Submitted:
06 September 2023
Posted:
07 September 2023
Read the latest preprint version here
Abstract
Lipofuscin is indigestible garbage that accumulates in the autophagic vesicles and cytosol of post-mitotic cells with age. Drs. Brunk and Terman postulated that lipofuscin accumulation is the main or at least a major driving factor in aging. They even posited that the evolution of memory is the reason why we get lipofuscin at all, as stable synaptic connections must be maintained over time, meaning that the somas of neurons must also remain in the same locale. In other words, they cannot dilute out their garbage over time through cell division. Mechanistically, their position certainly makes sense given that rendering a large percentage of a post-mitotic cell’s lysosomes useless must almost certainly negatively affect that cell and the surrounding microenvironment. Here, I explore the possibility that the accumulation of lipofuscin may to some extent exacerbate every other kind of age-related damage. I do not think that lipofuscin removal will reverse/prevent all forms of aging, just the major component facing us currently. In this piece, I will review what is known about lipofuscin accumulation from evolutionary and mechanistic standpoints and discuss a method of removing it from single cells in vitro.
Keywords:
Anti-aging
; Hydra vulgaris
; TFEB
; telomerase
; FluidFM
Introduction:
Biological aging is a complex molecular process that takes place over time in all organisms. However, organisms have evolved mechanisms to repair various forms of age-related damage. For example, DNA repair enzymes exist that can fix damage in nuclear DNA. Mitophagy enables the degradation of damaged mitochondria. The immune system, when one is young at least, eliminates at least some proportion of one’s senescent cells.
There are many theories about why we age, but one stands out to me as being the most plausible based on the evolutionary and mechanistic evidence. That is the “garbage catastrophe theory of aging.” Drs. Brunk and Terman postulated years ago that the problem of aging can essentially be summed up as a “garbage disposal issue [1].” The main idea is that basically old molecules are sometimes damaged in ways that prevent the lysosomes from breaking them down properly, and over time these damaged, old molecules accumulate inside the lysosomes. Eventually, the lysosomes become full of this indigestible garbage, i.e., “lipofuscin”, and cannot perform their normal function - then there is a garbage back-up and the cell starts to decline health-wise.
There are two arguments for lipofuscin removal being the most important goal of anti-aging science currently. One is evolutionary and one is mechanistic.
Evolutionary argument:
In nature, there are only a handful of organisms that can be said to be essentially biological immortal. Hydra vulgaris (i.e., magnipapillata) is one of such organisms, and the reason for this might be that its indigestible garbage is essentially released from its body over time. It has three cell lineages - ectodermal epithelial, interstitial, and endodermal epithelial. All the epithelial cells in its body column are stem cells that continuously divide - displacing cells toward its extremities. The cells at the extremities slough off eventually [2,3]. In terms of the interstitial lineage, the differentiated cells that its stem cells produce are closely associated with epithelial cells and so are continuously displaced as well. This is a convenient way to dispose of lipofuscin - i.e., through dilution and cell shedding. However, continuous replacement of neurons may not allow for the stable inter-neuronal interactions required for long-term memory [4]. Lobsters continually grow throughout life; their fully differentiated cells express telomerase, allowing them to keep dividing as needed [5]. This includes the cells of their central nervous system, which allows for adult neurogenesis [6]. They also shed their shells periodically. Thus, the same logic appears to apply to them. However, their growth process does not appear to be fast enough to prevent lipofuscin from accumulating over time [7]. Notably, lipofuscin accumulation in eyestalk ganglia [8] is used as a gauge of biological age in lobsters (and myocardial lipofuscin accumulation can be used a marker of chronological age in humans [9]). Lobsters can retain memories, but only for a short time span [10]. Naked mole rats are also very long lived, and it has been shown that they have unusually active autophagic systems [11,12] (in addition to better anti-cancer defenses [13]). Even with better autophagy, naked mole rats still do accumulate lipofuscin in their post-mitotic tissues [14].
It appears as though all animals that age, e.g., flies [15], worms [16,17], lobsters, naked mole rats, mice [18], non-human primates [19], and humans [20] accumulate lipofuscin in their post-mitotic tissues. None of the aforementioned organisms seem to possess any evolutionarily “built-in” ways of exporting the lipofuscin that accumulates in their post-mitotic cells from their bodies, presumably because that would be an unnecessary expenditure of energy in light of procreation.
Export from the post-mitotic cells themselves is possible through exocytosis [21], extracellular vesicle secretion, or secretory autophagy. That part is not too energetically costly. However, from in vitro studies, it does not seem as though lipofuscin is exported from post-mitotic cells very often [22]. It also has only rarely been observed in vivo [23].
More importantly, when exported, there would ultimately be nowhere for the garbage to go except to be picked up by tissue-resident or circulating phagocytes, which themselves become bloated with lipofuscin. (Transfer of lipofuscin to tissue-resident phagocytes through tunneling nanotubes [TNTs] or partial cell fusion is also theoretically possible.)
Lipofuscin within aged tissue-resident macrophages is perhaps mostly derived from damaged molecules generated by their own, internal metabolic processes - rather than the phagocytosis of extracellular, lipid-saturated debris or efferocytosis in the context of aged, lipofuscin-laden cells [24].
Crucially, I have seen no evidence in the literature that tissue-resident or circulating phagocytes efficiently leave the body through migration to the gastrointestinal tract, urogenital tract, skin, or lungs, except possibly when there are infections in those areas.
Mechanistic argument:
Lipofuscin is broadly a complex amalgam of highly oxidized cross-linked macromolecules, including proteins, lipids, sugars, and metal cations. It varies in composition between species, individuals, cell types, and plausibly even cells of the same type [20,25,26,27].
While it was originally widely believed that lipofuscin is inert, it may in fact permeabilize or otherwise destabilize lysosomes and promote apoptosis or necrosis [20,28,29,30,31] Even if the damaged molecules are mostly sequestered within lysosomes and are not actively harmful to the cell, the fact that many lysosomes become full of garbage and therefore are almost surely unable to perform their normal functions nearly as well just logically seems as though it must be a major problem for the cell. If a critical threshold is reached in enough cells in a tissue, e.g., the brain, it clearly would be problematic. The cells may try to produce more lysosomes - but will eventually reach capacity.
Along these lines, lipofuscin accumulation decreases the ability of cells to adapt to amino acid starvation [32] and increases their susceptibility to oxidative stress [33]. Increases in the dietary intake of metal cations such as Fe2+, which plays a key role in the formation of lipofuscin, augments lipofuscin accumulation [34,35,36] and speeds up aging [37]. Manganese acts similarly [38]. Furthermore, artificial lipofuscin loading into human cells results in a significant loss of cellular viability [39,40] - although it is unclear how similar artificial lipofuscin is to real, age-related lipofuscin. Another study showed that the dietary intake of artificial lipofuscin shortens the lifespan of Drosophila melanogaster [41].
Dr. Aubrey de Grey postulated that we can co-opt enzymes from soil bacteria and fungi and install them in our cells to degrade our lipofuscin [42]. However, I do not believe this will be feasible, as lipofuscin is quite heterogeneous in terms of composition between species, individuals, cell types, and plausibly even cells of the same type - as mentioned before.
Thus, it would likely require an inordinate number of microbial enzymes to degrade the majority of our lipofuscin, almost all of which would still have to be discovered, evolved, or rationally-designed - and many of them may be toxic to our cells. There are some molecular species that seem to be major contributors to lipofuscin or lipofuscin’s toxic effects at least in some cell types, like the fluorophore A2E (retina) [43] and the oxysterol 7-ketocholesterol (throughout the body - especially atherosclerotic plaques and the retina) [44], but we cannot neglect other organs or tissues, or they will fail and we will die anyway. It is worth looking into to see if there are major lipofuscin constituents present across a wide variety of cell types that would be very good targets. But it is unlikely, at least in my mind, that a small number of enzymes will be sufficient to degrade the majority of our lipofuscin deposits.
With regard to short-lived species, like mice and rats, lipofuscin may not have enough time to accumulate to pathological levels before they die of cancer. It is estimated that 50-90% of aged mice die of cancer [45]. Even still, it was shown that in the cerebral cortex neurons of lamina Vb in 630-700-day old rats, lipofuscin occupied 23% of the soma volume [46,47]. This could still certainly have a negative physiological effect. Unsurprisingly, we do see a cognitive decline in mice with age [48]. However, even the oldest mice do not develop age-related neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, or amyotrophic lateral sclerosis [49]. Ostensibly, they simply do not live long enough for sufficient build-up of lipofuscin in their neurons. Other conditions like age-related macular degeneration and sarcopenia do occur in mice, but whether their most severe cases are as bad as the most severe human cases is unclear to me. It is also possible that some mouse tissues accumulate lipofuscin more rapidly than others due to cell type differences in metabolic rates, etc. Some types of human neurons, for example, have not accumulated much lipofuscin by the time others are nearly full [50].
With regard to humans, it has been demonstrated that multiple neuronal cell subtypes become densely packed with lipofuscin granules with age [51,52,53]. In large motor neurons of centenarians, lipofuscin constitutes up to 75% of total cytoplasmic volume [54]. Other post-mitotic cell types also accumulate substantial amounts of lipofuscin [9,55,56]. Lipofuscin-laden lysosomes are often much larger than typical lysosomes. The typical size of a lysosome in a fed, unaged cell is ~100 nm-500 nm in diameter [57]. In contrast, lipofuscin granules are generally 1-5 microns in diameter [54].
Dysfunctional mitochondria are a hallmark of aging [58]. With lipofuscin accumulation, damaged mitochondria - sometimes with mtDNA mutations, sometimes just with damage to their membranes, proteins, lipids, and DNA - may not be recycled as rapidly and may then start to accumulate. Tau may begin to build up inside neurons due to the garbage back-up, eventually becoming hyper-phosphorylated and forming neurofibrillary tangles. Similarly, β-amyloid that is normally degraded may continue to persist, building to levels that lead to plaque formation. Hopefully, if lipofuscin is removed, plaques that have already formed could regress - in line with Le Chatelier’s Principle. In other words, if lipofuscin is cleared and β-amyloid in solution is taken up and autophagocytosed, insoluble plaques may start to dissipate back into solution as well - at which point the β-amyloid in solution would again be taken up and degraded. Transthyretin amyloid could also be another amyloid that forms in the extracellular spaces of our bodies as a result of lipofuscin accumulation in various tissues. Finally, if one could remove the lysosomes from laden macrophages stuck at plaques in artery walls, the plaques may eventually regress, given a healthy diet and sufficient exercise as well [59].
Furthermore, with the extracellular matrix (ECM) not being properly cared for by its resident cells due to lipofuscin accumulation, other damage such as fragmentation, glycation, elastocalcinosis, and cross-linking could rapidly reach pathological levels. After extensive lipofuscin removal, hopefully much of this will damage prove to be reversible. If healthy proteostasis is restored within our parenchymal cells, ECM turnover efficiency could revert to youthful levels [60] - although it may not be able to reverse all the damage that has accumulated in elderly individuals. Certain cross-links may be cleavable by endogenous, secreted enzymes; after treatment, said cross-link–degrading enzymes could be secreted at the appropriate levels.
Glucosepane is a cross-link that becomes prevalent with age in human tissues that is uncleavable - or at least not cleaved very efficiently/often. Exogenously administered cross-link–breaking enzymes could be helpful [61]. However, there are almost certainly a multitude of indigestible cross-links that accumulate with age. Just like with lipofuscin, an unfeasible number of enzymes might be required to address the problem in this way. Fortunately, even if there are some cross-links that can’t be degraded by endogenous enzymes - if the ECM turnover efficiency is restored through extensive lipofuscin removal - molecules bound together by uncleavable cross-links could be excised and endocytosed by tissue-resident cells or phagocytosed by (bioengineered or regular) tissue-resident macrophages.
Senescent cells may start to accumulate if tissue-resident immune cells are rendered inert by lipofuscin accumulation and the non-functionality of the parenchymal cells around them also caused by lipofuscin accumulation, leading to dilapidation of the ECM. Here I refer to irreversibly senescent cells, which have suffered DNA damage and can no longer function properly. However, many cells that show signs of senescence like the senescence-associated secretory phenotype [62] may be reversibly senescent. They may have entered into that state due to epigenetic damage brought on by lipofuscin accumulation. Perhaps this epigenetic state would revert back to a youthful state if their lipofuscin can be removed.
Additionally, cancer may be more likely to initiate or progress if many microenvironments throughout the body are corrupted by lipofuscin accumulation. Along those lines, stem cell niche corruption could prevent them from replicating efficiently to replenish tissues.
Blood stem cells from aged individuals can still function normally if they are “rejuvenated” ex vivo - and then transplanted into a young niche [63]. It appears as though every compound that I’ve seen used to “rejuvenate” aged stem cells (e.g., CASIN and rapamycin) actually decreases false lipofuscin in the cells through the stimulation of autophagy [64,65]. From a theoretical perspective, one could imagine that if a stem cell is slowly-dividing, it could build up lipofuscin over time even with some replication. Alternatively, a rapidly-dividing stem cell could be restrained by a niche full of lipofuscin, and then start to accumulate lipofuscin itself. Lipofuscin removal from the stem cells/their niches may make them better able to degrade damaged telomerase components and generate new ones, but it is still possible that a gene vector encoding telomerase could be needed to lengthen the telomeres of our stem cells - preventing them from becoming cancerous or senescent.
A paper published recently in Nature Aging that suggests that epigenetic damage is mostly reversible at least [66]. The hallmarks of aging that it related to were mitochondrial dysfunction, nutrient sensing, and stem cell composition. If lipofuscin is removed, damaged mitochondria will be recycled and nutrient sensing processes should go back to normal. Stem cells in this study likely show a younger epigenetic age because they divide frequently and thus dilute out their lipofuscin. That relates to my hypothesis about why Yamanaka factors rejuvenate tissues [67]; I believe it may be because they transiently induce a pluripotent stem cell state. Thus, cells that normally wouldn’t divide start to divide transiently, and thereby dilute out their lipofuscin. Sox2, one of the four Yamanaka factors, also initially stimulates autophagy [68] - which decreases “false” lipofuscin. False lipofuscin is intracellular garbage that can be degraded by cellular machinery if said machinery is simply activated more robustly. True lipofuscin is that which is indigestible by cells no matter how they are transcriptionally manipulated - namely the junk that must be removed. Perhaps, however, some age-related epigenetic changes will not revert after lipofuscin removal. If so, the aforementioned rejuvenation technique, known as partial reprogramming, could help to reverse them. However, partial reprogramming may be dangerous; it poses a serious risk of causing cancer through teratoma formation.
Finally, lipofuscin accumulation also explains the downward spiral of functionality that is seen in aging - i.e., the rapid acceleration in decline starting around 60-70 years of age [69]. That is because the decline in autophagy probably increases the rate at which lipofuscin is formed by allowing aggregates to stay around longer and develop further oxidative damage. Also, lipofuscin accumulation may lead to more free radical production [70], thus accelerating its accumulation as lipofuscin is heavily comprised of oxidatively damaged biomolecules. Furthermore, eventually, when autophagy levels have decreased substantially in many cells in a tissue, the rate of accumulation of other forms of age-related damage probably accelerates as well - and could further accelerate lipofuscin accumulation.
Testing the importance of lipofuscin accumulation in aging in vitro now:
TFEB is the master regulator of autophagy (and lysosomal biogenesis) [71]. TFEB overexpression increases lysosomal acidity and boosts lysosomal hydrolase levels in lysosomes. It was shown that TFEB overexpression can shrink lipofuscin deposits - but this work was done in the context of a transgenic mouse model with overproduction of an aggregating protein, which I would define as “false” lipofuscin [72]. Namely, if the protein ceased to be produced and the cells weren’t already completely overwhelmed, it could be degraded over a reasonable amount of time by endogenous lysosomal hydrolases.
TFEB could still be of use for true lipofuscin, however. It also induces the exocytosis of lysosomal contents through fusion of the lysosomal and plasma membranes. One problem with boosting endogenous exocytosis processes is that they may lose potency with age; oxidatively warped lipids from lipofuscin may insert themselves into the membranes of the lysosomes that contain them and prevent efficient fusion with the plasma membrane. The main problem, however, is that the two most well-known inducers of lysosomal exocytosis, Ca2+ and TFEB, do not induce a substantial amount of exocytosis - even in youthful cells [73,74].
Notably, a small molecule was shown to induce some lipofuscin exocytosis in monkey retinal pigment epithelial cells, which are important in macular degeneration. However, this small molecule, remofuscin, is a potent and reversible inhibitor of the H+/K+ ATPase [75]. Proton pump inhibitors increase lysosomal pH and activate TFEB - so remofuscin’s effects may not be additive with TFEB overexpression.
In order to test the theory that lipofuscin is currently the main issue with regard to age-related disease in a way that would be more definitive, we could utilize a recently developed organelle sampling FluidFM approach [76] to remove lipofuscin-laden lysosomes from single skeletal muscle cells taken from biopsies of elderly humans [77,78,79]. By doing so, we could observe whether lipofuscin removal reverses the age-related phenotype of these cells. We could also determine how long they live in culture compared to untreated single skeletal muscle cells.
Along these lines, in one study, it was shown that at ~day 25, rat hippocampal neurons in vitro have an aged phenotype, including dysfunctional mitochondria, higher reactive oxygen species production, and at least one feature of senescence [80]. It was also shown that defective autophagy was linked to this senescence-like phenotype - and that aged neurons in culture accumulate lipofuscin [81]. Thus, if the post-mitotic cells’ lipofuscin were removed from the cells and new, functional lysosomes were introduced exogenously or generated through TFEB, their aged phenotype may reverse.
Cells from elderly patients may require a more extensive initial session of lipofuscin removal. That is because once the garbage back-up is cleared at the end-point, i.e., the lysosomal level, oxidatively damaged mitochondria would then be mitophagocytosed and cytoplasmic lipofuscin granules would possibly be engulfed by autophagosomes and delivered to new lysosomes. Thus, new lysosomes might be quickly overwhelmed again - and they should be exported as well. Perhaps three rounds of lysosomal removal in quick succession would be appropriate for the first treatment session. The elderly cell may also have autophagosomes that have been present for too long in the cytoplasm. Their outer membranes may have become corrupted by oxidatively warped lipids; thus, they may not even be able to fuse with new lysosomes. They might have to be exported as well.
It is important to keep in mind that while primary cell cultures often die after a few days-weeks [82], the cause of death may not be related to normal aging. It may instead be related to infection or hyperosmolality due to evaporation of the media [83]. These factors should be mitigated or negated if possible. This is perhaps especially tricky when it comes to single-cell manipulations.
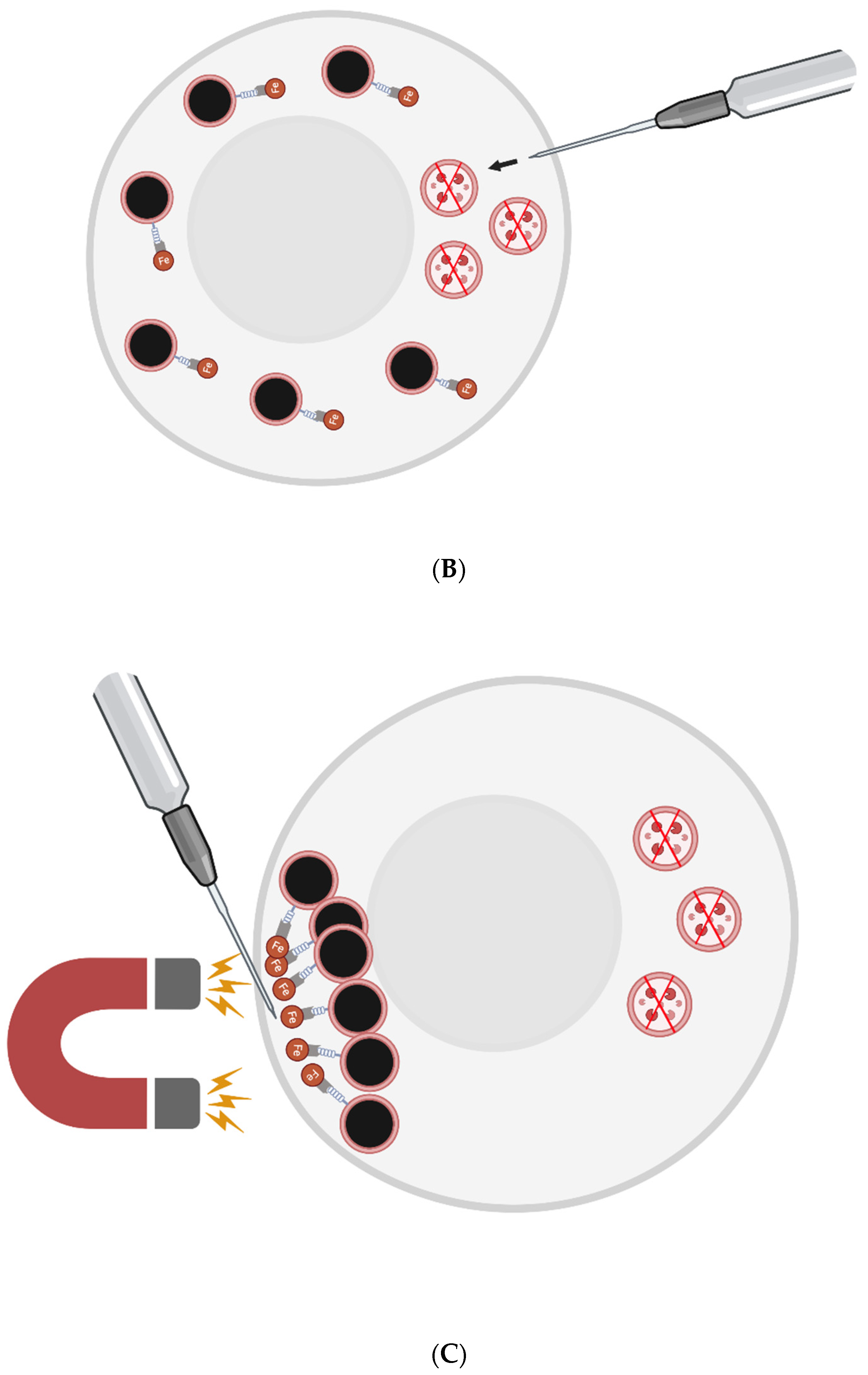
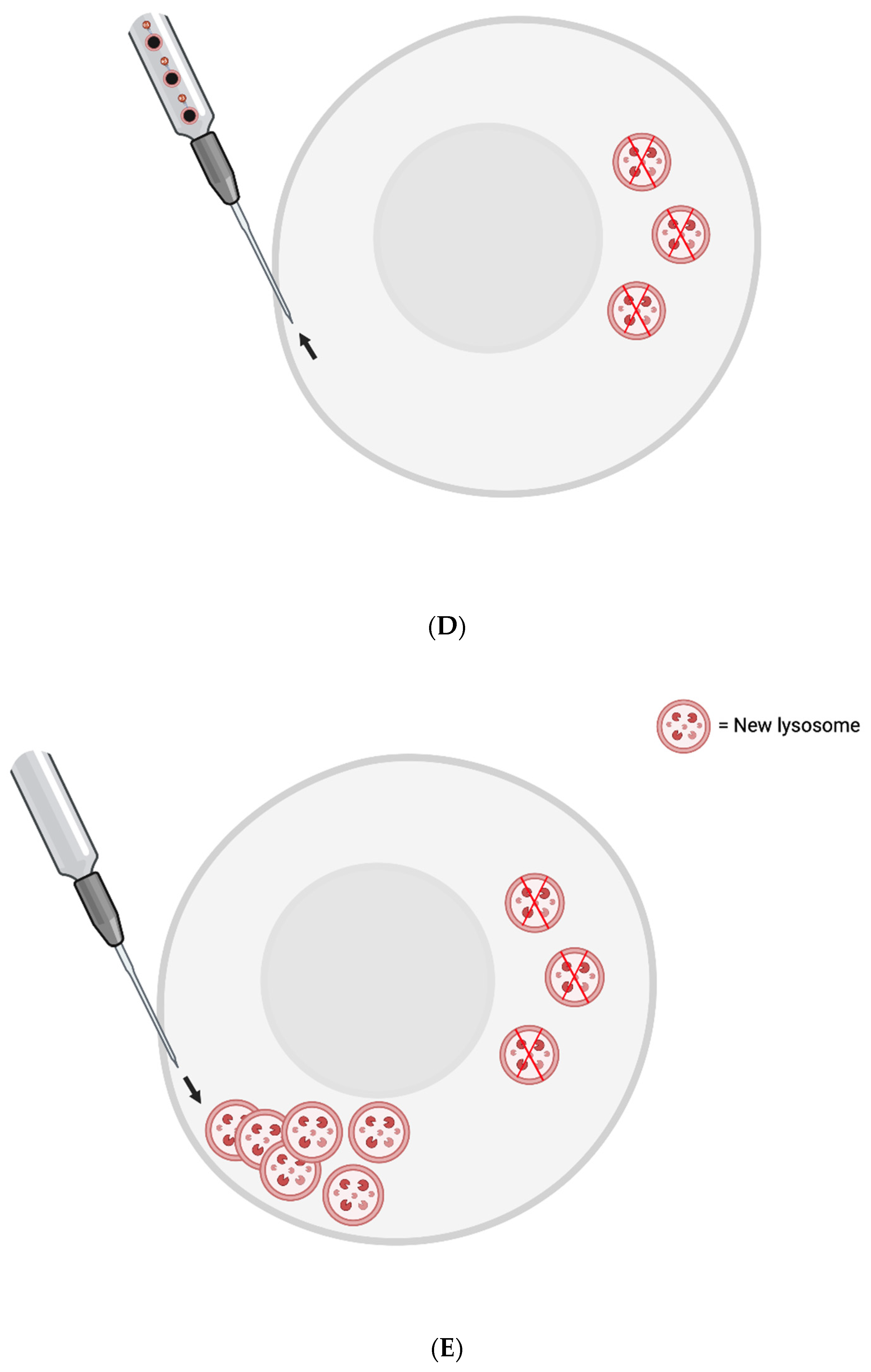
The FluidFM-based approach involves a small needle that penetrates the cell membrane and can inject content into or aspirate content from a single cell [76]. Single skeletal muscle cells from elderly patients could be injected with magnetic nanoparticles (MNPs) conjugated with a nanobodies that target LAMP-1. Then, a magnetic field could be applied to cluster the lysosomes to one small area at the plasma membrane. They would then be removed by the needle. New lysosomes could then be injected to replenish the cell’s stores - or perhaps generated via transient, induced TFEB overexpression.
To prevent cell death, one may need to co-inject some number of new lysosomes with the nanobody-conjugated MNPs. The new lysosomes would first be pre-incubated with nanobodies that are not conjugated to MNPs, however - to sterically block the relevant LAMP-1 site.
Figure 1: FluidFM for Lipofuscin Removal
Conclusion:
It would be extremely interesting to put the “garbage catastrophe theory of aging” to the test with organelle-sampling FluidFM - using single skeletal muscle cells (in large replicate) from elderly humans. If in vitro testing reveals lipofuscin accumulation to be a critical factor in the aging of such cells, we will have to eliminate it in situ or remove it from the body somehow. Through synthetic biology, I believe we can clear lipofuscin from our tissues.
Ethics approval and consent to participate
N/A
Consent for publication
N/A
Availability of data and material
N/A
Competing interests
The author declares that he has no competing interests.
Authors' contributions
M.R. wrote the paper.
Funding
Funding not received for the study.
Acknowledgements
Thank you to my family and friends. The figures in this piece were created with BioRender.com.
References
- Terman A and Brunk UT. Lipofuscin. The International Journal of Biochemistry & Cell Biology 2004;36(8):1400–1404. [CrossRef]
- Siebert S, Farrell JA, Cazet JF, et al. Stem Cell Differentiation Trajectories in Hydra Resolved at Single-Cell Resolution. Science 2019;365(6451):eaav9314. [CrossRef]
- Murad R, Macias-Muñoz A, Wong A, et al. Coordinated Gene Expression and Chromatin Regulation during Hydra Head Regeneration. Genome Biology and Evolution 2021;13(12):evab221. [CrossRef]
- Terman A, Brunk UT. Is aging the price for memory? Biogerontology (2005) 6:205–210. [CrossRef]
- Klapper W, Kühne K, Singh KK, Heidorn K, Parwaresch R, Krupp G. Longevity of lobsters is linked to ubiquitous telomerase expression. FEBS Letters (1998) 439:143–146. [CrossRef]
- Beltz BS, Sandeman DC. Regulation of life-long neurogenesis in the decapod crustacean brain. Arthropod Structure & Development (2003) 32:39–60. [CrossRef]
- Peregrim I. Why we age — a new evolutionary view. Biologia (2017) 72:475–485. [CrossRef]
- Sheehy M, Shelton P, Wickins J, Belchier M, Gaten E. Ageing the European lobster Homarus gammarus by the lipofuscin in its eyestalk ganglia. Mar Ecol Prog Ser (1996) 143:99–111. [CrossRef]
- Kakimoto Y, Okada C, Kawabe N, Sasaki A, Tsukamoto H, Nagao R, Osawa M. Myocardial lipofuscin accumulation in ageing and sudden cardiac death. Sci Rep (2019) 9:3304. [CrossRef]
- Karavanich C, Atema J. Individual recognition and memory in lobster dominance. Animal Behaviour (1998) 56:1553–1560. [CrossRef]
- Zhao S, Lin L, Kan G, Xu C, Tang Q, Yu C, Sun W, Cai L, Xu C, Cui S. High autophagy in the naked mole rat may play a significant role in maintaining good health. Cell Physiol Biochem (2014) 33:321–332. [CrossRef]
- Triplett JC, Tramutola A, Swomley A, Kirk J, Grimes K, Lewis K, Orr M, Rodriguez K, Cai J, Klein JB, et al. Age-related changes in the proteostasis network in the brain of the naked mole-rat: Implications promoting healthy longevity. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease (2015) 1852:2213–2224. [CrossRef]
- Hadi F, Kulaberoglu Y, Lazarus KA, Bach K, Ugur R, Beattie P, Smith ESJ, Khaled WT. Transformation of naked mole-rat cells. Nature (2020) 583:E1–E7. [CrossRef]
- Edrey YH, Hanes M, Pinto M, Mele J, Buffenstein R. Successful aging and sustained good health in the naked mole rat: a long-lived mammalian model for biogerontology and biomedical research. ILAR J (2011) 52:41–53. [CrossRef]
- Panno JP, Nair KK. Effects of increased lifespan on chromatin condensation in the adult male housefly. Mech Ageing Dev (1986) 35:31–38. [CrossRef]
- Clokey GV, Jacobson LA. The autofluorescent “lipofuscin granules” in the intestinal cells of Caenorhabditis elegans are secondary lysosomes. Mech Ageing Dev (1986) 35:79–94. [CrossRef]
- Houthoofd K, Braeckman BP, Lenaerts I, Brys K, De Vreese A, Van Eygen S, Vanfleteren JR. Ageing is reversed, and metabolism is reset to young levels in recovering dauer larvae of C. elegans. Exp Gerontol (2002) 37:1015–1021. [CrossRef]
- Goyal VK. Lipofuscin pigment accumulation in the central nervous system of the mouse during aging. Exp Gerontol (1982) 17:89–94. [CrossRef]
- Gilissen EP, Staneva-Dobrovski L. Distinct Types of Lipofuscin Pigment in the Hippocampus and Cerebellum of Aged Cheirogaleid Primates. The Anatomical Record (2013) 296:1895–1906. [CrossRef]
- Moreno-García A, Kun A, Calero O, Medina M, Calero M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Frontiers in Neuroscience (2018) 12: https://www.frontiersin.org/article/10.3389/fnins.2018.00464 [Accessed March 31, 2022]. [CrossRef]
- Gray DA and Woulfe J. Lipofuscin and Aging: A Matter of Toxic Waste. Science of Aging Knowledge Environment 2005;2005(5):re1–re1. [CrossRef]
- Terman A, Brunk UT. Is Lipofuscin Eliminated from Cells? Investigative Ophthalmology & Visual Science (1999) 40:2463–2464.
- Wang L, Xiao C-Y, Li J-H, Tang G-C, Xiao S-S. Transport and Possible Outcome of Lipofuscin in Mouse Myocardium. Adv Gerontol 2022;12:247–63. [CrossRef]
- Burns JC, Cotleur B, Walther DM, Bajrami B, Rubino SJ, Wei R, Franchimont N, Cotman SL, Ransohoff RM, Mingueneau M. Differential accumulation of storage bodies with aging defines discrete subsets of microglia in the healthy brain. eLife (2020) 9:e57495. [CrossRef]
- Boellaard JW and Schlote W. Ultrastructural Heterogeneity of Neuronal Lipofuscin in the Normal Human Cerebral Cortex. Acta Neuropathol 1986;71(3–4):285–294. [CrossRef]
- Sohal RS, Wolfe LS. Chapter 11 Lipofuscin: characteristics and significance. In: Swaab DF, Fliers E, Mirmiran M, Van Gool WA, Van Haaren F, editors. Progress in Brain Research, vol. 70, Elsevier; 1986, p. 171–83. [CrossRef]
- Sheehy MRJ. Individual variation in, and the effect of rearing temperature and body size on, the concentration of fluorescent morphological lipofuscin in the brains of freshwater crayfish, Cherax cuspidatus (Crustacea: Parastacidae). Comparative Biochemistry and Physiology Part A: Physiology 1990;96:281–6. [CrossRef]
- Brunk UT, Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med (2002) 33:611–619. [CrossRef]
- Kurz T, Terman A, Gustafsson B, et al. Lysosomes and oxidative stress in aging and apoptosis. Biochimica et Biophysica Acta (BBA) - General Subjects 2008;1780(11):1291–1303. [CrossRef]
- Gabandé-Rodríguez E, Keane L, Capasso M. Microglial phagocytosis in aging and Alzheimer’s disease. Journal of Neuroscience Research 2020;98(2):284–298. [CrossRef]
- Pan C, Banerjee K, Lehmann GL, et al. Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proceedings of the National Academy of Sciences 2021;118(47):e2100122118. [CrossRef]
- Terman A, Dalen H, Brunk UT. Ceroid/lipofuscin-loaded human fibroblasts show decreased survival time and diminished autophagocytosis during amino acid starvation☆. Experimental Gerontology 1999;34(8):943–957. [CrossRef]
- Terman A, Abrahamsson N, Brunk UT. Ceroid/lipofuscin-loaded human fibroblasts show increased susceptibility to oxidative stress. Exp Gerontol 1999;34(6):755–770. [CrossRef]
- Lv Z, Jiang H, Xu H, et al. Increased iron levels correlate with the selective nigral dopaminergic neuron degeneration in Parkinson’s disease. J Neural Transm 2011;118(3):361–369. [CrossRef]
- Maccarinelli F, Pagani A, Cozzi A, et al. A novel neuroferritinopathy mouse model (FTL 498InsTC) shows progressive brain iron dysregulation, morphological signs of early neurodegeneration and motor coordination deficits. Neurobiology of Disease 2015;81:119–133. [CrossRef]
- Bhoiwala D, Song Y, Cwanger A, et al. High iron diet causes elevation of retinal iron levels and RPE autofluorescence. Investigative Ophthalmology & Visual Science 2015;56(7):4203.
- Mangan D. Iron: an underrated factor in aging. Aging 2021;13(19):23407–23415. [CrossRef]
- hgami N, Yajima I, Iida M, et al. Manganese-mediated acceleration of age-related hearing loss in mice. Sci Rep 2016;6(1):36306. [CrossRef]
- Höhn A, Grune T. Lipofuscin: formation, effects and role of macroautophagy. Redox Biol 2013;1(1):140–144. [CrossRef]
- von Zglinicki T, Nilsson E, Döcke WD, et al. Lipofuscin accumulation and ageing of fibroblasts. Gerontology 1995;41 Suppl 2:95–108. [CrossRef]
- Tsakiri EN, Iliaki KK, Höhn A, et al. Diet-derived advanced glycation end products or lipofuscin disrupts proteostasis and reduces life span in Drosophila melanogaster. Free Radic Biol Med 2013;65:1155–1163. [CrossRef]
- de Grey AD. Appropriating Microbial Catabolism: A Proposal to Treat and Prevent Neurodegeneration. Neurobiology of aging 2006;27(4):589–595. [CrossRef]
- Sparrow JR, Parish CA, Hashimoto M, et al. A2E, a Lipofuscin Fluorophore, in Human Retinal Pigmented Epithelial Cells in Culture. Investigative Ophthalmology & Visual Science 1999;40(12):2988–2995.
- Anderson A, Campo A, Fulton E, et al. 7-Ketocholesterol in Disease and Aging. Redox Biology 2020;29:101380. [CrossRef]
- Seluanov A, Gladyshev VN, Vijg J, et al. Mechanisms of cancer resistance in long-lived mammals. Nat Rev Cancer 2018;18(7):433–441. [CrossRef]
- Samorajski T, Ordy JM, Rady-Reimer P. Lipofuscin pigment accumulation in the nervous system of aging mice. The Anatomical Record (1968) 160:555–573. [CrossRef]
- Brizzee KR, Johnson FA. Depth distribution of lipofuscin pigment in cerebral cortex of albino rat. Acta Neuropathol (1970) 16:205–219. [CrossRef]
- Yanai S, Endo S. Functional Aging in Male C57BL/6J Mice Across the Life-Span: A Systematic Behavioral Analysis of Motor, Emotional, and Memory Function to Define an Aging Phenotype. Frontiers in Aging Neuroscience 2021;13. [CrossRef]
- Lutz CM, Osborne MA. Optimizing mouse models of neurodegenerative disorders: are therapeutics in sight? Future Neurology 2014;9(1):67–75. [CrossRef]
- Double KL, Dedov VN, Fedorow H, et al. The comparative biology of neuromelanin and lipofuscin in the human brain. Cell Mol Life Sci 2008;65(11):1669–1682. [CrossRef]
- Mann DM, Yates PO, Stamp JE. The relationship between lipofuscin pigment and ageing in the human nervous system. J Neurol Sci (1978) 37:83–93. [CrossRef]
- Goyal VK. Lipofuscin pigment accumulation in human brain during aging. Experimental Gerontology 1982;17(6):481–487. [CrossRef]
- Benavides SH, Monserrat AJ, Fariña S, et al. Sequential histochemical studies of neuronal lipofuscin in human cerebral cortex from the first to the ninth decade of life. Archives of Gerontology and Geriatrics 2002;34(3):219–231. [CrossRef]
- Yin D. Biochemical basis of lipofuscin, ceroid, and age pigment-like fluorophores. Free Radical Biology and Medicine 1996;21(6):871–888. [CrossRef]
- Wing GL, Blanchard GC, Weiter JJ. The topography and age relationship of lipofuscin concentration in the retinal pigment epithelium. Investigative Ophthalmology & Visual Science (1978) 17:601–607.
- Dayan D, Abrahami I, Buchner A, Gorsky M, Chimovitz N. Lipid pigment (lipofuscin) in human perioral muscles with aging. Experimental Gerontology (1988) 23:97–102. [CrossRef]
- Xu H, Ren D. Lysosomal Physiology. Annu Rev Physiol (2015) 77:57–80. [CrossRef]
- López-Otín C, Blasco MA, Partridge L, et al. The Hallmarks of Aging. Cell 2013;153(6):1194–1217. [CrossRef]
- JACC: Cardiovascular Imaging, 1205. [CrossRef]
- Gumpenberger M, Wessner B, Graf A, et al. Remodeling the Skeletal Muscle Extracellular Matrix in Older Age—Effects of Acute Exercise Stimuli on Gene Expression. Int J Mol Sci 2020;21(19):7089. [CrossRef]
- Furber JD. Extracellular glycation crosslinks: prospects for removal. Rejuvenation Res 2006;9(2):274–278. [CrossRef]
- Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34(23–24):1565–1576. [CrossRef]
- Guidi N, Marka G, Sakk V, et al. An Aged Bone Marrow Niche Restrains Rejuvenated Hematopoietic Stem Cells. STEM CELLS 2021;39(8):1101–1106. [CrossRef]
- Landspersky T, Saçma M, Rivière J, et al. Autophagy in Mesenchymal Progenitors Protects Mice against Bone Marrow Failure after Severe Intermittent Stress. Blood 2022;139(5):690–703. [CrossRef]
- Zhang X, Chen W, Gao Q, et al. Rapamycin Directly Activates Lysosomal Mucolipin TRP Channels Independent of MTOR. PLOS Biology 2019;17(5):e3000252. [CrossRef]
- Kabacik S, Lowe D, Fransen L, et al. The Relationship between Epigenetic Age and the Hallmarks of Aging in Human Cells. Nat Aging 2022;2(6):484–493. [CrossRef]
- Lu Y, Brommer B, Tian X, et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020;588(7836):124–129. [CrossRef]
- Wang S, Xia P, Ye B, et al. Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 2013;13(5):617–625. [CrossRef]
- Kang, Y.-K., Min, B., Eom, J. & Park, J. S. Different phases of aging in mouse old skeletal muscle. Aging (Albany NY) 14, 143–160 (2022). [CrossRef]
- Vida C, de Toda IM, Cruces J, et al. Role of macrophages in age-related oxidative stress and lipofuscin accumulation in mice. Redox Biol 2017;12:423–437. [CrossRef]
- Settembre C, Di Malta C, Polito VA, et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011;332(6036):1429–1433. [CrossRef]
- Wang H, Wang R, Carrera I, et al. TFEB Overexpression in the P301S Model of Tauopathy Mitigates Increased PHF1 Levels and Lipofuscin Puncta and Rescues Memory Deficits. eNeuro 2016;3(2). [CrossRef]
- Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. Journal of Cell Biology (2002) 159:625–635. [CrossRef]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, et al. Transcriptional Activation of Lysosomal Exocytosis Promotes Cellular Clearance. Dev Cell (2011) 21:421–430. [CrossRef]
- Julien S, Schraermeyer U. Lipofuscin can be eliminated from the retinal pigment epithelium of monkeys. Neurobiol Aging (2012) 33:2390–2397. [CrossRef]
- Gäbelein CG, Feng Q, Sarajlic E, Zambelli T, Guillaume-Gentil O, Kornmann B, Vorholt JA. Mitochondria transplantation between living cells. PLOS Biology (2022) 20:e3001576. [CrossRef]
- Beregi E, Regius O, Hüttl T, Göbl Z. Age-related changes in the skeletal muscle cells. Z Gerontol 1988;21:83–6.
- Hütter E, Skovbro M, Lener B, Prats C, Rabøl R, Dela F, et al. Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell 2007;6:245–56. [CrossRef]
- Triolo M, Hood DA. Manifestations of Age on Autophagy, Mitophagy and Lysosomes in Skeletal Muscle. Cells (2021) 10:1054. [CrossRef]
- Dong W, Cheng S, Huang F, Fan W, Chen Y, Shi H, He H. Mitochondrial dysfunction in long-term neuronal cultures mimics changes with aging. Med Sci Monit (2011) 17:BR91–BR96. [CrossRef]
- Moreno-Blas D, Gorostieta-Salas E, Pommer-Alba A, Muciño-Hernández G, Gerónimo-Olvera C, Maciel-Barón LA, Konigsberg M, Massieu L, Castro-Obregón S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging (Albany NY) (2019) 11:6175–6198. [CrossRef]
- Li Z. “5.43 - In Vitro Micro-Tissue and -Organ Models for Toxicity Testing.,” In: Moo-Young M, editor. Comprehensive Biotechnology (Second Edition). Burlington: Academic Press (2011). p. 551–563. [CrossRef]
- Potter SM, DeMarse TB. A new approach to neural cell culture for long-term studies. Journal of Neuroscience Methods (2001) 110:17–24. [CrossRef]
Figure 1.
A) Single aged skeletal muscle cells are injected via the FluidFM needle with MNPs conjugated to nanobodies against LAMP-1 and perhaps new lysosomes with sterically-blocked LAMP-1. B) The nanobodies conjugated to MNPs find their LAMP-1 targets on the surface of pre-existing lysosomes. C) Magnetism is used to cluster at least the majority of the pre-existing lysosomes in one area of the cell (i.e., near the needle tip - opposite the region where new lysosomes were injected). D) The needle is used to aspirate the indigestible garbage-filled lysosomes. E) Another set of new lysosomes are injected into the cell where the pre-existing lysosomes were extracted.
Figure 1.
A) Single aged skeletal muscle cells are injected via the FluidFM needle with MNPs conjugated to nanobodies against LAMP-1 and perhaps new lysosomes with sterically-blocked LAMP-1. B) The nanobodies conjugated to MNPs find their LAMP-1 targets on the surface of pre-existing lysosomes. C) Magnetism is used to cluster at least the majority of the pre-existing lysosomes in one area of the cell (i.e., near the needle tip - opposite the region where new lysosomes were injected). D) The needle is used to aspirate the indigestible garbage-filled lysosomes. E) Another set of new lysosomes are injected into the cell where the pre-existing lysosomes were extracted.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



