G.R.; Medsger, T.A. , Jr Severe Restrictive Lung Disease in Systemic Sclerosis. Arthritis Rheum. 1994, 37, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.; Knight, C.; Lunt, M.; Black, C.M.; Silman, A.J. Predictors of End Stage Lung Disease in a Cohort of Patients with Scleroderma. Ann. Rheum. Dis. 2003, 62, 146–150. [Google Scholar] [CrossRef]
- Bouros, D.; Wells, A.U.; Nicholson, A.G.; Colby, T.V.; Polychronopoulos, V.; Pantelidis, P.; Haslam, P.L.; Vassilakis, D.A.; Black, C.M.; du Bois, R.M. Histopathologic Subsets of Fibrosing Alveolitis in Patients with Systemic Sclerosis and Their Relationship to Outcome. Am. J. Respir. Crit. Care Med. 2002, 165, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Yoo, B.; Lee, J.S.; Kim, E.K.; Lim, C.M.; Lee, S.D.; Koh, Y.; Kim, W.S.; Kim, W.D.; Colby, T.V.; et al. The Major Histopathologic Pattern of Pulmonary Fibrosis in Scleroderma Is Nonspecific Interstitial Pneumonia. Sarcoidosis Vasc. Diffuse Lung Dis. 2002, 19, 121–127. [Google Scholar]
- Fischer, A.; Swigris, J.J.; Groshong, S.D.; Cool, C.D.; Sahin, H.; Lynch, D.A.; Curran-Everett, D.; Gillis, J.Z.; Meehan, R.T.; Brown, K.K. Clinically Significant Interstitial Lung Disease in Limited Scleroderma: Histopathology, Clinical Features, and Survival. Chest 2008, 134, 601–605. [Google Scholar] [CrossRef]
- Winstone, T.A.; Assayag, D.; Wilcox, P.G.; Dunne, J.V.; Hague, C.J.; Leipsic, J.; Collard, H.R.; Ryerson, C.J. Predictors of Mortality and Progression in Scleroderma-Associated Interstitial Lung Disease: A Systematic Review. Chest 2014, 146, 422–436. [Google Scholar] [CrossRef]
- Beon, M.; Harley, R.A.; Wessels, A.; Silver, R.M.; Ludwicka-Bradley, A. Myofibroblast Induction and Microvascular Alteration in Scleroderma Lung Fibrosis. Clin. Exp. Rheumatol. 2004, 22, 733–742. [Google Scholar]
- Savarino, E.; Bazzica, M.; Zentilin, P.; Pohl, D.; Parodi, A.; Cittadini, G.; Negrini, S.; Indiveri, F.; Tutuian, R.; Savarino, V.; et al. Gastroesophageal Reflux and Pulmonary Fibrosis in Scleroderma: A Study Using PH-Impedance Monitoring: A Study Using PH-Impedance Monitoring. Am. J. Respir. Crit. Care Med. 2009, 179, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Appel, J.Z., 3rd; Lee, S.M.; Hartwig, M.G.; Li, B.; Hsieh, C.-C.; Cantu, E., 3rd; Yoon, Y.; Lin, S.S.; Parker, W.; Davis, R.D. Characterization of the Innate Immune Response to Chronic Aspiration in a Novel Rodent Model. Respir. Res. 2007, 8, 87. [Google Scholar] [CrossRef]
- Christmann, R.B.; Wells, A.U.; Capelozzi, V.L.; Silver, R.M. Gastroesophageal Reflux Incites Interstitial Lung Disease in Systemic Sclerosis: Clinical, Radiologic, Histopathologic, and Treatment Evidence. Semin. Arthritis Rheum. 2010, 40, 241–249. [Google Scholar] [CrossRef]
- de Souza, R.B.C.; Borges, C.T.L.; Capelozzi, V.L.; Parra, E.R.; Jatene, F.B.; Kavakama, J.; Kairalla, R.A.; Bonfá, E. Centrilobular Fibrosis: An Underrecognized Pattern in Systemic Sclerosis. Respiration 2009, 77, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Venalis, P.; Kumánovics, G.; Schulze-Koops, H.; Distler, A.; Dees, C.; Zerr, P.; Palumbo-Zerr, K.; Czirják, L.; Mackevic, Z.; Lundberg, I.E.; et al. Cardiomyopathy in Murine Models of Systemic Sclerosis: Histopathologic Features of Cardiomyopathy in SSc. Arthritis Rheumatol. 2015, 67, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Meune, C. Primary Myocardial Involvement in Systemic Sclerosis: Evidence for a Microvascular Origin. Clin. Exp. Rheumatol. 2010, 28, S48–53. [Google Scholar]
- Hachulla, A.-L.; Launay, D.; Gaxotte, V.; de Groote, P.; Lamblin, N.; Devos, P.; Hatron, P.-Y.; Beregi, J.-P.; Hachulla, E. Cardiac Magnetic Resonance Imaging in Systemic Sclerosis: A Cross-Sectional Observational Study of 52 Patients. Ann. Rheum. Dis. 2009, 68, 1878–1884. [Google Scholar] [CrossRef]
- Hesselstrand, R.; Scheja, A.; Akesson, A. Mortality and Causes of Death in a Swedish Series of Systemic Sclerosis Patients. Ann. Rheum. Dis. 1998, 57, 682–686. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A.; Vlachoyiannopoulos, P.G.; Haidich, A.-B.; Medsger, T.A., Jr; Lucas, M.; Michet, C.J.; Kuwana, M.; Yasuoka, H.; van den Hoogen, F.; Te Boome, L.; et al. Mortality in Systemic Sclerosis: An International Meta-Analysis of Individual Patient Data. Am. J. Med. 2005, 118, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.D.; Medsger, T.A. , Jr Severe Organ Involvement in Systemic Sclerosis with Diffuse Scleroderma. Arthritis Rheum. 2000, 43, 2437–2444. [Google Scholar] [CrossRef]
- Kahan, A.; Coghlan, G.; McLaughlin, V. Cardiac Complications of Systemic Sclerosis. Rheumatology (Oxford) 2009, 48 Suppl 3, iii45–8. [Google Scholar] [CrossRef]
- Allanore, Y.; Meune, C.; Kahan, A. Outcome Measures for Heart Involvement in Systemic Sclerosis. Rheumatology (Oxford) 2008, 47 Suppl 5, v51–3. [Google Scholar] [CrossRef]
- Ferri, C.; Valentini, G.; Cozzi, F.; Sebastiani, M.; Michelassi, C.; La Montagna, G.; Bullo, A.; Cazzato, M.; Tirri, E.; Storino, F.; et al. Systemic Sclerosis: Demographic, Clinical, and Serologic Features and Survival in 1,012 Italian Patients. Medicine (Baltimore) 2002, 81, 139–153. [Google Scholar] [CrossRef]
- Purevsuren, M.; Uehara, M.; Ishizuka, M.; Suzuki, Y.; Shimbo, M.; Kakuda, N.; Ishii, S.; Sumida, H.; Miyazaki, M.; Yamashita, T.; et al. Native T1 Mapping in Early Diffuse and Limited Systemic Sclerosis, and Its Association with Diastolic Function. J. Cardiol. 2023, 82, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, B.H.; Ridolfi, R.L.; Salyer, W.R.; Hutchins, G.M. Myocardial Lesions of Progressive Systemic Sclerosis. A Cause of Cardiac Dysfunction. Circulation 1976, 53, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Belloli, L.; Carlo-Stella, N.; Ciocia, G.; Chiti, A.; Massarotti, M.; Marasini, B. Myocardial Involvement in Systemic Sclerosis. Rheumatology (Oxford) 2008, 47, 1070–1072. [Google Scholar] [CrossRef]
- Desai, C.S.; Lee, D.C.; Shah, S.J. Systemic Sclerosis and the Heart: Current Diagnosis and Management. Curr. Opin. Rheumatol. 2011, 23, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Tzelepis, G.E.; Kelekis, N.L.; Plastiras, S.C.; Mitseas, P.; Economopoulos, N.; Kampolis, C.; Gialafos, E.J.; Moyssakis, I.; Moutsopoulos, H.M. Pattern and Distribution of Myocardial Fibrosis in Systemic Sclerosis: A Delayed Enhanced Magnetic Resonance Imaging Study. Arthritis Rheum. 2007, 56, 3827–3836. [Google Scholar] [CrossRef]
- Meune, C.; Allanore, Y.; Devaux, J.-Y.; Dessault, O.; Duboc, D.; Weber, S.; Kahan, A. High Prevalence of Right Ventricular Systolic Dysfunction in Early Systemic Sclerosis. J. Rheumatol. 2004, 31, 1941–1945. [Google Scholar]
- Kahan, A.; Devaux, J.Y.; Amor, B.; Menkès, C.J.; Weber, S.; Venot, A.; Strauch, G. The Effect of Captopril on Thallium 201 Myocardial Perfusion in Systemic Sclerosis. Clin. Pharmacol. Ther. 1990, 47, 483–489. [Google Scholar] [CrossRef]
- Duboc, D.; Kahan, A.; Maziere, B.; Loc’h, C.; Crouzel, C.; Menkès, C.J.; Amor, B.; Strauch, G.; Guérin, F.; Syrota, A. The Effect of Nifedipine on Myocardial Perfusion and Metabolism in Systemic Sclerosis. A Positron Emission Tomographic Study. Arthritis Rheum. 1991, 34, 198–203. [Google Scholar] [CrossRef]
- Kahan, A.; Jy, D.; Amor, B.; Menkes, C.-J.; Weber, S.; Guerin, F.; Venot, A.; Strauch, G. Pharmacodynamic Effect of Nicardipine on Left Ventricular Function in Systemic Sclerosis. J. Cardiovasc. Pharmacol. 1990, 15, 249–253. [Google Scholar] [CrossRef]
- Lekakis, J.; Mavrikakis, M.; Emmanuel, M.; Prassopoulos, V.; Papazoglou, S.; Papamichael, C.; Moulopoulou, D.; Kostamis, P.; Stamatelopoulos, S.; Moulopoulos, S. Cold-Induced Coronary Raynaud’s Phenomenon in Patients with Systemic Sclerosis. Clin. Exp. Rheumatol. 1998, 16, 135–140. [Google Scholar]
Figure 2.
Keratinocytes and adipocytes as key modifiers of skin pathophysiology in systemic sclerosis (SSc). This concept map illustrates the multifaceted roles of keratinocytes and adipocytes in the pathogenesis of SSc skin involvement. Dysfunctional keratinocytes upregulate various disease-associated molecules, contributing to dermal fibroblast activation and fibrosis. Keratinocyte-specific Fli1 deficiency leads to epithelial cell activation, inducing dermal fibrosis, and thymus dysfunction. While the interaction with skin microbiota remains unclear in SSc, it may modulate keratinocyte function and immunity. Adipocytes contribute to dermal fibrosis through adipocyte-to-myofibroblast transition and altered adipokine production, collectively driving SSc skin pathology.
Figure 2.
Keratinocytes and adipocytes as key modifiers of skin pathophysiology in systemic sclerosis (SSc). This concept map illustrates the multifaceted roles of keratinocytes and adipocytes in the pathogenesis of SSc skin involvement. Dysfunctional keratinocytes upregulate various disease-associated molecules, contributing to dermal fibroblast activation and fibrosis. Keratinocyte-specific Fli1 deficiency leads to epithelial cell activation, inducing dermal fibrosis, and thymus dysfunction. While the interaction with skin microbiota remains unclear in SSc, it may modulate keratinocyte function and immunity. Adipocytes contribute to dermal fibrosis through adipocyte-to-myofibroblast transition and altered adipokine production, collectively driving SSc skin pathology.
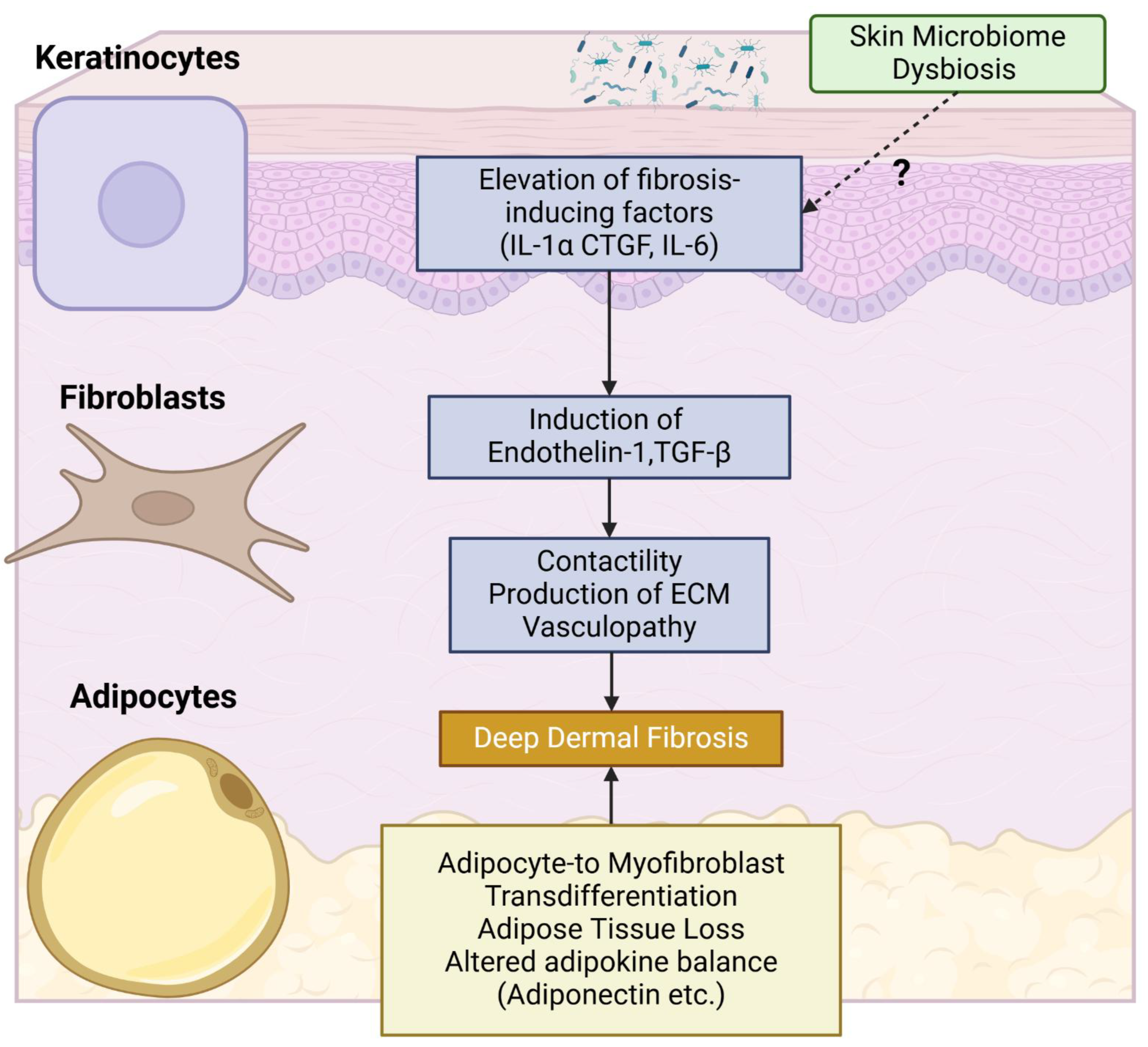
Figure 3.
Pathophysiological mechanisms of esophageal involvement in systemic sclerosis (SSc). This schematic illustrates the multi-factorial pathogenesis of gastrointestinal (GI) manifestations in SSc. The cascade is initiated by Immune Abnormality, including the presence of anti-muscarinic acetylcholine receptor M3 antibodies, and Vasculopathy. Both of these pathological processes contribute to disturbed enteric nervous system function, ultimately resulting in esophageal hypomotility. Disturbed enteric nervous system induces smooth muscle atrophy, further contributing to esophageal hypomotility and leading to fibrosis of esophagus. Esophageal stratified squamous epithelia can produce inflammatory and fibrotic cytokines, further promoting fibrosis. Functional Heartburn may also occur, potentially linked to inflammatory cytokines produced by the esophageal stratified squamous epithelia. These interconnected pathways highlight the complex interplay of immune, vascular, epithelial, and neural factors in SSc-related GERD.
Figure 3.
Pathophysiological mechanisms of esophageal involvement in systemic sclerosis (SSc). This schematic illustrates the multi-factorial pathogenesis of gastrointestinal (GI) manifestations in SSc. The cascade is initiated by Immune Abnormality, including the presence of anti-muscarinic acetylcholine receptor M3 antibodies, and Vasculopathy. Both of these pathological processes contribute to disturbed enteric nervous system function, ultimately resulting in esophageal hypomotility. Disturbed enteric nervous system induces smooth muscle atrophy, further contributing to esophageal hypomotility and leading to fibrosis of esophagus. Esophageal stratified squamous epithelia can produce inflammatory and fibrotic cytokines, further promoting fibrosis. Functional Heartburn may also occur, potentially linked to inflammatory cytokines produced by the esophageal stratified squamous epithelia. These interconnected pathways highlight the complex interplay of immune, vascular, epithelial, and neural factors in SSc-related GERD.
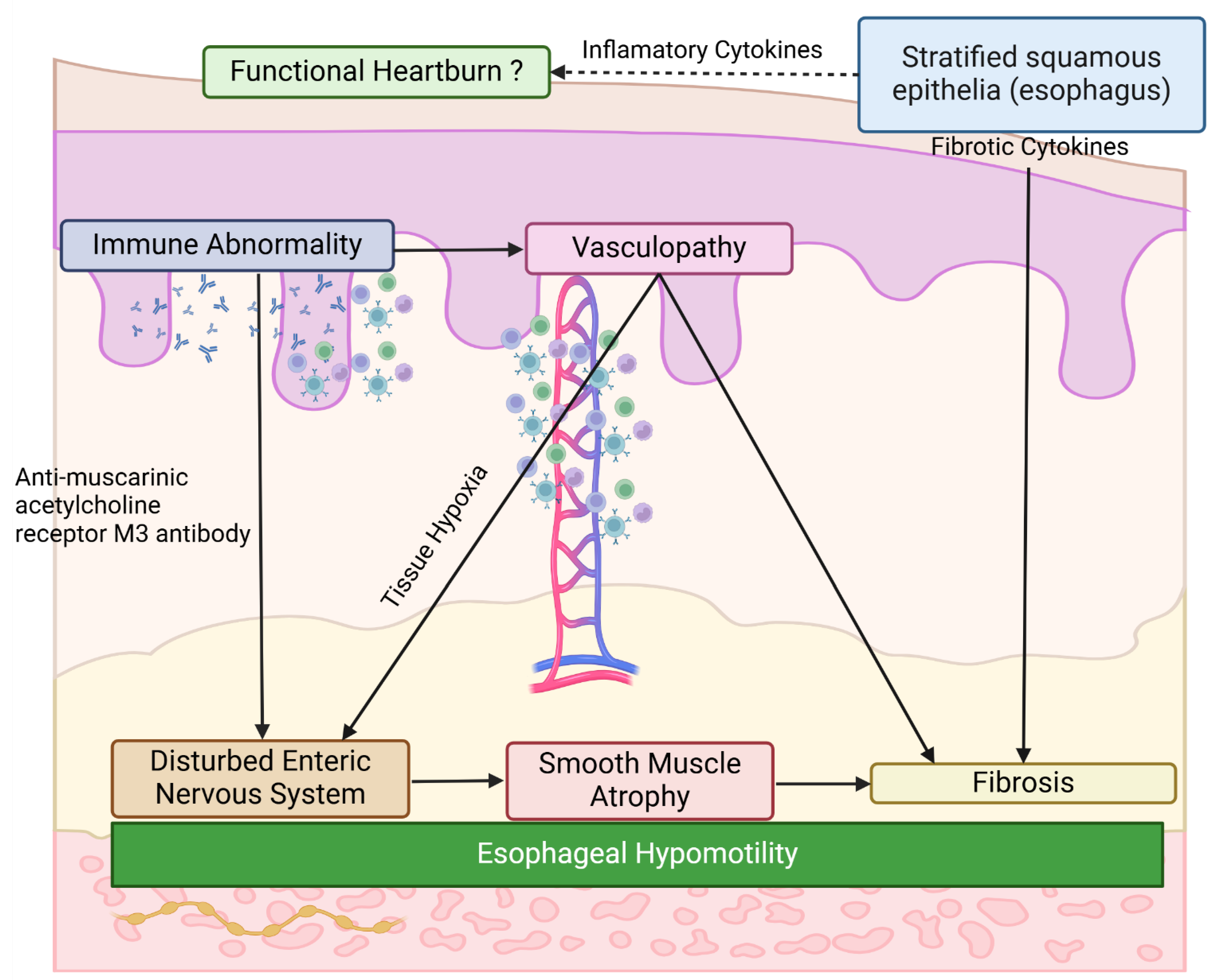
Figure 4.
Schematic diagram of the complex pulmonary pathology in SSc, encompassing both ILD and PH. [Upper panel: Interstitial Lung Disease (ILD)] SSc-ILD pathogenesis is driven by the common SSc-specific pathological cascade, leading to distinct histological stages of nonspecific interstitial pneumonia (NSIP), and in some cases, usual interstitial pneumonia (UIP). Gastroesophageal reflux disease (GERD) acts as an organ-specific modifier, potentially exacerbating ILD progression through microaspiration and contributing to centrilobular fibrosis (CLF). [Lower panel: Pulmonary Hypertension (PH)] SSc-PH encompasses pulmonary arterial hypertension (PAH) due to pulmonary arteriolar fibrosis (Group 1 WHO classification), as well as PH related to cardiac involvement (Group 2) or ILD (Group 3). SSc-PAH is characterized by pulmonary arteriole occlusion and may coexist with pulmonary veno-occlusive disease (PVOD)-like changes.
Figure 4.
Schematic diagram of the complex pulmonary pathology in SSc, encompassing both ILD and PH. [Upper panel: Interstitial Lung Disease (ILD)] SSc-ILD pathogenesis is driven by the common SSc-specific pathological cascade, leading to distinct histological stages of nonspecific interstitial pneumonia (NSIP), and in some cases, usual interstitial pneumonia (UIP). Gastroesophageal reflux disease (GERD) acts as an organ-specific modifier, potentially exacerbating ILD progression through microaspiration and contributing to centrilobular fibrosis (CLF). [Lower panel: Pulmonary Hypertension (PH)] SSc-PH encompasses pulmonary arterial hypertension (PAH) due to pulmonary arteriolar fibrosis (Group 1 WHO classification), as well as PH related to cardiac involvement (Group 2) or ILD (Group 3). SSc-PAH is characterized by pulmonary arteriole occlusion and may coexist with pulmonary veno-occlusive disease (PVOD)-like changes.
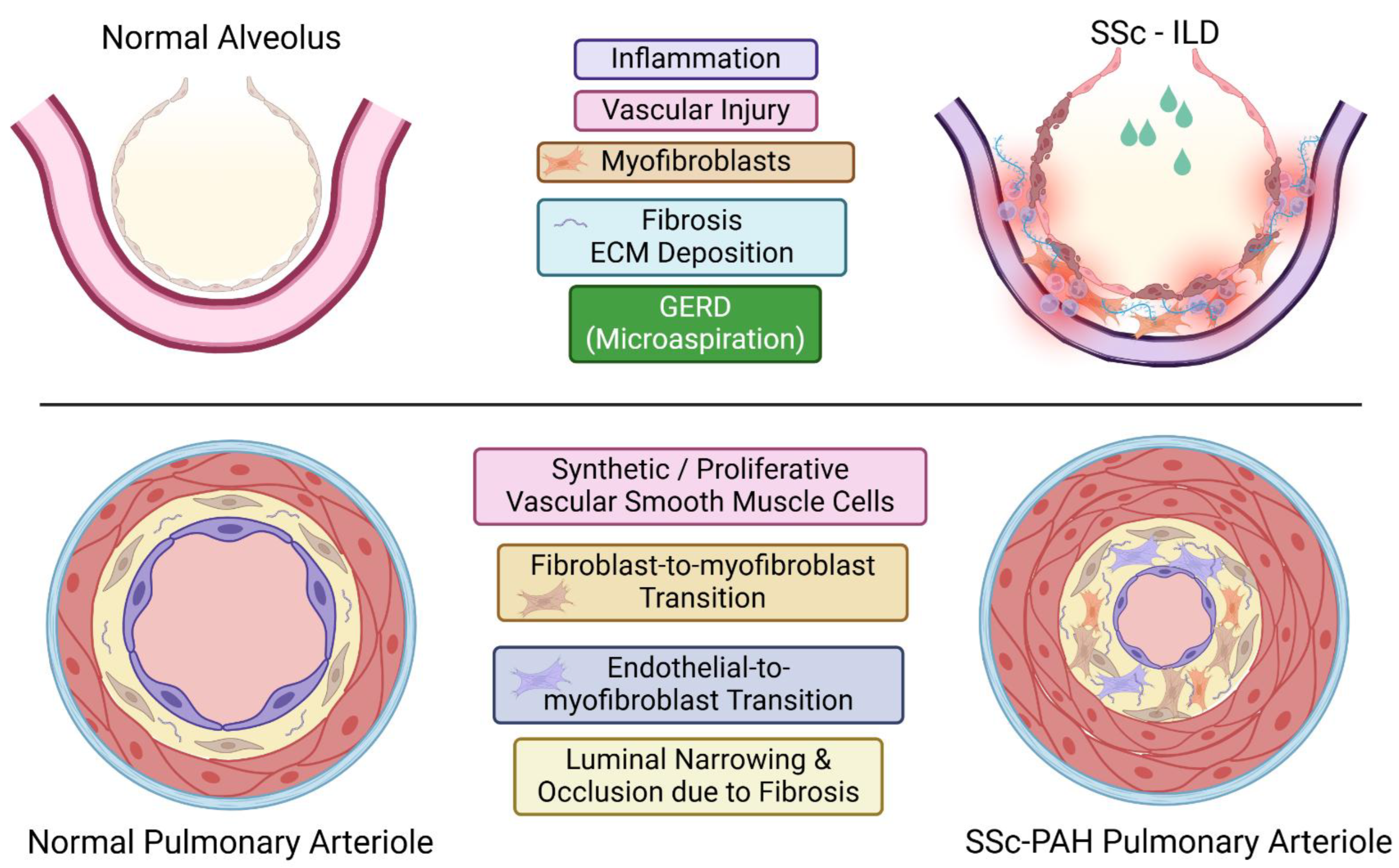
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



