
Submitted:
31 March 2025
Posted:
01 April 2025
You are already at the latest version
Abstract
Background/Objectives: Atherosclerosis and diabetes are largely recognized as inflammatory disorders and several inflammatory markers are used in clinical settings. Here we investigate the link of interleukin-6 (IL-6), C-Reactive Protein (CRP) and fibrinogen (FIB) with predisposing factors for atherosclerosis and diabetes; NT-pro-BNP, glycemia, insulin resistance, HDL and LDL respectively.
Methods: the literature was collected using PubMed database.
Results: CRP provides complementary information to NT-pro-BNP for high risk for major adverse cardiac events (MACE) in post-MI patients. CRP predicts cardiovascular and mortality in type 2 diabetic subjects. CRP binds to LDL triggering structural changes in HDL. CRP is also found in atheromatous plaque.
IL-6 is a predictor of incident heart failure (HF) and IL-6 inhibition reduces HF hospitalization. IL-6 is related to a greater body mass index, NT–pro-BNP levels and insulin resistance. By blocking IL-6 may be a strategy for insulin resistance treatment. IL-6 levels correlate with endothelial dysfunction and carotid intima-media thickness, activating LDL-Receptor transcription.
The FIB-to-albumin ratio in HF is positively correlated with NT-pro-BNP. Diabetic patients and insulin resistance condition could be related to hyperfibrinogenemia. The glycation level in the fibrinogen molecule is 2–3-fold higher in type 2 diabetic than in non-diabetic individuals, forming fibrin clots with enhanced resistance to fibrinolysis. Finally, because FIB is independently associated with the expression of a more atherogenic lipoprotein subfraction profile, it should be included in the assessment of coronary risk factors.
Conclusions: CRP, IL-6 and FIB could be active players and potential targets for antiatherosclerotic and anti-diabetic therapies.
Keywords:
IL-6
; CRP
; fibrinogen
; cardiovascular disease
1. Introduction
All evidence indicates that atherosclerosis and diabetes prevalence is increasing worldwide. Atherosclerosis is largely recognized as a chronic inflammatory disorder caused by vascular and extravascular inflammatory factors [1], many of which have been identified as consolidated predictive markers in the progression of atherosclerosis and in plaque rupture [2,3]. Diabetes, in particular Type 2 (T2D), is a chronic metabolic disorder whose pathophysiology includes cytokine dysregulation and inflammation [4].
C-reactive protein (CRP), interleukin-6 (IL-6), and fibrinogen (FIB) are the most consolidated inflammatory factors used in clinical laboratories for the diagnosis, prognosis and follow-up of individuals affected by atherosclerosis and/or diabetes.
In this review we focus on findings from recent literature regarding CRP, IL-6, and FIB in relation to NT-pro-BNP, glycemia, insulin resistance, HDL and LDL, that are considered markers of heart failure, diabetes and atherosclerosis, respectively.
NT-pro-BNP is the N-terminal fragment of the B-type natriuretic propeptide. It derives from the peptide secreted predominantly by myocytes in the cardiac ventricles in response to volume expansion or pressure loading, then dissociating into the physiologically active BNP and NT-pro-BNP. Elevated NT-pro-BNP values indicate ventricular wall stress and volume overload as occurs in heart failure (consistent with severity based on NYHA classification) and left ventricular dysfunction [5].
Insulin deficiency, hyperglycemia and dyslipidemia determine the common features of T2D that are associated with long-term macrovascular (atherosclerosis, coronary heart disease, cardiomyopathy, cerebrovascular disease) and microvascular complications (retinopathy, nephropathy, and neuropathy) [4].
Atherosclerosis has been considered as chronic inflammation in the vascular system [6,7,8] and the accumulation of low-density lipoprotein (LDL) in the arterial intima and its subsequent oxidization (ox-LDL) are the initial steps of atherosclerosis process [9].
Unlike LDL, the high density lipoprotein cholesterol (HDL-C), which has anti-inflammatory properties, is reduced in atherosclerosis [10]. HDL best recognized ability is to promote the efflux of cholesterol from the cells and to be an effective antioxidant, inhibiting the oxidative modification of LDL. Moreover, HDL inhibits the expression of adhesion molecules in endothelial cells and reduce the recruitment of blood monocytes into the artery wall, protecting against the development of atherosclerosis [11,12]).
Here we intend to highlight if CRP, IL-6 and FIB are merely hallmarkers of atherosclerosis and inflammation in diabete, or whether they are instead active players in the pathogenesis of cardiovascular disease and potential targets for anti-atherosclerotic and anti-diabetic therapies.
2. CRP
CRP is a sensitive marker of inflammation [13]), that is synthesized by the liver under the stimulation by IL-6 [14]. It circulates at low concentrations in healthy individuals and increases dramatically in response to infections, tissue injury and inflammation [15]. CRP is also widely used for risk stratification of cardiovascular events [13]. CRP was thought to be a single, non glycosylated, multi subunit protein, arranged non covalently in a ring (MW ~120 kDa) [16]. Successively, CRP has been showed to exhist in three distinct isoforms: a pentamer of five identical globular subunits p(CRP), a monomer m(CRP) resulting from a conformational change when the subunits are dissociated from the pentamer, and a transitional isoform where the pentamer remains intact but is partially changed to express mCRP structural characteristics (pCRP*). Some studies have shown that the pro-inflammatory pCRP* and mCRP isoforms facilitate complement binding, immune cell activation, activate platelets, monocytes and endothelial cells [17], directly mediating the inflammatory reactions and the innate immune response in the context of localized tissue injury [18,19]. Moreover, the development of mCRP assays tested in recent years has suggested that mCRP is potentially a more accurate marker of atherosclerotic disease than CRP [20].
The role of CRP in host defense has been thought to be largely due to its ability to bind phosphocholine (PC) activating the classical complement cascade, enhancing phagocytosis and promoting the removal of damaged or apoptotic cells [21]. Furthermore, CRP can dissociate on the membranes of activates platelets [22], monocytes [19], endothelial cells [23] and microparticles [18], all of which are abundant in phosphocholin, thus providing the requisite binding site for CRP [24]. Interestingly, the dissociation of pCRP to mCRP appears to be dependent upon phospholipase A2 (PLA2) that, generating LPC, facilitates CRP binding [23].
2.1. CRP and Vascular Injury
Since CRP has been shown to be stable in the frozen sample, numerous studies have permitted to establish its vascular risk [25]. According to them, CRP stimulates the release of pro-inflammatory cytokines, VCAM-1, ICAM-1, and E-selectin from endothelial cells and monocytes, inducing endothelial dysfunction and monocyte adhesion to the endothelium [26,27]. In addition CRP decreases endothelial NO synthase (eNOS) mRNA and enzyme activity i.e., conversion of l-arginine to l-citrulline in cultured human endothelial cells [28,29] and reduces angiogenesis promoting NO-dependent apoptosis in the endothelial cells. These results support clinical observations of an inverse correlation between CRP levels and endothelial vasoreactivity in patients with coronary artery disease (CAD) [30], suggesting its implication in the destabilization of atherosclerotic plaque [31,32].
Other authors observed that CRP behaves as pro-coagulant downregulating Prostacyclin2, NO metabolism and altering the fibrinolytic system [33,34]. In fact, CRP infusion in healthy volunteers results in a significant increase in serum levels of prothrombin fragment 1 and 2, D-dimer, and plasminogen activator inhibitor-1 [35].
2.2. CRP and Cardiovascular Diseases
Several clinical studies reported the role of CRP with acute and chronic inflammation in cardiovascular disease [36,37,38,39]. Interestingly, an important study on 22.962 subjects showed that patients with CRP ranging from normal (⩽3 mg/l) to elevated (>3 mg/l) had the minimum hazard ratio (HR) for all-cause mortality increased of 6.7-fold (p<0.001) relative to cases whose CRP remained normal [40].
Furthermore a meta-analysis on 160.309 patients without a previous history of cardiovascular disease showed that increased high sensitive (hs) CRP levels were associated with increased risk of CAD, ischaemic stroke and cardiovascular death, of 37%, 27% and 55%, respectively, and that the magnitude of such risk was comparable with other traditional cardiovascular risk factors, including total cholesterol (16%), non-HDL cholesterol (28%) and arterial systolic blood pressure (35%) [41].
2.3. CRP and NT-pro-BNP
As concern NT-pro-BNP, previous studies have revealed that combined assays of NT-pro-BNP and hs-CRP improve the risk stratification in patients with CAD [42,43]. In fact, the activation of inflammation plays an important role not only in the pathogenesis of atherosclerosis, but also in the initiation of acute coronary syndrome [44]. Elevated levels of NT-pro-BNP, on the other hand, is released as a result of both ischemia and necrosis of myocardial cells and could predict subsequent coronary events [45,46,47]. A recent study found a strong association between NT-pro-BNP and hs-CRP, indicating a close relationship between myocardial stretch and inflammatory pathways in the setting of MI [48]. Moreover, it suggests that the enhanced vascular inflammation and activation of neurohumoral axis may play synergistic roles in the process of atherosclerosis. Therefore, combined use of NT-pro-BNP and hs-CRP provides complementary information that increases the prediction of high risk of MACE development in post-MI patients.
2.4. CRP, Glycemia and Insulin Resistance
Increased CRP levels have been linked to excess body weight since adipocytes produce tumor necrosis factor α (TNF-α) and IL-6, which are pivotal factors for CRP stimulation. Furthermore, it is known that hepatocytes of T2D patients produce elevated levels of CRP compared to health subjects. It has been documented the presence of high CRP levels in individuals with insulin resistance [49,50,51]. Insulin resistence is a reduced physiological response of peripheral tissues to the action of insulin that represents one of the major causes of TD2.
In a Japanese cohort, an association between insulin levels and inflammation was observed in populations with T2D and hyperglycemia [52,53,54]. Furthermore, a meta analysis including six prospective cohort studies involving 22.322 T2D patients confirmed that CRP predicts cardiovascular and all-cause mortality in T2D patients [55].
Recently, it was observed that the genetic ablation of CRP gene confers resistance to obesity and insulin resistance in rats. Glucose clamp studies revealed enhanced hepatic insulin signalling and actions, while deficiency of CRP gene enhanced the weight-reducing effect of central injected leptin [56]. Elevated CRP level is thought to induce IR through possible mechanisms that include promotion of thrombogenic agent production, activation of complement cascade, enhancement of endothelial adhesion molecule expression, and reduction of eNOS [28,57,58]. Strategies to support a relative reduction in serum insulin levels may reduce inflammation and its health consequences, including prediabetes and T2DM [59].
2.5. CRP, LDL and HDL
CRP binds to LDL and it is also found in atheromatous plaque so that its causal role in CAD has been proposed [60,61,62]. Furthermore, CRP was showed to be a better cardiovascular marker than LDL [63]. Above all, the recent EXAMINE trial, that enrolled T2D patients with recent acute coronary syndrome to assess the risk of MACE (major adverse cardiovascular events) by utilizing hsCRP and LDL-C, observed that hsCRP levels were associated with recurrent cardiovascular events. Furthermore, this association appears to be independent to the achieved LDL-C levels [64].
By in vitro study, Bian et al. found that CRP directly increased the transcytosis of LDL across endothelial cells and increased LDL retention in vascular walls. These actions were associated with generation of reactive oxygen species, activation of protein PKC and Src kinase, and translocation of caveolar or soluble forms of the N-ethylmaleimide-sensitive factor attachment protein [65].
In addition, CRP promotes the expression of LOX-1, which plays an important role in the damaging effects of ox-LDL on endothelial function [66].
In light of these data we can consider CRP a valid inflammatory marker for risk detection and in cardiovascular prognosis and it can be considered an interesting new target for intervention.
As concern HDL, a recent population-based cohort study including 6.554 participants from the China Health and Retirement Longitudinal Study (CHARLS) have showed a correlation between the hs-CRP/HDL-C ratio and the likelihood of developing CVD. Inflammation leads to structural changes in HDL and in a decrease in HDL-related proteins, enzymes, and transfer proteins that are involved in HDL metabolism and function undergoing to significant alterations [67]. Reverse cholesterol transport, antioxidant, anti-inflammatory, endothelial/vasodilatory, antithrombotic, and cytoprotective actions are the primary biological attributes of HDL-C [68].
Recently, a study enrolling 8581 adults from the National Health and Nutrition Examination Survey 2015–2018 reported that the hs-CRP/HDL-C ratio was a significant risk factor for CVD among US adults, in which hypertension, diabetes, hypercholesterolemia, and obesity played important mediating roles [69].
3. IL-6
IL-6 is a small glycoprotein first isolated by Hirano and colleagues [70]: this protein consists of 212 amino acids with a mass of 21–26 kDa. IL-6 is a member of the family of similar cytokines, that comprises IL-6, IL-11, IL-30, IL-31 and non-IL molecules including oncostatin M, leukemia inhibitor factor(LIF), ciliary neutrophic factor, cardiotrophin 1 and cardiotrophin like cytochine. They are characterized by sharing the common receptor subunit glycoprotein 130 (gp/130) and by the structure of four helices with an up-up down-down topology [71].
IL-6 is normally expressed in low amount in healthy individuals, while it is produced by many cell types under numerous stimuli [72], such as infections, trauma or cardiovascular diseases [73,74,75,76]. IL-6 increases in serum with age [77], which in turn can be positively correlated with insulin resistance [78]. Furthermore, IL-11 and IL-30 have also been implicated in cardiovascular disease, particularly cardiac fibrosis and coronary artery disease [79].
However, IL6 is the most widely used cytokine in clinical practice of Cardiovascular Disease Centers [80].
IL-6 exhibits a high degree of pleiotropic activities ranging from participation in the innate immune response [81], to the induction of acute phase proteins, CRP, several complement system proteins, and the coagulation cascade [50,81].
At cellular level IL-6 activates signaling through the Jak-STAT pathway, whereby the IL-6r–gp130 heterodimer activates members of the Janus-activated protein kinases (Jak), Jak1, Jak2 and Tyk2. These proteins then phosphorylate and activate the signal transducer and activator of transcription (STAT) in a multitude of cell types [82,83]. As a result of STAT- signalling, IL6 induce transcription of a family of proteins called the suppressors of cytokine signaling (SOCS) [84].
3.1. IL-6 and Vascular Injury
Among the high degree of IL-6 pleiotropic activities, IL-6 is related to tissue fibrosis and vascular endothelial injury, promoting angiogenesis and increasing vascular permeability by stimulating the proliferation and migration of circulating endothelial progenitor cells [85]. It also participates in the proliferation and migration of smooth muscle cells [86] that lead to extracellular matrix remodeling and consequently, to the development of CAD, stroke and peripheral arterial disease [87].
Furthermore IL-6 induces FIB production in hepatocytes, which increases blood clotting and promotes thrombosis [88].
3.2. IL-6 and Cardiovascular Disease
According to some authors, IL-6 seems to be a marker more sensitive and specific than CRP in vascular inflammation, thus showing a stronger association with cardiovascular disease [80,89]. Takeda et al. found that IL-6 administration exacerbated atherosclerosis in ApoE-/- mice fed with normal or high-fat diet, while the administration of IL-6 inhibitor (Am80) prevent atherosclerosis suppressing scavenger receptor expression and foam cell formation [90].
Other authors observed that IL-6 is responsible for the development and rupture of atherosclerotic plaques [91] determining the intensity of plaque inflammation and its vulnerability [92]. Therefore IL-6 seems to be an effective independent index of increased mortality in unstable CAD and could characterize subjects who benefit from an initial invasive strategy. In fact, increased preoperative IL-6 levels are predictors of both early graft occlusion and late cardiovascular events after coronary artery bypass grafting [93].
In the myocardial infarction process IL-6 induces cardiomyocyte apoptosis by reducing myocardial contractility and promoting the collection of inflammatory cells in the injured myocardium [94], that play an important role in the onset of ventricular remodeling [95,96] and myocardial infarction death [96]. In ischemia and hypoxia, IL-6 binds to its receptor-coupled protein gp130 by transducing signals into cells through JAK/STAT3, thus leading to cardiomyocyte hypertrophy and abnormal endothelium-dependent vasodilation [97], muscle atrophy [98] and left ventricular dysfunction [99].
3.3. IL-6 and NT-pro-BNP
Elevated NT-pro-BNP and IL-6 levels were observed in a large population of patients with HF [100]. Recently Alogna et al. measured IL-6 in 374 patients with HF and preserved ejection fraction (pEF), even in association with the severity of symptomatology. They found that patients with the highest IL-6 levels had greater body mass index, higher NT–pro-BNP, CRP, TNFα levels, worse renal function, lower hemoglobin levels and were more likely to suffer from diabetes. Although cardiac structure and function measured at rest were similar, patients with pEF and highest IL-6 concentrations had more severely impaired peak oxygen consumption. These findings support the hypothesis that therapies that inhibit IL-6 in patients with HF and pEF may improve clinical status [101].
Also in CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) study, IL-6 levels not only predicted incident HF, but inflammation inhibition in the IL-1β to IL-6 pathway reduced rates of HF hospitalization, an effect most pronounced among those with the greatest reductions in IL-6 and CRP [102]. In particular, a specific targeting of IL-1β (interleukin-1β) with the monoclonal antibody canakinumab results in a significant reduction of IL-6 and CRP. By contrast, the Cardiovascular Inflammation Reduction Trial [103] showed that the use of a nonspecific approach, such as a low-dose methotrexate, an agent that had no impact on circulating levels of IL-1β, IL-6 and hsCRP, had no benefit in terms of cardiovascular event reduction. Therefore focused cytokine inhibition and not broad spectrum anti-inflammatory therapy are likely to be crucial for atheroprotection.
3.4. IL-6, Glycemia and Insulin Resistance
IR is one of the major hallmarks for pathogenesis and etiology of T2D and it is directly interlinked with various inflammatory responses [104]. A low-grade of chronic inflammation in obesity is characterized by an increased systemic level of cytokines including IL-6, that represents a risk factor of the subsequent development of insulin resistance and T2D [105,106]. In T2D, hyperglycaemia is due to the failure of pancreatic beta-cells to compensate for peripheral insulin resistance [107].
The mechanism by which IL-6 induces insulin resistance is various and complex [78]. Among its actions, IL-6 prevents the metabolism of non-oxidative glucose and suppresses the lipoprotein lipase that consecutively increases the plasma levels of triglycerides [108]. In particular, IL-6 activates the SOCS proteins [109,110] which may block the cytokine-mediated transcriptional factor related to insulin receptor, such as transducer and activator of transcription STAT3, STAT5B belonging to the STAT family of transcription factors [111]. SOCS proteins have negative effects on insulin action while IL-6 can activate these SOCS proteins [112].
If IL-6 is implicated in causing insulin resistance and hyperglicemia in T2D patients, remains yet equivocal [113]. When glucose infusion rate was used to correlate IL-6 to insulin sensitivity during a clamp in T2D patients (and in matched healthy controls), no relationship was found, however a strong relationship was found between IL-6 and and BMI, suggesting that the increased IL-6 in T2D patients is strongly related to fat mass and not insulin response [113]. In support of this findings, Vozarova et al. demonstrated that, studing an ethnic population susceptible to insulin resistance, IL-6 was negatively correlated to insulin action and positively correlated to adiposity [114]. Obesity and/or insulin resistance is associated with the release of IL-6 from adipose tissue [115].
Another study investigated the effects of IL-6 on insulin-stimulated glucose metabolism in T2D patients by 1 h intravenous infusion with recombinant human IL-6, followed by 3 h of hyperinsulinemic-isoglycemic clamp. IL-6 infusion did not change glucose infusion rate during the clamp, however it enhanced phosphorylation of STAT3 in skeletal muscle without changing SOCS3 expression [116]. At the same time, other works report that IL-6 activates the suppressor of cytokine signaling (SOCS) proteins, as SOCS 1, SOCS 3 proteins which suppresses the activity of tyrosine kinase by significantly competing with STAT [109,110,117] which may block the cytokine-mediated transcriptional factor activation of insulin receptor [111].
In conclusion IL-6 could be considered as an important biomarker for the development of insulin resistance [112]. Therefore, by blocking IL-6 may be an effective strategy for the treatment of insulin resistance and type 2 diabetes.
3.5. IL-6, LDL and HDL
Several trials suggest that anti-inflammatory approaches targeting IL-6 signaling can reduce cardiovascular risk. However, it remains unknown whether targeting IL-6 signaling could reduce risk additively to LDL-C lowering. Recently, Georgakis et al divided 408. 225 White British individuals in UK Biobank into groups characterized by lifelong exposure to downregulated IL-6 signaling, lower LDL-C, or both and assessed the genetic scores for IL-6 signaling downregulation and LDL-C lowering. They found that, individuals with both genetic scores both for IL-6 signaling and LDL-C lower than the median, were at lower odds of cardiovascular disease (OR, 0.92; 95% CI, 0.90–0.95) compared with individuals with all or one genetic score above the median, suggesting that genetically downregulated IL-6 signaling and genetically lowered LDL-C are associated with additively lower lifetime risk of cardiovascular disease [118].
IL-6 levels predict future cardiovascular risk and correlate with endothelial dysfunction and carotid intima-media thickness [119,120]. In this regard, an vitro study in endothelial cells by Lubrano et al showed that modified LDL up-regulate IL-6 [121], that in turn induces the expression of macrophage scavenger receptors involved in the uptake of modified LDL, thus promoting the formation of foam cells [122] and establishing an inflammatory cycle in the plaque. Moreover another study in HepG2 cells has demonstrated that IL-6 activates LDL-R transcription by enhancing the binding of SREBP-1a and SREBP-2 as well as the binding of Sp1 and Sp3 to their cognate DNA sequence in repeat 2 and repeat 3 of the LDL-receptor (LD-R) promoter. Consequently, the LDL-R activity on the surface of liver cells is enhanced, leading to an increased uptake of LDL from the circulation. These data are consistent with the hypothesis that hypocholesterolemia after myocardial injury, surgery, or infection is partly due to an enhanced catabolism of LDL in the liver by IL-6. [123]. Large epidemiological studies have shown that low plasma levels of HDL-C are associated with increased incidence of coronary heart disease (CHD). In a population of 1044 community dwelling older Italian subjects from the InChianti study, Zuliani et al. provided the first epidemiological evidence that IL-6 is one of the factors that contribute to low HDL-C levels independently from the effects of a large number of possible confounders (triglycerides, fasting insulin, diabetes, hypertension, BMI, waist circumference), and life style habits (smoking, alcohol intake, physical activity) [124]. Successively, another Italian study of 429 patients with stable chest pain who underwent coronary computer tomography showed that low HDL cholesterol and high IL-6 are independent predictors of high risk of coronary diseases [125].
4. FIB
FIB is a large glycoprotein (340 kDa) made up of two identical units, each consisting of three polypeptides: Aα (610 aa, 67 kDa), Bβ (461 kDa, 56 kDa) and γ (411 aa, 48 kDa). The protein formation steps are quite complex: from RNA transcription, six chains are formed, which will assemble into a hexameric complex linked by disulfide bridges [126,127].
FIB is constitutively expressed mainly in hepatocytes and is regulated by acute-phase proteins, mainly by IL-6 produced by monocytes, macrophages, and endothelial cells, while IL-1β and TNF-α suppress its synthesis [127,128]. The production of fibrinogen is also increased by glucocorticosteroids. It is considered an acute phase protein, and its biosynthesis increases during inflammation until it exceeds 7 g/L [128].
4.1. FIB and Vascular Injury
The main role of fibrinogen is its involvement in the blood coagulation cascade, where plasma fibrinogen is converted into an insoluble fibrin clot in the presence of thrombin. The cleavage sites for thrombin are located in the E-region, the central part of molecule [129]. Within the fibrinogen sequence, lysine residues are located in close proximity to thrombin cleavage sites and polymerization motifs; fibrin cross-linking is accomplished by the formation of covalent bonds between glutamine and lysine residues within the α- and γ-chains in the presence of factor XIII (FXIII) [130].
Post-translational modifications of fibrinogen including limited proteolysis, alterations of N-glycosaminoglycans, amino acid phosphorylation, tyrosine sulfation, glycation, nitration and acetylation may play an important role in the pathophysiology of blood coagulation [131,132]. An in vitro study of isolated fibrinogen and plasma from patients with diabetes mellitus shows that fibrinogen glycation and the presence of glucose impair fibrin polymerization [133]. Moreover, fibrinogen nucleotide polymorphism(s) such as γ' fibrinogen polymorphism led to the formation of blood clots that are very resistant to fibrinolysis, which increased the risk of CVD [134,135,136].
4.2. FIB and Cardiovascular Disease
The increased fibrinogen plasma concentration directly activates many mechanisms, which, consequently, may intensify the progression of atherosclerosis [137]. This seems to affect CVD risk more than increased levels of serum cholesterol. In addition, fibrinogen is associated with an increased risk of future myocardial infarction, independently from other CVD risk factors, [138]; a 1 g/L increase in fibrinogen corresponds to a 142% increase of the risk of CAD, 146% increase in the risk of stroke, 176% increase of the risk of death from other vascular events, and 103% increase of the risk of death from nonvascular events, regardless of sex and age [139]. Furthermore, especially when combined with assessment of D'dimer or albumin concentration, plasma fibrinogen concentration is a valuable biomarker of primary and secondary CVD risk [140,141]. According to some authors, increased fibrinogen was independently associated with all-cause mortality and long-term cardiac mortality among patients with CAD undergoing percutaneous coronary intervention, especially those with diabetes mellitus and pre-diabetes [142] and independently predicted mortality in critically ill patients with acute exacerbation of chronic heart failure [143].
4.3. FIB and NT-pro-BNP
Elevated levels of fibrinogen and decreased levels of albumin are risk factors of thrombosis events. [144,145,146] and are common in patients with HF. The fibrinogen-to-albumin ratio (FAR) was developed to improve diagnostic sensitivity and specificity for predicting poor long-term outcomes of acute HF. According to a recent study, in patients with heart failure FAR was positively correlated with NT-pro-BNP, and this association was stronger than with fibrinogen, albumin, PAR, and PLR [147].
4.4. FIB, Glycemia and Insulin Resistance
A cohort study of 5237 patient investigate the impact of high FIB on cardiovascular outcomes in patients with stable CAD and pre-diabetes mellitus (pre-DM) or diabetes mellitus.
When patients were stratified by both glucose metabolism status and Fib levels, high Fib was associated with a higher risk of MACEs in pre-DM (HR 1.66, 95% CI 1.02–2.71, P < 0.05) and in DM (HR 2.28, 95% CI 1.42–3.66, all P < 0.05). After adding the combination of Fib and glucose status to the Cox model, the C-statistic was increased by 0.015 (0.001–0.026). This study suggests that Fib may provide incremental value in the cardiovascular risk stratification of pre-DM and DM patients [148]. Furthermore a relationship between plasma fibrinogen and elevated insulin levels, as well as the different parameters of the insulin resistance syndrome has been described. Raynaud et al studying 62 nondiabetic, nonhypertensive patients, found that only insulin sensitivity appeared to account for the ability to predict fibrinogen values. Thus, they hypothesized that the state of insulin resistance rather than hyperinsulinemia per se was related to hyperfibrinogenemia, in connection with some factors like free fatty acids or tumor necrosis factor-α, which have been implicated in the pathogenesis of insulin resistance [149].
Another study investigated if altered response to insulin contributes to hyperfibrinogenemia in T2D; they measured fibrinogen fractional (FSR) and absolute synthesis rates (ASR) using a leucine isotopic model in type 2 diabetic men (n = 7; age = 51 ± 3 years; BMI = 26.7 ± 1 kg/m2) compared with matched nondiabetic subjects under basal conditions and following a 4-h euglycemic-, euaminoacidemic-hyperinsulinemic clamp. Basal fibrinogen concentration (+35%, P < 0.05) and ASR (+35%, P < 0.05) were greater in the diabetic subjects. Following clamp, fibrinogen FSR and ASR were acutely increased by insulin when euglycemia and euaminoacidemia are maintained in T2D, instead they were unchanged in the control subjects. This study suggests that enhanced fibrinogen production by insulin is likely to be a key alteration contributing to cardiovascular risk in T2D. Unchanged fibrinogen production in nondiabetic individuals suggests a role of plasma amino acids in regulating fibrinogen production in humans [150].
Additionally, increased glucose concentration induces oxidative stress and the generation of highly reactive products, which is known to induce structural modifications and functional impairments of various proteins, including fibrinogen [151]. It has been observed that the glycation level in the fibrinogen molecule was 2–3-fold higher in T2D than in non-diabetic individuals [152,153,154,155].
4.5. FIB, LDL and HDL
In 1996 Halle M showed that, clinically healthy nonsmoking men with serum FIB concentrations >2.90 g/L, have a significantly unfavorable LDL subfraction profile that was independent from other coronary risk factors, such as BMI, age, IR, total cholesterol, serum triglycerides, uric acid, and BP [156] (Halle M, 1996).
Therefore, in addition to its effect on coagulation, FIB could influence atherogenesis by worsening the LDL subfraction profile. Because FIB is independently associated with the expression of a more atherogenic lipoprotein subfraction profile, it should be included in the assessment of coronary risk factors, particularly in patients with dyslipoproteinemia.
The mortality risk predictive capacity of fibrinogen to HDL-cholesterol ratio (FHR) in AMI patients has been not yet well investigated. Recently Jai et al. explored this aspect in a retrospective study involving 13,221 patients with acute myocardial infarct (AMI) from the Cardiorenal Improvement II cohort (NCT05050877) [157] AMI patients with increased baseline FHR values had higher all-cause and cardiovascular mortality, regardless of established CVD risk factors suggesting FHR as a valuable tool for evaluating mortality risk in AMI patients.
Instead, as concern HDL, plasma fibrinogen concentration is inversely related to serum HDL cholesterol concentration [158].
5. Conclusions
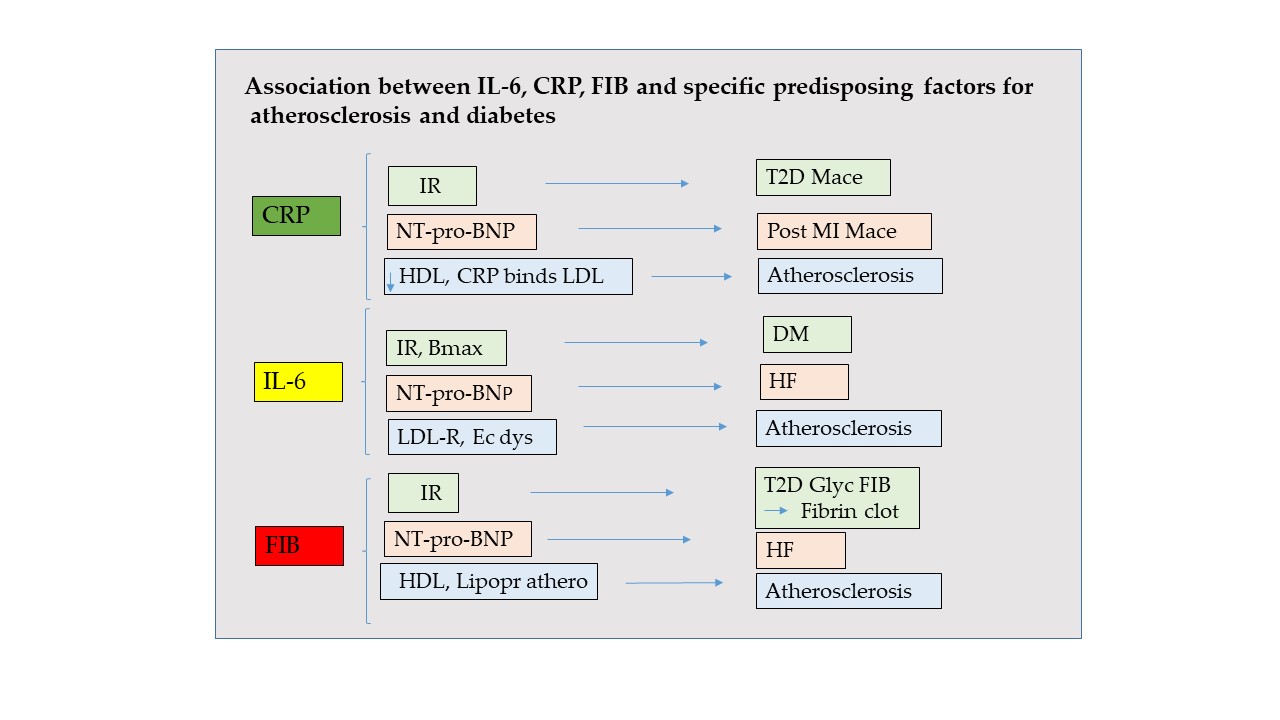
This review provides an update on the relation between the role of consolidated inflammatory markers in atherosclerosis and diabetes. CRP, IL-6 and FIB are not merely hallmarkers of these common diseases, but play synergistic roles being active players in the pathogenesis of them. CRP provides complementary information to NT-pro-BNP for high risk of MACE in post-MI patients and predicts cardiovascular events and mortality in type T2D. CRP binds to LDL and leads to structural changes in HDL. CRP is also found in atheromatous plaque.
IL-6 predicts HF incidence, and inflammatory IL6 inhibition by the monoclonal antibody canakinumab reduces HF hospitalization. IL-6 is related to greater BMI, NT–pro-BNP and insulin resistance. IL-6 blockade may be a strategy for insulin resistance treatment. IL-6 levels predict future cardiovascular risk and correlate with endothelial dysfunction and carotid intima-media thickness, activating LDL-R transcription.
The fibrinogen-to-albumin ratio in HF is positively correlated with NT-pro-BNP. Diabetic patients and insulin resistance condition could be related to hyperfibrinogenemia. The glycation level in the FIB molecule is 2–3-fold higher in T2D than in non-diabetic individuals, forming fibrin clots with enhanced resistance to fibrinolysis. Finally, because FIB is independently associated with the expression of a more atherogenic lipoprotein subfraction profile, it should be included in the assessment of coronary risk factors.
Author Contributions
Conceptualization, V.L. and S.B.; writing—review and editing, V.L., S.B., L.S., A.P., contributed as first author to the manuscript; V.L. and S.B contributed as last author to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
References
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Rader, D.J. Inflammatory markers of coronary risk. N. Engl. J. Med. 2000, 343, 1179–1182. [Google Scholar] [CrossRef]
- Morrow, D.A.; Braunwald, E. Future of biomarkers in acute coronary syndromes: moving toward a multimarker strategy. Circulation 2003, 108, 250–252. [Google Scholar] [CrossRef]
- Velikova, T.V.; Kabakchieva, P.P.; Assyov, Y.S.; Georgiev, T.А. Targeting Inflammatory Cytokines to Improve Type 2 Diabetes Control. Biomed. Res. Int. 2021, 7297419. [Google Scholar] [CrossRef]
- Markousis-Mavrogenis, G.; Tromp, J.; Ouwerkerk, W.; Devalaraja, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.S.; van der Harst, P.; Lang, C.C.; et al. The clinical significance of interleukin-6 in heart failure: results from the BIOSTAT-CHF study. Eur. J. Heart Fail. 2019, 21, 965–973. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2019, 8563845. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal. Transduct. Target Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Malekmohammad, K.; Bezsonov, E.E.; Rafieian-Kopaei, M. Role of Lipid Accumulation and Inflammation in Atherosclerosis: Focus on Molecular and Cellular Mechanisms. Front. Cardiovasc. Med. 2021, 8, 707529. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Vergeer, M.; Holleboom, A.G.; Kastelein, J.J.; Kuivenhoven, J.A. The HDL hypothesis: does high-density lipoprotein protect from atherosclerosis? J. Lipid Res. 2010, 51, 2058–73. [Google Scholar] [CrossRef]
- Barter, P.J.; Nicholls, S.; Rye, K.A.; Anantharamaiah, G.M.; Navab, M.; and Fogelman, A.M. Anti-inflammatory Properties of HDL. Circulation Research 2004, 95, 764–772. [Google Scholar] [CrossRef]
- Fotakis, P.; Kothari, V.; Thomas, D.G.; Westerterp, M.; Molusky, M.M.; Altin, E.; Abramowicz, S.; Wang, N.; He, Y.; Heinecke, J.W.; Bornfeldt, K.E.; Tall, A.R. Anti-Inflammatory Effects of HDL (High-Density Lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e253–e272. [Google Scholar] [CrossRef]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: a critical update. J. Clin. Invest. 2003, 111, 1805–1812. [Google Scholar] [CrossRef]
- Lelubre C, Anselin S, Zouaoui Boudjeltia K, Biston P, Piagnerelli, M. Interpretation of C-reactive protein concentrations in critically ill patients. Biomed. Res. Int. 2013, 124021. [CrossRef]
- Wu, Y.; Potempa, L.A.; El Kebir, D.; Filep, J.G. C-reactive protein and inflammation: conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar] [CrossRef]
- Pepys, M.B. The Pentraxins 1975-2018: Serendipity, Diagnostics and Drugs. Front. Immunol. 2018, 9, 2382. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Kiefer, J.; Braig, D.; Loseff-Silver, J.; Potempa, L.A.; Eisenhardt, S.U.; Peter, K. Dissociation of C-Reactive Protein Localizes and Amplifies Inflammation: Evidence for a Direct Biological Role of C-Reactive Protein and Its Conformational Changes. Front. Immunol. 2018, 9, 1351. [Google Scholar] [CrossRef]
- Ji, S.R.; Wu, Y.; Zhu, L.; Potempa, L.A.; Sheng, F.L.; Lu, W.; Zhao, J. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRP(m). FASEB J. 2007, 21, 284–294. [Google Scholar] [CrossRef]
- Braig, D.; Nero, T.L.; Koch, H.G.; Kaiser, B.; Wang, X.; Thiele, J.R.; Morton, C.J.; Zeller, J.; Kiefer, J.; Potempa, L.A.; et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat. Commun. 2017, 8, 14188. [Google Scholar] [CrossRef]
- Melnikov, I.; Kozlov, S.; Saburova, O.; Avtaeva, Y.; Guria, K.; Gabbasov, Z. Monomeric C-Reactive Protein in Atherosclerotic Cardiovascular Disease: Advances and Perspectives. Int. J. Mol. Sci. 2023, 24, 2079. [Google Scholar] [CrossRef]
- Du Clos, T.W. C-reactive protein as a regulator of autoimmunity and inflammation. Arthritis. Rheum. 2003, 48, 1475–1477. [Google Scholar] [CrossRef]
- Eisenhardt, S.U.; Habersberger, J.; Murphy, A.; Chen, Y.C.; Woollard, K.J.; Bassler, N.; Qian, H.; von Zur Muhlen, C.; Hagemeyer, C.E.; et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ. Res. 2009, 105, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.R.; Habersberger, J.; Braig, D.; Schmidt, Y.; Goerendt, K.; Maurer, V.; Bannasch, H.; Scheichl, A.; Woollard, K.J.; von Dobschütz, E.; et al. Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation: in vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy. Circulation 2014, 130, 35–50. [Google Scholar] [CrossRef]
- Volanakis, J.E.; Wirtz, K.W. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers. Nature 1979, 281, 155–157. [Google Scholar] [CrossRef]
- Pearson, T.A.; Mensah, G.A.; Alexander, R.W.; Anderson, J.L.; Cannon, R.O.; Criqui, M.; Fadl, Y.Y.; Fortmann, S.P.; Hong, Y.; Myers, G.L.; et al. Centers for Disease Control and Prevention; American Heart Association. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003, 107, 499–511. [Google Scholar] [CrossRef]
- Pasceri, V.; Cheng, J.S.; Willerson, J.T.; Yeh, E.T. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation 2001, 103, 2531–1234. [Google Scholar] [CrossRef] [PubMed]
- Pasceri, V.; Willerson, J.T.; Yeh, E.T. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation 2000, 102, 2165–2168. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.K.; Devaraj, S.; Yuhanna, I.; Shaul, P.; Jialal, I. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation 2002, 106, 1439–1441. [Google Scholar] [CrossRef]
- Grad, E.; Danenberg, H.D. C-reactive protein and atherothrombosis: Cause or effect? Blood Rev. 2013, 27, 23–29. [Google Scholar] [CrossRef]
- Fichtlscherer, S.; Rosenberger, G.; Walter, D.H.; Breuer, S.; Dimmeler, S.; Zeiher, A.M. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation 2000, 102, 1000–1006. [Google Scholar] [CrossRef]
- Verma, S.; Wang, C.H.; Li, S.H.; Dumont, A.S.; Fedak, P.W.; Badiwala, M.V.; Dhillon, B.; Weisel, R.D.; Li, R.K.; Mickle, D.A.; et al. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation 2002, 106, 913–919. [Google Scholar] [CrossRef]
- Nabata, A.; Kuroki, M.; Ueba, H.; Hashimoto, S.; Umemoto, T.; Wada, H.; Yasu, T.; Saito, M.; Momomura, S.I.; Kawakami, M. C-reactive protein induces endothelial cell apoptosis and matrix metalloproteinase-9 production in human mononuclear cells: Implications for the destabilization of atherosclerotic plaque. Atherosclerosis 2008, 196, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.W.; Qamirani, E.; Ren, Y.; Kuo, L. C-reactive protein impairs coronary arteriolar dilation to prostacyclin synthase activation: role of peroxynitrite. J. Mol. Cell Cardiol. 2009, 47, 196–202. [Google Scholar] [CrossRef]
- Ji, Y.; Fish, P.M.; Strawn, T.L.; Lohman, A.W.; Wu, J.; Szalai, A.J.; Fay, W.P. C-reactive protein induces expression of tissue factor and plasminogen activator inhibitor-1 and promotes fibrin accumulation in vein grafts. J. Thromb. Haemost. 2014, 12, 1667–177. [Google Scholar] [CrossRef]
- Bisoendial, R.J.; Kastelein, J.J.; Levels, J.H.; Zwaginga, J.J.; van den Bogaard, B.; Reitsma, P.H.; Meijers, J.C.; Hartman, D.; Levi, M.; Stroes, E.S. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ. Res. 2005, 96, 714–716. [Google Scholar] [CrossRef]
- Luan, Y.Y.; Yao, Y.M. The Clinical Significance and Potential Role of C-reactive Protein in Chronic Inflammatory and Neurodegenerative Diseases. Front. Immunol. 2018, 9, 1302. [Google Scholar] [CrossRef]
- Arroyo-Espliguero, R.; Viana-Llamas, M.C.; Silva-Obregón, A.; Avanzas, P. The Role of C-reactive Protein in Patient Risk Stratification and Treatment. Eur. Cardiol. 2021, 16, e28. [Google Scholar] [CrossRef] [PubMed]
- Kao, P.C.; Shiesh, S.C.; Wu, T.J. Serum C-reactive protein as a marker for wellness assessment. Ann. Clin. Lab. Sci. 2006, 36, 163–169. [Google Scholar]
- Koenig, W.; Sund, M.; Fröhlich, M.; Fischer, H.G.; Löwel, H.; Döring, A.; Hutchinson, WL.; Pepys, MB. C-reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation 1999, 99, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Currie, C.J.; Poole, C.D.; Conway, P. Evaluation of the association between the first observation and the longitudinal change in C-reactive protein, and all-cause mortality. Heart 2008, 94, 457–462. [Google Scholar] [CrossRef]
- Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; e, al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef]
- Huang, P.H.; Lu, T.M.; Wu, T.C.; Lin, F.Y.; Chen, Y.H.; Chen, J.W.; Lin, S.J. Usefulness of combined high-sensitive C-reactive protein and N-terminal-probrain natriuretic peptide for predicting cardiovascular events in patients with suspected coronary artery disease. Coron. Artery. Dis. 2008, 19, 187–93. [Google Scholar] [CrossRef]
- Kim, H.; Yang, D.H.; Park, Y.; Han, J.; Lee, H.; Kang, H.; Park, H.S.; Cho, Y.; Chae, S.C.; Jun, J.E.; et al. Incremental prognostic value of C-reactive protein and N-terminal proB-type natriuretic peptide in acute coronary syndrome. Circ. J. 2006, 70, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Rymer, J.A.; Newby, L.K. Failure to Launch: Targeting Inflammation in Acute Coronary Syndromes. JACC Basic Transl. Sci. 2017, 2, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef] [PubMed]
- Li, K.P.; Zhang, H.Y.; Xu, X.D.; Ming, Y.; Li, T.J.; Song, S.T. Recombinant human brain natriuretic peptide attenuates myocardial ischemia-reperfusion injury by inhibiting CD4(+) T cell proliferation via PI3K/AKT/mTOR pathway activation. Cardiovasc. Ther. 2020, 2020, 1389312–10.1155. [Google Scholar] [CrossRef]
- Chan, M.Y.; Neely, M.L.; Roe, M.T.; Goodman, S.G.; Erlinge, D.; Cornel, J.H.; Winters, K.J.; Jakubowski, J.A.; Zhou, C.; Fox, K.A.A.; et al. Temporal biomarker profiling reveals longitudinal changes in risk of death or myocardial infarction in non-ST-segment elevation acute coronary syndrome. Clin. Chem. 2017, 63, 1214–1226. [Google Scholar] [CrossRef]
- Cao, Y.X.; Li, S.; Liu, H.H.; Zhang, M.; Guo, Y.L.; Wu, N.Q.; Zhu, C.G.; Dong, Q.; Sun, J.; Dou, K.F.; et al. Prognostic Value of N-Terminal Pro-B-Type Natriuretic Peptide and High-Sensitivity C-Reactive Protein in Patients With Previous Myocardial Infarction. Front. Cardiovasc. Med. 2022, 9, 797297. [Google Scholar] [CrossRef]
- Freeman, D.J.; Norrie, J.; Caslake, M.J.; Gaw, A.; Ford, I.; Lowe, G.D.O.; O’Reilly, D.S.J.; Packard, C.J.; Sattar, N. C-reactive protein is an independent predictor of risk for the development of diabetes in the West of Scotland Coronary Prevention Study. Diabetes 2002, 51, 1596–1600. [Google Scholar] [CrossRef]
- Sproston, N.R.; and Ashworth, J.J. Role of C-reactive protein at sites of inflammation and infection Frontiers in Immunology 2018, 9, 754–754. [CrossRef]
- Stanimirovic, J.; Radovanovic, J.; Banjac, K.; Obradovic, M.; Essack, M.; Zafirovic, S.; Gluvic, Z.; Gojobori, T.; Isenovic, E.R. Role of C-Reactive Protein in Diabetic Inflammation. Mediators Inflamm. 2022, 17, 3706508. [Google Scholar] [CrossRef]
- Gelaye, B.; Revilla, L.; Lopez, T.; Suarez, L.; Sanchez, S.E.; Hevner, K.; Fitzpatrick, A.L.; Williams, M.A. Association between insulin resistance and c-reactive protein among Peruvian adults. Diabetol. Metab. Syndr. 2010, 2, 30. [Google Scholar] [CrossRef]
- Kim, G.R.; Choi, D.W.; Nam, C.M.; Jang, S.I.; Park, E.C. Synergistic association of high-sensitivity C-reactive protein and body mass index with insulin resistance in non-diabetic adults. Sci. Rep. 2020, 18417. [Google Scholar] [CrossRef] [PubMed]
- Uemura, H.; Katsuura-Kamano, S.; Yamaguchi, M.; Bahari, T.; Ishizu, M.; Fujioka, M.; Arisawa, K. Relationships of serum high-sensitivity C-reactive protein and body size with insulin resistance in a Japanese cohort. PLoS One 2017, 12, e0178672. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Tian, M.; Wang, L.; Qian, H.; Zhang, S.; Pang, H.; Liu, Z.; Fang, L.; Shen, Z. C-reactive protein for predicting cardiovascular and all-cause mortality in type 2 diabetic patients: A meta-analysis. Cytokine 2019, 117, 59–64. [Google Scholar] [CrossRef]
- Yang, M.; Qiu, S.; He, Y.; Li, L.; Wu, T.; Ding, N.; Li, F.; Zhao, A.Z.; Yang, G. Genetic ablation of C-reactive protein gene confers resistance to obesity and insulin resistance in rats. Diabetologia 2021, 64, 1169–1183. [Google Scholar] [CrossRef]
- Fukuchi, Y.; Miura, Y.; Nabeno, Y.; Kato, Y.; Osawa, T.; Naito, M. Immunohistochemical detection of oxidative stress biomarkers, dityrosine and N(epsilon)-(hexanoyl)lysine, and C-reactive protein in rabbit atherosclerotic lesions. J. Atheroscler. Thromb. 2008, 15, 185–192. [Google Scholar] [CrossRef]
- Jarva, H.; Jokiranta, T.S.; Hellwage, J.; Zipfel, P.F.; Meri, S. Regulation of complement activation by C-reactive protein:Targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8–11. J. Immunol. 1999, 163, 3957–3962. [Google Scholar] [CrossRef] [PubMed]
- Kanmani, S.; Kwon, M.; Shin, M.K.; Kim, M.K. Association of C-Reactive Protein with Risk of Developing Type 2 Diabetes Mellitus, and Role of Obesity and Hypertension: A Large Population-Based Korean Cohort Study. Sci. Rep. 2019, 9, 4573. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Cliff, W.J.; Schoefl, G.I.; Higgins, G. Coronary C-reactive protein distribution: its relation to development of atherosclerosis. Atherosclerosis 1999, 145, 375–379. [Google Scholar] [CrossRef]
- Danesh, J.; Wheeler, J.G.; Hirschfield, G.M.; Eda, S.; Eiriksdottir, G.; Rumley, A.; Lowe, G.D.; Pepys, M.B.; Gudnason, V. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N. Engl. J. Med. 2004, 350, 1387–1397. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Cook, N.R.; Bradwin, G.; Buring, J.E. Non-HDL cholesterol, apolipoproteins A-I and B100, standard lipid measures, lipid ratios, and CRP as risk factors for cardiovascular disease in women. JAMA 2005, 294, 326–333. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N. Engl. J. Med. 2002, 347, 1557–1565. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Morrow, D.A.; Cannon, C.P.; Liu, Y.; Bergenstal, R.; Heller, S.; Mehta, C.; Cushman, W.; Bakris, GL.; Zannad, F.; et al. High-sensitivity C-reactive protein, low-density lipoprotein cholesterol and cardiovascular outcomes in patients with type 2 diabetes in the EXAMINE (Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care) trial. Diabetes Obes. Metab. 2018, 20, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Bian, F.; Yang, X.; Zhou, F.; Wu, P.H.; Xing, S.; Xu, G.; Li, W.; Chi, J.; Ouyang, C.; Zhang, Y.; et al. C-reactive protein promotes atherosclerosis by increasing LDL transcytosis across endothelial cells. Br. J. Pharmacol. 2014, 171, 2671–2684. [Google Scholar] [CrossRef]
- Li, L.; Roumeliotis, N.; Sawamura, T.; Renier, G. C-reactive protein enhances LOX-1 expression in human aortic endothelial cells: relevance of LOX-1 to C-reactive protein-induced endothelial dysfunction. Circ. Res. 2004, 95, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Grunfeld, C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In Feingold, K.R.; Anawalt, B., Ed.; Blackman, M.R. eds. Endotext South Dartmouth (MA): MDText.com, Inc.; March 7, 2022. [Google Scholar]
- Kosmas, C.E.; Martinez, I.; Sourlas, A.; Bouza, K.V.; Campos, F.N.; Torres, V.; Montan, P.D.; Guzman, E. High-density lipoprotein (HDL) functionality and its relevance to atherosclerotic cardiovascular disease. Drugs. Context 2018, 7, 212525. [Google Scholar] [CrossRef]
- Li, J.; Ma, H. Associations of the hs-CRP/HDL-C ratio with cardiovascular disease among US adults: Evidence from NHANES 2015-2018. Nutr. Metab. Cardiovasc. Dis. 2024, 103814. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Taga, T.; Yamasaki, K.; Matsuda, T.; Yasukawa, K.; Hirata, Y.; Yawata, H.; Tanabe, O.; Akira, S.; Kishimoto, T. Molecular cloning of the cDNAs for interleukin-6/B cell stimulatory factor 2 and its receptor. Ann. N Y. Acad. Sci. 1989, 557, 167–178, discussion 178–180. [Google Scholar] [CrossRef]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold. Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Mantovani, A.; Bussolino, F.; Dejana, E. Cytokine regulation of endothelial cell function. FASEB J. 1992, 6, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Rus, H.G.; Vlaicu, R.; Niculescu, F. Interleukin-6 and interleukin-8 protein and gene expression in human arterial atherosclerotic wall. Atherosclerosis, 1996, 127, 263–271. [Google Scholar] [CrossRef]
- Su, J.H.; Luo, M.Y.; Liang, N.; et al. Interleukin-6: A Novel Target for Cardio-Cerebrovascular Diseases. Front. Pharmacol. 2021, C12, 745061. [Google Scholar] [CrossRef] [PubMed]
- Puzianowska-Kuźnicka, M.; Owczarz, M.; Wieczorowska-Tobis, K.; Nadrowski, P.; Chudek, J.; Slusarczyk, P.; Skalska, A.; Jonas, M.; Franek, E.; Mossakowska, M. Interleukin-6 and C-reactive protein, successful aging, and mortality: the Pol Senior study. Immun. Ageing 2016, 13, 21. [Google Scholar] [CrossRef]
- Abbatecola, A.M.; Ferrucci, L.; Grella, R.; Bandinelli, S.; Bonafe, M.; Barbieri, M.; Corsi, A.M.; Lauretani, F.; Franceschi, C.; Paolisso, G. Diverse effect of inflammatory markers on insulin resistance and insulin-resistance syndrome in the elderly. J. Am. Geriatr. Soc. 2004, 52, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ye, D.; Wang, Z.; Pan, H.; Lu, X.; Wang, M.; Xu, Y.; Yu, J.; Zhang, J.; Zhao, M.; Xu, S.; Pan, W.; Yin, Z.; Ye, J.; Wan, J. The Role of Interleukin-6 Family Members in Cardiovascular Diseases. Front. Cardiovasc. Med 2022, 9, 818890. [Google Scholar] [CrossRef]
- Lubrano, V.; Cocci, F.; Battaglia, D.; Papa, A.; Marraccini, P.; Zucchelli, G.C. Usefulness of high-sensitivity IL-6 measurement for clinical characterization of patients with coronary artery disease. J. Clin. Lab. Anal. 2005, 19, 110–114. [Google Scholar] [CrossRef]
- Geiger, T.; Andus, T.; Klapproth, J.; Hirano, T.; Kishimoto, T.; Heinrich, P.C. Induction of rat acute-phase proteins by interleukin 6 in vivo. Eur. J. Immunol. 1988, 18, 717–721. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef]
- Carey, L.; Febbraio, M.A. Interleukin-6 and insulin sensitivity: friend or foe? Diabetologia 2004, 47, 1135–1142. [Google Scholar] [CrossRef]
- Starr, R.; Willson, T.A.; Viney, E.M.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; Hilton, D.J. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect Biol. 2018, 10, a028456. [Google Scholar] [CrossRef]
- Viedt, C.; Vogel, J.; Athanasiou, T.; et al. Monocyte chemoattractant protein-1 induces proliferation and interleukin-6 production in human smooth muscle cells by differential activation of nuclear factor-kappaB and activator protein-1. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.; Lacey, B.; Sherliker, P.; et al. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Kerr, R.; Stirling, D.; Ludlam, C.A. Interleukin 6 and haemostasis. Br. J. Haematol. 2001, 115, 3–12. [Google Scholar] [CrossRef]
- Cesari, M.; Penninx, B.W.; Newman, AB; et al. Inflammatory markers and cardiovascular disease (The Health, Aging and Body Composition [Health ABC] Study). Am. J. Cardiol. 2003, 92, 522–528. [Google Scholar] [CrossRef]
- Takeda, N.; Manabe, I.; Shindo, T.; et al. Synthetic retinoid Am80 reduces scavenger receptor expression and atherosclerosis in mice by inhibiting IL-6. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Zamani, P.; Schwartz, G.G.; Olsson, A.G.; Rifai, N.; Bao, W.; Libby, P.; Ganz, P.; Kinlay, S. Inflammatory Biomarkers, Death, and Recurrent Nonfatal Coronary Events after an Acute Coronary Syndrome in the MIRACL Study. J. Am. Heart Assoc. 2013, 2, e003103–10.1161. [Google Scholar] [CrossRef]
- Lindmark, E.; Diderholm, E.; Wallentin, L.; Siegbahn, A. Relationship between interleukin 6 and mortality in patients with unstable coronary artery disease: effects of an early invasive or noninvasive strategy. JAMA 2001, 286, 2107–2113. [Google Scholar] [CrossRef]
- Hedman, A.; Larsson, P.T.; Alam, M.; Wallen, N.H.; Nordlander, R.; Samad, B.A. CRP, IL-6 and endothelin-1 levels in patients undergoing coronary artery bypass grafting. Do preoperative inflammatory parameters predict early graft occlusion and late cardiovascular events? Int. J. Cardiol. 2007, 120, 108–114. [Google Scholar] [CrossRef]
- Souza, J.R.; Oliveira, R.T.; Blotta, M.H.; Coelho, O.R. Serum levels of interleukin-6 (Il-6), interleukin-18 (Il-18) and C-reactive protein (CRP) in patients with type-2 diabetes and acute coronary syndrome without ST-segment elevation. Arq. Bras. Cardiol. 2008, 90, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ. Res. 2002, 91, 988–998. [Google Scholar] [CrossRef]
- Huang, M.; Yang, D.; Xiang, M.; Wang, J. Role of interleukin-6 in regulation of immune responses to remodeling after myocardial infarction. Heart Fail. Rev. 2015, 20, 25–38. [Google Scholar] [CrossRef]
- Tsutamoto, T.; Hisanaga, T.; Wada, A.; Maeda, K.; Ohnishi, M.; Fukai, D.; Mabuchi, N.; Sawaki, M.; Kinoshita, M. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J. Am. Coll. Cardiol. 1998, 31, 391–398. [Google Scholar] [CrossRef]
- Tsujinaka, T.; Fujita, J.; Ebisui, C.; Yano, M.; Kominami, E.; Suzuki, K.; Tanaka, K.; Katsume, A.; Ohsugi, Y.; Shiozaki, H.; Monden, M. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J. Clin. Invest. 1996, 97, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.C.; Hilfiker, A.; Kaminski, K.; Hilfiker-Kleiner, D.; Guener, Z.; Klein, G.; Podewski, E.; Schieffer, B.; Rose-John, S.; Drexler, H. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. FASEB J. 2003, 17, 2118–2120. [Google Scholar] [CrossRef]
- Markousis-Mavrogenis, G.; Tromp, J.; Ouwerkerk, W.; Devalaraja, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.S.; van der Harst, P.; Lang, C.C.; et al. The clinical significance of interleukin-6 in heart failure: results from the BIOSTAT-CHF study. Eur. J. Heart Fail. 2019, 21, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Alogna, A.; Koepp, K.E.; Sabbah, M.; Espindola Netto, J.M.; Jensen, M.D.; Kirkland, J.L.; Lam, C.S.P.; Obokata, M.; Petrie, M.C.; Ridker, P.M.; Sorimachi, H.; Tchkonia, T.; Voors, A.; Redfield, M.M.; Borlaug, B.A. Interleukin-6 in Patients with Heart Failure and Preserved Ejection Fraction. JACC Heart Fail. 2023, 11, 1549–1561. [Google Scholar] [CrossRef]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, PM. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Ridker, P.M. Anticytokine Agents: Targeting Interleukin Signaling Pathways for the treatment of atherothrombosis. Circ. Res. 2019, 124, 437–450. [Google Scholar] [CrossRef]
- Rehman, K.; Akash, M.S.H.; Liaqat, A.; Kamal, S.; Qadir, MI.; Rasul, A. Role of Interleukin-6 in Development of Insulin Resistance and Type 2 Diabetes Mellitus. Crit. Rev. Eukaryot. Gene. Expr. 2017, 27, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Kroke, A.; Möhlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bao, W.; Liu, J.; Ouyang, Y.Y.; Wang, D.; Rong, S.; Xiao, X.; Shan, Z.L.; Zhang, Y.; Yao, P.; Liu, LG. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E745–751. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, Y.; Naka, T.; Fujimoto, M.; Kohzaki, H.; Morita, Y.; Narazaki, M.; Okumura, K.; Saitoh, H.; Nakagawa, R.; Uchiyama, Y.; Akira, S.; Kishimoto, T. Signal transducer and activator of transcription (STAT)-induced STAT inhibitor 1 (SSI-1)/suppressor of cytokine signaling 1 (SOCS1) inhibits insulin signal transduction pathway through modulating insulin receptor substrate 1 (IRS-1) phosphorylation. J. Exp. Med. 2001, 193, 263–269. [Google Scholar] [CrossRef]
- Emanuelli, B.; Peraldi, P.; Filloux, C.; Sawka-Verhelle, D.; Hilton, D.; Van Obberghen, E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J. Biol. Chem. 2000, 275, 15985–15991. [Google Scholar] [CrossRef]
- Krebs, D.L.; Hilton, D.J. SOCS: physiological suppressors of cytokine signaling. J. Cell. Sci. 2000, 113, 2813–2819. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Inflammatory mechanisms in the regulation of insulin resistance. Mol. Med. 2008, 14, 222–231. [Google Scholar] [CrossRef]
- Carey, AL. Interleukin-6 and tumor necrosis factor-alpha are not increased in patients with Type 2 diabetes: evidence that plasma interleukin-6 is related to fat mass and not insulin responsiveness. Diabetologia 2004, 47, 1029–1037. [Google Scholar] [CrossRef]
- Vozarova, B.; Weyer, C.; Hanson, K.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes. Res. 2001, 9, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Lyngso, D.; Simonsen, L.; Bulow, J. Interleukin-6 production in human subcutaneous abdominal adipose tissue: the effect of exercise. J. Physiol. 2002, 543, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Harder-Lauridsen, N.M.; Krogh-Madsen, R.; Holst, J.J.; Plomgaard, P.; Leick, L.; Pedersen, B.K.; Fischer, C.P. Effect of IL-6 on the insulin sensitivity in patients with type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E769–E778. [Google Scholar] [CrossRef] [PubMed]
- Hwa, V.; Nadeau, K.; Wit, J.M.; Rosenfeld, R.G. STAT5b deficiency: lessons from STAT5b gene mutations. Best. Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 61–75. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Malik, R.; Burgess, S.; Dichgans, M. Additive Effects of Genetic Interleukin-6 Signaling Downregulation and Low-Density Lipoprotein Cholesterol Lowering on Cardiovascular Disease: A 2×2 Factorial Mendelian Randomization Analysis. J. Am. Heart Assoc. 2022, 11, e023277. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Pirillo, A.; Norata, G.D. Vascular inflammation and low-density lipoproteins: is cholesterol the link? A lesson from the clinical trials. Br. J. Pharmacol. 2017, 174, 3973–3985. [Google Scholar] [CrossRef]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef]
- Lubrano, V.; Gabriele, M.; Puntoni, MR.; Longo, V.; Pucci, L. Relationship among IL-6, LDL cholesterol and lipid peroxidation. Cell. Mol. Biol. Lett. 2015, 20, 310–22. [Google Scholar] [CrossRef]
- Schuett, H.; Luchtefeld, M.; Grothusen, C.; Grote, K.; Schieffer, B. How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb. Haemost. 2009, 102, 215–222. [Google Scholar]
- Gierens, H.; Nauck, M.; Roth, M.; Schinker, R.; Schürmann, C.; Scharnagl, H.; Neuhaus, G.; Wieland, H.; März, W. Interleukin-6 stimulates LDL receptor gene expression via activation of sterol-responsive and Sp1 binding elements. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1777–1783. [Google Scholar] [CrossRef]
- Zuliani, G.; Volpato, S.; Blè, A.; Bandinelli, S.; Corsi, AM.; Lauretani, F.; Paolisso, G.; Fellin, R.; Ferrucci, L. High interleukin-6 plasma levels are associated with low HDL-C levels in community-dwelling older adults: the InChianti study. Atherosclerosis 2007, 192, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Caselli, C.; De Graaf, M.A.; Lorenzoni, V.; Rovai, D.; Marinelli, M.; Del Ry, S.; Giannessi, D.; Bax, J.J.; Neglia, D.; Scholte, A.J. HDL cholesterol, leptin and interleukin-6 predict high risk coronary anatomy assessed by CT angiography in patients with stable chest pain. Atherosclerosis 2015, 241, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W.; Siebenlist, K.R.; Meh, D.A. The structure and biological features of fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 2001, 936, 11–30. [Google Scholar] [CrossRef]
- Neerman-Arbez, M.; Casini, A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int. J. Mol. Sci. 2018, 19, 192. [Google Scholar] [CrossRef]
- Vilar, R.; Fish, R. J, Casini, A, et al: Fibrinogen in human disease: both friend and foe. Haematologica 2020, 105, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Bridge, K.I.; Philippou, H.; Ariens, R. Clot properties and cardiovascular disease. Thromb. Haemost. 2014, 112, 901–908. [Google Scholar] [CrossRef]
- Wang, W. Identification of respective lysine donor and glutamine acceptor sites involved in factor XIIIa-catalyzed fibrin alpha chain cross-linking. J. Biol. Chem. 2011, 286, 44952–44964. [Google Scholar] [CrossRef]
- de Vries, J.J.; Snoek, C.J.M.; Rijken, D. C, et al: Effects of Post-Translational Modifications of Fibrinogen on Clot Formation, Clot Structure, and Fibrinolysis: A Systematic Review. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 554–569. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef]
- Luzak, B.; Boncler, M.; Kosmalski, M.; Mnich, E.; Stanczyk, L.; Przygodzki, T.; Watala, C. Fibrinogen Glycation and Presence of Glucose Impair Fibrin Polymerization-An In Vitro Study of Isolated Fibrinogen and Plasma from Patients with Diabetes Mellitus. Biomolecules 2020, 10, 877. [Google Scholar] [CrossRef]
- Farrell, D.H. γ' Fibrinogen as a novel marker of thrombotic disease. Clin. Chem. Lab. Med. 2012, 50, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Uitte de Willige, S.; de Visser, MC.; Houwing-Duistermaat, J.J.; Rosendaal, F.R.; Vos, H.L.; Bertina, R.M. Genetic variation in the fibrinogen gamma gene increases the risk for deep venous thrombosis by reducing plasma fibrinogen gamma' levels. Blood 2005, 106, 4176–4183. [Google Scholar] [CrossRef]
- Kannel, W.B.; Wolf, P.A.; Castelli, W.P.; D’Agostino, R.B. Fibrinogen and risk of cardiovascular disease. JAMA 1987, 258, 1183–1186. [Google Scholar] [CrossRef]
- Kryczka, K.E.; Kruk, M.; Demkow, M.; Lubiszewska, B. Fibrinogen and a Triad of Thrombosis, Inflammation, and the Renin-Angiotensin System in Premature Coronary Artery Disease in Women: A New Insight into Sex-Related Differences in the Pathogenesis of the Disease. Biomolecules 2021, 11, 1036. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hennekens, C.H.; Ridker, P.M.; Stampfer, M.J. A prospective study of fibrinogen and risk of myocardial infarction in the Physicians' Health Study. J. Am. Coll. Cardiol. 1999, 33, 1347–1452. [Google Scholar] [CrossRef]
- Danesh, J.; Lewington, S.; Thompson, S.G.; Lowe, G.D.; Collins, R.; Kostis, J.B.; Wilson, A.C.; Folsom, A.R.; Wu, K.; Benderly, M.; et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA 2005, 294, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zheng, Y.Y.; Tang, J.N.; Yang, X.M.; Guo, Q.Q.; Zhang, J.C.; Cheng, M.D.; Song, F.H.; Wang, K.; Zhang, Z.L.; et al. D-Dimer to Fibrinogen Ratio as a Novel Prognostic Marker in Patients After Undergoing Percutaneous Coronary Intervention: A Retrospective Cohort Study. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620948586. [Google Scholar] [CrossRef]
- Zhang, D.P.; Mao, X.F.; Wu, T.T.; Chen, Y.; Hou, X.G.; Yang, Y.; Ma, X.; Zhang, J.Y.; Ma, Y.T.; Xie, X.; et al. The Fibrinogen-to-Albumin Ratio Is Associated With Outcomes in Patients With Coronary Artery Disease Who Underwent Percutaneous Coronary Intervention. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620933008. [Google Scholar] [CrossRef]
- Yuan, D.; Jiang, P.; Zhu, P.; Jia, S.; Zhang, C.; Liu, Y.; Liu, R.; Xu, J.; Tang, X.; Zhao, X.; et al. Prognostic value of fibrinogen in patients with coronary artery disease and prediabetes or diabetes following percutaneous coronary intervention: 5-year findings from a large cohort study. Cardiovasc. Diabetol. 2021, 20, 143. [Google Scholar] [CrossRef]
- Meng, Z.; Zhao, Y.; He, Y. Fibrinogen Level Predicts Outcomes in Critically Ill Patients with Acute Exacerbation of Chronic Heart Failure. Dis. Markers. 2021. [Google Scholar] [CrossRef]
- Xu, Q.; Zhu, C.; Zhang, Q.; Hu, Z.; Ji, K.; Qian, L. Association between fibrinogen-to-albumin ratio and prognosis of patients with heart failure. Eur. J. Clin. Invest. 2023, 53, e14049. [Google Scholar] [CrossRef] [PubMed]
- Isordia-Salas, I.; Galván-Plata, M.; Leaños-Miranda, A.; Aguilar-Sosa, E.; Anaya-Gómez, F.; Majluf-Cruz, A.; Santiago-Germán, D. Proinflammatory and prothrombotic state in subjects with different glucose tolerance status before cardiovascular disease. J. Diabetes. Res. 2014, 631902. [Google Scholar] [CrossRef]
- Kumar, S.; Chapagain, A.; Nitsch, D.; Yaqoob, M. Proteinuria and hypoalbuminemia are risk factors for thromboembolic events in patients with idiopathic membranous nephropathy: an observational study. BMC. Nephrol. 2012, 13, 107. [Google Scholar] [CrossRef]
- Wang, L.; Cong, H.L.; Zhang, J.X.; Li, X.M.; Hu, Y.C.; Wang, C.; Lang, J.C.; Zhou, B.Y.; Li, T.T.; Liu, C.W.; Yang, H.; Ren, L.B.; Qi, W.; Li, W.Y. Prognostic performance of multiple biomarkers in patients with acute coronary syndrome without standard cardiovascular risk factors. Front. Cardiovasc. Med. 2022, 9, 916085. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Wu, N.Q.; Shi, H.W.; Dong, Q.; Dong, Q.T.; Gao, Y.; Guo, Y.L.; Li, J.J. Fibrinogen is associated with glucose metabolism and cardiovascular outcomes in patients with coronary artery disease. Cardiovasc. Diabetol. 2020, 19, 36. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, E.; Pérez-Martin, A.; Brun, J.; Aïssa-Benhaddad, A.; Fédou, C.; Mercier, J. Relationships between fibrinogen and insulin resistance. Atherosclerosis 2000, 150, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Barazzoni, R.; Kiwanuka, E.; Zanetti, M.; Cristini, M.; Vettore, M.; Tessari, P. Insulin acutely increases fibrinogen production in individuals with type 2 diabetes but not in individuals without diabetes. Diabetes 2003, 52, 1851–1856. [Google Scholar] [CrossRef]
- Perween, S.; Abidi, M.; Faizy, A.F.; Moinuddin. Post-translational modifications on glycated plasma fibrinogen: A physicochemical insight. Int. J. Biol. Macromol. 2019, 126, 1201–1212. [Google Scholar] [CrossRef]
- Austin, G.E.; Mullins, R.H.; Morin, L.G. Non-enzymic glycation of individual plasma proteins in normoglycemic and hyperglycemic patients. Clin. Chem. 1987, 33, 2220–2224. [Google Scholar] [CrossRef]
- Rheeder, P.; Jerling, J.C.; Loots, D.T.; van der Westhuizen, F.H.; Gottsche, L.T.; Weisel, J.W. Glycation of fibrinogen in uncontrolled diabetic patients and the effects of glycaemic control on fibrinogen glycation. Thromb. Res. 2007, 120, 439–446. [Google Scholar]
- Pieters, M.; Covic, N.; Loots du, T.; van der Westhuizen, F.H.; van Zyl, D.G.; Rheeder, P.; Jerling, J.C.; Weisel, J.W. The effect of glycaemic control on fibrin network structure of type 2 diabetic subjects. Thromb. Haemost. 2006, 96, 623–629. [Google Scholar] [PubMed]
- Pieters, M.; van Zyl, D.G.; Rheeder, P.; Jerling, J.C.; Loots, D.T.; van der Westhuizen, F.H.; Gottsche, L.T.; Weisel, J.W. Glycation of fibrinogen in uncontrolled diabetic patients and the effects of glycaemic control on fibrinogen glycation. Thromb. Res. 2007, 120, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Halle, M.; Berg, A.; Keul, J.; and Baumstark, W.M. Association Between Serum Fibrinogen Concentrations and HDL and LDL Subfraction Phenotypes in Healthy Men. Arterioscler. Thromb. Vasc. Biol. 1996, 16. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Wu, W.; Lu, H.; Liu, J.; Chen, S.; Liang, G.; Zhou, Y.; Yu, S.; Qiao, L.; Chen, J.; Tan, N.; Liu, Y.; Chen, J. Fibrinogen to HDL-Cholesterol ratio as a predictor of mortality risk in patients with acute myocardial infarction. Lipids Health Dis. 2024, 23, 86. [Google Scholar] [CrossRef]
- Kaptoge, S.; White, I.R.; Thompson, S.G.; Wood, A.M.; Lewington, S.; Lowe, G.D.; Danesh, J. Associations of plasma fibrinogen levels with established cardiovascular disease risk factors, inflammatory markers, and other characteristics: individual participant meta-analysis of 154,211 adults in 31 prospective studies: the fibrinogen studies collaboration. Fibrinogen Studies Collaboration. Am. J. Epidemiol. 2007, 166, 867–179. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



