
Submitted:
14 February 2023
Posted:
16 February 2023
You are already at the latest version
Abstract
Development of K-Ras independence may explain failure of targeted therapy for pancreatic cancer (PC). In this paper active N as well as K-Ras was shown in all human cell lines tested. In a cell line dependent on mutant K-Ras, it was shown that depleting K-Ras reduced total Ras activity, while cell lines described as independent had no significant decline in total Ras activity. Knockdown of N-Ras showed it had an important role in controlling the relative level of oxidative metabolism but only K-Ras depletion caused a decrease in G2 cyclins, proteasome inhibition reversed this and other targets of APC/c were also decreased by K-Ras depletion. K-Ras depletion did not cause an increase in ubiquitinated G2 cyclins but instead caused exit from G2 phase to slow relative to completion of S-phase, suggesting mutant K-Ras may inhibit APC/c prior to anaphase but stabilizes G2 cyclins independently of this. We propose that during tumorigenesis, cancer cells expressing wild type N-Ras protein are selected because the protein protects cancer cells from the deleterious effects of cell cycle independent induction of cyclins by mutant K-Ras. Mutation independence results when N-Ras activity becomes adequate to drive cell division even in cells where K-Ras is inhibited. Keywords: Pancreatic Ductal Adenocarcinoma, K-Ras, N-Ras, G2 cyclins.
Keywords:
Pancreatic Ductal Adenocarcinoma
; K-Ras
; N-Ras
; G2 cyclins
1. Introduction
Despite a growing understanding of the molecular changes that result in pancreatic cancer, the disease remains largely intractable to chemotherapy and prognosis is amongst the worst for any malignancy; with 5 year survival of approximately 10%[1].
The KRAS gene is mutated in most cases of Pancreatic Ductal Adenocarcinoma (PDAC), with estimates of over 90%[2]. Furthermore, mutant K-Ras is found in pre-neoplastic lesions such as Pancreatic Intraepithelial Neoplasias (PanINs)[3] and Intraductal Papillary Mucinous Neoplasias (IPMN)[4]. This has led to the assumption that mutation of KRAS is a common initiating event in pancreatic tumorigenesis, which is supported by work on murine models[5]. Activation of mutant KRAS under its own promoter in pancreatic progenitor cells using Cre recombinase under the pdx promoter results in PanIN like lesions and eventually cancer[5]. These progenitor (bud) cells give rise to islets as well as both acinar and ductal epithelial cells. Expression of mutant K-Ras directly in ductal[6] or acinar[7] cells is not usually sufficient to induce PanIN or PDAC development in mice. Activation of mutant K-Ras under its own promoter, using Cre recombinase under an acinar specific (elastase) promoter can reproduce pancreatic tumorigenesis, but only if K-Ras is expressed during development or following cerulean-induced pancreatitis[8]. This may require the induction of Acinar Ductal Metaplasia (ADM) via increased expression of the Hippo signalling protein YAP1[9]. Suggesting that the best established role of K-Ras mutation, induction of Cyclin D1 in G1 cells, is inadequate in itself to promote carcinogenesis. Rise and fall of cyclin levels drives the cell cycle. In metazoans there is a sequential rise of G1 cyclins (e.g Cyclin D1) in G1, followed by rise of G2 cyclins (e.g. cyclin B1) in G2. Although, recent evidence suggests that the cyclins are redundant in respect of cell cycle progression (rise and fall of any single cyclin type or any combination of cyclins would achieve progression) [10]. In quiescent cells Ras activity causes production of G1 cyclins to start this process. However, once cells are cycling, level of cyclin D1 is determined by its protein stability and not by transcriptional regulation via Ras[11].
Targeting K-Ras has a long tradition in pancreatic cancer research, but agents have struggled to make progress from the laboratory into clinical practice, recent progress with KRAS-G12C inhibitors offers light at the end of the tunnel, and these have been licensed in the treatment of lung cancer, but not yet pancreatic cancer[12]. The lack of an effective small molecule inhibitor of mutant K-Ras in general, might in part be explained by the ability of cancer cells to survive having lost K-Ras once tumours are established[13]. K-Ras loss in cell lines and murine models can be compensated for by other mutations (including over-expression of the YAP1 gene)[14]. Some cell lines which retain oncogenic K-Ras have become independent of the mutation, although others remain addicted to the oncogene[15]. Independence of K-Ras often being associated with YAP1 overexpression and a consequent expression profile consistent with a quasi-mesenchymal phenotype[16].
The progression from pre-neoplastic lesions to carcinoma following K-Ras mutation requires Epidermal Growth Factor Receptor (EGFR) activity[17] and oncogenic K-Ras increases the level of this receptor[18]. Mutation of p53 relieves the requirement for EGFR activation[17, 19], furthermore inflammation in the absence of TP53 leads to pancreatic cancer in mice without the requirement for K-Ras mutation[20].
This is in contrast to other cancer types where K-Ras mutation seems to negate the need for stimulation of EGFR[21, 22]. Co-dependence on EGFR and mutant K-Ras may reflect the fact that they can activate the cell cycle via cyclin D expression independently[23] and this is supported further by the observation that inhibition of various forms of cyclin dependent kinase (including CDK1 and 2) is equivalent in action to inhibition of mutant K-Ras in cancers that are addicted to K-Ras[24].
K-Ras dependency in cell lines is associated with an expression pattern that is also closely associated with EGFR inhibition[15]. Other forms of Ras (N and H-Ras) are rarely mutated in PDAC[25], nevertheless both are expressed[26] and they may increase EGFR levels and responsiveness to EGF[27]. Knockdown of N and H-Ras may therefore promote K-Ras addiction. Which may explain why high expression of N and H-Ras are associated with poorer prognosis[28]. Although, it has been suggested that K-Ras activates both N and H-Ras via the AKT target endothelial nitric oxide synthase (eNOS)[29], in which case they would lie downstream of K-Ras and not be independent of it. Singh et al provided evidence that the K-Ras addiction expression profile is negatively associated with epithelial- mesenchymal transformation (EMT)[15], such that addicted cells appear more epithelial. Only one of the 9 K-Ras dependent cell lines tested by this group was reported to have an expression profile predictive of K-Ras independence[15]; this was the pancreatic line Suit-2, which is well known to be more mesenchymal than other pancreatic cell lines[30]. We set out to characterise how mutant K-Ras interacts with wild type Ras isoforms to control proliferation in this cell line.
2. Materials and Methods
Cell Culture
Suit-2, PANC-1, MIA-Pa-Ca-2, BxPC3 and HeLa cell lines were cultured in RPMI 1640 with 10% FBS and 1% L-glutamine (100mM/L). These cell lines were genotyped using the PowerPlex 16HS system (Promega, UK). Primary mouse pancreatic cancer cell lines were obtained from Professor Michael Schmid (Liverpool University) who isolated them from tumours arising in KrasLSL-G12D/+, p53R172H/+, Pdx1-Cre mice (KPC) using a protocol described by Olive et al., 2009[31]. After 3-4 passages, primary KPC derived murine pancreatic cancer cells were grown in DMEM medium supplemented with 10% fetal bovine serum, 1% Amphotericin B, penicillin (100 U/mL), streptomycin (100 µg/mL) and L-glutamine (10 mmol/L) (all from Sigma Aldrich, UK). Low passage (<10) KPC cells were used for these experiments. Confluent T75 flask of cells was used to generate cell lysates. Cells were washed and recovered in PBS and then cells were scraped off the bottom of the flask using a cell scraper The cell suspension was spun at 1000xg for 10 minutes in a bench top centrifuge. The cell pellet was stored at -80oC until cell lysates were made for western blotting.
siRNA Treatment
Cells were seeded at 0.5x105 cells per well of a 6 well plate. The cells were transfected using Lipofectamine-2000 following the manufacturer’s protocol. siRNA was purchased from GE Healthcare (Lafayette, CO, USA), 10 nmol of human KRAS (J-005069-10-0050), HRAS (L-004142-00-0010) or CCND1 (L-003210-00-0010) siRNA was used and/or 100 nmol human NRAS (L-003919-00-0010) siRNA. The cells were harvested 48hrs after siRNA treatment and stored as a cell pellet until required at -80oC.
Western Blot Analysis
Lysates were prepared from pellets using RIPA buffer and sonication, following standard protocols. Western Blots were run on precast Any KD gels (Bio-Rad Labororatories Ltd, Hercules, CA, USA) in accordance to standard protocols and transferred using the Bio-Rad Turbo blotter high molecular weight program. K-Ras (F234, sc-30, 1:500), N-Ras (F155, sc-31, 1:1000), Cyclin A (H-432, sc-751, 1:1000) , B (H-433, sc-752, 1:1000) and E (C-19, sc198, 1:1000) all 1:1000 were all purchased from Santa Cruz Biotechnology (Heidelberg, Germany) and Cyclin D (ab137875, 1:1000) was purchased from AbCam (Cambridge, UK). RNA-induced silencing complex (RISC) free control was from GE healthcare/Dharmacon Lafayette, CO, USA (D-001600-01-05). The membranes were blocked in 5% milk Phosphate Buffered Saline with 0.1% Tween (PBST 0.1%) for three hours before being incubated with primary antibody in 5% milk, PBST 0.1% at 4o C overnight. Six, ten minute washes in PBST 0.1% were followed by incubation of the membrane with an HRP conjugated secondary anti-mouse antibody (sc-2357). Following incubation with a secondary antibody, six ten minute washes with PBST 0.1% were carried out and the membrane was visualised using chemo-luminescence and developed (Western lightning plus ECL Life Technologies, Paisley, #31985-062).
Active Ras Pulldown
After 48hr of siRNA treatment, cells were lysed and active Ras was pulled down using Raf as a substrate, following the instructions provided in the Active Ras pulldown kit (Life Technologies, Paisley, UK).
Ubiquitin Pulldown Assay
After 24hrs of siRNA treatment the cells were treated with Bortezomib (Santa Cruz Biotechnology, sc-217785, 10nM) or DMSO diluted in 500µl of RPMI media. The cells were harvested 48hrs after siRNA treatment and ubiquitinated proteins pulled down using a polyubiquitin affinity column (Life Technologies, Paisley, UK) following the manufacturer’s protocol.
Seahorse Analysis
Cells were seeded in a seahorse 24 well plate (Seahorse Bioscience, Copenhagen, DK) at 6000 cells/well. The cells were then treated with siRNA as above but only 15µl of the siRNA mix was added to each well. The cells were then left for 48hrs at 37O C, 5% CO2. The cells were run on the Seahorse Bioanalyzer (Seahorse Biosciences) following manufacturer’s protocols supplied for an aerobic stress test.
Seahorse data were reported as OCR/ECAR which gives a measure of relative oxidative against non-oxidative phosphorylation. OCR/ECAR was obtained for each test prior to addition of any drug and then the value was divided by OCR/ECAR for a control that was run in parallel (to normalise to the plate). At least 4 usable technical repeats were carried out for each experiment and each experiment was done in triplicate. Data was considered usable only if the stress test showed the predicted response to the drugs (i.e. if any of the agents had given no response or the opposite response to that expected with Antimycin A + Rotenone, Oligomycin, or FCCP then the data for that run was considered unusable, this was binary, it either worked or it didn’t, and in all cases was the consequence of a technical problems such as a reagent not being added or being expired and not due to variability in the assay).
FACS Analysis
Cells were treated with siRNA as above and left for 48hrs they were then harvested and spun at 500x g in a bench top centrifuge, before being washed with PBS and re-spun. The cell pellet was then resuspended in 70% ethanol and left at 4oC for at least 16hrs. The samples were then spun at 500x g for 5 minutes and the supernatant was discarded. The sample was resuspended in 500µl Propidium Iodide (PI) (50 µg/ml) and 50µl of RNase (10 mg/ml diluted 1:10 in PBS). The sample was then gently mixed before being transferred to a FACS tube. A minimum of 50,000 cells per treatment were analyzed on a BD LSR Fortessa flow cytometer. Data were analyzed with appropriate gating to exclude debris, doublets and clumps using the software FlowJo version 7 (TreeStar, Inc., Ashland, OR). Cell cycle analysis was performed on the FlowJo cell cycle analysis platform using the Dean Jett Fox model.
Edu Analysis
Cells were labelled with 10 nmol Edu (Life Technologies, Paisley, UK) at either 24,26,28,30 or 32hrs post siRNA treatment. The cells were then grown until 48hrs post siRNA treatment when they were harvested and stained using Alexa Fluor 647 (Click-iT® EdU Alexa Fluor® 647 Flow Cytometry Assay Kit, Life Technologies, C10424) and PI following the manufacturer’s protocols.
Transcriptional Analysis
The cells were treated with siRNA and harvested as described above. The cell pellet was lysed and mRNA extracted using a RNAeasy Protect Mini Kit (Qiagen Ltd, Manchester, UK) following the manufacturer’s protocol. cDNA was made using a QuantiTech Reverse Transcription Kit (Qiagen Ltd, Manchester, UK) again following the protocol supplied by the manufacturer. The qPCR was then done using K-Ras, Cyclin D and GAPDH primers (Qiagen) using the Roche cyber green system on the Roche light cycler following the manufacturer’s instructions.
Statistics
All statistical analysis was performed using Statistical Package for the Social Sciences software (version 21.0 for Windows; SPSS, Inc, Chicago, Ill). Repeat measure data was tested using an independent sample T-test (significance quoted is 2-tailed).
3. Results
Western blotting confirmed that N-Ras, H-Ras and K-Ras were expressed in all of the pancreatic cancer cell lines tested (Figure 1a). All of the cell lines had active Ras when tested for activity using binding to Raf and a pan-Ras antibody (Figure 1a). BxPC3 cell line, which is a wild type K-Ras pancreatic cell line, appears to have the lowest levels. To test whether this activity was due to K-Ras alone each of the isoforms was specifically knocked down with siRNA; the K-Ras dependent cell line Suit-2 showed the most marked decrease in active Ras when K-Ras was knocked down. In the other cell lines any decline in active Ras following K-Ras depletion was marginal. The non-pancreatic cell line HeLa showed the greatest decline in Ras activity when N-Ras was specifically targeted (Figure 1a). When the specific activity of each Ras isotype was tested in the K-Ras dependent cell line (Suit-2) using specific K, H and N-Ras antibodies (Figure 1b), there was evidence of N as well as K-Ras activity, H-Ras activity was not seen, although this could reflect the sensitivity of the assay. Knockdown of K-Ras did not cause any noticeable change in N-Ras or appearance of detectable H-Ras activity, suggesting that the residual Ras activity seen in Suit-2 after K-Ras knockdown was largely the basal level of N-Ras activity. The K-Ras wild type cell line BxPC3 had substantial N-Ras activity, while N-Ras activity was also detectable (albeit at a relatively low level) in the K-Ras independent cell line Panc-1, which has the same p.G12D mutation as Suit-2 (Figure 1c). As neither of these cell lines is dependent on a K-Ras mutation, neither exhibits a loss of total Ras activity following K-Ras depletion (Figure 1a) and both have at least some N-Ras activity, it is reasonable to assume isotype agnostic Ras activity can support cell division in these cell lines. By contrast, the primary mouse pancreatic cancer cell line isolated from tumours arising in KrasLSL-G12D/+, p53R172H/+, Pdx1-Cre mice (KPC) mouse model[5] had little or no detectable N-Ras Raf binding activity (Figure 1c). The KPC mice express mutant Ras in progenitor cells of the pancreas, which differentiate into the cells of the mature pancreas while the human cell lines have spontaneous K-Ras mutations occurring (presumably) in differentiated cells. In both forms of tumorigenesis the mutation must be tolerated in pre-cancerous lesions. In the case of spontaneous mutations toleration can result from selection (most cells will die with the select few adapted or mutated to accept the change), while in the progenitor cells toleration requires an innate characteristic, as most cells must survive in order for the pancreas to form. On this basis, the innate toleration might not require expression of other (wild type) Ras isoforms, in contrast to a selected toleration.
Effect on Cyclins and the Cell Cycle of Knocking down Ras Species
siRNA-mediated depletion of K-Ras in Suit-2 cells resulted in reduction of the main G2 cyclins (A2 and B1) with comparatively little effect on G1 cyclins (D1 and E) (Figure 2a). Depletion of N-Ras, either alone or in combination with depletion of K-Ras, had little effect on the levels of any of the cyclins (Figure 2a).
Regulation of G2 cyclins is usually considered to be via the Anaphase Promoting Complex (APC/c)[32], although previous reports of EMT control via the G2 cyclin, A2, implied an alternative regulatory mechanism[33]. To see if inhibition of the APC/c complex could be responsible for the K-Ras mediated stabilisation of G2 cyclins, other targets of APC/c were analysed; the levels of both survivin and geminin exhibited an equivalent relationship with K and N-Ras depletion (Figure 2b). The same relationship between survivin and K-Ras depletion has been reported previously[34].
Activation of the APC/c complex following K-Ras depletion would be expected to drive cells out of G2. The cell cycle pattern following K-Ras depletion did not show a large decline in the G2 population, median for 4 replicates without K-Ras depletion was 19.78% (IQR 12.8, 23.74) while with K-Ras depletion it was 18.1% (IQR 16.7%, 19.7%). This difference is non-significant by the Mann-Whitney U-Test (P=0.686), the largest effect of K-Ras depletion was a decline in S-phase, which without K-Ras depletion was 14.3% (IQR 12.8%, 16.5%) while with K-Ras depletion it was 6.8% (IQR 6.7%, 11.7%), although even this difference did not reach significance on a Mann-Whitney U-Test (P=0.057) (Figure 3a). In 5-ethynyl-2’-deoxyuridine (EdU) labelling experiments, at the start of labelling all G2 cells will be unlabelled and only labelled cells will enter G2 from S-phase. At first unlabelled cells will enter G1 from G2 then after a period of time labelled and unlabelled cells will enter G1. There should be a decline in the proportion of cells in G2 that are unlabelled over time and the proportion of unlabelled cells in G2 should decline (as each unlabelled G2 cell becomes 2 unlabelled G1 cells) but this decline should slow over time as more of the G2 cells entering G1 become labelled. With the Suit-2 cell line we note that the majority of unlabelled G2 cells have entered G1 within 18 hours of labelling (Figure 3b and 3c), but the proportion of G2 cells that are unlabelled then remains fairly constant (Figure 3c), suggesting that while labelled cells can pass easily out of G2 at the same rate that they enter it, unlabelled cells transition much more slowly. As the total proportion of unlabelled cells that are in G2 is still declining (Figure 3d), there must also be even slower transition of unlabelled cells through S-phase than the transition from unlabelled cells from G2 into G1. With knockdown of K-Ras the proportion of unlabelled cells in G2 is much higher after 18 hours, confirming that the knockdown slows cell cycle progression and this includes the transition from G2 to G1. The cycle is slowed, but not stopped and the proportion of G2 cells that are unlabelled does gradually decline (Figure 3c). However, the proportion of unlabelled cells that are in G2 (Figure 3d) does not decline, if anything increasing, suggesting that more unlabelled cells are passing through S-phase than are entering G1 from G2. This strongly suggests that K-Ras depletion does not speed exit from G2, but slows exit from G2 relative to entry into S-phase. This indicates that the decline in G2 cyclins after K-Ras depletion is not associated with anaphase transition.
K-Ras Depletion Reduces G2 Cyclin Levels by a Proteasome Mediated Reduction in G2 Cyclin Transcription
The APC/c complex targets G2 cyclins to the proteasome via ubiquitination[32]. Therefore inhibition of the proteasome with Bortezomib should result in accumulation of APC/c targets and the increase in protein should be the result of an increase in ubiquitinated forms of the target. Consistent with this G2 cyclins levels are increased with Bortezomib treatment and this is most pronounced following K-Ras depletion; suggesting that the stabilisation of G2 cyclins by K-Ras is via reduction in protein degradation by the proteasome (Figure 4a). However, the level of ubiquitinated G2 cyclin decreased rather than increased after K-Ras depletion (Figure 4b).
If K-Ras depletion does not cause an increase in the proteasomal degradation of G2 cyclins, the question arises as to whether their regulation is at the transcriptional level. Quantitative Reverse Transcriptase Real Time PCR (qRT-PCR) of mRNA from G2 cyclins following K-Ras depletion shows that cyclin B1 was down regulated when K-Ras levels were depleted (Figure 5). This reduction could be explained via reduced G1 to S transition resulting from failure to up-regulate cyclin D1, however, cyclin D1 knockdown did not have as significant an impact on cyclin B1 transcription levels as K-Ras depletion. Reduction in cyclin A2 was less clear than for cyclin B1 and not significantly different from the reduction in cyclin A2 seen when cyclin D was depleted (Figure 5).
Role of Wild Type N-Ras in Cell Lines with Mutant K-Ras
N-Ras has been shown to be active in all the human cell lines used in this study (Figure 1). Previous studies have shown that depleting wild type N and H-Ras in K-Ras mutant cell lines reduces EGFR responsiveness [27]. Mutant K-Ras is known to reduce oxidative phosphorylation in cancer cells relative to glycolysis[35]; this was confirmed by metabolic analysis shown in Figure 6. Depletion of K-Ras increases oxygen consumption rate (OCR) relative to extra cellular acidification rate (ECAR). Although, N-Ras depletion alone has little effect, a slight increase in OCR/ECAR ratio when compared to cells treated with control siRNA, when both K and N-Ras are depleted the increase in OCR is significantly less than with depletion of K-Ras alone (Figure 6d). Further assessment of the data indicates that the increase in OCR/ECAR with K-Ras depletion seems to be due to an increase in maximum mitochondrial activity (Figure 6a) but with no increase in efficiency of respiration (Figure 6e), the OCR/ECAR increase at baseline being mainly due to a decrease in glycolysis rate (Figure 6b). In contrast N-Ras depletion causes a reduction in both maximal levels of mitochondrial activity (Figure 6a) and a decrease in the efficiency of respiration (Figure 6e). This suggests that mutant K-Ras reduces both maximal respiration and efficiency of respiration, compensated for in part by an increase in glycolysis, but also by a K-Ras dependent activity of N-Ras which increases efficiency of respiration and glycolysis.
4. Discussion
The role of activating mutations of K-Ras in tumorigenesis and in maintenance of cancer is far from clear. Our work confirms that K-Ras depletion reduces the level of APC/c targets (Figure 2)[34], which would fit with mutant K-Ras increasing G2 cyclins by inhibiting APC/c or just in general inhibiting proteasomes. This concept was initially supported by the observation that proteasome inhibition negates the decline in G2 cyclins following K-Ras depletion (Figure 4). Inhibition of the proteasome has previously been shown to increase the APC/c targets via an increase in the level of ubiquitinated protein[36]. However, with K-Ras depletion bortezomib treatment failed to deliver any increase in ubiquitination of the G2 cyclins; in contrast to the effect of bortezomib following cyclin D1 depletion (where the expected increase in ubiquitinated cyclin B1 was seen). An observed decline in cyclin B1 transcript levels following loss of K-Ras is evidence that a function of mutant K-Ras is to increase at least this cyclin at a transcriptional level (Figure 5). Transcriptional control of cyclin B1 by K-Ras must be dependent on preventing degradation of a protein or proteins by the proteasome (Bortezomib increases Cyclin B1 level but not ubiquitinated Cyclin B1 level after K-Ras depletion, Figure 4), but the affected proteins are as yet undefined.
The effect of mutant K-Ras increasing cyclins (G2 or otherwise) would be predicted to be to increase the rate of progression from G1, through S-phase and G2, but would if anything inhibit anaphase. Depletion of K-Ras would (in this purely cell cycle centric model) drive cells out of G2 and cause an accumulation of cells in G1.
During progression cells can become independent of K-Ras in respect of cell survival and cell division. Even Suit-2 (which was previously described as K-Ras dependent) actually doesn’t need the continued expression of the oncogene for survival although its growth rate is severely restricted following K-Ras depletion. K-Ras mutation does drive quiescent cells to divide via G1 cyclins, EGFR contributes to the increase in G1 cyclins and in early cancers it is essential for tumour progression but further mutations relieve this requirement[17]. Our data also shows that in Suit-2 cells cyclin D1 levels are unaffected by K-Ras depletion and so transcriptional activation of cyclin D1 by K-Ras is probably of little importance in an established tumour. K-Ras can promote EMT which is important for tumorigenesis, but again may not be important to maintain an established cancer and indeed our data supports a K-Ras activity that drives up G2 cyclins, which if anything would act to oppose EMT, given the previous reports of cyclin A2’s role in stabilising an epithelial phenotype[33]. Given the reduced growth rate in Suit-2 after K-Ras depletion it is reasonable to assume that it is K-Ras driven G2 cyclin production that supports maintenance of cancer cell division, but this is not linked to any inhibition of exit from G2 (as would be expected if cyclin levels were maintained at a high level) given that the relative level of exit from G2 compared to exit from G1 is actually decreased by knockdown of mutant K-Ras (Figure 3). It is convenient to think of two forms of G2 cyclin regulation, one form being cycle dependent, G2 cyclins rise in G2 because it is G2 phase and fall at anaphase (because it is anaphase) and another form of regulation which is independent of the cell cycle, in the sense that the G2 cyclins rise due to processes that in a normal cell occur in G2 and fall due to processes that occur at anaphase in a normal cell (but which do not in themselves define G2 or anaphase).
At some point in the history of all our human cancer cell lines N-Ras became active at a basal level. This N-Ras is presumably acting in a wild-type, cell cycle dependent, manner. N-Ras activity is not necessary for the increase in G2 cyclins or indeed other APC/c targets (Figure 2). N-Ras activity does impact on other functions of K-Ras such as inhibition of oxidative phosphorylation (Figure 6d). N-Ras depletion on its own has little effect on the OCR/ECAR ratio but when both K and N-Ras are lost the OCR/ECAR ratio is lower than when K-Ras alone is depleted. This can be explained by a K-Ras dependent activity of N-Ras which increases both glycolysis and respiration (see Figure 6). This suggests that during tumorigenesis the deleterious effects of K-Ras are offset by selection of cells which can produce wild-type N-Ras and that K-Ras mutation causes an N-Ras activity that compensates for the mutation. In a mouse tumour where mutant K-Ras is endogenously expressed alongside mutant p53, N-Ras basal activity does not appear to be necessary for tumour maintenance (Figure 1b). This may be due to the artificial nature of the genesis of the mouse derived tumour, which may not have progressed through all of the aberrations and alterations that are normally associated with the step wise tumour development seen naturally.
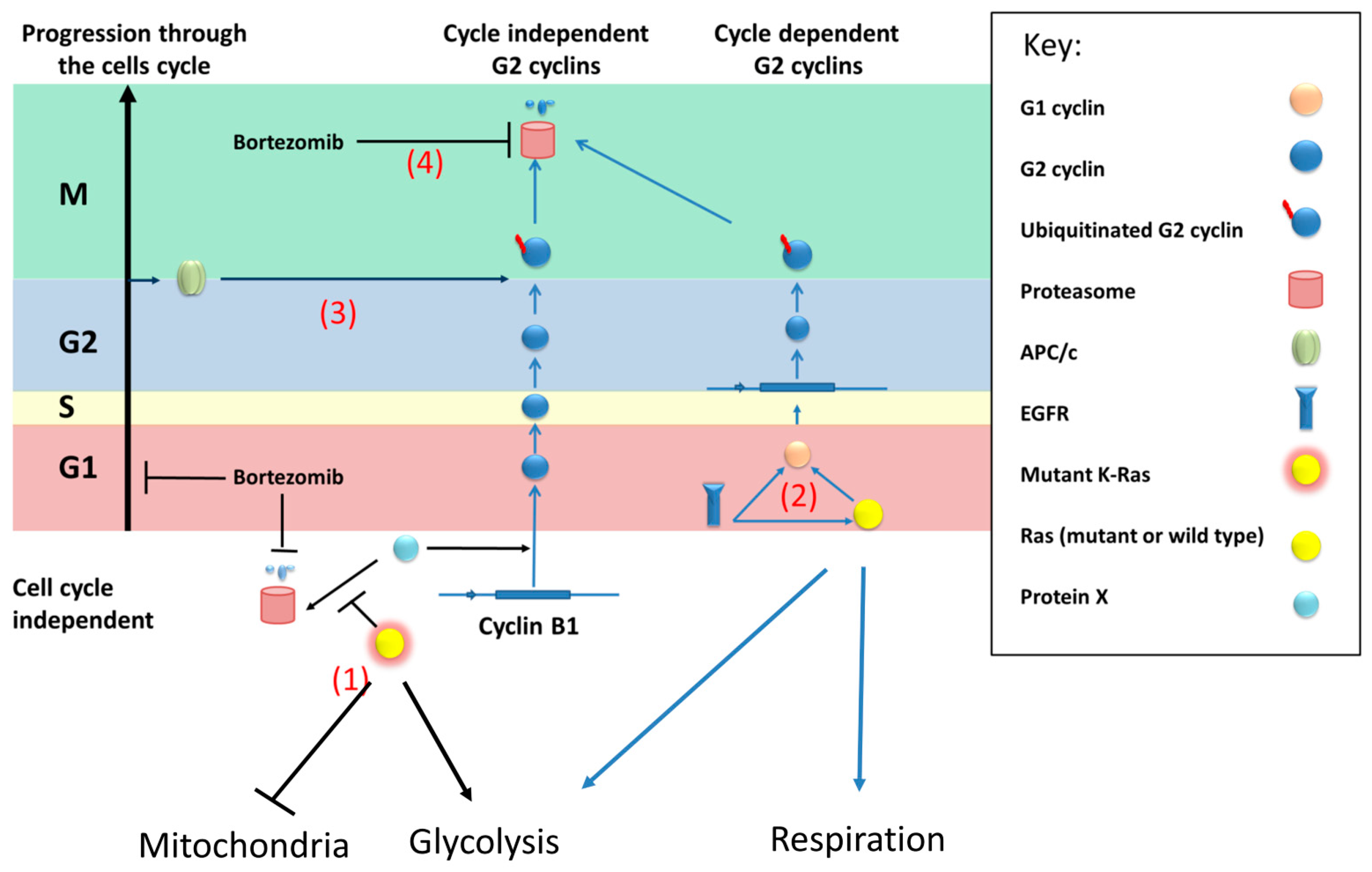
We therefore propose a model (Figure 7) where an unknown protein (referred to as protein X) activates transcription of cyclin B1 (and possibly other G2 cyclins) in a cell cycle independent fashion. This protein(s) is normally degraded by the proteasome but mutant K-Ras directly or indirectly prevents this degradation, for example by inhibiting its ubiquitination.
Given the importance of the tumor microenvironment in PDAC tumorigenesis and progression, it should be emphasized as a limitation that this current study is based solely on in vitro data of monocultured PDAC tumour cells.
5. Conclusions
In summary we have observed that mutant K-Ras promotes an increase in G2 cyclins that is not accompanied by any inhibition of exit from G2. This appears to be at least in part via a form of transcriptional regulation which is proteasome related. We have shown that activation of wild-type N-Ras is a common event in pancreatic cancer cell lines, which impacts on their metabolism. N-Ras acts in an antagonistic fashion in regulation of oxidative phosphorylation and N-Ras does not influence the transcriptional regulation of G2 cyclins.
Author Contributions
“Conceptualization, R.F. and W.G. methodology, R.F., KA., W.G., A.E.; formal analysis, R.F., A.E., W.G.; investigation, R.F., K.A., J.A., A.W., L.B., W.G.; resources, V.S., W.G.; data curation, R.F., W.G. writing—original draft preparation, R.F., A.E., W.G.; writing—review and editing, K.A., V.S., J.A., E.C.; supervision, K.A. V.S., E.C., W.G..; project administration, V.S., W.G.; funding acquisition, W.G. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research was funded by This work was funded by the European Union via the Framework 7 Canceralia consortium and Cancer Research UK and the NIHR which supported the Liverpool Experimental Cancer Medicine Centre (C18616/A25153).
Institutional Review Board Statement
“Not applicable” for studies not involving humans or animals.
Informed Consent Statement
Not applicable for studies not involving humans.
Data Availability Statement
Raw data available on request, processed data included in manuscript.
Acknowledgments
We would also like to acknowledge Professor Michael Schmid for the KPC mouse cell line.
Conflicts of Interest
The authors declare no conflict of interest.”
References
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin 2021;71:7-33.
- Sidaway P. Pancreatic cancer: TCGA data reveal a highly heterogeneous disease. Nat Rev Clin Oncol 2017;14:648. [CrossRef]
- Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia 2005;7:17-23. [CrossRef]
- Amato E, Molin MD, Mafficini A, Yu J, Malleo G, Rusev B, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol 2014;233:217-27. [CrossRef]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer cell 2003;4:437-50.
- Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res 2003;63:2005-9.
- Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res 2003;63:2016-9.
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer cell 2007;11:291-302. [CrossRef]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014;158:171-84. [CrossRef]
- Basu S, Greenwood J, Jones AW, Nurse P. Core control principles of the eukaryotic cell cycle. Nature 2022;607:381-6. [CrossRef]
- Sa G, Hitomi M, Harwalkar J, Stacey AW, Gc GC, Stacey DW. Ras is active throughout the cell cycle, but is able to induce cyclin D1 only during G2 phase. Cell Cycle 2002;1:50-8. [CrossRef]
- Liu J, Kang R, Tang D. The KRAS-G12C inhibitor: activity and resistance. Cancer Gene Ther 2021. [CrossRef]
- Muzumdar MD, Chen PY, Dorans KJ, Chung KM, Bhutkar A, Hong E, et al. Survival of pancreatic cancer cells lacking KRAS function. Nature communications 2017;8:1090. [CrossRef]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2019;179:1239. [CrossRef]
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with "K-Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer cell 2009;15:489-500. [CrossRef]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014;158:185-97. [CrossRef]
- Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer cell 2012;22:318-30. [CrossRef]
- Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer cell 2012;22:304-17. [CrossRef]
- Lampson BL, Kendall SD, Ancrile BB, Morrison MM, Shealy MJ, Barrientos KS, et al. Targeting eNOS in pancreatic cancer. Cancer Res 2012;72:4472-82. [CrossRef]
- Swidnicka-Siergiejko AK, Gomez-Chou SB, Cruz-Monserrate Z, Deng D, Liu Y, Huang H, et al. Chronic inflammation initiates multiple forms of K-Ras-independent mouse pancreatic cancer in the absence of TP53. Oncogene 2017;36:3149-58. [CrossRef]
- Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba, II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. Journal of the National Cancer Institute 2005;97:339-46. [CrossRef]
- Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757-65. [CrossRef]
- Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. Journal of cell science 2008;121:3853-7. [CrossRef]
- Kazi A, Chen L, Xiang S, Vangipurapu R, Yang H, Beato F, et al. Global Phosphoproteomics Reveal CDK Suppression as a Vulnerability to KRas Addiction in Pancreatic Cancer. Clin Cancer Res 2021;27:4012-24. [CrossRef]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399-405. [CrossRef]
- Omerovic J, Hammond DE, Clague MJ, Prior IA. Ras isoform abundance and signalling in human cancer cell lines. Oncogene 2008;27:2754-62. [CrossRef]
- Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov 2013;3:112-23. [CrossRef]
- Martinez-Useros J, Li W, Georgiev-Hristov T, Fernandez-Acenero MJ, Borrero-Palacios A, Perez N, et al. Clinical Implications of NRAS Overexpression in Resectable Pancreatic Adenocarcinoma Patients. Pathol Oncol Res 2019;25:269-78. [CrossRef]
- Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature 2008;452:646-9. [CrossRef]
- Ikenaga N, Ohuchida K, Mizumoto K, Akagawa S, Fujiwara K, Eguchi D, et al. Pancreatic cancer cells enhance the ability of collagen internalization during epithelial-mesenchymal transition. PLoS One 2012;7:e40434. [CrossRef]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009;324:1457-61. [CrossRef]
- Manchado E, Eguren M, Malumbres M. The anaphase-promoting complex/cyclosome (APC/C): cell-cycle-dependent and -independent functions. Biochem Soc Trans 2010;38:65-71. [CrossRef]
- Bendris N, Cheung CT, Leong HS, Lewis JD, Chambers AF, Blanchard JM, et al. Cyclin A2, a novel regulator of EMT. Cell Mol Life Sci 2014. [CrossRef]
- Tecleab A, Sebti SM. Depletion of K-Ras promotes proteasome degradation of survivin. Cell Cycle 2013;12:522-32. [CrossRef]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656-70. [CrossRef]
- Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nature cell biology 2006;8:700-10. [CrossRef]
Figure 1.
Total and active Ras isoforms in cancer cell lines a)Western blot analysis of total Ras in K-Ras mutation addicted (Suit-2) and independent cell lines (Panc1, BXPC3, HeLa). For each cell line upper panels show all Ras protein isotypes and the below panel active Ras isolated by pulldown with Raf, followed by panels of the total levels of each individual isotype. Cells were treated as shown with K-Ras siRNA or Non-targeting control as indicated (RF= RISC Free, NT = Non-Targeting, KD = Knock Down). K-Ras depletion only depletes active Ras in the mutant Ras dependent cell line Suit-2. b) Western blot analysis of Suit-2 showing the presence of N-Ras in upper panels and active N-Ras in the lower panel. Cells were treated as shown with K-Ras siRNA or Non-targeting control. c) Total Ras isoforms (left) or active Ras isolated by Raf pulldown (right). Cell line name is given above the blot.
Figure 1.
Total and active Ras isoforms in cancer cell lines a)Western blot analysis of total Ras in K-Ras mutation addicted (Suit-2) and independent cell lines (Panc1, BXPC3, HeLa). For each cell line upper panels show all Ras protein isotypes and the below panel active Ras isolated by pulldown with Raf, followed by panels of the total levels of each individual isotype. Cells were treated as shown with K-Ras siRNA or Non-targeting control as indicated (RF= RISC Free, NT = Non-Targeting, KD = Knock Down). K-Ras depletion only depletes active Ras in the mutant Ras dependent cell line Suit-2. b) Western blot analysis of Suit-2 showing the presence of N-Ras in upper panels and active N-Ras in the lower panel. Cells were treated as shown with K-Ras siRNA or Non-targeting control. c) Total Ras isoforms (left) or active Ras isolated by Raf pulldown (right). Cell line name is given above the blot.
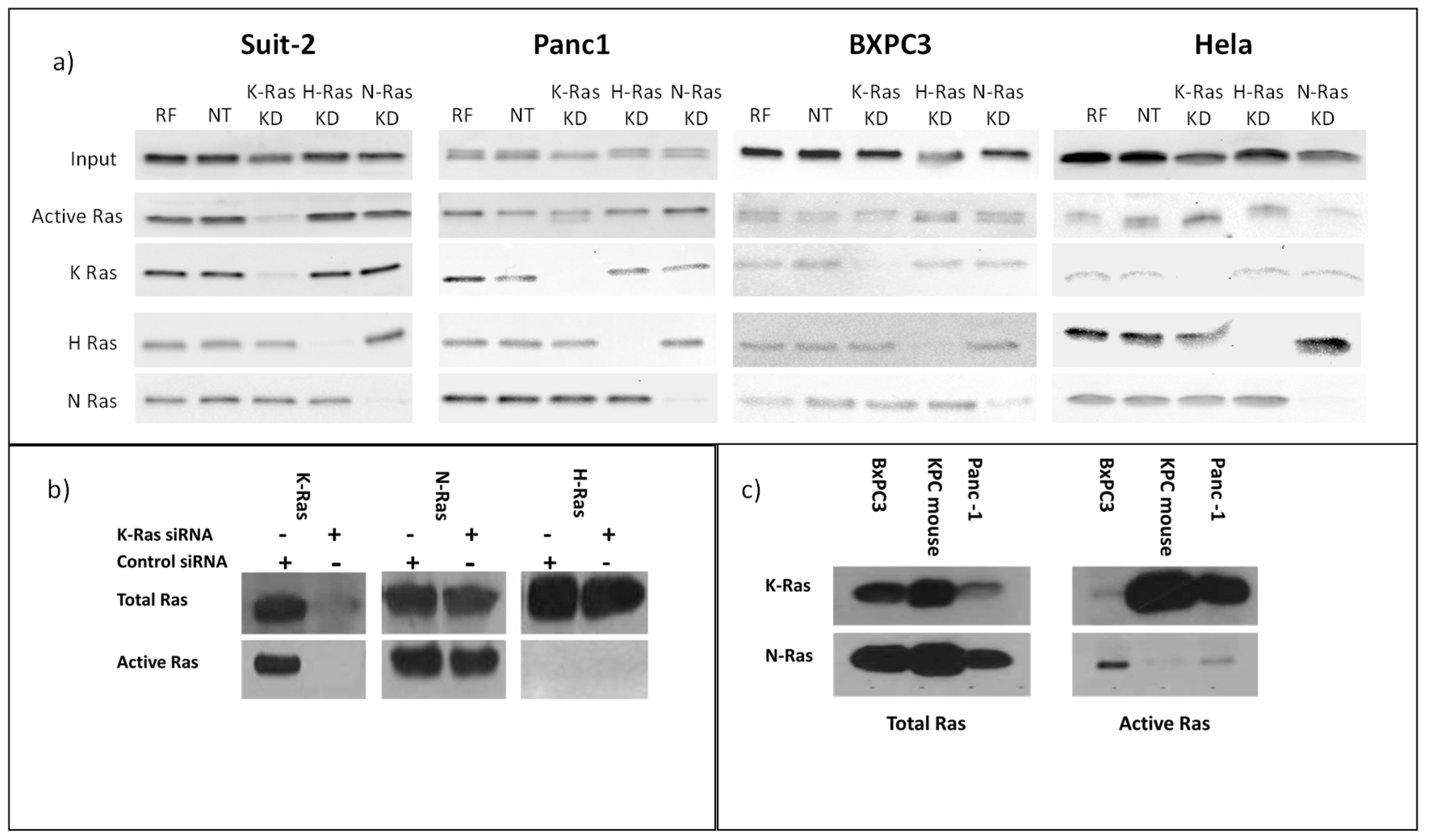
Figure 2.
Effect of K-Ras depletion on G2 cyclins and other APC/c targets. a) Western blot analysis of Suit-2cells treated with K-Ras, N-Ras or Non-targeting siRNA showing the levels of cyclins. b) Other targets of the APC/c complex and their levels following siRNA treatment.
Figure 2.
Effect of K-Ras depletion on G2 cyclins and other APC/c targets. a) Western blot analysis of Suit-2cells treated with K-Ras, N-Ras or Non-targeting siRNA showing the levels of cyclins. b) Other targets of the APC/c complex and their levels following siRNA treatment.
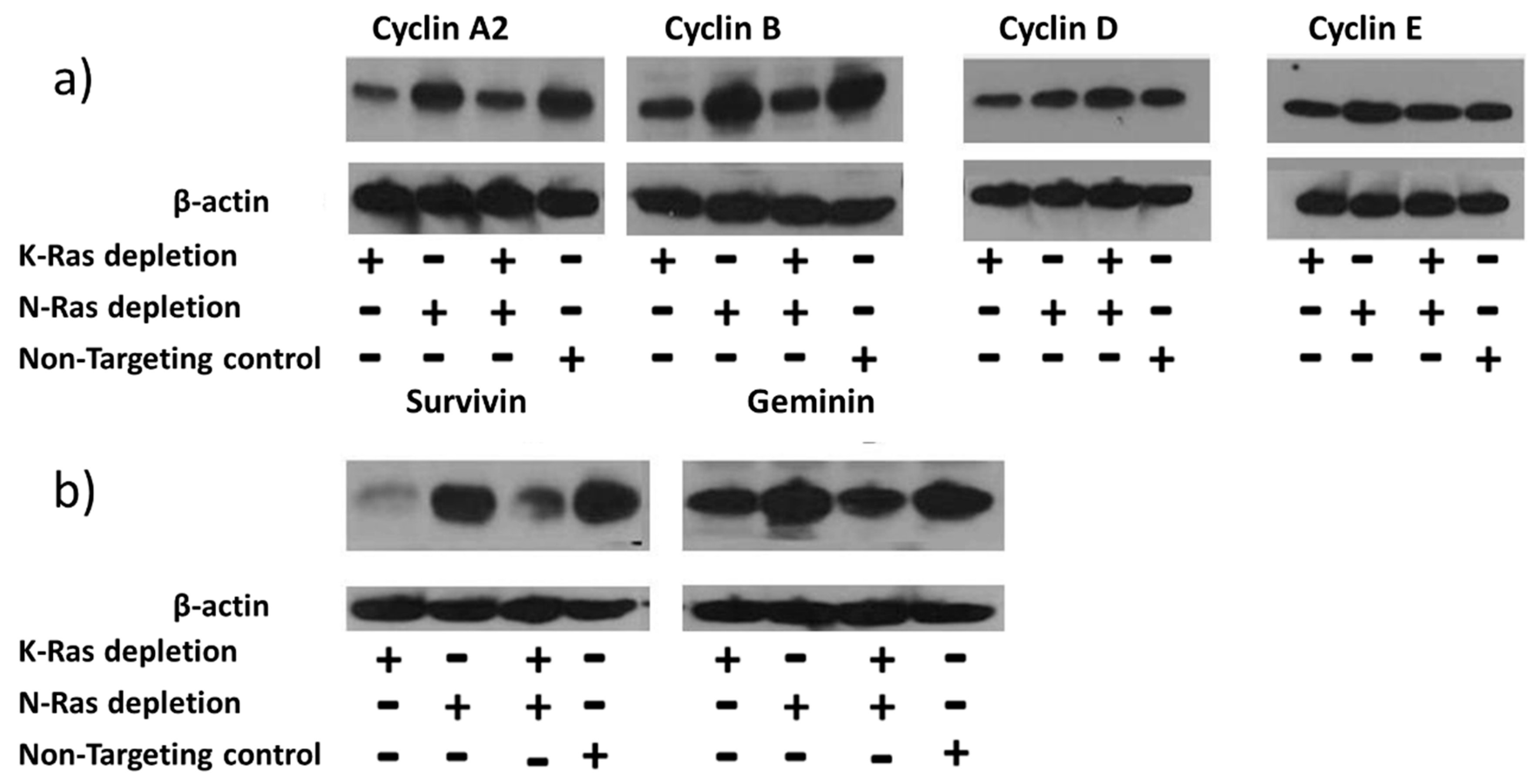
Figure 3.
Cell cycle analysis following K-Ras depletion. a) Representative FACS analysis of the cell cycle using PI staining showing reduced S-phase but little change in G2 level. b) An example of Edu analysis of the cell cycle showing that K-Ras depletion increases the percentage of unlabelled cells in G2. c-d) Graphs showing how unlabelled G2 cells change with length of Edu labelling. c) After 18 hours of labelling nearly 40% of K-Ras depleted G2 cells are still unlabelled, but this declines with labelling time as labelled cells enter from S-phase and unlabelled cells move towards G1. Without K-Ras depletion, the proportion of unlabelled cells seem to be stable (so any entry of cells from S-phase is compensated for by disproportionate exit of G2 of labelled cells compared to unlabelled). d) With K-Ras depletion the percentage of total unlabelled cells that are in G2 remains almost constant. Suggesting unlabelled cells are leaving G2 and entering G1 at a slower rate than unlabelled G1 cells are entering S-phase. Without K-Ras depletion the unlabelled cells from G2 accumulate in G1 over time, suggesting that cells are exiting G2 at an equivalent, or greater rate, than cells are leaving G1.
Figure 3.
Cell cycle analysis following K-Ras depletion. a) Representative FACS analysis of the cell cycle using PI staining showing reduced S-phase but little change in G2 level. b) An example of Edu analysis of the cell cycle showing that K-Ras depletion increases the percentage of unlabelled cells in G2. c-d) Graphs showing how unlabelled G2 cells change with length of Edu labelling. c) After 18 hours of labelling nearly 40% of K-Ras depleted G2 cells are still unlabelled, but this declines with labelling time as labelled cells enter from S-phase and unlabelled cells move towards G1. Without K-Ras depletion, the proportion of unlabelled cells seem to be stable (so any entry of cells from S-phase is compensated for by disproportionate exit of G2 of labelled cells compared to unlabelled). d) With K-Ras depletion the percentage of total unlabelled cells that are in G2 remains almost constant. Suggesting unlabelled cells are leaving G2 and entering G1 at a slower rate than unlabelled G1 cells are entering S-phase. Without K-Ras depletion the unlabelled cells from G2 accumulate in G1 over time, suggesting that cells are exiting G2 at an equivalent, or greater rate, than cells are leaving G1.
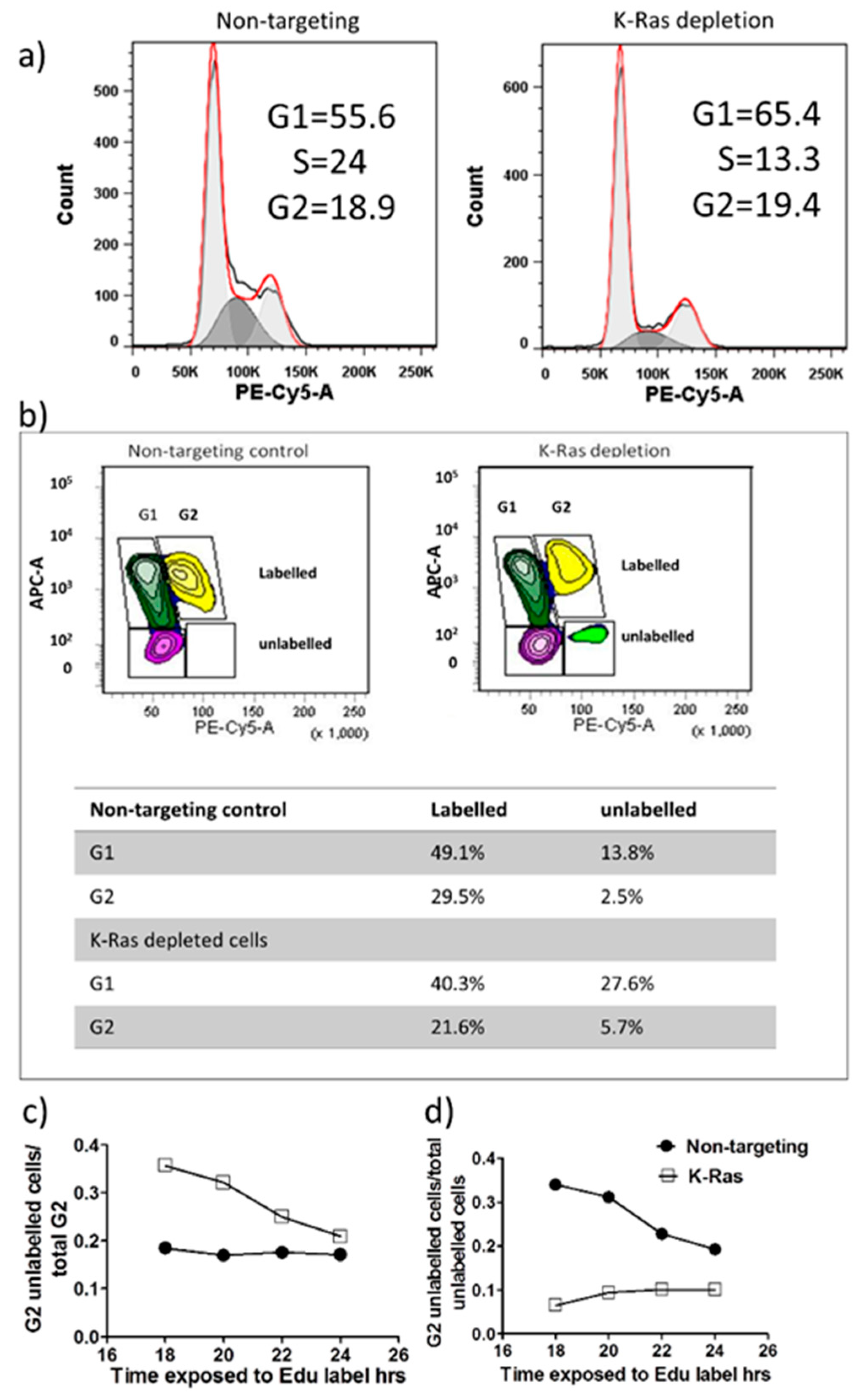
Figure 4.
Effect of K-Ras depletion on G2 cyclins with or without Bortezomib treatment. a) Western blot showing the effect of K-Ras depletion on total cyclin B in Suit-2 cells and how bortezomib treatment alters the effect. b) Western blot showing the effect of bortezomib and siRNA in combination. K-Ras depletion decreases ubiquitinated cyclin B and bortezomib treatment decreases it further. Cyclin D depletion also decreases ubiquitinated cyclin B, but bortezomib treatment restores levels.
Figure 4.
Effect of K-Ras depletion on G2 cyclins with or without Bortezomib treatment. a) Western blot showing the effect of K-Ras depletion on total cyclin B in Suit-2 cells and how bortezomib treatment alters the effect. b) Western blot showing the effect of bortezomib and siRNA in combination. K-Ras depletion decreases ubiquitinated cyclin B and bortezomib treatment decreases it further. Cyclin D depletion also decreases ubiquitinated cyclin B, but bortezomib treatment restores levels.
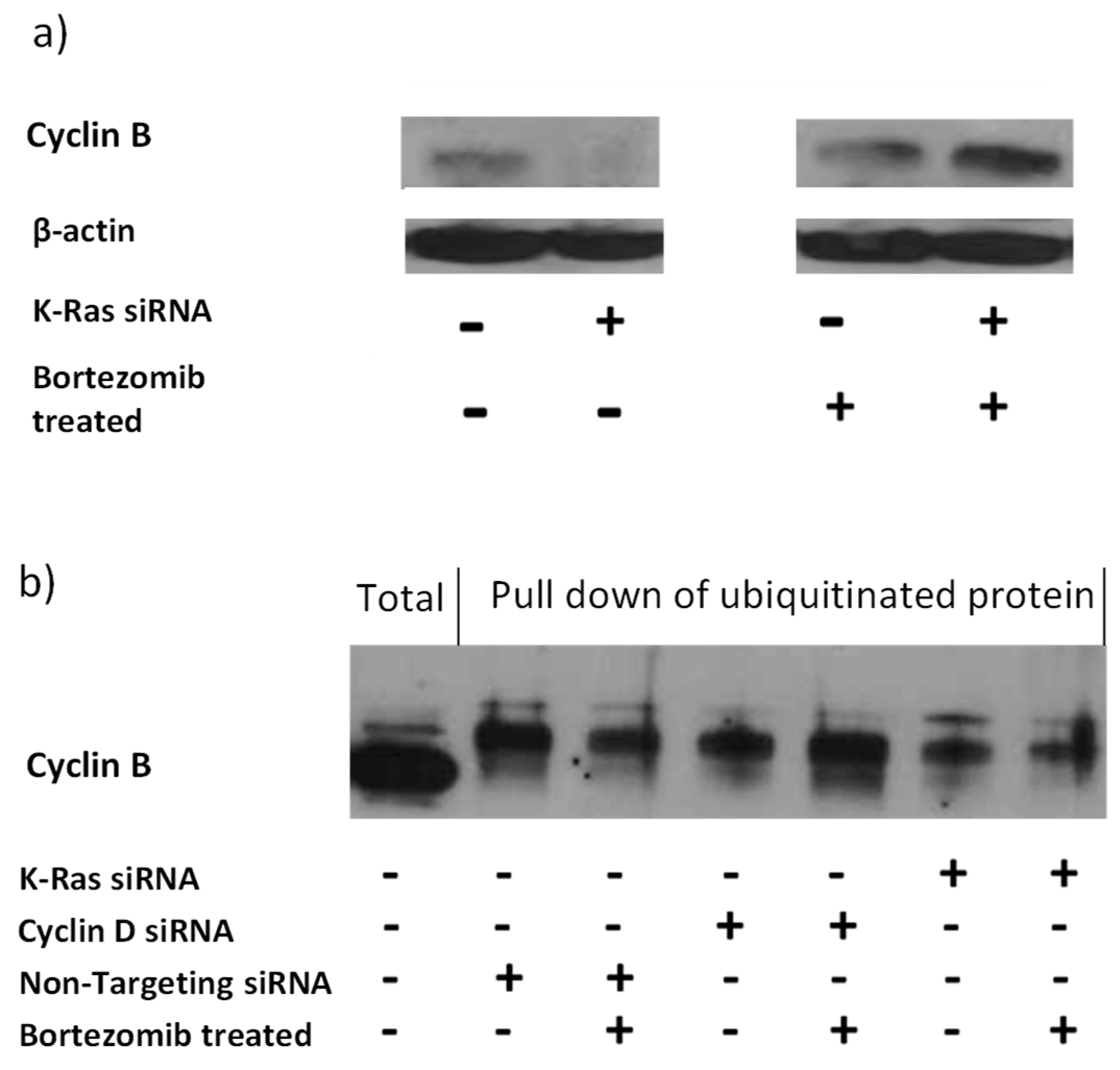
Figure 5.
Effect of K-Ras depletion on G2 cyclin transcription. Graph showing an example of the decrease observed in G2 cyclin transcript when K-Ras or cyclin D is depleted. Cyclin B transcript levels after depletion with K-Ras are significantly lower than in non-targeting controls and after depletion with cyclin D (P < 0.001, Mann Whitney U test, standard deviation used as error bar). Cyclin A levels are also significantly lower after depletion of K-Ras than with no depletion but there was an equivalent decrease after cyclin D depletion.
Figure 5.
Effect of K-Ras depletion on G2 cyclin transcription. Graph showing an example of the decrease observed in G2 cyclin transcript when K-Ras or cyclin D is depleted. Cyclin B transcript levels after depletion with K-Ras are significantly lower than in non-targeting controls and after depletion with cyclin D (P < 0.001, Mann Whitney U test, standard deviation used as error bar). Cyclin A levels are also significantly lower after depletion of K-Ras than with no depletion but there was an equivalent decrease after cyclin D depletion.
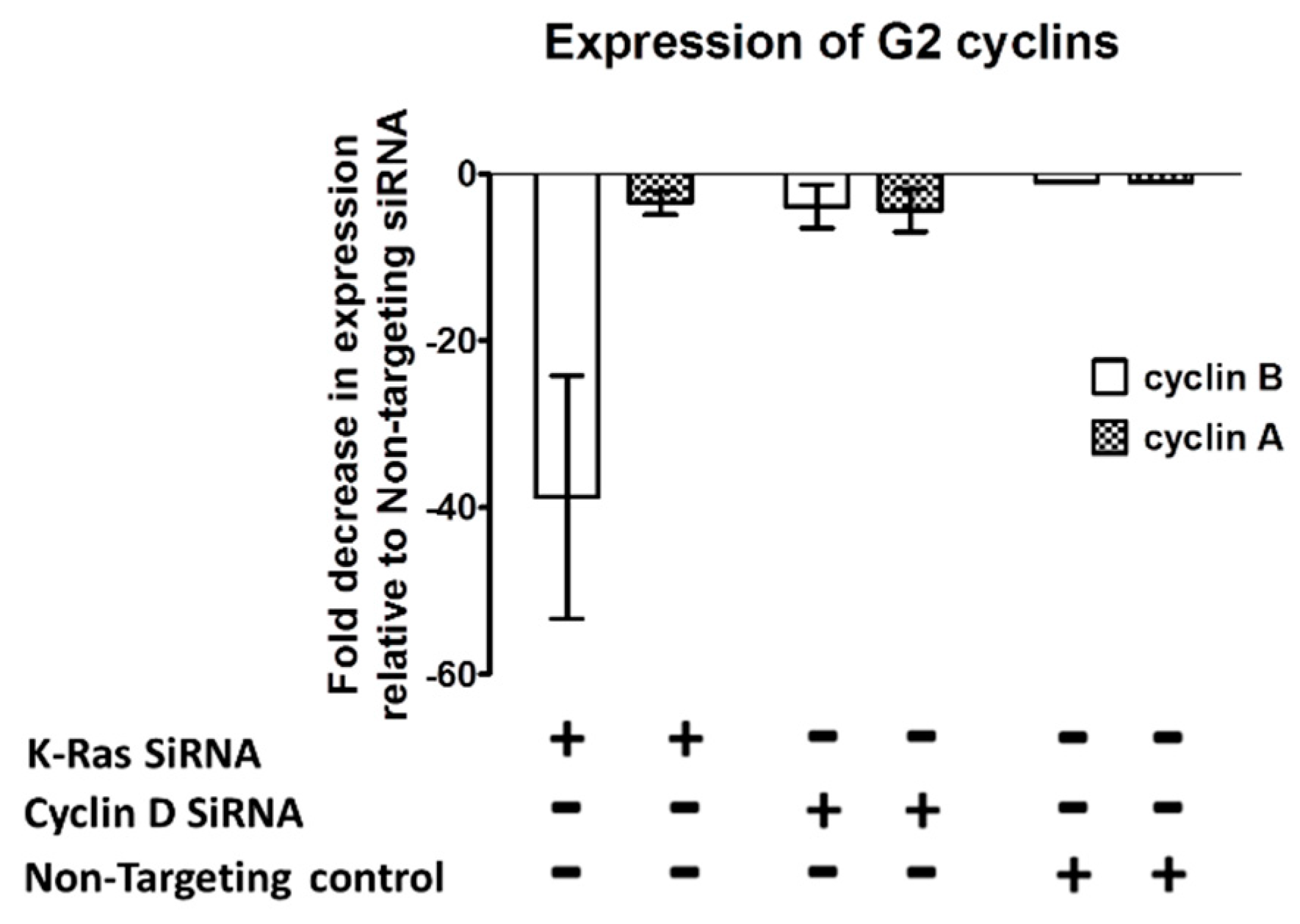
Figure 6.
Effects of K-Ras and N-Ras depletion on oxidative phosphorylation. a) Representative Sea Horse analysis showing oxygen consumption rate (OCR) measured over time with the treatments shown. Oligomycin reducing oxygen consumption to just the result of proton leak and FCCP uncoupling mitochondria to give maximal oxygen consumption. K-Ras knockdown caused an increase in uncoupled oxygen consumption while N-Ras knockdown caused a reduction in maximal and basal oxygen consumption as well as a decrease in proton leak from mitochondria. Knockdown of N-Ras with K-Ras restores basal oxygen consumption with a smaller increase in maximal oxygen consumption and no reduction in proton leak. b) The same experiment but showing Extra Cellular Acidification Rate (ECAR). K-Ras knockdown causes a reduction in ECAR, N-Ras knockdown alone causes a smaller ECAR reduction which is equivalent to the ECAR reduction seen when N-Ras and K-Ras are knocked down. c) OCR/ECR is a measure of relative levels of respiration and glycolysis. K-Ras knockdown causes an increase in relative levels of respiration, repeated experiments showed this to be significantly greater than the increase seen with both K-Ras and N-Ras knockdown. N-Ras knockdown alone had little overall effect (d). e) When OCR/ECAR is adjusted to the maximum increase in active mitochondrial activity (the difference from maximal to proton leak oxygen consumption) N-Ras knockdown was shown to reduce mitochondrial efficiency (Mann-Whitney U test, P < 0.001, standard deviation used as error bar), K-Ras knockdown had less effect on efficiency and combining K-Ras with N-Ras knockdown restored mitochondrial efficiency.
Figure 6.
Effects of K-Ras and N-Ras depletion on oxidative phosphorylation. a) Representative Sea Horse analysis showing oxygen consumption rate (OCR) measured over time with the treatments shown. Oligomycin reducing oxygen consumption to just the result of proton leak and FCCP uncoupling mitochondria to give maximal oxygen consumption. K-Ras knockdown caused an increase in uncoupled oxygen consumption while N-Ras knockdown caused a reduction in maximal and basal oxygen consumption as well as a decrease in proton leak from mitochondria. Knockdown of N-Ras with K-Ras restores basal oxygen consumption with a smaller increase in maximal oxygen consumption and no reduction in proton leak. b) The same experiment but showing Extra Cellular Acidification Rate (ECAR). K-Ras knockdown causes a reduction in ECAR, N-Ras knockdown alone causes a smaller ECAR reduction which is equivalent to the ECAR reduction seen when N-Ras and K-Ras are knocked down. c) OCR/ECR is a measure of relative levels of respiration and glycolysis. K-Ras knockdown causes an increase in relative levels of respiration, repeated experiments showed this to be significantly greater than the increase seen with both K-Ras and N-Ras knockdown. N-Ras knockdown alone had little overall effect (d). e) When OCR/ECAR is adjusted to the maximum increase in active mitochondrial activity (the difference from maximal to proton leak oxygen consumption) N-Ras knockdown was shown to reduce mitochondrial efficiency (Mann-Whitney U test, P < 0.001, standard deviation used as error bar), K-Ras knockdown had less effect on efficiency and combining K-Ras with N-Ras knockdown restored mitochondrial efficiency.
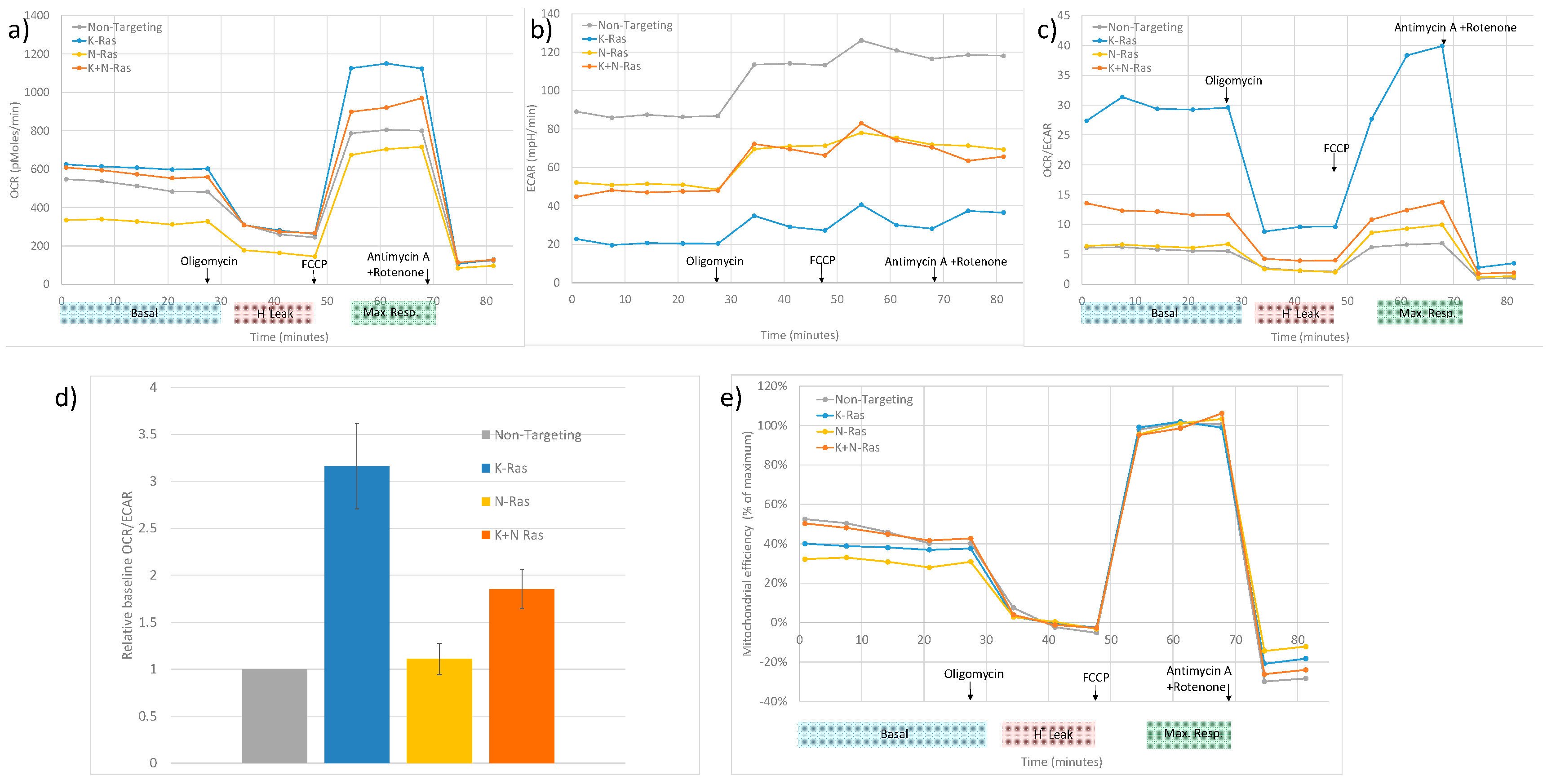
Figure 7.
Model of a compensatory mechanism of wild type Ras expression in cells with mutant K-Ras. (1) Mutant K-Ras causes reduced mitochondrial activity (possibly by reducing the number of mitochondria) and promotes production of G2 cyclins by inhibition of the proteolysis of an unknown transcriptional activator (x) of cyclin B1. (2) Wild-type (or mutant) Ras will increase transcription of G2 cyclins via G1 cyclins in a cell cycle dependent fashion. EGFR will activate this process but can also increase G1 cyclins by directly binding to their promoters compensating for the non-cyclic levels linked to the mutant protein. Furthermore, based on results from N-Ras knockdown, wild type (N-Ras) increases glycolysis and the efficiency of respiration. (3) G2 cyclins will be degraded via the APC/c complex and so in the absence of mutant Ras the level of the G2 cyclins will be limited by the proportion of cells in G2. (4) Bortezomib will inhibit the proteasome causing stabilisation of ubiquitinated G2 cyclins, but will reduce the production of ubiquitinated protein by restricting entry to (and so exit from) G2.
Figure 7.
Model of a compensatory mechanism of wild type Ras expression in cells with mutant K-Ras. (1) Mutant K-Ras causes reduced mitochondrial activity (possibly by reducing the number of mitochondria) and promotes production of G2 cyclins by inhibition of the proteolysis of an unknown transcriptional activator (x) of cyclin B1. (2) Wild-type (or mutant) Ras will increase transcription of G2 cyclins via G1 cyclins in a cell cycle dependent fashion. EGFR will activate this process but can also increase G1 cyclins by directly binding to their promoters compensating for the non-cyclic levels linked to the mutant protein. Furthermore, based on results from N-Ras knockdown, wild type (N-Ras) increases glycolysis and the efficiency of respiration. (3) G2 cyclins will be degraded via the APC/c complex and so in the absence of mutant Ras the level of the G2 cyclins will be limited by the proportion of cells in G2. (4) Bortezomib will inhibit the proteasome causing stabilisation of ubiquitinated G2 cyclins, but will reduce the production of ubiquitinated protein by restricting entry to (and so exit from) G2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



