Submitted:
08 August 2020
Posted:
08 August 2020
You are already at the latest version
Abstract
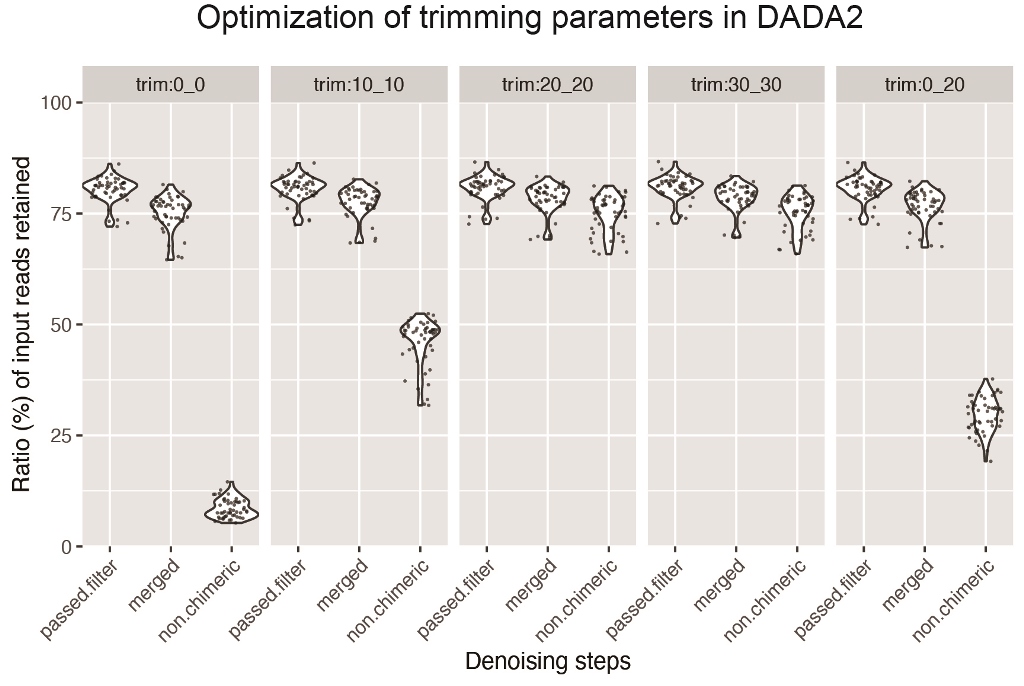
The bacterial composition of oral samples has traditionally been determined by PCR amplicon sequencing of 16S rRNA genes. Recent amplicon sequence variant (ASV)-based analyses of 16S rRNA genes differ from that based on operational taxonomic unit (OTU) clustering in the way it deals with sequences having potential errors. However, little information is available on its application in oral microbiome studies. Here, we conducted ASV-based analysis of oral microbiome samples using QIIME 2. We investigated the optimal parameters for sequence denoising, using DADA2, and found the trimming of the first 20 nucleotides from 5′-end of both paired reads avoided excessive sequence loss during chimera removal. Truncating reads at positions 240–245 allowed the removal of low-quality sequences while maintaining sufficient length to merge matching paired ends. Taxonomic assignment, using the naïve Bayes classifier trained with the V3-V4 region of reference 16S rRNA sequences in the extended human oral microbiome database (eHOMD), resulted in bacterial compositions similar to those of OTU-based analyses. Contrary to OTU-based clustering, ASV-based analysis showed taxonomic abundance at the genus or species level to not differ significantly in tongue microbiomes, regardless of brushing. QIIME 2 can, therefore, be a standard pipeline for ASV-based analysis of oral microbiomes.
Keywords:
tongue microbiome
; salivary microbiome
; amplicon sequence variant (ASV)
; operational taxonomical unit (OTU)
; denoising
; DADA2
; taxonomic classifier
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



