
Submitted:
31 March 2025
Posted:
02 April 2025
You are already at the latest version
Abstract
Background: Several mitochondrial abnormalities such as defective energy production, depletion of energy stores, Ca2+-accumulation, generation of reactive oxygen species and impaired intracellular signaling are associated with cardiac dysfunction during the development of different heart diseases. Methods: A narrative review was compiled by a search for applicable literature in MEDLINE via PubMed. Results: Mitochondria generate ATP through the processes of electron transport and oxidative phosphorylation, which is used as energy for cardiac contractile function. Mitochondria in fact are the key subcellular organelle for the regulation of intracellular Ca2+ concentration and are considered to serve as a buffer to maintain Ca2+ homeostasis in cardiomyocytes. However, during the development of heart disease, excessive accumulation of intracellular Ca2+ results in mitochondria Ca2+-overload, which, in turn, impairs mitochondrial energy production and induces cardiac dysfunction. Mitochondria also generate reactive oxygen species (ROS), including superoxide and hydroxyl radicals as well as H2O2, a well-known oxidant that promotes lipid peroxidation and subsequent disturbance of Ca2+ homeostasis, cellular damage and death. Conclusion: Oxidative stress plays a critical role in mitochondrial disruption during the pathogenesis of different cardiac pathologies.
Keywords:
mitochondria
; oxidative stress
; Ca2+-handling defects
; cell death
; cardiac dysfunction
; heart disease
1. Introduction
Oxidative stress and intracellular Ca2+-overload are intimately involved in different cardiac pathologies including heart failure, diabetic cardiomyopathy and ischemia-reperfusion injury [1,2,3,4,5,6,7]. Such defects in cardiomyocyte Ca2+-handling have been attributed to subcellular remodeling during the development of heart disease [8,9,10,11,12,13,14]. Mitochondria are the major source of ATP production through oxidative phosphorylation and electron transport required for cardiac function [15,16,17,18] and are regarded as multifunctional organelles involved in cardiomyocyte function and integrity. Indeed, mitochondria regulate key processes including mitophagy, apoptosis, redox balance and Ca2+-homeostasis [19,20,21,22,23,24]. There is unequivocal evidence that mitochondria are a high source of reactive oxygen species (ROS), such as superoxide and hydroxyl radicals as well as the oxidant, H2O2, which promote lipid peroxidation leading to a dysregulation of cation homeostasis, cellular damage and cell death [25,26]. In addition, the excessive production of ROS can lead to an accumulation of 4-hydroxynonenal inside the mitochondria, which is a product of lipid peroxidation and reactive aldehyde, and is far more harmful than ROS [27]. Furthermore, the occurrence of oxidative stress that is accompanied by a depletion of antioxidant enzymes and other redox-regulating molecules, which exacerbates the imbalance between ROS generation and detoxification, contributing to the acceleration of myocardial abnormalities in structure and function.
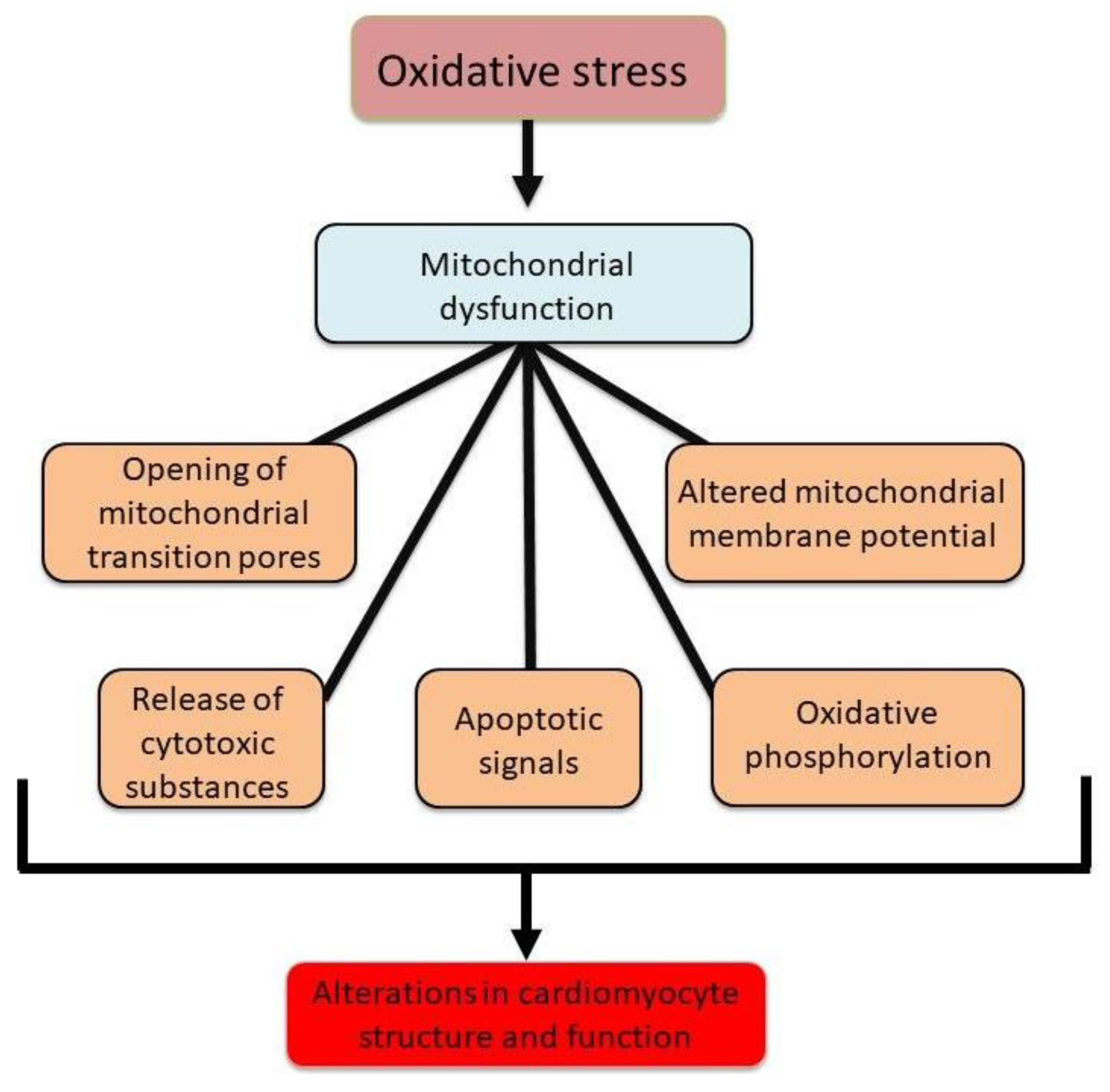
Mitochondria are known to accumulate a considerable amount of Ca2+ and thus are considered as a Ca2+ reservoir/sink designed to maintain the intracellular concentration of free Ca2+ ([Ca2+]i) within optimal range [15,16,28]. However, during the development of different cardiac diseases, an excessive amount of intracellular Ca2+ results in mitochondria Ca2+-overload that subsequently harms mitochondrial energy production [15,16,29]. Taken together, it can be seen from Figure 1 that mitochondrial dysfunction in different types of heart disease and pathophysiological conditions is a key component in the pathogenesis of cardiac dysfunction. This functional decline of the heart is strongly associated with excessive ROS generation, which originates from multiple sources, including NADPH oxidase, monoamine oxidase, and mitochondrial respiratory complexes I, II, and III. Notably, NADPH oxidase 4 (NOX4) has been identified to be present in mitochondria mainly and serves as a principal driver of oxidative stress in heart failure [30]. Figure 2 summarizes the alterations in mitochondrial ROS generating systems.
It should be mentioned that endothelium-associated xanthine oxidase (XO), which is known to generate superoxide anion, is activated by angiotensin II and NADPH oxidase activity in endothelial cells [31]. This surplus of superoxide anions generated through these pathways induces widespread damage to cellular macromolecules, including DNA, proteins, lipids, and carbohydrates, ultimately resulting in mitochondrial dysfunction and irreversible cytotoxicity [32]. Indeed, the interplay between ROS and mitochondrial components creates a self-amplifying cycle of oxidative damage, further exacerbating mitochondrial dysfunction, contractile impairment and overall progression of heart failure [33,34].
Oxidative stress represents a state of redox disequilibrium characterized by the excessive generation of ROS, including superoxide radicals, hydrogen peroxide, and hydroxyl radicals, alongside a concurrent reduction in endogenous antioxidant capacity [35]. The accumulation of mitochondrial ROS accompanied with a variety of different factors including heightened inflammatory response, formation of advanced glycation end-products and lipid peroxidation all collectively exacerbate oxidative stress (Figure 3).
The deleterious effects of oxidative stress are not limited to mitochondrial impairment, but also contribute to the pathological remodeling of the myocardium through the upregulation of pro-inflammatory cytokines and activation of fibroblasts in the extracellular matrix [36,37]. These mechanisms collectively drive interstitial fibrosis and increased myocardial stiffness, which are hallmarks of heart failure progression. It should be mentioned that Nrf2 (nuclear factor erythroid 2-related factor 2) is a transcription factor that regulates antioxidant responses and plays a critical role in cellular defense against oxidative stress, inflammation, and apoptosis. It is a key regulator of several genes for several endogenous antioxidants such as superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase, which are involved in protecting against the development of oxidative damage and mitochondrial dysfunction, making it a promising therapeutic target in cardiovascular diseases [38]. In fact, numerous studies suggest that Nrf2 activation is a crucial cardioprotective mechanism against the adverse effects of ischemic myocardial injury.
The prolonged exposure of the heart to high levels of circulating vasoactive hormones including angiotensin II and catecholamines in chronic myocardial infarction have been shown to induce Ca2+-handling abnormalities that have been linked to the occurrence of mitochondrial Ca2+-overload, mitochondrial dysfunction, and the generation of oxidative stress all leading to an impairment of cardiovascular function [39,40,41]. Both Ca2+ and oxidative stress are considered to induce conformational alterations in the mitochondrial cristae embedded F1/F0, ATP synthase and permit the formation of membrane permeability transition pores (MPTP) for releasing solutes and proteins, including cytochrome C, apoptosis-inducing factor and Smac/DIABLO, from the mitochondrial matrix [42,43,44]. If the MPTP remains in the open state, the cardiomyocyte is unable to sustain its ATP levels ultimately leading to mitochondrial stress, cell death and cardiac dysfunction [45].
Taken together, Figure 4 demonstrates the critical role of mitochondria in alterations in cardiomyocyte structure and function through modulating energy metabolism, formation of the MPTP and inducing apoptotic signals. In view of the importance of mitochondria in normal cell function, this narrative review is intended to describe the role of defects in mitochondrial energy generation, increased ROS production, dysregulation of Ca2+-handling and mitochondrial Ca2+overlaod as well as cell apoptosis in cardiac dysfunction in different pathophysiological conditions such as heart failure, diabetic cardiomyopathy and ischemia-reperfusion injury. Accordingly, appropriate literature searched on MEDLINE via PubMed by using the search terms: mitochondrial dysfunction, cardiac ischemia-reperfusion injury, diabetic cardiomyopathy, heart failure, reactive oxygen species, oxidative stress, Ca2+-handling, intracellular Ca2+-overload and combination thereof was conducted and the articles cited in this review were those selected to provide support of our hypothesis.
2. Evidence of Involvement of ROS and Ca2+-Overload in Cardiac Mitochondria
Mitochondria play a pivotal role in cellular redox signaling by generating ROS as by-products of oxidative phosphorylation [46,47,48]. However, not all oxidants have a role in signal transduction as it appears that this is dependent upon the cell type and animal species. Furthermore, low concentrations of oxidants or exposure for a transient period stimulate the signal transduction mechanisms for both cardiomyocyte function and gene expression for cell survival, while high concentrations of oxidants and/or exposure for a prolonged period of time produces oxidative stress and subsequent harmful outcomes [8,49].
Figure 4.
Mitochondrial abnormalities due to oxidative stress leading changes in cardiac function and structure.
Figure 4.
Mitochondrial abnormalities due to oxidative stress leading changes in cardiac function and structure.
The impairment of mitochondrial function by ROS generating systems and oxidants has been reported [50,51]. In this regard, normal rat hearts perfused with an ROS generating system, xanthine (X) plus xanthine oxidase (XO) has been shown to decrease mitochondrial state 3, uncoupled respiration and ADP-to-O ratio without any changes in the state 4 respiration (Figure 5). On the other hand, perfusion with a well known oxidant, H2O2 increased mitochondrial state 4 respiration and decreased the ADP-to-O ratio as well as mitochondrial state 3 and uncoupled respiration (Figure 5) [50]. The role of ROS and oxidants in mitochondrial impairment of oxidative stress was further demonstrated by the observations that the changes in mitochondrial function due to X plus XO were attenuated or prevented by the presence of SOD plus CAT, whereas those by H2O2 were attenuated by the presence of CAT plus mannitol, but not by CAT alone (Table 1) [50]. The impact of oxidant effect on [Ca2+]i is demonstrated by the data presented in Table 2. It was observed that the H2O2-induced increase in [Ca2+]i is concentration dependent (Table 2A) [51]. In contrast, incubation of cardiomyocytes with CAT before exposure to H2O2 attenuated the H2O2-induced increase in [Ca2+]i . It should be noted that mannitol did not exert any effect on the H2O2-induced increase in [Ca2+]i (Table 2B) [51]. Taken together, it can be inferred that the formation of H2O2 in different cardiac pathologies can induce changes in Ca2+-homeostasis in cardiomyocytes and induce cardiac contractile dysfunction.
3. Development of Mitochondrial Ca2+-Overload due to Oxidative Stress
Mitochondria participate in I/R-injury due to oxidative stress and dysregulation of Ca2+-homeostasis [52]. In addition, following I/R, cardiomyocytes accumulate high levels of peroxides, leading to mitochondrial dysfunction and induction of ferroptosis and exacerbation of ROS production and oxidative stress [53]. The oxidative stress induced abnormalities in Ca2+-handling are known to lead to mitochondrial Ca2+-overload resulting in impaired mitochondrial production of energy [54,55,56]. Interestingly, mitochondrial ATPase inhibitory factor-1, which is increased under conditions of oxidative stress has been reported to disturb mitochondrial Ca2+-handling; however, the loss of mitochondrial Ca2+ uniporter (mCUP) has been reported to trigger arrhythmias attributed to a probable effect on SR Ca2+-handling [57,58]. Interestingly, in I/R-injury, Ca2+-influx in to mitochondria is considered to occur through the mCUP; however, deletion of the mCUP has been reported to result in an increase in mitochondrial Ca2+, suggesting that some other mechanism may also be involved in Ca2+-influx [59]. It should also be mentioned that a defect in the cross talk between mitochondrial function and control of ryanodine-receptor-mediated SR Ca2+-release has been linked to an increase in the risk of arrhythmia in heart disease [60]. Clearly, targeting Ca2+-homeostasis in cardiomyocytes and mitochondrial Ca2+-overload due to oxidative stress would be seen as beneficial in attenuating calcium dysregulation in heart disease, including myocardial infarction, heart failure and cardiomyopathies [61].
The sensitivity of mitochondria to Ca2+ concentrations is critical, as both excessive and deficient Ca2+ levels can impair mitochondrial oxidative phosphorylation. High-glucose conditions in cardiomyocytes have been shown to reduce mCUP expression, decrease mitochondrial Ca2+ levels, and alter glucose and lipid metabolic profiles, further compromising cardiac function [62]. Mitochondrial Ca2+-overload, in turn, contributes to oxidative stress, which exacerbates mitochondrial dysfunction and creates a vicious cycle of cellular injury. This cascade ultimately leads to apoptosis or necrosis, further impairing both systolic and diastolic heart function [63]. In the context of diabetic cardiomyopathy, the role of mCUP and its regulatory subunit, mitochondrial calcium uptake protein 1 (MICU1), has emerged as a critical factor in Ca2+-transport. It has been shown that in diabetic mice, there is an upregulation of MICU1 expression in the heart, accompanied by a downregulation of MCU and associated regulatory proteins, such as EMRE, a key mCUP subunit. This imbalance leads to compromised mitochondrial Ca2+- uptake, diminished mitochondrial function, and consequently reduced cardiac performance.
Mitochondria Ca2+ accumulation serves as a key trigger of mitochondrial dysfunction, especially when it occurs in the presence of additional stressors such as oxidative or nitrosative stress [64]. Ca2+ signaling has emerged as a critical modulator of mitochondrial function, with evidence indicating that Ca2+ contributes to the initiation of mitochondrion-dependent apoptosis [65]. Mitochondria serve as both ATP producers and crucial intracellular Ca2+ buffers. The mCUP, located on the inner mitochondrial membrane, plays a pivotal role in mediating Ca2+ influx into the mitochondrial matrix. Under normal physiological conditions, even modest fluctuations in Ca2+ levels are sufficient to activate dehydrogenases like FoF1-ATP, promoting ATP synthesis. However, under pathophysiological conditions, these processes are disrupted. Inositol trisphosphate receptors (IP₃Rs) are essential for maintaining intracellular calcium homeostasis. The release of Ca²⁺ from IP₃Rs functions as a second messenger, orchestrating various intracellular processes and inter-organellar communication in both physiological and pathological contexts. Over activation of IP₃Rs has been linked to the pathogenesis of several cardiac disorders, including ischemia, diabetes-induced arrhythmias, and cardiac hypertrophy. Dysregulated Ca²⁺ signaling within cytosolic, mitochondrial, and nucleoplasmic compartments contributes to the progression of these diseases [66]. Interestingly, an adaptive mechanism through which mitochondria mitigate ROS-induced damage is apoptosis induction. This process involves an increase in mitochondrial outer membrane permeability, leading to solute and water influx into the matrix, loss of membrane potential, cessation of ATP synthesis, and excessive mitochondrial calcium uptake—culminating in complete mitochondrial failure [67].
4. Mitochondrial Metabolic Alterations and Mitochondrial Dynamics
It is well established that mitochondrial dysfunction is a hallmark of cardiovascular diseases (CVDs), manifesting as impaired oxidative phosphorylation, excessive ROS production, altered calcium signaling, and disrupted metabolic homeostasis. Conditions such as ischemia-reperfusion injury, hypertension and diabetic cardiomyopathy are associated with compromised mitochondrial energetics and structural integrity, culminating in cardiac contractile dysfunction [68,69,70,71,72,73,74]. The heart is an energetically demanding organ, necessitating a continuous and substantial supply of adenosine triphosphate (ATP) to sustain contractile function. Despite its limited ATP reserves, the heart maintains an exceptionally efficient bioenergetic system, predominantly driven by mitochondria, which constitute nearly 30% of cardiomyocyte volume. These organelles facilitate ATP generation through oxidative phosphorylation, orchestrating substrate oxidation, electron transport, and ATP synthesis to meet the heart’s metabolic demands [75,76,77,78,79]. Mitochondrial function is intricately linked to substrate availability, with ATP synthesis reliant on the oxidation of fatty acids, glucose, ketone bodies, and amino acids. It should be mentioned that biologically active amines such as spermine and agmatine have distinct roles in mitochondrial function that differentiate them from other amines; notably, spermine undergoes oxidative deamination by amine oxidases, producing ROS, which may further exacerbate the opening of the MPTP and contribute to apoptosis [44].
In the healthy myocardium, fatty acid oxidation contributes approximately 60–70% of ATP production, while glucose metabolism accounts for 20–30% [80,81,82,83]. The efficiency of ATP synthesis per unit of oxygen is higher for glucose than for fatty acids, a critical factor under hypoxic or ischemic conditions [84,85,86,87]. The intrinsic compensatory mechanisms that regulate intracellular calcium and the antioxidant defense system, which typically maintain mitochondrial substrate oxidation and ATP generation, become insufficient in the context of chronic cardiac dysfunction [88,89,90]. It should be mentioned that insulin resistance in diabetes reduces glucose transporter expression and pyruvate dehydrogenase activity, shifting myocardial energy reliance towards fatty acid β-oxidation. This metabolic shift increases oxygen consumption while decreasing ATP yield efficiency, predisposing mitochondria to oxidative stress and lipotoxic damage [91,92,93,94]. Prolonged metabolic perturbations, including excessive fatty acid uptake and β-oxidation inefficiencies, promote lipid accumulation, mitochondrial dysfunction, and cardiomyocyte apoptosis. These maladaptive changes contribute to myocardial energy deficits, compromised contractility, and heightened susceptibility to heart failure [95,96,97,98,99,100]. In addition, the chronic dysregulation of glycolipid metabolism in diabetes leads to both excessive ROS production and impaired ROS clearance. Mitochondria serve as the primary source of ROS in diabetic cardiomyocytes, and their dysfunction perpetuates a vicious cycle of oxidative damage. This process severely compromises cardiomyocyte function and survival by exacerbating metabolic disturbances, energy depletion, and oxidative stress-driven apoptosis [101]. However, it should be noted that chronic hyperglycemia during diabetes, further increases mitochondrial ROS production and impairs endogenous antioxidant defense mechanisms thus leading to excessive apoptosis and myocardial dysfunction [102,103]. A schematic diagram indicating the role of oxidative stress in inducing mitochondrial metabolic changes associated with depression in energy stores, cellular death and lipid deposits in cardiomyocytes and subsequent cardiac dysfunction in diseased hearts is shoen in Figure 6.
It should be pointed out that mitochondrial generated oxidative stress, signal transduction, metabolic reprogramming, regulation of iron and cell death depend on the mitochondrial quality control (MQC) system that includes mitochondrial dynamics (fission and fusion cycles) and mitochondrial biogenesis to maintain structural integrity and cardiac function [104,105,106]. Thus, targeting mitochondrial bioenergetics and metabolic flexibility represents a promising therapeutic strategy for mitigating CVD progression and preserving cardiac function. The mitochondrial ribosomal protein S5 (MRPS5/uS5m) is essential for maintaining mitochondrial protein translation and oxidative phosphorylation. Loss of MRPS5 in the developing heart leads to embryonic lethality, while postnatal loss impairs oxidative phosphorylation and mitochondrial protein synthesis, contributing to cardiac hypertrophy and heart failure [107]. Mitochondrial dynamics, including fusion and fission processes, are integral to maintaining mitochondrial function and integrity [108]. Fusion proteins such as Mfn-1, Mfn-2, and OPA-1 are essential for mitochondrial stability, as their inhibition leads to dilated cardiomyopathy, contractile dysfunction, increased apoptosis, and mitochondrial fragmentation [109]. Deletion of these fusion proteins results in abnormal mitochondrial morphology, ventricular wall thickening, and eccentric hypertrophy. Conversely, excessive mitochondrial fission disrupts mitochondrial mass, impairs oxidative phosphorylation, and results in ATP deficits, mitochondrial permeabilization, cytochrome C release, and apoptosis. The absence of dynamin-related protein 1 (Drp1), a key fission protein, results in lethal dilated cardiomyopathy [110], further highlighting the critical balance between fusion and fission in maintaining cardiac mitochondrial function.
Alterations in mitochondrial ultrastructure and bioenergetics are widely observed in heart failure patients, particularly in the later disease stages. These changes include reductions in the activities of respiratory chain complexes (I–IV) and impaired oxidative phosphorylation capacity. Interestingly, in cases of chronic hypertrophy without systolic dysfunction, mitochondrial function appears to be preserved or even enhanced in both animal models and human studies [111,112,113,114,115,116,117,118,119,120,121,122]. Initial stages of cardiac hypertrophy are often characterized by an increase in oxidative phosphorylation activity, which gradually declines as the disease progresses toward heart failure [16,123]. A reduction in the expression of key oxidative phosphorylation components has been linked to mitochondrial respiratory deficits in heart failure and cardiomyopathies [124,125]. It is pointed out that in ventricular fibrillation, mitochondrial damage activates the mitochondrial apoptotic pathway, characterized by the release of cytochrome C into the cytosol, a reduction in caspase-9 levels, and the subsequent activation of caspase-3. This cascade coincides with significant impairments in LV function. Notably, cytochrome C "leaks" into the bloodstream, and its concentration is inversely proportional to survival outcomes [126]. Taken together, these findings underline the critical role of mitochondria during cardiac resuscitation by modulating both energy metabolism and apoptotic signaling pathways, positioning mitochondrial-targeted therapies as promising strategies for enhancing outcomes during cardiac resuscitation [126].
5. Novel Interventions Targeting Mitochondria in Different Cardiac Pathologies
In view of the fact that mitochondria constitute 30-40% of the cardiomyocyte [127], and that mitochondria are considered as the key determinants of cardiac injury and dysfunction [128,129,130,131,132], mitochondria represent a viable target for therapeutic intervention. For example, the regulation of mitochondrial Ca2+- uptake and the interplay between various molecules and pathways offer promising avenues for therapeutic intervention. Mitochondria have been suggested to exhibit a cardioprotective role due to the presence of KATP channels [133]. In this regard, cardioprotection via hypoxic preconditioning or exposure to the ATP-dependent K+- channel opener diazoxide increases mitochondrial resistance to oxidative damage. Thus, targeting the MPTP, either by direct inhibition or modulation of mitochondrial stressors, represents a promising therapeutic approach for conditions such as I/R-injury [65,134]. Interestingly, the reperfusion injury salvage kinase (RISK) pathway for cardioprotection involves the prevention of the opening of the membrane permeability transition pore and subsequent attenuation of cell death [135]. Thus, this pathway has emerged as an important cardioprotective target in I/R-injury.
In reperfusion-induced injury is a significant challenge during cardiac surgery, coronary thrombosis treatment, and stroke management. Preventing MPTP opening, either directly with agents like cyclosporine A or indirectly by reducing oxidative stress or Ca2+- overload, represents a potential therapeutic strategy to mitigate reperfusion injury. Additionally, mice deficient in Cyclophilin D (CyP-D), a critical component of the MPTP, are protected from ischemia/reperfusion injury in the heart, further substantiating the role of MPTP in mediating cellular injury [136]. In less severe cellular insults, the MPTP may open transiently, leading to mitochondrial swelling sufficient to trigger cytochrome C release and activation of the apoptotic pathway, rather than necrosis. However, CyP-D knockout mice do not exhibit enhanced protection against a broad range of apoptotic stimuli, suggesting that the MPTP is not universally involved in apoptosis [136]. Recently, it has been suggested that circadian rhythm may play an important role in the control of I/R-injury [137]. MiRNAs regulate mitochondrial apoptosis through an effect on mitochondrial fission and fusion, generation of ROS and dysregulating Ca2+-homeostasis [138]. Interestingly, it has been suggested that mitochondrial transplantation has the potential to exert beneficial actions in I/R-injury; however, clinical application is limited [139]. On the other hand, mitochondrial regeneration can be seen to increase oxidative phosphorylation and decrease oxidative stress and thus may be of clinical value under conditions of ischemic insult of the heart [140]. In fact, targeting MQC has emerged as a promising target in mitigating hypoxia-related cardiac dysfunction [141].
In the context of diabetic cardiomyopathy, the role of mCUP and its regulatory subunit, mitochondrial calcium uptake protein 1 (MICU1), has emerged as a critical factor in Ca2+- transport. Studies have shown that in diabetic mice, there is an upregulation of MICU1 expression in the heart, accompanied by a downregulation of MCU and associated regulatory proteins, such as EMRE, a key mCUP subunit. This imbalance leads to compromised mitochondrial Ca2+- uptake, diminished mitochondrial function, and, consequently, reduced cardiac performance. Importantly, restoring MCU expression has been shown to ameliorate both mitochondrial and cardiac dysfunction, highlighting the therapeutic potential of restoring mitochondrial Ca2+ homeostasis in diabetic cardiomyopathy treatment [142,143]. Distinct isoforms of IP₃Rs, IP₃R1 and IP₃R2, exhibit different roles in cardiac pathology. IP₃R1 is particularly involved in cardiac ischemia and arrhythmias associated with diabetes, while IP₃R2 is implicated in sepsis-induced cardiomyopathy and hypertrophy [66]. Thus, IP₃Rs have been shown to play pivotal roles in various forms of cell death, such as apoptosis, pyroptosis, and ferroptosis, underlining their multifaceted involvement in cardiac disease. Targeting IP₃Rs, either through genetic manipulation or pharmacological inhibition using IP₃R antagonists, has emerged as a promising therapeutic strategy to mitigate IP₃R-related pathologies, offering potential for therapeutic intervention in CVD [66].
6. Conclusion
From the foregoing discussion, it is evident that mitochondria are not only involved in energy production, but are also the major source of oxidative stress production as well as intracellular Ca2+- accumulation. In fact, the development of oxidative stress and the occurrence of mitochondrial Ca2+- overload are the main mechanisms for induction of energy store depletion and cardiac dysfunction. It is noteworthy that ROS generated by the activation of sarcolemmal NADPH oxidase 2 as well as extra-mitochondrial (endothelial cells, serum, cytosol) xanthine oxidase are also considered to promote the generation of mitochondrial oxidative stress during the development of heart disease [7,8,144,145,146,147]. It should be mentioned that mitochondria are known to contain different components for the production of ROS such as the electron transport chain, NADPH oxidase 4 and monoamine oxidase, in addition to endogenous antioxidants such as SOD, CAT and glutathione peroxidase. Particularly, it may also be noted that there occurs accumulation of different vasoactive hormones such as angiotensin II and endothelin (activators of NADPH oxidase 4) as well as catecholamines and serotonin (substrates for oxidation by MAO) in cardiomyocytes of diseased hearts. Furthermore, the occurrence of mitochondrial Ca2+- overload has been associated with the activation of MAO and the induction of defects in electron transport system in mitochondria in ischemic heart disease.
There is now a wealth of information that has demonstrated that mitochondrial Ca2+- overload and increased generation of ROS are central features in cardiac dysfunction in different cardiac pathologies including heart failure, diabetic cardiomyopathy and ischemia-reperfusion injury. Although mitochondria accumulate high amounts of Ca2+ and thus serve as an intracellular Ca2+ reservoir, abnormalities in the processes involved in energy production through oxidative phosphorylation produce an oxidative stress that impacts the structural and functional integrity of the cell. Indeed, ROS-induced ROS production by mitochondria exacerbates ROS generation and severity of oxidative stress. The mitochondria-generated ROS as a consequence of mitochondrial Ca2+-overload leads to further deterioration of mitochondrial function. Accordingly, the mitochondria present viable therapeutic target for the prevention of cardiac dysfunction in at-risk population. Therefore, the development of specific interventions that are effective in attenuating mitochondrial metabolic alterations as well as the development of novel antioxidants that target mitochondrial ROS generating systems could be highly beneficial.
Author Contributions
“Conceptualization, N.S.D.; resources, P.S.T.; P.O.; writing—original draft preparation, P.S.T.; P.O.; writing—review and editing, N.S.D. All authors have read and agreed to the published version of the manuscript.”
Funding
This project did not receive any external funding.
Data Availability Statement
The original contributions presented in this study are included in the article.
Acknowledgments
The infrastructural support of this article was provided by the St. Boniface Hospital Albrechtsen Research Centre.
Conflicts of Interest
The authors have no conflict of interest.
References
- Bolli, R.; Marban, E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 1999, 79, 609–634. [Google Scholar] [CrossRef]
- Bolli, R.; Jeroudi, M.O.; Patel, B.S.; Aruoma, O.I.; Halliwell, B.; Lai, E.K.; McCay, P.B. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial “stunning” is a manifestation of reperfusion injury. Circ Res 1989, 65, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res 2000, 47, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Reimer, K.A. The cell biology of acute myocardial ischemia. Annu Rev Med 1991, 42, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Piper, H.M.; Meuter, K.; Schafer, C. Cellular mechanisms of ischemia reperfusion injury. Ann Thorac Surg 2003, 75, S644–S648. [Google Scholar] [CrossRef]
- Dridi, H.; Santulli, G.; Bahlouli, L.; Miotto, M.C.; Weninger, G.; Marks, A.R. Mitochondrial calcium overload plays a causal role in oxidative stress in the failing heart. Biomolecules 2023, 13, 1409. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Ostadal, P.; Tappia, P.S. Involvement of oxidative stress and antioxidants in modification of cardiac dysfunction due to ischemia-reperfusion injury. Antioxidants 2025, 14, 340. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of oxidative stress in the development of subcellular defects and heart disease. Biomedicines 2022, 10, 393. [Google Scholar] [CrossRef]
- Kukreja, R.C.; Weaver, A.B.; Hess, M.L. Sarcolemmal Na-K-ATPase: inactivation by neutrophil-derived free radicals and oxidants. Am J Physiol Heart Circ Physiol 1990, 259, H1330–H1336. [Google Scholar] [CrossRef]
- Ostadal, P.; Elmoselhi, A.B.; Zdobnicka, I.; Lukas, A.; Elimban, V.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced changes in Na-K ATPase isoform expression in rat heart. Antioxid Redox Signal 2004, 6, 914–923. [Google Scholar] [CrossRef]
- Saini, H.K.; Dhalla, N.S. Defective calcium handling in cardiomyocytes isolated from hearts subjected to ischemia-reperfusion. Am J Physiol Heart Circ Physiol 2005, 288, H2260–H2270. [Google Scholar] [CrossRef]
- Saini, H.K.; Elimban, V.; Dhalla, N.S. Attenuation of extracellular ATP response in cardiomyocytes isolated from hearts subjected to ischemia reperfusion. Am J Physiol Heart Circ Physiol 2005, 289, H614–H623. [Google Scholar] [CrossRef]
- Temsah, R.M.; Netticadan, T.; Chapman, D.; Takeda, S.; Mochizuki, S.; Dhalla, N.S. Alterations in sarcoplasmic reticulum function and gene expression in ischemic reperfused rat heart. Am J Physiol Heart Circ Physiol 1999, 277, H584–H594. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, R.; Ronca-Testoni, S.; Yu, G.; Galbani, P.; Ronca, G.; Mariani, M. Effect of ischemia and reperfusion on cardiac ryanodine receptors sarcoplasmic reticulum Ca2+ channels. Circ Res 1994, 74, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R. The role of mitochondria in ischemic heart disease. J Cardiovasc Pharmacol 1996, 28 Suppl 1, S1–S10. [Google Scholar]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J Mol Cell Cardiol 2001, 33, 1065–1089. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R. Mitochondrial function in the heart. Annu Rev Physiol 1979, 41, 485–506. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Dhalla, N.S. Status of mitochondrial oxidative phosphorylation during the development of heart failure. Antioxidants 2023, 12, 1941. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol Cell Biol 2018, 19, 713–730. [Google Scholar]
- Lopez-Crisosto, C.; Pennanen, C.; Vasquez-Trincado, C.; Morales, P.E.; Bravo-Sagua, R.; Quest, A.F.G.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum–mitochondria communication in cardiovascular pathophysiology. Nat Rev Cardiol 2017, 14, 342–360. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and mitophagy in cardiovascular disease. Circ Res 2017, 120, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Budde, H.; Hassoun, R.; Tangos, M.; Zhazykbayeva, S.; Herwig, M.; Varatnitskaya, M.; Sieme, M.; Delalat, S.; Sultana, I.; Kolijn, D.; et al. The interplay between S-glutathionylation and phosphorylation of cardiac troponin I and myosin binding protein C in end-stage human failing hearts. Antioxidants 2021, 10, 1134. [Google Scholar] [CrossRef]
- Leichert, L.I.; Gehrke, F.; Gudiseva, H.V.; Blackwell, T.; Ilbert, M.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Jakob, U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci 2008, 105, 8197–8202. [Google Scholar] [CrossRef]
- Delbridge, L.M.D.; Mellor, K.M.; Taylor, D.J.; Gottlieb, R.A. Myocardial stress and autophagy: Mechanisms and potential therapies. Nat Rev Cardiol 2017, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med 2005, 38, 1278–1295. [Google Scholar] [CrossRef] [PubMed]
- Tani, M. Effects of anti-free radical agents on Na, Ca2+, and function in reperfused rat hearts. Am J Physiol Heart Circ Physiol 1990, 259, H137–H143. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Parini, A. Cardiac monoamine oxidases: at the heart of mitochondrial dysfunction. Cell Death Dis 2020, 11, 54. [Google Scholar] [CrossRef]
- Ferrari, R.; Pedersini, P.; Bongrazio, M.; Gaia, G.; Bernocchi, P.; Di Lisa, F.; Visioli, O. Mitochondrial energy production and cation control in myocardial ischaemia and reperfusion. Basic Res Cardiol 1993, 88, 495–512. [Google Scholar] [CrossRef]
- Long, X.; Goldenthal, M.J.; Wu, G.M.; Marin-Garcia, J. Mitochondrial Ca2+-flux and respiratory enzyme activity decline are early events in cardiomyocyte response to H2O2. J Mol Cell Cardiol 2004, 37, 63–70. [Google Scholar] [CrossRef]
- Stevenson, M.D.; Canugovi, C.; Vendrov, A.E.; Hayami, T.; Bowles, D.E.; Krause, K.H.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates inflammation in ischemic heart failure: Role of soluble epoxide hydrolase. Antioxid Redox Signal 2019, 31, 39–58. [Google Scholar] [CrossRef]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler Thromb Vasc Biol 2007, 27, 943–948. [Google Scholar] [CrossRef]
- Dia, M.; Gomez, L.; Thibault, H.; Tessier, N.; Leon, C.; Chouabe, C.; Ducreux, S.; Gallo-Bona, N.; et al. Reduced reticulum-mitochondria Ca2+ transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res Cardiol 2020, 115, 74. [Google Scholar] [CrossRef]
- Stowe, D.F.; Camara, A.K.S. Mitochondrial reactive oxygen species production in excitable cells: Modulators of mitochondrial and cell function. Antioxid Redox Signal 2009, 11, 1373–1414. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival anti-oxidant. Antioxid Redox Signal 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Scialò, F.; Sriram, A.; Stefanatos, R.; Spriggs, R.V.; Loh, S.H.Y.; Martins, L.M.; Sanz, A. Mitochondrial complex I derived ROS regulate stress adaptation in Drosophila melanogaster. Redox Biology, 2020, 32, 101450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dhalla, N.S. The Role of pro-inflammatory cytokines in the pathogenesis of cardiovascular disease. Int J Mol Sci 2024, 25, 1082. [Google Scholar] [CrossRef]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res 2009, 81, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, J.; Dmitrii, G.; Zhang, X.A. Potential targets of natural products for improving cardiac ischemic injury: The role of Nrf2 signaling transduction. Molecules 2024, 29, 2005. [Google Scholar] [CrossRef]
- Qin, F.; Yan, C.; Patel, R.; Liu, W.; Dong, E. Vitamins C and E attenuates apoptosis, β-adrenergic receptor desensitization, and sarcoplasmic reticular Ca2+ ATPase downregulation after myocardial infarction. Free Radic Biol Med 2006, 40, 1827–1842. [Google Scholar] [CrossRef]
- Shao, Q.; Ren, B.; Elimban, V.; Tappia, P.S.; Takeda, N.; Dhalla, N.S. Modification of sarcolemmal Na+-K+-ATPase and Na+/Ca2+ exchanger in heart failure by blockade of renin-angiotensin system. Am J Heart Physiol Circ Physiol 2005, 288, H2637–H2646. [Google Scholar] [CrossRef]
- Li, Q.; Pogwizd, S.M.; Prabhu, S.D.; Zhou, L. Inhibiting Na+-K+ ATPase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS ONE 2014, 9, e93928. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.T.; Mani, K.; Nam, Y.J.; Kitsis, R.N. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res 2004, 95, 957–970. [Google Scholar] [CrossRef]
- Bertero, E.; Popoiu, T.A.; Maack, C. Mitochondrial calcium in cardiac ischemia/reperfusion injury and cardioprotection. Basic Res Cardiol 2024, 119, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Grancara, S.; Ohkubo, S.; Artico, M.; Ciccariello, M.; Manente, S.; Bragadin, M.; Toninello, A.; Agostinelli, E. Milestones and recent discoveries on cell death mediated by mitochondria and their interactions with biologically active amines. Amino Acids 2016, 48, 2313–2326. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Pinton, P. Modulation of mitochondrial permeability transition pores in reperfusion injury: Mechanisms and therapeutic approaches. Eur J Clin Invest 2025, 55, e14331. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Ferko, M.; Alanova, P.; Janko, D.; Opletalova, B.; Andelova, N. Mitochondrial peroxiredoxins and monoamine oxidase-A: Dynamic regulators of ROS signaling in cardioprotection. Physiol Res 2024, 73, 887–900. [Google Scholar] [CrossRef]
- Popoiu TA, Maack, C., Bertero, E. Mitochondrial calcium signaling and redox homeostasis in cardiac health and disease. Front Mol Med 2023, 3, 1235188. [CrossRef]
- Tappia, P.S.; Dent, M.R.; Dhalla, N.S. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic Biol Med 2006, 41, 349–361. [Google Scholar] [CrossRef]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. Am J Physiol Heart Circ Physiol 2007, 292, H1986–H1994. [Google Scholar] [CrossRef]
- Wang, X.; Takeda, S.; Mochizuki, S.; Jindal, R.; Dhalla, N.S. Mechanisms of hydrogen peroxide-induced increase in intracellular calcium in cardiomyocytes. J Cardiovasc Pharmacol Ther 1999, 4, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chang, X.; Zhao, D.; He, Y.; Dong, G.; Gao, L. Mitochondria and myocardial ischemia/reperfusion injury: Effects of Chinese herbal medicine and the underlying mechanisms. J Pharm Anal 2025, 15, 101051. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, X.; Li, X.; Wang, K. Molecular therapy of cardiac ischemia-reperfusion injury based on mitochondria and ferroptosis. J Mol Med 2023, 101, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Juhaszova, M.; Aon, M.A.; Zorov, D.B.; Sollott, S.J. Mitochondrial Ca2+, redox environment and ROS emission in heart failure: Two sides of the same coin? J Mol Cell Cardiol 2021, 151, 113–125. [Google Scholar] [CrossRef]
- Pavez-Giani, M.G.; Sanchez-Aguilera, P.I.; Bomer, n.; Miyamota, S.; Booij, H.G.; Giraldo, P.; Oberdorf-Maass, S.U.; Nijholt, K.T.; Yurista, S.R.; Milting, H.; et al. ATPase inhibitory factor-1 disrupts mitochondrial Ca2+ handling and promotes pathological cardiac hypertrophy through CaMKIIδ. Int J Mol Sci 2021, 22, 4427. [Google Scholar] [CrossRef]
- Bertero, E.; Nickel, A.; Kohlhaas, M.; Hohl, M.; Sequeira, V.; Brune, C.; Schwemmlein, J.; Abeber, M.; Schuh, K.; Kutschka, I.; et al. Loss of mitochondrial Ca2+ uniporter limits inotropic reserve and provides trigger and substrate for arrhythmias in Barth syndrome cardiomyopathy. Circulation 2021, 144, 1694–1713. [Google Scholar] [CrossRef]
- Donoso, P.; Sanchez, G.; Bull, R.; Hidalgo, C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci 2011, 16, 553–567. [Google Scholar] [CrossRef]
- Tokuhisa, T.; Yano, M.; Obayashi, M.; Noma, T.; Mochizuki, M.; Oda, T.; Okuda, S.; Doi, M.; Liu, J.; Ikeda, Y.; et al. AT1 receptor antagonist restores cardiac ryanodine receptor function, rendering isoproterenol-induced failing heart less susceptible to Ca2+-leak induced by oxidative stress. Circ J 2006, 70, 777–786. [Google Scholar] [CrossRef]
- Murphy, E.; Eisner, D.A. How does mitochondrial Ca2+ change during ischemia and reperfusion? Implications for activation of the permeability transition pore. J Gen Physiol 2025, 157, e202313520. [Google Scholar] [CrossRef]
- Hamilton, S.; Terentyeva, R.; Clements, R.T.; Belevych, A.E.; Terentyev, D. Sarcoplasmic reticulum-mitochondria communication; implications for cardiac arrhythmia. J Mol Cell Cardiol 2021, 156, 105–113. [Google Scholar] [CrossRef]
- Morciano, G.; Rimessi, A.; Patergnani, S.; Vitto, V.A.M.; Danese, A.; Kahsay, A.; Palumbo, L.; Bonora, M.; et al. Calcium dysregulation in heart diseases: Targeting calcium channels to achieve a correct calcium homeostasis. Pharmacol Res 2022, 177, 106119. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Juarez, J.; Suarez, J.; Cividini, F.; Scott, B.T.; Diemer, T.; Dai, A.; Dillmann, W.H. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am J Physiol Cell Physiol 2016, 311, C1005–C1013. [Google Scholar] [CrossRef]
- Uchi, J.; Ryu, S.Y.; Jhun, B.S.; Hurst, S.; Sheu, S.S. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antiox Redox Signal 2014, 21, 987–1006. [Google Scholar] [CrossRef]
- Duchen, M.R. Roles of mitochondria in health and disease. Diabetes 2004, 53, S96–S102. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Csordás, G.; Hajnóczky, G. Mitochondrial Ca2+ signaling and cardiac apoptosis. Biol Signals Recept 2001, 10, 200–223. [Google Scholar] [CrossRef] [PubMed]
- Piamsiri, C.; Fefelova, N.; Pamarthi, S.H.; Gwathmey, J.K.; Chattipakorn, S.C.; Chattipakorn, N.; Xie, L.H. Potential roles of IP3 receptors and calcium in programmed cell death and implications in cardiovascular diseases. Biomolecules 2024, 14, 1334. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J Hypertens 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.S.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 2007, 177, 1029–1036. [Google Scholar] [CrossRef]
- Stein, L.R.; Imai, S.-I. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab 2012, 23, 420–428. [Google Scholar]
- Chen, W.W.; Birsoy, K.; Mihaylova, M.M.; Snitkin, H.; Stasinski, I.; Yucel, B.; Bayraktar, E.C.; Carette, J.E.; Clish, C.B.; Brum-melkamp, T.R.; et al. Inhibition of ATPIF1 ameliorates severe mitochondrial respiratory chain dysfunction in mammalian cells. Cell Rep 2014, 7, 27–34. [Google Scholar] [CrossRef]
- Wrogemann, K.; Nylen, E.G. Mitochondrial calcium overloading in cardiomyopathic hamsters. J Mol Cell Cardiol 1978, 10, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Lee, S.-L.; Shah, K.R.; Elimban, V.; Suzuki, S.; Jasmin, G. Behaviour of subcellular organelles during the development of congestive heart failure in cardiomyopathic hamsters (UM-X7. 1). In Cardiomyopathic Heart; New York Raven Press Ltd.: New York, NY, USA, 1994; pp. 1–14. [Google Scholar]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases—From pathophysiology to treatment. Ann Transl Med 2018, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Marcillat, O.; Giulivi, C.; Ernster, L.; Davies, K.J. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem 1990, 265, 16330–16336. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Williams, M. Mitochondrial function and dysfunction: An update. J Pharmacol Exp Ther 2012, 342, 598–607. [Google Scholar] [CrossRef]
- Van Der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell biology of the mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial function, biology, and role in disease: A scientific statement from the American Heart Association. Circ Res 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ Res 2012, 111, 1198–1207. [Google Scholar] [CrossRef]
- Williamson, J.R. Mitochondrial heart function. Ann Rev Physiol 1979, 41, 485–506. [Google Scholar] [CrossRef]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J 2002, 21, 1616–1627. [Google Scholar] [CrossRef]
- Glatz, J.F.; Nabben, M.; Young, M.E.; Schulze, P.C.; Taegtmeyer, H.; Luiken, J.J. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim Biophys Acta Mol Basis Dis 2020, 1866, 165579. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.W.; Fahy, E.; Zhang, B.; Glenn, G.M.; Warnock, D.E.; Wiley, S.; Murphy, A.N.; Gaucher, S.P.; Capaldi, R.A.; Gibson, B.W.; et al. Characterization of the human heart mitochondrial proteome. Nat Biotechnol 2003, 21, 281–286. [Google Scholar] [CrossRef]
- Zhao, Q.; Sun, Q.; Zhou, L.; Liu, K.; Jiao, K. Complex Regulation of mitochondrial function during cardiac development. J Am Heart Assoc 2019, 8, e012731. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Rutter, J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev 2013, 27, 2615–2627. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol 2014, 12, 34. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Investig 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart-an engine out of fuel. N Engl J Med 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat Rev Cardiol 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med 2005, 38, 1278–1295. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Q.; Feng, X.; Liu, Y.; Zhou, Y. Mitochondrial dysfunction in cardiovascular diseases: Potential targets for treatment. Front Cell Dev Biol 2022, 10, 841523. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann Med 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Calbet, J.A.L.; Martín-Rodríguez, S.; Martin-Rincon, M.; Morales-Alamo, D. An integrative approach to the regulation of mitochondrial respiration during exercise: Focus on high-intensity exercise. Redox Biol 2020, 35, 101478. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail Rev 2013, 18, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, M.; Rohrbach, S.; Niemann, B. Heart and mitochondria: Pathophysiology and implications for cardiac surgeons. Thorac Cardiovasc Surg 2018, 66, 11–19. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat Rev Cardiol 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Borghetti, G.; von Lewinski, D.; Eaton, D.M.; Sourij, H.; Houser, S.R.; Wallner, M. Diabetic cardiomyopathy: Current and future therapies. Beyond glycemic control. Front Physiol 2018, 9, 1514. [Google Scholar] [CrossRef]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J Cell Physiol 2019, 234, 21630–21641. [Google Scholar] [CrossRef]
- Paolisso, P.; Bergamaschi, L.; Saturi, G.; D’Angelo, E.C.; Magnani, I.; Toniolo, S.; Stefanizzi, A.; Rinaldi, A.; Bartoli, L.; Angeli, F.; et al. Secondary prevention medical therapy and outcomes in patients with myocardial infarction with non-obstructive coronary artery disease. Front Pharmacol 2020, 10, 1606. [Google Scholar] [CrossRef]
- Wang, X.L.; Gao, P.; Zou, Y.Z. The relationship between mitochondrial oxidative stress and diabetic cardiomyopathy. Chin Clin. Pharmacol Therap 2021, 33, 1080–1085. [Google Scholar]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med 2020, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, I.; Nuti, R.; Picchioni, T.; Dotta, F.; Palazzuoli, A. Molecular dysfunction and phenotypic derangement in diabetic cardiomyopathy. Int J Mol Sci 2019, 20, 3264. [Google Scholar] [CrossRef] [PubMed]
- Biala, A.K.; Dhingra, R.; Kirshenbaum, L.A. Mitochondrial dynamics: Orchestrating the journey to advanced age. J Mol Cell Cardiol 2015, 83, 37–43. [Google Scholar] [CrossRef]
- Chang, X.; Li, Y.; Cai, C.; Wu, F.; He, J.; Zhang, Y.; Zhong, J.; Tan, Y.; Liu, R.; Zhu, H.; Zhou, H. Mitochondrial quality control mechanisms as molecular targets in diabetic heart. Metabolism 2022, 137, 155313. [Google Scholar] [CrossRef]
- Tokuyama, T.; Yanagi, S. Role of mitochondrial dynamics in heart diseases. Genes 2023, 14, 1876. [Google Scholar] [CrossRef]
- Gao, F.; Liang, T.; Lu, Y.W.; Fu, X.; Dong, X.; Pu, L.; Hong, T.; et al. A defect in mitochondrial protein translation influences mitonuclear communication in the heart. Nat Commun 2023, 14, 1595. [Google Scholar] [CrossRef] [PubMed]
- Gabillard-Lefort, C.; Thibault, T.; Lenaers, G.; Wiesner, R.J.; Mialet-Perez, J.; Baris, O.R. Heart of the matter: Mitochondrial dynamics and genome alterations in cardiac aging. Mech Ageing Dev 2025, 224, 112044. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W. 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Qin, Y.; Li, A.; Liu, B.; Jiang, W.; Gao, M.; Tian, X.; Gong, G. Mitochondrial fusion mediated by fusion promotion and fission inhibition directs adult mouse heart function toward a different direction. FASEB J 2020, 34, 663–675. [Google Scholar] [CrossRef]
- Jarreta, D.; Orús, J.; Barrientos, A.; Miró, O.; Roig, E.; Heras, M.; Moraes, C.T.; Cardellach, F.; Casademont, J. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc Res 2000, 45, 860–865. [Google Scholar] [CrossRef]
- Buchwald, A.; Till, H.; Unterberg, C.; Oberschmidt, R.; Figulla, H.R.; Wiegand, V. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur Heart J 1990, 11, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Doenst, T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 2011, 90, 234–242. [Google Scholar] [CrossRef]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol 2017, 7, 170007. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD+/NADH redox state and diabetic cardiomyopathy. Antioxid Redox Signal 2019, 30, 375–398. [Google Scholar] [CrossRef] [PubMed]
- Mongirdien, A.; Liuiz, A.; Karˇciauskait, D.; Mazgelyt, E.; Liekis, A.; Sadauskien, I. Relationship between oxidative stress and left ventricle markers in patients with chronic heart failure. Cells 2023, 12, 803. [Google Scholar] [CrossRef]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative Stress as a mechanism for functional alterations in cardiac hypertrophy and heart failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef]
- Li, Y.Y.; Chen, D.; Watkins, S.C.; Feldman, A.M. Mitochondrial abnormalities in tumor necrosis factor-α–induced heart failure are associated with impaired DNA repair activity. Circulation 2001, 104, 2492–2497. [Google Scholar] [CrossRef]
- Ozcan, C.; Bienengraeber, M.; Hodgson, D.M.; Mann, D.L.; Terzic, A. Mitochondrial tolerance to stress impaired in failing heart. J Mol Cell Cardiol 2003, 35, 1161–1166. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Sharov, V.; Riddle, J.M.; Kono, T.; Lesch, M.; Goldstein, S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol 1992, 24, 1333–1347. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.-I.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Sobel, B.E.; Spann, J.F., Jr.; Pool, P.E.; Sonnenblick, E.H.; Braunwald, E. Normal oxidative phosphorylation in mitochondria from the failing heart. Circ Res 1967, 21, 355–364. [Google Scholar] [CrossRef]
- Sordahl, L.; McCollum, W.; Wood, W.; Schwartz, A.; Peterzan, M.A.; Lygate, C.A.; Neubauer, S.; Rider, O.J.; Gong, G.; Liu, J.; et al. Mitochondria and sarcoplasmic reticulum function in cardiac hypertrophy and failure. Am J Physiol Content 1973, 224, 497–502. [Google Scholar] [CrossRef]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic therapy with elamipretide (MTP-131), a novel mitochondria-targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circ Heart Fail 2016, 9, e002206. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, I.M.; Radhakrishnan, J.; Gazmuri, R.J. Targeting mitochondria for resuscitation from cardiac arrest. Crit Care Med 2008, 36, S440–S446. [Google Scholar] [CrossRef]
- Rudokas, M.W.; McKay, M.; Toksoy, Z.; Eisen, J.N.; Bögner, M.; Young, L.H.; Akar, F.G. Mitochondrial network remodeling of the diabetic heart: implications to ischemia related cardiac dysfunction. Cardiovasc Diabetol 2024, 23, 261. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim Biophys Acta Mol Basis Dis 2020, 1866, 165768. [Google Scholar] [CrossRef]
- Dongworth, R.K.; Hall, A.R.; Burke, N.; Hausenloy, D.J. Targeting mitochondria for cardioprotection: examining the benefit for patients. Future Cardiol 2014, 10, 255–272. [Google Scholar] [CrossRef]
- Yang, Y.; Owusu, F.B.; Wu, H.; Zhang, X.; Li, R.; Liu, Z.; Zhang, S.; Leng, L.; Wang, Q. Mitochondria as therapeutic targets for natural products in the treatment of cardiovascular diseases. J Ethnopharmacol 2025, 6, 119588. [Google Scholar] [CrossRef]
- Moe, G.W.; Marín-García, J. Role of cell death in the progression of heart failure. Heart Fail Rev 2016, 21, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Peart, J.N.; Gross, G.J. Sarcolemmal and mitochondrial KATP channels and myocardial ischemic preconditioning. J Cell Mol Med 2002, 6, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Yellon, D.M.; Beikoghli Kalkhoran, S.; Davidson, S.M. The RISK pathway leading to mitochondria and cardioprotection: how everything started. Basic Res Cardiol 2023, 118, 22. [Google Scholar] [CrossRef]
- Halestrap, A.P. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 2006, 34, 232–237. [Google Scholar] [CrossRef]
- Rabinovich-Nikitin, I.; Kirshenbaum, L.A. Circadian regulated control of myocardial ischemia-reperfusion injury. Trends Cardiovasc Med 2024, 34, 1–7. [Google Scholar] [CrossRef]
- Zhao, Y.; Ponnusamy, M.; Dong, Y.; Zhang, L.; Wang, K.; Li, P. Effects of miRNAs on myocardial apoptosis by modulating mitochondria related proteins. Clin Exp Pharmacol Physiol 2017, 44, 431–440. [Google Scholar] [CrossRef]
- Hayashida, K.; Takegawa, R.; Shoaib, M.; Aoki, T.; Choudhary, R.C.; Kuschner, C.E.; Nishikimi, M.; Miyara, S.J.; et al. Mitochondrial transplantation therapy for ischemia reperfusion injury: a systematic review of animal and human studies. J Transl Med 2021, 19, 214. [Google Scholar] [CrossRef]
- Lotz, C.; Herrmann, J.; Notz, Q.; Meybohm, P.; Kehl, F. Mitochondria and pharmacologic cardiac conditioning-at the heart of ischemic injury. Int J Mol Sci 2021, 22, 3224. [Google Scholar] [CrossRef]
- Guo, Z.; Tian, Y.; Liu, N.; Chen, Y.; Chen, X.; Yuan, G.; Chang, A.; Chang, X.; et al. Mitochondrial stress as a central player in the pathogenesis of hypoxia-related myocardial dysfunction: New insights. Int J Med Sci 2024, 21, 2502–2509. [Google Scholar] [CrossRef]
- Suarez, J.; Cividini, F.; Scott, B.T.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Suarez, J.A.; et al. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J Biol Chem 2018, 293, 8182–8195. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, W.H. Diabetic cardiomyopathy. Circ Res 2019, 124, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Mota, K.O.; Elimban, V.; Shah, A.K.; de Vasconcelos, C.M.L.; Bhullar, S.K. Role of vasoactive hormone-induced signal transduction in cardiac hypertrophy and heart failure. Cells 2024, 13, 856. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, N.; Yi, D.; Xiao, Y.; Li, X.; Shao, B.; Wu, Z.; Bai, J.; Shi, X.; Wu, C.; et al. ROS-mediated ferroptosis and pyroptosis in cardiomyocytes: An update. Life Sci 2025, 123565. [Google Scholar] [CrossRef]
- Daiber, A.; Hahad, O.; Andreadou, I.; Steven, S.; Daub, S.; Münzel, T. Redox-related biomarkers in human cardiovascular disease - classical footprints and beyond. Redox Biol 2021, 42, 101875. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Dhalla, N.S. Angiotensin II-induced signal transduction mechanisms for cardiac hypertrophy. Cells 2022, 11, 3336. [Google Scholar] [CrossRef]
Figure 1.
Abnormalities associated with mitochondrial dysfunction leading to changes in cardiomyocyte structure and function in different cardiac pathologies.
Figure 1.
Abnormalities associated with mitochondrial dysfunction leading to changes in cardiomyocyte structure and function in different cardiac pathologies.
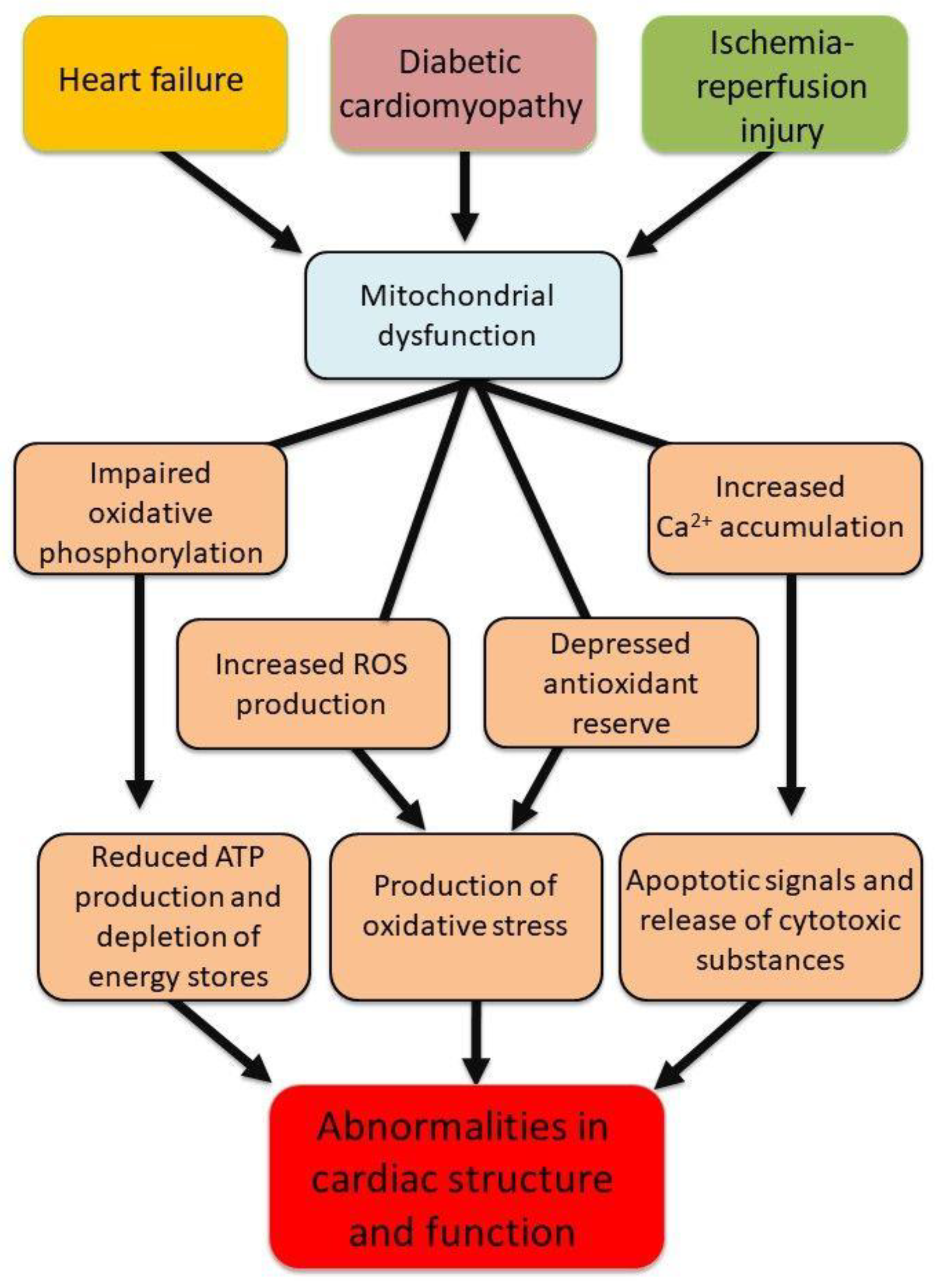
Figure 2.
Alterations in different ROS generating systems in mitochondria due to pathological stimulus. Abbreviation: ROS = reactive oxygen species.
Figure 2.
Alterations in different ROS generating systems in mitochondria due to pathological stimulus. Abbreviation: ROS = reactive oxygen species.
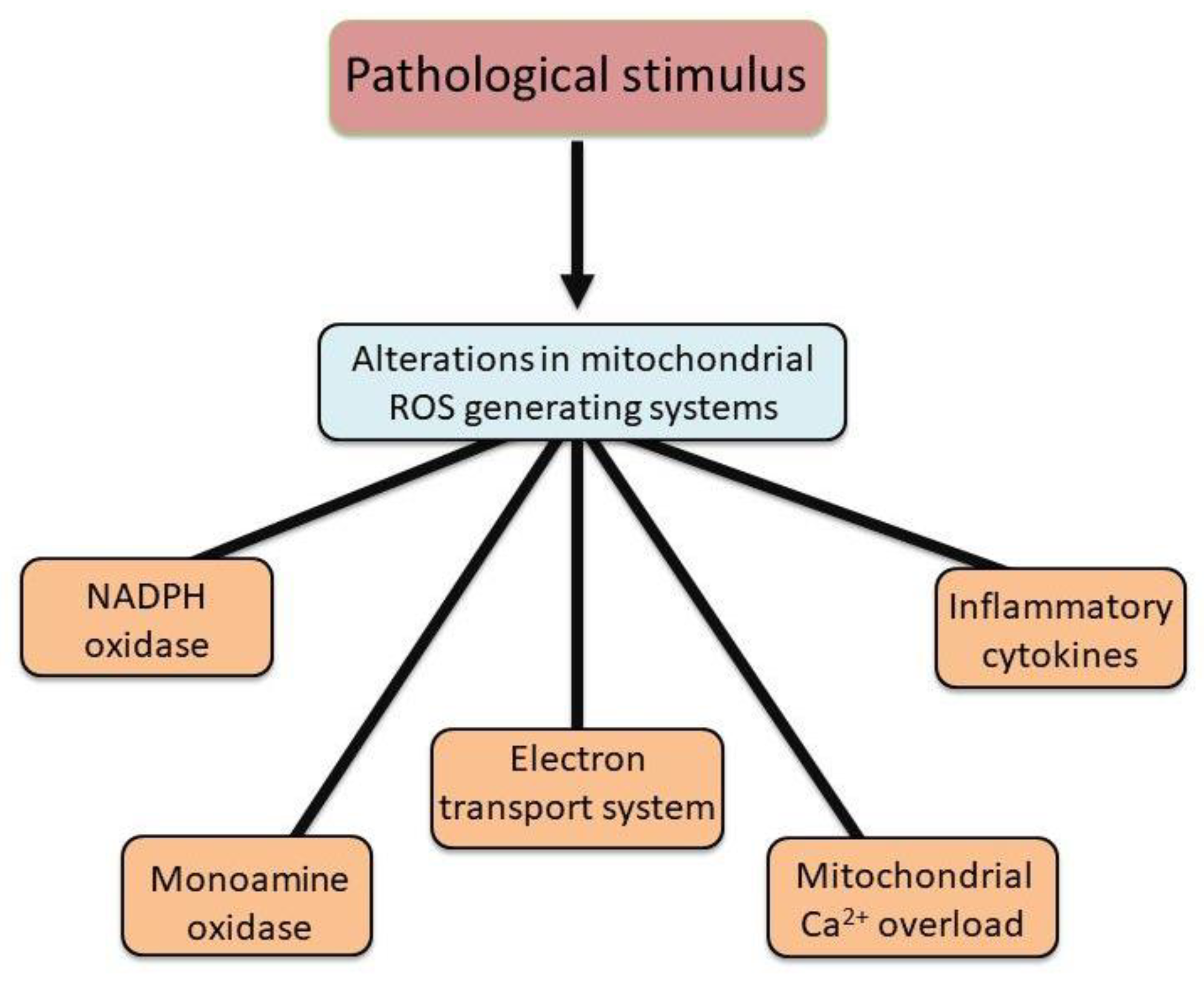
Figure 3.
Different factors involved in the development of oxidative stress in diseased heart.
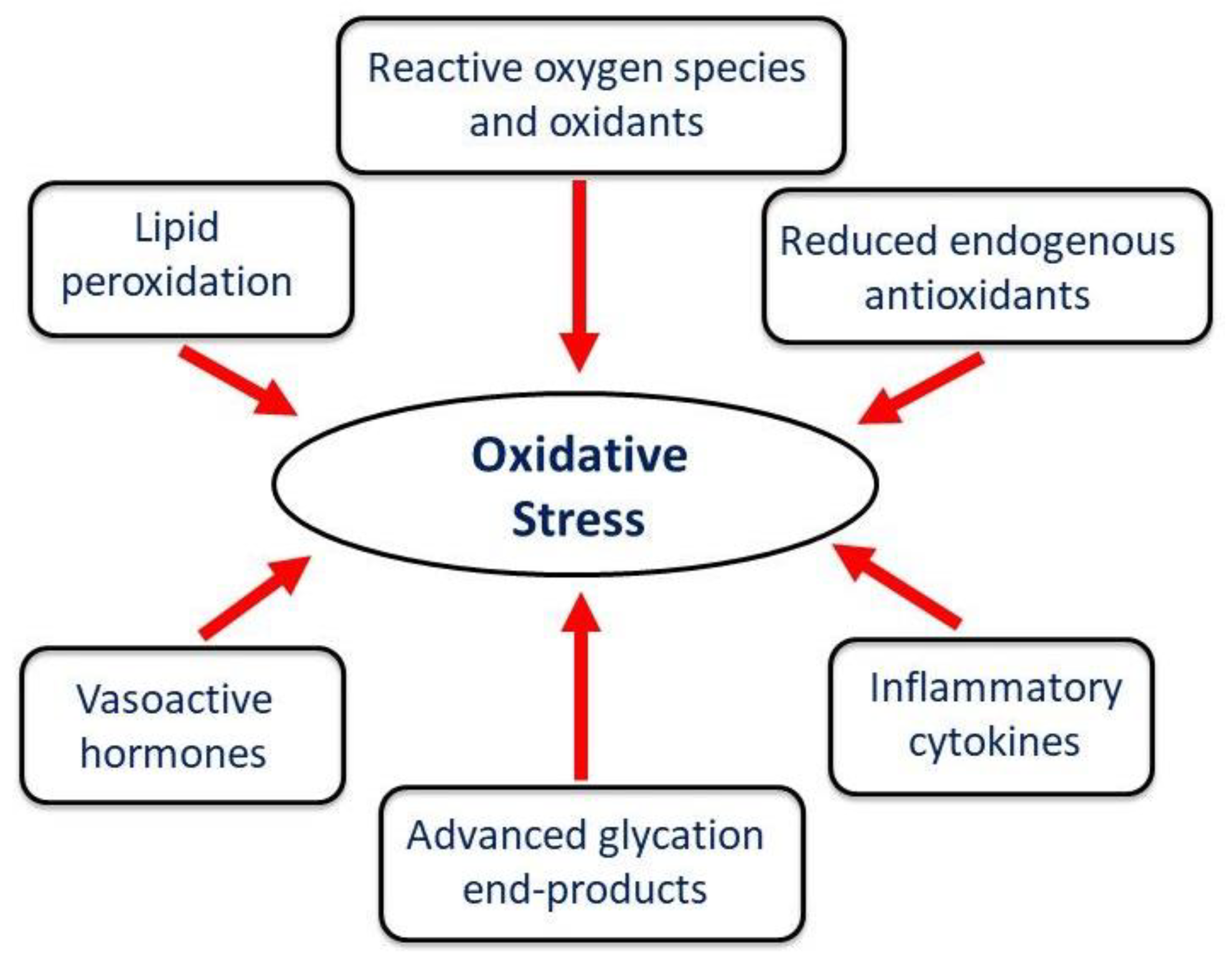
Figure 5.
Mitochondrial respiration and oxidative phosphorylation activities of rat hearts perfused with xanthine +xanthine oxidase or H2O2. Hearts were perfused with 2 mM X and 60 mU/ml XO or with 100 µM H2O2 for 30 min. Data are taken from our paper [50]. Values are mean ± SE of 3 experiments. * = p < 0.05. Abbreviations: X = xanthine; XO = xanthine oxidase; H2O2 = hydrogen peroxide.
Figure 5.
Mitochondrial respiration and oxidative phosphorylation activities of rat hearts perfused with xanthine +xanthine oxidase or H2O2. Hearts were perfused with 2 mM X and 60 mU/ml XO or with 100 µM H2O2 for 30 min. Data are taken from our paper [50]. Values are mean ± SE of 3 experiments. * = p < 0.05. Abbreviations: X = xanthine; XO = xanthine oxidase; H2O2 = hydrogen peroxide.
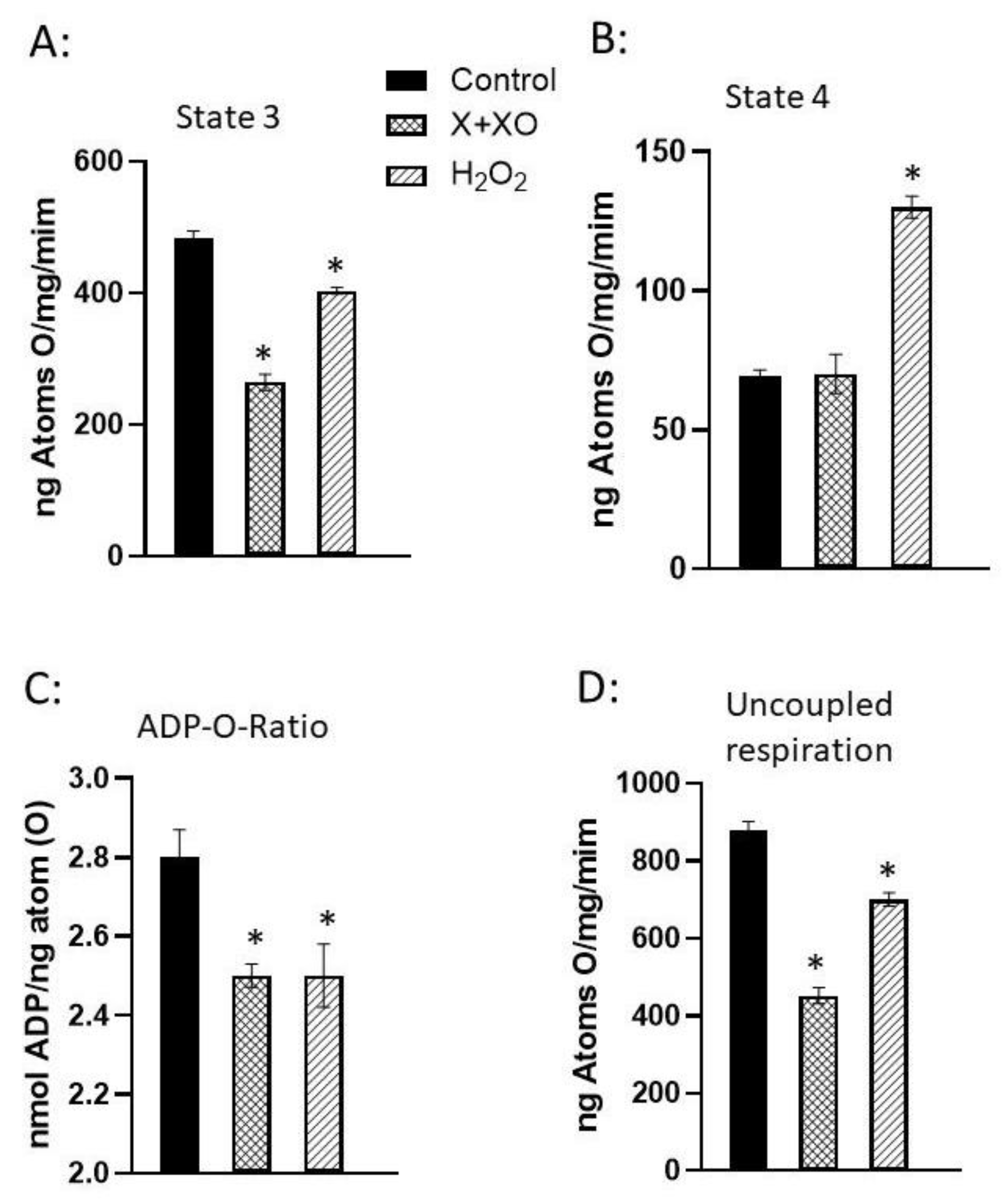
Figure 6.
Involvement of oxidative stress in inducing changes in mitochondrial metabolism for the occurrence of cardiac dysfunction due to pathological stimulus. .
Figure 6.
Involvement of oxidative stress in inducing changes in mitochondrial metabolism for the occurrence of cardiac dysfunction due to pathological stimulus. .
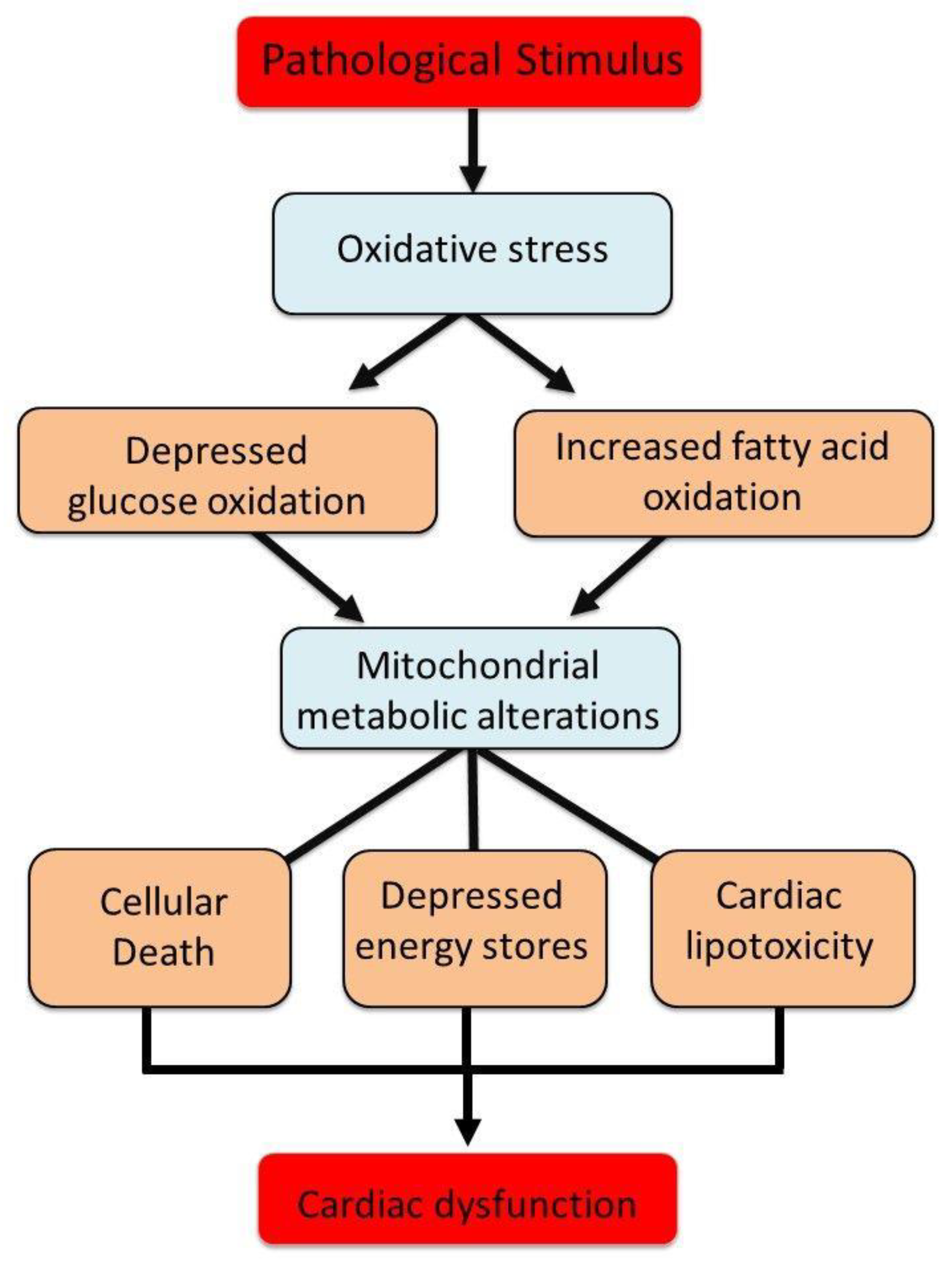

Table 1.
Modification of ROS-induced mitochondrial oxidative phosphorylation by antioxidants.
| ADP-to-O Ratio (nmol ADP/ ng atom O) | Uncoupled Respiration (ng atoms O/min/mg protein) | |
|---|---|---|
| A. X+XO Effects | ||
| Control | 3.0.6±0.15 | 575±9 |
| X+XO | 2.55±0.07* | 196±7* |
| X+XO+SOD+CAT | 2.81±2.0.04# | 426±30*# |
| B. H2O2 Effects | ||
| Control | 3.13±0.09 | 543±29 |
| H2O2 | 2.37±0.03* | 153±5* |
| H2O2+CAT | 2.52±0.04* | 170±7* |
| H2O2+CAT+MAN | 2.84±0.11# | 195±12*# |
Mitochondria isolated from unperfused hearts were incubated with 0.3 mM xanthine (X) and 11 mU xanthine oxidase (XO) for 3 min at 37°C. For antioxidant treatment, mitochondria were exposed for 2 min in the presence of 50 U/ml SOD and 50 U/ml CAT before exposing to X plus XO for 2 min. To study the effects of H2O2, mitochondria were incubated with 30 µM concentration of H2O2 for 3 min. The effect of CAT (8 mU/ml) or mannitol (20 mM) was examined by pretreatment of mitochondria for 2 min before exposing to 20 µM H2O2 for 3 min. All these preparations were washed twice and resuspended in a buffer to measure respiratory activities. *p < 0.05 vs. control; # p < 0.05 vs. respective value in the presence of X+XO or H2O2 alone. Values are means ± SE of 8 experiments. Data are from our paper Makazan et al [50].
Table 2.
Modification of H2O2-induced increase in intracellular Ca2+ concentration by antioxidants.
| Increase in [Ca2+]i in cardiomyocytes (% of control) |
|
|---|---|
|
A. H2O2-induced [Ca2+]i Control |
100 |
| 0.25 mM | 141±11* |
| 0.5 mM | 168±17* |
| 0.75 mM | 216±12* |
| 1.0 mM | 240±23* |
|
B. Antioxidants on H2O2-induced [Ca2+]i Control |
52.8±4.7 |
| CAT | 14.6±2.0* |
| MAN | 48.9±5.6 |
| CAT+MAN | 8.7±2.5* |
Concentration-dependent effects of H2O2 on rat cardiomyocyte [Ca2+]¡. Data shown in (A) were recorded 10 minutes after incubation of Fura-2-loaded cells (10~/mL) with different concentrations of H2O2. Fura-2-loaded cardiomyocytes (10~ cells/mL) were treated with 10 µg/ml catalase (CAT), 20 mM mannitol (MAN) or both and blank buffer (control) for 10 minutes before exposure to 0.5 mM H2O2. The concentration of Ca2+ in the incubation medium was 1 mM. Fluorescent signals were recorded 10 minutes after the addition of H2O2 (B). Control value for [Ca2+]i is 120.9±8.1 mM. Data are expressed as means ±SEM of 6-8 experiments. *p < 0.05 vs. control. The data are taken from our paper Wang et al [51].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



