
Submitted:
14 September 2022
Posted:
22 September 2022
You are already at the latest version
Abstract
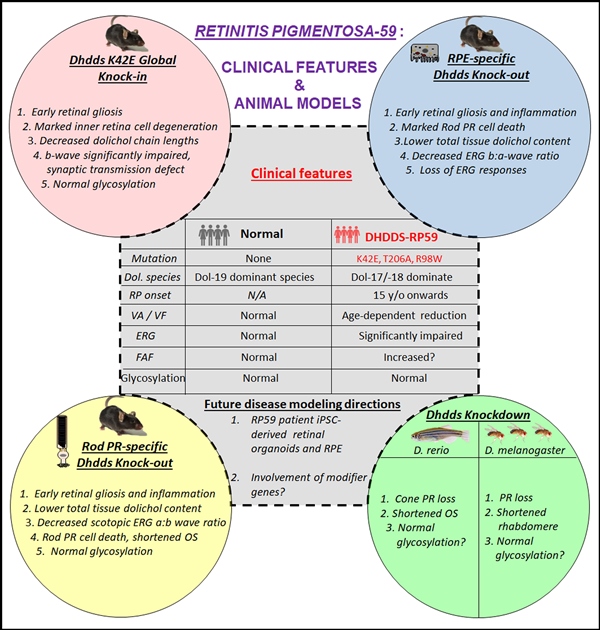
Retinitis pigmentosa-59 (RP59) is a rare, recessive form of RP, caused by mutations in the gene encoding DHDDS (dehydrodolichyl diphosphate synthase). DHDDS forms a heterotetrameric complex with Nogo-B Receptor (NgBR; gene NUS1) to form a cis-prenyltransferase (CPT) enzyme complex, which is required for synthesis of dolichol, which in turn is required for protein N-glycosylation as well as other glycosylation reactions in eukaryotic cells. Herein, we review the published phenotypic characteristics of RP59 models extant, with an emphasis on their ocular phenotypes, based primarily upon knock-in of known RP59-associated DHDDS mutations as well as cell type- and tissue-specific knockout of DHDDS alleles in mice. We also briefly review findings in RP59 patients with retinal disease and other patients with DHDDS mutations causing epilepsy and other neurologic disease. We discuss these findings in the context of addressing “knowledge gaps” in our current understanding of the underlying pathobiology mechanism of RP59, as well as their potential utility for developing therapeutic interventions to block the onset, or to dampen the severity or progression, of RP59.
Keywords:
cis-prenyltransferase
; DHDDS
; dolichol
; Nogo-B receptor
; retinal degeneration
; RP59
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



