
Submitted:
27 June 2023
Posted:
28 June 2023
Read the latest preprint version here
Abstract
The technique of using drugs to target latent virus reservoirs has been introduced to reawaken dormant viruses such that the immune system can attack them, but further tests have shown this method fails. In this study, the author attempted to analyze whether drugs can be used to reawaken dormant virus reservoirs and proposed the use of viral proteins to activate the sleeping virus. The results show that the amino acid sequence ARG of Gag proteins of HTLV-1, HTLV-2, STLV-1 and STLV-2 match their primer binding site GGGGGCTCG in the 3'-to-5' direction and that the amino acid sequence SPR of Gag proteins of HIV-1, HIV-2, SIV and FIV match their primer binding site GGCGCCCGA in the 3'-to-5' direction. The author hence believes that the latency-reversing drugs are involved in the process of the transcription of cancer genes, and because the virus genome they reawaken contains the same NF-κB binding sites, the drugs indirectly reawaken dormant retrovirus infection. Related studies showed that the genomic Gag/Gag-Pol complex recruits the LysRS/tRNA complex, the selective packaging of the tRNA primer requires HIV-1 Gag and Gag-Pol, and an interaction between LysRS and Gag is observed in vitro. In contrast, Gag proteins can more reliably be used to directly reawaken dormant HIV infection, which recruits human uncharged tRNA to serve as the reverse transcription primer.
Keywords:
HIV
; HTLV
; NF-κB
; tRNA
; Gag
1. Introduction
Acquired immunodeficiency syndrome (AIDS) is a disease caused by human immunodeficiency virus (HIV). The virus attacks immune system cells in the body and then uses their machinery to make copies of itself. However, some HIV-infected immune cells enter a state in which they do not produce new virus, which is called the resting or latent state. These cells form a latent HIV reservoir in which HIV can hide for years, resulting in the avoidance of HIV therapy. At any time, these cells can become active again and start to make more copies of the virus [1]. Scientists have used this opportunity to develop methods to target these latent reservoirs and make them active such that they can be identified and targeted by HIV therapy. However, scientists at Johns Hopkins reported compounds they hoped would 'wake up' dormant reservoirs of HIV inside the immune system, but T cells have failed to achieve this effect in laboratory tests using white blood cells collected directly from patients infected with HIV [2]. Hence, further investigation is needed to determine the applicability of this method. In this study, the author attempted to mathematically analyze whether latency-reversing drugs can reawaken the sleeping retrovirus.
2. Methods
Because HIV uses the host’s NF-κB signaling pathway to activate viral transcription [3], the author designed the following experiment. First, the author prepared several T cells and HIV-1 RNAs. The HIV genome contains at least nine genes, including Gag, Pol and Env [4]. The IGF1R gene is located on human chromosome 15, which contains at least 21 exons, such as ENSE00003838363 and ENSE00001316091 [5]. Using mathematical models can help understand phenomena in biology. In mathematics, the genome can be defined as a set of elements by listing the elements between curly brackets and separated by commas:
where denotes the set of HIV genomes and represents the set of IGF1R genes. The IGF1R gene is one of the known target genes of androgen receptor activation [6]. Hence, the process of transcription can be written in the following form:
where the domain of is the set of genes that RNA polymerase II will transcribe, and represents the androgen. The CRISPR‒Cas9 enzyme [7,8] then copies the enhancer of the IGF1R gene into the promoter-proximal region of HIV-1 RNAs. Subsequently, T cells are infected with the modified virus, and the form can then be rewritten as follows:
Androgens are then injected into the T cells. After the IGF1R gene is transcribed by RNA polymerase II [9], HIV will also 'wake up' [10]. Python is one of the most popular programming languages [11] and can be used to write scripts that can check the accuracy of mathematical formulas:

The set represents the HIV genome, and the set represents the IGF1R gene. The string is then defined to represent androgen, whereas represents the function of RNA polymerase II, which returns the IGF1R gene and HIV genome applied to the string . The result of is then printed to verify whether the virus was activated. As a result, the Python program returns True, which indicates that dormant HIV infection is reawakened.

Can this possibly mean that androgen reawakens sleeping HIV? The answer is that androgen reawakens both the IGF1R gene and HIV genome and not only the retrovirus. In fact, even the Python program returns a result of False.
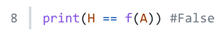
The collection of elements returned by the method includes the HIV set, which does not mean that the two sets are equal. Mathematically, their relationship can be expressed as follows:
The author will not actually copy the enhancer of the IGF1R gene into the virus due to related studies: the promoter-proximal (enhancer) region of the HIV-1 long terminal repeat contains two adjacent NF-κB binding sites that play a central role in mediating inducible HIV-1 gene expression [3,12,13].
Several studies claim that AZD5582 can reawaken sleeping HIV and SIV, but the effectiveness rate was found to be only 42% [14,15,16]. Most importantly, the novel small-molecule IAP inhibitor AZD5582 has been used for the treatment of cancer and reportedly causes cIAP1 degradation and thus induces apoptosis in the MDA-MB-231 breast cancer cell line at subnanomolar concentrations in vitro [17].
Latency-reversing drugs are involved in the process of transcription of cancer genes, and the virus genome reawakened by these drugs contains NF-κB binding sites; thus, the drugs do not directly reawaken dormant retrovirus infection. This method may not be easily understood by some readers; thus, the author provides another example:
To ensure that babies are not carried away by the wrong parents in a hospital (nucleus) with three newborn babies, Adam (HIV genome), Bob (IGF1R gene), and Claire (cancer gene), babies and their parents are given wristbands with their corresponding names (enhancer region). Adam is a naughty boy who secretly made a copy of Claire’s wristband (NF-κB binding sites) and placed it on his hand. Because nurses (RNA polymerase II) cannot recognize the appearance of babies, babies can only be identified by their wristbands. When Claire’s parents (NF-κB) wanted to take their child away from the hospital, the parents handed over their wristband to the nurses and asked them to find their child. Because both Adam and Claire had Claire’s name written on their wristbands, both babies were taken away by the parents.
Based on this example, it feels like the previous studies were attempting to use Claire’s wristband (NF-κB binding sites) to find Adam (HIV genome), which is clearly inappropriate. More importantly, the mutation rate of HIV-1 is extremely high [18]; if Claire’s name on the wristband mutates to Clara or Clark (another cancer gene), the drugs that target Claire will have no effect on the mutated virus.
In addition to AZD5582, many studies claim that latency-reversing drugs, including ciapavir [19], bryostatin-1 [20], disulfiram [21], ingenol-B [22], and prostratin [23], can be used to reawaken sleeping HIV. These latency-reversing drugs have also been used for the treatment of cancer: disulfiram inhibits prostate cancer cell growth [24], bryostatin-1 exhibits potent antitumor activity in vitro and in vivo in human tumor xenografts [25], semisynthetic ingenol compounds show potent antitumor activity on all cancer cell lines evaluated [26], and prostratin exerts a potential anticancer effect through SIK3 inhibition [27].
One type of latency-reversing drug approach will not work in different patients infected with different types of mutated viruses unless multiple drugs are used at the same time. However, the mutation rate of HIV-1 is extremely high, which means that scientists have to constantly develop new drugs for new viruses. More importantly, the virus uses the NF-κB pathway to enhance its expression, which does not mean that the virus must have an NF-κB primer binding site. If the primer binding site is mutated into the enhancer of other genes unrelated to NF-κB, latency-reversing drugs will have no effect on patients. The author thus believes that instead of using Claire, Clara and Clark’s wristbands to find Adam indirectly, it would be better to use Adam’s wristbands to find Adam directly. In other words, the use of viral proteins can more reliably reawaken dormant retroviruses.
Viral RNA is specifically packaged into virions, not IGF1R or cancer RNA; thus, the virus can accurately identify viral RNA. Therefore, viral proteins carry information that can identify viral RNA, just as the androgen receptor activates the IGF1R gene. It is possible that a certain viral protein has a similar function to NF-κB or androgen receptor, which can be used to identify viral RNA directly.
It is well known that HIV recruits human uncharged tRNA to serve as the reverse transcription primer [28], and tRNA serves as the physical link between the mRNA and the amino acid sequences of proteins [29]. The author hence believes that uncharged tRNA serves as the physical link between the promoter and the protein receptors, which are recruited by RNA polymerase II. To determine which viral proteins match the primer binding site, a Python program was written to match all proteins with their own gene sequences and display them graphically.
3. Results
Latent HIV can synthesize a 5'-3' RNA chain by transcribing the existing 3'-5' complementary DNA strand after cellular infection [30,31]. The author uses the x-axis to represent the protein and the y-axis to represent the primer binding site. Negative numbers indicate that the protein or tRNA may have rotated 180 degrees (which did not happen) or been bound in the 3'-to-5' direction (if both values are negative).
The author also expanded the analysis to include other retroviruses, including Deltaretroviruses (HTLV and STLV) and Lentiviruses (HIV, SIV, and FIV). The gene data was sourced from the GenBank database at the NCBI. The author used the following sequences for the analysis: NC_001436 (HTLV-1) [32], NC_001488 (HTLV-2) [33], NC_000858 (STLV-1) [34], NC_001815 (STLV-2) [35], NC_001802 (HIV-1) [36], NC_001722 (HIV-2) [37], NC_001549 (SIV) [38], and NC_001482 (FIV) [39].
Having 2 amino acid sequences of the matching points leads to many possibilities, and it is thus impossible to confirm which protein matches the primer binding site. When there are 4 amino acid sequences, no matching target can be found. However, when there are 3 amino acid sequences, there is exactly one perfect matching region. Different types of retroviruses are represented by different patterns and colors, and their sequences around the primer binding site are matched with their own proteins, as shown in Figure 1.
As shown in Figure 1, inside the red box, the coordinates of 8 different viruses appear at the same time and are extremely close, which means that they represent the same protein. Other locations contained either only Deltaretroviruses or only Lentiviruses, and the spacing between the different color coordinates was too large, indicating that they were not even the same protein and were therefore excluded. If the virus amino acid sequences of the protein mutated, its primer binding site remained the same, which means that it was not the matching target.
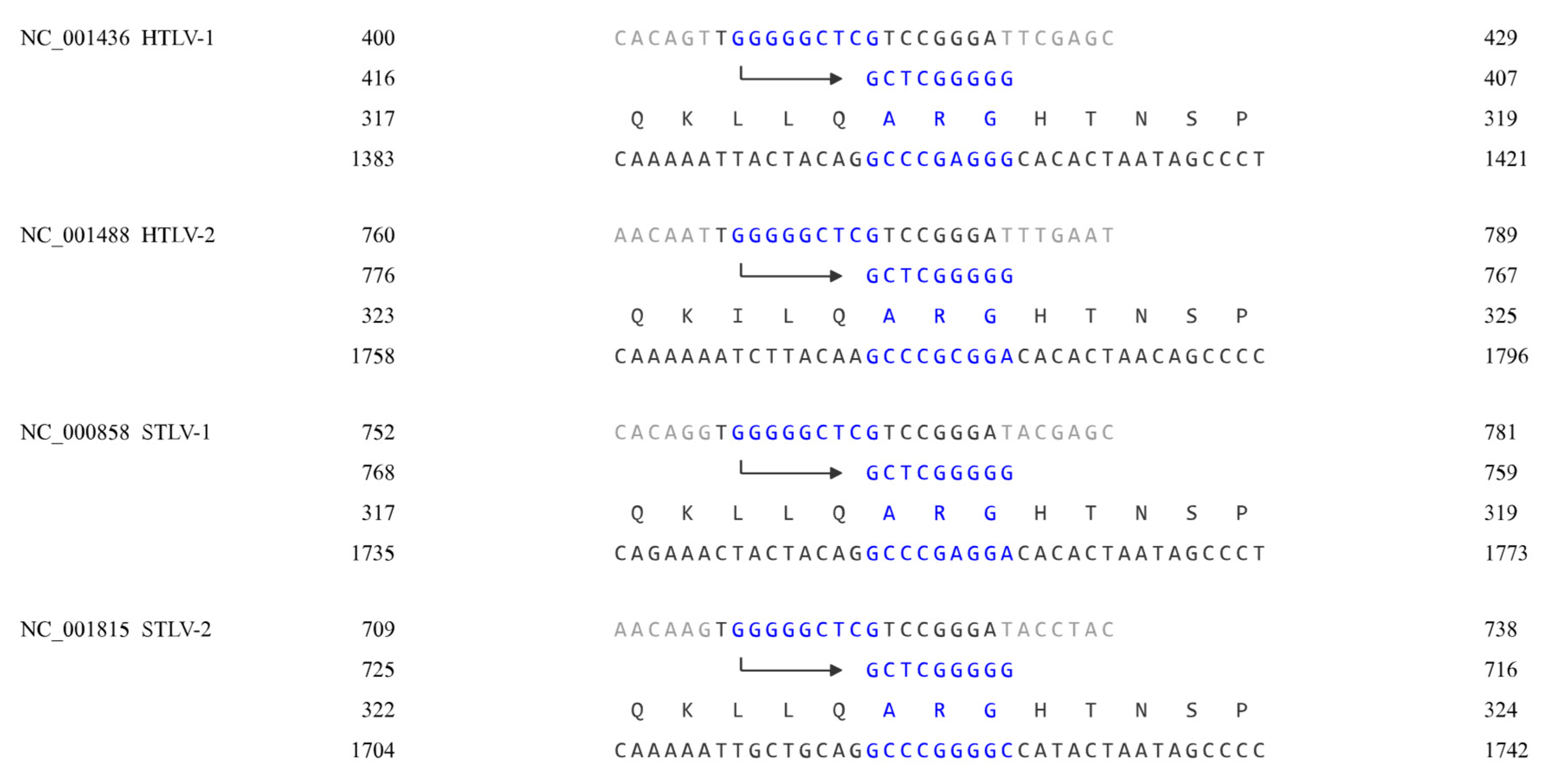
In the GenBank database, the primer binding site of the HTLV-2 (NC_001488) genome is approximately nt 766 to 783, and that of the HIV-1 (NC_001802) genome is approximately nt 182 to 199. Their primer binding sites start with TGG and end with GGGA, and after aligning the sequences, their matching points can be found in the same position, as shown in Figure 2.
As shown in Figure 2, the sequences on the second line represent the 3'-5' complementary DNA strand that gag proteins match with, and the arrow indicates the 3'-5' direction. The Gag proteins of the viruses match the same primer binding site, even though the viruses are highly different.
To determine whether this finding is a coincidence, the author analyzed the probability. Because viruses of the same type, Deltaretrovirus or Lentivirus, have the same primer binding site, one virus can be considered a mutation from another. The author used the HTLV-1 and HIV-1 genomes as templates and used Pairwise Sequence Alignment (EMBOSS Needle) to compare the genetic similarity of different viruses. The similarity of the NC_001488 (HTLV-2), NC_000858 (STLV-1) and NC_001815 (STLV-2) genomes to the NC_001436 (HTLV-1) genome was 59.2%, 89.3% and 61.0%, respectively. The similarity of the NC_001722 (HIV-2), NC_001549 (SIV) and NC_001482 (FIV) genomes to the NC_001802 (HIV-1) genome was 51.1%, 54.1% and 49.1%, respectively. The similarity of six viruses can be written as
The average probabilities of the amino acid sequences A (GCT, GCC, GCA, GCG), R (CGT, CGC, CGA, CGG, AGA, AGG), G (GGT, GGC, GGA, GGG), S (TCT, TCC, TCA, TCG, AGT, AGC) and P (CCT, CCC, CCA, CCG) remaining unchanged after a mutation are 3/63, 5/63, 3/63, 5/63, and 3/63, respectively. Thus, the average probabilities of the amino acid sequences ARG and SPR remaining unchanged after a mutation are 11/189 and 13/189, respectively. Therefore, the average probability that 3 amino acid sequences of six viruses remain unchanged after a mutation can be represented by as follows:
Assuming that each gene sequence has the same probability of mutation, the number of amino acid sequence mutations increases with increases in the diversity of the viruses. The probability that 3 amino acid sequences of different viruses match the same primer binding site is
The result shows that the probability is approximately 3.67636×10-9, which is extremely small; thus, it can be determined that Gag proteins can match the primer binding site.
Related studies showed that the genomic Gag/Gag-Pol complex recruits the LysRS/tRNA complex [40], the selective packaging of the tRNA primer requires HIV-1 Gag and Gag-Pol [41], and an interaction between LysRS and Gag is observed in vitro [42]. Since HIV-1 initiates the process of reverse transcription by using tRNA(Lys) to bind to the genomic RNA at the primer binding site [43], it has been proven that the gag protein matches the primer binding site.
In HIV-1, Gag/LysRS interaction depends on Gag sequences within the C-terminal domain (CTD) of CA around amino acids 283-363 [44] and motif 1 of LysRS around amino acids 208-259 [42]. It should be noted that the amino acid sequence SPR of the Gag protein is located at amino acids 148-150 within the N-terminal domain (NTD) of CA, specifically at the NTD-NTD interface 1.
4. Discussion
Because the primer binding sites of different viruses are extremely stable, other amino acid sequences of Gag proteins may also match these sites, but there is no sufficient information to confirm this hypothesis at present. The current information demonstrates that drugs fail to reawaken dormant HIV infection, and it is more reliable to use viral proteins to directly reawaken dormant retroviruses. Compared with the use of latency-reversing drugs to activate NF-κB primer binding sites, the use of Gag proteins can directly reawaken retrovirus without considering the mutation of the NF-κB primer binding site at adjacent locations, and the pattern exists in both Deltaretrovirus and Lentivirus, which means that the treatment can be used for most patients without the need to take multiple drugs at the same time. More importantly, the CRISPR‒Cas9 enzyme can be used to modify the amino acid sequences of viral proteins to avoid uncharged tRNAs it recruits to match the NF-κB primer binding site, which results in avoiding the activation of cancer genes and reducing unknown risks to patients. It is also possible to design a new NF-κB, which has amino acid sequences of Gag proteins, to reawaken dormant HIV infection.
5. Conclusions
Latency-reversing drugs are involved in the transcription of cancer genes, and the virus genomes that they reawaken contain the same NF-κB primer binding sites; thus, the drugs are not directly reawaken dormant HIV infection. The amino acid sequence ARG of Gag proteins of HTLV-1, HTLV-2, STLV-1 and STLV-2 match their primer binding site GGGGGCTCG in the 3'-to-5' direction, and the amino acid sequence SPR of Gag proteins of HIV-1, HIV-2, SIV and FIV match their primer binding site GGCGCCCGA in the 3'-to-5' direction. Related studies showed that the genomic Gag/Gag-Pol complex recruits the LysRS/tRNA complex, the selective packaging of the tRNA primer requires HIV-1 Gag and Gag-Pol, and an interaction between LysRS and Gag is observed in vitro. In contrast, Gag proteins can more reliably be used to directly reawaken dormant HIV infection, which recruits human uncharged tRNA to serve as the reverse transcription primer.
Authors Contributions: S.C. wrote the manuscript.
Funding: None.
Ethics approval and consent to participate: Not applicable.
Consent to publish: The author gives consent for the publication of identifiable details, which can include photographs and details within the text to be published in the above Journal and Article.
Availability of data and materials: The datasets were produced by Python3, and the tool is available at https://github.com/rheast/genome. Pairwise Sequence Alignment (EMBOSS Needle) was used to identify regions of similarity between two biological sequences with the tool available at https://www.ebi.ac.uk/Tools/psa/. Nucleotides were downloaded from the NCBI database at https://www.ncbi.nlm.nih.gov/nuccore/. The sample nucleotides correspond to the accession numbers NC_001436, NC_001488, NC_000858, NC_001815, NC_001802, NC_001722, NC_001549 and NC_001482.
Acknowledgments: None.
Competing interests: There are no conflicts of interest.
Abbreviations
| NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| IGF1R | Insulin-like growth factor 1 receptor |
| CRISPRs | Clustered regularly interspaced short palindromic repeats |
| IAP | Inhibitor of apoptosis protein |
| cIAP1 | Cellular inhibitor of apoptosis protein 1 |
| SIK3 | Salt-inducible kinase 3 |
| HTLV | Human T-lymphotropic virus |
| STLV | Simian T-lymphotropic virus |
| HIV | Human immunodeficiency virus |
| SIV | Simian immunodeficiency virus |
| FIV | Feline immunodeficiency virus |
| LysRS | Lysyl-tRNA synthetase |
| PBS | primer binding site |
| CTD | C-terminal domain |
| NTD | N-terminal domain |
| Gag | Group-specific antigen |
| CA | Capsid |
References
- National Institute of Health. What is a Latent HIV Reservoir? NIH. 2021 August 4. https://hivinfo.nih.gov/understanding-hiv/fact-sheets/what-latent-hiv-reservoir.
- Siliciano RF. Drugs fail to reawaken dormant HIV infection. Johns Hopkins Medicine. 2014 March 24. https://hopkinsmedicine.org/news/media/releases/drugs_fail_to_reawaken_dormant_hiv_infection.
- Hiscott J, Kwon H, Génin P. Hostile takeovers: viral appropriation of the NF-kB pathway. The Journal of clinical investigation. 2001 Jan 15;107(2):143-51. [CrossRef]
- Seibert SA, Howell CY, Hughes MK, Hughes AL. Natural selection on the gag, pol, and env genes of human immunodeficiency virus 1 (HIV-1). Molecular biology and evolution. 1995 Sep 1;12(5):803-13. [CrossRef]
- Abbott AM, Bueno R, Pedrini MT, Murray JM, Smith RJ. Insulin-like growth factor I receptor gene structure. Journal of Biological Chemistry. 1992 ;267(15):10759-63. 25 May. [CrossRef]
- Pandini G, Mineo R, Frasca F, Roberts Jr CT, Marcelli M, Vigneri R, Belfiore A. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer research. 2005 Mar 1;65(5):1849-57. [CrossRef]
- Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Human molecular genetics. 2014 Sep 15;23(R1):R40-6. [CrossRef]
- Bak RO, Gomez-Ospina N, Porteus MH. Gene editing on center stage. Trends in Genetics. 2018 Aug 1;34(8):600-11. [CrossRef]
- Aleksic T, Gray N, Wu X, Rieunier G, Osher E, Mills J, Verrill C, Bryant RJ, Han C, Hutchinson K, Lambert AG. Nuclear IGF1R interacts with regulatory regions of chromatin to promote RNA polymerase II recruitment and gene expression associated with advanced tumor stage. Cancer research. 2018 Jul 1;78(13):3497-509. [CrossRef]
- Ott M, Geyer M, Zhou Q. The control of HIV transcription: keeping RNA polymerase II on track. Cell host & microbe. 2011 Nov 17;10(5):426-35. [CrossRef]
- Van Rossum G. An introduction to Python for UNIX/C programmers. Proceedings of the NLUUG najaarsconferentie (Dutch UNIX users group). 1993 Nov.
- Kwon H, Pelletier N, DeLuca C, Genin P, Cisternas S, Lin R, Wainberg MA, Hiscott J. Inducible expression of IκBα repressor mutants interferes with NF-κB activity and HIV-1 replication in Jurkat T cells. Journal of biological chemistry. 1998 Mar 27;273(13):7431-40. [CrossRef]
- Quinto I, Mallardo M, Baldassarre F, Scala G, Englund G, Jeang KT. Potent and stable attenuation of live-HIV-1 by gain of a proteolysis-resistant inhibitor of NF-κB (IκB-αS32/36A) and the implications for vaccine development. Journal of biological chemistry. 1999 Jun 18;274(25):17567-72. [CrossRef]
- National Institute of Health. NIH-supported scientists reverse HIV and SIV latency in two animal models. NIH. 2020 January 22. https://www.nih.gov/news-events/news-releases/nih-supported-scientists-reverse-hiv-siv-latency-two-animal-models.
- Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, Mattingly C, Ho PT, Schoof N, Cammon CG, Tharp GK. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature. 2020 Feb;578(7793):160-5. [CrossRef]
- McBrien JB, Mavigner M, Franchitti L, Smith SA, White E, Tharp GK, Walum H, Busman-Sahay K, Aguilera-Sandoval CR, Thayer WO, Spagnuolo RA. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature. 2020 Feb;578(7793):154-9. [CrossRef]
- Hennessy EJ, Adam A, Aquila BM, Castriotta LM, Cook D, Hattersley M, Hird AW, Huntington C, Kamhi VM, Laing NM, Li D. Discovery of a novel class of dimeric Smac mimetics as potent IAP antagonists resulting in a clinical candidate for the treatment of cancer (AZD5582). Journal of medicinal chemistry. 2013 Dec 27;56(24):9897-919. [CrossRef]
- Cuevas JM, Geller R, Garijo R, López-Aldeguer J, Sanjuán R. Extremely high mutation rate of HIV-1 in vivo. PLoS biology. 2015 Sep 16;13(9):e1002251. [CrossRef]
- Pache L, Marsden MD, Teriete P, Portillo AJ, Heimann D, Kim JT, Soliman MS, Dimapasoc M, Carmona C, Celeridad M, Spivak AM. Pharmacological activation of non-canonical NF-κB signaling activates latent HIV-1 reservoirs in vivo. Cell Reports Medicine. 2020 Jun 23;1(3):100037. [CrossRef]
- Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nature medicine. 2014 Apr;20(4):425-9. [CrossRef]
- Spivak AM, Andrade A, Eisele E, Hoh R, Bacchetti P, Bumpus NN, Emad F, Buckheit III R, McCance-Katz EF, Lai J, Kennedy M. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1–infected adults on antiretroviral therapy. Clinical infectious diseases. 2014 Mar 15;58(6):883-90. [CrossRef]
- Darcis G, Kula A, Bouchat S, Fujinaga K, Corazza F, Ait-Ammar A, Delacourt N, Melard A, Kabeya K, Vanhulle C, Van Driessche B. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+ JQ1 and ingenol-B+ JQ1 to potently reactivate viral gene expression. PLoS pathogens. 2015 Jul 30;11(7):e1005063. [CrossRef]
- Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, Siliciano JD, Siliciano RF. Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. The Journal of clinical investigation. 2015 ;125(5):1901-12. 1 May. [CrossRef]
- Lin J, Haffner MC, Zhang Y, Lee BH, Brennen WN, Britton J, Kachhap SK, Shim JS, Liu JO, Nelson WG, Yegnasubramanian S. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. The Prostate. 2011 Mar 1;71(4):333-43. [CrossRef]
- Philip PA, Rea D, Thavasu P, Carmichael J, Stuart NS, Rockett H, Talbot DC, Ganesan T, Pettit GR, Balkwill F, Harris AL. Phase I study of bryostatin 1: assessment of interleukin 6 and tumor necrosis factor α induction in vivo. JNCI: Journal of the National Cancer Institute. 1993 Nov 17;85(22):1812-8. [CrossRef]
- Silva VA, Rosa MN, Tansini A, Lima JP, Jones C, Pianowski LF, Reis RM. Cytotoxic activity of semi-synthetic ingenol derived from Euphorbia tirucalli on a large panel of human cancer cell lines. 2013; e13559-e13559. [CrossRef]
- Alotaibi D, Amara S, Johnson TL, Tiriveedhi V. Potential anticancer effect of prostratin through SIK3 inhibition. Oncology letters. 2018 Mar 1;15(3):3252-8. [CrossRef]
- Duchon AA, St. Gelais C, Titkemeier N, Hatterschide J, Wu L, Musier-Forsyth K. HIV-1 exploits a dynamic multi-aminoacyl-tRNA synthetase complex to enhance viral replication. Journal of virology. 2017 Oct 13;91(21):e01240-17. [CrossRef]
- Weiner AM, Maizels N. tRNA-like structures tag the 3'ends of genomic RNA molecules for replication: implications for the origin of protein synthesis. Proceedings of the National Academy of Sciences. 1987 Nov;84(21):7383-7. [CrossRef]
- Hu WS, Temin HM. Retroviral recombination and reverse transcription. Science. 1990 Nov 30;250(4985):1227-33. [CrossRef]
- Negroni M, Buc H. Copy-choice recombination by reverse transcriptases: reshuffling of genetic markers mediated by RNA chaperones. Proceedings of the National Academy of Sciences. 2000 Jun 6;97(12):6385-90. [CrossRef]
- Petropoulos, C. Retroviral taxonomy, protein structures, sequences, and genetic maps. Retroviruses; Cold Spring Harbor Laboratory Press, Cold Spring Harbor: NY, 1997; pp. 757–805. [Google Scholar]
- Shimotohno K, Takahashi Y, Shimizu N, Gojobori T, Golde DW, Chen IS, Miwa M, Sugimura T. Complete nucleotide sequence of an infectious clone of human T-cell leukemia virus type II: an open reading frame for the protease gene. Proceedings of the National Academy of Sciences. 1985 May;82(10):3101-5. [CrossRef]
- Saksena NK, Hervé V, Sherman MP, Durand JP, Mathiot C, Müller M, Love JL, Leguenno B, Sinoussi FB, Dube DK, Poiesz BJ. Sequence and phylogenetic analyses of a new STLV-I from a naturally infected tantalus monkey from Central Africa. Virology. 1993 Jan 1;192(1):312-20. [CrossRef]
- Van Brussel M, Salemi M, Liu HF, Gabriëls J, Goubau P, Desmyter J, Vandamme AM. The Simian T-Lymphotropic Virus STLV-PP1664 fromPan paniscusIs Distinctly Related to HTLV-2 but Differs in Genomic Organization. Virology. 1998 Apr 10;243(2):366-79. [CrossRef]
- Martoglio B, Graf R, Dobberstein B. Signal peptide fragments of preprolactin and HIV-1 p-gp160 interact with calmodulin. The EMBO journal. 1997 Nov 15;16(22):6636-45. [CrossRef]
- Kirchhoff F, Jentsch KD, Bachmann B, Stuke A, Laloux C, Luke W, Stahl-Hennig C, Schneider J, Nieselt K, Eigen M, Hunsmann G. A novel proviral clone of HIV-2: biological and phylogenetic relationship to other primate immunodeficiency viruses. Virology. 1990 Jul 1;177(1):305-11. [CrossRef]
- Fomsgaard A, Hirsch VM, Allan JS, Johnson PR. A highly divergent proviral DNA clone of SIV from a distinct species of African green monkey. Virology. 1991 ;182(1):397-402. 1 May. [CrossRef]
- Olmsted RA, Hirsch VM, Purcell RH, Johnson PR. Nucleotide sequence analysis of feline immunodeficiency virus: genome organization and relationship to other lentiviruses. Proceedings of the National Academy of Sciences. 1989 Oct;86(20):8088-92. [CrossRef]
- Jin D, Musier-Forsyth K. Role of host tRNAs and aminoacyl-tRNA synthetases in retroviral replication. Journal of Biological Chemistry. 2019 Apr 5;294(14):5352-64. [CrossRef]
- Mak J, Jiang M, Wainberg MA, Hammarskjöld ML, Rekosh D, Kleiman L. Role of Pr160gag-pol in mediating the selective incorporation of tRNA (Lys) into human immunodeficiency virus type 1 particles. Journal of virology, 1994, 68(4): 2065-2072. [CrossRef]
- Kovaleski BJ, Kennedy R, Hong MK, Datta SA, Kleiman L, Rein A, Musier-Forsyth K. In vitro characterization of the interaction between HIV-1 Gag and human lysyl-tRNA synthetase. Journal of Biological Chemistry, 2006, 281(28): 19449-19456. [CrossRef]
- Rhim H, Park J, Morrow CD. Deletions in the tRNA(Lys) primer-binding site of human immunodeficiency virus type 1 identify essential regions for reverse transcription. Journal of virology 65.9 (1991): 4555-4564. [CrossRef]
- Bell NM, Lever AM. HIV Gag polyprotein: processing and early viral particle assembly. Trends in microbiology 21.3 (2013): 136-144. [CrossRef]
Figure 1.
Coordinates of matched points.
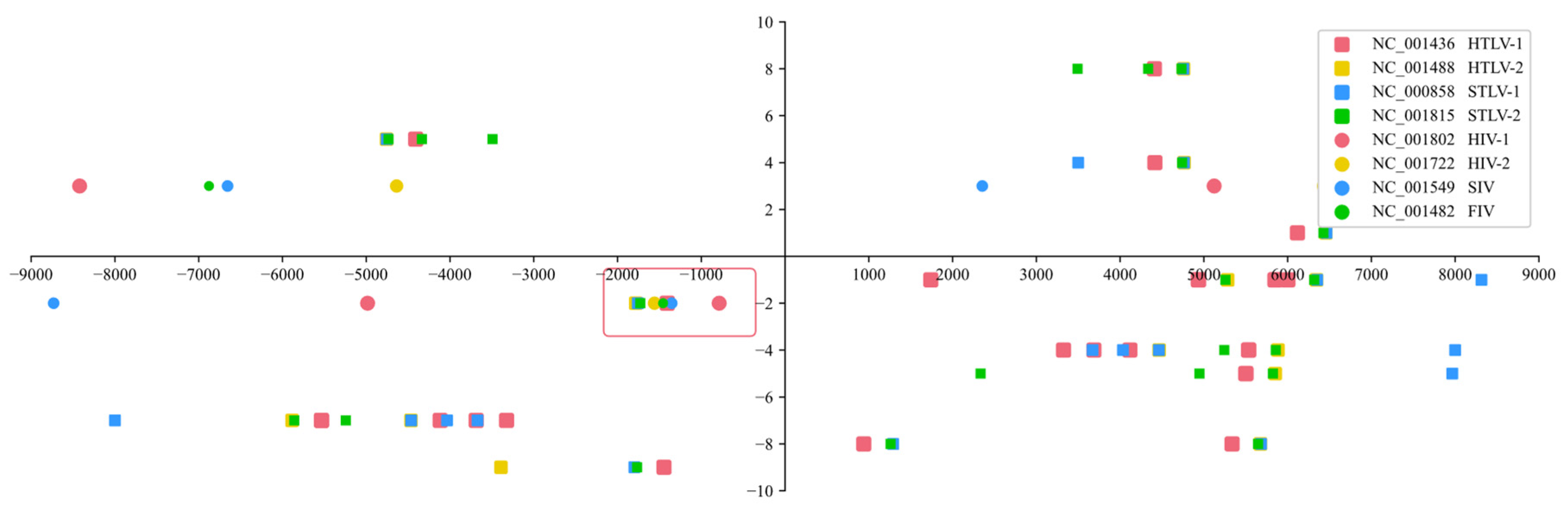
Figure 2.
Deltaretrovirus and Lentivirus.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



