
Submitted:
26 January 2024
Posted:
29 January 2024
You are already at the latest version
Abstract
FacioScapuloHumeral Dystrophy (FSHD) is one of the most prevalent inherited muscle disorders, and is linked to the inappropriate expression of the DUX4 transcription factor in adult muscles. The deregulated molecular network causing FSHD skeletal muscle dysfunction and pathology is still not well understood. It has been shown that the hypoxia response factor HIF1α is critically disturbed in FSHD and has a major role in DUX4 induced cell death. In this study, we further explore the relationship between DUX4 and HIF1α. We found that the DUX4 and HIF1α link differed according to the stage of myogenic differentiation and was conserved between human and mouse muscle. Furthermore, we found that HIF1α knock-down in a mouse model of DUX4 local expression exacerbated DUX4-mediated muscle fibrosis. Our data indicate that the suggested role of HIF1α in DUX4 toxicity is complex and that targeting HIF1α might be challenging in the context of FSHD therapeutic approaches.
Keywords:
FSHD
; DUX4
; HIF1α
; myogenesis and skeletal muscle
1. Introduction
FacioScapuloHumeral muscular Dystrophy (FSHD) is a dominant hereditary disease characterized by a progressive and often left/right asymmetric skeletal muscle weakness that initially affects facial muscles, and progresses through a rostro-caudal pattern. FSHD lowers quality of life and approximately 30% of patients become wheelchair-bound [1,2]. FSHD involves complex genetic and epigenetic components leading to activation in skeletal muscle of DUX4, a gene which encodes a potent transcription factor [3,4,5,6,7,8]. DUX4 is normally expressed in germline and early embryogenesis, where it plays a role in zygotic genome activation and appears involved in placentation [9,10,11]. The FSHD epigenetic defect is located in the 4q35 chromosome region at the macrosatellite D4Z4 repeat array, which is hypermethylated in unaffected individuals and therefore in a closed chromatin conformation. FSHD results from the hypomethylation and epigenetic derepression of the D4Z4 repeat array, and thus, a more permissive chromatin structure, which allows DUX4 gene transcription from the distal-most D4Z4 unit [1,3,12]. DUX4 RNAs extend from the distal D4Z4 unit to the flanking pLAM region where they acquire an intron and an exon with a polyadenylation signal (PAS) allowing for production of a stable mRNA that can be translated to generate toxic DUX4 protein. This PAS sequence is only present on the permissive distal 4qA allele, but not on 4qB [13,14].
DUX4 protein toxicity likely results from disturbances of at least four different cell signaling pathways. First, despite its expression pattern in rare and short bursts, DUX4 transcriptional activity causes a large deregulation of gene expression activating germ-line specific genes [5] but inhibiting genes involved in myogenesis [15,16] and oxidative stress response [17,18,19,20] (reviewed in [8]). Second, based on single cell RNA sequencing (sc-RNAseq) and microarray data, Banerji and Zammit have determined that repression of a PAX7 target gene signature is a reliable biomarker of the pathology and is associated with disease progression [1,15,16]. PAX7 is a myogenic transcription factor normally expressed in satellite cells, which can be activated for regeneration, a process altered by DUX4 in FSHD. The single PAX7 homeodomain is similar to those of DUX4, and PAX7 can rescue DUX4-induced cytotoxicity in mouse muscle cells [17]. DUX4 and PAX7 can bind similar DNA elements and compete on reporter target gene activation [15]. Third, Jagannathan et al. found a large discrepancy between the proteomic and transcriptomic (RNAseq) landscapes in DUX4 expressing muscle cells, highlighting disturbances of post-transcriptional processes such as RNA and protein quality control pathways [21,22]. Finally, other studies suggested that DUX4 interaction with specific protein partners could participate in FSHD-associated pathological processes, such as interference with regeneration [23,24].
The molecular map of FSHD-associated interaction signaling established by Banerji et al. [25] and based on a meta-analysis of microarray data sets from FSHD muscle biopsies [25] demonstrated that the hypoxia response pathway was critically perturbed in FSHD, among other pathways such as the WNT pathway. In FSHD, PAX7 repression was also associated with induction of hypoxia-response genes [15]. Accordingly, Tsumagari et al. had independently described HIF1α-signaling network as one of the over-represented pathways among FSHD dysregulated genes [26]. HIF1α is the master regulator of physiological adaptive mechanisms in response to hypoxia [27]; it induces expression of multiple effector genes that modulate various cellular processes such as glucose metabolism and oxidative stress. In skeletal muscle, hypoxia modulates not only muscle fiber type profile but also myogenesis, regeneration and vascularization. Activation of HIF1α can also be involved in pathological conditions independently from hypoxia. This “pseudohypoxia” was mostly described in cancer [28]. In FSHD, available data suggest that the primary genetic defect per se could cause HIF1α pathway disturbance through a putative DUX4-HIF1α axis (as we reviewed in [29]). Indeed, HIF1α was identified as necessary for DUX4 toxicity by Lek et al in a genome-wide CRISPR-Cas9 screen performed to identify genes whose loss-of-function could allow survival of myoblasts expressing DUX4 [30]. However, the DUX4-HIF1α axis requires clarification, particularly by taking into account variations of HIF1α expression and effects during myogenic differentiation [31].
If one considers a putative pathological contribution of HIF1α to FSHD it is noteworthy that HIF1α promotes a metabolic switch in favor of anaerobic glycolysis, the metabolism favoured in early embryogenesis, where DUX4 and mouse Dux have a function in zygotic genome activation [9,10,32]. Interestingly, most stem cell types reside in hypoxic niches where HIF1α controls pluripotency gene expression, promotes glycolytic metabolism and inhibits mitochondrial biogenesis. An aberrant DUX4/HIF1α activation could therefore contribute to metabolic disturbances in adult FSHD muscle cells. In addition, HIF1α modulates myogenic differentiation [31], a process which occurs during skeletal muscle regeneration and is disturbed in FSHD [33,34]. DUX4-induced HIF1α pathway misregulation could therefore participate in the FSHD-associated defect in adult myogenesis. Accordingly, we showed that DUX4 suppressed HIF1α-mediated precocious differentiation of human myoblasts [31]. Moreover, Heher et al. showed that hypoxia aggravates the hypotrophic FSHD myotube phenotype, this effect being due to a DUX4-mediated metabolic mis-adaptation, leading to an exacerbated oxidative stress, particularly in conditions of varying O2 tension [19].
From a therapeutic perspective, Lek et al. showed that pharmacological HIF1α signaling inhibitors could improve DUX4-associated muscle phenotypes in FSHD-like zebrafish embryos. However, this approach could prove deleterious at later muscle development stages because of the negative effect of indirect HIF1α inhibitors on protein synthesis or on HIF1α contribution to the myogenic program. In addition the effects of a specific HIF1α knockdown have not been investigated in a mature muscle, nor in a murine model of DUX4 expression.
The present study aimed to fill the gap of knowledge regarding (i) the link between DUX4 and HIF1α during human muscle cell differentiation [35], (ii) its conservation in murine models of DUX4 expression in vitro [36] and in vivo [37], (iii) the effect of a targeted HIF1α knock-down on DUX4-mediated muscle lesion.
2. Results
2.1. Effect of DUX4 on HIF1α expression and nuclear protein level at different stages of human myoblast differentiation in vitro
We used the LHCN-M2-iDUX4 cell line with doxycycline (DOX)-inducible DUX4 expression, engineered from the immortalized human myoblast line LHCN-M2 [35,38]. Because of its high toxicity the impact of DUX4 induction was first evaluated by cell viability assays (MTT and CCK8) under a standard oxygen partial pressure (21% PO2), 24h after DOX addition to the culture medium. These tests were performed in LHCN-M2-iDUX4 myoblasts (Figure S1B and D) as well as on LHCN-M2 to test DOX toxicity per se (Figure S1A and C). DUX4 induction had no effect on cell viability up to 62.5 ng/ml DOX. From 125 ng/ml and above, a significant decrease of viability was observed in both the MTT and CCK8 tests (Figure S1B and D). DOX exposure per se had no effect on myoblast viability in the absence of DUX4 expression as verified in LHCN-M2 cells (Figure S1A and C). The production of the DUX4 protein was also confirmed by immunofluorescence (IF) in LHCN-M2-iDUX4 myoblasts (Figure S1E). After addition of 15.6 or 31.2 ng/ml of DOX to the culture medium for 24h, we detected 32% (± 4%) and 56% (± 6%) of DUX4-positive (DUX4+) nuclei, respectively. This percentage rose to 80% with 62.5 and up to 250 ng/ml of DOX but was not significantly different among these higher doses. The dose of 62.5 ng/ml of DOX was therefore selected for further experiments because it showed the best DUX4 induction with no effect on cell viability.
The impact of DUX4 on HIF1α mRNA and protein level at a standard PO2 of 21% was then studied in LHCN-M2-iDUX4 cells in proliferation (myoblasts), as well as in early (myocytes) and late (myotubes) differentiation stages.
In proliferating myoblasts, DUX4 induction nearly halved the proportion of HIF1α-positive (HIF1α+) nuclei with 44 % (± 3%) HIF1α+ nuclei in control cells but only 26% (±5%) in the presence of DUX4 (Figure 1A-C). Accordingly, HIF1α mRNA level was halved in proliferating myoblasts expressing DUX4 (Figure 1D) and only 17.8% of HIF1α+ nuclei presented DUX4 IF labeling (Figure 1E-F).
In myocytes with DUX4 induction, the percentage of HIF1α+ nuclei trended toward a decrease without reaching statistical significance (p=0.051, t-test) (Figure 1G-I) while HIF1α mRNA level was not significantly changed (Figure 1J). At this stage, 8% of HIF1α+ nuclei presented DUX4 IF labelling (Figure 1K-L).
2.3. Effect of DUX4 on HIF1α target genes at different stages of human myoblast differentiation in vitro
To further determine the impact of DUX4 on the HIF1α pathway at 21% PO2 we studied the effect of DUX4 expression on two direct HIF1α transcriptional targets: VEGF and PDK1. PDK1 mRNA level was decreased twofold in proliferating myoblasts expressing DUX4, but VEGF mRNA level was not significantly changed compared to non-induced cells (Figure 2A). In agreement with our results obtained at the RNA level, western blotting showed a significant decrease in PDK1 protein abundance in proliferating myoblasts after induction of DUX4 expression (Figure 2B-C). In myocytes expressing DUX4, the mRNA level of both HIF1α target genes was not significantly modified (Figure 2D) but PDK1 protein relative abundance was reduced, as observed in myoblasts (Figure 2E-F). In myotubes, similarly to the results obtained for HIF1α, the mRNA level of its target genes PDK1 and VEGF were significantly increased 3- and 4-fold, respectively (Figure 2G). However, PDK1 protein level was significantly decreased upon DUX4 expression (Figure 2H-I).
2.4. Effect of DUX4 on HIF1α pathway in murine myoblasts and myocytes in vitro.
Since many DUX4-mediated signaling alterations are conserved between human and mouse [25], we investigated whether the effect of DUX4 expression on HIF1α mRNA level observed in human myoblasts was conserved in murine muscle cells. To this aim, we used C2C12-iDUX4 cells derived from mouse C2C12 myoblasts and harboring a DOX-inducible DUX4 gene [17]. LHCN-M2 iDUX4 and C2C12-iDUX4 at a standard PO2 of 21% were both induced using increasing DOX doses (Figure 3A-C). In both cell lines, DUX4 expression decreased the proportion of HIF1α+ nuclei. However, the basal percentage of HIF1α+ nuclei in mouse myoblasts (73% ± 4%) was higher than in human myoblasts (44% ± 3%) (Figure 3C). Moreover, in human myoblasts, DUX4 induction by low DOX doses was sufficient to decrease the percentage of HIF1α+ nuclei. Indeed, 15.6 ng/ml of DOX resulted in a significant decrease in the percentage of HIF1α+ nuclei (32% vs 44% in non-induced controls), while in mouse myoblasts, the reduction of HIF1α+ nuclei could only be seen from 31.2 ng/ml of DOX (Figure 3C). As expected, in mouse myoblasts and myocytes, 62.5 ng/ml of DOX induced the mRNA level of the DUX4 footprint gene Wfdc3 (Figure 3D and 3I). In myoblasts, though the change in Hif1α expression did not reach statistical significance, we observed decreased levels of Hif1α target genes Vegf and Pdk1 in DUX4 expressing cells (Figure 3E), consistent with the reduced percentage of HIF1α+ nuclei presented in Figure 3C. In murine myocytes, DUX4 induction significantly decreased the percentage of HIF1α+ nuclei (Figure 3H). However, as observed in the human cell model (Figure 2D), no significant changes were observed in Hif1a, Vegf and Pdk1 mRNA levels (Figure 3J).
2.5. Characterization of HIF1α pathway modifications upon DUX4 expression in an adult mouse muscle in vivo
To investigate the potential link between DUX4 and the HIF1α pathway in a mature muscle in vivo, we used the DUX4 IMEP mouse model that we had previously developed [37]. In this model, a DUX4 expression plasmid (pCIneo-DUX4) is injected in the mouse Tibialis Anterior (TA) hindlimb muscle followed by electroporation (IMEP) leading to local DUX4 expression and myopathy. First, a dose–response analysis was performed with increasing amounts of pCIneo-DUX4, using the backbone control plasmid (pCIneo) or a saline solution as negative controls. TA muscles were harvested 7, 14 and 21 days post IMEP and frozen (Figure S2A). Cryosections were stained using Hematoxylin-Eosin-Heindehain blue (HEB). The quantification of muscle damage characterized at day 7 by extracellular matrix expansion (fibrosis) and atrophic myofibres (Figure S2B) was reported to total muscle section and performed as described in [37]. At 14 and 21 days after pCIneo-DUX4 electroporation, the TA muscle of DUX4-IMEP mice no longer exhibited these histological features but presented many fibers with centrally located nuclei suggesting muscle regeneration (Figure S2B). Upon quantification of the damaged area, we found no statistical difference between the saline and the pCIneo control plasmid groups, therefore, we pooled data from both groups into a single control group. The lowest pCIneo-DUX4 dose causing a significant increase of the damaged area (median of 20%) compared to the control group (median of 7%) was 5 µg (p<0.05, ANOVA on Ranks followed by Dunn's post hoc test; Figure S2C). To check DUX4 biological activity, we quantified the mRNA level of its mouse target gene Wfdc3. We found no statistical difference in Wfdc3 mRNA level between the saline and the pCIneo control plasmid groups, therefore, we pooled data from both groups into a single control group. A significant increase in Wfdc3 mRNA level was detected by RT-qPCR at days 1, 3, 7 and 14 post-injection, confirming DUX4 expression in the injected TA (Figure S2D). However, we could no longer detect an increase of Wfdc3 mRNA level at 21 days post-injection. We then investigated Hif1α pathway in this model at 1, 3, 7 and 14 days post-injection with the lowest dose of pCIneo-DUX4 causing a significant increase of the damaged TA muscle area (5 µg). No significant difference was detected in mRNA levels of Hif1α and its target gene Pdk1 at any timepoint. However, at one day post-injection only, a significant increase of Vegf mRNA level was observed in the control mice injected with pCIneo as compared to saline. This increase was not detected with pCIneo-DUX4 injection (Figure S2E)
In contrast to the data that we obtained in human myotubes, DUX4 expression did not affect the HIF1α pathway in mouse adult myofibers in the DUX4 IMEP model at the investigated times post injection and by using a 5-µg dose of DUX4 expression plasmid. The first hypothesis that would explain those divergent results was that DUX4 could influence the HIF1α pathway in human but not in murine muscle cells. However, we have shown that DUX4 could decrease the number of HIF1α+ nuclei in murine as well as in human myoblasts (Figure 3). We therefore investigated whether HIF1α dysregulation could constitute an early event following DUX4 expression. To this aim, and to respect the ethical principle of reduction of animal experimentation, we first selected the most relevant acute timepoints in a model in vitro. Uninducible C2C12 murine myoblasts were transfected with pCIneo-DUX4 because this method was closer to the conditions used in the DUX4 IMEP model in vivo as compared to DUX4 inducible cell models (Figure 4A). To evaluate the kinetic of DUX4 target gene transcription following the transfection, we quantified, by RT-qPCR, the mRNA levels of two DUX4 target genes, Wfdc3 and Zscan4 (Figure 4B). We could detect a significant increase of Zscan4 expression at 5h and 6h post-transfection of 4- and 17-fold, respectively. There was also an 8-fold increase in Wfdc3 mRNA level at 6h only. The Hif1α pathway was therefore investigated at these timepoints in the DUX4 IMEP model. To increase the model sensitivity, we also increased the dose of pCIneo-DUX4 up to 20µg to detect a highly significant (compared to 5µg) increase of the muscle lesion area (median of 26%) (p<0.001, ANOVA on Ranks followed by Dunn's post hoc test; Figure 4C-D and S2C). At 6h, 1 day and 7 days post-injection, a significant increase of Wfdc3 mRNA was detected, confirming DUX4 expression in the injected TA (Figure 4E). Concerning Hif1α mRNA level, an increase was observed in TA 6h after injection of pCIneo-DUX4 and pCIneo. At 1 day post injection, Hif1α expression was only increased in the pCIneo-DUX4 group, as compared to the control groups injected with the pCIneo plasmid or the saline solution. However, the expression of target genes Pdk1 and Vegf was not significantly modified whatever experimental group and time point (Figure 4F).
2.7. Effect of a targeted Hif1α knockdown on DUX4-mediated muscle lesions in vivo.
Since DUX4 deregulated Hif1α mRNA level in mouse TA muscle, we studied the involvement of Hif1α in DUX4-mediated muscle damages in vivo through loss-of-function experiments by using siRNAs targeting Hif1α transcripts (siHIF1α) in the DUX4 IMEP model. We first checked siHIF1α efficiency through a dose- response analysis (Figure S3) one day post-injection, when morphological alterations are not yet observable (as we described in [37]). We found that injection of 2µg of siHIF1α allowed a significant two-fold downregulation of Hif1α mRNA level as compared to the TA injected with the control siRNA (siCTL) or saline (Figure 5A and S3). To evaluate the implication of Hif1α in muscle lesions induced by DUX4, we first used as an outcome measure the global quantification of the damaged surface area in the TA muscle of DUX4 IMEP mice at 7 days, namely when a local muscle lesion is observable. At this time point, the lesion is characterized by an extra-cellular matrix (ECM) expansion (fibrosis) and a high number of atrophic fibres [37]. Here, the pCIneo-DUX4 DNA was injected alone or in combination with 2µg siCTL or siHIF1α (Figure 5B-H). A significant 10% increase of muscle lesion area was observed in the siHIF1α group, as compared to the siCTL and saline groups (Figure 5B-C). The muscle lesion in the siHIF1α mouse group was characterized by an exacerbated ECM expansion (as shown by HEB staining, Figure 5B-C) . However, concerning the diameter of remaining myofibers, TA cross-sectional area (CSA) and fiber size distribution were similar in the siHIF1α and the siCTL groups, and were not significantly different from the control DUX4 IMEP mice having not received any siRNA (Figure 5D-F). We also evaluated the kinetic of DUX4 target gene transcription following the IMEP procedure. Within the damaged TA muscle at 7 days, we did not detect any significant difference in Wfdc3 mRNA levels between all groups (Figure 5G). Similarly, we did not observe any difference for the mRNAs of Hif1α or its target genes Pdk1 and Vegf among the groups (Figure 5H).
3. Discussion
3.1. DUX4-induced HIF1 pathway disturbances depend on the differentiation state of human muscle cells.
The hypoxic response pathway was described as critically disturbed in FSHD muscles [15,25]. HIF1α, key driver of this response, was presented as one of the main actors of DUX4-induced myoblast death [30]. In the present study, we confirmed that HIF1α nuclear protein level was altered in human muscle cells upon DUX4 expression. However, these alterations differed according to the stage of myogenic differentiation. Indeed, in proliferating myoblasts, HIF1α was downregulated at the mRNA and protein level upon DUX4 expression but in contrast, DUX4 induced HIF1α and its pathway in myotubes. At the intermediate differentiation stage (myocytes), HIF1α expression was not significantly modified by DUX4 expression. Previous data had suggested that DUX4-mediated cell death required induction of the HIF1α pathway in another model of DUX4 inducible human myoblast (iDUX4-MB135). Indeed, Lek et al. showed HIF1α protein stabilization and nuclear localization upon DUX4 expression as well as a colocalization of HIF1α in nuclei with high DUX4 immunolabeling suggesting that a threshold of DUX4 expression was necessary to trigger HIF1α stabilization [30]. In our study, this positive relationship was observed in DUX4-expressing human LHCN-M2-iDUX4 myotubes, but not in proliferating myoblasts. The use of different muscle cell lines is not expected to strongly influence the DUX4/HIF1α axis, contrarily to culture conditions. Indeed, addition of dexamethasone to the culture medium (as per Lek et al.) was found to lower DUX4 expression in FSHD myoblasts [39]. Similarly, dexamethasone treatment of human myoblasts for 24h decreased expression of VEGF, a HIF1α target gene [40]. In contrast, in the present study, we cultured myoblasts in a medium lacking dexamethasone, based on the original publication which described the inducible LHCN-M2-iDUX4 cell line [18].
The different regulation of HIF1α upon DUX4 expression in myoblasts and myotubes could partly be explained by distinct basal levels of HIF1α according to the differentiation stages. Indeed, basal Hif1α protein level was shown to be higher in murine myoblasts compared to myotubes. This is unlikely due to different gene expression levels since no change in mRNA abundance was seen [41]. We recently confirmed that the percentage of HIF1α+ nuclei decreased during human myoblast differentiation [31]. In the same study, we showed that DUX4 suppressed HIF1α-mediated precocious differentiation of human myoblasts. Several studies of transcription profiles or myocyte fusion index have shown that myogenesis impairment and defects in regeneration were typical features of FSHD muscle biopsies or cell cultures [1,15,18,26,34,42], Our data suggest that the DUX4-mediated disturbances of the HIF1α pathway in myoblasts and myotubes could contribute to these pathological characteristics of FSHD muscle cells.
In keeping with the idea that DUX4 mediated HIF1α inhibition in proliferating myoblasts, we observed a particularly low percentage of DUX4+/HIF1α + nuclei at this stage. In myotubes, this percentage increased but only reached half of HIF1α+ nuclei co-expressing DUX4. Direct or indirect mechanistic hypotheses might be suggested to explain this discrepancy: (I) a direct DUX4-mediated transcription activation of the HIF1A gene. In that case, we expect different kinetics between DUX4 protein synthesis in the cytoplasm, its nuclear translocation, activation of HIF1A gene expression, HIF1α protein synthesis and nuclear translocation (as we described for other DUX4 target genes in [43]). Moreover the kinetics of nuclear/cytoplasm shuttling or protein turnover could also differ. Therefore, the expression dynamics and an asynchronous regulation of DUX4 and HIF1α nuclear location and half-life could explain why both proteins were detected either individually in separate nuclei or together in identical nuclei [43]. Indirect mechanisms may also be suggested: (II) DUX4 could activate HIF1α pathway in satellite cells by disturbing the ability of PAX7 to regulate its transcriptional network [15]. (III) The HIF1α protein could also be stabilized by Reactive Oxygen Species (ROS) through an inhibition of prolyl hydroxylase enzyme (PHDs) activities [44]. Indeed, several clinical and experimental studies indicated both systemic and muscle-specific oxidative stress in FSHD [19,42,45,46,47,48,49]. A recent study also showed high mitochondrial ROS in FSHD myogenic cells [19]. DUX4 could either favor ROS generation or disrupt anti-oxidant processes leading to global ROS increase that could stabilize HIF1α protein even in myotube nuclei that lack DUX4.
3.2. DUX4-induced PDK1 disturbances involved HIF1α-dependent and -independent processes.
We further investigated the effect of DUX4 on the HIF1α pathway through the analysis of its target gene expression. VEGF and PDK1 were globally repressed in proliferating human myoblasts, unchanged in myocytes and induced in myotubes expressing DUX4. Those results are in keeping with variation of HIF1α mRNA and protein levels at the same stages of the myogenic differentiation and suggested a regulatory switch during the differentiation process. The turning point seems to occur at the myocyte stage, namely around the first two days of differentiation in culture. This transition could involve HIF1α-dependent changes metabolic . Indeed, HIF1α is known to up-regulate genes encoding enzymes that promote a glycolytic metabolism [50]. It is well established that most stem cell types reside in hypoxic niches where HIF1α controls pluripotency gene expression, promotes glycolytic metabolism and inhibits mitochondrial biogenesis. However, when myoblasts differentiate to myocytes that fuse into multinucleated myofibers, a reconfiguration of metabolic programs toward OXPHOS occurs, especially via an increased mitochondrial biogenesis and activity [51]. DUX4 was shown to disturb this process via its target gene activation or repression to orchestrate a transcriptome characteristic of a less-differentiated cell state [52]. Therefore, since DUX4 expression activates embryonic genes, and the embryo metabolism is glycolytic, an abnormal activation of glycolysis through HIF1α could contribute to metabolic disturbances in FSHD muscle cells. Accordingly, Heher et al. reported that DUX4-induced changes in oxidative metabolism impaired muscle cells in FSHD and that this phenomenon was amplified when metabolic adaptation to varying O2 tension was required [19].
On the other hand, HIFα -independent processes may also be involved in DUX4-mediated PDK1 disturbances at the protein level. Indeed, whatever the differentiation stage, DUX4 induced a decrease of PDK1 protein abundance. At the opposite, in myotubes, PDK1 mRNA level was induced by DUX4, suggesting a post-translation regulation involving factors independent of HIF1α transcriptional activity. Importantly, PDK1 is a metabolic regulator mainly located in the mitochondria [53]. Actually, evidence for mitochondrial dysfunction was found in FSHD muscle where impaired energy metabolism was linked to alterations in mitochondrial ultrastructure and subcellular distribution [45]. Furthermore, a dynamic transcriptomic analysis identified that suppression of PGC1α/ ERRα axis, a critical component of the mitochondrial biogenesis pathway, was associated to the myogenesis defect in FSHD [33]. Transcriptomic data from FSHD muscle showed enrichment for disturbed mitochondrial pathways. The alteration of mitochondrial ROS metabolism was correlated with mitochondrial membrane polarization and myotube hypotrophy. DUX4-induced modifications of mitochondrial function occurred before mitochondrial ROS generation and affected hypoxia signaling via complex I [19]. Therefore, mitochondrial dysfunction along with the DUX4-mediated mitochondrial ROS production could lead to PDK1 post-transcriptional deregulation e.g. protein degradation through the proteasome or autophagy [54].
3.3. The DUX4/HIF1a axis is conserved in murine models in vitro and in vivo
Our data indicate that the DUX4-HIF axis highlighted in human muscle cells is similar in murine myoblasts and mature muscle in mice. This point is of interest because most therapeutic strategies need testing in vivo in preclinical models. Here, we used the DUX4-IMEP mouse model in which a DUX4 expression plasmid is injected and electroporated into a TA muscle, inducing a dose-dependent local myopathy. In this model, Hif1α mRNA level was increased in both the pCIneo-DUX4 and pCIneo IMEP mice 6 h post injection. The increase in the control plasmid group was likely linked to damage induced by the IMEP procedure. Indeed, an activation of Hif1α signaling was observed in injured TA muscle, increasing from day 1 to day 7 after a cardiotoxin injection [55]. One day post injection the increased Hif1α expression was only maintained in the pCIneo-DUX4 group, meaning that this increase was DUX4-related. In summary, DUX4 expression in murine mature muscle induces an early and transient Hif1α overexpression. However, no modification could be detected in expression of Hif1α target genes Pdk1 and Vegf. We cannot exclude that this could result from limitations of the IMEP model (e.g. variability among animals, local transgene expression levels). However, given that the model sensitivity was sufficient to highlight significant changes of Hif1α expression, our data suggested that both Hif1α-dependent and independent factors could influence Pdk1 and Vegf expression levels. One hypothesis concerning these interfering factors is an activation of the muscle regeneration process [56]. Interestingly, Vegf was shown to play a role in skeletal myofiber regeneration in vivo. Concerning Pdk1, other transcription factors are known to regulate Pdk1 expression such as C-Myc or the Wnt pathway [57,58,59,60].
3.4. Targeted Hif1α knock down in mice exacerbates DUX4-induced muscle fibrosis.
In the IMEP model, muscle alterations caused by one local boost of DUX4 expression constitute an easy read-out through a semi-automated histological quantification of the damaged area by color-thresholding [37]. Moreover, targeting the TA in this local FSHD model is pertinent as it is one of the most affected leg muscle in patients [61]. Here, to specifically evaluate involvement of Hif1α in the development of DUX4 induced muscle lesions, we performed in this model a loss of function study with siRNAs against Hif1α mRNA (siHIF1α). The amount of siRNA we used caused about 2-fold Hif1α mRNA reduction. However, HIF1α knockdown did not reduce the proportion of atrophic fibers in the DUX4 IMEP mice. At the opposite, we observed a significant increase of muscle lesion area characterized by an exacerbated extra-cellular matrix expansion. The extension of muscle lesion with Hif1α knock down could be explained by (I) a synergistic and negative effect of DUX4 and siHIF1α on the myofiber itself or (II) a loss of the critical role of HIF1α in muscle regeneration. Indeed, a role of HIF1α in the regeneration process was first described in murine myoblasts [62,63] and in Hif1α KO mice [64,65]. Moreover, we recently showed that HIF1α is necessary for early myogenic differentiation of human myoblasts [31].
The exacerbation of muscle lesion observed here upon HIF1α knockdown is contrasting with the results by Lek et al [33] obtained with FDA approved HIF signaling inhibitors. In these studies, FSHD-like zebrafish models (i.e. either single cell embryos injected with DUX4 mRNA or transgenic fish eggs with inducible DUX4 expression) were used and an improvement of muscle structure and function was observed. However, most of the drugs used were indirect inhibitors of HIF1α and had multiple effects, notably on protein turnover. and only short-term effects were investigated. For a chronic treatment of muscle disorders, the negative effect of indirect HIF1α inhibitors on protein synthesis should be avoided given the risk to exacerbate muscle atrophy. In addition, as we mentioned in [29], drugs developed to interfere with HIF1α expression or activity were developed in order to induce targeted cancer cell death. This highlights the importance of the context of use when considering the action on HIF1α pathway. While DUX4 was induced at an early embryonic stage in the zebrafish models, our results were obtained by inducing DUX4 expression directly in mature muscle fibers in our mouse model. Both DUX4 and HIF1 α are known to have specific activities during these two different development stages. This could explain the discrepancies observed in the results obtained. Because of its critical role in many skeletal muscle mechanisms, HIF1α remains challenging to target in a therapeutic perspective.
In conclusion, our study has set the basis for further investigations on the role of HIF1α in relationship with DUX4 in FSHD (Figure 6). We found that this link differed according to the muscle cell differentiation stage. Our results also suggested that this axis was conserved between human and mouse. Finally, we found that HIF1α silencing in an FSHD mouse model unexpectedly expanded the DUX4-mediated muscle damaged area, particularly through an exacerbation of fibrosis in the lesion sites. This indicated that the DUX4-HIF1α axis was not as simple as expected and that targeting HIF1α might be challenging in the context of FSHD therapeutic approaches. Finally, given (I) the regeneration defects in FSHD [66,67,68], (II) the role of HIF1α in this process [29,31], (III) the impact of DUX4 expression on the HIF1α pathway depending on the differentiation state, further investigations, especially in cellular actors of muscle regeneration e.g. satellite cells, appear critical for a better understanding of FSHD-associated muscle regeneration disturbances. Moreover, regarding the pivotal role of HIF1α in muscle metabolism, it will be important to clarify whether metabolic disturbances could contribute to the development of muscle dysfunction in FSHD. Such studies would provide more in-depth mechanistic insights into the FSHD pathogenic network and could suggest additional therapeutic targets.
4. Materials and Methods
4.1. Cell culture
Immortalized human myoblast cell lines (LHCN-M2 and LHCN-M2 iDUX4) were kindly provided by Prof. M.Kyba (Lillehei Heart Institute, University of Minnesota, Minneapolis). Cells were cultured in proliferation medium DMEM F12 (BioWest) supplemented with 20% FBS (Biowest), and 1% Penicillin/Streptomycin (P/S, Thermofisher) at 37 °C in a 5% CO2 and atmospheric O2 levels (standard PO2 of 21%). For myogenic differentiation, cells were cultured on matrigel coated dishes (Corning) in proliferation medium until 100% confluence. Cells were then washed once with PBS and differentiated for two days for myocytes and four days for myotubes using differentiating medium (DMEM/F12 (Corning Cellgro), supplemented with human insulin 10 µg/ml (Sigma), bovine apo-transferrin 100 µg/ml (Sigma) and 1% Penicillin/Streptomycin (P/S, Thermofisher).
Immortalized mouse myoblast cell lines (C2C12 and C2C12-iDUX4) were kindly provided by Prof. M. Kyba. They were cultured in proliferation medium DMEM high glucose (BioWest) supplemented with 10% FBS (Biowest), and 1% Penicillin/Streptomycin (P/S, Thermofisher) at 37 °C in a 5% CO2 atmosphere.
4.2. Viability test
For the Vybrant® MTT Cell Proliferation Assay Kit (ThermoFisher), LHCN-M2 iDUX4 et LHCN-M2 cells were seeded in a 96 well-plate and induced for 24h with doxycycline. Cells were then incubated for 2h with MTT 1.2 mM reagent diluted in proliferation medium at 37°C. After that step, medium was replaced by DMSO to solubilize the formazan product and the plate was incubated for 10 min at 37°C under agitation. The absorbance was then measured by a spectrophotometer (VERSA max-SoftMax Pro) at 540 nm.
For Cell Counting Kit-8 (Sigma), LHCN-M2 iDUX4 and LHCN-M2 myoblasts were seeded in 96 well-plate and induced for 24h with doxycycline. Cells were then incubated for 1h with CCK-8 solution diluted in proliferation medium at 37°C. Absorbance was then measured by a spectrophotometer (VERSA max-SoftMax Pro) at 450 nm.
4.3. Myoblast transfection
105 C2C12 mouse cells were seeded in 6-well plates and transfected 24 hours later in Opti-MEM (Invitrogen, CA, USA) with 5µl of Lipofectamin 2000 (Invitrogen) and 1600 ng of DNA vector according to the manufacturer's instructions.
4.4. Immunofluorescence
Cells previously seeded in 6-well plate on glass slide, were fixed with 4% paraformaldehyde/PBS for 10 min, permeabilized with 0.5% TritonX-100/PBS for 10 min, then incubated with blocking solution (5% normal goat serum (Biowest), TritonX-100/PBS) for 1h at room temperature. Cells were then incubated with primary antibodies (anti-DUX4 9A12 MABD116; 1:100, Merck ([43]); HIF1α ab179483, 1:500, Abcam) at 4 °C overnight. They were subsequently rinsed in PBS and incubated with secondary antibodies Alexa 555 Goat anti-rabbit IgG (1:500, Biotium) and Alexa 488 Goat anti-mouse IgG (1:500, Biotium) at room temperature for 1 h. Immunolabelled cells were rinsed in PBS and mounted with EverBrite Mounting Medium with DAPI (Biotium) for nuclear staining. Pictures were taken with a Nikon Eclipse 80i microscope and merged using NIS-Elements software.
4.5. qPCR
RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s directions. Total RNA was then treated with DNAse I (amplification grade, ThermoFisher). cDNAs were synthetized using Maxima First Strand cDNA Synthesis kit (ThermoFisher). All qPCRs were performed in triplicates using SYBR Green FastStart Essential DNA Green Master (Roche) and corresponding primers (Eurogentec) (See Supplementary materials). Cycling conditions were as follows: initial denaturation step at 95 °C for 10 min, followed by 40 cycles of 15 s at 95 °C and 60 s at primer Tm. qPCR results were analyzed with LightCycler 96 software (Roche). Quantifications were performed using the 2-∆∆Ct method.
4.6. Western Blot
Cells were lysed using RIPA buffer. Proteins were separated on 12% SDS-PAGE gels for 3h at 100V and transferred to nitrocellulose membrane for 1h45 at 260 mA. Membrane blocking was performed using 5% non fat dry milk diluted in TBST-T. Primary (PDK1, ab110025, Abcam) and secondary HRP conjugated antibody (NA931, ECL) were diluted in 5% skim milk in TBST and incubated overnight at 4 °C and 1 hour at room temperature respectively. HRP signal was visualized using Supersignal West Femto max. sensitivity kit (Thermo Scientific) and the Fusion FX7 spectra (Vilber)
4.7. Ethics statement
All animal experiments met the Belgian national standard requirements regarding animal care and were conducted in accordance with the Ethics and Welfare Committee of the University of Mons (reference number LE018/02).
4.8. IMEP mouse model
Female C57BL/6 mice, aged between 8 and 12 weeks, were purchase from Charles River laboratories (France). Mice were housed in a conventional animal colony and maintained at 35–40% relative humidity with a constant room temperature (21 °C) and natural day/night light cycle (12–12 h). Food and water were provided ad libitum and animals were subjected to an adaptation period of 7 days before experiments. IMEP model was generated as previously described [37]. Briefly, tibialis anterior (TA) muscles were injected with 40 µg of hyaluronidase. Each TA was then injected with either naked plasmid DNA alone or complemented with siRNAs targeting HIF1α RNA (siHIF; Qiagen, #1027416, FlexiTube) or control (siCTL; Qiagen, #1027280, “all star negative control”). As in [31] a mix of 4 siRNAs directed against the HIF1α mRNA were used. Redundancy experiments using several distinct siRNAs targeting different sequences of the same mRNA prevent sequence-derived off-target effects. SiRNA were electroporated using an EMKA stimulator. Mice were checked daily and then sacrificed by an intraperitoneal injection of Nembutal (Kela).
4.9. Tissue preparation and histology
At the indicated euthanasia time points, right and left TAs were removed, embedded in OCT compound (VWR) and frozen in liquid nitrogen-cooled isopentane. 8 µm thick cryostat sections from proximal and medial of TA were cut using a Leica cryotome and sections were stained with Hematoxylin–Eosin–Heidenhain blue (HEB) to evaluate the percentage of muscle lesions. HEB staining consists in a basic Hematoxylin–Eosin coloration followed by a 45-s incubation in Heidenhain’s Blue staining (mix of orange G and Aniline Blue, Sigma-Aldrich, USA), allowing an intense blue labeling of fibrotic fibers and collagenous tissues. Slides were then scanned using the NanoZoomer-SQ Digital slide scanner (Hamamatsu Photonics). Images were processed as described in [37]. Transgene expression and lesion distribution were characterized in [37]. Each myofibre cross-section areas (CSA) were measured on the whole muscle section by using the Image J software. Total CSA was calculated. Myofibres were also classified in clusters according to their CSA. Myofiber CSA distribution was presented as well as the cumulative percentage of myofibres in each cluster.
5. Statistical analysis
Normality tests (Shapiro-Wilk) were performed on each data sets to assess the data distribution and thus appropriate statistic tests could be chosen. Differences were considered statistically significant at a p-value < 0.05. All data were represented as mean +/- SEM or boxplot (5 and 95th percentile) for parametric or non-parametric statistical tests, respectively. Statistical analyses were done using GraphPad Prism software, version 8.02 and SigmaPlot software, version 14.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization: Thuy-Hang Nguyen, Alexandre Legrand, Anne-Emilie Declèves, Philipp Heher, Alexandra Belayew, Christopher R. S. Banerji, Peter S. Zammit and Alexandra Tassin; Data curation: Thuy-Hang Nguyen, Sihame Bouhmidi, Lise Paprzycki, Maelle Limpens and Alexandra Tassin; Funding acquisition: Thuy-Hang Nguyen, Peter S. Zammit, Philipp Heher and Alexandra Tassin; Investigation: Thuy-Hang Nguyen, Sihame Bouhmidi, Maelle Limpens, Lise Paprzycki, and Alexandra Tassin; Supervision: Alexandre Legrand, Anne-Emilie Declèves, Alexandra Belayew, Peter S. Zammit and Alexandra Tassin; Writing – original draft: Thuy-Hang Nguyen, Alexandra Belayew and Alexandra Tassin; Writing – review & editing, Alexandre Legrand, Anne-Emilie Declèves, Philipp Heher, Alexandra Belayew, Christopher R. S. Banerji, Peter S. Zammit and Alexandra Tassin.
Funding
T.-H.N. and M.L. held a FRIA doctoral fellowship (FC 29703 and FC 47057) from the National Fund for Scientific Research (F.R.S – FNRS), Belgium. L.P. had a UMONS-UNAMUR doctoral fellowship. PH was mainly funded by the Medical Research Council (MR/P023215/1) and then by an Erwin Schroedinger post-doctoral fellowship awarded by the Austrian Science Fund (FWF, J4435-B), supported by Friends of FSH Research (Project 936270) and the FSHD Society (FSHD-Fall2020-3308289076) and currently the Medical Research Council (MR/S002472/1).This scientific study was funded by several patient associations: the French non-profit organization AMIS FSH (France) whose objective is to heal and support patients suffering from Facio Scapulo Humeral Dystrophy, Association Belge contre les Maladies neuro-Musculaires (ABMM, Belgium) and Association Française contre les Myopathies (AFM Telethon France). This work was also supported by the Fonds de la Recherche Scientifique - FNRS (Belgium) under Grant(s) n° Equipment UN07220F. CRSB was supported by the Turing-Roche Strategic Partnership.
Acknowledgments
We would like to thank Pr. M. Kyba for providing the human and mouse myoblast lines. We thank V. Jenart for excellent technical help.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Banerji, C.R.S.; Zammit, P.S. Pathomechanisms and Biomarkers in Facioscapulohumeral Muscular Dystrophy: Roles of DUX4 and PAX7. EMBO Mol. Med. 2021, 13, e13695. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Tawil, R. Facioscapulohumeral Dystrophy. Curr. Neurol. Neurosci. Rep. 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Mattéotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a Candidate Gene of Facioscapulohumeral Muscular Dystrophy, Encodes a Transcriptional Activator of PITX1. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.L.F.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 Activates Germline Genes, Retroelements and Immune-Mediators: Implications for Facioscapulohumeral Dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef]
- Snider, L.; Geng, L.N.; Lemmers, R.J.L.F.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral Dystrophy: Incomplete Suppression of a Retrotransposed Gene. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Vanderplanck, C.; Ansseau, E.; Charron, S.; Stricwant, N.; Tassin, A.; Laoudj-Chenivesse, D.; Wilton, S.D.; Coppée, F.; Belayew, A. The FSHD Atrophic Myotube Phenotype Is Caused by DUX4 Expression. PLOS ONE 2011, 6, e26820. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-Family Transcription Factors Regulate Zygotic Genome Activation in Placental Mammals. Nat. Genet. 2017, 49, 941–945. [Google Scholar] [CrossRef]
- Hendrickson, P.G.; Doráis, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, J.-W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved Roles of Mouse DUX and Human DUX4 in Activating Cleavage-Stage Genes and MERVL/HERVL Retrotransposons. Nat. Genet. 2017, 49, 925–934. [Google Scholar] [CrossRef]
- Bosnakovski, D.; Gearhart, M.D.; Ho Choi, S.; Kyba, M. Dux Facilitates Post-Implantation Development, but Is Not Essential for Zygotic Genome Activation†. Biol. Reprod. 2021, 104, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Gabriëls, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide Sequence of the Partially Deleted D4Z4 Locus in a Patient with FSHD Identifies a Putative Gene within Each 3.3 Kb Element. Gene 1999, 236, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R. Chromosome 4q DNA Rearrangements Associated with Facioscapulohumeral Muscular Dystrophy. Nat. Genet. 1992, 2, 26–30. [Google Scholar] [CrossRef] [PubMed]
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD Associated DNA Rearrangements Are Due to Deletions of Integral Copies of a 3.2 Kb Tandemly Repeated Unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Banerji, C.R.S.; Panamarova, M.; Hebaishi, H.; White, R.B.; Relaix, F.; Severini, S.; Zammit, P.S. PAX7 Target Genes Are Globally Repressed in Facioscapulohumeral Muscular Dystrophy Skeletal Muscle. Nat. Commun. 2017, 8, 2152. [Google Scholar] [CrossRef] [PubMed]
- Banerji, C.R.S. PAX7 Target Gene Repression Associates with FSHD Progression and Pathology over 1 Year. Hum. Mol. Genet. 2020, 29, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Xu, Z.; Gang, E.J.; Galindo, C.L.; Liu, M.; Simsek, T.; Garner, H.R.; Agha-Mohammadi, S.; Tassin, A.; Coppée, F.; et al. An Isogenetic Myoblast Expression Screen Identifies DUX4-Mediated FSHD-Associated Molecular Pathologies. EMBO J. 2008, 27, 2766–2779. [Google Scholar] [CrossRef] [PubMed]
- Bosnakovski, D.; Gearhart, M.D.; Toso, E.A.; Ener, E.T.; Choi, S.H.; Kyba, M. Low Level DUX4 Expression Disrupts Myogenesis through Deregulation of Myogenic Gene Expression. Sci. Rep. 2018, 8, 16957. [Google Scholar] [CrossRef] [PubMed]
- Heher, P.; Ganassi, M.; Weidinger, A.; Engquist, E.N.; Pruller, J.; Nguyen, T.H.; Tassin, A.; Declèves, A.-E.; Mamchaoui, K.; Banerji, C.R.S.; et al. Interplay between Mitochondrial Reactive Oxygen Species, Oxidative Stress and Hypoxic Adaptation in Facioscapulohumeral Muscular Dystrophy: Metabolic Stress as Potential Therapeutic Target. Redox Biol. 2022, 51, 102251. [Google Scholar] [CrossRef]
- Celegato, B.; Capitanio, D.; Pescatori, M.; Romualdi, C.; Pacchioni, B.; Cagnin, S.; Viganò, A.; Colantoni, L.; Begum, S.; Ricci, E.; et al. Parallel Protein and Transcript Profiles of FSHD Patient Muscles Correlate to the D4Z4 Arrangement and Reveal a Common Impairment of Slow to Fast Fibre Differentiation and a General Deregulation of MyoD-Dependent Genes. PROTEOMICS 2006, 6, 5303–5321. [Google Scholar] [CrossRef]
- Jagannathan, S.; Ogata, Y.; Gafken, P.R.; Tapscott, S.J.; Bradley, R.K. Quantitative Proteomics Reveals Key Roles for Post-Transcriptional Gene Regulation in the Molecular Pathology of Facioscapulohumeral Muscular Dystrophy. eLife 2019, 8, e41740. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.M.; Petek, L.M.; Miller, D.G. Endogenous DUX4 Expression in FSHD Myotubes Is Sufficient to Cause Cell Death and Disrupts RNA Splicing and Cell Migration Pathways. Hum. Mol. Genet. 2015, 24, 5901–5914. [Google Scholar] [CrossRef] [PubMed]
- Ansseau, E.; Eidahl, J.O.; Lancelot, C.; Tassin, A.; Matteotti, C.; Yip, C.; Liu, J.; Leroy, B.; Hubeau, C.; Gerbaux, C.; et al. Homologous Transcription Factors DUX4 and DUX4c Associate with Cytoplasmic Proteins during Muscle Differentiation. PloS One 2016, 11, e0146893. [Google Scholar] [CrossRef] [PubMed]
- DeSimone, A.M.; Leszyk, J.; Wagner, K.; Emerson, C.P. Identification of the Hyaluronic Acid Pathway as a Therapeutic Target for Facioscapulohumeral Muscular Dystrophy. Sci. Adv. 2019, 5, eaaw7099. [Google Scholar] [CrossRef] [PubMed]
- Banerji, C.R.S.; Knopp, P.; Moyle, L.A.; Severini, S.; Orrell, R.W.; Teschendorff, A.E.; Zammit, P.S. β-Catenin Is Central to DUX4-Driven Network Rewiring in Facioscapulohumeral Muscular Dystrophy. J. R. Soc. Interface 2015, 12, 20140797. [Google Scholar] [CrossRef] [PubMed]
- Tsumagari, K.; Chang, S.-C.; Lacey, M.; Baribault, C.; Chittur, S.V.; Sowden, J.; Tawil, R.; Crawford, G.E.; Ehrlich, M. Gene Expression during Normal and FSHD Myogenesis. BMC Med. Genomics 2011, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Spivak-Kroizman, T.R.; Powis, G. HIF-1 Regulation: Not so Easy Come, Easy Go. Trends Biochem. Sci. 2008, 33, 526–534. [Google Scholar] [CrossRef]
- Macklin, P.S.; Yamamoto, A.; Browning, L.; Hofer, M.; Adam, J.; Pugh, C.W. Recent Advances in the Biology of Tumour Hypoxia with Relevance to Diagnostic Practice and Tissue-Based Research. J. Pathol. 2020, 250, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-H.; Conotte, S.; Belayew, A.; Declèves, A.-E.; Legrand, A.; Tassin, A. Hypoxia and Hypoxia-Inducible Factor Signaling in Muscular Dystrophies: Cause and Consequences. Int. J. Mol. Sci. 2021, 22, 7220. [Google Scholar] [CrossRef]
- Lek, A.; Zhang, Y.; Woodman, K.G.; Huang, S.; DeSimone, A.M.; Cohen, J.; Ho, V.; Conner, J.; Mead, L.; Kodani, A.; et al. Applying Genome-Wide CRISPR-Cas9 Screens for Therapeutic Discovery in Facioscapulohumeral Muscular Dystrophy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Nguyen, T.-H.; Paprzycki, L.; Legrand, A.; Declèves, A.-E.; Heher, P.; Limpens, M.; Belayew, A.; Banerji, C.R.S.; Zammit, P.S.; Tassin, A. Hypoxia Enhances Human Myoblast Differentiation: Involvement of HIF1α and Impact of DUX4, the FSHD Causal Gene. Skelet. Muscle 2023, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- De Iaco, A.; Verp, S.; Offner, S.; Grun, D.; Trono, D. DUX Is a Non-Essential Synchronizer of Zygotic Genome Activation. Dev. Camb. Engl. 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Banerji, C.R.S.; Panamarova, M.; Pruller, J.; Figeac, N.; Hebaishi, H.; Fidanis, E.; Saxena, A.; Contet, J.; Sacconi, S.; Severini, S.; et al. Dynamic Transcriptomic Analysis Reveals Suppression of PGC1α/ERRα Drives Perturbed Myogenesis in Facioscapulohumeral Muscular Dystrophy. Hum. Mol. Genet. 2019, 28, 1244–1259. [Google Scholar] [CrossRef] [PubMed]
- Barro, M.; Carnac, G.; Flavier, S.; Mercier, J.; Vassetzky, Y.; Laoudj-Chenivesse, D. Myoblasts from Affected and Non-Affected FSHD Muscles Exhibit Morphological Differentiation Defects. J. Cell. Mol. Med. 2010, 14, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 Recruits P300/CBP through Its C-Terminus and Induces Global H3K27 Acetylation Changes. Nucleic Acids Res. 2016, 44, 5161–5173. [Google Scholar] [CrossRef]
- Bosnakovski, D.; Xu, Z.; Gang, E.J.; Galindo, C.L.; Liu, M.; Simsek, T.; Garner, H.R.; Agha-Mohammadi, S.; Tassin, A.; Coppée, F.; et al. An Isogenetic Myoblast Expression Screen Identifies DUX4-Mediated FSHD-Associated Molecular Pathologies. EMBO J. 2008, 27, 2766–2779. [Google Scholar] [CrossRef] [PubMed]
- Derenne, A.; Tassin, A.; Nguyen, T.H.; De Roeck, E.; Jenart, V.; Ansseau, E.; Belayew, A.; Coppée, F.; Declèves, A.-E.; Legrand, A. Induction of a Local Muscular Dystrophy Using Electroporation in Vivo: An Easy Tool for Screening Therapeutics. Sci. Rep. 2020, 10, 11301. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.-H.; Mouly, V.; Cooper, R.N.; Mamchaoui, K.; Bigot, A.; Shay, J.W.; Di Santo, J.P.; Butler-Browne, G.S.; Wright, W.E. Cellular Senescence in Human Myoblasts Is Overcome by Human Telomerase Reverse Transcriptase and Cyclin-Dependent Kinase 4: Consequences in Aging Muscle and Therapeutic Strategies for Muscular Dystrophies. Aging Cell 2007, 6, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.N.; Khawaja, H.; Chen, Y.-W. Culture Conditions Affect Expression of DUX4 in FSHD Myoblasts. Molecules 2015, 20, 8304–8315. [Google Scholar] [CrossRef]
- Pirkmajer, S.; Filipovic, D.; Mars, T.; Mis, K.; Grubic, Z. HIF-1α Response to Hypoxia Is Functionally Separated from the Glucocorticoid Stress Response in the in Vitro Regenerating Human Skeletal Muscle. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 299, R1693–R1700. [Google Scholar] [CrossRef]
- Dehne, N.; Kerkweg, U.; Otto, T.; Fandrey, J. The HIF-1 Response to Simulated Ischemia in Mouse Skeletal Muscle Cells Neither Enhances Glycolysis nor Prevents Myotube Cell Death. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 293, R1693–R1701. [Google Scholar] [CrossRef] [PubMed]
- Winokur, S.T.; Barrett, K.; Martin, J.H.; Forrester, J.R.; Simon, M.; Tawil, R.; Chung, S.-A.; Masny, P.S.; Figlewicz, D.A. Facioscapulohumeral Muscular Dystrophy (FSHD) Myoblasts Demonstrate Increased Susceptibility to Oxidative Stress. Neuromuscul. Disord. NMD 2003, 13, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.-W.; Mercier, J.; Coppée, F.; Belayew, A. DUX4 Expression in FSHD Muscle Cells: How Could Such a Rare Protein Cause a Myopathy? J. Cell. Mol. Med. 2013, 17, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Hagen, T. Oxygen versus Reactive Oxygen in the Regulation of HIF-1α: The Balance Tips. Biochem. Res. Int. 2012, 2012, 436981. [Google Scholar] [CrossRef] [PubMed]
- Turki, A.; Hayot, M.; Carnac, G.; Pillard, F.; Passerieux, E.; Bommart, S.; Raynaud de Mauverger, E.; Hugon, G.; Pincemail, J.; Pietri, S.; et al. Functional Muscle Impairment in Facioscapulohumeral Muscular Dystrophy Is Correlated with Oxidative Stress and Mitochondrial Dysfunction. Free Radic. Biol. Med. 2012, 53, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Passerieux, E.; Hayot, M.; Jaussent, A.; Carnac, G.; Gouzi, F.; Pillard, F.; Picot, M.-C.; Böcker, K.; Hugon, G.; Pincemail, J.; et al. Effects of Vitamin C, Vitamin E, Zinc Gluconate, and Selenomethionine Supplementation on Muscle Function and Oxidative Stress Biomarkers in Patients with Facioscapulohumeral Dystrophy: A Double-Blind Randomized Controlled Clinical Trial. Free Radic. Biol. Med. 2015, 81, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Wilson, V.D.; Thomas, C.; Passerieux, E.; Hugon, G.; Pillard, F.; Andrade, A.G.; Bommart, S.; Picot, M.-C.; Pincemail, J.; Mercier, J.; et al. Impaired Oxygen Demand during Exercise Is Related to Oxidative Stress and Muscle Function in Facioscapulohumeral Muscular Dystrophy. JCSM Rapid Commun. 2018, 1, 1–13. [Google Scholar] [CrossRef]
- Sasaki-Honda, M.; Jonouchi, T.; Arai, M.; Hotta, A.; Mitsuhashi, S.; Nishino, I.; Matsuda, R.; Sakurai, H. A Patient-Derived iPSC Model Revealed Oxidative Stress Increases Facioscapulohumeral Muscular Dystrophy-Causative DUX4. Hum. Mol. Genet. 2018, 27, 4024–4035. [Google Scholar] [CrossRef]
- Karpukhina, A.; Galkin, I.; Ma, Y.; Dib, C.; Zinovkin, R.; Pletjushkina, O.; Chernyak, B.; Popova, E.; Vassetzky, Y. Analysis of Genes Regulated by DUX4 via Oxidative Stress Reveals Potential Therapeutic Targets for Treatment of Facioscapulohumeral Dystrophy. Redox Biol. 2021, 43, 102008. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1, O(2), and the 3 PHDs: How Animal Cells Signal Hypoxia to the Nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef]
- Fortini, P.; Ferretti, C.; Iorio, E.; Cagnin, M.; Garribba, L.; Pietraforte, D.; Falchi, M.; Pascucci, B.; Baccarini, S.; Morani, F.; et al. The Fine Tuning of Metabolism, Autophagy and Differentiation during in Vitro Myogenesis. Cell Death Dis. 2016, 7, e2168–e2168. [Google Scholar] [CrossRef] [PubMed]
- Knopp, P.; Krom, Y.D.; Banerji, C.R.S.; Panamarova, M.; Moyle, L.A.; den Hamer, B.; van der Maarel, S.M.; Zammit, P.S. DUX4 Induces a Transcriptome More Characteristic of a Less-Differentiated Cell State and Inhibits Myogenesis. J. Cell Sci. 2016, 129, 3816–3831. [Google Scholar] [CrossRef]
- Wang, X.; Shen, X.; Yan, Y.; Li, H. Pyruvate Dehydrogenase Kinases (PDKs): An Overview toward Clinical Applications. Biosci. Rep. 2021, 41, BSR20204402. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; Dias, I.H.K.; Debelec, B.; Vucetic, M.; Fladmark, K.E.; Basaga, H.; Ribaric, S.; Milisav, I.; Cuadrado, A. Redox Control of Protein Degradation. Redox Biol. 2015, 6, 409–420. [Google Scholar] [CrossRef]
- Drouin, G.; Couture, V.; Lauzon, M.-A.; Balg, F.; Faucheux, N.; Grenier, G. Muscle Injury-Induced Hypoxia Alters the Proliferation and Differentiation Potentials of Muscle Resident Stromal Cells. Skelet. Muscle 2019, 9, 18. [Google Scholar] [CrossRef]
- Arsic, N.; Zacchigna, S.; Zentilin, L.; Ramirez-Correa, G.; Pattarini, L.; Salvi, A.; Sinagra, G.; Giacca, M. Vascular Endothelial Growth Factor Stimulates Skeletal Muscle Regeneration in Vivo. Mol. Ther. 2004, 10, 844–854. [Google Scholar] [CrossRef]
- Li, Z.; Van Calcar, S.; Qu, C.; Cavenee, W.K.; Zhang, M.Q.; Ren, B. A Global Transcriptional Regulatory Role for C-Myc in Burkitt’s Lymphoma Cells. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 8164–8169. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gao, P.; Liu, Y.-C.; Semenza, G.L.; Dang, C.V. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol. Cell. Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt Signaling Directs a Metabolic Program of Glycolysis and Angiogenesis in Colon Cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef]
- Lee, M.; Chen, G.T.; Puttock, E.; Wang, K.; Edwards, R.A.; Waterman, M.L.; Lowengrub, J. Mathematical Modeling Links Wnt Signaling to Emergent Patterns of Metabolism in Colon Cancer. Mol. Syst. Biol. 2017, 13, 912. [Google Scholar] [CrossRef]
- Olsen, D.B.; Gideon, P.; Jeppesen, T.D.; Vissing, J. Leg Muscle Involvement in Facioscapulohumeral Muscular Dystrophy Assessed by MRI. J. Neurol. 2006, 253, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Resmini, G.; Ghiroldi, A.; Piccoli, M.; Bergante, S.; Tettamanti, G.; Anastasia, L. Activation of the Hypoxia-Inducible Factor 1a Promotes Myogenesis through the Noncanonical Wnt Pathway, Leading to Hypertrophic Myotubes. FASEB J. 2017, 31, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Vlaminck, B.; Toffoli, S.; Ghislain, B.; Demazy, C.; Raes, M.; Michiels, C. Dual Effect of Echinomycin on Hypoxia-Inducible Factor-1 Activity under Normoxic and Hypoxic Conditions. FEBS J. 2007, 274, 5533–5542. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, S.; Wang, C.; Kuang, S. The Hypoxia-Inducible Factors HIF1α and HIF2α Are Dispensable for Embryonic Muscle Development but Essential for Postnatal Muscle Regeneration. J. Biol. Chem. 2017, 292, 5981–5991. [Google Scholar] [CrossRef] [PubMed]
- Scheerer, N.; Dehne, N.; Stockmann, C.; Swoboda, S.; Baba, H.A.; Neugebauer, A.; Johnson, R.S.; Fandrey, J. Myeloid Hypoxia-Inducible Factor-1α Is Essential for Skeletal Muscle Regeneration in Mice. J. Immunol. Baltim. Md 1950 2013, 191, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, L.; Giacalone, F.; Ragozzino, E.; Saccone, V.; Tiberio, F.; De Bardi, M.; Picozza, M.; Borsellino, G.; Lattanzi, W.; Guadagni, E.; et al. Non-Myogenic Mesenchymal Cells Contribute to Muscle Degeneration in Facioscapulohumeral Muscular Dystrophy Patients. Cell Death Dis. 2022, 13, 793. [Google Scholar] [CrossRef] [PubMed]
- Ganassi, M.; Zammit, P.S. Involvement of Muscle Satellite Cell Dysfunction in Neuromuscular Disorders: Expanding the Portfolio of Satellite Cell-Opathies. Eur. J. Transl. Myol. 2022, 32. [Google Scholar] [CrossRef]
- Ganassi, M.; Muntoni, F.; Zammit, P.S. Defining and Identifying Satellite Cell-Opathies within Muscular Dystrophies and Myopathies. Exp. Cell Res. 2022, 411, 112906. [Google Scholar] [CrossRef]
Figure 1.
Differential effect of DUX4 on HIF1α expression and protein level in human LHCN-M2-iDUX4 muscle cells depends on the stage of differentiation. LHCN-M2-iDUX4 myoblasts were cultured and seeded as described in [31] at a standard PO2 of 21%. DUX4 expression was induced by the addition of 62,5 ng/ml of doxycycline (DOX) to the culture medium. For differentiation cells at confluence were switched to differentiation medium for two (myocytes) or four days (myotubes). Cells were fixed in 4% PAF and immunofluorescence (IF) was performed with antibodies directed against HIF1α or DUX4 and appropriate secondary antibodies coupled to Alexa Fluor (s). A, G, M. Experiment time courses. B, H, N. Representative fields showing HIF1α positive (HIF1α+) nuclei (red IF). DAPI was used to visualize nuclei (blue). Scale bar = 100µm. C, I, O. Quantification of HIF1α+ nuclei normalized to the total number of nuclei (DAPI staining). Mean ± SEM, **p<0.01, T-test. D, J, P. Relative HIF1α mRNA level normalized to RPLP0. Mean ± SEM, *p<0.05, **p<0.01, T-test. N=4 for myoblasts, N=3 for myocytes and myotubes. E, K, Q. Proportion of DUX4+ nuclei among HIF1α+ nuclei. F, L, R. Representative field showing HIF1α+ (red IF) and DUX4+ (green IF). Nuclei were stained with DAPI (blue). All experiments were performed on 3 independent cultures, each at least in triplicate.
Figure 1.
Differential effect of DUX4 on HIF1α expression and protein level in human LHCN-M2-iDUX4 muscle cells depends on the stage of differentiation. LHCN-M2-iDUX4 myoblasts were cultured and seeded as described in [31] at a standard PO2 of 21%. DUX4 expression was induced by the addition of 62,5 ng/ml of doxycycline (DOX) to the culture medium. For differentiation cells at confluence were switched to differentiation medium for two (myocytes) or four days (myotubes). Cells were fixed in 4% PAF and immunofluorescence (IF) was performed with antibodies directed against HIF1α or DUX4 and appropriate secondary antibodies coupled to Alexa Fluor (s). A, G, M. Experiment time courses. B, H, N. Representative fields showing HIF1α positive (HIF1α+) nuclei (red IF). DAPI was used to visualize nuclei (blue). Scale bar = 100µm. C, I, O. Quantification of HIF1α+ nuclei normalized to the total number of nuclei (DAPI staining). Mean ± SEM, **p<0.01, T-test. D, J, P. Relative HIF1α mRNA level normalized to RPLP0. Mean ± SEM, *p<0.05, **p<0.01, T-test. N=4 for myoblasts, N=3 for myocytes and myotubes. E, K, Q. Proportion of DUX4+ nuclei among HIF1α+ nuclei. F, L, R. Representative field showing HIF1α+ (red IF) and DUX4+ (green IF). Nuclei were stained with DAPI (blue). All experiments were performed on 3 independent cultures, each at least in triplicate.
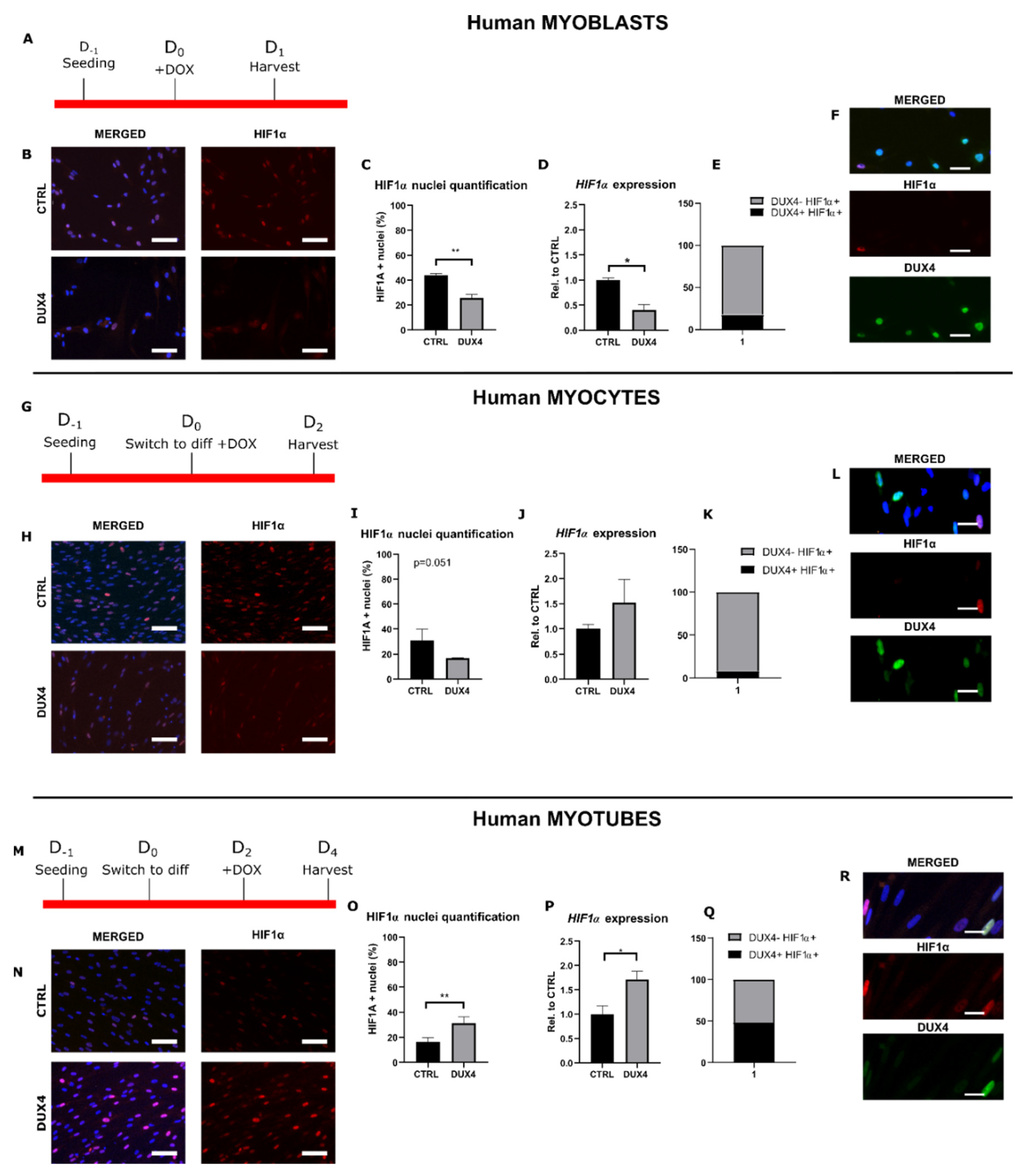
Figure 2.
Effect of DUX4 induction on HIF1α target genes in human LHCN-M2-iDUX4 muscle cells. Cell culture, induction of DUX4 expression by doxycycline and myogenic differentiation were performed at a standard PO2 of 21% as in Figure 1. A, D, G. Expression levels of PDK1 and VEGF mRNAs. Quantifications were performed by RT-qPCR and normalized to RPLP0. Mean ± SEM, **p<0.01, ***p<0.001, T-test. N=4 for myoblasts, N=3 for myocytes and myotubes. B, E, H. PDK1 protein level determined by Western blot. Densitometry signal was normalized to total protein stained by Ponceau red. N=6 for myoblasts and myocytes, n=3 for myotubes. C, F, I. Representative Western blot and Ponceau red staining for PDK1 detection .
Figure 2.
Effect of DUX4 induction on HIF1α target genes in human LHCN-M2-iDUX4 muscle cells. Cell culture, induction of DUX4 expression by doxycycline and myogenic differentiation were performed at a standard PO2 of 21% as in Figure 1. A, D, G. Expression levels of PDK1 and VEGF mRNAs. Quantifications were performed by RT-qPCR and normalized to RPLP0. Mean ± SEM, **p<0.01, ***p<0.001, T-test. N=4 for myoblasts, N=3 for myocytes and myotubes. B, E, H. PDK1 protein level determined by Western blot. Densitometry signal was normalized to total protein stained by Ponceau red. N=6 for myoblasts and myocytes, n=3 for myotubes. C, F, I. Representative Western blot and Ponceau red staining for PDK1 detection .
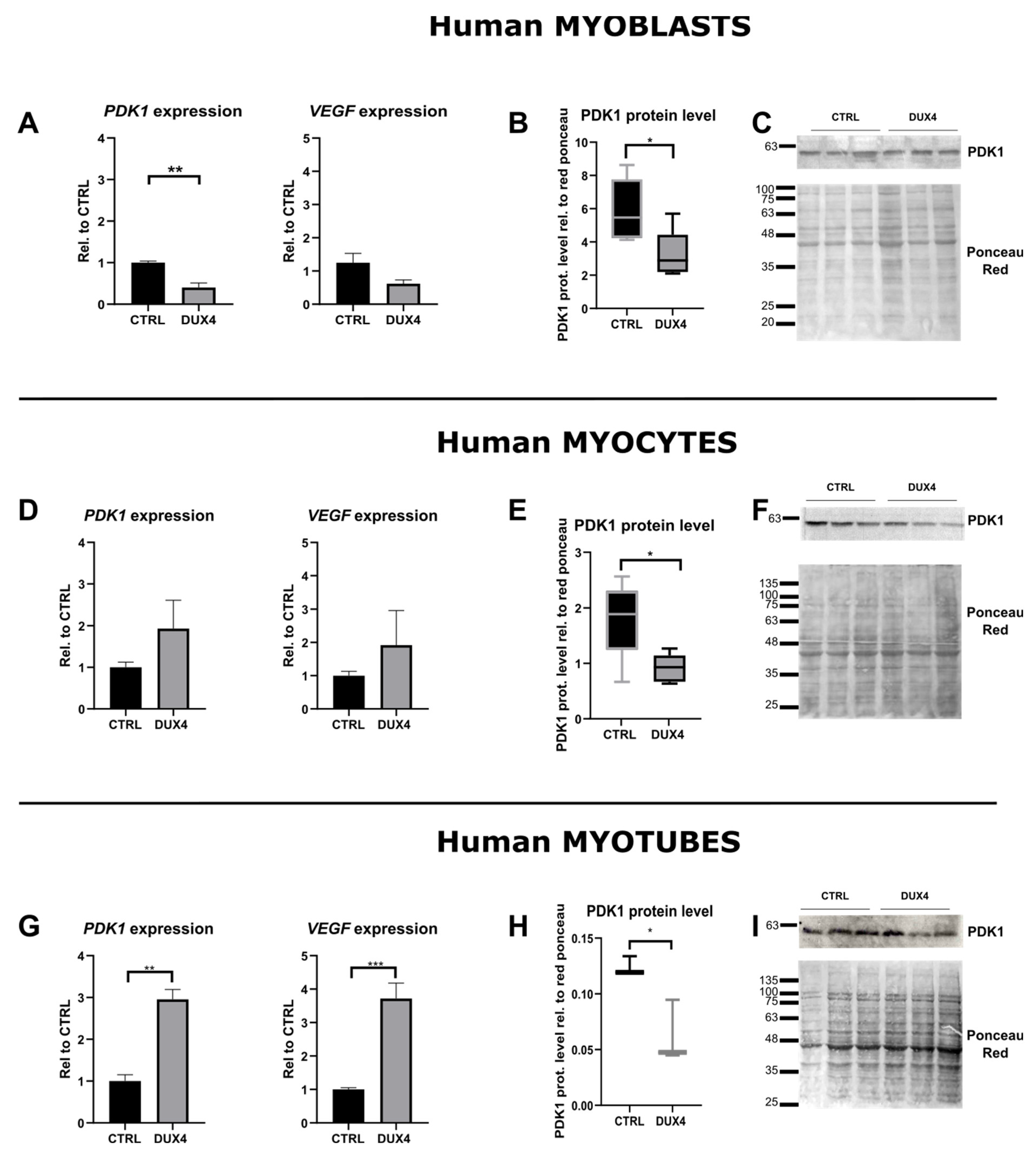
Figure 3.
Effect of DUX4 on the number of HIF1α-positive nuclei (HIF1α+) in mouse (C2C12-iDUX4) vs human (LHCN-M2-iDUX4 ) muscle cells . (A-E) C2C12-iDUX4 murine myoblasts: 25 000 or 200 000 cells were seeded per well in 24-well or 6-well plates, respectively and grown at a standard PO2 of 21%. 24h later, DUX4 expression was induced for 24 h with increasing doses of doxycycline (DOX, ng/ml). HIF1α was detected by immunofluorescence (IF). For comparison, LHCN-M2-iDUX4 myoblasts were cultured as in Figure 1 and DUX4 expression was induced for 24 h with increasing doses of DOX. (F-J) C2C12-iDUX4 murine myocytes: 750 000 cells were seeded per well in 6-well plates. 24h later, cells were switched to the differentiation medium for two days. DUX4 expression was induced with 62.5 ng/ml of DOX for 48h. HIF1α was detected by IF. A, F. Experiment time courses. B, G. Representative fields showing HIF1α+ nuclei (red IF). Nuclei were stained with DAPI (blue). Scale bar= 100µm. C. Quantification of HIF1α+ nuclei normalized to the total number of nuclei (DAPI staining) in LHCN-M2-iDUX4 myoblasts and C2C12-iDUX4 myoblasts. Mean ± SEM, **p<0.01, ***p<0.01, One way ANOVA with Holm Sidak post hoc test vs the control (DOX : 0 ng/ml). H. Quantification of HIF1α+ nuclei normalized to the total number of nuclei (DAPI staining) in C2C12 iDUX4 myocytes. Mean ± SEM, ***p<0.001, T-test. D, I. Expression level of Wfdc3 mRNA. Quantifications were performed by RT-qPCR and normalized to RPLP0. Mean ± SEM, *p<0.05, ***p<0.001, T-test. E, J. Expression levels of Pdk1 and Vegf mRNAs. Quantifications were performed by RT-qPCR and normalized to RPLP0. Mean ± SEM, *p<0.05, T-test. The experiments were performed on 3 independent cultures, each in triplicate (n=3).
Figure 3.
Effect of DUX4 on the number of HIF1α-positive nuclei (HIF1α+) in mouse (C2C12-iDUX4) vs human (LHCN-M2-iDUX4 ) muscle cells . (A-E) C2C12-iDUX4 murine myoblasts: 25 000 or 200 000 cells were seeded per well in 24-well or 6-well plates, respectively and grown at a standard PO2 of 21%. 24h later, DUX4 expression was induced for 24 h with increasing doses of doxycycline (DOX, ng/ml). HIF1α was detected by immunofluorescence (IF). For comparison, LHCN-M2-iDUX4 myoblasts were cultured as in Figure 1 and DUX4 expression was induced for 24 h with increasing doses of DOX. (F-J) C2C12-iDUX4 murine myocytes: 750 000 cells were seeded per well in 6-well plates. 24h later, cells were switched to the differentiation medium for two days. DUX4 expression was induced with 62.5 ng/ml of DOX for 48h. HIF1α was detected by IF. A, F. Experiment time courses. B, G. Representative fields showing HIF1α+ nuclei (red IF). Nuclei were stained with DAPI (blue). Scale bar= 100µm. C. Quantification of HIF1α+ nuclei normalized to the total number of nuclei (DAPI staining) in LHCN-M2-iDUX4 myoblasts and C2C12-iDUX4 myoblasts. Mean ± SEM, **p<0.01, ***p<0.01, One way ANOVA with Holm Sidak post hoc test vs the control (DOX : 0 ng/ml). H. Quantification of HIF1α+ nuclei normalized to the total number of nuclei (DAPI staining) in C2C12 iDUX4 myocytes. Mean ± SEM, ***p<0.001, T-test. D, I. Expression level of Wfdc3 mRNA. Quantifications were performed by RT-qPCR and normalized to RPLP0. Mean ± SEM, *p<0.05, ***p<0.001, T-test. E, J. Expression levels of Pdk1 and Vegf mRNAs. Quantifications were performed by RT-qPCR and normalized to RPLP0. Mean ± SEM, *p<0.05, T-test. The experiments were performed on 3 independent cultures, each in triplicate (n=3).
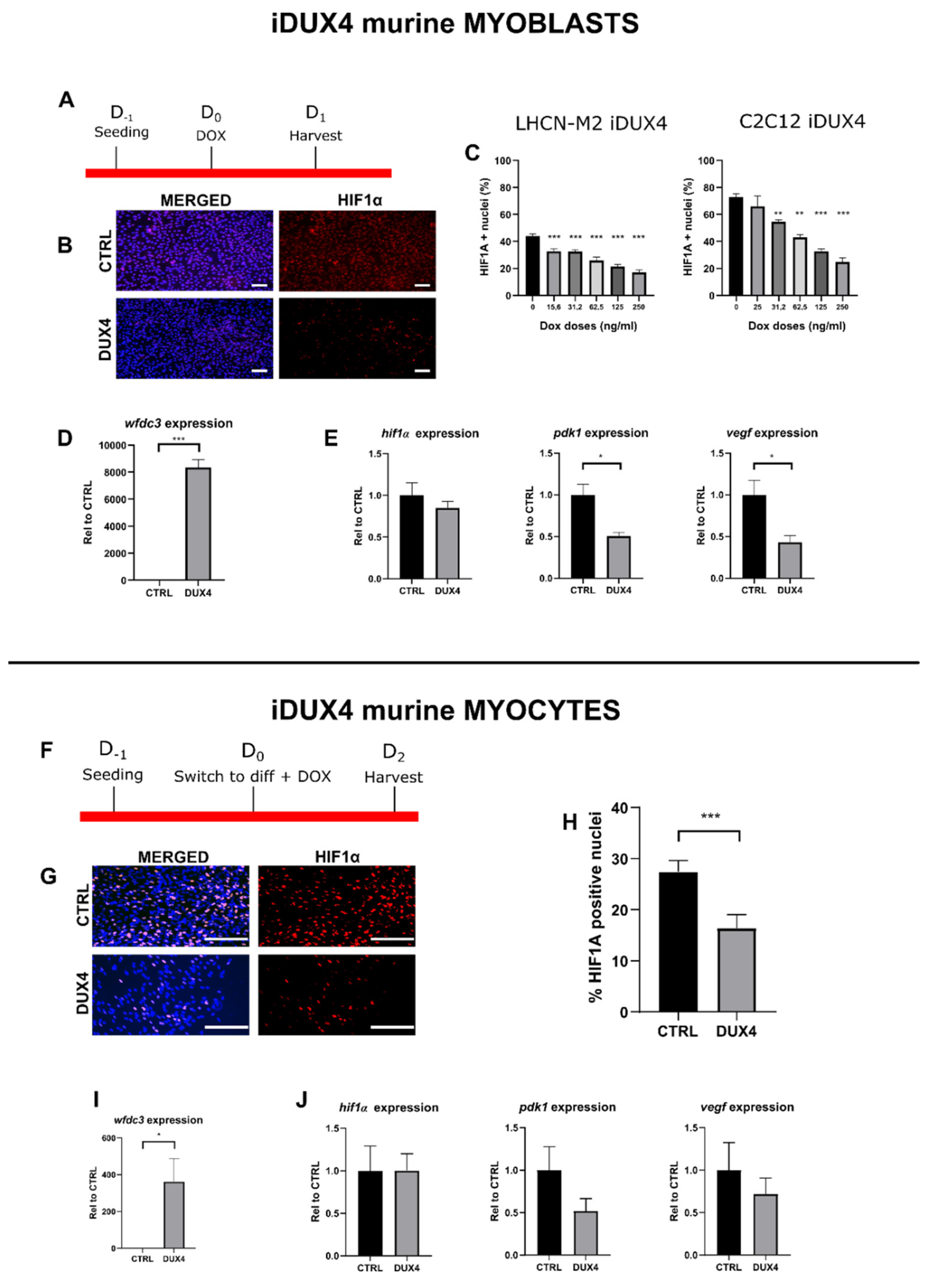
Figure 4.
Early effects of DUX4 expression on HIF1α pathway in vivo in the DUX4 IMEP mouse model with a high dose of DUX4 expression. A, C. Experiment time courses. B. mRNAs of DUX4 target genes Wfdc3 and Zscan4 were quantified by RT-qPCR in C2C12 myoblasts 4, 5 and 6 h post-transfection with pCIneo-DUX4. Quantifications were normalized to Rlpl0. N=5 for control group and n=4 for 4 h, 5 h, and 6 h groups, results presented as boxplots * p<0.05, Kruskal Wallis followed by a Dunn’s post-hoc test. D. Representative sections of TA electroporated with saline solution (Left), 20 µg of pCIneo (Middle) or pCIneo-DUX4 plasmid (Right). Scale = 100µm. E. Effect of DUX4 induction on the level of Wfdc3 mRNA in the IMEP model. mRNA levels were quantified by RT-qPCR and normalized to Rplp0. Results presented as boxplots,*p<0.05, Kruskal Wallis followed by a Dunn’s post-hoc test, N=8 for each group. F. Effect of DUX4 induction on the level of Hif1α pathway mRNAs Hif1α, Pdk1 and Vegf in the IMEP model. mRNA levels were quantified by RT-qPCR and normalized to Rplpl0. Results presented as boxplots, *p<0.05, Kruskal Wallis followed by a Dunn’s post-hoc test. N=8 for each group.
Figure 4.
Early effects of DUX4 expression on HIF1α pathway in vivo in the DUX4 IMEP mouse model with a high dose of DUX4 expression. A, C. Experiment time courses. B. mRNAs of DUX4 target genes Wfdc3 and Zscan4 were quantified by RT-qPCR in C2C12 myoblasts 4, 5 and 6 h post-transfection with pCIneo-DUX4. Quantifications were normalized to Rlpl0. N=5 for control group and n=4 for 4 h, 5 h, and 6 h groups, results presented as boxplots * p<0.05, Kruskal Wallis followed by a Dunn’s post-hoc test. D. Representative sections of TA electroporated with saline solution (Left), 20 µg of pCIneo (Middle) or pCIneo-DUX4 plasmid (Right). Scale = 100µm. E. Effect of DUX4 induction on the level of Wfdc3 mRNA in the IMEP model. mRNA levels were quantified by RT-qPCR and normalized to Rplp0. Results presented as boxplots,*p<0.05, Kruskal Wallis followed by a Dunn’s post-hoc test, N=8 for each group. F. Effect of DUX4 induction on the level of Hif1α pathway mRNAs Hif1α, Pdk1 and Vegf in the IMEP model. mRNA levels were quantified by RT-qPCR and normalized to Rplpl0. Results presented as boxplots, *p<0.05, Kruskal Wallis followed by a Dunn’s post-hoc test. N=8 for each group.
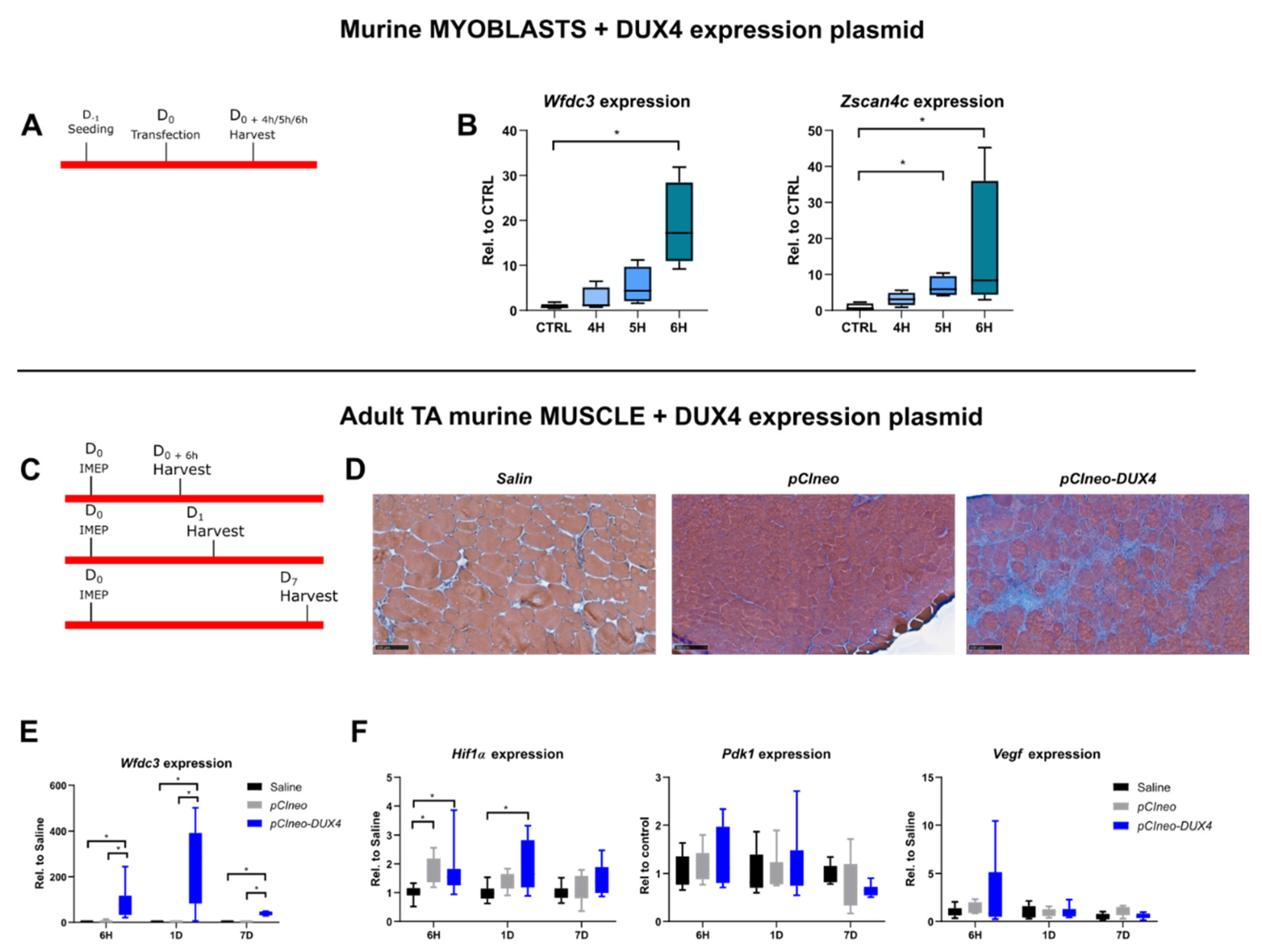
Figure 5.
Involvement of HIF1α pathway in DUX4-induced muscle damage. A. Efficiency of siRNA directed against Hif1α mRNA (siHIF1α). The TA muscle was electroporated with either saline solution, siCTL or siHIF1α. TA muscles were harvested 1 day after the IMEP procedure. Hif1α mRNA level was quantified by RT-qPCR and normalized to Rplp0. Mean ± SEM, *p<0.05, ***p<0.001, Kruskal Wallis followed by a Dunn’s post-hoc test. N=10 per group. B. Representative cryosections of TA electroporated with 20 µg of pCIneo-DUX4 plasmid in combination or not with 2µg of siCTL or siHIF1α. TA muscles were harvested 7 days after the IMEP procedure. Muscle sections were stained with HEB. Top: global view of the muscle sections; Scale = 500µm. Bottom: magnification of the damaged area; Scale = 100µm. C. The percentage of lesion area was evaluated on muscle stained with HEB as represented in B. Data presented as scatter plots with Mean ± SEM, *p<0.05, One way ANOVA followed by Holm-Sidak post-hoc test. N=8 per group. D. Myofibre cross-section areas (CSA) were measured on the whole muscle section by using the Image J software. Mean ± SEM, ANOVA One Way: NS. N=8 per group. E. Fibre size distribution. Myofibres were classified in clusters according to their area. Chi-square: NS. N=8 per group. F. Cumulative percentage of myofibres in clusters. G. Wfdc3 mRNA level was quantified by RT-qPCR in the IMEP model. Quantifications were normalized to Rplp0. Data presented as boxplots, Kruskal Wallis followed by a Dunn’s post-hoc test : NS. N=8 per group. H. Effect of DUX4 induction on levels of Hif1α pathway mRNAs Hif1α, Pdk1 and Vegf in the IMEP model. mRNA levels were quantified by RT-qPCR and normalized to Rplpl0. Data presented as boxplots, Kruskal Wallis followed by a Dunn’s post-hoc test : NS. N=8 per group.
Figure 5.
Involvement of HIF1α pathway in DUX4-induced muscle damage. A. Efficiency of siRNA directed against Hif1α mRNA (siHIF1α). The TA muscle was electroporated with either saline solution, siCTL or siHIF1α. TA muscles were harvested 1 day after the IMEP procedure. Hif1α mRNA level was quantified by RT-qPCR and normalized to Rplp0. Mean ± SEM, *p<0.05, ***p<0.001, Kruskal Wallis followed by a Dunn’s post-hoc test. N=10 per group. B. Representative cryosections of TA electroporated with 20 µg of pCIneo-DUX4 plasmid in combination or not with 2µg of siCTL or siHIF1α. TA muscles were harvested 7 days after the IMEP procedure. Muscle sections were stained with HEB. Top: global view of the muscle sections; Scale = 500µm. Bottom: magnification of the damaged area; Scale = 100µm. C. The percentage of lesion area was evaluated on muscle stained with HEB as represented in B. Data presented as scatter plots with Mean ± SEM, *p<0.05, One way ANOVA followed by Holm-Sidak post-hoc test. N=8 per group. D. Myofibre cross-section areas (CSA) were measured on the whole muscle section by using the Image J software. Mean ± SEM, ANOVA One Way: NS. N=8 per group. E. Fibre size distribution. Myofibres were classified in clusters according to their area. Chi-square: NS. N=8 per group. F. Cumulative percentage of myofibres in clusters. G. Wfdc3 mRNA level was quantified by RT-qPCR in the IMEP model. Quantifications were normalized to Rplp0. Data presented as boxplots, Kruskal Wallis followed by a Dunn’s post-hoc test : NS. N=8 per group. H. Effect of DUX4 induction on levels of Hif1α pathway mRNAs Hif1α, Pdk1 and Vegf in the IMEP model. mRNA levels were quantified by RT-qPCR and normalized to Rplpl0. Data presented as boxplots, Kruskal Wallis followed by a Dunn’s post-hoc test : NS. N=8 per group.
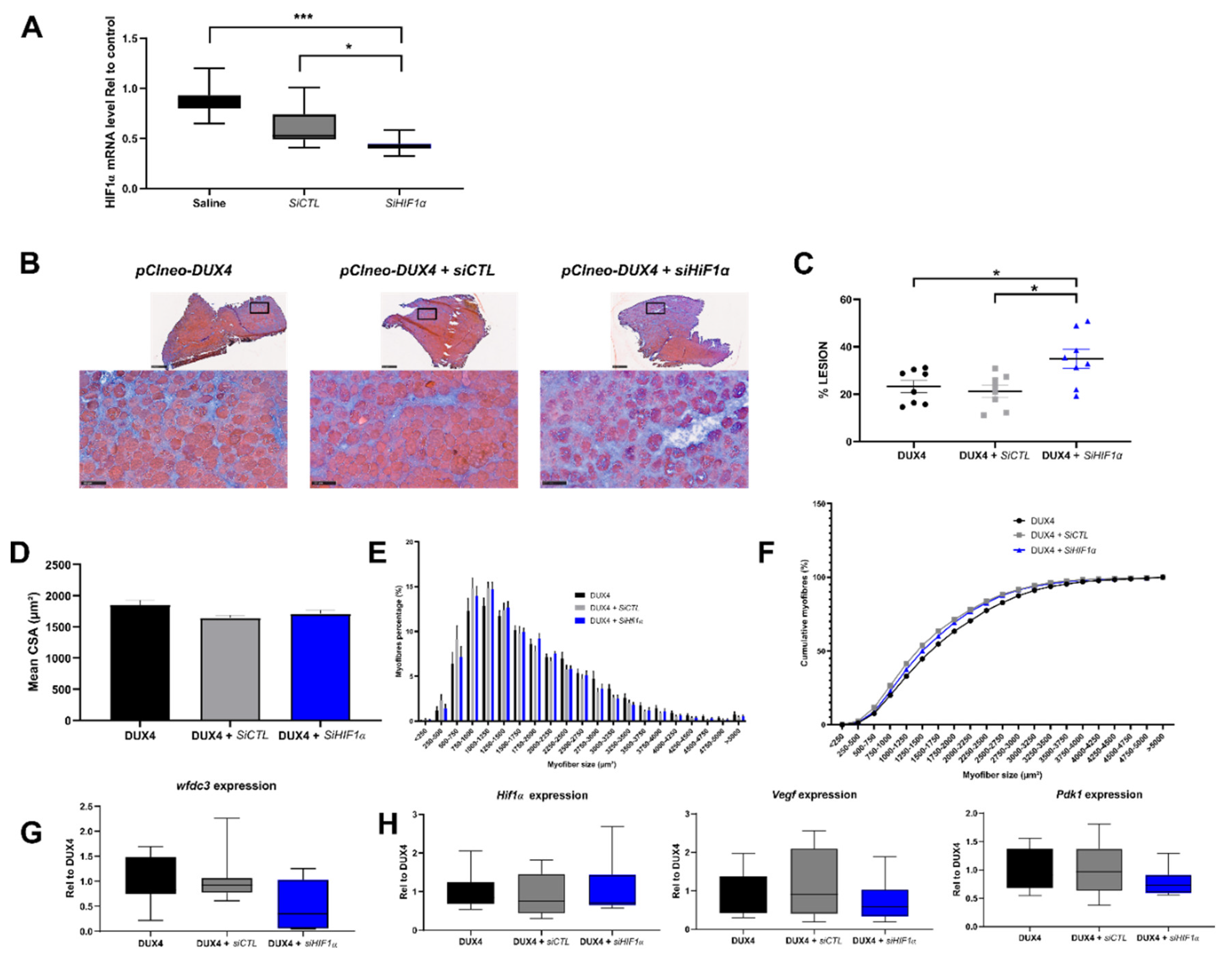
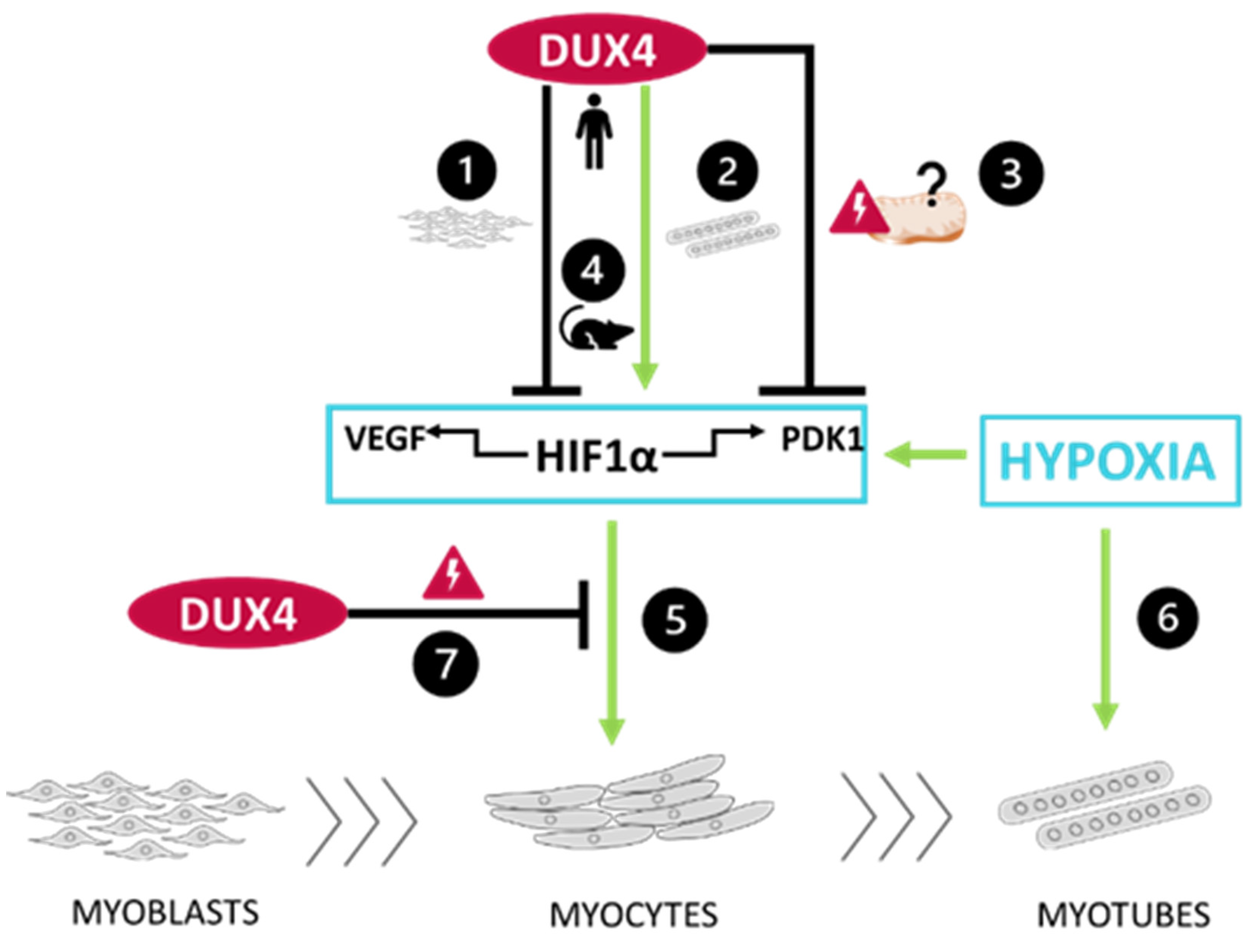
Figure 6.
Schematic conclusions ❶ DUX4 inhibits HIF1α pathway in proliferating myoblasts but ❷ induces it in late differentiation into myotubes. Data regarding the two HIF1α target genes VEGF and PDK1 are consistent with those results. However, ❸ DUX4 decreases PDK1 protein level whatever the differentiation stages, likely due to the DUX4-induced mitochondrial dysfunction. ❹ The DUX4-HIF1α axis is conserved in mouse myoblasts (as well as is adult muscle in vivo). Moreover, as we described in [31] in the context of adult myogenesis, hypoxia ❺ increases early myogenic differentiation in a HIF1α-dependent way and ❻ induces myocyte fusion independently of HIF1α. Finally, ❼ DUX4 represses HIF1α effect in early myoblast differentiation.
Figure 6.
Schematic conclusions ❶ DUX4 inhibits HIF1α pathway in proliferating myoblasts but ❷ induces it in late differentiation into myotubes. Data regarding the two HIF1α target genes VEGF and PDK1 are consistent with those results. However, ❸ DUX4 decreases PDK1 protein level whatever the differentiation stages, likely due to the DUX4-induced mitochondrial dysfunction. ❹ The DUX4-HIF1α axis is conserved in mouse myoblasts (as well as is adult muscle in vivo). Moreover, as we described in [31] in the context of adult myogenesis, hypoxia ❺ increases early myogenic differentiation in a HIF1α-dependent way and ❻ induces myocyte fusion independently of HIF1α. Finally, ❼ DUX4 represses HIF1α effect in early myoblast differentiation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



