Submitted:
24 December 2022
Posted:
03 January 2023
You are already at the latest version
Abstract
The HER2 overexpression occurs in 10–20% of breast cancer patients. HER2+ tumors are characterized by increase in Ki67, early relapse and increased metastasis. There is little known about factors influencing early stages of HER2- tumorigenesis and diagnostic markers. Previously, it was shown that deletion of NEDD9 in mouse models of HER2 cancer interferes with tumor growth, but the role of NEDD9 upregulation is currently unexplored. We report that NEDD9 is overexpressed in a significant subset of HER2+ breast cancers and correlates with limited response to anti-HER2 therapy. To investigate the mechanisms through which NEDD9 influences HER2-dependent tumorigenesis, we generated MMTV-Cre-NEDD9 transgenic mice. The analysis of mammary glands shows extensive ductal epithelium hyperplasia, increased branching, and terminal end bud expansion. The addition of oncogene Erbb2 (neu) leads to development of early hyperplastic benign lesions earlier (~16 weeks) with a significantly shorter latency than the control mice. Similarly, NEDD9 upregulation in MCF10A-derived acini leads to hyperplasia like DCIS. This phenotype is associated with activation of ERK1/2 and AURKA kinases leading to an increased proliferation of luminal cells. These findings indicate that NEDD9 is setting permissive conditions for HER2-induced tumorigenesis, thus identifying this protein as a potential diagnostic marker for early detection.
Keywords:
HER2
; breast cancer
; NEDD9 adaptor protein
; hyperplasia
; AURKA
1. Introduction
The HER2 (Human Epidermal Growth Factor Receptor 2) amplification or overexpression occurs in 10–20% of breast carcinomas [1,2]. A high level of Ki67 expression, early relapse rate, and increased metastasis characterize HER2-positive (+) tumors [3]. The localized HER2+disease, confined to the breast tissue, has a 5-year survival of 97.3%, while regional disease with lymph node positivity is 82.8%, and stage IV disease with metastases to distant organs is 38.8% [1]. This statistic highlights the importance of studies addressing the disease's initiation to improve early detection and, thus, survival.
Many advances have been made in treating HER2-positive breast cancer since the introduction of targeted monoclonal antibody therapies [4]. These include trastuzumab, pertuzumab-antibody-based therapies, and lapatinib, neratinib, and pyrotinib - the kinase inhibitors. However, many patients become resistant to treatment and relapse [5,6]. Identifying novel therapeutic targets is critical to combat the resistance and improve outcomes.
HER2 (also known as v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 (Erbb2), and neu) normally functions as a receptor tyrosine kinase (RTK) that can be activated by dimerization with other EGFR family members. Phosphorylation of the intracellular domain at (Tyr1144, Tyr1201, Tyr1226/1227, or Tyr1253) enables signaling through MAPK, AKT, and JNK resulting in increased proliferation and survival [1,3]. Minimal increases in levels of HER2 allow for altered growth of mammary glands [4], setting the stage for early breast lesions leading to DCIS [5,6]. Disease progression correlates with increased HER2 expression. Mouse models have been developed to analyze mammary gland transformation using rodent neu protein [7]. MMTV-neu mouse model expressing an unactivated form of rat Erbb2 gene (homolog of HER2) similar to human cancers characterized by long latency (29-48 weeks). This model allows for the analysis of early transformation events [8], starting from hyperplasia/dysplasia and moving toward adenocarcinoma [9]. Long latency indicates additional events are required to enable transformation since unactivated Erbb2 is primarily found in monomeric/inactive form [10]. Little is known about early cooperating events in HER2-amplified tumors. The mutations in the p53 tumor suppressor, upregulation of heterodimerization partners such as HER3 and EGFR, and activation of downstream targets, such as Src kinase, were often found in HER2+ breast cancer patients and promoted tumorigenesis [11].
NEDD9 (Neural Precursor Cell Expressed Developmentally Down-regulated 9) is an adaptor protein highly expressed in epithelial cells and is critical for the activation of multiple kinases, including Src[12], FAK[13], AURKA[14], and integrins [8]. NEDD9, also known as Cas-L or HEF1, is a member of the CAS (Crk-Associated substrate) family of adaptor/scaffold proteins that function in cell signaling [9]. Other CAS proteins, such as p130Cas, are also expressed in HER2+ breast cancer [15]. As shown by whole-body gene knockout, endogenous NEDD9 is required for tumor growth in MMTV-neu transgenic mice [16]. NEDD9 is involved in cell division, migration, and invasion regulation, identifying this Cas-L family member as a potential cooperating oncogene in HER2-driven carcinogenesis [17,18,19,20]. However, specific processes affected by NEDD9 during the early stages preceding transformation are currently not well defined.
Here we report that increased NEDD9 expression tightly correlates with the expression of HER2 protein in breast cancer patient biopsies and anti-HER2 therapy response. Additionally, survival analysis of breast cancer (BC) patient cohorts in patient microarray datasets showing high expression of NEDD9 correlates with poorer overall survival. The conditional knock-in mouse model of NEDD9 in the mammary gland is characterized by mammary epithelial hyperplasia and promotes the early onset of Erbb2/Neu-driven tumorigenesis. Mechanistically, NEDD9 upregulation led to increased branching morphogenesis and expansion of luminal epithelial cells. In HER2+ human breast cancer cell lines, NEDD9 was upregulated compared to control and correlates with proliferation. The upregulation of NEDD9 also correlates with limited response to anti-HER2-targeted therapies, suggesting its involvement in sustainable downstream signaling. Our findings suggest that NEDD9 plays a crucial role in HER2+ breast cancer initiation, progression, and drug response; thus, targeting NEDD9 might provide new treatment strategies for BC patients and expose new vulnerabilities that would improve patient survival.
2. Materials and Methods
2.1. Generation of conditional NEDD9 knock IN (Floxed-STOP-NEDD9) transgenic strain and locus insertion.
A targeting vector was utilized for homologous recombination at the Rosa26 locus of the murine genome. The cDNA of human NEDD9 was cloned under CAAG ubiquitously expressed promoter (CMV immediate early enhancer/ chicken b-actin promoter fusion), separated by a stop codon that is flanked by loxP Cre recombinase recognition sites (Figure 2A). GenOway, Inc. performed vector design, mouse embryo injection, and transplantation into a C57BL/6 background. The PCR screening for homologous recombination at the 5’ end of the targeting vector was performed using a set of designed primers: GX6043/44. Once homozygous floxed NEDD9 knock-in mice were generated, the transgenic line was cryopreserved and stored at the Jackson Laboratory. Due to the significant impact of genetic background on tumorigenesis, the NEDD9 transgene was transferred to the FVB background using speed congenic technology executed by Charles Rivers Laboratory and WVU Transgenic Animal Facility. The successful transfer of NEDD9 transgene was confirmed by PCR analysis, as outlined below.
2.2. Generation of FVB/J-MMTV-Cre-NEDD9+/+ mice for mammary gland specific upregulation of NEDD9.
The targeting vector was utilized for homologous recombination at the Rosa26 locus of the murine genome. The cDNA of human NEDD9 (OriGene Technologies, Inc., Rockville, MD, USA) was cloned under CAAG ubiquitously expressed promoter (CMV immediate early enhancer/ chicken b-actin promoter fusion), separated by a stop cassette that is flanked by LoxP Cre recombinase recognition sites (Figure 2A). GenOway, Inc., France, performed vector design, mouse embryo injection, and transplantation into a C57BL/6 background. This PCR is performed using a reverse primer (GX6044: 5’GCAGTGAGAAGAGTACCACCATGAGTCC3’) hybridizing in an exogenous sequence added just upstream of the CAGG promoter and a forward primer (GX6043: 5’AAGACGAAAAGGGCAAGCATCTTCC3’) hybridizing upstream of the targeting vector homology sequence (Figure 2B). Because of its localization, this primer set allows unequivocal and specific detection of the Rosa26 Knock-in locus. Once homozygous floxed NEDD9 knock-in mice were generated, the transgenic line was cryopreserved and stored at the Jackson Laboratory, Bar Harbor, ME, USA. Due to the significant impact of genetic background on tumorigenesis, the NEDD9 transgene was transferred to the FVB/J background using speed congenic technology executed by Charles Rivers Laboratories (Wilmington, MA, USA) and WVU Transgenic Animal Facility. The successful transfer of NEDD9 transgene was confirmed by PCR analysis, as outlined.
2.3. Generation of FVB/J-MMTV-Cre-NEDD9+/+ mice for mammary gland-specific upregulation of NEDD9.
The FVB/J-Floxed-STOP-NEDD9fx/fx mice were crossed with FVB/J-MMTV-Cre mice expressing Cre recombinase under the mouse mammary tumor virus (MMTV) promoter, specific for mammary gland epithelium. This line was obtained via a gracious gift from Dr. J Michael Ruppert (West Virginia University, Morgantown, USA)[21]. The resultant offspring were genotyped using a set of designed primers to detect wild-type, recombined, and Cre-induced (Stop-cassette excised) NEDD9 cDNA. Homozygous FVB/J-MMTV-Cre-NEDD9+/+ mice were used to analyze normal mammary gland development and for other crosses with mammary tumor models. The upregulation of NEDD9 in the mammary epithelium was confirmed by western blot analysis and immunohistochemistry.
2.4. PCR screening strategy for the genotyping of the homozygous NEDD9fx/fx knock-in mice.
The homozygous animals were identified using a PCR specific to the wild-type Rosa26 allele. The primer pair 027-ROSA-GX2942 / 028-ROSA-GX2943 has been designed and validated by genOway for the specific detection of the wild-type allele, producing 304bp product. The forward primer 027-ROSA-GX2942 (5’CAATACCTTTCTGGGAGTTCTCTGC3’) hybridizes within the 5’ homology arm. The reverse primer 028-ROSA-GX2943 (5’CTGCATAAAACCCCAGATGACTACC3’) is located within the Rosa26 locus in a region, which is deleted in knock-in mice, and thus, no PCR product is produced.
2.5. PCR screening strategy for the genotyping of the induced NEDD9 knock-in line (with removed transcription stop cassette).
Mouse DNA was isolated from 0.3-0.5cm tail fragment using the PrepEase Genomic DNA Isolation Kit (Affymetrix Inc., Cleveland, OH, USA) as per manufactures instructions. This PCR was performed using 61976cre-PUG1 (5’CTGCTCTATGACGGTCAGGATGTCTCC3’) and 61977cre-PUG1 (5’GGGCAACGTGCTGGTTATTGTGC3’) primers enabling the detection of the Cre-mediated excised Rosa26 Knock-in allele. The primer 61977cre-PUG1 is located within the NEDD9 transgene. The primer 61976cre-PUG1 hybridizes within the CAAG promoter. The Cre-excised recombined allele produced 296 bp product, while the non-excised recombined allele produced 3314 bp.
2.6. PCR screening strategy for the genotyping of the homozygous NEDD9/Cre/Erbb2 mice.
The FVB/J-MMTV-Cre-NEDD9+/+ mice were crossed with FVB/J-MMTV-Erbb2/neu mouse model for mammary tumors in humans purchased from The Jackson Laboratory (Cat#002376), Bar Harbor, ME, USA. This strain expresses unactivated wild-type cDNA of rat Erbb2 (neu). The resultant strain expressing all three transgenes: Cre, NEDD9, and Erbb2 (neu), was confirmed by PCR analysis using target-specific primers as outlined above. The triple transgene female mice, along with controls: MMTV-Cre, MMTV-Cre-NEDD9+/+, and MMTV-neu, were used in studies outlined here. Mice were genotyped using the following primers: human NEDD9 cDNA: 5’TCCCAGAGTGTGCCGAGGAA3’, 5’GGGCCTTTTGCTGATGAGGG3’; ROSA: 5’CAATACCTTTCTGGGAGTTCTCTGC3’, 5’CTGCATAAAACCCCAGATGACTACC3’; Erbb2: 5’CGGAACCCACATCAGGCC3’, 5’TTTCCTGCAGCAGCCTACGC3’; Cre: 5’GGTTCTGATCTGAGCTCTGAGTG3’, 5’CATCACTCGTTGCATCGACCG3’ using PCR RANGER mix (Meridian BioScience Inc., Memphis, TN, USA) resulted in the following band sizes, an indication of the presence of the NEDD9 (knock-in) 553bp (base pairs), ROSA (wild type) 304bp, Cre 900bp, Erbb2 600bp.
2.7. Generation of Mouse Embryonic Fibroblasts with NEDD9 knock-in.
Mouse embryonic fibroblasts (MEFs) were generated from embryos harvested at day 12-14 (E12-14) as previously described[22,23]. Tissue was minced using #10 scalpel blades and incubated for 15 minutes in 0.25% Trypsin-EDTA at 37°C. Full medium, Dulbecco’s modified Eagle’s (DMEM) medium supplemented with 15% fetal bovine serum (FBS) and antibiotic-antimycotic was added to neutralize the trypsin. Cells were pelleted by centrifugation, resuspended in a complete medium, plated, and passaged. All cell culture reagents were purchased from ThermoFisher Scientific, Waltham, MA, USA. Cells were cryopreserved for later use. As per manufacturer instructions, MEF cells were infected with adenovirus expressing GFP-tagged Cre-recombinase (green fluorescent protein) and Ad-Cre-GFP (Vector Biolabs, Malvern, PA, USA). Control and Ad-Cre-GFP positive cells 72h post-infection were lysed and probed by western for NEDD9 and GAPDH protein levels as previously described [24].
2.8. Quantification of Branching Density in Mammary Gland Whole-Mounts.
The fifth inguinal mammary gland was removed from the mouse, according to[25], and placed on a positively charged slide (StatLab, McKinney, TX, USA). Subsequently fixed in Carnoy’s solution (Fisher Scientific, Pittsburgh, PA, USA) overnight and stained in a Carmine solution prepared as previously described [26]. Following staining, tissue was dehydrated with increasing ethanol, 70-100% - 15 minutes each. To clear tissue was placed in xylene for 12-72 hours. Once the tissue became transparent, it was mounted using Permount Mounting Medium (Fisher Scientific, Pittsburgh, PA, USA). Images were captured using an Olympus VS120 Slide Scanner microscope (Olympus Lifescience, Center Valley, PA, USA) with 10X U Plan S Apo/0.75 NA objective. Quantification of mammary gland branching and terminal end buds was conducted as previously described [25] using five mice per genotype and five randomly selected standard size fields of view per mouse.
2.9. Cell Culture, Plasmids, and other Reagents.
Cell lines BT-474, SK-BR-3, AU565, and MCF10A were purchased from and authenticated by American Type Culture Collection (Manassas, VA, USA) and cultured based on the manufacturer’s recommendations. JIMT-1 was purchased and authenticated by Addexbio (San Diego, CA, USA). JIMT-1-Br3 was generously provided by Dr. Paul Lockman (West Virginia University School of Pharmacy, WV, USA)[27]. MCF10A-HER2-WT and MCF10A-Empty control was generously provided by Dr. Yehenew Agazie (Department of Biochemistry, West Virginia University School of Medicine, WV, USA) was grown in medium as previously described [28]. The cell lines have been authenticated every 10-20 passages and only low passage cells (2-6) were used for experiments. Cell medium, supplements including Horse serum, EGF, penicillin and streptomycin, antibiotic/antimycotic, TrypLE, trypsin were purchased from ThermoFisher Scientific, Waltham, MA, USA, FBS (fetal bovine serum) was purchased from VWR (Radnor, PA, USA), insulin, cholera toxin, hydrocortisone (Sigma-Aldrich, St. Louis, MO, USA).
2.10. Lentivirus constructs, Cell Infection and Transfection Reagents.
The cDNA for mouse NEDD9 (OriGene Technologies, Inc., Rockville, MD, USA) was subcloned (XhoI/EcoRI) into pLUTz (tet-inducible) lentivirus vectors ([29], gift from Dr. A. Ivanov, West Virginia University School of Medicine, WV, USA) to produce pLUTz-ms-NEDD9. The construct was transfected into Lenti-X™ 293T cells (Takara Bio, San Jose, CA, USA) based on the manufacturer’s recommendations using Turbofect transfection reagent (ThermoFisher Scientific, Waltham, MA, USA). The lentivirus particles were prepared for infection using established protocols [30]; concentrated using ultracentrifugation and 100ul (~108 MOI) added to 50% confluent cells twice a day for 3 days with polybrene 5-10ug/ml (Sigma-Aldrich, St. Louis, MO, USA). Infected cells were subjected to zeocin (InvivoGen US, San Diego, CA, USA) selection (200ug/ml) for 2 weeks with medium changed every three days.
2.11. Lentivirus constructs, Cell Infection and Transfection Reagents.
The cDNA for mouse NEDD9 (OriGene Technologies, Inc., Rockville, MD, USA) was subcloned (XhoI/EcoRI) into pLUTz (tet-inducible) lentivirus vectors ([29], gift from Dr. A. Ivanov, West Virginia University School of Medicine, WV, USA) to produce pLUTz-ms-NEDD9. The construct was transfected into Lenti-X™ 293T cells (Takara Bio, San Jose, CA, USA) based on the manufacturer’s recommendations using Turbofect transfection reagent (ThermoFisher Scientific, Waltham, MA, USA). The lentivirus particles were prepared for infection using established protocols [30]; concentrated using ultracentrifugation and 100ul (~108 MOI) added to 50% confluent cells twice a day for 3 days with polybrene 5-10ug/ml (Sigma-Aldrich, St. Louis, MO, USA). Infected cells were subjected to zeocin (InvivoGen US, San Diego, CA, USA) selection (200ug/ml) for 2 weeks with medium changed every three days.
2.12. Tumor Micro Array and Patient Data.
The Cancer Genome Atlas (TCGA, https://www.cancer.gov) breast cancer data and Kaplan-Meier Plotter (https://kmplot.com) web-based tools were used to evaluate genomic DNA and RNA-seq expression of NEDD9 in HER2+ breast cancer as previously described [31,32]. The specific selection criteria used for analysis provided in Supplementary Figure S1. High-density breast cancer tissue microarrays BR2082 and BR1008 were used in this study. Diagnostics, stage and HER2 positivity scoring data provided in Supplementary Table S1. The biopsies were collected with full donor consent by US Biomax Inc./TissueArray Derwood, MD, USA. The HER2+ cases were selected for further analysis.
2.13. Immunohistochemistry and Scoring Procedures.
Immunohistochemistry (IHC) was done according to the manufacturer's recommendations (US Biomax Inc. Derwood, MD, USA) using previously validated anti-NEDD9 antibodies (clone 2G9)[33] and HER2. Manual scoring of staining intensity [negative (0), weak (1+), moderate (2+), or strong (3+)], as well as location and cell types was completed by an independent pathologist from US Biomax, Inc. Each core was scanned by the Aperio Scanning System (Leica Biosystems Deer Park, IL, USA) as previously described [24]. The total number of positive cells and the intensity of NEDD9 staining were computed by Aperio Image- Scope10.1 software based on the digital images taken (x20) from each core and normalized to the background control and normal adjacent tissue.
2.14. Fluorescent immunohistochemistry and Hematoxylin & Eosin (H&E) staining.
Briefly, deparaffinization and rehydration of 5um sections were performed as follows: 1) three incubations for 3 minutes in xylene, 2) three incubations for 2 minutes in 100% ethanol, 3) 2 min each in 95, 80, and 70% ethanol 4) 5 minutes in 1x TBS (Tris-Buffered Saline). All chemicals, BSA and buffers were purchased from Fisher Scientific, Pittsburgh, PA, USA. Antigen retrieval was performed using citrate buffer, pH 8.0 at 98C, for 20 minutes as previously reported[34]. Sections were subsequently blocked using 5% Bovine Serum Albumin (BSA), 1xTBS solution. Sections were stained with anti-Ki67-AlexaFluro-555 conjugated antibody (BD Biosciences, Franklin Lakes, NJ, USA), anti-cytokeratin 8[35] (1:100 dilution, TROMA-I was deposited to the DSHB (Developmental Studies Hybridoma Bank) by Drs. Brulet, P. & Kemler, R.), and anti-cytokeratin5 (Poly19055, BioLegend San Diego, CA, USA) antibodies, dilution 1:500, overnight incubation as previously reported[36]. Secondary antibodies diluted, 1:10000, included AlexaFluor-488, 555, and 647 (ThermoFisher Scientific, Waltham, MA, USA). The sections were mounted with ProLong Gold DAPI-containing media (ThermoFisher Scientific, Waltham, MA, USA). Images were captured using an Olympus VS120 Slide Scanner microscope with 10X U Plan S Apo/0.75 NA objective (Olympus Lifescience, Center Valley, PA, USA).
For H&E staining slides post deparaffinization and rehydration were incubated with Hematoxylin (Fisher Scientific, Pittsburgh, PA, USA) for 45 seconds, rinsed with water for 30 seconds, followed by Eosin for 1 minute, rinsed and dehydrated in 95% ethanol, followed by three incubations for 1 minute in 100% ethanol and three incubations for 1 minute in xylene. Slides were mounted using Richard-Allan Scientific Mounting Medium (ThermoFisher Scientific, Waltham, MA, USA). Samples were evaluated by an experienced, blinded to identity of slides pathologist.
2.15. Histopathology Evaluation and Grading.
Histopathology assessments were done by a board-certified veterinary anatomic pathologist (MJH) blinded to the identity of the samples. Hematoxylin and eosin-stained slides were used to assess the mouse mammary glands (4th inguinal gland was used) for MIN, DCIS, and mammary tumors (adenoma, carcinoma). The slides were digitally scanned at 40X magnification using an Aperio slide scanner and the total number of lesions were counted per mammary gland. A total of 32 slides from 16-week-old females with four genotypes (Cre, Cre-NEDD9, Cre-neu, Cre-neu-NEDD9) were evaluated. The average area per slide was 100-120mm2. Low-grade MIN lesions were characterized by glands lined by 1-2 layers of atypical epithelial cells with enlarged nuclei, clumped chromatin, variable nuclear size and shape, and an increased mitotic rate. High-grade MIN lesions were typified by loss of lumens and thickening of the epithelium by multiple layers of disorganized epithelial cells with increased pleomorphism. Ductal proliferative lesions (DCIS) were observed as marked expansion and filling of mammary ducts with a monomorphic population of epithelial cells, causing marked enlargement of the gland, without invasion of the adjacent basement membrane, or formation of a palpable mass. DCIS lesions can be considered high-grade MIN lesions [37,38].
2.16. Western blotting
Western blotting was performed using standard procedures [24]. Primary antibodies used were anti-NEDD9 (2G9), -phospho-ERK1/2, -ERK1/2, -phospho-Aurora Kinase A, -HER2, -phospho-Src, FAK (Cell Signaling Technology, Danvers, MA USA), anti-phospho-FAK (ThermoFisher Scientific, Waltham, MA, USA), anti-Src (Sigma-Aldrich, St. Louis, MO, USA), anti-Aurora Kinase A (BD Biosciences, Franklin Lakes, NJ, USA). The dilution was based on manufacturer’s recommendation. Secondary anti-mouse and anti-rabbit Horseradish peroxidase (HRP)-conjugated antibodies (Jackson ImmunoResearch Laboratories, Inc. West Grove, PA) were diluted 1:10,000 followed by chemiluminescence-based detection with Luminata Forte Western HRP substrate (Sigma-Aldrich, St. Louis, MO, USA) and were quantified using ImageJ software (ImageJ – National Institute of Health, https://imagej.nih.gov).
2.17. MTT-based Proliferation and Viability Assay.
Cell Proliferation and IC50 values were assessed via MTT (ThermoFisher Scientific, Waltham, MA, USA) [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide] assay. Briefly, 1 x 105 cells/well were plated in a 96-well plate. At 24-, 48-, 72-, and 96-hour time points, 10uL of MTT (5mg/mL) was added to each well, incubated at 37C for 4 hours, then dimethyl sulfoxide (DMSO)was added, and absorbance was measured using Cytation5 imaging system (Agilent Technologies, Santa Clara, CA, USA) at 540nm. The results were reported as a fold of change over control at each time point.
2.18. Drug Treatment and Viability Assay.
Cells were plated at 2.5x103, 5.0x103, 10x103 density per 96-well plate in triplicates. For each cell line, two different shRNA targeting NEDD9 (ThermoFisher Scientific, Waltham, MA, USA) and scramble shRNA non-targeting control were used as previously described [39]. Cells were treated with various concentrations of lapatinib (ThermoFisher Scientific, Waltham, MA, USA) for 48 hours. CyQUANT Cell Proliferation Assay (ThermoFisher Scientific, Waltham, MA, USA) was utilized per the manufacturer’s protocol to quantify the number of viable cells per well using Biotek Cytation Plate Reader (Agilent Technologies, Santa Clara, CA, USA) excitation/emission 480nm/520nm. Percent viability was calculated by setting the control cell line to 100%. One-way ANOVA was used to calculate statistical significance in two independent experiments with three technical replicas.
2.19. Acini Formation, Imaging and Quantification Procedures.
The Acini/Morphogenesis assay was performed as previously described [39]. Briefly, 105 cells/ml cells were resuspended in DMEM/F12 medium, supplemented with 2% horse serum, 10 μg/ml insulin, 1 ng/ml cholera toxin, 100 μg/ml hydrocortisone, 1x antibiotic-antimycotic. Eight-well cell culture chamber slides (ThermoFisher Scientific, Waltham, MA, USA, USA) were coated with 45ul of Matrigel (ThermoFisher Scientific, Waltham, MA, USA, USA) per well. The cells were mixed 1:1 with DMEM/F12 medium containing 4% Matrigel and was added to Matrigel-coated slide. The medium was replaced every 3-4 days. The acini formation was documented daily via microscopy total of 10-14 days. The images from five randomly selected fields of view of standard size were collected with 10X U Plan S Apo/0.75 NA objective using Echo Rebel microscope (Discover Echo, San Diego, CA, USA). After 14 days acini were fixed and immunofluorescent analysis was conducted as previously described [39]. Briefly, acini were fixed using 3% paraformaldehyde for 20 minutes followed by washing with 1x PBS (Phosphate Buffered Saline). Permeabilization using 1xPBS containing 0.5% Triton X-100 for 10 minutes at 4C. Rinsed three times using 1xPBS/Glycine for 10 minutes each. Acini were mounted with ProLong Gold DAPI-containing media (ThermoFisher Scientific, Waltham, MA, USA, USA). A total of 10 fields with acini per condition were imaged using Ti2Nikon Spinning Disk Confocal microscope (Nikon Instruments Inc. Melville, NY, USA) equipped with CSU-W1 Yokogawa spinning head (Yokogawa America, Sugar Land, TX, USA) ; z-stack of images taken at 10mm per step up to 200mm total were processed using NIS software and 3D projection images quantified. The total number of cells per acini was quantified using DAPI. Bright-field images were used to measure size/diameter of the acini. Minimum 50 acini per condition in 10 randomly selected fields of view of standard size were quantified in three independent experiments with two biological replicas.
2.20. Kaplan–Meier Analysis.
Kaplan–Meier (KM) analysis to evaluate the prognostic value of NEDD9 in breast cancer was done using publicly available microarray database from breast cancer patients as outlined at https://kmplot.com/analysis/index.php?p=service&cancer=breast. The relapse-free survival (RFS) was calculated in a cohort of HER2-positive (n=1273, HER2+ status - array). All patients treated and untreated were included. The following analysis parameters were used: Gene-NEDD9 (JetSet best probe set (202149_at), Gene-ERBB2 (JetSet best probe set (216836_s_at). The HER2+ patients treated with any anti-HER2 therapy were split by the auto-select best cutoff, unless indicated otherwise in the text. The data were censored at the threshold; the redundant samples were removed. The biased arrays were removed; The p-value was calculated using the log-rank test and plotted in R. The specific parameters used outlined in Supplementary materials.
2.21. Statistical analysis.
Statistical comparisons were made using a two-tailed Student’s t-test, or one-way or two-way analysis of variance (ANOVA) when more than two samples were compared. P ≤ 0.05 was considered to be significant (*). Experimental values were reported as the means with ±S.E.M (standard error of the mean), p values were reported as adjusted, and statistical significance calculations were made using Prism9 software (GraphPad Software, Inc., San Diego, CA, USA). All experimental data sets reported here were collected from multiple independent experiments with multiple technical and biological replicas.
3. Results
3.1. NEDD9 expression correlates with disease progression, outcomes, and HER2+ pathological score.
NEDD9 expression was previously documented to correlate with poor prognosis in triple-negative breast cancers [24,31]. However, the impact of NEDD9 expression on HER2+ breast cancers (BC) is currently unknown. The breast cancer tissue microarrays (TMAs) with 110 breast patient biopsies were analyzed for NEDD9 expression using a validated antibody to address this gap. The results of quantitative immunohistochemistry staining were then correlated with the pathological stage of the disease and the HER2+ score (0+, 1+, 2+, 3+) assigned by a pathologist (Figure 1A-C). When compared to a normal/non-cancer adjacent tissue (NAT), expression of NEDD9 increased from benign to invasive ductal carcinoma (IDC) and metastatic lesions (lymph node-Met/LN; Figure 1A-B). NEDD9 was significantly upregulated in tumor biopsies and correlated with HER2 pathological score (Figure 1C). The clinical relevance of these findings is supported by the in silico analysis of publicly available microarray data from TCGA (Kaplan-Meier Plotter) [32,40]. The results show a strong correlation between NEDD9 mRNA and relapse-free survival (RFS) in HER2+ breast cancers. In HER2+ tumors, higher expression of NEDD9 correlates with lower RFS (Figure 1D; HR=1.29, p=0.02). The RFS analysis of ERBB2 shows similar findidings (Figure 1 D). Higher expression of ERBB2 was associated with worsen RFS (HR=0.131, p=0.04). Overall, this data suggests that NEDD9 is elevated at the protein and RNA levels in HER2+ breast cancers and that increased levels of NEDD9 correlate with lower RFS selectively in the HER2+ subset of BC patients.
In agreement with low RFS, the NEDD9-high patients also demonstrate lower response rates to anti-HER2 therapy in HER2+ breast cancer patients (Figure 1E-F, https://www.rocplot.org) as documented by lower pCR (pathological Complete Response) and RFS 5-year survival post treatment. The area under the curve (any anti-HER2 therapy, AUC=0.612, p= 0.0067; and AUC=0.719, p= 0.004) criteria shows significant influence of NEDD9 expression on pCR and relapse-free 5-year survival. The graphs show better treatment outcomes when using anti-HER2 therapies in NEDD9-low patients. This trend remains true in HER2 breast cancers classified based on molecular subtype as defined by St. Gallen criteria [41]. Moreover, based on ROC plotter analysis NEDD9 outperforms HER2/ERBB2 (probe#216836) in predicting anti-HER2-therapy response and survival post therapy in HER2+BCs (any anti-HER2 therapy, pCR AUC=0.539, p= 0.2 ns-non significant; RFS AUC=0.658, p= 0.031). Trastuzumab was the only treatment with all breast cancer patients included independently of HER2 status that provided better correlation with HER2 expression (pCR AUC=0.629, p= 0.00084). Thus, understanding the role NEDD9 plays in HER2-driven BCs is critical for uncovering early events in disease onset and potentially identifying patients with a higher risk of relapse and drug resistance.
Figure 1.
NEDD9 is Overexpressed in HER2+ Breast Cancers and correaltes with disease progression, poor survival and therapy response. (A). Representative images of immunohistochemical staining of tissue microarray (TMA, n=110 including 32 normal/non-cancer tissue) with anti-NEDD9 and -HER2 antibodies, developed with DAB-(brown), hematoxylin-nuclei (blue); invasive ductal carcinoma (IDC), metastatic invasive ductal carcinoma, lymph nodes (Mets-LN). Scale bar-300mm. The de-identified tumor biopsy information is shown in Suppl. Table S1. (B). Quantification of intensity of DAB staining and positivity (percentage of cells stained positively in the same core stained by anti-HER2 or -NEDD9) by automated AperioScope imaging tool as in (A); Patient stratified based on diagnosis (B) NAT – 50, IDC -47, Mets/LN-13 or HER2+ pathological score as in (C). NAT-56, +1(14), +2(8), +3(32). One-way ANOVA with Dunnett's multiple comparisons test, p-values as indicated in figure panel. (D) Kaplan–Meier patient survival plots were generated by the Kaplan–Meier Plotter (http://www.kmplot.com) to determine the effect of NEDD9 or HER2 expression (microarray data Affy ID: 202149_at) on the relapse free survival (RFS) of 882 patients with HER2+ (array) breast cancer. Difference between expression levels shown by hazard ratio and p-values provided in each Kaplan-Meier plot. Hazard Ratios and p-value as indicated in figure panel (E). pROC assessment of anti-HER2 therapy response and NEDD9 expression levels in HER2+ breast cancer patients (HER2-array) Input settings for response – pathological complete response, HER2 status – positive, Treatment – anti-HER2 Therapy. Responders N=80, Non-responders=77. AUC=0.612, p=6.7x10-3. pROC assessment of anti-HER2 therapy response and NEDD9 expression levels in HER2+ breast cancer patients (HER2-array) Input settings for response – relapse-free survival at 5 years, HER2 status – positive, Treatment – anti-HER2 Therapy. Responders N=26, Non-responders=18. AUC=0.719, p=4.0x10-3. pROC assessment of anti-HER2 therapy response and Erbb2 expression levels in HER2+ breast cancer patients (HER2-array) Input settings for response –pathological complete response, HER2 status – positive, Treatment – anti-HER2 Therapy. Responders N=80, Non-responders=77. AUC=0.539, p=0.2. pROC assessment of anti-HER2 therapy response and Erbb2 expression levels in HER2+ breast cancer patients (HER2-array) Input settings for response – relapse-free survival at 5 years, HER2 status – positive, Treatment – anti-HER2 Therapy. Responders N=26, Non-responders=18. AUC=0.658, p=3.1x10-2.
Figure 1.
NEDD9 is Overexpressed in HER2+ Breast Cancers and correaltes with disease progression, poor survival and therapy response. (A). Representative images of immunohistochemical staining of tissue microarray (TMA, n=110 including 32 normal/non-cancer tissue) with anti-NEDD9 and -HER2 antibodies, developed with DAB-(brown), hematoxylin-nuclei (blue); invasive ductal carcinoma (IDC), metastatic invasive ductal carcinoma, lymph nodes (Mets-LN). Scale bar-300mm. The de-identified tumor biopsy information is shown in Suppl. Table S1. (B). Quantification of intensity of DAB staining and positivity (percentage of cells stained positively in the same core stained by anti-HER2 or -NEDD9) by automated AperioScope imaging tool as in (A); Patient stratified based on diagnosis (B) NAT – 50, IDC -47, Mets/LN-13 or HER2+ pathological score as in (C). NAT-56, +1(14), +2(8), +3(32). One-way ANOVA with Dunnett's multiple comparisons test, p-values as indicated in figure panel. (D) Kaplan–Meier patient survival plots were generated by the Kaplan–Meier Plotter (http://www.kmplot.com) to determine the effect of NEDD9 or HER2 expression (microarray data Affy ID: 202149_at) on the relapse free survival (RFS) of 882 patients with HER2+ (array) breast cancer. Difference between expression levels shown by hazard ratio and p-values provided in each Kaplan-Meier plot. Hazard Ratios and p-value as indicated in figure panel (E). pROC assessment of anti-HER2 therapy response and NEDD9 expression levels in HER2+ breast cancer patients (HER2-array) Input settings for response – pathological complete response, HER2 status – positive, Treatment – anti-HER2 Therapy. Responders N=80, Non-responders=77. AUC=0.612, p=6.7x10-3. pROC assessment of anti-HER2 therapy response and NEDD9 expression levels in HER2+ breast cancer patients (HER2-array) Input settings for response – relapse-free survival at 5 years, HER2 status – positive, Treatment – anti-HER2 Therapy. Responders N=26, Non-responders=18. AUC=0.719, p=4.0x10-3. pROC assessment of anti-HER2 therapy response and Erbb2 expression levels in HER2+ breast cancer patients (HER2-array) Input settings for response –pathological complete response, HER2 status – positive, Treatment – anti-HER2 Therapy. Responders N=80, Non-responders=77. AUC=0.539, p=0.2. pROC assessment of anti-HER2 therapy response and Erbb2 expression levels in HER2+ breast cancer patients (HER2-array) Input settings for response – relapse-free survival at 5 years, HER2 status – positive, Treatment – anti-HER2 Therapy. Responders N=26, Non-responders=18. AUC=0.658, p=3.1x10-2.

3.2. Generation of conditional NEDD9 knock-in (KI) transgenic mouse model.
Previously it was shown that deletion of NEDD9 via gene knock-out (KO) in mice overexpressing wild-type (unactivated) Erbb2 (neu) leads to a drastic decrease in HER2-driven tumorigenesis with only 10-15% of mice being able to form tumors in their lifetime. Note that this tumor incidence (~13.9%) corresponds to naturally occurring spontaneous mammary gland tumors documented in this strain [32], suggesting that NEDD9 is critical for the oncogenic activity of HER2 and without it no tumors above naturally set background can arise. To determine the role of NEDD9 in HER2-induced tumorigenesis, we generated a NEDD9 overexpression transgenic mouse model to mimic pathological conditions observed in human HER2+ BCs.
The targeting vector containing a cDNA sequence for human NEDD9 under the CAG promoter (hybrid promoter consisting of the cytomegalovirus (CMV) enhancer fused to the chicken beta-actin promoter) was utilized for homologous recombination at the Rosa26 locus of the murine genome. The promoter was followed by a stop cassette flanked by LoxP Cre recombinase recognition sites. This design allows inducible, tissue/cell type-specific expression of an extra copy of NEDD9 (Figure 2A). The vector design and C57BL/6 mouse embryo injection/transplantation were carried out in collaboration with GenOway (France). The PCR screening for homologous recombination at the 5’ end of the targeting vector was performed using a set of designated primers as outlined in materials and methods. The conditional (floxed, fx) NEDD9 transgene was next transferred into the FVB background using speed congenics services provided by Charles Rivers Laboratories (USA). The resultant homozygous strain FVB/J-LoxP-STOP-LoxP-NEDD9, called NEDD9fx/fx, was confirmed by PCR (Figure 2B) and Southern blotting. To test the effectiveness of the transgenic construct, mouse embryonic fibroblasts (MEFs) were isolated from NEDD9fx/fx E14 embryos. The MEFs were infected with pre-packaged adenovirus expressing Cre recombinase fused with GFP (green fluorescent protein, Ad-Cre-GFP). The western blot analysis of control (GFP only) and Cre-GFP infected cells for NEDD9 shows upregulation of its expression, thus confirming the functionality of inserted whole body transgene upon Cre expression (Figure 2C).
Figure 2.
Design and expression of Cre inducible NEDD9 knock-in mouse model. (A). Design of Cre recombinase-inducible NEDD9 cDNA knock-in vector for homologous recombination at the Rosa26 locus (top). In the presence of Cre recombinase the STOP cassette is resected out of the genome due to flanking LoxP sites (middle) resulting in CAAG promoter driven NEDD9 expression (bottom). (B). Genotyping results in triplicate indicating the presence of either the heterozygote (fx/wt) (M1-3), homozygous (fx/fx) (M4-6) or wild type Rosa26 loci (wt/wt) (M7-9). (C) Schematic outline of adenovirus Cre delivery into mouse embryonic fibroblasts (MEFs) isolated from NEDD9fx/fx mice (n=3). MEFs were incubated with GFP (-) or advenoviral GFP-Cre (+) virus particles. Western blot analysis of MEF’s lysates with anti-NEDD9, -GAPDH is a loading control. (D) Mating scheme of mice used in study. (E) Representative genotyping results of mice (genotypes as indicated in D) using genomic DNA and target-specific primers, showing presences of Rosa 26 loci (WT), NEDD9 (N9), Cre Recombinase (Cre), Erbb2 (neu). (F) Western blot analysis of lysates prepared from mammary gland tissue of MMTV-Cre-control or MMTV-Cre-NEDD9 mice (as in E, n=3) with anti-NEDD9, -GAPDH is a loading control. .
Figure 2.
Design and expression of Cre inducible NEDD9 knock-in mouse model. (A). Design of Cre recombinase-inducible NEDD9 cDNA knock-in vector for homologous recombination at the Rosa26 locus (top). In the presence of Cre recombinase the STOP cassette is resected out of the genome due to flanking LoxP sites (middle) resulting in CAAG promoter driven NEDD9 expression (bottom). (B). Genotyping results in triplicate indicating the presence of either the heterozygote (fx/wt) (M1-3), homozygous (fx/fx) (M4-6) or wild type Rosa26 loci (wt/wt) (M7-9). (C) Schematic outline of adenovirus Cre delivery into mouse embryonic fibroblasts (MEFs) isolated from NEDD9fx/fx mice (n=3). MEFs were incubated with GFP (-) or advenoviral GFP-Cre (+) virus particles. Western blot analysis of MEF’s lysates with anti-NEDD9, -GAPDH is a loading control. (D) Mating scheme of mice used in study. (E) Representative genotyping results of mice (genotypes as indicated in D) using genomic DNA and target-specific primers, showing presences of Rosa 26 loci (WT), NEDD9 (N9), Cre Recombinase (Cre), Erbb2 (neu). (F) Western blot analysis of lysates prepared from mammary gland tissue of MMTV-Cre-control or MMTV-Cre-NEDD9 mice (as in E, n=3) with anti-NEDD9, -GAPDH is a loading control. .

3.3. Production and analysis of mammary gland-specific expression of NEDD9.
To assess the specific role of NEDD9 overexpression in mammary tumorigenesis and mammary gland development, we crossed the NEDD9fx/fx mice with a strain expressing Cre recombinase under the control of the mouse mammary tumor virus promoter, MMTV-Cre [21]. The offspring were genotyped using a set of primers described in the material and methods. The resulting mice overexpress NEDD9 protein in the mammary gland epithelial cells. Homozygous MMTV-Cre-NEDD9+/+ mice and appropriate controls (MMTV-Cre and NEDD9fx/fx) were used to analyze mammary gland development and crossed with mammary tumor models. The upregulation of NEDD9 in the mammary epithelium was confirmed by western blot and immunocytochemistry analysis (Figure 2F). Mice overexpressing NEDD9 in mammary epithelium develop normally and produce/nurse healthy offspring. In conclusion, we developed a mouse model of conditional NEDD9 expression that can be successfully used to upregulate NEDD9 in a tissue-specific manner to enable further evaluation of NEDD9 function in vivo under normal and pathological conditions.
3.4. NEDD9 overexpression alters mammary gland architecture by increasing mammary gland budding and branching morphogenesis and cooperates with HER2.
Previously it was shown that whole-body NEDD9 knock-out did not affect mammary gland development [16], while the impact of overexpression is currently unknown. To address this gap in our knowledge, we compared mammary gland architecture between MMTV-Cre and MMTV-Cre-NEDD9+/+ mature nulliparous mice. The whole mammary gland mounts were prepared from 16 weeks old female mice (Figure 3A-B). We found that upon NEDD9 overexpression, the buds-to-branch ratio increased. There were no significant differences between the groups in the secondary and tertiary branches (Figure 3C), while more terminal end buds (TEBs) were observed in NEDD9 overexpressing glands compared to MMTV-Cre (Figure 3D). The changes in the branch-to-bud ratio are indicative of increased invasion and/or proliferation during mammary gland development. Thus, overexpression of NEDD9 in normal mammary epithelium might lead to early hyperplasia.
3.5. Cooperation between NEDD9 and HER2 promotes the early initiation of tumorigenesis.
The MMTV-Cre-NEDD9+/+ and MMTV-Cre mice were further crossed with the MMTV-Erbb2/neu (JAX Strain #002376) model to assess the impact of NEDD9 upregulation on oncogene-driven carcinogenesis. The MMTV-neu model develops physiologically relevant, spontaneous mammary tumors. Similarly to many human HER2+ breast cancers, it overexpresses the wild-type form of Erbb2, which develops slowly, starting from foci of hyperplastic, dysplastic mammary epithelium and progressing to aggressive metastatic carcinoma[42]. The first lesions appear at 4-6 months, with a median incidence of 205 days. Both virgin and non-virgin female mice develop tumors. The MMTV-Cre-Erbb2 and MMTV-Cre-Erbb2-NEDD9+/+ mice were successfully produced, and nulliparous females were examined for mammary gland development and early lesions at the ~16 weeks. This time point was selected based on early preneoplastic lesions detected in Cre-NEDD9+/+ female mice (Figure 3A-D) and previously published reports on MMTV-Erbb2/neu mice [43]. We found that mice expressing MMTV-Cre-Erbb2-NEDD9+/+ had more tertiary branches and TEBs when compared to MMTV-Cre-Erbb2/neu mice (Figure 3E-H).
3.6. NEDD9 overexpression is associated with mammary intra-epithelia neoplasia.
In agreement with TEBs expansion, the analysis of mammary glands showed a significant increase in the number of mice with mammary intra-epithelial neoplasia (MIN) lesions associated with NEDD9 overexpression (Figure 4A-B, Table 1). The MIN was observed at age ~16 weeks and characterized by glands lined by 1-2 layers of atypical epithelial cells with variable nuclear size and shape. The loss of a noticeable lumen and thickening of the epithelium by multiple layers of disorganized cells with increased pleomorphism and mitotic rate, consistent with high-grade MIN lesions (Figure 4A). Since mammary intraepithelial neoplasia, including DCIS, is associated with an increased risk of breast cancer [33], these results suggest that NEDD9 upregulation might pre-dispose to neoplastic transformation. Similarly, the MIN lesions were found at higher frequency in triple transgene mice with NEDD9 than Erbb2 alone (Figure 4B, Table 1). These findings suggest that overexpression of NEDD9 increases the frequency of preneoplastic events in the Erbb2 model.
3.7. NEDD9 overexpression causes hyperproliferation of luminal cells.
The terminal end buds are responsible for the production of mature luminal and myoepithelial cells leading to ductal tree formation[44,45]. To determine if TEB expansion and development of the MIN lesions is due to increased proliferation, the immunohistochemistry with Ki67 antibody was performed (Figure 5A). The upregulation of NEDD9 in the mammary gland led to an increased number of Ki67-positive cells in ducts normalized to the total number of cells (Figure 5B). Next, the immunofluorescent staining with antibodies against known markers of luminal epithelial (cytokeratin 8) and basal myoepithelial (cytokeratin 5) cells was conducted (Figure 5C). The NEDD9 overexpressing mice had a significantly increased number of luminal epithelial cells (Figure 5D, top). No significant differences in myoepithelial cells (Figure 5D, bottom) were observed. These data agree with a previously published report documenting a reduction in luminal progenitors and mature cells in NEDD9 knock-out mice [16]. Overall, these results suggest NEDD9 plays a crucial role in luminal cell proliferation and expansion of the ductal compartment, which is a primary source of human cancer.
3.8. NEDD9 is overexpressed in human HER2+ breast cancer cell lines.
Similarly to TMA findings (Figure 1), NEDD9 protein was increased in a panel of HER2+ breast cancer cell lines (BT-474, SK-BR-3, JIMT1, and JIMT1-Br3) when compared to non-transformed MCF10A cells (Figure 6A-B). Next, we tested the impact of NEDD9 expression on sensitivity to lapatinib, standard of care HER2-targeting drug. The HER2+ breast cancer cell lines that have been previously reported as sensitive (SKBR-3, AU565) or resistant to lapatinib (JIMT-1)[47] were used. In agreement with ROC data (Figure 1E), depletion of NEDD9 by shRNAs (Figure 6C) in resistant cells - JIMT1 improved response to lapatinib. Similarly, a decrease in NEDD9 led to increased sensitivity to lapatinib in AU549 and BT474 cells (Figure 6D), suggesting that the combination of anti-HER2 therapies with NEDD9 depletion might lead to improved outcomes.
3.9. NEDD9 overexpression in normal mammary epithelial cells causes increased proliferation.
To evaluate our findings on NEDD9-driven MIN/DCIS increase in the human model, we overexpress NEDD9 in human mammary epithelial cells - MCF10A, alone or in combination with HER2 or empty vector controls (Figure 7A). We found that MCF10A cells overexpressing NEDD9 significantly increased proliferation in a 2D assay (Figure 7B). The cells grown in a 3D Matrigel matrix form duct-like structures (acini), polarized spheroids that consist of a single layer and have a hollow center. It was previously shown that upon HER2 upregulation/activation, the acini enlarge via excessive proliferation, loss of apical polarity, and lack apoptosis [40]. A similar phenomenon was observed in MCF10A cells that overexpress exogenous NEDD9 alone or in combination with HER2 (Figure 7C-D), confirming the critical role of NEDD9 in the regulation of proliferation in non-transformed cells. It was shown that NEDD9 interacts with and activates the mitotic kinase AURKA [48]. Hence, we analyzed the activation of AURKA in our experimental model. The level of active, phosphorylated AURKA, was significantly increased upon NEDD9 overexpression. Interestingly, overexpression of HER2 stabilized total AURKA but did not increase its activity. The phosphorylation of ERK1/2 was also significantly increased (Figure 7E) upon overexpression of NEDD9 indicative of MAPK signaling activation. The SRC and FAK – classical targets of HER2 downstream signal transduction were not significantly affected in MCF10A-NEDD9 cells compared to control (Figure 7F), suggesting non-canonical AURKA-driven activation of proliferation in human cells with upregulated NEDD9.
4. Discussion
HER2-positive breast cancer subtype shows a high expression of the HER2 kinase and is associated with proliferation-related genes characterized by high K67/mitotic index and aggressive growth [49]. HER2 signaling function is enabled via ligand-bound heterodimerization with HER3 or EGFR [50] that leads to phosphorylation and docking of multiple signaling adaptor molecules, thus activating several downstream pathways including proliferation (ERK1/2), invasion (FAK/Src) and survival (AKT) [51]. A few adaptor molecules were documented to be key signaling hubs in HER2 oncogenic activity, including Grb2 [52], p130Cas [15], and NEDD9 [16]. The knockout of murine Nedd9 led to a reduction in luminal progenitors, HER2 signaling, and tumor growth [53]. While deletion of NEDD9 is not observed in human breast cancers, the upregulation of NEDD9 has been linked to tumor progression and metastasis in various cancers, including breast, liver, colon, pancreatic, ovarian, lung, and brain [20,54,55,56,57]. However, the impact of the upregulation of NEDD9 on HER2+ breast cancers has not been explored. Here, we report that NEDD9 protein (TMA, Figure 1A-C, Figure 6A-C) and mRNA (TCGA, Figure 1D) expression is elevated in HER2+ human breast cancer patient biopsies and established cell lines. The NEDD9 upregulation correlates with disease progression, HER2 expression, low anti-HER2 therapy response, and Relapse Free Survival (RFS).
To determine the impact of NEDD9 on HER2 carcinogenesis and disease progression, we generated a mouse model overexpressing NEDD9 specifically within the mouse mammary gland. We analyzed the effect of NEDD9 upregulation in MMTV-neu (rodent homolog of HER2, unactivated). The MMTV-neu mice form tumors spontaneously at ~29-48 weeks [58]. The long latency is thought to be due to the need for additional mutations to enable transformation, including p53, which is often observed in HER2+ breast cancers [59]. The upregulation of NEDD9 resulted in higher rates and earlier formation of preneoplastic, benign lesions, such as mammary intraepithelial neoplasia (MIN) and DCIS. These findings suggest that the upregulation of NEDD9 might play a key role in the transition from normal to preneoplastic and neoplastic lesions [60].
To understand better the effects of NEDD9 overexpression within the MMTV-neu mouse model, we evaluated the mouse mammary gland for changes, specifically at the morphological and cellular levels. NEDD9 overexpression was able to increase the number of tertiary branches as well as terminal end buds which suggested NEDD9 plays a role in proliferation. The expansion of the ductal tree and proliferation at the leading edge of the terminal end buds point to changes in mammary gland development. [60]. We found that the proliferation of luminal (cytokeratin 8+) cells was increased while the number of myoepithelial cells (Keratin 5+) had decreased. The knockout of murine Nedd9 in the MMTV-neu model was shown to reduce the number of luminal progenitors [16] but did not affect mammary gland architecture. In agreement with earlier findings, the overexpression of NEDD9 led to the expansion of luminal cells. Additionally, it also significantly increased mammary gland morphogenesis. Taken together, our results suggest that NEDD9 might serve as a predictive biomarker of preneoplastic, an early stage in HER2-driven carcinogenesis.
To evaluate our findings in the context of human cancer, we analyzed the expression of NEDD9 in multiple HER2+ human breast cell lines. The protein expression of HER2 shows some variability among cell lines, with the lowest expression in JIMT1, HER2-therapy drug-resistant cells. Similarly, four of the five HER2+ cell lines had increased expression of NEDD9 protein when compared to non-transformed MCF10A cells. Although AU565 and SK-BR-3 are isogenic cell lines isolated from the same patient and have a similar mutation landscape like p53 mutant (p53 R175H), they vary in HER2/NEDD9 levels and differ in downstream signaling. Unlike SK-BR-3, AU565 is poorly responsive to EGF and has higher HER3 expression that might enable direct MAPK/AKT activation without NEDD9 [62,63,64]. To further explore the effect of NEDD9 upregulation on non-transformed breast epithelial cells, we overexpressed both proteins in MCF10A cells. The parental cell line has low levels of NEDD9 and HER2. The 2D proliferation assay shows an increase in proliferation upon overexpression of NEDD9 alone or in combination with HER2. Similarly, the MCF10A-NEDD9 and MCF10A-NEDD9-HER2 cells, when grown in 3D Matrigel matrix, formed significantly larger acini as documented by increased diameter and number of cells.
Previously it was shown that NEDD9 plays a role in tumor formation when ablated in vivo [16]. In our study, NEDD9 overexpression leads to an increase in neoplasia events as well as morphological and cellular changes which impact proliferation and cell lineage fate. Uncovering this role of NEDD9 supports further investigation of pre-tumorigenic events to successfully administer the earlier patient intervention.
Recent studies confirm that HER2 molecular subtype yields the best clinical and therapeutic response by anti-HER2 therapies, Resistance to anti-HER2 therapies has been studied at great length. Some factors of resistance currently are: 1) changes in the binding sites or to tyrosine kinase receptor domain, 2) Overexpression of HER2 receptor, 3) dimerization with other receptors for activity, 4) alternate activation of downstream signaling pathways[61]. Increased NEDD9 expression correlation with non-responders; additionally, Area Under Curve (AUC) in sensitivity/specificity plots indicates that NEDD9 is a strong biomarker for HER2+ therapy response. AUC with the best cut-off point shows how separated responders and non-responders groups are and yields predictive value for example, estrogen receptor-positive breast cancer (ER+) patients show a response to tamoxifen when ESR1 expression is elevated [62].
5. Conclusions
A few adaptor molecules were reported to be critical downstream effectors of the HER2 oncogene. In this study, we outline the key role of the Cas-family scaffolding protein - NEDD9 in the initiation and progression of HER2-driven tumors. NEDD9 is elevated in HER2+ cancers and correlates with resistance to anti-HER2 therapy and low Relapse Free Survival (RFS) rates post-treatment. The NEDD9 induces higher rates and earlier formation of preneoplastic, benign lesions, such as mammary intraepithelial neoplasia (MIN) and DCIS. The increase in proliferation correlates with increased activity of MAPK and AURKA proliferation control pathways. Thus, depletion of NEDD9 protein might provide significant benefits in treating therapy resistant HER2+ breast cancers, and expression of NEDD9 serve as a prognostic marker for therapy response.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Information: Original blots.
Author Contributions
Conceptualization, E.NP. and M.LP.; methodology, E.NP. and M.LP., R.S., R.J.I., M.J.H.; software, E.NP. and M.LP.; validation, E.NP., R.S. and M.L.P.; formal analysis, E.NP., R.S and M.LP., and R.J.I; investigation, M.LP., R.S., R.J.I., M.J.H.; resources, E.N.P.; data curation, E.NP and M.L.P.; writing—original draft preparation, M.L.P.; writing—review and editing, E.N.P.; visualization, E.NP. and M.LP.; supervision, E.N.P.; project administration, E.N.P.; funding acquisition, E.N.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIH-NCI CA148671 to E.N.P. WVU HSC Office of Graduate Research and Education, department of Biochemistry and WVU Cancer Institute provided administrative support and funding to R.J.I and M.L.P. Imaging was performed in the West Virginia University Animal Models & Imaging Facility, supported by NIH grants P20RR016440, P30GM103488, U54GM104942, P20GM103434, and P20GM103434.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of West Virginia University (protocol #1604002251 and August 30, 2022).
Informed Consent Statement
Not applicable. Only deidentifies human tissue samples have been used in the study.
Data Availability Statement
Publicly available datasets were analyzed in this study. Data can be found at: https://www.cbioportal.org, https://kmplot.com/analysis/index.php?p=service&cancer=breast, and https://www.rocplot.org/site/treatment.
Acknowledgments
We would like to thank WVU HSC Office of Graduate Research and Education for great administrative support. Special acknowledgement to Dr. Michael J. Ruppert for assistance with review of the manuscript and constructive feedback. We are grateful to Drs. Alexey Ivanov and Paul Lockman for providing reagents.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cancer Facts & Figures 2022. 2022, American Cancer Society: Atlanta.
- Giaquinto, A.N., et al., Breast Cancer Statistics, 2022. Ca-a Cancer Journal for Clinicians, 2022. 72(6): p. 524-541. [CrossRef]
- Loibl, S. and L. Gianni, HER2-positive breast cancer. Lancet, 2017. 389(10087): p. 2415-2429. [CrossRef]
- Arteaga, C.L., et al., Treatment of HER2-positive breast cancer: current status and future perspectives. Nature Reviews. Clinical Oncology, 2012. 9(1): p. 16-32. [CrossRef]
- Muthuswamy, S.K., Trastuzumab resistance: all roads lead to SRC. Nature Medicine, 2011. 17: p. 416. [CrossRef]
- Peiro, G., et al., Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. (1532-1827 (Electronic)). [CrossRef]
- Belsches-Jablonski, A.P., et al., Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene, 2001. 20: p. 1465. [CrossRef]
- Shagisultanova, E., et al., Preclinical and clinical studies of the NEDD9 scaffold protein in cancer and other diseases. Gene, 2015. 567(1): p. 1-11. [CrossRef]
- Ahn, J., V. Sanz-Moreno, and C.J. Marshall, The metastasis gene NEDD9 product acts through integrin β3 and Src to promote mesenchymal motility and inhibit amoeboid motility. Journal of Cell Science, 2012. 125(7): p. 1814. [CrossRef]
- Muller, W.J., et al., Synergistic interaction of the Neu proto-oncogene product and transforming growth factor alpha in the mammary epithelium of transgenic mice. Molecular and Cellular Biology, 1996. 16(10): p. 5726-5736. [CrossRef]
- Hsu, J.L. and M.-C. Hung, The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer metastasis reviews, 2016. 35(4): p. 575-588. [CrossRef]
- Sima, N., et al., The Overexpression of Scaffolding Protein NEDD9 Promotes Migration and Invasion in Cervical Cancer via Tyrosine Phosphorylated FAK and SRC. PLOS ONE, 2013. 8(9): p. e74594. [CrossRef]
- Izumchenko, E., et al., NEDD9 Promotes Oncogenic Signaling in Mammary Tumor Development. Cancer Research, 2009. 69(18): p. 7198-7206. [CrossRef]
- Pugacheva, E.N., et al., HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell, 2007. 129(7): p. 1351-1363. [CrossRef]
- Cabodi, S., et al., p130Cas as a New Regulator of Mammary Epithelial Cell Proliferation, Survival, and HER2-Neu Oncogene–Dependent Breast Tumorigenesis. Cancer Research, 2006. 66(9): p. 4672-4680. [CrossRef]
- Little, J.L., et al., A requirement for Nedd9 in luminal progenitor cells prior to mammary tumorigenesis in MMTV-HER2/ErbB2 mice. Oncogene, 2013. 33: p. 411. [CrossRef]
- Dai, J., et al., Downregulation of NEDD9 by apigenin suppresses migration, invasion, and metastasis of colorectal cancer cells. (1096-0333 (Electronic)). [CrossRef]
- Li, Y., et al., HEF1, a novel target of Wnt signaling, promotes colonic cell migration and cancer progression. Oncogene, 2011. 30: p. 2633. [CrossRef]
- Speranza, M.C., et al., NEDD9, a novel target of miR-145, increases the invasiveness of glioblastoma. Oncotarget, 2012. 3(7): p. 723-34. [CrossRef]
- Wang, H., et al., NEDD9 overexpression is associated with the progression of and an unfavorable prognosis in epithelial ovarian cancer. Human Pathology, 2014. 45(2): p. 401-408. [CrossRef]
- Li, G., et al., Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development, 2002. 129(17): p. 4159-4170. [CrossRef]
- Zhong, J., et al., NEDD9 stabilizes focal adhesions, increases binding to the extra-cellular matrix and differentially effects 2D versus 3D cell migration. PloS one, 2012. 7(4): p. e35058-e35058. [CrossRef]
- Jozefczuk, J., K. Drews, and J. Adjaye, Preparation of Mouse Embryonic Fibroblast Cells Suitable for Culturing Human Embryonic and Induced Pluripotent Stem Cells. Jove-Journal of Visualized Experiments, 2012(64). [CrossRef]
- Ice, R.J., et al., NEDD9 Depletion Destabilizes Aurora A Kinase and Heightens the Efficacy of Aurora A Inhibitors: Implications for Treatment of Metastatic Solid Tumors. Cancer Research, 2013. 73(10): p. 3168-3180. [CrossRef]
- Stanko, J.P. and S.E. Fenton, Quantifying Branching Density in Rat Mammary Gland Whole-mounts Using the Sholl Analysis Method. J Vis Exp, 2017(125). [CrossRef]
- Stanko, J.P. and S.E. Fenton, Quantifying Branching Density in Rat Mammary Gland Whole-mounts Using the Sholl Analysis Method. Journal of visualized experiments : JoVE, 2017(125): p. 55789. [CrossRef]
- Lyle, L.T., et al., Alterations in Pericyte Subpopulations Are Associated with Elevated Blood–Tumor Barrier Permeability in Experimental Brain Metastasis of Breast Cancer. Clinical Cancer Research, 2016. 22(21): p. 5287-5299. [CrossRef]
- Zhou, X. and Y.M. Agazie, Molecular mechanism for SHP2 in promoting HER2-induced signaling and transformation. The Journal of biological chemistry, 2009. 284(18): p. 12226-12234. [CrossRef]
- Addison, J.B., et al., Functional Hierarchy and Cooperation of EMT Master Transcription Factors in Breast Cancer Metastasis. Molecular Cancer Research, 2021. 19(5): p. 784-798. [CrossRef]
- Tiscornia, G., O. Singer, and I.M. Verma, Production and purification of lentiviral vectors. Nat Protoc, 2006. 1(1): p. 241-5. [CrossRef]
- Whately, K.M., et al., Nuclear Aurora-A kinase-induced hypoxia signaling drives early dissemination and metastasis in breast cancer: implications for detection of metastatic tumors. Oncogene, 2021. 40(37): p. 5651-5664. [CrossRef]
- Lánczky, A. and B. Győrffy, Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J Med Internet Res, 2021. 23(7): p. e27633. [CrossRef]
- Pugacheva, E.N. and E.A. Golemis, The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nature Cell Biology, 2005. 7(10): p. 937-U18. [CrossRef]
- Whately, K.M., et al., Nuclear Aurora-A kinase-induced hypoxia signaling drives early dissemination and metastasis in breast cancer: implications for detection of metastatic tumors. Oncogene, 2021. 40(37): p. 5651-5664. [CrossRef]
- Brulet, P., et al., Monoclonal antibodies against trophectoderm-specific markers during mouse blastocyst formation. Proc Natl Acad Sci U S A, 1980. 77(7): p. 4113-7. [CrossRef]
- Wu, M., et al., Gli Transcription Factors Mediate the Oncogenic Transformation of Prostate Basal Cells Induced by a Kras-Androgen Receptor Axis. J Biol Chem, 2016. 291(49): p. 25749-25760. [CrossRef]
- Cardiff, R.D., et al., The mammary pathology of genetically engineered mice: the consensus report and recommendations from the Annapolis meeting. Oncogene, 2000. 19(8): p. 968-988. [CrossRef]
- Cardiff, R.D., D. Moghanaki, and R.A. Jensen, Genetically Engineered Mouse Models of Mammary Intraepithelial Neoplasia. Journal of Mammary Gland Biology and Neoplasia, 2000. 5(4): p. 421-437. [CrossRef]
- Jones, B.C., et al., Dual Targeting of Mesenchymal and Amoeboid Motility Hinders Metastatic Behavior. Mol Cancer Res, 2017. 15(6): p. 670-682. [CrossRef]
- Muthuswamy, S.K., et al., ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nature Cell Biology, 2001. 3(9): p. 785-792. [CrossRef]
- Györffy, B., et al., An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Research and Treatment, 2010. 123(3): p. 725-731. [CrossRef]
- Mihály, Z. and B. Győrffy Improving Pathological Assessment of Breast Cancer by Employing Array-Based Transcriptome Analysis. Microarrays, 2013. 2, 228-242. [CrossRef]
- Guy, C.T., et al., Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci U S A, 1992. 89(22): p. 10578-82. [CrossRef]
- Andrechek, E.R., et al., Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America, 2000. 97(7): p. 3444-3449. [CrossRef]
- Williams, J.M. and C.W. Daniel, Mammary ductal elongation: differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev Biol, 1983. 97(2): p. 274-90. [CrossRef]
- Macias, H. and L. Hinck, Mammary gland development. Wiley Interdiscip Rev Dev Biol, 2012. 1(4): p. 533-57. [CrossRef]
- O'Brien, N.A., et al., Activated phosphoinositide 3-kinase/AKT signaling confers Resistance to trastuzumab but not lapatinib. Mol Cancer Ther, 2010. 9(6): p. 1489-502. [CrossRef]
- Pugacheva, E.N. and E.A. Golemis, The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nature Cell Biology, 2005. 7(10): p. 937-946. [CrossRef]
- Fragomeni, S.M., A. Sciallis, and J.S. Jeruss, Molecular Subtypes and Local-Regional Control of Breast Cancer. Surgical oncology clinics of North America, 2018. 27(1): p. 95-120. [CrossRef]
- Berger, M.B., J.M. Mendrola, and M.A. Lemmon, ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS Letters, 2004. 569(1-3): p. 332-336. [CrossRef]
- Bazley, L.A. and W.J. Gullick, The epidermal growth factor receptor family. Endocrine-Related Cancer Endocr Relat Cancer, 2005. 12(Supplement_1): p. S17-S27. [CrossRef]
- Beigbeder, A., F.J.M. Chartier, and N. Bisson, MPZL1 forms a signalling complex with GRB2 adaptor and PTPN11 phosphatase in HER2-positive breast cancer cells. Scientific Reports, 2017. 7(1): p. 11514. [CrossRef]
- Little, J.L., et al., A requirement for Nedd9 in luminal progenitor cells prior to mammary tumorigenesis in MMTV-HER2/ErbB2 mice. Oncogene, 2014. 33(4): p. 411-20. [CrossRef]
- Jin, Y., et al., NEDD9 promotes lung cancer metastasis through epithelial–mesenchymal transition. International Journal of Cancer, 2014. 134(10): p. 2294-2304. [CrossRef]
- Speranza, M.C., et al., NEDD9, a novel target of miR-145, increases the invasiveness of glioblastoma. Oncotarget; Vol 3, No 7: July 2012, 2012. [CrossRef]
- Li, Y., et al., HEF1, a novel target of Wnt signaling, promotes colonic cell migration and cancer progression. Oncogene, 2011. 30(23): p. 2633-43. [CrossRef]
- Iida, J., et al., Role for chondroitin sulfate glycosaminoglycan in NEDD9-mediated breast cancer cell growth. Experimental Cell Research, 2015. 330(2): p. 358-370. [CrossRef]
- Guy, C.T., et al., Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proceedings of the National Academy of Sciences, 1992. 89(22): p. 10578-10582. [CrossRef]
- Zelazny, E., et al., Cooperating Oncogenic Events in Murine Mammary Tumorigenesis: Assessment of ErbB2, Mutant p53, and Mouse Mammary Tumor Virus. Experimental and Molecular Pathology, 2001. 70(3): p. 183-193. [CrossRef]
- Williams, J.M. and C.W. Daniel, Mammary ductal elongation: Differentiation of myoepithelium and basal lamina during branching morphogenesis. Developmental Biology, 1983. 97(2): p. 274-290. [CrossRef]
- Tortora, G., Mechanisms of Resistance to HER2 Target Therapy. JNCI Monographs, 2011. 2011(43): p. 95-98. [CrossRef]
- Mihály, Z., et al., A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Research and Treatment, 2013. 140(2): p. 219-232. [CrossRef]
Figure 3.
NEDD9 overexpression alters mammary gland architecture. (A-B). Representative images of mammary gland whole mounts prepared from MMTV-Cre and MMTV-Cre-NEDD9. scale bar-500mm, Inset – 400x, S – Secondary Branch, T – Tertiary Branch, TEB – Terminal End Bud. (C). Quantification of secondary and tertiary branches (fields of view= 5, n=5 per genotype). (D) Quantification of terminal end buds (TEBs) (fields of view= 5, n=5 per genotype). non-parametric student t-test, ±S.E.M, ns- not significant, TEBs - p<0.0001. (E-F) Representative images of mammary gland whole mounts prepared from MMTV-Cre-Erbb2 and MMTV-Cre-Erbb2-NEDD9 mice, scale bar-500um. Inset – 400x, S – Secondary Branch, T – Tertiary Branch, TEB – Terminal End Bud). (G). Quantification of secondary and tertiary branches and (H). terminal end buds (TEBs) (fields of view= 5, n=5 per genotype). Non-parametric student t-test, ±S.E.M, ns- not significant, p<0.0001 for tertiary branches and TEBs.
Figure 3.
NEDD9 overexpression alters mammary gland architecture. (A-B). Representative images of mammary gland whole mounts prepared from MMTV-Cre and MMTV-Cre-NEDD9. scale bar-500mm, Inset – 400x, S – Secondary Branch, T – Tertiary Branch, TEB – Terminal End Bud. (C). Quantification of secondary and tertiary branches (fields of view= 5, n=5 per genotype). (D) Quantification of terminal end buds (TEBs) (fields of view= 5, n=5 per genotype). non-parametric student t-test, ±S.E.M, ns- not significant, TEBs - p<0.0001. (E-F) Representative images of mammary gland whole mounts prepared from MMTV-Cre-Erbb2 and MMTV-Cre-Erbb2-NEDD9 mice, scale bar-500um. Inset – 400x, S – Secondary Branch, T – Tertiary Branch, TEB – Terminal End Bud). (G). Quantification of secondary and tertiary branches and (H). terminal end buds (TEBs) (fields of view= 5, n=5 per genotype). Non-parametric student t-test, ±S.E.M, ns- not significant, p<0.0001 for tertiary branches and TEBs.
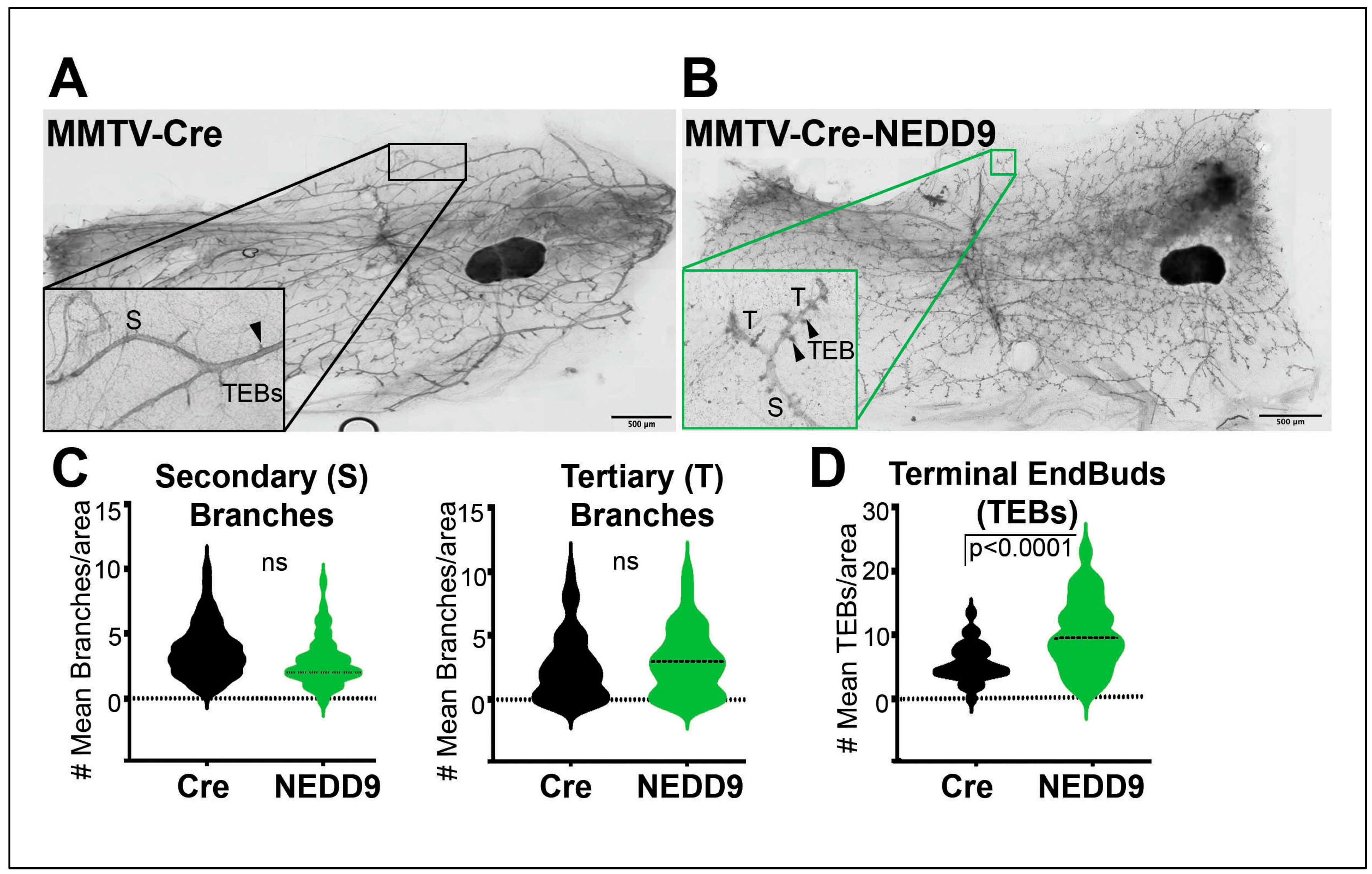
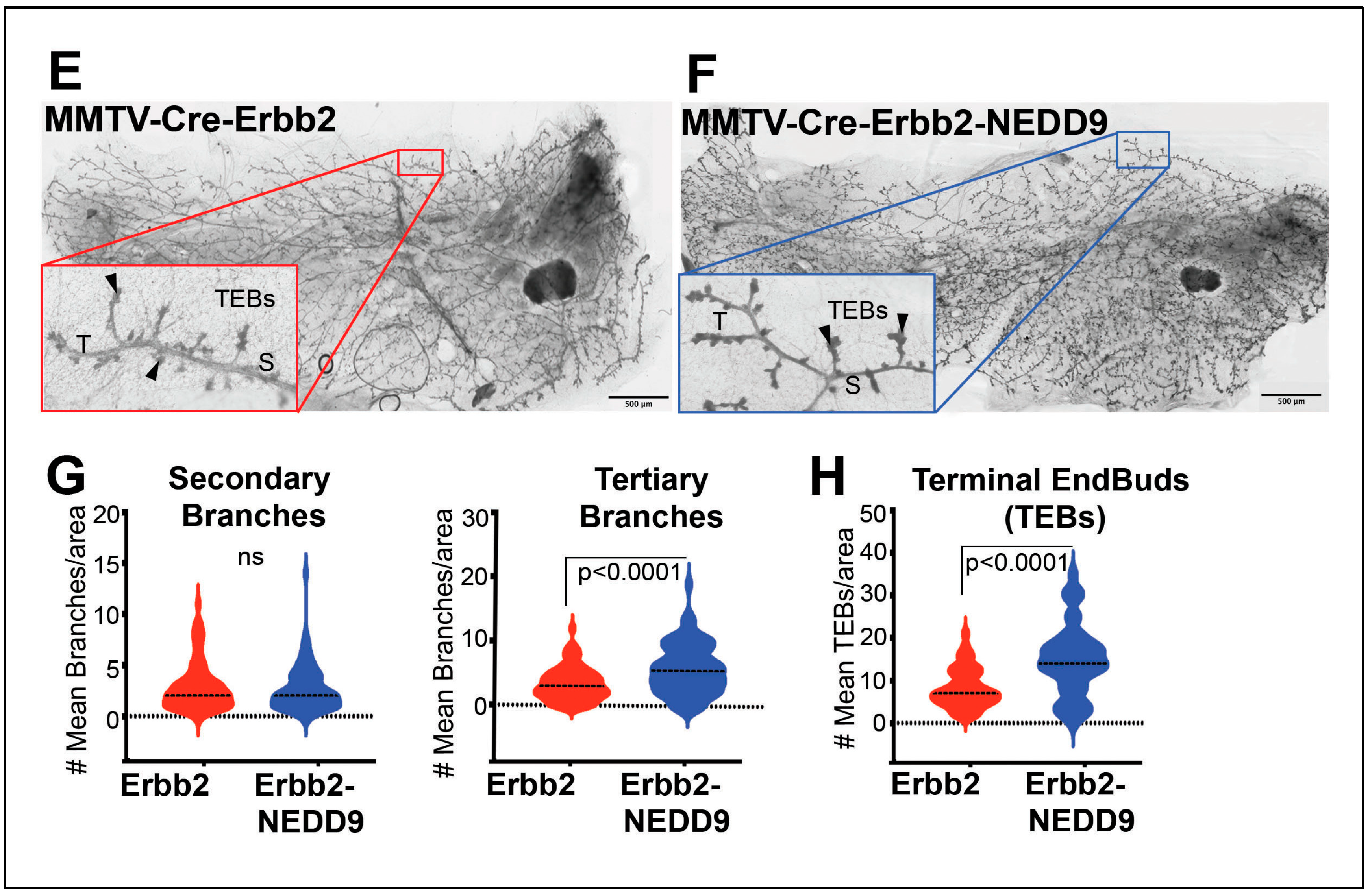
Figure 4.
NEDD9 overexpression is associated with mammary intra-epithelia neoplasia. (A). Representative images of mouse mammary glands stained with hematoxylin and eosin (H&E), genotypes as indicated in the figure. Scale bar – 20mm, Insets - 200x. A total of 40 slides from 16-week-old nulliparous female mice with four genotypes were quantified. The average area per slide was 100-120mm2. MIN was determined by lesions characterized by glands lined by 1-2 layers of atypical epithelial cells with enlarged nuclei, clumped chromatin, variable nuclear size and shape, and an increased mitotic rate. (B). Quantification of mice with MIN per group (n=10/genotype). White-no MIN detected, MIN+ positive (colored). Fisher’s exact test, p*=0.0137, number reported in Table 1.
Figure 4.
NEDD9 overexpression is associated with mammary intra-epithelia neoplasia. (A). Representative images of mouse mammary glands stained with hematoxylin and eosin (H&E), genotypes as indicated in the figure. Scale bar – 20mm, Insets - 200x. A total of 40 slides from 16-week-old nulliparous female mice with four genotypes were quantified. The average area per slide was 100-120mm2. MIN was determined by lesions characterized by glands lined by 1-2 layers of atypical epithelial cells with enlarged nuclei, clumped chromatin, variable nuclear size and shape, and an increased mitotic rate. (B). Quantification of mice with MIN per group (n=10/genotype). White-no MIN detected, MIN+ positive (colored). Fisher’s exact test, p*=0.0137, number reported in Table 1.
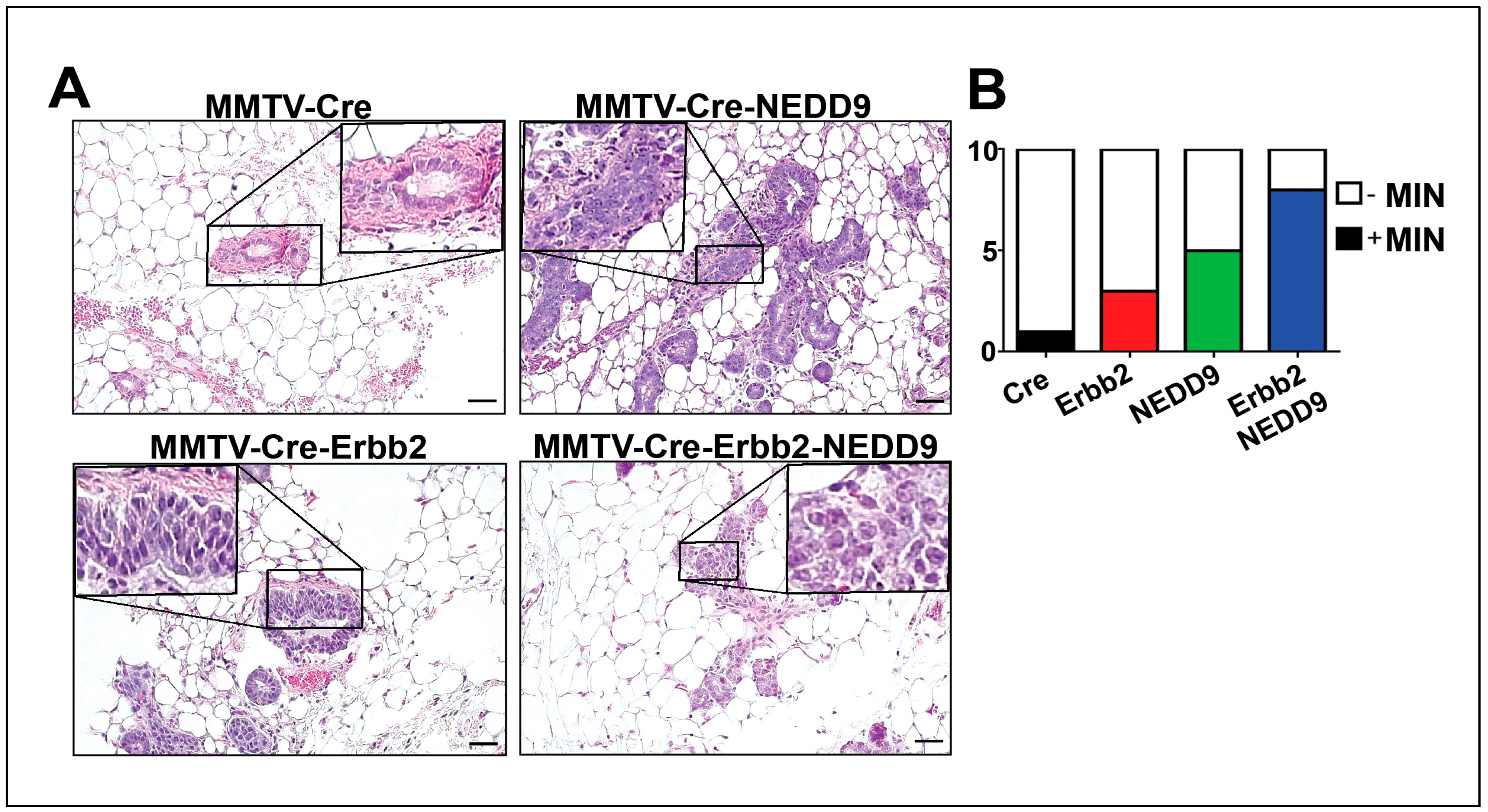
Figure 5.
NEDD9 overexpression increases proliferation of luminal cells. (A). Representative mammary glands images (3D confocal projections, cropped from whole mammary gland scanned) from n=3-6 mice per genotypes as indicated in figure, Fluorescent-IHC staining with anti-Ki67 (red) and DAPI - nuclei (blue), scale bar-5mm. (B). Quantification of number of positive cells/duct normalized to area (n=5 ducts/mouse, n=5 per genotype). Data presented as mean ±S.E.M. p-value indicated in figure panels. One-way ANOVA, Tukey’s multiple comparisons: Cre vs. Cre-Erbb2 is non-significant (ns); Cre vs. Cre-NEDD9 is **p=0.009; Cre vs. Cre-Erbb2-NEDD9 is ***p<0.0001. Cre-Erbb2 vs. Cre-NEDD9 is *p=0.0002, or vs. Cre-Erbb2-NEDD9 is ***p <0.0001. Cre-NEDD9 vs. Cre-Erbb2-NEDD9 is *p=0.05. (C) Representative mammary glands images (3D confocal projections, cropped from whole mammary gland scanned) from n=3-6 mice per genotypes as indicated in figure, Fluorescent-IHC staining with anti-cytokeratin 8 (green) or cytokeratin 5 (red) and DAPI - nuclei (blue), scale bar-5mm. (D). Quantification of the number of luminal (K8+) and basal (K5+) cells/duct normalized to area/mammary gland ducts (n=5 ducts/mouse, n=5 per genotype). One-way ANOVA, Tukey’s multiple comparisons ±S.E.M. p-value indicated in figure panels. ns - not significant. Cre vs. Cre-Erbb2-NEDD9 is *p=0.01; Cre-NEDD9 vs. Cre-Erbb2 is *p=0.02 or vs. Cre-Erbb2 is*** p<0.0001.
Figure 5.
NEDD9 overexpression increases proliferation of luminal cells. (A). Representative mammary glands images (3D confocal projections, cropped from whole mammary gland scanned) from n=3-6 mice per genotypes as indicated in figure, Fluorescent-IHC staining with anti-Ki67 (red) and DAPI - nuclei (blue), scale bar-5mm. (B). Quantification of number of positive cells/duct normalized to area (n=5 ducts/mouse, n=5 per genotype). Data presented as mean ±S.E.M. p-value indicated in figure panels. One-way ANOVA, Tukey’s multiple comparisons: Cre vs. Cre-Erbb2 is non-significant (ns); Cre vs. Cre-NEDD9 is **p=0.009; Cre vs. Cre-Erbb2-NEDD9 is ***p<0.0001. Cre-Erbb2 vs. Cre-NEDD9 is *p=0.0002, or vs. Cre-Erbb2-NEDD9 is ***p <0.0001. Cre-NEDD9 vs. Cre-Erbb2-NEDD9 is *p=0.05. (C) Representative mammary glands images (3D confocal projections, cropped from whole mammary gland scanned) from n=3-6 mice per genotypes as indicated in figure, Fluorescent-IHC staining with anti-cytokeratin 8 (green) or cytokeratin 5 (red) and DAPI - nuclei (blue), scale bar-5mm. (D). Quantification of the number of luminal (K8+) and basal (K5+) cells/duct normalized to area/mammary gland ducts (n=5 ducts/mouse, n=5 per genotype). One-way ANOVA, Tukey’s multiple comparisons ±S.E.M. p-value indicated in figure panels. ns - not significant. Cre vs. Cre-Erbb2-NEDD9 is *p=0.01; Cre-NEDD9 vs. Cre-Erbb2 is *p=0.02 or vs. Cre-Erbb2 is*** p<0.0001.

Figure 6.
NEDD9 is overexpressedin Human HER2+ Breast Cells and supports resitance to lapatinib. (A). Western blot (WB) analysis of MCF10A, BT-474, SK-BR-3, AU565, JIMT-1 and JIMT-1-Br3 (brain metastases subline of JIMT1) using anti-NEDD9, -HER2 and -GAPDH as loading control. (B). WB-based quantification of NEDD9 (green) and HER2 (red) protein expression in cell lines as in A, n=3 indepndent experiments. Parson correlation coefficient r=0.972, with expeption of JIMT1 where expression of NEDD9 and HER2 do not correlate . (C). Western blot analysis of lysates of AU565, JIMT1 and BT474 cells treated with with scramble (-) non-targeting control or two different shRNAs against NEDD9 (shNEDD9) using anti-NEDD9 and -GAPDH as loading control treated. (D). Cell viability assesment using Cyquant dye with different concentrations of lapatinib as indicated in the figure. Two-way ANOVA, Tukey’s multiple comparison +/- SEM. AU565, JIMT1, BT474 *p<0.0001 (DMSO vs.shNEDD-1 or -2).
Figure 6.
NEDD9 is overexpressedin Human HER2+ Breast Cells and supports resitance to lapatinib. (A). Western blot (WB) analysis of MCF10A, BT-474, SK-BR-3, AU565, JIMT-1 and JIMT-1-Br3 (brain metastases subline of JIMT1) using anti-NEDD9, -HER2 and -GAPDH as loading control. (B). WB-based quantification of NEDD9 (green) and HER2 (red) protein expression in cell lines as in A, n=3 indepndent experiments. Parson correlation coefficient r=0.972, with expeption of JIMT1 where expression of NEDD9 and HER2 do not correlate . (C). Western blot analysis of lysates of AU565, JIMT1 and BT474 cells treated with with scramble (-) non-targeting control or two different shRNAs against NEDD9 (shNEDD9) using anti-NEDD9 and -GAPDH as loading control treated. (D). Cell viability assesment using Cyquant dye with different concentrations of lapatinib as indicated in the figure. Two-way ANOVA, Tukey’s multiple comparison +/- SEM. AU565, JIMT1, BT474 *p<0.0001 (DMSO vs.shNEDD-1 or -2).
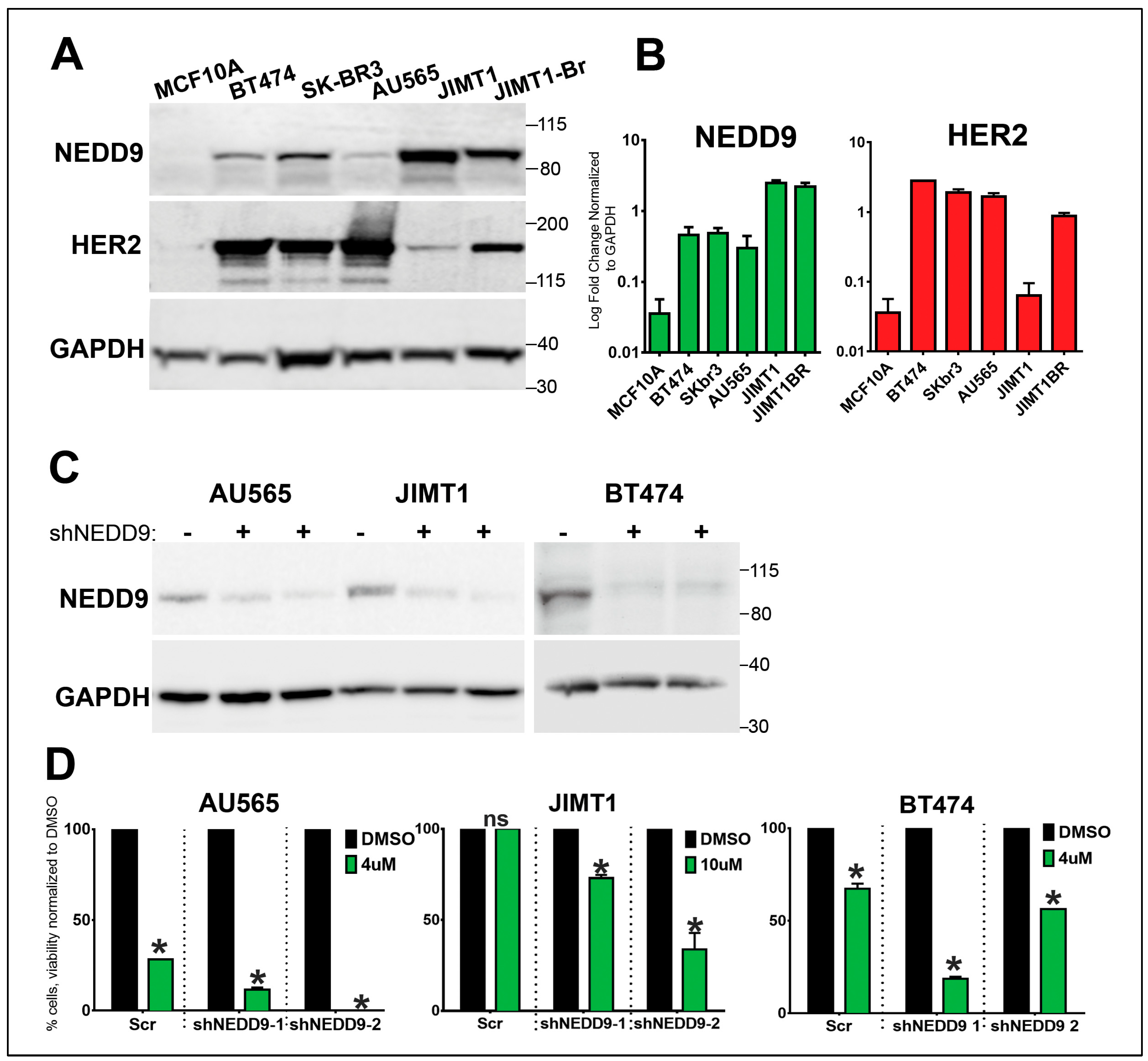
Figure 7.
NEDD9 expression increases proliferation through alteration of Aurora A Kinase expression. (A). Western blot analysis of MCF10A cells overexpressing NEDD9, HER2 or both with anti-NEDD9, -HER2, -pERK1/2, -ERK1/2 and -GAPDH as a loading control. n=3 independent experiments. (B). MTT proliferation assay using cells as in 7A measured at 24, 48, 72, and 96 hours. Two-way ANOVA +/- SEM, Tukey’s multiple comparisons; vector vs. NEDD9 or HER2+NEDD9 *p<0.001 at 96h; vector vs. HER2 is ns. (C). Representative brightfield images of mammary acini using cells as in (A), inset nuclei (blue) – DAPI (3D confocal projections every 10mm total of 200mm), scale bar – 20mm. (D). Quantification of number of cells per acini using confocal imaging (n=10 standard fields of view per each subline) in 3 independent experiments. One-way ANOVA +/- SEM. Tukey’s multiple pairwise comparisons: vector vs. NEDD9 or HER2+NEDD9 – *p<0.001; HER2 vs NEDD9 or HER2+NEDD9 – *p<0.001. (E). Western blot analysis of MCF10A cells as in (A) with anti- pAURKA, -AURKA, -GAPDH is loading control. (F). Western blot analysis of MCF10A cells with anti-pFAK, -FAK, -pSrc, -Src, and GAPDH is loading control.
Figure 7.
NEDD9 expression increases proliferation through alteration of Aurora A Kinase expression. (A). Western blot analysis of MCF10A cells overexpressing NEDD9, HER2 or both with anti-NEDD9, -HER2, -pERK1/2, -ERK1/2 and -GAPDH as a loading control. n=3 independent experiments. (B). MTT proliferation assay using cells as in 7A measured at 24, 48, 72, and 96 hours. Two-way ANOVA +/- SEM, Tukey’s multiple comparisons; vector vs. NEDD9 or HER2+NEDD9 *p<0.001 at 96h; vector vs. HER2 is ns. (C). Representative brightfield images of mammary acini using cells as in (A), inset nuclei (blue) – DAPI (3D confocal projections every 10mm total of 200mm), scale bar – 20mm. (D). Quantification of number of cells per acini using confocal imaging (n=10 standard fields of view per each subline) in 3 independent experiments. One-way ANOVA +/- SEM. Tukey’s multiple pairwise comparisons: vector vs. NEDD9 or HER2+NEDD9 – *p<0.001; HER2 vs NEDD9 or HER2+NEDD9 – *p<0.001. (E). Western blot analysis of MCF10A cells as in (A) with anti- pAURKA, -AURKA, -GAPDH is loading control. (F). Western blot analysis of MCF10A cells with anti-pFAK, -FAK, -pSrc, -Src, and GAPDH is loading control.


Table 1.
Analysis of Mammary Intraepithelial Neoplasia (MIN) in mice.
| Genotype | MIN+, N (%) | MIN-, N(%) |
|---|---|---|
| MMTV-Cre | 1 (10%) | 9 (90%) |
| MMTV-Cre-Erbb2 | 3 (30%) | 7 (70%) |
| MMTV-Cre-NEDD9 | 5 (50%) | 5 (50%) |
| MMTV-Cre-Erbb2-NEDD9 | 8 (80%) | 2 (20%) |
1 MIN+ mammary intraepithelial neoplasia positive, or – negative mice. N=10 per each genotype. Fisher’s Exact test statistics p*= 0.0137.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



