
Submitted:
08 August 2019
Posted:
11 August 2019
You are already at the latest version
Abstract
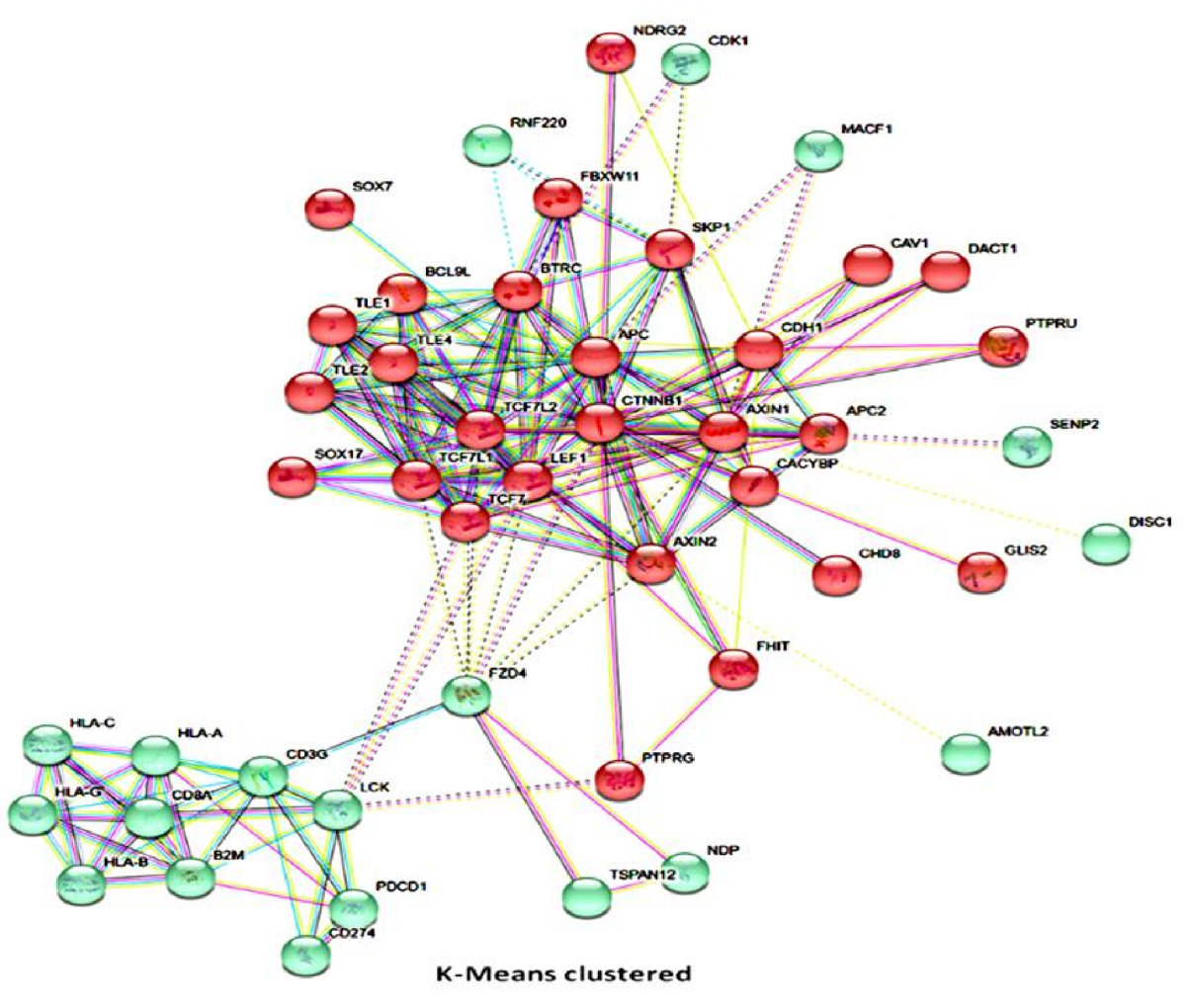
Background: Accurate prediction of patients’ response to therapy is clinically indispensable, howbeit challenging. With increased understanding of the human genome and malignancies, there is the renaissance of in silico pharmacogenomics with renewed interest in drug response predictability based on gene-drug interaction. Objective: Evidence-based transcript-proteome profiling is essential for synthesizing clinically applicable algorithms for predicting response to anticancer therapy, including immune checkpoint blockade (ICBT); thus, saving physicians’ time, reducing polypharmacy, and curtailing unnecessary treatment expense. In this study, we tested and validated the hypothesis that a selected proteomic signature in ICBT-naïve patients is sufficient for the prediction of response to ICBT. Methods: Using a multimodal approach consisting of computational pharmacogenomics, transcript-proteome analytics, mathematical modeling, and machine learning systems; we delineated therapy-sensitivity and stratified patients into graduated response groups based on their proteomic profile. Protein expression levels in our cohort tissue specimens were evaluated based on T cell- and non-T cell- inflamed phenotypes by immunohistochemistry. Results: We established β-catenin, PDL1, CD3 and CD8 expression-based ICBT response model. Statistical regression models validated the predictive association between our predefined algorithms and therapeutic outcome. Interestingly, our 4-gene prediction classifier was constitutively independent of tumor tissue origin, correctly stratified patients into high-, low-, and non- responders pre-treatment, with high prediction accuracy, and exhibited good association with patients’ performance status and prognosis (p < 0.01). Conclusion: Our findings demonstrate the possibility of accurate proteomics based ICBT response prediction and provide a putative basis for drug response prediction based on selective proteome profile in untreated cancer patients.
Keywords:
pharmacogenomics
; immune checkpoint blockade
; immunotherapy
; drug response prediction
; algorithm
; mathematical model
; precision medicine
; personalized medicine
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



