
Submitted:
11 March 2023
Posted:
14 March 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Abstract: The SARS-CoV-2 pandemic has significantly impacted world health and economic status. In response, much work has been undertaken to provide effective, safe vaccines, antibodies and antiviral drugs with which to address this pandemic. Treatment of a pandemic population presents multiple challenges in addition to the primary issue of drug efficacy and safety, such as large scale drug manufacture and distribution, drug stability, oral dosing and pharmacoeconomic considerations. Ideally, these factors must be addressed if new candidate drugs are to be advanced for treatment of large (pandemic) populations. Subsequently, new antivirals have reached the market but choices are few. According to the NIH Covid Treatment Guidelines, only three small molecule antiviral drugs are available to treat COVID-19 disease. As such, a significant part of the research towards discovery of new antiviral drugs has focused on screening and evaluation of ‘repurposed drugs’ or previously approved or clinical stage drugs. Yet, in spite of this increased research activity, one promising clinical stage candidate drug has received little attention regarding its potential as a monotherapy or component of combination therapy for treatment of COVID-19 disease. Tucaresol, with documented human safety and pharmacokinetic data, is an orally active, stable, small molecule amenable to large scale manufacture by a proprietary two-step synthesis developed by us. Tucaresol functions as a host-targeted antiviral by selective protection/reconstitution of CD4+ T helper cells as demonstrated in HIV patients and SIV macaques. In view of similarities between HIV and SARS-CoV-2, especially with respect to host CD4+ T helper cells, and the suitability of Tucaresol for facile treatment of pandemic populations, Tucaresol is presented herein for treatment of mild-to-moderate COVID-19 patients but may also be useful for treatment of advanced disease accompanied by lymphopenia.
Keywords:
Tucaresol
; COVID-19
; SARS-CoV-2
; HIV
; T helper cell
; CD4 receptor
Background
With 675 million cases of COVID-19 world-wide, SARS-CoV-2 has significantly impacted the health and economic status of the world population. Although successful as regards significant protection to the general population against COVID-19 disease, vaccines are not 100% protective or a cure. This vaccine induced protection is further eroded by multiple viral mutations competing to evade vaccine induced immune defense (typical of cold viruses) along with poor vaccine compliance, apparently more so with the mRNA vaccines. Sought after herd immunity is unlikely, and COVID-19 will probably become endemic with the need for annual vaccination. Indeed, the CEO of Pfizer, a significant provider of COVID-19 mRNA vaccine (Comirnaty) and antiviral drug (Paxlovid), recently opined that there is a threat of “constant waves of COVID-19” [1]. Additionally, large scale immunization associated with a pandemic is hampered by practical considerations such as the need to refrigerate most vaccines, the need to wait days to achieve protective immunity and the need to provide one or more booster doses of vaccine in order to achieve protective immunity. Monoclonal antibodies also improve immune defense but can be rendered ineffective by viral mutations, as has been the case with the recent omicron coronavirus variant. In fact, clinical monoclonal antibodies are ineffective against the most recent omicron BQ and XBB sub variants, including the most transmissible yet XBB. 1.5 sub variant. Antibody therapy is not conveniently administered, is expensive and is therefore not practical for widespread use. Therefore, in view of the widespread, pandemic occurrence of the mutating SARS-CoV-2 virus, much effort is in progress to bring forward as quickly as possible orally active (convenient dosing), readily manufactured antiviral drugs (accessible, stable and affordable to treat large populations) with significant efficacy and good safety profiles. In spite of best efforts to rapidly develop small molecule antiviral drugs, according to the NIH Covid Treatment Guidelines, last up-dated April 29, 2022, only three small molecule antiviral drugs are available for treatment of COVID-19 disease: Veklury, Paxlovid and Lagevrio. Of these three drugs, only Veklury is approved by the US FDA for treatment of COVID-19 disease.
Veklury (remdesivir, Gilead) is a broad-spectrum antiviral nucleoside monophosphate prodrug that inhibits viral RNA chain elongation. When administered within 5 days of symptom onset, it reduces the risk of hospitalization of patients with mild-to-moderate COVID-19 disease by 90% [2]. Veklury was first approved for the treatment of COVID-19 in May 2020 for patients requiring hospitalization. It was later approved in January 2022 for treatment of mild-to-moderate COVID-19 (i.e., non-hospitalized patients). Notably, however, Veklury does not offer the convenience of oral dosing and is significantly more expensive than the two other FDA supported antivirals.
On December 22, 2021, the FDA issued an EUA (Emergency Use Authorization; not an FDA approval) for Paxlovid (nirmatrelvir and ritonavir tablets, Pfizer) as the first oral antiviral drug for treatment of mild-to-moderate COVID-19 disease in infected patients at high risk for progression to severe COVID-19 disease. Nirmatrelvir is a protease inhibitor that inhibits the main (3C-like) protease of SARS-CoV-2 and subsequent viral replication. Ritonavir retards catabolism of nirmatrelvir by inhibition of cytochrome P450 3A4. When orally administered within 5 days of symptom onset, it reduces risk of hospitalization or death by 88% [3]. However, due to the need for ritonavir to extend the half-life of nirmatrelvir, Paxlovid must be prescribed with caution for individuals requiring multiple prescription drugs since ritonavir may interfere with their metabolism by cytochrome P450, which could subsequently result in a too high concentration of the co-prescribed drugs. Additionally, it was recently reported that patients who recover from COVID-19 after treatment with Paxlovid might experience a return of symptoms or test positive again for COVID-19 even after previously testing negative for the virus [4]. These rebounds have occurred 2 to 8 days after initial recovery. According to the US CDC, there is no evidence that additional treatment with Paxlovid is required for rebound cases but the CDC recommends patients re-isolate for at least 5 days (viral rebound is not limited to Paxlovid and has been reported, for example, to occur in HIV patients treated with indinavir monotherapy or HAART triple therapy [5]). A phase 2/3 clinical trial failed to achieve statistical significance in a post-exposure protection protocol wherein adults were prophylactically administered Paxlovid and exposed to virus through someone they live with, such as a family member [6].
Only one day after Paxlovid’s approval, the FDA issued an EUA for Lagevrio (molnupiravir capsules, Merck) as the second oral antiviral drug for the treatment of mild-to-moderate COVID-19 disease in infected patients at risk for progression to severe COVID-19 disease when other authorized treatment options are not available or clinically appropriate. Lagevrio is the prodrug of an older nucleoside antiviral drug, β-d-N4-hydroxycytidine, that introduces errors (mutations) into the replicating virus RNA chain as a result of incorrect base pairing. However, β-d-N4-hydroxycytidine is also mutagenic to mammalian cells, and based on findings from animal reproduction studies Lagevrio is therefore not recommended for use during pregnancy [7,8]. When orally administered within 5 days of symptom onset, Lagevrio reduces the risk of hospitalization by 30% [9].
Crucially, the clinical effectiveness of these three antiviral drugs is predicated upon a relatively brief period of time (i.e., a maximum of 5 days between the appearance of symptoms and commencement of antiviral treatment). And although starting antiviral therapy as soon as possible after infection is typical for acute viral infections (e.g., the recommendation to administer Tamiflu antiviral within 2 days upon appearance of symptom(s) of viral influenza), the particular challenge for successful treatment with these COVID-19 antivirals is noteworthy: since (i) the first disease symptom(s) may be indistinguishable from a cold or allergies, which can result in a delay for seeking treatment, and (ii) a drug prescription should be preceded by a positive COVID-19 test(s), further constraining the narrow time frame. However, in order to gain easier access to the two antiviral drugs in the US, the FDA just removed the requirement, February 2023, that individuals test positive in order to qualify for Paxlovid or Lagevrio.
The NIH Covid treatment guidelines that recommended the three antivirals for treatment of COVID-19 disease also recommended against some candidate antivirals where the clinical trial results were ambiguous for multiple reasons, including a too small patient sample or too little information regarding the novel SARS-CoV-2 virus upon which to guide the best choice of therapy and/or trial design. In order to address these issues, some potential therapeutics have been tested multiple times. For example, more than 150 hydroxychloroquine trials have been undertaken [10] with the outcome being a recommendation against the use of hydroxychloroquine, chloroquine, azithromycin, ivermectin, nitazoxanide, lopinavir/ritonavir or other HIV protease inhibitors. Not mentioned in the NIH guidelines is the pyrazine antiviral favipiravir, which had the potential for being a promising candidate given that it is active against RNA (e.g., human influenza) viruses, but it demonstrated ambiguous clinical activity against SARS-CoV-2 [11]. Indeed, the challenges regarding the discovery/development of an antiviral drug compared to an antibiotic or antifungal drug help explain why only a few SARS-CoV-2 antivirals have successfully navigated clinical trials [12].
Antiviral chemotherapy has routinely experienced slow development as a consequence of the unique properties of a virus. A virus cannot exist outside of the cells it will infect since it cannot reproduce by itself, but once inside the target cell the infected cell can produce thousands of copies of the virus. Viruses offer significantly fewer druggable (biochemical) targets for development of selective antivirals that do not target host functional proteins and subsequently are not too toxic. Therefore, the main thrust of SARS-CoV-2 drug development has been the evaluation of ‘repurposed drugs’ that were previously approved either for treatment of viral infections other than SARS-CoV-2 or (and perhaps less ideally) those approved for other diseases. Drug repurposing can be cost effective because it can significantly reduce the time required for drug approval by: (i) reducing the time required for drug safety assessment, (ii) increasing the likelihood of an effective trial design that demonstrates drug efficacy and (iii) facilitating the procurement of requisite investment capital with advanced knowledge of both drug toxicity and (to a lesser extent) drug efficacy.
Efforts directed towards the discovery of SARS-CoV-2 antivirals have included evaluation of libraries of FDA approved drugs, as exemplified by the evaluation of a library of 12,000 FDA approved or clinical stage (candidate) drugs [13]. Screening of this large library identified 100 inhibitors of viral replication that included 21 known drugs. More focused drug repurposing screens included a virtual screen of 6,218 FDA approved or clinical stage (candidate) drugs [14], wherein the compounds were screened against SARS-CoV-2 main protease and RNA-dependent RNA polymerase. This focused screening resulted in 15 and 23 potential repurposed drugs, respectively, of which three exhibited activity in human lung (Calu-3) cells. Interestingly, omipalisib, an FDA approved cancer (PI3K/mTOR inhibitor) drug exhibited 200 fold higher activity than remdesivir in Calu-3 cells. Pertinent to this article is the observation that three drug combinations displayed “strong synergistic effects in inhibiting SARS-CoV-2” [15], thereby improving antiviral efficacy and reducing the risk of toxicity of each component of the drug combination. Multiple examples of compound screens in various cell-based or virtual assays have been published since last year. A mention of one compound class, polyphenolic plant compounds, merits note since multiple members of this class have been or are under clinical evaluation as treatments for COVID-19 disease [16].
Polyphenolic compounds, polyphenols, are plant secondary metabolites of diverse structure and molecular weight. They contain at least 2 phenol moieties that impart antioxidant properties along with the associated health benefits. In fact, these antioxidant properties, and subsequently documented anti-inflammatory and antiviral properties, attracted interest regarding their potential therapeutic activity against SARS-CoV-2 as noted elsewhere [17], where the authors list 30 clinical trials regarding pure polyphenols and polyphenolic extracts registered in the US National Library of Medicine. Similar to repurposed drugs approved for human use, many of the polyphenols are available for human use as nutraceuticals or supplements. Polyphenol clinical trials include the red wine antioxidant resveratrol, tannic acid and tannins, the flavones quercetin and hesperidin and the major constituent of turmeric, curcumin. The generally observed clinical improvement was restoration of the pro/anti-inflammatory (Th1/Th2) balance as supported by an improved cytokine profile. Additionally, multiple polyphenols have been shown to potentially bind to SARS-CoV-2 protein targets such as the main protease by virtual (e.g., computer docking) studies or in-vitro enzyme inhibition assays (e.g., IC50; main and papain-like protease of SARS-CoV-2).
In summary, screening of thousands of repurposed drugs that were previously approved for treatment of viral infections other than SARS-CoV-2 or treatment of other diseases have identified promising compounds but it remains that only three small molecule antiviral drugs are available for treatment of COVID-19 disease. Of course, with completion of ongoing COVID-19 clinical trials of repurposed drugs, along with continued evaluation of clinical stage candidate drugs, a few more orally active antiviral drugs may become available. However, the three available antivirals have issues that demonstrate a need and opportunity for improved (i.e., second generation) orally active drugs for treatment of COVID-19 disease.
Possibly inspired by the success of HIV protease inhibitors, of which eleven drugs are FDA approved, multiple SARS-CoV-2 main protease inhibitors are under development. One protease inhibitor candidate drug with FDA fast track designation, EDP-235 (Enanta Pharmaceuticals), is currently in a phase 2 clinical trial. Unlike Paxlovid and Molnupiravir, which require twice-daily doses, EDP-235 is targeted as a once-daily oral drug that does not require co-administration of Ritonavir [18]. Interestingly, Glecaprevir, a component of the hepatitis C drug Mavyret (identified jointly by Enanta and AbbVie), is a high affinity macrocyclic inhibitor of SARS-CoV-2 main protease [19]. As such, is EDP-235 repurposed macrocyclic Glecaprevir that is being developed as an antiviral drug against SARS-CoV-2? This is not unreasonable, since Enanta has two issued compound ownership (COM) US patents (8,648,037; and 9,220,748). More likely EDP-235 is a macrocyclic spiropyrrolidine derived structure cited in US patent 11,319,325 and assigned last year to Enanta for treatment of coronavirus infection. A confounding factor regarding little information about the structure of EDP-235 may be a patent infringement suit concerning Paxlovid, filed by Enanta June 21, 2022 against Pfizer and prompted by Enanta’s US patent 11,358,953, issued June 14, 2022. Ensitrelvir (S-217622, Shionogi & Co.) is another SARS-CoV-2 main protease inhibitor that is centered about a 1,3,5-triazinane scaffold. It is currently in a phase 2b/3 clinical trial. In the prior phase 2a trial, Ensitrelvir was orally administered once daily for 5 days, resulting in rapid clearance of the virus. The drug was well tolerated in patients with mild-to-moderate COVID-19 disease. However, birth defects were observed during animal safety studies with doses of Ensitrelvir that were higher than used in the clinical trial [20]. Based on animal safety studies, Molnupiravir poses similar birth defect risks and is not recommended for use during pregnancy. Contingent upon successful completion of clinical trials, a similar limitation for use during pregnancy may be placed upon Ensitrelvir. PBI-0451 (Pardes Biosciences) is an acyclic peptide mimetic main protease inhibitor with an electrophilic cyano group mimic of the substrate peptide scissile bond. PBI-0451 recently successfully completed a phase 1 safety, tolerability and pharmacokinetic study. A phase 2 double blind trial is in progress whereby patients with mild-to-moderate disease are treated twice daily with PBI-0451 for 5 days. VV116 (Junshi Biosciences) is an orally active analog of Veklury (remdesivir). A cohort study was recently undertaken in omicron variant infected SARS-CoV-2 patients treated twice daily with oral VV116 within 5 days of the first disease symptom, along with a placebo group. Those who received VV116 demonstrated a significant decrease in the time of viral shedding relative to the control group [21]. Most recently, in July 2022, Chinese regulatory authorities approved its first ‘home grown’ COVID-19 antiviral pill, Azvudine (Genuine Biotech), which was already approved in China for treatment of HIV. It is an analog of the first drug approved by the FDA for treatment of HIV, Zidovudine (AZT). Both drugs are reverse transcriptase inhibitors. SIM0417 (Jiangsu Simcere Pharmaceutical Group) is a small molecule main protease inhibitor currently in a phase 2 trial in combination with ritonavir, presumably to inhibit cytochrome P450 3A4.
The above overview provides examples of candidate drugs currently in development with documented human data suggesting varying degrees of promise. But a candidate drug in development does not ensure a commercial product. In fact, the large patient population that requires treatment in a pandemic setting adds more criteria regarding the definition of an ideal candidate drug for use during a pandemic, as further discussed below.
Tucaresol, Clinical Studies
In view of the important role played by T cells regarding patient survivability during an HIV infection, as reflected by the appearance of secondary infections that may accompany HIV, and with regard to the selective interaction (binding) of Tucaresol with CD4+ T helper cells, it is appropriate to consider a proof-of-concept (phase 2) clinical trial employing Tucaresol for treatment of patients with mild-to-moderate (non-hospitalized) COVID-19 disease, either in a monotherapy or preferably combination antiviral drug therapy setting. However, another option is treatment of patients with severe (hospitalized) COVID-19 disease accompanied by lymphopenia. In the case of the earlier evaluation by GlaxoSmithKline of Tucaresol for treatment of HIV infected patients in a multicenter, placebo controlled phase 2 trial, a two dose, 25 mg and 50 mg daily, 3 month combination therapy study with HAART (Highly Active Antiretroviral Therapy) was planned with 45 patients already receiving HAART and a study start date of 2004-11-01. This trial appears lengthy with a study completion verification date of 2008-10-13, an issuance of “History of Changes for Study: NCT00343941” and notification that “Final trial results were not reported” [23]. However, demonstration of significant modulation of T cell activity by Tucaresol during a phase 1b/2a trial in HIV patients was published in 2004 as a GlaxoSmithKline and University of Milan collaboration [24]. This clinical trial consisted of 4 groups of HIV positive patients, a total of 24 patients, in which half of the patients received HAART prior to the trial and the other half were HAART naive. This was a 16 week pulse dose escalation protocol in which Tucaresol was administered as one 25 mg dose during week 1, 25 mg/day for 4 days during week 4, 50 mg/day for 4 days during week 8 and 100 mg/day for 4 days during week 12 with time between doses to permit drug wash out. One of the 4 groups received only Tucaresol while the second group received Tucaresol and HAART at the same time. Groups 3 and 4, already on HAART prior to the trial, received Tucaresol according to the dose schedule above. The difference between the two groups is that one group consists of patients that are immunologic non-responders as evidenced by a below average CD4+ T helper cell count. Following administration of Tucaresol, increases in percentages of memory T lymphocytes (CD4+/CD45RO+) were observed in all patients, including those patients treated only with Tucaresol. Following administration of Tucaresol, a significant increase in memory T cells was seen on week 12 in the group already on HAART and on week 13 in the group of immunologic non-responders; P<0.05 in both cases. Any significant P value is of note since each group consists of only 6 patients. More significant was a sustained increase in percentages of naive T lymphocytes (CD4+/CD45RA+) observed in all patients concomitantly with administration of Tucaresol. Also, an increase in naive CD4+ T cells was observed at approximately week 8 in all groups following the third Tucaresol administration. Env-stimulated perforin-expressing CD8+ T cells were increased in all groups. Perforin-expressing p24-stimulated CD8+ T cells were similarly amplified. As regards HIV plasma viremia, there were no changes in HIV RNA levels, including those patients treated only with Tucaresol, except a decrease in HIV RNA in patients starting simultaneous treatment with Tucaresol and HAART. Therefore, while treatment with Tucaresol alone did not eliminate viremia, it prevented a significant increase in HIV viremia. Administration of Tucaresol was associated with increased interleukin 12 and decreased interleukin 10. In particular, the reduction of interleukin 10 upon administration of Tucaresol was significant at weeks 8, 12 and 16 in the patient group already on HAART; P<0.001. High interleukin 10 expression, similar to interleukin 6 expression, can predict poor clinical outcomes in COVID-19 patients [25]. The above phase 2 (planned) clinical trial was preceded by a phase 1 dose escalating safety trial in 24 HIV infected patients already on HAART (ClinicalTrials.gov registry number; NCT00006209).
In conclusion, in patients on HAART with proper viral suppression, treatment with Tucaresol resulted in: (i) stimulation of cytotoxic T lymphocyte activity, (ii) generation of naive T cells and (iii) did not result in any adverse effects or increase in patient viral load.
Tucaresol, Preclinical Studies
The three patents granted to Glaxo Wellcome Inc. (US 5,508,310; 5,892,151; and 6,096,786) demonstrated the host targeted antiviral activity of Tucaresol by exemplification of the elimination of simian immunodeficiency virus (SIV) in a macaque, as well as reduction of SIV in the second animal. Two of four macaques of average weight 2.5 kg with established SIV infection were injected ip; 30 mg/kg Tucaresol every other day for nine days (five injections). Viral load was measured on day 11. No virus was detected in the first animal while a ten-fold reduction in SIV was observed in the second animal.
Human pharmacokinetic and pharmacodynamic data, along with other human preclinical data, is available in three publications from Wellcome Research Laboratories, later Glaxo Wellcome R & D Ltd. [26,27,28]. Interestingly, these studies were not targeted towards clinical evaluation of Tucaresol at a dose of up to 100 mg/daily as noted above but instead were directed to the maximum tolerated dose of Tucaresol, since the preclinical studies were initially focused on Tucaresol as a potential treatment for sickle cell disease. Therefore, in the first administration of Tucaresol into humans, the pharmacokinetics and pharmacodynamics were studied in healthy male volunteers following oral doses of 200 mg - 3,600 mg. Tucaresol is well tolerated with some gastrointestinal discomfort at oral doses of 1,200 mg or more. There were no clear effects on routine hematology or biochemistry, platelet aggregation, resting or exercise heart rates or blood pressure. Lymphadenopathy was observed in a few individuals at high dose Tucaresol (>2 grams), 7-10 days after beginning administration, which suggests stimulation of the immune system. At the time of peak whole blood concentration following administration of 3,600 mg of Tucaresol, approximately 70% of the candidate drug is present in the blood, indicating good oral bioavailability. Food intake and gender apparently have no significant effect on orally administered Tucaresol.
Additional information regarding preclinical evaluation of Tucaresol, including studies undertaken in rodents and larger animals, is available in a Doctor of Medicine thesis based on work at Wellcome Research Labs [29]. Similar to the three publications noted above, this work was focused on significantly higher doses of Tucaresol as a potential treatment for sickle cell anemia. Animal toxicology studies included acute oral and iv mouse studies, 14 days oral subacute rabbit studies and a one month oral dose ranging Wistar rat study. The latter study included a hematology, biochemistry and pathology profile. A one month oral toxicology study was undertaken with cynomolgus monkeys. Also, teratogenicity (rats, rabbits) and Ames mutagenicity tests were undertaken. Pharmacokinetic studies were performed with a single oral dose of Tucaresol in rats, single oral carbon-14 labeled dose in rabbits, three iv carbon-14 labeled doses over three days in two beagles and three iv doses over three days in two cynomolgus monkeys. Another pharmacokinetic study was done with carbon-14 labeled Tucaresol administered orally to two monkeys and administered iv to another two monkeys.
As noted above, Tucaresol is a benzoic acid substituted benzaldehyde small molecule with a unique mechanism of action based on a selective transient covalent bond (Schiff base) formed between its aldehyde moiety and amino groups present on CD4+ T helper cells (lysine side chains). This T helper cell Schiff base formation with Tucaresol induces sodium/potassium ion channel activation and subsequent tyrosine phosphorylation of cytoplasmic signaling proteins. This costimulatory signal transduction pathway converges with the signaling events resulting from virus peptide fragment binding (within the context of MHC II) to the T cell receptor. Tucaresol co-stimulation favors enhancement of a Th1 proinflammatory cytokine response by effectively increasing interleukin 2, interleukin 12 and interferon-gamma while diminishing a Th2 anti-inflammatory cytokine response with reduction of interleukin 4 [30]. As such, Tucaresol mimics the transient covalent bond (Schiff base) formation that occurs between the ‘specialized’ carbonyls (likely arising from glycated proteins) from MHC (major histocompatibility complex) class II bound virus peptide fragment present at the surface of APCs (Antigen Presenting Cells) and amino groups (lysine side chains) from CD4+ T helper cells [31]. This mimetic activity of Tucaresol enhances antigen-specific T cell activation and cell mediated immunity via the increased Th1 proinflammatory cytokine response.
Perhaps one reason Tucaresol has not received much attention as a clinical stage candidate drug for treatment of SARS-CoV-2 is the perception that the aldehyde function will covalently bind nonspecifically off-target to lysine(s) present in multiple proteins thereby resulting in drug toxicity. However, this concern was addressed in 2019 with FDA approval of a 2-hydroxybenzaldehyde analog of Tucaresol, Voxelotor (GBT440, Global Blood Therapeutics), for the treatment of sickle cell disease. Therapy is achieved by high dose stoichiometric binding (and reversible Schiff base formation) with hemoglobin via a mechanism first observed with Tucaresol. Lack of significant toxicity of Voxelotor was supported by the recent $5.4 billion acquisition of Global Blood Therapeutics by Pfizer. Interestingly, it was published this year that the time spent by Voxelotor correctly bound to hemoglobin (the residence time) was “dramatically enhanced” by a hydroxyl group ortho to the aldehyde moiety of Voxelotor, thereby facilitating a favorable equilibration of the drug correctly bound to hemoglobin [32]. The lack of toxicity reported for Tucaresol in animal and human studies cited above is in agreement with this lack of toxicity observed with Voxelotor.
The above overview of the mechanism of Tucaresol suggests that it offers a unique tool for treatment of viral infections. In summary, Tucaresol represents a hybrid or bridge between antiviral drugs and vaccines. Similar to low molecular weight antiviral drugs, Tucaresol is a small molecule, orally active, relatively fast acting chemical entity while similar to vaccines, Tucaresol will enhance both the antibody (humoral) and T cell (cell mediated) immune response, via interaction with important CD4+ T helper cells. Antiviral drugs are best administered for therapeutic purposes or treatment of active infections ideally as soon as possible while vaccines are best administered for prophylactic purposes especially when the virus is rapidly pathogenic such as Ebola virus. However, Tucaresol may be useful in both a therapeutic setting, for example administered in combination with a virucidal drug, or in a prophylactic setting, for example administered at the same time as immunization with a vaccine and for a few days thereafter, to provide immune protection during the period of immune priming, first dose of vaccine, and commencement of boosting of the immune response, second dose of vaccine.
Discussion
Only three small molecule antiviral drugs are available for treatment of COVID-19 disease: Veklury, Paxlovid and Lagevrio. These medications represent the first generation COVID-19 disease antivirals with which to address the current pandemic. However, experience with older antivirals against other viral infections teaches that a broad array of readily available antiviral medicines is required to address SARS-CoV-2 ‘head on’. Furthermore, a forceful assault on COVID-19 disease not only requires a broad choice of effective medicines but also a combination of two or more effective antivirals in order to have a significant impact upon viral infections. Combination therapy (‘drug cocktails’) have demonstrated superior performance regarding treatment of HIV and hepatitis C. One reason for the proven success of combination antiviral therapy is that, because viruses generally replicate faster than bacteria, viruses are generally more prone to develop drug resistance. However, combination therapy with two or more antiviral drugs with distinctly different mechanisms of action, different biochemical or druggable targets, will protect the infected organism longer than when the virus only has to circumvent one viral replication drug inhibition pathway. That is, the best combination therapy regimen is one that offers two or more distinctly different viral replication inhibition mechanisms that make it as difficult as possible for the virus to subjugate the host cell. As such, one recommendation to achieve as distinct as possible a mechanistic difference for the virus to overcome is to treat the infection with one drug that directly inhibits viral replication and, at the same time, treat the infection with a host-targeted drug that denies the virus access to a component of the host that it usurps in order to replicate and prevail. Another advantage of a host-targeted drug is that it will not be directly affected by viral mutation(s) since it does not directly inactivate the virus. Combination therapy with Tucaresol offers such an opportunity, as demonstrated above in the HIV clinical trial results reported for combination therapy with HAART (zidovudine, lamivudine, nevirapine) and Tucaresol [33]. Addition of Tucaresol to the HAART triple drug combination does not impose an impediment to the treatment protocol, since Tucaresol adds only a small amount of active ingredient, less than 100 mg daily per patient, to the drug cocktail. The ease of synthesis of Tucaresol, especially per our recent proprietary protocol, ensures avoidance of supply shortages that can occur when more complex antiviral drugs are required to address a pandemic infection. Furthermore, any requirement for large amounts of Tucaresol should not be cost prohibitive since the synthetic protocol does not require use of expensive reagents such as precious metal catalysts. Tucaresol’s lack of optical centers negates loss of product (inactive enantiomer) or need for a stereospecific synthesis.
With respect to selection of a combination therapy regimen for treatment of mild-to-moderate COVID-19 disease with one of the three approved antiviral drugs, the best choice may be combination therapy with oral Paxlovid and Tucaresol. Veklury does not offer the convenience of an oral dosing regimen, while Molnupiravir is significantly less efficacious than Paxlovid against mild-to-moderate COVID-19 disease. Indeed, addition of Tucaresol to the Paxlovid treatment regimen, five days b.i.d. after the appearance of the first symptom of COVID-19 disease, could be conveniently accomplished with a 25 mg daily dose of Tucaresol for five days formulated along with the daily dose of Ritonavir that accompanies Paxlovid (perhaps a combination 125 mg Ritonavir/Tucaresol tablet?). The potential advantage of early treatment of mild-to-moderate COVID-19 disease is that co-administration of Tucaresol with Paxlovid would offer immediate protection of the adaptive immune response from induction of viral lymphopenia so that the T cell component would be better able to ‘mop up’ residual virus not inactivated by the five day treatment with Paxlovid. Indeed, the decline in CD4+ T helper cells begins almost immediately with the first symptom(s) of infection by SARS-Cov-2 following a kinetic profile that resembles infection with HIV [34]. In fact, two recent publications [35,36], correlate with increased immune dysfunction that may be expected to accompany CD4+ T helper cell depletion. Reinfection with COVID-19 significantly increases the risk of health issues, hospitalization and death. Furthermore, this risk increases with each additional COVID-19 infection. Subsequently, February 2023, it was reported that norovirus infections in people 65+ reached levels not seen in over a decade. Similarly, increases in RSV and influenza were observed at the beginning of the winter respiratory disease season attributed to “less population immunity due to the (COVID-19) pandemic. Another potential advantage to co-administration of Tucaresol with Paxlovid may be protection from viral rebound (as noted above) to not only occur with Paxlovid but with the HIV protease inhibitor Indinavir or HAART [37]. Viral rebound associated with Paxlovid recently received renewed attention since US President Biden experienced it after infection with SARS-CoV-2. It was reported by NBC News [38] that more people are reporting viral rebound after treatment with Paxlovid, such that approximately 5% of treated patients are reporting rebound compared to the 1 - 2% of patients observed in the Pfizer Paxlovid clinical trial. It was also reported that Molnupiravir exhibits a similar percent of viral rebound. Recently, August 2022, Pfizer was ordered by the FDA to conduct an additional trial by September, 2023 to determine if a second five day treatment with Paxlovid would diminish viral rebound. Two recent studies suggest that COVID rebound is “more prevalent and pronounced among users of Paxlovid” [39], although it can occur among those infected with SARS-CoV-2. One theory is that the adaptive immune response does not activate quickly enough and so treatment with Tucaresol could protect or better facilitate a more robust response to residual virus not inactivated by the five day treatment with Paxlovid. Some support for this theory was published seven years ago [40] regarding a mathematical model that demonstrated “a range in CTL (CD8+ T cell) response strengths where a (AIDS) patient may show either viral rebound or post-treatment control, depending upon the size of the latent (virus) reservoir at treatment termination” of HIV infection.
Nonetheless, with increasing use of Paxlovid as monotherapy, concern increases regarding the possible appearance of a Paxlovid resistant strain of SARS-CoV-2 virus, as reflected in two commentaries published in Science [41,42]. One concern is that studies from different labs identified drug resistant mutations in the virus protease that render the virus less susceptible to Paxlovid. These mutations are also present in circulating viral variants found in infected people. Fortunately, a significantly Paxlovid resistant virus has not been reported in general circulation which may be accounted for reasons that include the relatively short five day dose schedule. However, both commentaries note that the usual approach to prevent or at least delay virus drug resistance is by means of combination therapy or a drug cocktail. Apparently Pfizer is not pursuing combination therapy with Paxlovid, but instead recently entered into a collaboration with Clear Creek Bio Inc. to identify an oral inhibitor that targets the other essential SARS-CoV-2 protease, Papain-like protease, instead of the main protease, Mpro [45].
Of course, other candidate antivirals are progressing through clinical trials that, for reasons similar to those discussed above concerning Paxlovid, justify exploration of combination therapy with Tucaresol. Two examples are Ensitrelvir (Shionogi), recently approved in Japan as Xocova, and EDP-235 (Enanta). Like Paxlovid, both candidate drugs are orally active protease inhibitors with clinical data. The advantage of Ensitrelvir and EDP-235 is that they require one daily dose for five days and do not require Ritonavir. This regimen would permit convenient formulation with Tucaresol as it also requires one daily dose. Another opportunity exists wherein Tucaresol is combined with an approved repurposed drug. Indeed, there are examples where approved drugs have been in repeated clinical trials as monotherapy for COVID-19 disease. As noted above, for example, hydroxychloroquine has been tested in more than 150 trials [44]. In such cases it is possible that Tucaresol, with its unique mechanism of action, may result in a significant, synergistic antiviral activity in combination with an appropriate repurposed drug that did not previously display significant activity as a monotherapy. Potential repurposed drugs for evaluation with Tucaresol, based on prior interest as monotherapy for COVID-19 disease, include: favipiravir, azvudine, hydroxychloroquine, nitazoxanide, clofazimine, ivermectin, colchicine, antibiotics such as azithromycin, cefepime and ceftazidime and polyphenolic natural (plant) products such as tannic acid and tannins, resveratrol, quercetin, hesperidin and curcumin.
Finally, it should be mentioned that there is significant concern regarding future viral pandemics that may join or follow COVID-19 disease. Potential pandemics may include SARS-CoV, MERS-CoV, influenza, rift valley fever, lassa, nipah, hendra, ebola, marburg, yellow fever, dengue, zika and west nile viruses. Indeed, during any given year, an outbreak of a highly pathogenic virus may take place with the potential to become a pandemic. Recently for example, February 23, 2023, the WHO announced that there was a high risk, at the national level, of the first Marburg virus disease outbreak in Equatorial Guinea, Central Africa. Marburg virus, like Ebola virus, is a member of the filovirus family that causes hemorrhagic fever with high fatality. Therefore, with multiple viruses of pandemic potential in the background, it is highly desirable to have access to as many drugs as possible with activity against more than one pathogenic virus. This is where Tucaresol may become particularly important because, in theory, Tucaresol should display activity against any virus that targets CD4+ T helper cells, either directly (HIV or SARS-CoV-2) or indirectly, and subsequently induces a reduction in T cells including lymphopenia. In fact, lymphopenia is a common characteristic of filovirus the extent of which, especially with regard to depletion of T cells during infection, is an important determinant regarding the survivability of patients with these often fatal viruses. Therefore, access to large quantities of Tucaresol may become particularly desirable and would be achievable by means of our two step proprietary synthesis. Additionally, with potential activity of Tucaresol against other viruses of pandemic potential, exploration of the activity of analogs of Tucaresol becomes more important; especially since few analogs have been reported in the scientific literature. One example is the synthesis and evaluation of the long alkyl chain (hexadecyl) ester of Tucaresol as a vaccine adjuvant [45]. Our work with Tucaresol and analogs continues [46]. Additionally, after review of our article a request was received for permission to publish the version 2 preprint in the African Journal of Pharmaceutical Sciences (SvedbergOpen publishers) under the terms of a Creative Licence (by 4.0). Publication target date is March, 2023 [47].
Author Contributions
Christopher Penney Project conceptualization, Drafting of original manuscript, Review and editing, Literature collection and analysis, Project administration. Boulos Zacharie Project conceptualization, Review and editing, Literature collection and analysis, Project administration, Methodology. Jean-Simon Duceppe Review and editing, Methodology, Experimentation. All authors have read and agreed to the published version of the manuscript.
Funding
The authors declare that no grants were received in support of this work.
Institutional Review Board Statement
No animal studies or human trials were undertaken for this project by the authors thereby negating the need for animal (ethics) committee, IRB and human ethics committee approvals
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We gratefully acknowledge the skillful participation and efforts of Paulo Bouca, Elizabeth Cooper and Robert Rando during the early and/or middle days of ChemThera Sciences. The literature contributions and ‘competitive intelligence’ monitoring of Dr Alan Cameron are most appreciated. Also appreciated are informative conversations with Maged Butros this year regarding opportunities for this project. We also gratefully acknowledge the excellent technical assistance of Jaysen Penney regarding the final preparation and formatting of this manuscript.
Conflicts of Interest
The authors declare that there is no conflict of interest.
References
- Dunleavy, K. Pfizer reveals plan to make its products available to 45 poor countries at not-for-profit cost. FIERCE Pharma. May 25, 2022.
- Dunleavy, K. Gilead touts analysis showing COVID antiviral Veklury performs best when given early in illness. FIERCE Pharma. April 25, 2022.
- Coronavirus (COVID-19) update: FDA authorizes first oral antiviral for treatment of COVID-19. December 23, 2021.
- Becker, Z, Dunleavy K, Kansteiner F, Liu A. COVID-19 tracker. FIERCE Pharma. August 12, 2022.
- Havlir DV, Hellmann NS, Petropoulos CJ, Whitcomb JM, Collier AC, Hirsch MS, Tebas P, Sommadossi JP, Richman DD. Drug susceptibility in HIV infection after viral rebound in patients receiving indinavir-containing regimens. JAMA. 2000, 283, 229-234.
- Kansteiner, F. Paxlovid’s post-exposure protection flop hardly spells the end for Pfizer’s COVID franchise: analysts. FIERCE Pharma. , 2022. 2 May.
- Zhou S, Hill CS, Sarkar S, Tse LV, Woodburn BMD, Schinazi RF, Sheahan TP, Baric RS, Heise MT, Swanstrom R. β-d-N4-hydroxycytidine inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J. Infect. Dis. 2021, 224, 415–419. [CrossRef] [PubMed]
- Kabinger F, Stiller C, Schmitzová J, Dienemann C, Kokic G, Hillen HS, Höbartner C, Cramer P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746.
- Coronavirus (COVID-19) update: FDA authorizes additional oral antiviral for treatment of COVID-19 in certain adults. FDA News Release. December 23, 2021.
- Robinson PC, Liew DFL, Tanner HL, Grainger JR, Dwek RA, Reisler RB, Steinman L, Feldmann M, Ho LP, Hussell T, et al. COVID-19 therapeutics: challenges and directions for the future. Proc. Natl. Acad. Sci. U.S.A. 2022;119, e2119893119. [CrossRef]
- Vegivinti CTR, Evanson KW, Lyons H, Akosman I, Barrett A, Hardy N, Kane B, Keesari PR, Pulakurthi YS, Sheffels E, et al. Efficacy of antiviral therapies for COVID-19: a systematic review of randomized controlled trials. BMC Infect. Dis. 2022, 22, 107. [CrossRef] [PubMed]
- Trémolières F. Antivirals in the time of COVID-19 – not as easy as it looks! Infect Dis Now. 2021; 51, 2-6.
- Riva L, Yuan S, Xin Y, Martin-Sancho L, Matsunaga N, Pache L, Burgstaller-Muehlbacher S, De Jesus PD, Teriete P, et al. Discovery of SARS-CoV-2 antivirals through large scale drug repositioning. Nature. 2020; 586, 113-119.
- Jang WD, Jeon S, Kim S, Lee SP. Drugs repurposed for COVID-19 by virtual screening of 6218 drugs and cell-based assay. Proc. Natl. Acad. Sci. U.S.A. 2021; 118,e2024302118.
- Ibid.
- Gligorijevic N, Radomirovic M, Nedic O, Stojadinovic M, Khulal U, Stanic-Vucinic D, Velickovic TC. Molecular mechanisms of possible action of phenolic compounds in COVID-19 protection and prevention. Intl. J. Mol. Sci. 2021; 22,12385. [CrossRef]
- Ibid.
- Viera J. Enanta Pharmaceuticals receives FDA fast track designation for EDP-235, its oral 3CL protease inhibitor specifically designed for the treatment and prevention of COVID-19. Press Release. March 29, 2022.
- Shamsi A, Mohammad T, Anwar S, AlAjmi MF, Hussain A, Rehman MT, Islam A, Hassan MI. Glecaprevir and Maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: possible implication in COVID-19 therapy. Biosci. Rep. 2020 June;40, BSR20201256. [CrossRef]
- Notice regarding the media coverage about S-217622, a therapeutic drug for COVID-19. Press Release. April 13, 2022.
- Shen Y, Ai J, Lin N, Zhang H, Li Y, Wang H, Wang S, Wang Z, Li T, Sun F, et al. An open prospective cohort study of VV116 in Chinese participants infected with SARS-CoV-2 omicron variants. Emerg. Microbes Infect. 2022, 11, 1518–1523. [CrossRef] [PubMed]
- Davanzo GG, Codo AC, Brunetti NS, Boldrini V, Knittel TL, Monterio LB, De Moraes D, Ferrari AJR, De Souza GF, Muraro SP, et al. SARS-CoV-2 uses CD4 to infect T helper lymphocytes. medRxiv. 2020. 2020. [CrossRef]
- National Library of Medicine (U.S.). (2004 - 2008) Tucaresol as add-on to HAART (highly active antiretroviral therapy) in chronic HIV-1 infected adults. Identifier NCT00343941. 0034. Available online: https://clinicaltrials.gov/ct2/show/ NCT00343941.
- Gori A, Trabattori D, Bandera A, Sarsella M, Marchetti G, Gazzola L, Biasin M, Rhodes J, McDade H, Panebianco R, et al. Immunomodulation induced by Tucaresol in HIV infection: results of a 16 week pilot phase I/II trial. Antivir. Ther. 2004, 9, 603-614.
- Lu L, Zhang H, Dauphars DJ, He YW. A potential role of interleukin 10 in COVID-19 pathogenesis. Trends Immunol. 2021 Jan;42,3-5. [CrossRef]
- Peck RW, Wootton R, Wiggs R, Layton G, Posner J. Effect of food and gender on the pharmacokinetics of Tucaresol in healthy volunteers. Br. J. Clin. Pharmacol. 1998, 46, 83-86.
- Rolan PE, Mercer AJ, Wootton R, Posner J. Pharmacokinetics and pharmacodynamics of Tucaresol, an antisickling agent in healthy volunteers. Br. J. Clin. Pharmacol. 1995, 39, 375–380. [CrossRef] [PubMed]
- Rolan PE, Parker JE, Gray SJ, Weatherley BC, Ingram J, Leavens W, Wootton R, Posner J. The pharmacokinetics, tolerability, and pharmacodynamics of Tucaresol (589C80; 4[2-formyl-3-hydroxyphenoxymethyl] benzoic acid), a potential anti-sickling agent, following oral administration to healthy subjects. Br. J. Clin. Pharmacol. 1993, 35, 419-425.
- Rolan, PE. The exploratory clinical development of tucaresol. M.D. dissertation, University of Adelaide, Australia, 1996. Retrieved from. Available online: https://hdl.handle.net/2440/38411.
- See ref. 23.
- Rhodes J, Chen H, Hall SR, Beesley JE, Jenkins DC, Collins P, Zheng B. Therapeutic potentiation of the immune system by costimulatory Schiff-base-forming drugs. Nature 1995; 337,71-75.
- Yang T, Cuesta A, Wan X, Craven GB, Hirakawa B, Khamphavong P, May JR, Kath JC, Lapek JD Jr, Niessen S, et al. Reversible lysine-targeted probes reveal residence time-based kinase selectivity. Nat Chem Biol. 2022 Sep;18,934-941.
- See ref. 23.
- Peng X, Ouyang J, Isnard S, Lin J, Fombuena B, Zhu B, Routy JP. Sharing CD4 + T cell loss: when COVID-19 and HIV collide on immune system. Front. Immunol. 2020. [CrossRef]
- Diamond, F. Repeat COVID-19 infection deadlier than the first go-round, says study. FIERCE Healthcare. November 17, 2022.
- Jetelina, K. Norovirus has entered the chat. Your Local Epidemiologist. February 23, 2023. 23 February.
- See ref. 5.
- Bendix, A. As more people report Covid rebounds after Paxlovid, experts insist cases are rare. NBC News. July 27, 2022.
- Dunleavy, K. FDA wants Pfizer to study COVID rebound cases with a longer course of Paxlovid. FIERCE Pharma. August 22, 2022.
- Conway JM, Perelson AS. Post-treatment control of HIV infection. Proc. Natl. Acad. Sci, U.S.A. April 13, 2015; 112, 5467-5472.
- Service RF. Bad news for Paxlovid? Coronavirus can find multiple ways to evade covid-19 drug. Science. June 29, 2022; 377(6602). [CrossRef]
- Lowe, D. Paxlovid Resistance: Is it just a matter of time now? Science. July 11, 2022.
- Taylor, NP. Pfizer’s search for Paxlovid’s COVID - busting sibling leads to low - profile antiviral biotech. FIERCE Biotech. December 6, 2022.
- See ref. 10.
- Collins KC, Schlosburg JE, Lockner JW, Bremer PT, Ellis BA, Janda KD. Lipid tucaresol as an adjuvant for methamphetamine vaccine development. Chem Commun (Camb). 2014 Apr 21, 50, 4079-81.
- To advance this project in a timely manner, we require additional financial support. We welcome an appropriate collaboration and all inquiries of interest.
- Penney CL, Zacharie B, Duceppe J-S. Tucaresol: A Unique Oral Candidate Drug Ideally Accessible for Treatment of Covid-19 Disease. African Journal of Pharmaceutical Sciences 2023; 3, 34 - 46.
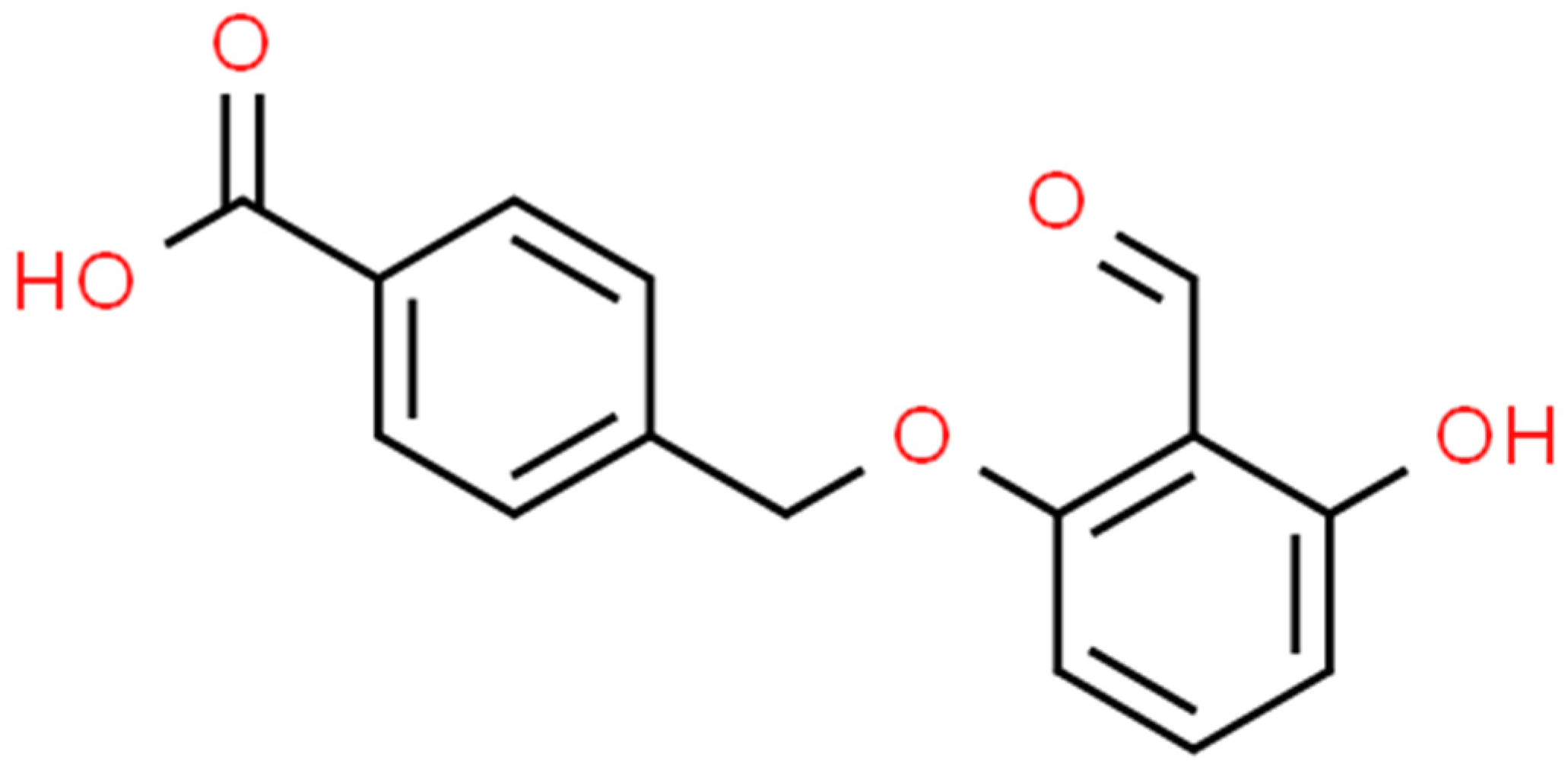
Figure 1.
Structure of Tucaresol (free acid).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



