
Submitted:
04 January 2023
Posted:
04 January 2023
You are already at the latest version
Abstract
Previous studies demonstrate the critical importance of non-coding RNAs interfacing with chromatin-modifying machinery resulting in promoter-enhancer-based gene regulation and raise the possibility that many other enhancer-like RNAs may operate via similar mechanisms. Critically, more than 80% of the disease-linked variations identified in genome-wide studies are located in the non-coding regions of genomes, especially non-coding RNA, suggesting non-coding RNAs are relevant to disease. Thus, a critical path forward for understanding non-coding RNAs' role, especially long non-coding RNAs, is to understand the genomic regions' transcriptional regulation, especially non-coding regions. Here, we developed a user-friendly R package called SomaGene for studying and identifying enhancer-like non-coding RNAs with enriched somatic mutations in the cancer genome. SomaGene accepts different genomic variants (whole genome/exome somatic point mutations, structural variations, copy number variations) to identify those RNAs that significantly mutated in diseases (e.g., cancer). It then uses multiple publicly available genomics and epigenetics datasets including ENCODE epigenomics annotations, FANTOM5 tissue-specific expression profiles, disease-associated genome-wide association SNPs, and tissue-specific eQTL pairs to identify those RNAs with potentially enhancer function. SomaGene, as a powerful R package, can provide the opportunity to cancer scientists to study the roles of non-coding RNAs in different cancer genomes.
Keywords:
somatic point mutations
; non-coding RNA
; biomarker discovery
; driver genes
; non-coding RNAs prioritization
; health data analytics
1. Introduction
Over the past years, attempts to associate mutated genomic regions to cancer development have been preferentially focused on protein-coding genes as their functional structure is well studied. Nevertheless, most parts of the genome are non-coding regions, representing the vast majority of transcripts, even though not translated, appeared to perform significant roles in cancer and genetic diseases, and their contribution to tumor initiation and progression has been known by genome-wide association and whole genome/exome studies (1-9). However, despite extensive studies on discovering the relationship between DNA sequence and genomic functions for protein-coding genes, this relationship for non-coding genomic regions is less understood. However, it is now well known that non-coding RNAs are able to contribute to various cellular or regulatory activities in the cell, such as regulating gene expression via interaction with other chromatin regulatory proteins (10) function as enhancers (11, 12), and regulate chromatin structure (13-16). Despite various studies on the impact of non-coding somatic mutations occurring in ncRNAs, the role of such non-coding RNAs has remained underexplored in cancer.
Moreover, the emerging of the next-generation sequencing technologies for identifying cancer driver mutations and cancer-associated genomic regions yields the comprehensive publicly available catalogs of mutations in various cancers provided by ICGC and TCGA consortia. The most widely used methods for prioritizing non-coding genes rely on modeling the mutation rates using mutational catalogs in cancer or viral genomes (6, 17-23). Although helping prioritize non-coding mutated regions such as Introns, UTRs, promoters, and mRNAs, the statistical methods modeling mutation rates fail to be fit for ncRNAs, in particular, long noncoding RNAs (lncRNA) as one of the most important classes of non-coding transcripts involved in many cellular processes which the length of these transcripts are highly variable (200 bp to 100 kb).
This paper introduces a new prioritization pipeline (Figure 1), which takes a set of genomic ranges (e.g., non-coding genes) and a comprehensive mutational catalog associated with various cancers. It provides a prioritized list of genomic ranges that are significantly and recurrently mutated in each cancer. Rather than focusing on modeling mutation rates at the nucleotide level, this method calculates a P-value for each genomic region (e.g., a non-coding gene) accounting to two factors: (1) the level of mutation recurrence in the region among samples and (2) the extent to which the mutations in the region are specific to the samples of the cancer of interest comparing to other cancers. This statistical test can be followed by annotating input genomic regions with various functional assays such as disease-related genomic variants and regulatory features (e.g., chromatin marks, ChromHMM predicted promoters enhancers). The whole pipeline is designed as an R package, SomaGene, to provide an integrative assessment of somatic mutations to obtain a list of candidate genomic regions potentially functional in developing cancer. SomaGene can analyze and prioritize any set of genomic regions such as coding and non-coding genes in cancers.
2. Implementation
Statistical framework
Let , be a set of mutational catalogs for cancers, and be the set of input genomic regions. Using all mutational catalogs, a p-value for the region in cancer is calculated using a one-sided Fisher’s exact test with the following contingency table:

Table 1.
Contingency table for Fisher’s exact test.
| #samples in that have a mutation in the region . | #samples in that do not have a mutation in the region . |
| #samples in all catalogs except that have a mutation in the region . | #samples in all catalogs except that do not have a mutation in the region . |
This Fisher’s exact test (which is applied for all regions of interest in ) is followed by a permutation test (typically for 1,000,000 random permutations - can be changed by the user) to estimate the probability that any of the associations emerges by chance. In the permutation process, the order of sample IDs in the original mutational catalogs is shuffled to generate a list of simulated datasets of mutations. Then the Fisher’s exact test is applied to each simulated dataset to obtain a list of simulated P-values for each (the number of simulated P-values equals the number of permutations). Finally, each is determined to be significant if its original P-value (obtained from Fisher’s exact test on the original data) is less than the permutations P-value at confidence interval 99%. Users can define the significance level for the Fisher’s exact test and the confidence interval for the permutation test. Using this statistical framework, the user can extract a significant list of genes (from an initial list) that compared to other cancers, are recurrently and specifically mutated in cancer .
Annotations scheme
Let be the set of input genomic regions and be a set of genomic annotation entries. can be indicated as one of five general types of annotation (e.g., tissue-specific histone marks, encode predicated promoters and enhancers, GWAS, eQTL) that attribute specific genomic features to a set of genomic regions. This package aides the user to interpret an arbitrary set of regions with an optional annotation (one of five general types).
In the case of annotation of type 1, which is a simple catalog of genomic regions such as the list of active enhancers by FANTOM5 (24) or genomic positions of disease-associated genomic variants by GWAS Catalog (25), the annotation output is a list designating the occurrence of overlap/the number of overlaps, of annotation entries with each regions.
The 2nd type of annotation assigns a category to each genomic annotation entries , (e.g., the annotation of chromatin state segmentation (26)) and the result for each region indicates the categories overlapping with along with the percentages of overlaps.
In the case of 3rd annotation, a catalog assigns a single score to each annotation entry (such as the list of genomic regions enriched for histone modifications (27)) and the annotation result for each regions will be an aggregated score and the percentage of its length that overlaps with annotation entries.
The 4th type of annotation assigns a group of IDs (which we call sub_ids) and their corresponding scores (which we call sub_scores) to each annotation entry (e.g., DNase clusters (28)). In this case, the output for each region will represent the overlapping sub_ids and the corresponding aggregated sub_scores.
The 5th type annotation is defined for chromatin interaction datasets (such as Hi-C). In this case, the user provides two commentaries, one for chromatin interactions and one for target genomic regions, such as . As a result, the package will identify the interactions between entries of original genomic regions and target genomic regions (see supplementary materials for more details). For instance, the user can investigate the interactions between a set of lncRNAs (entries of ) that interact with a group of protein-coding genes (entries of ) through a dataset of Hi-C interactions in breast cancer. More details about the calculation of overlaps, scores, and sub_scores are explained in the supplementary materials.
3. Application of SomaGene
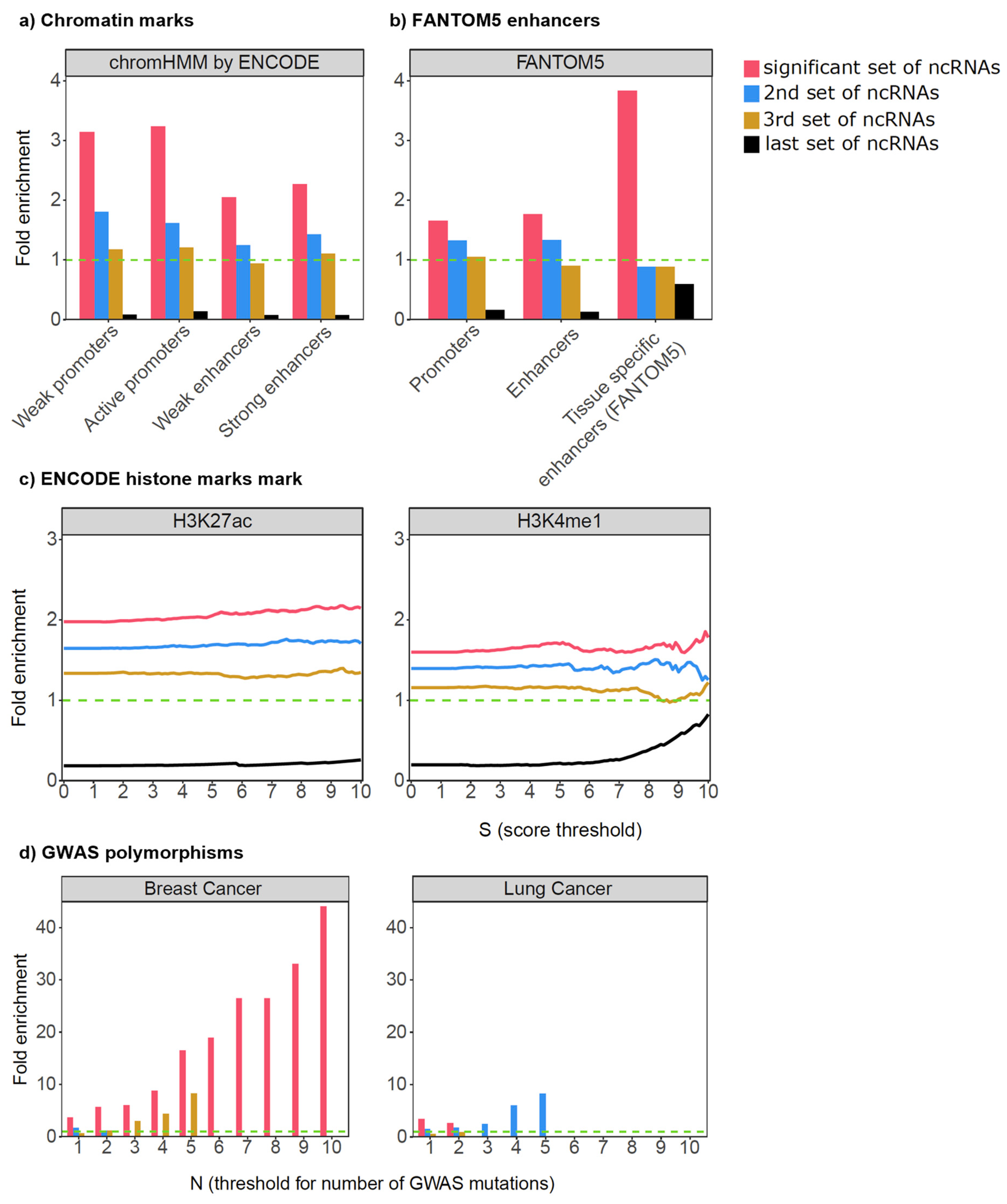
An application of SomaGene can be found in a recent comprehensive study aiming to prioritize non-coding RNA genes in the context of cancer Catalog (29). In this study, a list of 65,000 non-coding genes and the mutational catalogs of 19 cancer types downloaded from the ICGC consortia, were used as input to SomaGene for identifying those non-coding genes that significantly mutated in breast cancer samples. SomaGene identified 929 non-coding genes as significantly mutated genes in breast cancer samples (confidence interval 99% on 1,000,000 permutations provided by SomaGene). Interestingly, the candidate non-coding RNAs have significantly greater fraction of breast tissue related ENCODE enhancer and promoter marks (Figure 2a), FANTOM5 breast tissue differentially expressed enhancers (Figure 2b), ENCODE chromatin active histone marks (Figure 2c), and breast cancer associated genome-wide association SNPs (GWAS) (Figure 2d).
Also, we sorted non-coding RNAs based on their mutational p-value and repeated the enrichment analyses for the second, third and last set of non-coding RNAs (i.e., those non-coding RNAs that did not encompass any mutations). Interestingly, the enrichment for regulatory features have been also seen in the second and third lists of non-coding RNAs, but in much smaller fractions compared to the significant list of non-coding RNAs (Figure 2). As expected, no enrichment was seen for the last list of non-coding RNAs (Figure 2).
Finally, we used genomic and epigenetic annotations described in the above section and provided a prioritized list of non-coding RNAs with potential enhancer roles in breast cancer as well as five other cancers that can be accessed through our online resource http://ncrna.ictic.sharif.edu.
The medical and biological datasets are increasing rapidly. To analyse such big and complex data, artificial intelligence and integrative pipelines become most popular (5, 30-43). Here, we have developed SomaGene, a novel, user-friendly, interactive, open-access pipeline for identifying and annotating noncoding genes (or genomic coordinates) that significantly mutate in a cancer genome.
Supplementary Materials
Supplementary data are available at Bioinformatics online.
Author Contributions
HRR designed the study; MB, NR, MST, and HRR wrote and edited the manuscript with help from HAR. MB, NR, MH carried out all the analyses, including the statistical analyses, gene prioritization, annotation, and permutation under HAR and HRR supervision. MB generated all figures and tables under HRR supervision. All authors have read and approved the final version of the paper.
Data Availability Statement
The source code and a sample dataset can be accessed at https://github.com/bcb-sut/SomaGene. For more details about each parameter, please visit the https://github.com/bcb-sut/SomaGene/blob/master/README.md page.
Conflicts of Interest
The authors declare no competing financial interests.
Availability and Implementation
The SomaGene R package is freely accessible to the public at https://github.com/bcb-sut/SomaGene.
Tool Availability
The SomaGene source R package, a sample dataset, and instructions on how to run SomaGene are provided at https://github.com/bcb-sut/SomaGene and supplementary file entitled “user manual.”
References
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Research. 2012;22(9):1760-74. [CrossRef]
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, et al. The landscape of long noncoding RNAs in the human transcriptome. Nature Genetics. 2015;47:199. [CrossRef]
- Dashti H, Dehzangi, I., Bayati, M., Breen, J., Beheshti, A., Lovell, N. Integrative analysis of mutated genes and mutational processes reveals novel mutational biomarkers in colorectal cancer. BMC Bioinformatics. 2022;23(11):1-24. [CrossRef]
- Heidari R, Akbariqomi, M., Asgari, Y., Ebrahimi, D. A systematic review of long non-coding RNAs with a potential role in Breast Cancer. Mutation Research/Reviews in Mutation Research. 2021;787:108375. [CrossRef]
- Ghareyazi A, Mohseni, A., Dashti, H., Beheshti, A., Dehzangi, A., Rabiee, H. R. Whole-genome analysis of de novo somatic point mutations reveals novel mutational biomarkers in pancreatic cancer. Cancers. 2021;13(17):4376. [CrossRef]
- Bayati M, Rabiee, H. R., Mehrbod, M., Vafaee, F., Ebrahimi, D., Forrest, A. R. CANCERSIGN: a user-friendly and robust tool for identification and classification of mutational signatures and patterns in cancer genomes. Scientific reports. 2020;10(1):1-11. [CrossRef]
- Alinejad-Rokny H, Heng, J. I., & Forrest, A. R. Brain-enriched coding and long non-coding RNA genes are overrepresented in recurrent neurodevelopmental disorder CNVs. Cell Reports. 2020;33(4):108307. [CrossRef]
- Woodward KJ, Stampalia, J., Vanyai, H., Rijhumal, H., Potts, K., Taylor, F., ... & Heng, J. I. Atypical nested 22q11. 2 duplications between LCR 22B and LCR 22D are associated with neurodevelopmental phenotypes including autism spectrum disorder with incomplete penetrance. Molecular genetics & genomic medicine. 2019;7(2):e00507. [CrossRef]
- Poulton C, Baynam, G., Yates, C., Williams, S., Wright, H., ... & Heng, J. I. T. A review of structural brain abnormalities in Pallister-Killian syndrome. Molecular genetics & genomic medicine. 2018;6(1):92-8. [CrossRef]
- Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172(3):393-407. [CrossRef]
- Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143(1):46-58. [CrossRef]
- Kim T-K, Hemberg M, Gray JM. Enhancer RNAs: a class of long noncoding RNAs synthesized at enhancers. Cold Spring Harbor perspectives in biology. 2015;7(1):a018622. [CrossRef]
- Quinodoz S, Guttman M. Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends in cell biology. 2014;24(11):651-63. [CrossRef]
- Böhmdorfer G, Wierzbicki AT. Control of chromatin structure by long noncoding RNA. Trends in cell biology. 2015;25:623-32. [CrossRef]
- Alinejad-Rokny H, Ghavami Modegh, R., Rabiee, H. R., Ramezani Sarbandi, E., Rezaie, N., Tam, K. T., & Forrest, A. R. MaxHiC: A robust background correction model to identify biologically relevant chromatin interactions in Hi-C and capture Hi-C experiments. PLOS Computational Biology,. 2022;18(6):e1010241.
- Khakmardan S, Rezvani, M., Pouyan, A. A., Fateh, M. MHiC, an integrated user-friendly tool for the identification and visualization of significant interactions in Hi-C data. BMC genomics. 2020;21(1):1-10. [CrossRef]
- Rheinbay E, Parasuraman P, Grimsby J, Tiao G, Engreitz JM, Kim J, et al. Recurrent and functional regulatory mutations in breast cancer. Nature. 2017;547:55. [CrossRef]
- Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nature Genetics. 2014;46:1160. [CrossRef]
- Parhami P, Fateh, M., Rezvani, M. A comparison of deep neural network models for cluster cancer patients through somatic point mutations. Journal of Ambient Intelligence and Humanized Computing. 2022:1-16. [CrossRef]
- Alinejad-Rokny H, Anwar, F., Waters, S. A., Davenport, M. P., & Ebrahimi, D. Source of CpG depletion in the HIV-1 genome. Molecular biology and evolution. 2016;33(12):3205-12. [CrossRef]
- Ebrahimi D, Davenport MP. Insights into the motif preference of APOBEC3 enzymes. PloS one. 2014;9(1):e87679. [CrossRef]
- Lloyd SB, Lichtfuss, M., Amarasena, T. H., Alcantara, S., De Rose, R., Tachedjian, G., ... & Kent, S. J. High fidelity simian immunodeficiency virus reverse transcriptase mutants have impaired replication in vitro and in vivo. Virology. 2016;492:1-10. [CrossRef]
- Gooneratne SL, Ebrahimi, D., Bohn, P. S., Wiseman, R. W., O'Connor, D. H., ... & Kent, S. J. Linking pig-tailed macaque major histocompatibility complex class I haplotypes and cytotoxic T lymphocyte escape mutations in simian immunodeficiency virus infection. Journal of virology. 2014;88(24):14310-25. [CrossRef]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455. [CrossRef]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic acids research. 2017;45(D1):D896-D901. [CrossRef]
- Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nature Biotechnology. 2010;28:817. [CrossRef]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic Maps and Comparative Analysis of Histone Modifications in Human and Mouse. Cell. 2005;120(2):169-81. [CrossRef]
- Sabo PJ, Hawrylycz M, Wallace JC, Humbert R, Yu M, Shafer A, et al. Discovery of functional noncoding elements by digital analysis of chromatin structure. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(48):16837. [CrossRef]
- Rezaie N BM, Tahaei MS, Hamidi M, Khorasani S, Lovell NH, Breen J, Rabiee HR. Somatic point mutations are enriched in long non-coding RNAs with possible regulatory function in breast cancer. Communications Biology. 2022;5(1):1-13.
- Dashti H, Dehzangi, A., Bayati, M., Breen, J., Lovell, N., Ebrahimi, D. Integrative analysis of mutated genes and mutational processes reveals seven colorectal cancer subtypes. bioRxiv. 2020;2020.
- Javanmard R, JeddiSaravi, K. Proposed a new method for rules extraction using artificial neural network and artificial immune system in cancer diagnosis. Journal of Bionanoscience. 2013;7(6):665-72. [CrossRef]
- Mahmoudi MR, Akbarzadeh, H., Parvin, H., Nejatian, S., Rezaie, V. Consensus function based on cluster-wise two level clustering. Artificial Intelligence Review. 2021;54(1):639-65. [CrossRef]
- Niu H, Khozouie, N., Parvin, H., Beheshti, A., & Mahmoudi, M. R. An ensemble of locally reliable cluster solutions. Applied Sciences. 2020;10(5):1891. [CrossRef]
- Rajaei P, Jahanian, K. H., Beheshti, A., Band, S. S., Dehzangi, A. VIRMOTIF: A user-friendly tool for viral sequence analysis. Genes. 2021;12(2):186. [CrossRef]
- Shamshirband S, Fathi, M., Dehzangi, A., Chronopoulos, A. T., A review on deep learning approaches in healthcare systems: Taxonomies, challenges, and open issues. Journal of Biomedical Informatics. 2021;113:103627. [CrossRef]
- Alinejad-Rokny H, Sadroddiny, E., & Scaria, V. Machine learning and data mining techniques for medical complex data analysis. Neurocomputing. 2018;276(1). [CrossRef]
- Alinejad-Rokny H, Pourshaban, H., Orimi, A. G., & Baboli, M. M. Network motifs detection strategies and using for bioinformatic networks. Journal of Bionanoscience. 2014;8(5):353-9. [CrossRef]
- Parvin H, & Parvin, S. A classifier ensemble of binary classifier ensembles. International Journal of Learning Management Systems. 2013;1(2):37-47. [CrossRef]
- Esmaeili L, Behrouz Minaei-Bidgoli, and Mahdi Nasiri. Hybrid recommender system for joining virtual communities. Research Journal of Applied Sciences, Engineering and Technology. 2012;4(5):500-9.
- Hosseinpoor M, Parvin, H., Nejatian, S., Rezaie, V., Bagherifard, K., Dehzangi, A. Proposing a novel community detection approach to identify cointeracting genomic regions. Mathematical Biosciences and Engineering. 2020;17(3):2193-217. [CrossRef]
- Alinejad-Rokny H. Proposing on Optimized Homolographic Motif Mining Strategy Based on Parallel Computing for Complex Biological Networks. Journal of Medical Imaging and Health Informatics. 2016;6(2):416-24. [CrossRef]
- Parvin H, Seyedaghaee, N., & Parvin, S. A heuristic scalable classifier ensemble of binary classifier ensembles. Journal of Bioinformatics and Intelligent Control. 2012;1(2):163-70. [CrossRef]
- Parvin H, Helmi H, Minaei B, Shirgahi H. Linkage learning based on differences in local optimums of building blocks with one optima. International Journal of Physical Sciences. 2011;6(14):3419-25.
Figure 1.
Flow diagram of SomaGene pipeline. SomaGene accepts a VCF file including genomic variants and a list of genomic coordinates. After counting the number of mutated samples in each non-coding RNA, it then uses Fisher’s exact test described in the method section to identify mutational P-value for each non-coding RNA. To determine the significantly mutated ncRNAs (can be also a genomic coordinate) for each ncRNA, SomaGene calculates P-values for 1,000,000 random permutations of disease/non-disease (for example breast/non-breast) labels to estimate the 99% C.I. threshold of P-values. SomaGene then investigates the overlapping of non-coding RNAs with cancertissue-related regulatory features (e.g., ENCODE predicted chromHMM, H3K27ac), cancer-related GWAS SNPs, eQTL polymorphisms, FANTOM5 promoters, enhancers and tissue/cell related Hi-C interacting regions (if available). The user has the option to use the existing annotation files in SomaGene or enter their annotation files in a standard bed format.
Figure 1.
Flow diagram of SomaGene pipeline. SomaGene accepts a VCF file including genomic variants and a list of genomic coordinates. After counting the number of mutated samples in each non-coding RNA, it then uses Fisher’s exact test described in the method section to identify mutational P-value for each non-coding RNA. To determine the significantly mutated ncRNAs (can be also a genomic coordinate) for each ncRNA, SomaGene calculates P-values for 1,000,000 random permutations of disease/non-disease (for example breast/non-breast) labels to estimate the 99% C.I. threshold of P-values. SomaGene then investigates the overlapping of non-coding RNAs with cancertissue-related regulatory features (e.g., ENCODE predicted chromHMM, H3K27ac), cancer-related GWAS SNPs, eQTL polymorphisms, FANTOM5 promoters, enhancers and tissue/cell related Hi-C interacting regions (if available). The user has the option to use the existing annotation files in SomaGene or enter their annotation files in a standard bed format.
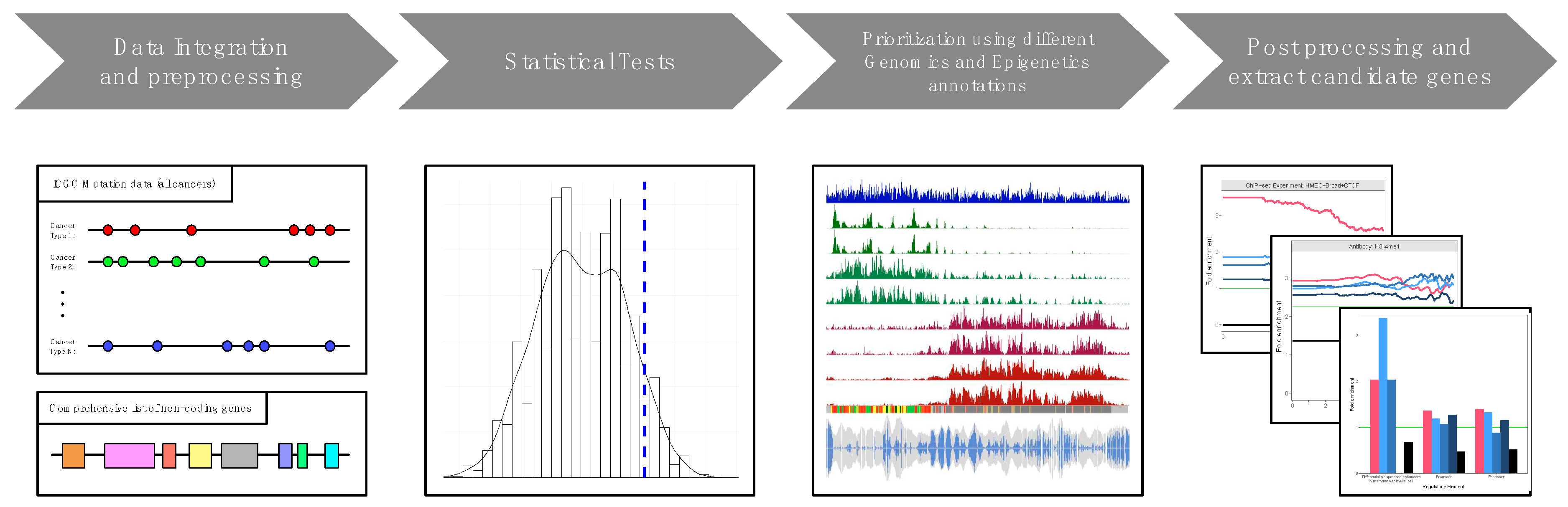
Figure 2.
Enrichment of regulatory features in the significant set of non-coding RNAs. a) Enrichment of breast-related promoters and enhancers identified by ENCODE (using chromatin segmentation by HMM). b) Enrichment of breast tissue differentially expressed enhancers identified by FANTOM5. c) Enrichemnt of ENCODE active histome marks. d) Enrichment of breast and lung cancer-associated GWAS SNPs. The enrichment is calculated by dividing the proportion of significantly mutated ncRNAs that overlap with each item by the proportion of all ncRNAs that overlap with that item. This enrichment is calculated for a significant set of ncRNAs (929 ncRNAs) shown in red color, 2nd set (blue), 3rd set (brown) of highly mutated ncRNAs. The enrichment was also calculated for the last set of ncRNAs that had no mutation in breast cancer samples. Each set of ncRNAs contains 929 elements.
Figure 2.
Enrichment of regulatory features in the significant set of non-coding RNAs. a) Enrichment of breast-related promoters and enhancers identified by ENCODE (using chromatin segmentation by HMM). b) Enrichment of breast tissue differentially expressed enhancers identified by FANTOM5. c) Enrichemnt of ENCODE active histome marks. d) Enrichment of breast and lung cancer-associated GWAS SNPs. The enrichment is calculated by dividing the proportion of significantly mutated ncRNAs that overlap with each item by the proportion of all ncRNAs that overlap with that item. This enrichment is calculated for a significant set of ncRNAs (929 ncRNAs) shown in red color, 2nd set (blue), 3rd set (brown) of highly mutated ncRNAs. The enrichment was also calculated for the last set of ncRNAs that had no mutation in breast cancer samples. Each set of ncRNAs contains 929 elements.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



