
Submitted:
07 April 2025
Posted:
07 April 2025
You are already at the latest version
Abstract
Turnersyndrome (TS) is a genetic chromosomal disorder including various manifestations depending on the karyotype;endocrine, gastrointestinal, respiratory, neurological, urogenital, musculoskeletal, and cardiovascular disorders contribute to increased morbidity and mortality. Liver function abnormalitiesare less well studiedand mostly associated withinsulinresistance, obesity, diabetes, hypogonadism, hypothyroidism, and autoimmune conditions. The association of liver pathology with architectural changes of various etiologies and the metabolicdysfunction-associated liver diseaseis of particular interest. Herein, we presentthree cases of adult women with TSand persistent elevation of liver enzymesdue toporto-sinusoidal vascular disorder(PSVD). In one case,the diagnosis of TS followedthe liver biopsy results.The absence of cardiometabolic risk factors, the low liver stiffness andthe cardiovascular malformationsmay predict this histological diagnosis. Liver function impairment in TS may derive from a broad spectrum of liver pathology, including PSVD, and requires careful evaluation,to decrease the risk of complications.
Keywords:
Turner syndrome
; porto-sinusoidal vascular disorder
; obliterative portal venopathy
; karyotype
; aminotransferases
; cirrhosis
; steatosis
; vascular liver disease
; liver biopsy
Introduction
Turner syndrome (TS) is a genetic chromosomal disorder of phenotypic females,consisting of one intact X chromosome and complete or partial absence of the second sex chromosome. Mosaicism of the X chromosome (45,XO/46,XX or 45,X/47,XXX) or Y chromosome variants and X chromosome abnormalities (isochromosome of either p or q arm, deletion of Xp or Xq, ring X and mosaicism)may also be encountered. It affects about 1 in 2000 or 2500 live female births, but the true prevalence is unknown, as many individuals with mild phenotype remain undiagnosed, while 99% of severely affected fetuses won’t survive to term. [1] The median age at diagnosis is 15 years and is characterized by three age peaks: prenatal period (during screening), between 5 and 20 years (due to short stature or delayed puberty), and between 30 and 40 years (due to infertility). [2] TSpresentation includesendocrine, gastrointestinal, respiratory, neurological, urogenital, musculoskeletal and cardiovascular disorders, that are associated with increased morbidity and mortality. [3] The severity of clinical presentation, morbidity and mortality is proportional to the karyotype, with 45,XO being the most severe affected karyotype,followed by isochromosome X. [1,4]
Liver function abnormalities are less well described in TS.They affect approximately 30-50%of patients, according to several population studies [5,6,7] and approximately 60-90%, according to matched control studies. [8,9,10] Several risk factors have been associated with liver disease, including isochromosome X karyotype, age, insulin resistance (IR), metabolic syndrome (MS), hypogonadism, hypothyroidism, autoimmune conditions and cardiovascular disease. [9,10] Patients with TS have a 6-fold increased risk of cirrhosis comparedwith the general population, [11] and liver disease seems to be the second most commoncause of death in adult women with TS. [12] MS isassociated with steatosis, with or without inflammation,which may progress to steatohepatitis with fibrosis, whileCVD (cardiovascular disease) is associated with hepatic vascular architectural changes. [13] Recently, the latter hasbeenincludedwithin the spectrum of porto-sinusoidal vascular disorder (PSVD), a group of hepatic vascular diseases in non-cirrhotic patients with lesions of portal vasculature and sinusoids, irrespective of the presence or absence of portal hypertension. [14,15] Liver biopsy is required for diagnosis, and the histological changes consist ofportal and periportal vascular alterations, abnormal vascular relationships, and nodular regenerative hyperplasia (NRH).Conditions associated with PSVD include hematological diseases, prothrombotic conditions, immunological disorders, drug-induced liver injury, repeated gastrointestinal infections, and genetic diseases, including TS. [15]
Herein, we present three cases of adult women with TS having a chronic and stable elevation of aminotransferases of unknown etiology, that was finally attributed to PSVD. In addition,we discuss the possible pathogenetic associations and present a review of the literature.
Cases Presentation
Case 1
A 22-year-old femalehas been followed at the Hepatology Unitdue to elevated serum aminotransferases since the age of 16years. Her medical history included a full-term 38-week labor, with normal birth weight,body length and head circumference (3400g, 51cm and 35cm, respectively), astigmatism, hypermetropia, strabismus and iron deficiency anemia.Menarche occurred at 12 years old, with normal menstrual cycle and complete pubertal development (Tanner stages: axillary hair AH=III, pubic hair PH=V, breasts B=V). She was only taking oral iron supplements and reportedno allergy/smoking history or alcohol use.Phenotypically, she had short stature (body height of 145 cm), broad short-appearing neck, low hairline at the back of the neck, deformity of external ears and broad chest and a BMI (Body Mass Index) of26 kg/m2. Laboratory investigation, except fromtransient liver enzymes elevation,showed hypochromic microcytic anemia and mild thrombocytosis.Hepatitis virus screening, autoantibodies, serum γ-globulins,immunoglobulin G (IgG),α1-antitrypsin, copper and ceruloplasminwere all negative (Table 1).Ultrasound scan of the heart, abdomen and pelviswas normal.Fibrosis 4 (FIB-4) score was 0.35 and liver stiffness measurement was6.9 kPa, indicating nosignificant fibrosis. Liver biopsy was indicative of PSVD and the genetic testing that followedshowed karyotype monosomy 45, XO, consistent with TS.
Case 2
A 30-year-old female, with a known mosaic TS (45,XO/46,XX, 80% and 20%, respectively)diagnosed at the age of 9 months, has been followed at the Hepatology Unit, due to elevated liver enzymes for the past 4 years.Her medical history included a late-term 41-week labor, with normal birth weight,body length and head circumference (2600g, 52cm and 35cm, respectively). She was diagnosed with bicuspid aortic valve and was operated for coarctation of aorta at the age of 9months. Moreover, she underwent total thyroidectomy due to thyroid cancer at the age of 11yearsand was diagnosed with astigmatism, hypermetropia, strabismus and dyslipidemia. Menarche occurred at 17years old, with irregular menstrual cycle and menorrhagia, and incomplete pubertal development (Tanner stages: AH=II, PH=IV, B=III). She was treated withrecombinant human growth hormone replacement therapy until puberty and took combined oral contraceptive pills for 10 years, until the age of27years.At present,she was treated with oral levothyroxine,hormone replacement therapy (HRT) (transdermalestradiol and oral progesterone), vitamin D supplements and omega-3 triglycerides. She had no known allergies and no smoking history or alcohol use. Phenotypically, she had short stature (body height of 153 cm), short neck with a webbed appearance, low hairline at the back of the neck and low-set ears and a BMI of17 kg/m2. Laboratory investigation showed increased liver enzymes, whereas hepatitis virus screening,autoantibodies, serum γ-globulins, IgG, α1-antitrypsin, copper and ceruloplasmin were all negative (Table 1). Liver ultrasound was normal. FIB-4 score was 0.88 and liver stiffness measurement was 2.6 kPa, indicating no significant fibrosis.
Case 3
A 23-year-old female diagnosed with classic TS (45,XO)at the age of 6 yearswas followed at the Endocrinology Unit and was referred to the Hepatology Unit due to transiently elevated hepatic enzymes for the past 4 years. Her medical history included a full-term labor, with normal birth weight,body length and head circumference (2800g, 50cm and 36 cm, respectively), mildconstriction of aorta,chronic autoimmune thyroiditisand operated eardrum perforation, leaving hearing loss in her left ear.She was treated with human recombinant growth hormone replacement therapy until the age of 14 years, while puberty and menarche were induced with hormone therapy. At present, she was treated with oral levothyroxine, HRT (combined transdermalestrogen and progesterone), and vitamin D supplements. She had a normal menstrual cycle and complete pubertal development (AH=III, PH=V, B=V). She reported no known allergies,history of smoking or alcohol use. Phenotypically, she had short stature (body height of 150cm), short, webbed neck, low hairline, ear deformity, shield-shaped thorax, widely spaced nipples and a normal BMI of 22.2kg/m2.Laboratory investigationdid not indicate chronic liver disease(Table 1).FIB-4 score was 0.57, liver stiffness measurement was 4.5 kPa and liver ultrasound showed no abnormal findings.
Diagnostic Assessment
Liver biopsy was performedin order toevaluatethe underlying pathophysiology of persistent aminotransferases elevation. In all cases, an adequate sample was obtained,including 12-17 portal tracts.Parenchymal architecture was abnormal, with irregular distribution of portal tracts, terminal hepatic venules, andmild non zonal sinusoidal dilatation. On reticulin stain, areas of hepatocellular regeneration without nodule formation were detected. A few portal tracts were hypervascularized, containing a large number of thin-walled blood vessels. Focally, aberrant periportal thin-walled vessels were also noted. Rare portal venules were stenosedsuggestive of obliterative portal venopathy, while few others were herniated, directly abutting into the neighboring liver parenchyma (Figure 1 and Figure 2). In cases 1 and 3, no portal inflammation or lesions indicative of cholangiopathy were noted.On the contrary, in case 2,there were mild portal chronic inflammation without interface activity and mild loss of interlobular bile ducts with associated mild chronic cholestasis, indicative of mild chronic cholangiopathy (Figure 2). Nosignificant lobular inflammation or steatosis was noted. In all cases, Masson trichrome staining highlighted mild sinusoidal and portal fibrosis without bridging septa,whileCD34 immunostaining indicated focal sinusoidal capillarization in zones 2 and/or 3.The overall histology,as described above,was within the spectrum of PSVD, including one specific (obliterative portal venopathy/portal vein stenosis) and one or several non-specific histological features. [14]
Discussion
In contrast to other TS manifestations, liver disease is less well studied and data regarding PSVD in TS are limited. Even though such cases have been described in the past as having NRH and/or obliterative portal venopathy, [5,7,16,17] no large case series exist.We present three cases of adult women with TS and unexplained liver enzyme increase, all being at low risk of developing advanced fibrosis and cirrhosis, according to FIB-4 and elastography measurements. On liver biopsy, overall histology was within the spectrum of PSVD. It is noteworthy that, on the contrary to the other two cases, in case 1, PSVD diagnosis guided the diagnosis of TS.
PSVD has recently been recognized as one of the disorders in the spectrum of liver diseaseassociated with TS. It is a rare, novel, umbrella clinicopathological entity, encompassing vascular lesions affecting portal venules and sinusoids, in the absence of cirrhosis. The term was introduced by the Vascular Liver Diseases Group to overcome limitations of the previously used “idiopathic non-cirrhotic portal hypertension”. [14,15] PSVD does not rely on the presence of portal hypertension and thereforeincludes asymptomatic patients at earlier stages of the disease. PSVD diagnosis requires an adequate liver biopsy excluding cirrhosis, and one of the following: (1) the presence of either one specific histological sign or one specific sign of portal hypertension or (2) the combination of one non-specific histological sign and one non-specific sign of portal hypertension). [14,15,18]
PSVD has been associated with a wide range of disorders, including autoimmuneand hematological diseases, drug exposure and various genetic syndromes,such asTS. [15] However, the exact pathogenetic mechanisms involved are not fully understood. It has been speculatedthat autoimmunity, vascular abnormalities, thrombotic and possibly genetic predisposition, maycontribute to the development of PSVD. [14,15,19]
Indeed, patients with TS face a 2-4 times increased risk of developing autoimmune disorders compared to the general population. [20] This has been attributed to thedysregulation of immunity tolerance and adaptation to antigen patterns, due to haploinsufficiency of X-linked genes. [21] In addition, TS patients are also at increased risk of autoimmune conditions linked to hepatobiliary pathology, such as celiac disease and inflammatory bowel diseases (Crohn’s disease and ulcerative colitis), with a ranging prevalence of0.67- 4%. [22] Markedly, patients with PSVD without the syndrome may also have altered immunity; increased activation of intrasinusoidal T lymphocytes may beresponsible for damaging the endothelial cell lining of portal venules and sinusoids. [23,24]
Liver parenchymal architectural changes in TS may be the consequence of a primary vascular involvement. [25] Congenital cardiovascular malformations including aortic coarctation (7-14%), bicuspid aortic valve (14-34%) and aortic dilation/aneurysm (3-42%)seem to progress rapidly. [1,26] Interestingly, in TS patients, aortic abnormalities may induce overt liver parenchymal architectural changes, including NRH, multiple focal nodular hyperplasia (FNH) and cirrhosis, that increase the risk of severe liver-related complications. [25,26] If a similar, rapid or slow, progression of vascular changes occurs in TS women, it could explain why liver involvement increases with age.
TS is also associated with venous thrombosis and increased number of circulating prothrombotic agents, such as von Willebrand factor or factor VIII. [27] This prothrombotic state may lead to repeated formation of micro-emboli inside the portal venule lumen, resulting inportal stenosis (obliterative portal venopathy). [27,28,29] NRH is known to result from impaired hepatic blood flow and is a specific histological feature of PSVD. [14]
TS-associated liver disease is often considered as a consequence of metabolic and endocrine disorders, such as obesity and MS. IR, central obesity, diabetes and hyperlipidemia are linked with a spectrum of liver injury, ranging from steatosis and steatohepatitis to cirrhosis. [30] Hypogonadism is frequently associated with metabolic dysfunction-associated liver disease (MASLD). [31] Hypothyroidism may drive hepatic steatosis due to impaired lipid metabolism and hypothyroidism-induced myopathy. [32]
Liver biopsy assessed by an expert hepato-pathologist seems to be a useful diagnostic and probably prognostic tool in TS patients with persistently abnormal liver tests. [33] TS-associated liver injury presents with three main types of histology: (1) portal inflammatory infiltrate and steatohepatitis, (2) biliary lesions with periductal fibrosis and (3) architectural changes. Roulot’ et al., in the largest clinicopathological studyin TS, [16] showed that patients without marked architectural changes do not experience liver-related complications and progressive liver disease. They proposed that cholangiopathy in TS patients without distorted liver tissue architecture may be related to altered blood supply and thus, correspond to one end of a spectrum of vascular anomalies, with marked architectural changes being on the other end. If this hypothesis is correct,two distinct, but not exclusive,major histology patterns can be recognized: (1) steatosis and steatohepatitis, likely linked to overweight, IR and MS, and (2) vascular abnormalities with architectural changes (with or without cholangiopathy),related to abnormal angiogenesis and vessel abnormalities (with or without autoimmune or thromboembolic disease).The first has low risk, whereas the second has increased risk of liver-related complications. Nevertheless, recent studies have shown that PSVD without portal hypertension is characterized by uncomplicated/benign course. [34,35] Bourcigaux et al., using both non-invasive tests and biopsy, reported that most patients (>90%) are at low risk of developing fibrosis [7] , although data suggest that TS women in general have a 6-fold increased risk of cirrhosis.
In our series, none of our patientshad obesity orMS. Two of them (cases 2 and 3) had constriction of aorta. Liver biopsy revealed either no or mild inflammation. The overall histology was within the spectrum of PSVD. Roulot et al., in accordance with our findings, reported that architectural hepatic changes are the principal lesion in TS patients without obesity or metabolic dysfunction and might be part of a general vascular disorder. [16] In Calanchini’s study, [5] half of the women with architectural hepatic changes had cardiac abnormalities and aortic dilatation. Recently,Bourcigaux et al. [7] supported the link between NRH and high aortic index.In our study, patients with cardiovascular malformations (cases 2 and 3) presented liver enzyme elevations at 25 years and 21 years of age, respectively, suggesting a slow progression rate of liver histological changes.Interestingly, the mild chronic cholangiopathy in one case(case 2)coexisted with PSVD whereas autoantibodies were all negative. This may indicate a relationship between chronic cholangiopathy and vascular abnormalities in TS.
According to a limited number of studies before 2019, approximately 20% of patients with obliterative portal venopathy have only liver biochemical abnormalities without any signs of portal hypertension at presentation. [36] Patients who presented with idiopathic noncirrhotic portal hypertension exhibited a mortality rate of 15-20% during a follow-up period of 8 years. [37,38,39]
Todate, data regarding the course of PSVD is rather sparse. However, according to two recent studies in adults, it seems that in the absence of portal hypertension, PSVD shows either slow or no disease progression. [34,35] The predisposingfactors of portal hypertension in PSVD have not yet beenidentified, however it is thought that prothrombotic and immunological disorders are the most decisive ones. [40]
Data regarding PSVD in TS are even more limited, and itscourse and prognosis is mostly unknown. Only a few cases have been described,having a poor clinical outcome, mainly due to complications of portal hypertension (variceal bleeding, ascites). [16,41]
Since liver biochemical abnormalities may be present in a significant subset of TS patients, Doppler ultrasound is recommended to detect signs of portal hypertension, and transient elastography to assess liver stiffness and evaluate the risk of advanced liver fibrosis and cirrhosis. [25] Liver biopsy may be considered, after assessing the patient’s history, imaging studies and clinical noninvasive fibrosis scores, such as FIB-4. [7] HRTis essential for sexual development and metabolic and bone health in TS, but concern has been raised regarding possible exacerbation of hepatic dysfunction. However, a trend towards a beneficial impact of HRT on TS-related liver disease has been observed in National Patient Registries, [8] retrospective, [42] and longitudinal studies. [43] Additionally, cardiometabolic risk factors management is of great significance in TS. [1] Improving metabolic parameters through lifestyle interventions (diet and physical activity), as well as maintaining a heathy body weight, not only reduces cardiovascular risk, but also helps preventing liver disease. [1,30,44]
Conclusions
A multisystemic approach is required for TS patients with liver pathology. Screening for liver function must be included in the follow-up of people with TS.Although PSVD is rather uncommon, it is important to be included in the differential diagnosis of people with TS and persistently elevated liver enzymes, especially when they have cardiovascular malformations but no cardiometabolicrisk factors. In this population,PSVD seems to be under-recognized,and may be the consequence of primary vascular involvement.
Conflicts of Interest statement
The authors declare no conflict of interest in relation to this manuscript.
Funding statement
No specific funding was received for this work.
Financial support statement
No specific funding was received for this work.
Ethics approval
Written informed consent was obtained from each patient included in the study. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the institution's human research committee.
Acknowledgments
We are thankful to the personnel of the Hepatology Department, Sotiria General Hospital and the Endocrine and Bone Metabolic Disorders Unit, Attikon University Hospital, Athens, Greece. We gratefully acknowledge the technical expertise of Dr DespoinaKarandrea, MsVassilikiSaltoyanni and Ms Georgia Lamari (Department of Pathology, Aretaieion Hospital, National and Kapodistrian University of Athens), in performing the histochemical and immunohistochemical stains. The opinions presented in this article are those of the authors, and do not necessarily represent those of their institutions.
Abbreviations
Body Mass Index, CVD; Cardiovascular Disease, FIB-4; Fibrosis 4 score, HRT; Hormone Replacement Therapy, IgG; Immunoglobulin G, IR; Insulin Resistance, MASLD; Metabolic dysfunction-Associated Steatotic Liver Disease, MS; Metabolic Syndrome, NRH; Nodular Regenerative Hyperplasia, PSVD; Porto-Sinusoidal Vascular Disorder, TS; Turner Syndrome.
References
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017, 177, G1–G70. [Google Scholar] [CrossRef] [PubMed]
- Gravholt, C.H.; Viuff, M.; Just, J.; Sandahl, K.; Brun, S.; van der Velden, J.; Andersen, N.H.; Skakkebaek, A. The Changing Face of Turner Syndrome. Endocr. Rev. 2022, 44, 33–69. [Google Scholar] [CrossRef]
- Gravholt, C.H.; Viuff, M.H.; Brun, S.; Stochholm, K.; Andersen, N.H. Turner syndrome: mechanisms and management. Nat. Rev. Endocrinol. 2019, 15, 601–614. [Google Scholar] [CrossRef]
- Pimblett, A.C.; La Rosa, C.; King, T.F.J.; Davies, M.C.; Conway, G.S. The Turner syndrome life course project: Karyotype-phenotype analyses across the lifespan. Clin. Endocrinol. 2017, 87, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Calanchini, M.; Moolla, A.; Tomlinson, J.W.; Cobbold, J.F.; Grossman, A.; Fabbri, A.; Turner, H.E. Liver biochemical abnormalities in Turner syndrome: A comprehensive characterization of an adult population. Clin. Endocrinol. 2018, 89, 667–676. [Google Scholar] [CrossRef]
- El-Mansoury, M.; Berntorp, K.; Bryman, I.; Hanson, C.; Innala, E.; Karlsson, A.; Landin-Wilhelmsen, K. Elevated liver enzymes in Turner syndrome during a 5-year follow-up study. Clin. Endocrinol. 2007, 68, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Bourcigaux, N.; Dubost, E.; Buzzi, J.-C.; Donadille, B.; Corpechot, C.; Poujol-Robert, A.; Christin-Maitre, S. Focus on Liver Function Abnormalities in Patients With Turner Syndrome: Risk Factors and Evaluation of Fibrosis Risk. J. Clin. Endocrinol. Metab. 2023, 108, 2255–2261. [Google Scholar] [CrossRef]
- Viuff, M.H.; Stochholm, K.; Grønbæk, H.; Berglund, A.; Juul, S.; Gravholt, C.H. Increased occurrence of liver and gastrointestinal diseases and anaemia in women with Turner syndrome – a nationwide cohort study. Aliment. Pharmacol. Ther. 2021, 53, 821–829. [Google Scholar] [CrossRef]
- Singh, I.; Noel, G.; Barker, J.M.; Chatfield, K.C.; Furniss, A.; Khanna, A.D.; Nokoff, N.J.; Patel, S.; Pyle, L.; Nahata, L.; et al. Hepatic abnormalities in youth with Turner syndrome. Liver Int. 2022, 42, 2237–2246. [Google Scholar] [CrossRef]
- Koulouri, O.; Ostberg, J.; Conway, G.S. Liver dysfunction in Turner's syndrome: prevalence, natural history and effect of exogenous oestrogen. Clin. Endocrinol. 2008, 69, 306–310. [Google Scholar] [CrossRef]
- Gravholt, C.H.; Juul, S.; Naeraa, R.W.; Hansen, J. Morbidity in Turner Syndrome. J. Clin. Epidemiology 1998, 51, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.M.; Jost, C.A.; Babovic-Vuksanovic, D.; Connolly, H.M.; Egbe, A. Long-Term Outcomes in Patients With Turner Syndrome: A 68-Year Follow-Up. J. Am. Hear. Assoc. 2019, 8, e011501. [Google Scholar] [CrossRef]
- Fedor, I.; Zold, E.; Barta, Z. Liver Abnormalities in Turner Syndrome: The Importance of Estrogen Replacement. J. Endocr. Soc. 2022, 6, bvac124. [Google Scholar] [CrossRef]
- De Gottardi, A.; Sempoux, C.; Berzigotti, A. Porto-sinusoidal vascular disorder. J. Hepatol. 2022, 77, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Rautou, P.-E.; Schouten, J.; Rubbia-Brandt, L.; Leebeek, F.; Trebicka, J.; Murad, S.D.; Vilgrain, V.; Hernandez-Gea, V.; Nery, F.; et al. Porto-sinusoidal vascular disease: proposal and description of a novel entity. Lancet Gastroenterol. Hepatol. 2019, 4, 399–411. [Google Scholar] [CrossRef]
- Roulot, D.; Degott, C.; Chazouillères, O.; Oberti, F.; Calès, P.; Carbonell, N.; Benferhat, S.; Bresson-Hadni, S.; Valla, D. Vascular involvement of the liver in Turner's syndrome. Hepatology 2004, 39, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Gioia, S.; Baiocchini, A.; D'Amati, G.; Tavano, D.; Ridola, L.; Nardelli, S.; de Felice, I.; Lapenna, L.; Merli, M.; Pellicelli, A.; et al. Porto-sinusoidal vascular disorder (PSVD): Application of new diagnostic criteria in a multicenter cohort of patients. Dig. Liver Dis. 2023, 56, 291–296. [Google Scholar] [CrossRef]
- Guido, M.; Alves, V.A.F.; Balabaud, C.; Bathal, P.S.; Bioulac-Sage, P.; Colombari, R.; Crawford, J.M.; Dhillon, A.P.; Ferrell, L.D.; Gill, R.M.; et al. Histology of portal vascular changes associated with idiopathic non-cirrhotic portal hypertension: nomenclature and definition. Histopathology 2018, 74, 219–226. [Google Scholar] [CrossRef]
- Ciriaci, N.; Bertin, L.; Rautou, P.-E. Genetic predisposition to porto-sinusoidal vascular disorder. Hepatology 2024. [Google Scholar] [CrossRef]
- Jørgensen, K.T.; Rostgaard, K.; Bache, I.; Biggar, R.J.; Nielsen, N.M.; Tommerup, N.; Frisch, M. Autoimmune diseases in women with Turner's Syndrome. Arthritis Rheum. 2010, 62, 658–666. [Google Scholar] [CrossRef]
- Lleo, A.; Moroni, L.; Caliari, L.; Invernizzi, P. Autoimmunity and Turner’s syndrome. Autoimmun. Rev. 2012, 11, A538–A543. [Google Scholar] [CrossRef] [PubMed]
- Gatti, S.; Gelzoni, G.; Catassi, G.N.; Catassi, C. The Clinical Spectrum of Inflammatory Bowel Disease Associated With Specific Genetic Syndromes: Two Novel Pediatric Cases and a Systematic Review. Front. Pediatr. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Tokushige, K.; Haruta, I.; Yamauchi, K.; Hayashi, N. Analysis of adhesion molecules in patients with idiopathic portal hypertension. J. Gastroenterol. Hepatol. 1999, 14, 364–369. [Google Scholar] [CrossRef]
- Kotani, K.; Kawabe, J.; Morikawa, H.; Akahoshi, T.; Hashizume, M.; Shiomi, S. Comprehensive Screening of Gene Function and Networks by DNA Microarray Analysis in Japanese Patients with Idiopathic Portal Hypertension. Mediat. Inflamm. 2015, 2015, 349215. [Google Scholar] [CrossRef]
- Roulot, D. Liver involvement in Turner syndrome. Liver Int. 2012, 33, 24–30. [Google Scholar] [CrossRef]
- Gravholt, C.H. Turner Syndrome and the Heart. Am. J. Cardiovasc. Drugs 2002, 2, 401–413. [Google Scholar] [CrossRef]
- Zilz, C.K.; Brenner, J.K.; Elnecave, R.H. Portal Vein Thrombosis and High Factor VIII in Turner Syndrome. Horm. Res. Paediatr. 2006, 66, 89–93. [Google Scholar] [CrossRef]
- Jobe, S.; Donohoue, P.; Di Paola, J. Deep Venous Thrombosis and Turner Syndrome. J. Pediatr. Hematol. 2004, 26, 272. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.B.; Silveira, T.R.; Bandinelli, E.; Röhsig, L. Portal vein thrombosis in children and adolescents: The low prevalence of hereditary thrombophilic disorders. J. Pediatr. Surg. 2004, 39, 1356–1361. [Google Scholar] [CrossRef]
- Hutchison, A.L.; Tavaglione, F.; Romeo, S.; Charlton, M. Endocrine aspects of metabolic dysfunction-associated steatotic liver disease (MASLD): Beyond insulin resistance. J. Hepatol. 2023, 79, 1524–1541. [Google Scholar] [CrossRef]
- Singeap, A.-M.; Stanciu, C.; Huiban, L.; Muzica, C.M.; Cuciureanu, T.; Girleanu, I.; Chiriac, S.; Zenovia, S.; Nastasa, R.; Sfarti, C.; et al. Association between Nonalcoholic Fatty Liver Disease and Endocrinopathies: Clinical Implications. Can. J. Gastroenterol. Hepatol. 2021, 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Piantanida, E.; Ippolito, S.; Gallo, D.; Masiello, E.; Premoli, P.; Cusini, C.; Rosetti, S.; Sabatino, J.; Segato, S.; Trimarchi, F.; et al. The interplay between thyroid and liver: implications for clinical practice. J. Endocrinol. Investig. 2020, 43, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Burt, A.D.; Gouw, A.S.H.; Callea, F.; Clouston, A.D.; Dienes, H.; Goodman, Z.D.; Kakar, S.; Kleiner, D.E.; Lackner, C.; Park, Y.N.; et al. Making Sense of ‘Porto-Sinusoidal Vascular Disorder’: What Does It Mean for the Pathologist and the Patient? Liver Int. 2024, 45, e16196. [Google Scholar] [CrossRef] [PubMed]
- Wöran, K.; Semmler, G.; Jachs, M.; Simbrunner, B.; Bauer, D.J.M.; Binter, T.; Pomej, K.; Stättermayer, A.F.; Schwabl, P.; Bucsics, T.; et al. Clinical Course of Porto-Sinusoidal Vascular Disease Is Distinct From Idiopathic Noncirrhotic Portal Hypertension. Clin. Gastroenterol. Hepatol. 2022, 20, e251–e266. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Q.; Zhong, Y.; Liu, D.; Liu, H.; Wang, L.; Du, Z.; Chen, M.; Zheng, Y.; Yang, Y. Clinical characteristics and natural history of porto-sinusoidal vascular disease: A cohort study of 234 patients in China. Liver Int. 2024, 44, 2329–2340. [Google Scholar] [CrossRef]
- Cazals-Hatem, D.; Hillaire, S.; Rudler, M.; Plessier, A.; Paradis, V.; Condat, B.; Francoz, C.; Denninger, M.-H.; Durand, F.; Bedossa, P.; et al. Obliterative portal venopathy: Portal hypertension is not always present at diagnosis. J. Hepatol. 2010, 54, 455–461. [Google Scholar] [CrossRef]
- Siramolpiwat, S.; Seijo, S.; Miquel, R.; Berzigotti, A.; Garcia-Criado, A.; Darnell, A.; Turon, F.; Hernandez-Gea, V.; Bosch, J.; Garcia-Pagán, J.C. Idiopathic portal hypertension: Natural history and long-term outcome. Hepatology 2013, 59, 2276–2285. [Google Scholar] [CrossRef]
- Hollande, C.; Mallet, V.; Darbeda, S.; Vallet-Pichard, A.; Fontaine, H.; Verkarre, V.; Sogni, P.; Terris, B.; Gouya, H.; Pol, S. Impact of Obliterative Portal Venopathy Associated With Human Immunodeficiency Virus. Medicine 2016, 95, e3081. [Google Scholar] [CrossRef]
- Schouten, J.N.L.; Van der Ende, M.E.; Koëter, T.; Rossing, H.H.M.; Komuta, M.; Verheij, J.; van der Valk, M.; Hansen, B.E.; Janssen, H.L.A. Risk factors and outcome of HIV-associated idiopathic noncirrhotic portal hypertension. Aliment. Pharmacol. Ther. 2012, 36, 875–885. [Google Scholar] [CrossRef]
- Guido, M.; Sarcognato, S.; Sonzogni, A.; Lucà, M.G.; Senzolo, M.; Fagiuoli, S.; Ferrarese, A.; Pizzi, M.; Giacomelli, L.; Colloredo, G. Obliterative portal venopathy without portal hypertension: an underestimated condition. Liver Int. 2015, 36, 454–460. [Google Scholar] [CrossRef]
- Kawabata, S.; Sakamoto, S.; Honda, M.; Hayashida, S.; Yamamoto, H.; Mikami, Y.; Inomata, Y. Liver transplantation for a patient with Turner syndrome presenting severe portal hypertension: a case report and literature review. Surg. Case Rep. 2016, 2, 68. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, M.; Hodgson, H.J.F.; Wass, J.A.H.; Conway, G.S. Hormone replacement therapy may improve hepatic function in women with Turner's syndrome. Clin. Endocrinol. 2001, 55, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, M.; Ruszała, A.; Januś, D.; Starzyk, J.B. Liver Biochemical Abnormalities in Adolescent Patients with Turner Syndrome. J. Clin. Res. Pediatr. Endocrinol. 2019, 11, 395–399. [Google Scholar] [CrossRef]
- Santi, M.; Flück, C.E.; Hauschild, M.; Kuhlmann, B.; Kuehni, C.E.; Sommer, G. Health behaviour of women with Turner Syndrome. Acta Paediatr. 2021, 110, 2424–2429. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Liver histopathology of cases 1 and 3. (A) Case 3: Zone 3 mild sinusoidal dilatation, H&E, x200; (B) Case 3: Portal tract with slit-like venule and abnormal periportal vessels, H&E, x400; (C) Case 1: Portal venule herniation (arrow), H&E, x400; (D) Case 3: Mild sinusoidal fibrosis (arrowheads), Masson trichrome, x400. PT; portal tract, THV; terminal hepatic venule.
Figure 1.
Liver histopathology of cases 1 and 3. (A) Case 3: Zone 3 mild sinusoidal dilatation, H&E, x200; (B) Case 3: Portal tract with slit-like venule and abnormal periportal vessels, H&E, x400; (C) Case 1: Portal venule herniation (arrow), H&E, x400; (D) Case 3: Mild sinusoidal fibrosis (arrowheads), Masson trichrome, x400. PT; portal tract, THV; terminal hepatic venule.
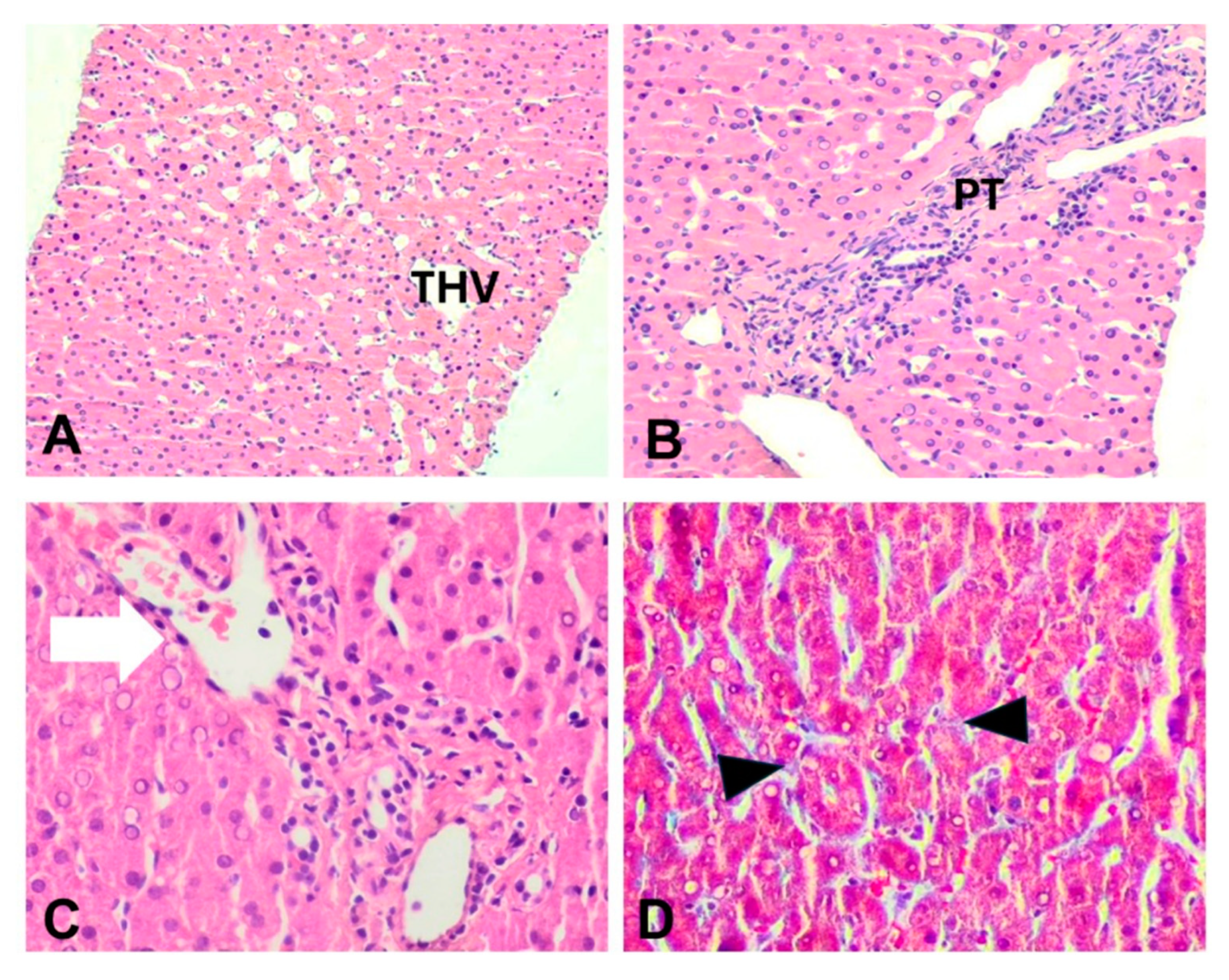
Figure 2.
Case 2: (A) Sinusoidal dilatation and irregular liver vascular architecture, H&E x40; (B) Hypovascularized portal tract, H&E, x200; (C) Sinusoidal capillarization, immunostaining CD34, x200; (D) Periportal intermediate keratin 7-positive hepatocytes (arrowheads) indicative of mild chronic cholestasis, keratin 7 immunostaining, x100. PT; portal tract, THV; terminal hepatic venule.
Figure 2.
Case 2: (A) Sinusoidal dilatation and irregular liver vascular architecture, H&E x40; (B) Hypovascularized portal tract, H&E, x200; (C) Sinusoidal capillarization, immunostaining CD34, x200; (D) Periportal intermediate keratin 7-positive hepatocytes (arrowheads) indicative of mild chronic cholestasis, keratin 7 immunostaining, x100. PT; portal tract, THV; terminal hepatic venule.
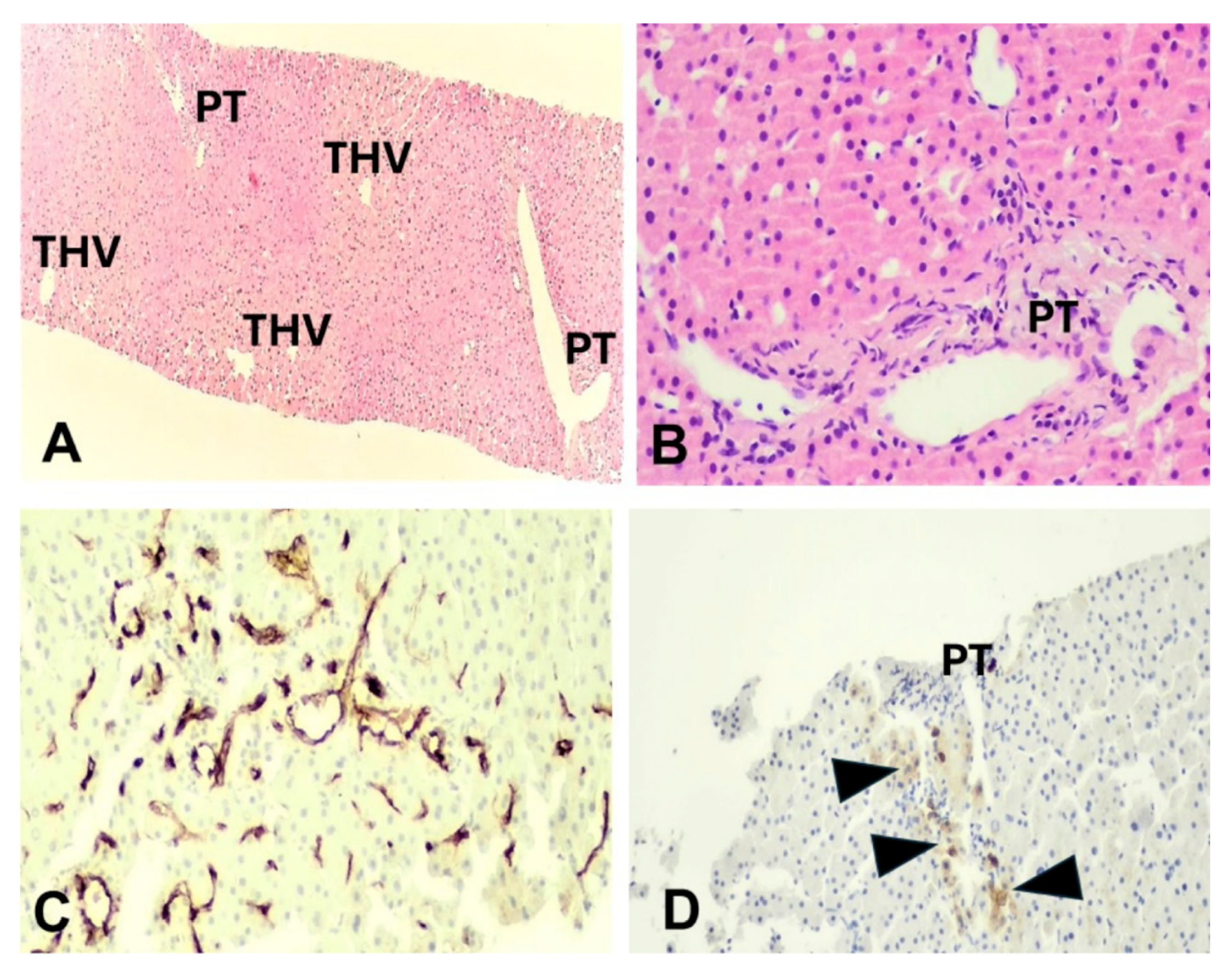

Table 1.
Clinical and laboratory findings of the three patients.
| Case 1 | Case 2 | Case 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Age(years) | 22 | 30 | 23 | ||||||
| Labor(weeks) | 38 | 41 | 40 | ||||||
| Menarche (years) | 12 | 17 | 12 (induction) | ||||||
|
Puberty (Tanner stages) |
AH=III, PH=V, B=V | AH=II, PH=IV, B=III | AH=III, PH=V, B=V | ||||||
| Body height (cm) | 145 | 153 | 150 | ||||||
| Medical history | astigmatism hypermetropia strabismus iron deficiency anemia |
astigmatism hypermetropia strabismus osteopenia dyslipidemia |
constriction of aorta chronic autoimmune thyroiditis gonadal dysgenesis osteopenia insulin resistance hearing loss |
||||||
| Karyotype test | 45,XO | 45,XO/46,XX (fragment 80%/20%) |
45,XO | ||||||
| Phenotype characteristics | short stature, broad short neck, low hairline, deformity of external ears, broad chest | short stature, short, webbed neck, low hairline, low-set ears | short stature, short, webbed neck, low hairline, ear deformity, shield-shaped thorax, widely spaced nipples | ||||||
| Ht (%) | 38 | 38.6 | 39.4 | ||||||
| Hb (g/dl) | 11.5 | 12.8 | 13.7 | ||||||
| WBC (cells/μl) | 7620 | 6930 | 4630 | ||||||
| PLTs (cells/μl) | 528000 | 190000 | 189000 | ||||||
| AST (5-32U/L) | 86 | 34 | 53 | ||||||
| ALT (5-31 U/L) | 106 | 37 | 130 | ||||||
| γ-GT (5-36 U/L) | 98 | 58 | 87 | ||||||
| ALP (35-120 U/L) | 125 | 149 | 71 | ||||||
| Total bilirubin (mg/dl) | 0,8 | 0.5 | 0.4 | ||||||
| Ferritin (ng/ml) | 13 | 120 | 32 | ||||||
| Glucose (mg/dl) | 81 | 92 | 93 | ||||||
| HBsAg | (-) | (-) | (-) | ||||||
| Anti-HBc | (-) | (-) | (-) | ||||||
| Anti-HBs | (+) | (+) | (+) | ||||||
| Anti-HCV | (-) | (-) | (-) | ||||||
| ANA | (-) | (-) | (-) | ||||||
| AMA | (-) | (-) | (-) | ||||||
| ASMA | (-) | (-) | (-) | ||||||
| IgG | N | N | N | ||||||
AH; Axillary hair, PH; Pubic hair, B; Breasts, Ht; Hematocrit, Hb; Hemoglobin, WBC; White Blood Cells, PLTs; Platelets, ALT; Alanine aminotransferase, AST; Aspartate aminotransferase, γ-GT; Gamma-glutamyl transpeptidase, ALP; Alkaline phosphatase, HBsAg; Hepatitis B surface antigen, Anti-HBc; Hepatitis B core antibodies, anti-HBs; Hepatitis B surface antibodies, anti-HCV; Hepatitis C virus antibodies, ANA; Anti-nuclear antibodies, AMA; Anti-mitochondrial antibodies, ASMA; Anti-smooth muscle antibodies, IgG; Immunoglobulin G, N; normal.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



