
Submitted:
09 February 2023
Posted:
10 February 2023
You are already at the latest version
Abstract
Expression analysis of small noncoding RNA (sRNA), including microRNA, piwi-interacting RNA, small rRNA-derived RNA, and tRNA-derived small RNA, is a novel and quickly developing field. Despite a range of proposed approaches, selecting and adapting a particular pipeline for transcriptomic analysis of sRNA remains a challenge. This paper focuses on the identification of the optimal pipeline configurations for each step of human sRNA analysis, including reads trimming, filtering, mapping, transcript abundance quantification and differential expression analysis. Based on our study, we suggest the following parameters for analysis of human sRNA in relation to categorical analyses with two groups of biosamples: (1) trimming with the lower length bound = 15 and the upper length bound = \(Read\ length - 40\% Adapter\ length\); (2) mapping on a reference genome with bowtie aligner with one mismatch allowed (-v 1 parameter); (3) filtering by mean threshold > 5; and (4) analyzing differential expression with DESeq2 with adjusted p-value < 0.05 or limma with p-value < 0.05 if there is very little signal and few transcripts.
Keywords:
sRNA analysis
; small RNA
; microRNA
; piRNA
; tRNA-derived small RNA
; RNA-seq
; small RNA fragments
; benchmarking
; differential expression analysis
Introduction
Small non-coding RNA (sRNA) less than 200 nucleotides in length are important regulatory molecules in the control of gene expression at both the transcriptional and the post-transcriptional level [1,2,3]. Research on sRNAs has accelerated over the past two decades and sRNAs have been utilized as markers of human diseases such as neurological conditions [4], cancer [5] and infertility [6,7,8], and in the identification of molecular biomarkers associating environmental exposures with health/disease outcomes [9,10]. Identification of sRNA in germ cells is of particular interest as they represent an additional source of parental hereditary information beyond DNA sequences and may have a potential role in programming offspring health [11,12]. Types of small RNA include, among others, microRNA (miRNA), piwi-interacting RNA (piRNA), small rRNA-derived RNA (rsRNA) and tRNA-derived small RNA (tsRNA) [2]. Next generation sequencing (NGS) has become the principal approach for global profiling of sRNA due to the steady decrease of sequencing costs, wider coverage and higher sensitivity than microarrays. However, bioinformatic analysis of NGS data for sRNA is prone to many challenges. For example, calculation of sRNA expression values from NGS reads may not reflect their absolute expression levels accurately [13,14]. In this study, we attempt to identify the optimal pipeline configurations for each step of sRNA analysis of human data, including read trimming, filtering, mapping, transcript abundance quantification, and differential expression (DE) analysis.
sRNA expression methods
All major pipelines for sRNA expression analysis may be classified into 3 groups. First, tools that only allow for a particular stage of expression analysis (namely, alignment). Second, the ones that allow for the more expanded analysis of a certain type of sRNA (piRNA or miRNA, for instance). The tools of the third group provide a means for the expanded analysis of 2 or more types of sRNA. Characteristics of some existing sRNA pipelines are presented in Table 1.
The two tools specialized in the mapping of small RNAs only, are similarly named: srnaMapper [15] and sRNAmapper [16]. Both have regular alignment options. srnaMapper has an additional ability to align reads without precise complementarity of the last few bases. This, however, may result in a reduction of mapping specificity and in a significant increase in the required computational resources.
Several tools in the second group were designed specifically for piRNA annotation of NGS-samples - namely, piPipes [17], PILFER [18], piClust [19], proTRAC [20]. Pipelines focused on miRNA detection include miRanalyzer [21] and sRNA workbench [22]. tsRFun [23] is focused on tsRNA analysis. Given that each of these tools is focusing on a single biotype of sRNA we do not consider them further in this study.
A number of tools are available for the analysis of several different types of sRNAs. First of all, these are sRNAtoolbox [24], sRNAnalyzer [25] and SPORTS [26], which have command-line based interfaces (CLI). Other tools have graphic-based user interface (GUI): sRNAPipe [27] (Galaxy-based pipeline) and iSmaRT [28]. Oasis 2 [29] offers only an online version, which wasn’t fully functional as of October 2022.
A comparison of the most popular pipelines is presented in Figure 1.
sRNAnalyzer [25] is a command-line interface pipeline with a text-based configuration file for the identification of miRNAs, piRNAs, small nucleolar RNA (snoRNA), and long non-coding RNA (lncRNA). The pipeline allows for processing the reads (upon adaptors removal), quality filtering, read mapping and counting. The preprocessing steps include adaptors removing and filtering by the minimum length of the read. For the analyzed sequences, sRNAnalyzer uses a ‘map and remove’ approach with a progressive alignment strategy to sequentially map the reads against various databases (only the reads unmapped to the current database will proceed to mapping to transcript sequences in the next database). Databases for the following species are available to be used with sRNAnalyzer: for human, mouse, rat, horse, macaque and plant. Moreover, they can be modified for samples of other species. sRNAnalyzer uses FASTQ files for input, while the output provides files with gene counts.
SPORTS1.0 [26] is a command line-based pipeline for the identification and quantification of miRNAs, piRNAs, tsRNA and rsRNA. SPORTS1.0 can be used with a wide range of species with available reference genome. Also, it is possible to substitute the default small RNA databases with custom databases provided by the user [30]. The pre-processing steps include adapter removing and filtering sequences by size and quality. The output is provided as gene counts and various visualization figures.
iSRAP [31] is another tool with a command line interface, focused on the annotation of sRNAs (miRNAs, piRNAs and also snoRNAs). A configuration file is needed to define options and optimize sRNA profiling in different datasets. This pipeline can be executed starting from either FASTQ or BAM alignment files as input. Output results are reported as PDF file and HTML documents, completed with graphical elements to illustrate the results.
One of the most popular instruments for small RNA annotation is a web-interface-based sRNAtoolbox [24]. It includes several tools: sRNAbench for the expression profiling of small RNAs and prediction of novel miRNAs from deep sequencing data; sRNAde for the DE analysis; sRNAblast for blast analysis of deep sequencing reads against a local database, and others. The adapter trimming, read quality and size filtering is available and optional in sRNAbench.
The above mentioned pipelines and others were recently reviewed in detail elsewhere [32].
Alignment-based tools
Several pipelines have been developed for RNA-seq data analysis [33,34,35]. Their recruitment for sRNA expression can potentially help to overcome the described ’map and remove’ approach problems, to avoid non-independent processing of different sRNA types.
The standard workflow for RNA-seq analysis includes several steps. First, preprocessing involves reads trimming to remove the adaptors or low-quality bases from reads ends and it may be carried out with specific tools, such as Trimmomatic [36] or cutadapt [37]. The next step is reads alignment and it can be done with a variety of tools, such as hisat2 [38], STAR [39], bowtie [40], bowtie2 [41].
At the next step, the mapped reads are summarized for a particular transcript annotation. Frequently used approaches for this procedure include featureCounts [42] from Rsubread, and HTSeq [43]. The output of this step is a count matrix that represents the expression values for all considered samples and genes or transcripts. Some approaches may combine alignment and quantification procedures. These include RSEM [44], which performs transcriptome alignment and produces expected counts for transcripts, and probabilistic pseudoaligners, such as Kallisto [45] and Salmon [46], which assign reads to transcripts based on their k-mer spectra pattern and result in estimated counts.
Finally, the obtained counts are normalized and, after filtering, inferred with statistical model for DE. Commonly used approaches for this step are DESeq2 [47], edgeR [48] and limma [49], or various Bayesian models [50,51]. As a result, the list of DE genes (i.e. gene signature) or transcripts is retrieved.
Adapting the described genome-alignment-based expression analysis workflow poses several challenges however. First, due to the very short length of sRNA transcripts, the influence of the samples and library preparation-related parameters (sequencing kit or adapters choice) on the result of preprocessing and trimming procedures is higher compared to mRNA reads preprocessing. Second, the small length of reads makes the alignment procedure more challenging [52]. Therefore, the performance of various aligning approaches needs to be evaluated specifically with respect to sRNA data applicability. Third, due to the significantly lower expression signal in sRNA data, the filtering of low-expressed transcripts aiming to reduce noise is important [53]. Finally, the choice of DE model was shown to have significant impact on the results [54,55,56,57]. Hence, quantitative estimation of the resulting expression signature needs to be conducted.
Thus, the objectives of the current study are as follows. First, to explore the optimal parameters for sRNA data specific preprocessing steps (such as the choice of sequencing kit or adapters and trimming threshold). Second, to assess the performance of aligning and summarize procedures of sRNA data. Third, to evaluate filtering procedures for lowly-expressed transcripts. And finally, to estimate the resulting expression signature quality after the DE inference.
Materials and Methods
Data sources
Small RNA sequencing data (SRA archives) from 7 published human studies were retrieved from GEO database and extracted to FASTQ reads using sra-toolkit [58] (see Table 2). Raw reads quality was explored by FastQC [59].
Dataset from Wong et al. article [60] (hereinafter referred to as "Wong") consists of 120 fastq-files for 30 blood plasma samples. In this study 3 small RNA library preparation kits (CleanTag, NEXTflex, QIAseq) and two RNA extraction methods (miRNeasy and MagnaZol) were compared. This dataset was used for small RNA seq data analysis benchmarking, but not for DE analysis.
A prospective case-control study from Huang et al. article [61] (hereinafter referred to as "Huang") was designed to identify the changes in expression of microRNA and mRNA, using 10 blood samples in dilated cardiomyopathy patients and 10 paired healthy control blood samples.
108 RNA sequencing samples from Delker et al. dataset [62] (hereinafter referred to as "Delker") from RNAlater preserved clinical biopsies (16 sessile serrated adenomas/polyps, 14 hyperlastic polyps, 14 adenomatous polyps, 34 uninvolved colon and 30 control colon samples) were used for small RNA seq data analysis benchmarking, and 2 contrasts (16 sessile serrated adenomoas/polyps vs 14 hyperlastic polyps ("Delker-P") and 15 right vs 15 left control colon ("Delker-C") samples) were used in DE analysis.
Six sperm samples were collected prospectively monthly from 17 healthy male participants for Morgan et al. study [63] (hereinafter referred to as "Morgan"). 87 human sperm samples with high and low rate of good quality embryos were analyzed in Hua et al. study [64] (hereinafter referred to as "Hua"). Semen of 13 lean and 10 obese individuals were analyzed in Donkin el.al study [65] (hereinafter referred to as "Donkin"). Pure fractions of motile spermatozoa collected from 12 young healthy individuals before, after 6 weeks of endurance training and after 3 months without exercise were analyzed in Ingerslev et al. [66] study (hereinafter referred to as "Ingerslev").
Preprocessing of RNA-seq data
All reads adapters were removed with cutadapt [37] following lab protocols [67,68,69]. Reads less 15 nt in length (lower bound) were removed since smaller reads length makes aligning and expression quantification difficult and not robust. To adjust the pipeline and derive optimal parameters, we used several trimming options for the upper bound. Reads without the upper read length bound and with differently varied thresholds were processed as follows (see Figure A):
-
where X = 10%, 20%, 30%,..., 100%
-
where X = 3, 6, 9,..., 30 nt
-
where X = 0.05, 0.1, 0.15,..., 0.5
Trimmed read length distributions were analyzed to infer the optimal threshold which can preserve the most sequencing data signal. One of the trimming strategies with suitable performance was chosen (see Results).
Processing of small RNA-seq data
sRNAs
The trimmed reads were processed with various alignment and pseudoalignment methods. Alignment methods include mapping on hg38 [70] reference genome with bowtie [40] (with 0 and 1 mismatch), hisat2 [38] and STAR [39] aligners.
Only one best alignment was determined using bowtie aligner for every read to be used in further analysis: bowtie -x genome_index -q in.fq -S out.sam -v {0,1} -m 100 -k 1 –best –strata
Hisat2 aligner was used with its standard parameters but without spliced alignments and softclip: hisat2 -x genome_index -U in.fq -S out.sam –no-spliced-alignment –no-softclip STAR aligner was also used with its standard parameters without allowing introns and mismatches: STAR –runMode alignReads –genomeDir genome_index –readFilesIn in.fq –outFileNamePrefix out –outFilterMismatchNmax 0 –alignIntronMax 0 –alignIntronMin 0 –genomeLoad LoadAndKeep
tRNA fragments
Given that several variants of the same fragment need to be considered, quantifying and assigning reads to tRNA fragments is a probabilistic procedure. Therefore, probabilistic aligner, such as kallisto, is a better choice for this part of the analysis.
Trimmed reads were mapped on hg38 reference genome with various described approaches and those transcripts that were assigned to tRNA by Rsubread were extracted and processed using several k-mer values kallisto [45] for probabilistic mapping on ITAS tsRNA fragments sequences.
sRNA tools
All samples, once trimmed and pre-processed as described above, were also processed using sRNA analysis tools SPORTS [26] and sRNAnalyzer [25] with the default parameters. All preprocessing steps including quality and size filtering, filtering for contaminates, were conducted using these tools.
SPORTS1.0 tool was used with default databases: reads from all analyzed human samples were sequentially "mapped and removed" to the snRNA databases (miRBase, rRNA database (collected from NCBI), GtRNAdb, piRNA database, Ensembl and Rfam). The only parameter used in command was -i for the path to the input file; -g, -m, -r, -t, -w for paths to genome, miRNA, rRNA, tRNA and piRNA files; -p for the number of involved processors. Summary text file was used as the list of gene counts for each sample.
sRNAnalyzer was launched with implemented databases "small RNA Databases" and "Human and Exogenous Databases". "NCBI Non-Human Databases" wasn’t used as redundant, and all samples were pre-filtered before starting sRNAnalyzer. The only modified file was DB_cofig.conf. Pipeline_config.yaml file was not modified. As output, "XX.feature" file with summary counts for each sample was used after analysis.
Filtering thresholds
Transcript filtering based on their expression values is an important step in DE analysis as sRNA transcripts with low coverage and low expression may add noise to the signal thus, decreasing robustness of the analysis.
Three filtering strategies were applied:
-
min filtering: expression value for a transcript in all samples is higher than the threshold (N)
-
mean filtering: mean expression value for a transcript in all samples is higher than the threshold (N)
-
median filtering: median expression value for a transcript in all samples is higher than the threshold (N)
Two threshold values, N = 5 and N = 10, were applied.
Differential expression
Transcripts from counts matrix were filtered using several thresholds: min count > {5,10}, mean count > {5,10}, median count > {5,10}. For DE analysis data filtered by mean and median count > 5 were used. DE analysis was conducted using DESeq2 [47], edgeR [48] and limma [49] packages. Datasets (from "Huang", "Hua", "Donkin" papers) or subdatasets from "Delker" and "Ingerslev" papers consisting of 2 groups were used. The following types of thresholds were obtained:
DE results in an expression signature, i.e. a list of transcripts with significant differences in expression between considered conditions. The following options are usually used for thresholds in the processing of initial signature:
- adjusted p-value threshold (preferable)
- combination of p-value and absolute fold change thresholds
Multiple testing correction is usually used in the DE analysis. However, due to the small signal in sRNA expression data, many studies (e.g. "Morgan", "Huang") use only not-adjusted p-value. Although it implies insufficient statistical power of the analysis, the results may suggest candidate transcripts for further exploration.
Expression signature quality evaluation
The number of DE transcripts cannot be used as a robust metric for DE analysis quality, as this approach doesn’t account for false positive results. To evaluate the expression signature quality we applied Hobotnica [71] approach.
In this process, the candidate expression signatures delivered after the application of differing preprocessing and processing procedures, such as differential model, or genome aligner, were inferred to the expression data to produce a distance matrix between samples. Next the distance matrix was compared with the expected samples relationship structure, given the known groups of samples. Results of expression signature quality evaluation presented as H-score value, characteristics of the quality of a candidate signature’s data separation. H-score is scaled from 0 (the worst) to 1 (the best).
Stages and metrics of benchmarking
We used metrics for each stage of RNA-seq data analysis from the biosampling and library preparation to the DE. They are described in Table 3. Lengths of input reads and adapters were used for the estimation of the upper bound of trimming. All reads from the analyzed datasets had suitable quality as assessed by the reads` quality threshold, an important metric for sRNA data. For the alignment-based pipelines the alignment rate was calculated as a ratio of aligned reads and input reads, and the assignment rate was calculated as a ratio of assigned reads and aligned reads. The assignment rate for sRNA-based and pseudoalignment-based pipelines was calculated as a ratio of assigned reads and input reads. After conducting a suitable pipeline, all expected sRNA biotypes should be returned. The filtering stage is important for removal of non-expressed transcripts, but it should retain sufficient transcript numbers for subsequent DE analysis. The metric for DE is the number of significant transcripts while the H-score characterizes the expression signature quality.
Different post DE downstream applications may be applied to the retrieved expression signatures. Among them are Connectivity Map [72,73], GO terms [74] and KEGG [75] pathways analysis, hallmark signatures inference [76], and genome browser analysis [77]. Although these approaches provide useful and valuable information, they cannot be used for the benchmark purposes, as appropriate metrics for the analysis quality cannot be robustly introduced [57].
Results
Input data
The nature of human biosamples and differences in their processing pipelines from library preparation to published fastq-files result in a variation between datasets. Thus, all mentioned metrics (such as biological object, lab kit, read and adapter length, reads quality) were taken into account at the first stage of analyses to choose suitable pipelines as presented in Table 2 and in further sections below.
Trimming
Based on the distribution and peaks of read length for various datasets we observe high variability between datasets and similar distribution and peaks across samples of the same dataset (for most datasets) (Supplemental Figure S1). However, the lower variability of reads length across samples was observed in "Hua", "Huang" and "Morgan" datasets. "Delker" reads prepared by NEBNext lab kit have peaks at 17, 22, and 32 nt. These peaks may indicate piRNA, miRNA and tsRNA, respectively. The same peaks are observed in the "Huang" dataset, and the "Wong" dataset (prepared by NEXTFlex). One strong peak at 23 nt is observed in the "Hua" dataset and the "Wong" dataset (prepared by Qiaseq). An additional 15 nt reads peak occurred in "Delker" dataset (prepared by TruSeq) and "Wong" dataset (prepared by CleanTag).
The results for various trimming strategies are presented in Figure .
The strategy of removing a fixed number of bases was inconsistent and not conserved across datasets and kit types, thus it was not feasible to observe an optimal trimming length value with sufficient number of reads retained for all observed data (Supplemental Figure S2). The strategy of removing a particular fraction of adapter based on read length/adapter length ratio exhibited similar pattern problem with even lower number of retained reads in most cases (Figure ).
The strategy of removing a given fraction of the adapter appeared to be the most conservative across all datasets and kits (Figure A). With of adapter length removal, a sufficient number of reads were retained after the trimming procedure (Figure B), and, for this reason, this approach was chosen as the preprocessing pattern for all the samples in all datasets.
Genome or transcriptome aligning
Reads were mapped with genome-alignment-based methods (bowtie, hisat2 and STAR) and prebuilt transcriptome alignment-based method (RSEM) demonstrated sufficient alignment rates (Supplemental Table S1). Comparison of alignment rates and the number of mapped reads is shown in Figure 3.
The highest values of alignment rates were observed for bowtie with 1 mismatch allowed (bowtie -v 1) (up to 97% for "Donkin" dataset and mean 89% for all datasets) and with no mismatches allowed (bowtie -v 0) as the second highest fraction of aligned reads (up to 86% for "Donkin" dataset and mean 74% for all). RNA-seq specific aligners, STAR and hisat2, and RSEM demonstrated lower mean values for aligning ratios (54%, 61% and 57% respectively). However, for some datasets hisat2 and STAR had higher alignment rates (74% for "Hua" dataset for hisat2 and 85% for "Huang" dataset for STAR). All alignment rates for RSEM didn’t exceed 70%.
Assigning
Assignment rates and input reads ratios are shown in Figure 4. In the process of assigning mapped reads to transcripts loci, all genome-alignment based approaches demonstrated similar ratio values of reads, successfully assigned to transcripts (Figure 4B and Supplemental Table S2). Bowtie with 1 mismatch demonstrated better results based on moderately higher alignment rate, although its assignment rate remained close to other approaches.
We observed large variation in assignment rates across datasets and across samples of several datasets, specifically for "Delker" and "Wong" data, that may be explained by different kits used for samples preparation within one dataset (Figure 4B–D). The IQR for "Wong" data for bowtie mapper with 1 mismatch (bowtie -v 1) ranged from 0.1 to 0.5. "Delker" data performed 0.4-0.7 IQR for the same pipeline (Figure 4B).
RSEM showed low assignment rate values for all datasets. The highest mean assignment rate (assigned/input) was 0.26 for "Morgan" data. This may be explained by the transcriptome assembly process (as a part of RSEM analysis), which was likely not optimized for short sRNA transcripts (Figure 4B).
Pseudoaligners kallisto and salmon do not have specific assignment rate, since reads are probabilistically aligned to transcripts, and therefore, the procedure combines alignment and assignment of reads. As expected, we observed large variation between datasets (Figure 4C,D, Supplemental Table S3). Kallisto probabilistic aligner demonstrated various ’aligning ratios’ for different kmer length. Kallisto with kmer length = 5 performed the highest assignment rate (assigned/input) for all datasets (all samples have assignment rate more then 0.5). Kmer length = 7 and 9 performed the lowest assignment rate (Supplemental Figure S3). These results suggest that kallisto pipeline with kmer length = 5 may provide many false positive reads. Salmon pseudoalignment results demonstrated no significant difference observed for various kmer lengths (Supplemental Figure S4).
sRNA biotypes distribution
Figure A,B demonstrate the distribution of sRNA expression values by sRNA biotypes in all pipelines and datasets, and the distribution of the numbers of expressed transcripts by sRNA biotypes in a similar manner, respectively. There is a large variability between datasets and pipelines for both, expression values and numbers of transcripts. Most datasets except "Huang" and "Wong" show relatively similar sRNA distribution across pipelines. There is also differences between alignment-based, pseudoalignment-based and sRNA-based pipelines within dataset, especially for "Donkin" and "Ingerslev" data. Due to the limitations of these pipelines, RSEM didn’t identify miRNA transcripts in all considered datasets; SPORTS didn’t identify piRNAs; and sRNAnalyzer didn’t identify tRNA transcripts. The distribution of the number of expressed transcripts by pipelines is shown in Supplemental Figure S5. Some pipelines, such as SPORTS, RSEM, and kallisto with kmer length 5, 7, and 9, performed worse than others, using the methods and criteria outlined in this study.
Filtering
Transcript numbers resulting from different filtering strategies (thresholds) per dataset and per pipeline are presented in the Supplemental Figure S6A and S6B, respectively. As expected, the min filtering approach resulted in to the lowest number of transcripts. The highest number of transcripts was observed with mean filtering with the threshold 5. Median filtering with the same threshold 5 and mean(counts) > 10 returned a slightly lower number of transcripts.
The mean(counts) > 5 appears to be the optimal filtering approach for expression processing based on the results of transcript numbers and distribution of sRNA biotypes. Using the mean(counts) > 10 and the median(counts) > 5 may be a preferred choice when high numbers of false-positive are found with mean filtering with the threshold = 5.
Differential expression
Results of the DE analysis were obtained using in-build models of packages DESeq2, edgeR and limma, two options of filtering (mean(counts) > 5 and median(counts) > 5) and two thresholds of significance, p-value<0.05 and q-value (FDR adjusted p-value) < 0.05. The numbers of DE transcripts with q-value < 0.05 per dataset and per pipeline are shown in Figure 6 and S7, and Supplemental Table S4.
DESeq2 delivered the highest number of significant DE transcripts with multiple testing correction filtering applied; the mean for all pipelines was 20.7 and 12.8, for the mean(counts) > 5 and the median(counts) > 5, respectively. EdgeR returned fewer significant transcripts; mean for all pipelines was 10.0, which was the same for both thresholds. The lowest number of significant transcripts was produced by limma; the mean for all pipelines was 6.4, which was the same for both thresholds (Supplemental Table S4).
DE analysis of the two groups of contrast samples revealed two types of datasets. "Delker", "Huang", "Hua" had a fairly large number of significantly DE transcripts, with the mean number for all methods by DESeq2 = 26.8, 44.5 and 68.5, respectively. The other group included "Donkin" and "Ingerslev" datasets, which had a very small number of significant transcripts; mean for all methods = 0.5 and 0.6, respectively. The mean by sRNAnalyzer with 2 allowed mismatches was 5 and 8, respectively.
We suggest that this grouping of datasets is predictable based on the biological differences between the analyzed samples. In both "Donkin" and "Ingerslev" datasets, we do not anticipate big differences between groups, because both obesity ("Donkin") and exercises ("Ingerslev") were not associated with sperm RNA expression by any known strong causative link. All other datasets were related to a distinct tissue type and disease state: "Delker" - different tissues and neoplasm vs no neoplasm, "Huang" - health and disease, "Hua" - embryo quality that may be directly related to sperm program.
Expression signature quality estimation
We measured the expression signature quality using the Hobotnica approach, which provides separation values (H-scores) from 0 (the worst) to 1 (the best) and are presented for all datasets, methods of filtering and DE analyses in Supplemental Table S5 and Supplemental Figure S8. Three packages for DE analysis, DESeq2, edgeR and limma demonstrated similar H-scores, ranging from 0.46 to 0.93 among all datasets and pipelines. Two datasets (controls "Delker-C" and "Huang") demonstrated good separation quality for genome-alignment-based and sRNA-based pipelines ("Delker-C": mean 0.93 and 0.79, respectively; "Huang": 0.76 and 0.75, respectively). Other datasets revealed similar and lower H-scores. Overall, genome-alignment-based methods usually produced signatures with higher H-scores (Figure 7). Also, DESeq2 with filtering demonstrated a significant difference between pipeline groups. Pipelines based on pseudoalignment provide smaller H-scores for all DE analysis methods (Supplemental Figures S8–S10). Limma delivered a higher H-score using DE transcripts without FDR p-value adjustment (Supplemental Figure S9).
Transfer RNA fragments analysis
For tsRNA expression, read alignment and quantification patterns were similar to other sRNA types. Reads assigned to mature tRNA by Rsubread were pseudoaligned by Kallisto to tsRNA sequences. High assignment rates are presented in Supplemental Table S6 and Supplemental Figure S11. The pipelines bowtie without mismatches allowed (bowtie -v 0) and hisat2 revealed the highest alignment rates across all estimates (mean = 97%), while STAR demonstrated the lowest assignment rates (mean = 79%). The ratio of assigned tsRNA reads and all input reads are shown in Supplemental Table S7. The hisat2 pipeline demonstrated the highest rates across all estimates (mean = 4.6%), while the lowest assignment rates were STAR (mean = 2.7%), and the two variations of bowtie - 3.3% of reads (Supplemental Figure S12).
A very small number of significant tsRNA was found using edgeR and limma across all datasets (up to 2.7 as mean for all pipelines with filtering and adjusted p-value < 0.05) and across all pipelines (up to 1.4 as mean for all datasets with filtering and adjusted p-value < 0.05) (Supplemental Table S8). The number of significant tsRNA using DESeq2 was greater; the mean for all pipelines was 32.0 and 2.5, with and the , respectively.
An H-score higher than was observed only for the gene signature obtained by limma with non-adjusted p-value < 0.05 (Supplemental Table S9). The highest H-score for an adjusted p-value signature was 0.68 ("Delker-C", filtering by mean(count)>5, DESeq2). H-scores higher than 0.6 was obtained by DESeq2 only. These values were lower than the ones observed for sRNA transcripts expression analysis, which may be explained by the lower signal of tsRNA compared to sRNA.
sRNA tools performance
SPORTS provided higher assignment rates than other pipelines for almost all datasets (Supplemental Table S10). For "Delker" and "Wong" data results were similar with the bowtie aligner rate. sRNAnalyzer without allowing mismatches demonstrated higher assignment rate then with allowing mismatches (mean = 0.28 versus 0.1 and 0.05 for sRNAnalyzer with 1 and 2 allowed mismatches, respectively), but less than SPORTS (mean = 0.67) (Figure 4). SPORTS and sRNAnalyzer signatures have a slightly lower H-score than alignment-based pipelines across all datasets (Figure 7B). But for "Donkin" and "Ingerslev", sRNA based pipelines managed to provide gene signatures with H-score higher than pseudoalignment or alignment based approaches (Figure 7A and Supplemental Table S5).
Overall performance and recommendation
Each stage of sRNA analysis, as indicated earlier, can be conducted employing various methods and different parameters setting for each method. Nevertheless, some sets of parameters or tools may produce better results than others, using metrics for each analytical stage of benchmarking. Thus, we suggest the following pipeline described in Table 4. Trimming procedure with the flexible upper bound () demonstrated more stable results assessed by reads after trimming across datasets. Bowtie aligner with 1 mismatch (bowtie -v 1) demonstrated the highest alignment rate for sRNAs across all datasets. Filtering by mean count higher than 5 and applying DESeq2 led to higher H-scores for obtained gene signatures.
Using main metrics/benchmarks we presented the results of the application of this pipeline for all datasets in Table 5.
Discussion
We used seven publicly available human datasets and metrics/benchmarks for each step of sRNA-seq data analysis from biosampling and library preparation to DE analysis in an effort to produce an optimized sRNA-seq pipeline. Based on our analysis, we suggest a pipeline that produces robust results of DE analysis of sRNA transcripts, at least for categorical factors and two-groups comparisons of biosamples.
Since each dataset was generated with different tissues, library preparation kits, and sequencing approaches (sequence machine and read length), large variations of data, as expected, were observed. We aimed to use existing tools to construct an optimal pipeline for quality sequencing data analysis despite the differences in input data.
To account for data variation in the original datasets, flexible trimming thresholds were applied. The input reads lengths were between 42 nt ("Donkin") and 150 nt ("Hua") across datasets. We suggest using 15 nt as a lower bound and as an upper bound of read length for trimming. This approach afforded good adapter removal and avoided significant loss of reads for a range of datasets.
There are many tools for assignment of trimmed reads to obtain usable expression data. Assigning processes can be conducted in different ways, and each has its strength and limitations. Alignment-based methods seem to be less specialized, and we suggest to use bowtie aligner which provides high assignment rate and H-score for sRNAs across datasets.
We tested 6 thresholds for data filtering. As expected, thresholds based on the minimum counts of transcripts (5 and 10) resulted in small numbers of transcripts and may result in the loss of important data (Supplemental Figure S6A,B). Filtering by mean(counts) > 5 and median(counts) > 5 provides more transcripts, so these two thresholds were used for further analysis. Median threshold is stricter for skewed distributed transcripts and provided less significant findings after DE analysis (Supplemental Figure S7). Gene signatures based on mean(counts) > 5 threshold had higher H-scores for alignment-based pipelines, so this cut-off is recommended for two groups analysis. Median (counts) > 5 threshold may be more useful for more complicated analysis such as for continuous data with many covariates, but this was not formally tested here.
For the DE analysis, three well established packages based on regression analysis (DESeq2, edgeR, limma) and two groups of contrast (categorical) factors have been used. To assess the quality of delivered expression signatures, the H-score metric of Hobotnica package was employed. We observed a similar medium quality for the data separation test using 3 packages across datasets. We suggest using DESeq2 with multiple test correction for data with strong and well-detected signals, and limma with no p-values adjustment for weaker signal data.
Tools designed specifically for small RNA analysis (such as SPORTS or sRNAnalyzer) may seem to be more suitable for sRNA seq data analysis. A disadvantage of the sRNA-specific tools is the ’map and remove’ approach, where the order of databases used to sequentially align reads can affect the analysis outcome, and different sRNA biotypes are not treated independently. sRNAnalyzer cannot analyze tRNA or rRNA, and SPORTS cannot analyze piRNA, as depicted in the Figure . These limitations and outcomes make bioinformatic analysis less flexible and informative.
It should be noted, that analyzed datasets demonstrated heterogeneity by processing steps. We suggest that the best pipeline depends on input data (cell/tissue type, library preparation kit, sequencing approaches), hypothesis of the study, and sRNA-seq data (type of factors, categorical or continuous, how many covariates for adjustment). In this study continuous factors and complex models with covariates were not analyzed. Nonetheless, for categorical factors and two groups of biosamples, our optimized pipeline for sRNA analysis recommends the following steps:
- Trimming with the lower length bound = 15 and the upper length bound = ;
- Mapping on a reference genome with bowtie aligner with one mismatch allowed (-v 1 parameter);
- Filtering by mean threshold > 5;
- DESeq2 for DE analysis with adjusted p-value < 0.05.
Although we investigated human microRNA, piRNA, tsRNA data we anticipate that our optimized pipeline may be employed with similar sRNA data from other organisms.
Conclusion
In this study, the effect of various factors that impact the expression analysis of human sRNA at different stages of data processing, were investigated. The optimal pipeline alternatives and their parameters were identified and an optimized pipeline for setting and running sRNA expression analysis are proposed. Assessing the resulting expression signatures with rank statistics-based inference suggests a way to estimate the quality of resulting signatures and performance of bioinformatical analysis for a particular biological data.
Author Contributions
Conceptualization, A.S. (Alexey Stupnikov) and O.S.; methodology, A.S. (Alexey Stupnikov); validation, V.B. and I.S.; formal analysis and investigation, V.B., I.S. and A.S. (Alexey Stupnikov); resources, O.S.; data curation, O.S. and V.B.; writing—original draft preparation, A.S. (Alexey Stupnikov) and V.B.; writing—review and editing, A.S. (Alexey Stupnikov), V.B., I.S., V.S., J.R.P., A.S. (Alexander Suvorov) and O.S.; visualization, V.B., I.S. and A.S. (Alexey Stupnikov); supervision, A.S. (Alexey Stupnikov); project administration, O.S and V.S.; funding acquisition, O.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MDPI | Multidisciplinary Digital Publishing Institute |
| DE | Differential expression |
| bowtie -v 1 | bowtie with one mismatch allowed |
| bowtie -v 0 | bowtie with no mismatches allowed |
| sRNA | small non-coding RNA |
| H-score | Hobotnica score |
References
- Storz, G. An expanding universe of noncoding RNAs. Science 2002, 296, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Peng, J.; Yi, C. The epitranscriptome of small non-coding RNAs. Non-coding RNA Research 2021, 6, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nature Reviews Genetics 2015, 16, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Penner-Goeke, S.; Binder, E.B. Epigenetics and depression. Dialogues in clinical neuroscience 2022. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nature reviews genetics 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.; Silva, J.V.; Howl, J.; Santos, M.A.; Fardilha, M. All you need to know about sperm RNAs. Human Reproduction Update 2022, 28, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Krawetz, S.A.; Kruger, A.; Lalancette, C.; Tagett, R.; Anton, E.; Draghici, S.; Diamond, M.P. A survey of small RNAs in human sperm. Human reproduction 2011, 26, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Oluwayiose, O.A.; Houle, E.; Whitcomb, B.W.; Suvorov, A.; Rahil, T.; Sites, C.K.; Krawetz, S.A.; Visconti, P.; Pilsner, J.R. Altered non-coding RNA profiles of seminal plasma extracellular vesicles of men with poor semen quality undergoing in vitro fertilization treatment. Andrology, 1111. [Google Scholar] [CrossRef]
- Marcho, C.; Oluwayiose, O.A.; Pilsner, J.R. The preconception environment and sperm epigenetics. Andrology 2020, 8, 924–942. [Google Scholar] [CrossRef]
- Kotsyfakis, M.; Patelarou, E. MicroRNAs as biomarkers of harmful environmental and occupational exposures: A systematic review. Biomarkers 2019, 24, 623–630. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Rassoulzadegan, M.; Tuorto, F.; Chen, Q. Sperm RNA code programmes the metabolic health of offspring. Nature Reviews Endocrinology 2019, 15, 489–498. [Google Scholar] [CrossRef]
- Cecere, G. Small RNAs in epigenetic inheritance: From mechanisms to trait transmission. Febs Letters 2021, 595, 2953–2977. [Google Scholar] [CrossRef] [PubMed]
- Micheel, J.; Safrastyan, A.; Wollny, D. Advances in Non-Coding RNA Sequencing. Non-coding RNA 2021, 7, 70. [Google Scholar] [CrossRef]
- Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics 2021, 11, 964. [Google Scholar] [CrossRef]
- Zytnicki, M.; Gaspin, C. srnaMapper: an optimal mapping tool for sRNA-Seq reads. BMC Bioinformatics 2022. [Google Scholar] [CrossRef]
- Roovers, E.F.; Rosenkranz, D.; Mahdipour, M.; Han, C.T.; He, N.; de Sousa Lopes, S.M.C.; van der Westerlaken, L.A.; Zischler, H.; Butter, F.; Roelen, B.A.; et al. Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell reports 2015, 10, 2069–2082. [Google Scholar] [CrossRef]
- Han, B.W.; Wang, W.; Zamore, P.D.; Weng, Z. piPipes: a set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome-and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics 2015, 31, 593–595. [Google Scholar] [CrossRef]
- Ray, R.; Pandey, P. piRNA analysis framework from small RNA-Seq data by a novel cluster prediction tool-PILFER. Genomics 2018, 110, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.; Park, J.C.; Kim, S. piClust: a density based piRNA clustering algorithm. Computational biology and chemistry 2014, 50, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, D.; Zischler, H. proTRAC-a software for probabilistic piRNA cluster detection, visualization and analysis. BMC bioinformatics 2012, 13, 1–10. [Google Scholar] [CrossRef]
- Hackenberg, M.; Rodríguez-Ezpeleta, N.; Aransay, A.M. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic acids research 2011, 39, W132–W138. [Google Scholar] [CrossRef]
- Stocks, M.B.; Mohorianu, I.; Beckers, M.; Paicu, C.; Moxon, S.; Thody, J.; Dalmay, T.; Moulton, V. The UEA sRNA Workbench (version 4.4): a comprehensive suite of tools for analyzing miRNAs and sRNAs. Bioinformatics 2018, 34, 3382–3384. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Chen, W.X.; Mei, S.Q.; Yang, Y.D.; Yang, J.H.; Qu, L.H.; Zheng, L.L. tsRFun: a comprehensive platform for decoding human tsRNA expression, functions and prognostic value by high-throughput small RNA-Seq and CLIP-Seq data. Nucleic acids research 2022, 50, D421–D431. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Puerta, E.; Lebrón, R.; Rueda, A.; Gómez-Martín, C.; Giannoukakos, S.; Jaspez, D.; Medina, J.M.; Zubkovic, A.; Jurak, I.; Fromm, B.; et al. sRNAbench and sRNAtoolbox 2019: intuitive fast small RNA profiling and differential expression. Nucleic acids research 2019, 47, W530–W535. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kim, T.K.; Baxter, D.; Scherler, K.; Gordon, A.; Fong, O.; Etheridge, A.; Galas, D.J.; Wang, K. sRNAnalyzer—a flexible and customizable small RNA sequencing data analysis pipeline. Nucleic acids research 2017, 45, 12140–12151. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Ko, E.A.; Sanders, K.M.; Chen, Q.; Zhou, T. SPORTS1. 0: a tool for annotating and profiling non-coding RNAs optimized for rRNA-and tRNA-derived small RNAs. Genomics, proteomics & bioinformatics 2018, 16, 144–151. [Google Scholar]
- Pogorelcnik, R.; Vaury, C.; Pouchin, P.; Jensen, S.; Brasset, E. sRNAPipe: a Galaxy-based pipeline for bioinformatic in-depth exploration of small RNAseq data. Mobile DNA 2018, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Panero, R.; Rinaldi, A.; Memoli, D.; Nassa, G.; Ravo, M.; Rizzo, F.; Tarallo, R.; Milanesi, L.; Weisz, A.; Giurato, G. iSmaRT: a toolkit for a comprehensive analysis of small RNA-Seq data. Bioinformatics 2017, 33, 938–940. [Google Scholar] [CrossRef]
- Rahman, R.U.; Gautam, A.; Bethune, J.; Sattar, A.; Fiosins, M.; Magruder, D.S.; Capece, V.; Shomroni, O.; Bonn, S. Oasis 2: improved online analysis of small RNA-seq data. BMC bioinformatics 2018, 19, 1–10. [Google Scholar] [CrossRef]
- Stupnikov, A.; Bezuglov, V.; Skakov, I.; Shtratnikova, V.; Pilsner, J.R.; Suvorov, A.; Sergeyev, O. ITAS: Integrated Transcript Annotation for Small RNA. Non-Coding RNA 2022, 8, 30. [Google Scholar] [CrossRef]
- Quek, C.; Jung, C.h.; Bellingham, S.A.; Lonie, A.; Hill, A.F. iSRAP–a one-touch research tool for rapid profiling of small RNA-seq data. Journal of extracellular vesicles 2015, 4, 29454. [Google Scholar] [CrossRef]
- Di Bella, S.; La Ferlita, A.; Carapezza, G.; Alaimo, S.; Isacchi, A.; Ferro, A.; Pulvirenti, A.; Bosotti, R. A benchmarking of pipelines for detecting ncRNAs from RNA-Seq data. Briefings in Bioinformatics 2019, 21, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome biology 2016, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular systems biology 2019, 15, e8746. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Bruno, V.M.; Rasko, D.A.; Cuomo, C.A.; Muñoz, J.F.; Livny, J.; Shetty, A.C.; Mahurkar, A.; Dunning Hotopp, J.C. Best practices on the differential expression analysis of multi-species RNA-seq. Genome biology 2021, 22, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 2009, 10, 1–10. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nature methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 2011, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nature biotechnology 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nature methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 2015, 43, e47–e47. [Google Scholar] [CrossRef]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic acids research 2015, 43, e140–e140. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef]
- Cho, H.; Davis, J.; Li, X.; Smith, K.S.; Battle, A.; Montgomery, S.B. High-resolution transcriptome analysis with long-read RNA sequencing. PloS one 2014, 9, e108095. [Google Scholar] [CrossRef]
- Stupnikov, A.; Glazko, G.V.; Emmert-Streib, F. Effects of subsampling on characteristics of RNA-seq data from triple-negative breast cancer patients. Chinese Journal of Cancer 2015, 34, 1–12. [Google Scholar] [CrossRef]
- Soneson, C.; Delorenzi, M. A comparison of methods for differential expression analysis of RNA-seq data. BMC bioinformatics 2013, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; Mason, C.E.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome biology 2013, 14, 1–13. [Google Scholar] [CrossRef]
- Assefa, A.T.; De Paepe, K.; Everaert, C.; Mestdagh, P.; Thas, O.; Vandesompele, J. Differential gene expression analysis tools exhibit substandard performance for long non-coding RNA-sequencing data. Genome biology 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Stupnikov, A.; McInerney, C.; Savage, K.; McIntosh, S.; Emmert-Streib, F.; Kennedy, R.; Salto-Tellez, M.; Prise, K.; McArt, D. Robustness of differential gene expression analysis of RNA-seq. Computational and structural biotechnology journal 2021, 19, 3470–3481. [Google Scholar] [CrossRef]
- https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software.
- S., A. FastQC: a quality control tool for high throughput sequence data.
- Y, W.R.K.; Meabh, M.; V, W.J.; A., S.D. A comparison of RNA extraction and sequencing protocols for detection of small RNAs in plasma. BMC Genomics 2019, 20.
- Huang, G.; Cao, M.; Huang, Z.; Xiang, Y.; Liu, J.; Wang, Y.; Wang, J.; Yang, W. Small RNA-sequencing identified the potential roles of neuron differentiation and MAPK signaling pathway in dilated cardiomyopathy. Biomedicine & Pharmacotherapy 2019, 114, 108826. [Google Scholar] [CrossRef]
- Kanth, P.; Hazel, M.W.; Boucher, K.M.; Yang, Z.; Wang, L.; Bronner, M.P.; Boylan, K.E.; Burt, R.W.; Westover, M.; Neklason, D.W.; et al. Small RNA sequencing of sessile serrated polyps identifies microRNA profile associated with colon cancer. Genes, Chromosomes and Cancer 2019, 58, 23–33. [Google Scholar] [CrossRef]
- Morgan, C.P.; Shetty, A.C.; Chan, J.C.; Berger, D.S.; Ament, S.A.; Epperson, C.N.; Bale, T.L. Repeated sampling facilitates within- and between-subject modeling of the human sperm transcriptome to identify dynamic and stress-responsive sncRNAs. Sci. Rep. 2020, 10, 17498. [Google Scholar] [CrossRef]
- Hua, M.; Liu, W.; Chen, Y.; Zhang, F.; Xu, B.; Liu, S.; Chen, G.; Shi, H.; Wu, L. Identification of small non-coding RNAs as sperm quality biomarkers for in vitro fertilization. Cell Discov. 2019, 5, 20. [Google Scholar] [CrossRef]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.R.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef]
- Ingerslev, L.R.; Donkin, I.; Fabre, O.; Versteyhe, S.; Mechta, M.; Pattamaprapanont, P.; Mortensen, B.; Krarup, N.T.; Barrès, R. Endurance training remodels sperm-borne small RNA expression and methylation at neurological gene hotspots. Clin. Epigenetics 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- https://international.neb.com/faqs/2017/07/17/how-should-my-nebnext-small-rna-library-be-trimmed.
- https://support.illumina.com/bulletins/2016/12/what-sequences-do-i-use-for-adapter-trimming.html.
- https://perkinelmer-appliedgenomics.com/wp-content/uploads/marketing/NEXTFLEX/miRNA/NEXTflex_Small_RNA_v3_Trimming_Instructions.pdf.
- https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/.
- Stupnikov, A.; Sizykh, A.; Budkina, A.; Favorov, A.; Afsari, B.; Wheelan, S.; Marchionni, L.; Medvedeva, Y. Hobotnica: exploring molecular signature quality [version 2; peer review: 2 approved]. F1000Research 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J. The Connectivity Map: a new tool for biomedical research. Nature reviews cancer 2007, 7, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Musa, A.; Ghoraie, L.S.; Zhang, S.D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Briefings in bioinformatics 2018, 19, 506–523. [Google Scholar] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. goseq: Gene Ontology testing for RNA-seq datasets. R Bioconductor 2012, 8, 1–25. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic acids research 2007, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC genome browser database. Nucleic acids research 2003, 31, 51–54. [Google Scholar] [CrossRef]
Figure 1.
Pipelines’ comparison for sRNAnalyzer, SPORTS and sRNAtoolbox.
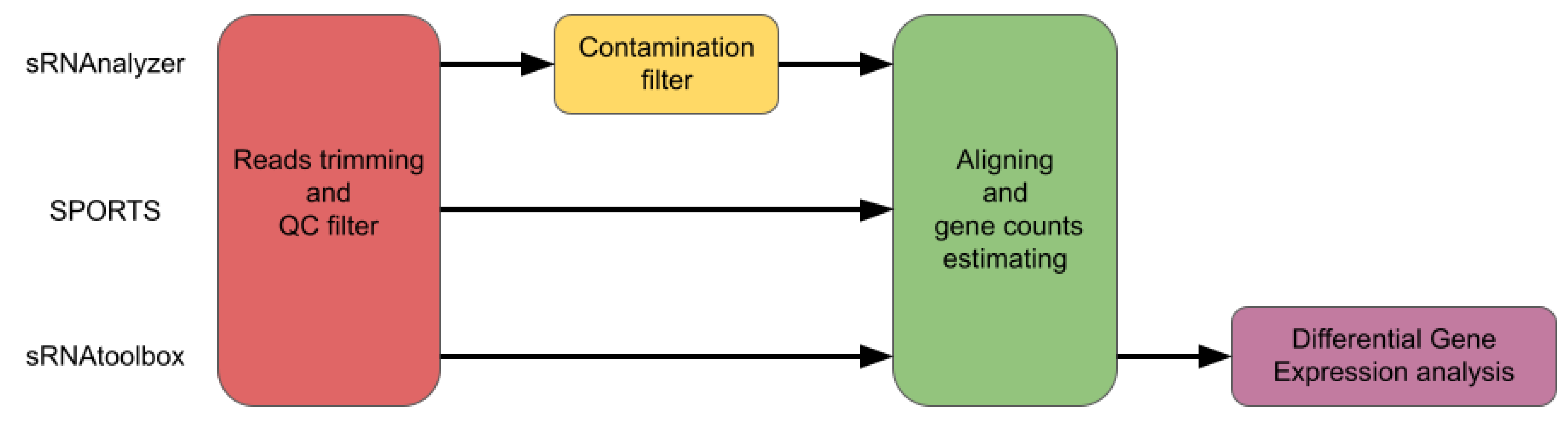
Figure 2.
(A) Percent of reads passing trimming for all datasets with various upper length bound. All reads were previously trimmed of adapters, and reads less than 15 nt were removed ( boxplot). Upper bound choosing strategy defined by color: - grey, - red, - green, - lightblue (B) Number of reads before (black border) and after (red border) trimming among datasets and adapters length (boxplot fill) (C) Mean read length after trimming.
Figure 2.
(A) Percent of reads passing trimming for all datasets with various upper length bound. All reads were previously trimmed of adapters, and reads less than 15 nt were removed ( boxplot). Upper bound choosing strategy defined by color: - grey, - red, - green, - lightblue (B) Number of reads before (black border) and after (red border) trimming among datasets and adapters length (boxplot fill) (C) Mean read length after trimming.
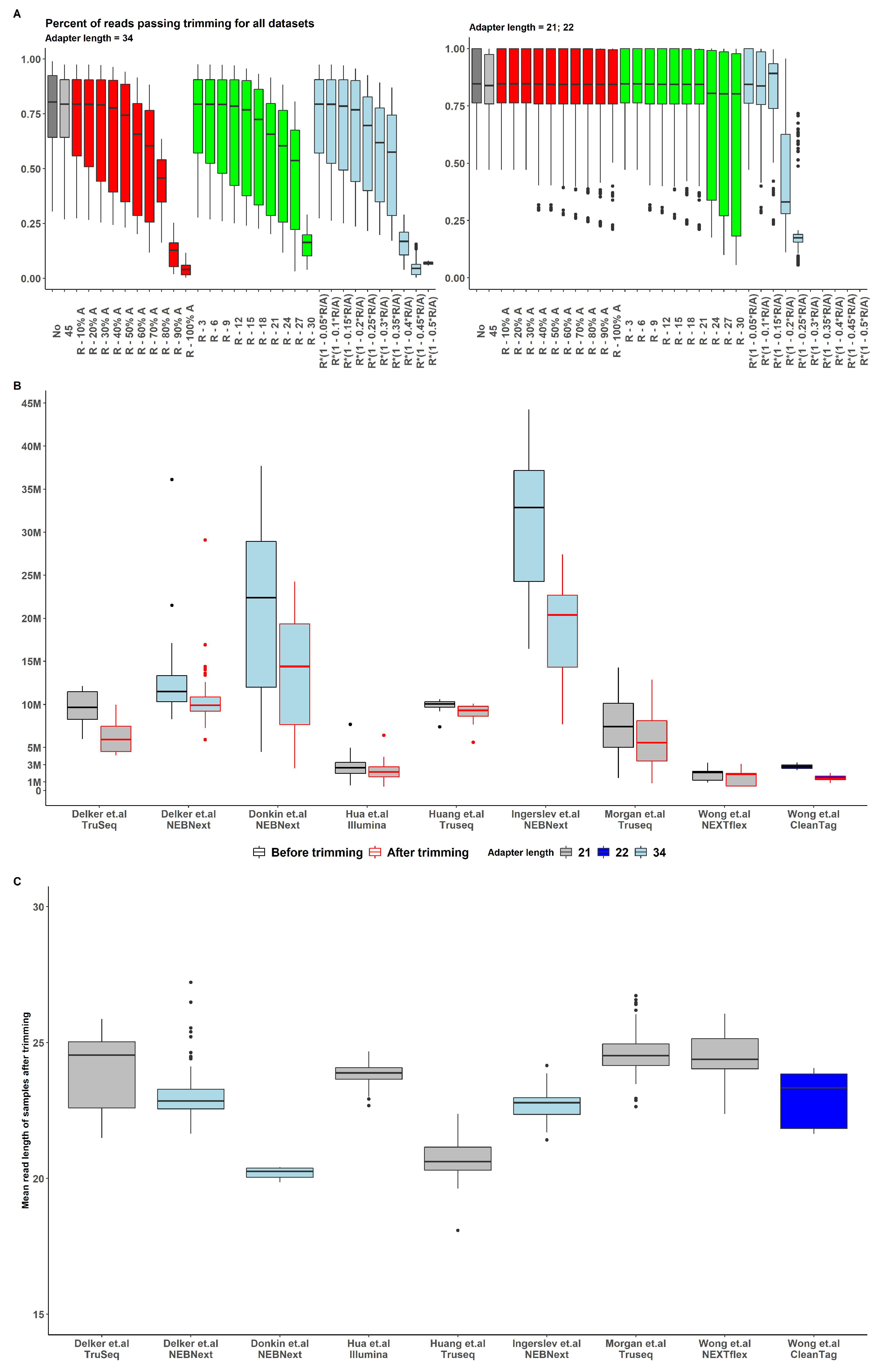
Figure 3.
(A) Percent of reads passing the genome alignment (for bowtie, hist2 and STAR genome aligners) or prebuilt transcriptome (for RSEM) (B) Number of reads passing the genome alignment (for bowtie, hist2 and STAR genome aligners) or prebuilt transcriptome (for RSEM).
Figure 3.
(A) Percent of reads passing the genome alignment (for bowtie, hist2 and STAR genome aligners) or prebuilt transcriptome (for RSEM) (B) Number of reads passing the genome alignment (for bowtie, hist2 and STAR genome aligners) or prebuilt transcriptome (for RSEM).
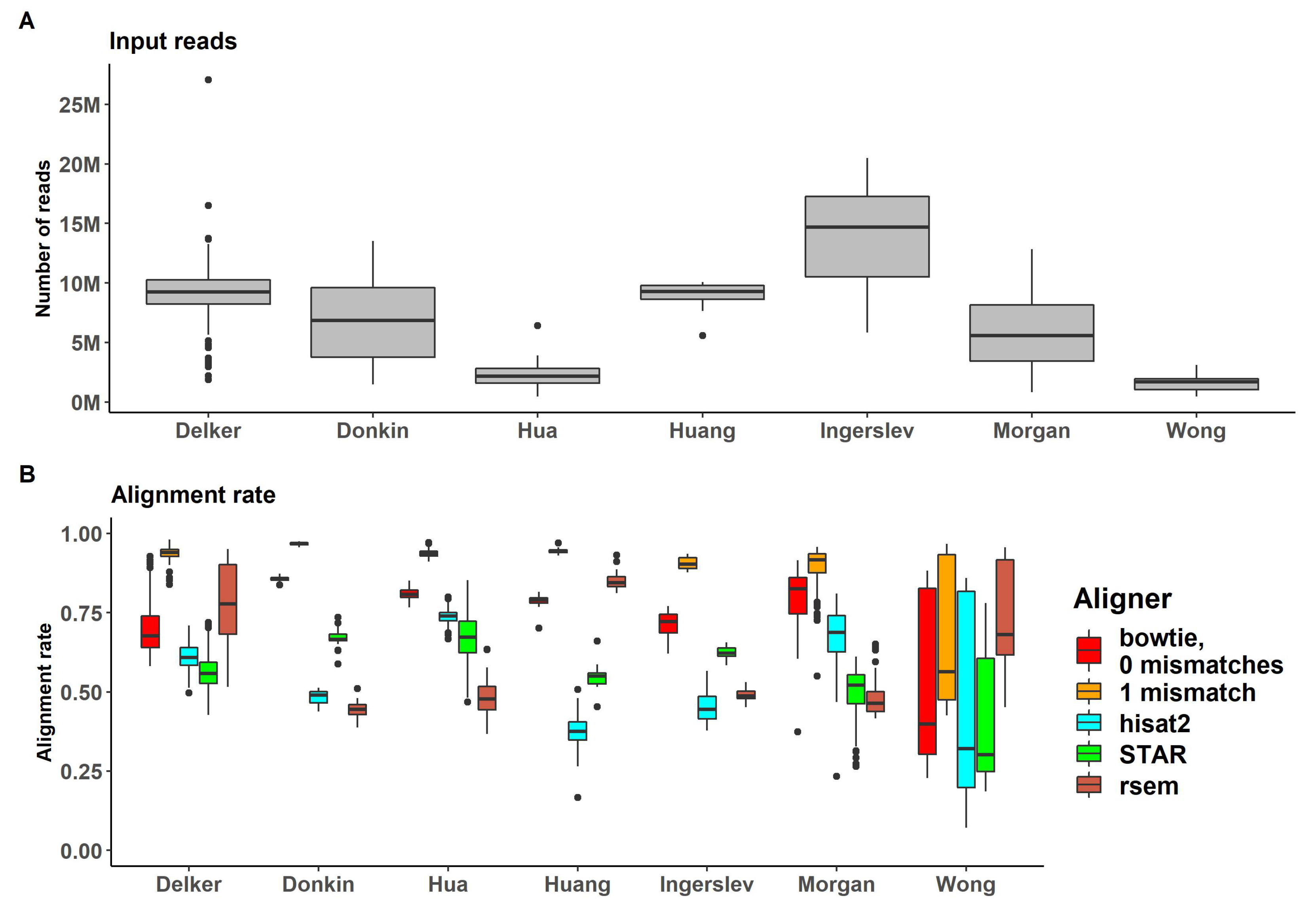
Figure 4.
Percent of reads processed by all used pipelines.
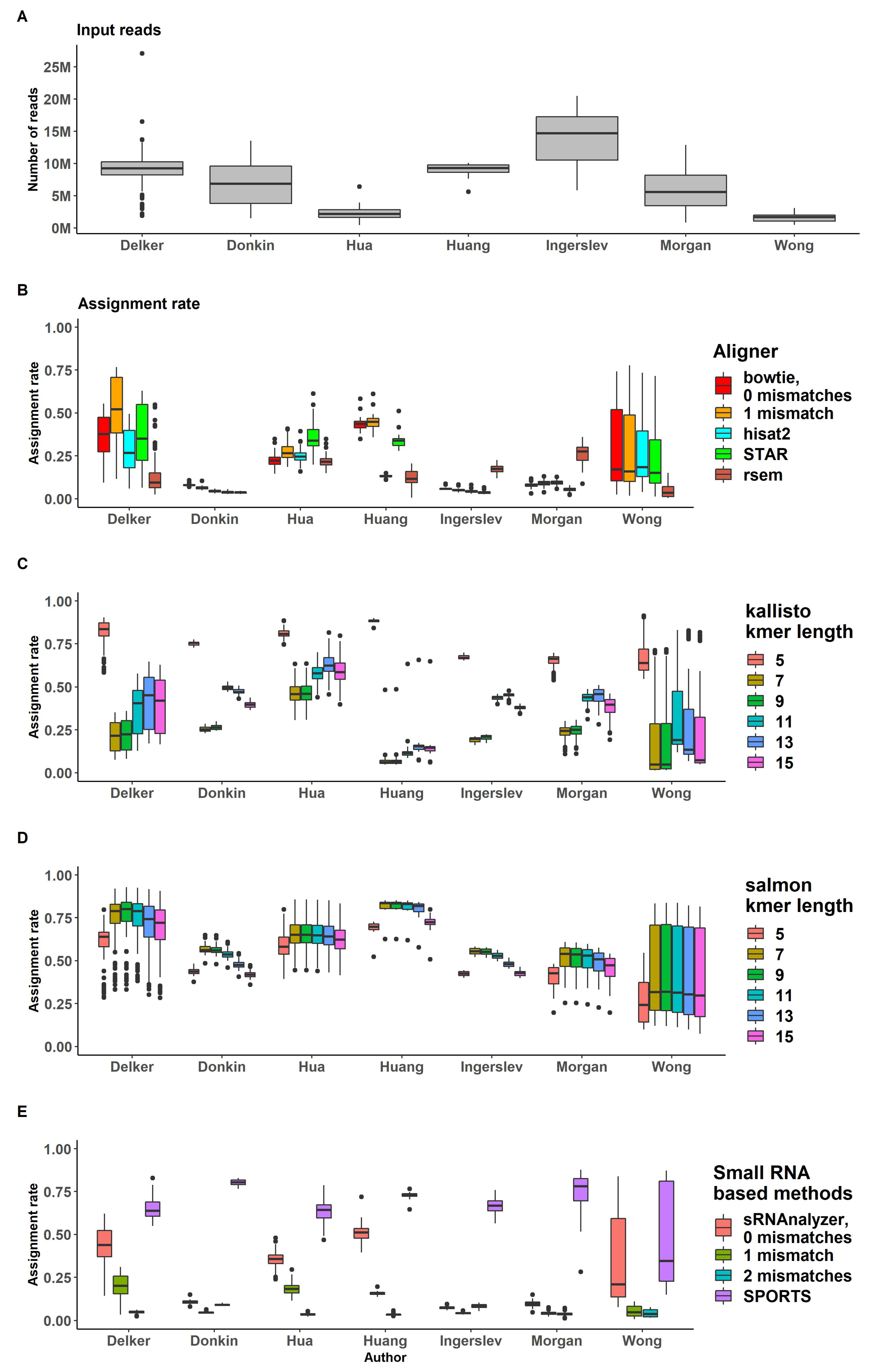
Figure 5.
(A) Distribution of sRNA types expression values by datasets and pipelines (B) Distribution of sRNA types transcripts by datasets and pipelines.
Figure 5.
(A) Distribution of sRNA types expression values by datasets and pipelines (B) Distribution of sRNA types transcripts by datasets and pipelines.

Figure 6.
Number of DE transcripts provided by DESeq2, edgeR and limma with p-value or adjusted p-value < 0.05 among data with different filtering.
Figure 6.
Number of DE transcripts provided by DESeq2, edgeR and limma with p-value or adjusted p-value < 0.05 among data with different filtering.
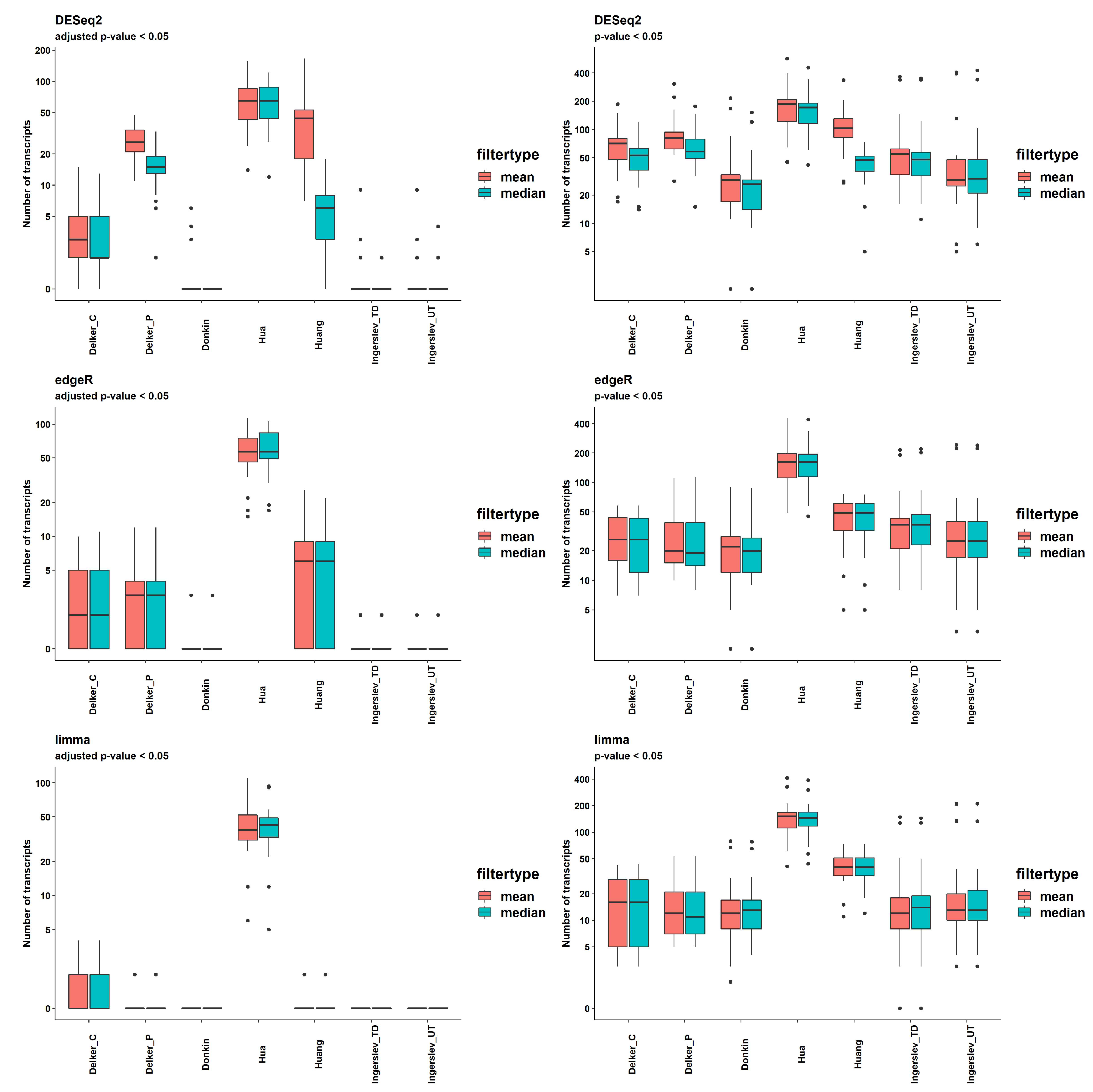
Figure 7.
Distribution of H-scores across pseudoalignment-based pipelines (kallisto and salmon), sRNA-based pipelines (SPORTS and sRNAnalyzer) and alignment-based pipelines (bowtie, hisat, STAR and rsem) for every dataset (A) and for all data (B).
Figure 7.
Distribution of H-scores across pseudoalignment-based pipelines (kallisto and salmon), sRNA-based pipelines (SPORTS and sRNAnalyzer) and alignment-based pipelines (bowtie, hisat, STAR and rsem) for every dataset (A) and for all data (B).
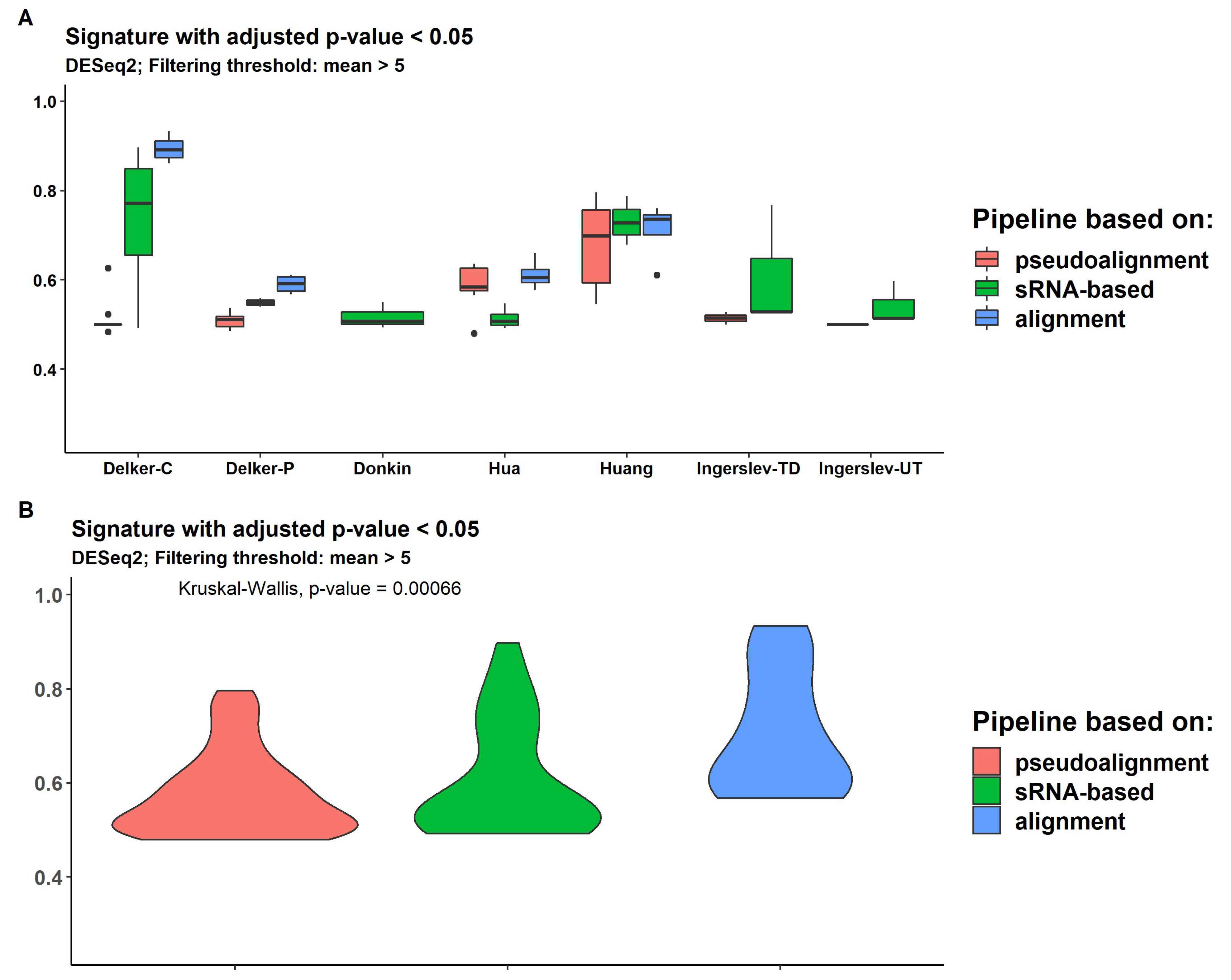

Table 1.
Main features of some existing sRNAs pipelines.
| name | year | last | status | interface | type | RNA classes | output |
|---|---|---|---|---|---|---|---|
| update | |||||||
| DSAP | 2010 | 2010 | not supported | GUI | map and | ncRNA (Rfam), miRNA | DE |
| for Solexa only | remove | transcripts | |||||
| Oasis 2 | 2018 | 2018 | not supported | online | map and | miRNA, piRNA, | transcripts |
| remove | small nucleolar RNA (snoRNA) | counts | |||||
| iSmaRT | 2017 | 2017 | supported | GUI | map and | miRNA, piRNA | DE |
| from ftp-server | remove | transcripts | |||||
| sRNAPipe | 2018 | 2021 | supported | GUI | map and | miRNA, piRNA, | transcripts |
| Galaxy-based | remove | rRNA, tRNAs, | counts | ||||
| transposable elements | |||||||
| iSRAP | 2015 | 2016 | supported | CLI | map and | miRNA, piRNA, snoRNA | DE |
| remove | transcripts | ||||||
| miARma-seq | 2016 | 2019 | not supported | CLI | map and | miRNA, snoRNA | DE |
| remove | transcripts | ||||||
| tsRFun | 2022 | 2022 | supported | online | map and | tsRNA | DE |
| remove | transcripts | ||||||
| piPipes | 2015 | 2016 | supported | CLI | map | piRNA | transcripts |
| counts | |||||||
| PILFER | 2018 | 2018 | supported | CLI | map | piRNA | transcripts |
| counts | |||||||
| miRanalyzer | 2011 | 2014 | not supported | online | map and | miRNA | DE |
| remove | transcripts | ||||||
| sRNA workbench | 2018 | 2018 | not supported | CLI | map | miRNA | alignment |
| sRNAtoolbox | 2022 | 2022 | supported | online | map and | miRNA, tRNA, | DE |
| remove | ncRNA, cDNA | transcripts | |||||
| sRNAnalyzer | 2017 | 2017 | supported | CLI | map and | miRNA, piRNA, tRNA, | transcripts |
| remove | snoRNA | counts | |||||
| SPORTS | 2018 | 2021 | supported | CLI | map and | miRNA, piRNA, rRNAs, | transcripts |
| remove | tRNAs, tRNA fragments | counts |
Table 2.
Description of the publicly available human datasets that have been used in this study for benchmarking.
Table 2.
Description of the publicly available human datasets that have been used in this study for benchmarking.
| GEO ID | Cite | Object | Total samples number | Raw reads | Library kit |
|---|---|---|---|---|---|
| (contrast groups) | length | ||||
| GSE118125 | [60] | Blood plasma | 30 | 76 | NEXTflex |
| CleanTag, Qiaseq | |||||
| GSE117841 | [61] | Blood | 20 (10 vs 10) | 50 | Truseq |
| GSE118504 | [62] | Colon Cancer | 108 (16 vs 14 and 15 vs 15) | 50 | NEBNext, Truseq |
| GSE159155 | [63] | Sperm | 98 | 50 | Truseq |
| GSE110190 | [64] | Sperm | 87 (64 vs 23) | 150 | Illumina |
| GSE74426 | [65] | Sperm | 23 (13 vs 10) | 42 | NEBNext |
| GSE109478 | [66] | Sperm | 24 (9 vs 9 and 9 vs 6) | 51 | NEBNext |
Table 3.
Metrics for benchmarking by stages of bioinformatic analysis.
| Stage | Metrics |
|---|---|
| Input data | Biological object, lab kit, read length, adapter length, reads quality |
| Trimming | Part of reads processed trimming, length threshold |
| Aligning (for genome alignment based pipelines) | Alignment rate |
| Assigning | Assignment rate, distribution by sRNA type |
| Filtering | Number of transcripts after trimming |
| DE | Number of significant transcripts |
| Expression signature quality evaluation | H-score |
Table 4.
Recommended pipeline for sRNA analyses.
| Stage | Pipeline command | Justification |
|---|---|---|
| Trimming | Read length: lower bound - 15 and | Retain sufficient and the same number of reads after |
| upper bound - | trimming for downstream analyses for all datasets | |
| Aligning | bowtie aligner with 1 mismatch allowed | The high alignment rate and H-score for all datasets |
| Assigning | ITAS [30] | Optimized annotation for small RNA |
| Filtering | mean count > 5 | Sufficient number of transcripts for the |
| downstream analysis and higher H-score | ||
| DE analysis | DESeq2 | Sufficient number of significant transcripts and |
| high H-score |
Table 5.
Results for bowtie -v 1 pipeline; * - assigned/aligned; ** - mean filtering; *** - DESeq2.
| Dataset | Biological object and | Alignment | Assignment* | Number of filtered | Number of | H-score |
|---|---|---|---|---|---|---|
| contrast | rate | rate | transcripts** | findings*** | ||
| "Hua" | Sperm | 0.94 | 0.29 | 901 | 71 | 0.66 |
| "Donkin" | Sperm | 0.97 | 0.07 | 966 | 0 | - |
| "Ingerslev" | Sperm untrained contrast | 0.9 | 0.06 | 1236 | 0 | - |
| detrained contrast | 0 | - | ||||
| "Morgan" | Sperm | 0.89 | 0.1 | - | - | - |
| "Delker" | colon cancer polyps contrast | 0.94 | 0.55 | 1142 | 21 | 0.6 |
| controls contrast | 5 | 0.86 | ||||
| "Huang" | Blood | 0.94 | 0.48 | 498 | 17 | 0.73 |
| "Wong" | Blood plasma | 0.68 | 0.38 | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



