
Submitted:
31 December 2022
Posted:
04 January 2023
You are already at the latest version
Abstract
Advances in nanomedicine bring the attention of researchers to the molecular targets which can play a major role in the development of novel therapeutic and diagnostic modalities for cancer management. The choice of a proper molecular target can decide on the efficacy of the treatment and endorse the personalized medicine approach. Gastrin-releasing peptide receptor (GRPR) is a G-protein-coupled membrane receptor, well known to be overexpressed in numerous malignancies including pancreatic, prostate, breast, lung, colon, cervical and gastrointestinal cancers. Therefore, many research groups express a deep interest in targeting GRPR with their nanoformulations. A broad spectrum of the GRPR ligands has been described in the literature, which allows tuning of the properties of the final formulation, particularly in the field of the ligand affinity to the receptor and internalization possibilities. Hereby the recent advances in the field of applications of various nanoplatforms which are able to reach the GRPR expressing cells are reviewed.
Keywords:
gastrin releasing peptide receptor
; GRPR
; bombesin receptor subtype 2
; BB2
; bombesin
; targeted therapy
; molecular imaging
; theranostics
; nanotechnology
; nanoparticle
; nanomedicine
1. Introduction
Despite many efforts, cancer remains a major challenge for medicine professionals and scientists around the world. According to data published by University of Oxford in cooperation with Global Change Data Lab, cancer is the second leading cause of death worldwide [1]. Therefore, there is a constant need for new therapeutic and diagnostic modalities which can improve the current situation. Nanomedicine is believed to bring about change in the field of oncology and revolutionize current cancer management strategies. Markedly, targeted delivery of biologically active compounds is gaining momentum due to the high hopes they raise concerning improved therapeutic efficacy, convenient imaging and staging of malignancies, and minimized side effects on patients. Many molecular targets have been discovered to date and most of the currently developed targeting strategies can be assigned to one of the three general categories: uncontrolled cell proliferation targeting, angiogenesis-associated targeting, and specific tumor cells targeting [2]. Due to the fact that the majority of cancer-associated deaths are caused by five particular cancer types (tracheal, bronchus, and lung; colon and rectum; stomach; breast; pancreatic), targeting the specific tumor cells is becoming particularly interesting. It can be successfully accomplished due to the specific receptors that are present or overexpressed in malignant, but not healthy cells. It is noteworthy, that as low as threefold overexpression of a receptor on cancer cells already allows exploitation of the targeted delivery mechanism and successful delivery of the cargo [3]. Examples of receptors that were found to be overexpressed in malignant tissues are epidermal growth factor receptor (EGFR), folate receptor (FR), somatostatin receptors (SSTRs), or gastrin-releasing peptide receptor (GRPR). The latter was found to be overexpressed in most of the abovementioned cancers that are rising the biggest death toll. Therefore, GRPR is attracting the particular attention of numerous research groups dealing with targeted nanomedicines, including those gathered in the Coordinated Research Project “Nanosized delivery systems for radiopharmaceuticals” by the International Atomic Energy Agency (IAEA) in 2014-2019 [4]. They have developed various strategies to exploit the potential of GRPR targeting with nanoformulations aiming at either cancer treatment, imaging, and diagnosis, or the combination of those two modalities. In their research, myriad of targeting ligands leading to different biological responses upon the interaction with the receptor is accompanied by nanoparticles of diverse shape, origin, and composition. Acknowledging the significant interest in GRPR targeting, hereby we have reviewed recent advances in the field of synthesis, characterization, and applications of various nanoplatforms which are able to reach the cells and tissues expressing gastrin-releasing peptide receptor- both in vitro and in vivo.
2. Gastrin-releasing peptide receptor - overview
Gastrin-releasing peptide receptor (GRPR, also known as the BB2 receptor, Figure 1) is a member of the mammalian bombesin receptor family, comprising three distinct heptahelical receptors, having a wide range of physiological and pathophysiological effects. The human GRPR gene is located on the X chromosome, and the GRPR protein contains 384 amino acids of an estimated molecular weight of 43 kDa [5,6]. It is a member of the G-protein-coupled-receptors (GPCR) family, being a class of transmembrane proteins responsible for regulating a wide range of functions through ligand-receptor interactions. The interactions are based on the activation of heterotrimeric G protein subunits α and β/γ, which bind to target proteins, thus initiating respective cellular signaling pathways. The regions responsible for agonist binding have been found to be Gln120, Pro198, Arg287, and Ala307 [7], while the antagonist binding is possible due to the presence of Thr296, Phe301 and Ser304 [8]. Endogenous ligand of GRPR is gastric-releasing peptide (GRP).
Because GRPRs are known to be coupled with phospholipase C, they have the ability to induce the breakdown of phosphoinositides and the subsequent generation of diacylglycerol. This stimulates the mobilization of cellular calcium and the activation of protein kinases C (PKC), triggering a variety of cellular changes (e.g., membrane and cytosolic proteins or indirect regulation of cell proliferation). The stimulation of the receptor is also responsible for the activation of phospholipases β1 and β2 as well as tyrosine kinases or phosphorylation of some proteins, leading to processes such as the appearance of focal adhesion plaques or actin proliferation. The kinases belonging to the Src family are also rapidly activated, which has been found to be strongly influential on the invasion and growth of certain types of cancer. It is worth noting that chronic stimulation of GRPR receptors was found to strongly impact a variety of processes, such as internalization of receptor-ligand complexes, own-regulation of cell surface receptors or receptor desensitization, all arising from a range of external or cellular stimuli [6].
GRPR activation entails the activation of phospholipases D and A2 as well as cyclic adenosine monophosphate (cAMP) increase in some tissues. It can also stimulate arresting translocation to the plasma membrane and has been found to enhance the growth of normal and neoplastic tissues. Moreover, the receptor activation has been found to stimulate the tumor cells to invade and migrate to healthy tissues via one of the Gα proteins or transesterification of the epidermal growth factor receptor (EGFR), which is said to influence the GRPR-stimulated DNA synthesis in tumor cells [6]. An overview of the molecular events upon activation of GRPR is depicted in Figure 2.
3. GRPR targeting and nanosystems
As GRPR is well acknowledged to be overexpressed in cancers, it attracts the attention of scientists dealing with novel delivery systems based on active targeting, where GRPR ligands are used as moieties able to lead to preferential accumulation of the nanoparticles in the sites of interest. These nanoparticles may act as drug delivery vehicles, so they deliver cytotoxic biological activity in the tumor site. On the other hand, detectable nanoparticles can be tracked in the human body using different imaging modalities, hence helping with cancer localization and staging. Finally, both these activities can be incorporated into one construct and act as theranostic, the idea derived from the personalized medicine approach, which connects oncological diagnostics and treatment at the same time. During the design of a targeted nanoformulation, fundamentally three vital points need to be initially considered: choice of the proper targeting ligand, type of nanoparticles, and the means of bringing those together (Figure 3). These three aspects need to be fine-tuned to allow the successful development of nanosystems with anticipated properties and activity, particularly with respect to the desired mode of operation: therapeutic, diagnostic, or combining both theranostic (Figure 3).
There are certain trends that are noticeable in GRPR nanotargeting research. This receptor is overexpressed in numerous cancers, i.e., breast, colon, glioblastoma, head and neck squamous cell, lung, neuroblastoma, pancreatic, and prostate [3]. However, the vast majority (ca. 70%) of the published studies concerning the use of nanosystems, focuses on applications in prostate or breast cancer research. It seems coherent, though, with the public health data pointing out that those malignancies are (next to the colon and rectum) in the top 3 of cancer prevalence by type worldwide [1]. However, when considering the type of nanoparticles exploited in the GRPR targeting research, almost half of the formulations are based on either gold nanostructures or liposomal/micellar vesicles, which are predominantly exploited in imaging and therapy, respectively. Those trends also seem well-grounded since out of 25 clinically approved nanoformulations (U.S. Food and Drug Administration (FDA) and/or European Medicines Agency (EMA)), ten were based on liposomes, proving the clinical potential of those structures [10]. Gold nanostructures maybe have not reached such clinical success yet, but the combination of relatively simple, cheap, and well-understood synthesis, beneficial biological half-life, convenient and stable modification due to universal gold-sulfur interactions, and wide range of activity (photothermal therapy, contrasting agent for photoacoustic imaging, radiosensitization, etc.) make them appear very promising and worth exploring.
3.1. Targeting ligands
Ligands of GRPR can fall into one of two groups: agonists and antagonists. Agonists share the receptor activation sequence (Trp-Ala-Val-Gly-His-Leu-Met-NH2) with GRP and activate the GRPR signaling pathways, leading to downstream effects such as cell growth, proliferation, increased motility, and invasiveness. It was pointed out though that these effects are not desired in clinically viable formulations. Moreover, some body of evidence suggests that antagonists may perform better as targeting ligands [11]. Despite those flaws, surprisingly, most of the research published in the field of nanostrategies for targeting this receptor concerns the use of agonists. One of the possible reasons might be that internalization of the nanoparticle delivering the cargo, together with the receptor upon its activation, may be beneficial for some drug delivery applications and is convenient to track in vitro. However, actual reasons for this state of the matter remain to be elucidated and may be much more trivial, e.g., easy access and broad awareness of bombesin (BBN), the best known GRPR ligand, which happens to be an agonist.
The wide range of agonistic ligands has been shown in the literature. The relatively easiest approach is to use the commercially available native sequence of bombesin [12] (Figure 4. a). Kulhari and co-workers exploited it in combination with biodegradable poly(lactic-co-glycolic acid) (PLGA) nanoparticles to successfully target both breast and prostate cancer cells and improve docetaxel (DTX) anticancer activity [13,14,15]. The same ligand was also used to target the [64Cu]CuS nanoparticles toward prostate cancer by Cai et al [16]. Interestingly, using native bombesin, Du and Li investigated if the synthetic strategy, namely pre/post-functionalization of nanostructured lipid carriers, affects the performance of the nanosystem for targeted lung cancer combination therapy [17].
Variations on the ligand structures start with exchanging single amino acids to engineer the sequence towards facilitated conjugation, for example, replacing the third amino acid from the native sequence with lysin [18,19,20], therefore providing additional -NH2 groups – such ligands are also commercially available [21] (Figure 4.b).
Nevertheless, much more sophisticated structures were also developed. One of the most prominent alterations in the bombesin architecture is to shorten the peptide chain down to an essential receptor binding sequence: Trp-Ala-Val-Gly-His-Leu-Met-NH2, which can lower the price of the custom peptide synthesis. These short peptide chains can also be further engineered to provide convenient means of condensation with nanoparticles [22,23,24], which will be discussed in more detail in the next subchapter. Frequently, the ligand is combined with poly(ethylene glycol) (PEG) to improve the pharmacokinetics of the construct and minimize the reticuloendothelial system uptake upon in vivo administration [25,26].
Adding extra functionality to the structure represents a very attractive strategy for bombesin structure alterations. An example of such modifications is introducing the radioisotope chelation functionality to the ligand. Two main chelators used in such research are 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and hydrazinonicotinic acid (HYNIC) and the isotopes used are 177Lu and 99mTc, respectively [27,28]. Other interesting idea is to combine bombesin binding motif with another peptide, creating heteromultimeric ligands, which can improve the construct targeting ability (e.g., RGD sequence against integrin αυβ3 [29,30], prostate-specific membrane antigen (PSMA) inhibiting peptides for targeting of prostate cancer) or cellular penetration (e.g., Tat(49–57), HIV-1 derived sequence [31,32]).
Regarding the antagonistic ligands, GRPR antagonists can be categorized into six classes, out of which five are peptides/peptoids while the last class comprises low molecular flavone derivatives [6]. Classes 2, 3, and 4 (in contrast to classes 1 and 5) are based on the modifications of the binding region of BBN. Class 2 are d-Phe12 analogs; class 3 focuses on modifications in positions 13-14 (or 26-27 when considering GRP sequence), while class 4 comprises analogs devoid of Met14 (desMet14). Among almost 100 papers hereby reviewed, only six dealt with the antagonist ligands. Lahooti and colleagues used a class 2 antagonist with d-Phe12 substitution to successfully target their ultrasmall paramagnetic iron oxide nanoparticles (USPION) to GRPR-positive breast cancer xenograft in vivo [33]. Particular attention was given to class 3 antagonist d-Phe-Gln-Trp-Ala-Val- NMeGly-His-Sta-Leu-NH2 (BBN-AA1) by the group of Accardo. They successfully incorporated this sequence into amphiphilic derivative monomer, which was used to form liposomes loaded with doxorubicin (DOX) [34]. The same monomer was also used to form sterically stabilized micelles as a targeted carrier of poorly water-soluble anticancer drug - gold(III) dithiocarbamate [35]. The BBN-AA1 sequence with some spacer and maleimide-reactive cysteine was used to obtain the kit for targeted DOX-loaded liposomes compliant with Good Manufacturing Practice (GMP), with satisfactory stability [36]. Li and co-workers exploited another class 3 antagonist sequence: [d-Phe6-Sta13-Leu14-NH2]bombesin(6-14) [37]. In their research, this sequence was further modified with Alexa Fluor 750 to obtain a carrier system for molecular imaging of oral squamous cell carcinoma. Tagliviani and co-workers exploited Demobesin-1 – GRPR peptide antagonist that falls into the class 4 analogs [38]. They have shown that upon adding a trioxatridecan-succinamic acid spacer, Demobesin-1 provided successful cell recognition functionality to the polyoxometalate clusters.
Accardo et al. analyzed the performance of seven different GRPR antagonists based on DOTA, and BBN derivatives coupled with PEG – formulations with a very simple design but offering an insight into the mechanisms by which they interact with GRPRs and hence provide varying targeting yield and diagnosis options [26]. From their systematic analysis, they concluded that most of the examined BBN derivatives serve equally well for targeting purposes (which was confirmed with gamma camera recordings), but those with NMeGly11 and Sta13-Leu14 would be most suitable for imaging in vivo since their plasma half-lives are over 15 days and the latter also provides a favorable absorption mostly into the tumor tissues and no other organs of the body.
As mentioned above, GRPR is the mammalian bombesin receptor family member, comprising three distinct receptors. Basically, each of these receptors is expressed in different neoplastic conditions and has its own set of ligands, however, there was the universal binding sequence discovered (d-Tyr6-Gln7-Trp8-Ala9-Val10-βAla11-His12- Phe13-Nle14) sometimes referred to as pan-bombesin. This finding is particularly attractive because with such a ligand it is possible to target tumors expressing various bombesin receptors patterns while improving the construct’s tumor-targeting efficiency and applicability. Heidari and co-workers used the peptide based on a universal binding sequence (Lys-Gly-Gly-Cys-Asp-Phe-Gln-Trp-Ala-Val-bAla-His-Phe-Nle) to target breast cancer in vitro and in vivo with gold nanorods for photothermal therapy [39] as well as imaging [40] and they pointed out its several advantages in comparison to native bombesin sequence, such as overcoming the issues related with receptor heterogeneity in tumors and ameliorating renal clearance due to the negatively charged hydrophilic aspartic acid residue. Similarly, Salouti et al. [41] and Jafari et al. [42] proposed the application of this peptide in breast cancer imaging using gold and superparamagnetic iron oxide nanoparticles, respectively. Pan-bombesin was also shown to improve prostate cancer imaging [43,44].
3.2. Ligand incorporation
One of the biggest challenges in the synthesis of particles targeting GRPR or any other receptor is the incorporation of the targeting ligand in the structure of the vehicle. Various approaches can be found in the literature. A state-of-the-art technique based on the formation of amide linkage between the ligand and the nanoparticle is most commonly used. Chemistry of 1-ethyl-3-(3-dimethyl aminopropyl)carbodiimide (EDC, EDAC) in combination with N-hydroxysuccinimide (NHS) [20] or sulfo-N-hydroxysuccinimide (sulfo-NHS) [39,40] is most commonly used to create the bond. Usually, this amide is formed between the N-terminus of the peptide ligands and carboxyls on the nanoparticles (Figure 5). Tang and co-workers successfully decorated their poly(acrylic acid)-functionalized lanthanide nanoparticles with BBN analog using EDC/sulfo-NHS chemistry [45]. It was even shown to be feasible to prepare ready-to-use kits with, e.g., stable NHS esters. EDC/NHS reactions are usually led in water, which is beneficial in terms of biocompatibility and purification procedures. However, the reaction, for instance, in tetrahydrofuran (THF) or dimethylformamide (DMF) is also possible as shown by Du and Li [17] and Mansour et al. [46], respectively.
Nevertheless, with no carboxylic groups available on the surface of the nanoparticle, nanoparticle’s surface, or no amine group available in the ligand, the other way around can be equally efficient [16,47]. Poly(amidoamine) (PAMAM) dendrimers were conjugated with DOTA-modified BBN using the amine group on the dendrimer and carboxylic group of the DOTA moiety [48,49]. Hajiramezanali and co-workers used this approach to exploit amine groups on chitosan and conjugate succinylated BBN derivative [50]. However, organic solvents and water-insoluble substrates are also used, such as 2-(1H-7-azabenzotriazol-1-yl)-1.1.3.3-tetramethyluroniumhexafluorophosphate) (HATU) [49,51,52] or N,N′-dicyclohexyl carbodiimide (DCC) [38,53] in DMF with the addition of N,N-diisopropylethylamine (DIPEA) [49]. Also, click chemistry such as thiol-maleimide [36,54,55] or copper-catalyzed azide-alkyne cycloaddition was useful [43,44,56,57]. Dash and colleagues used cysteine groups incorporated in the peptide structure to click the GRPR-binding peptide with maleimide-containing PEGylated magnetic reduced graphene oxide [58].
In some cases, the ligand can be incorporated into the structure of particle-forming substrates before the particle is formed [59]. Accardo et al. have used solid-phase peptide synthesis with Fmoc/tBu chemistry to synthesize the ligand peptide [DOTA-bAla]BBN(7-14) and combine it with the amphiphilic monomers [60]. Subsequently, these monomers were cleaved from the Rink amide resin, purified, and assembled into targeted supramolecular aggregates. A similar approach was adopted by Accardo and co-workers to prepare DOX-loaded targeted liposomes [34]. Kanazawa et al. used this approach to combine bombesin with stearic acid directly in solid-phase peptide synthesis, and they used it for polymeric micelles [61]. Yang and co-workers incorporated the GRPR binding motif of BBN into the structure of their engineered peptides able to form a stable coiled-coil nanostructure [62]. An interesting approach was also presented by Zhang and colleagues – they have genetically engineered Escherichia coli bacteria to express elastin-like peptides capable of coassembly into micelles [63]. One of the peptides was engineered to contain a GRP-derived binding sequence (Figure 6).
In some instances, the application of a linker is necessary due to the configuration of functional groups present in both ligand and particle; both homo– and heterobifunctional linkers are available. For example, Achilli et al. used glutaraldehyde as a homobifunctional amine-reactive crosslinker to functionalize human serum albumin (HSA) protein corona of biohybrid gold nanoparticles with [Lys1-Lys3(DOTA)]BBN [64]. Kim et al. tethered bombesin ligand to amine-modified poly(ethylene glycol) block of amphiphilic copolymer using a heterobifunctional linker, succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) [65]. Montet et al. have used amine and thiol-reactive succinimidyl iodoacetic acid to ligate the BBN derivative with amine-containing iron oxide nanoparticles [66]. Lee and co-workers used N-succinimidyl 3-(2-pyridyldithio)propionate (SPDP) to ligate cysteine-terminated derivative with glycol chitosan nanoparticles [67].
Also, noncovalent bonding may be useful in this context (Figure 7). Successful attachment of the BBN analog to gold nanoparticles can be achieved by exploiting the interaction between nitrogen atoms in free amine groups and gold surface [19,41]. Ocampo-Garcia et al. have developed a shelf-storage stable kit based on gold nanoparticles operating on this principle [18]. On the other hand, Chanda and co-workers used the thioctic-acid-modified bombesin derivative to exploit thiol-gold interactions for stable functionalization (stable even in a reducing environment of dithiothreitol). Hosta-Rigau et al. used additional cysteine to provide thiol for interaction with gold [69]. Combining functional linkers such as N-succinimidyl 3-(2-pyridyldithio)propionate (SPDP) and gold-sulfur interactions can be an interesting alternative for gold nanoparticles [23,67]. Li and colleagues exploited hydrogen bonds and π-π stacking to functionalize nano-graphene oxide with a Sta-BBN ligand [37]. Interestingly, Trujillo-Benítez and co-workers used the DOTA moiety incorporated in BBN derivative structure to achieve stable chelation of Sm atoms and therefore ligation of the ligand with metal oxide nanoparticles [30]. Young et al. have used one of the strongest known noncovalent interactions systems, biotin-streptavidin, to bind biotinylated bombesin to quantum dots decorated with streptavidin, resulting in efficient GRPR-selective fluorescent label operating in vivo [70].
Finally, a ligand can also be protected inside the nanoparticle. De Barros et al. postulate that due to natural plasma and tissue peptidases, peptides should be protected inside the nanocarrier, therefore they have encapsulated radiolabeled bombesin derivatives inside the long-circulating pH-sensitive liposomes (Figure 8). This approach led to the successful targeting of GRPR-positive breast cancer and Ehrlich tumor cells [71,72,73].
4. Targeted delivery of biologically active compounds
Table 1 summarizes the contemporary (c.a. 2009 to date) advances in the research concerning targeted delivery applications of nanocarriers functionalized with GRPR-avid moieties.
4.1. Micelles, liposomes, and other self-assembled structures
One of the most commonly used vehicles in drug delivery are supramolecular assemblies with a micellar or liposomal arrangement, following the patterns we can find in natural systems such as cell membranes. These materials offer a number of advantages, namely biocompatibility, a broad spectrum of functionalization possibilities, precisely controlled size, and convenient, still robust synthesis [60]. Therefore, it is no surprise that the first FDA-approved nanotechnology-based drug was a liposomal formulation. Since Doxil, liposomal DOX (Figure 9. a), was launched, researchers have continued the dynamic exploration of the field of liposomal and micellar formulations to search for further improvements, one of those being active targeting of the GRPR overexpressing tumors, such as prostate cancer [36] or small-cell lung cancer [55]. Much effort was shown in the area by Accardo and co-workers. They have examined multiple targeting ligand alternatives (as described above in Section 3.1) to choose the one that performs the best and used it in multiple studies regarding the development of self-assembled structures. Their most recent communication published in this matter complements their efforts in a form of the kit for industry-friendly prepared liposomal DOX targeted with the engineered amphiphilic ligand containing a bombesin active sequence and phospholipidic hydrocarbon chain (Figure 9. b) [36]. Their kit contains three vials: a) liposomal doxorubicin, b) BBN reactive freeze-dried phospholipid, and c) bombesin analog. The conjugate is efficiently formed in situ, with no loss of DOX or size alterations, and was shown to significantly inhibit tumor growth in comparison to non-targeted liposomal DOX.
Among the methods enabling liposome preparation one can mention, e.g., solvent evaporation method with subsequent film rehydration or simple emulsification and solvent evaporation. However, more sophisticated methods are also developed – Maqbool et al. used supercritical carbon dioxide technology to produce their albendazole-loaded liposomes. They have successfully synthesized structures sizes of ca. 160 nm size. Although functionalization with palmitic acid-anchored BBN(6-14) led to a slight increase in size, BBN-boosted specific uptake significantly improved liposomes cytotoxicity in the in vitro prostate cancer model in comparison to free drug.
Micellar vesicles also can be obtained by simple procedures. Kanazawa et al. prepared poly(ethylene glycol)-polycaprolactone (PEG-PCL) block polymeric micelles by creating water-in-oil (w/o) emulsions based on ultrapure water and drugs solutions in organic solvents [61]. The aim was to design a glioma-targeting nanocarrier with the ability to cross the blood-brain barrier and deliver the therapeutic of choice to the GRPR-overexpressing cells of the tumor. Therefore, micelles were functionalized with stearoyl-modified BBN and Tat(49–57), as well as loaded either with coumarin or camptothecin (CPT). Both formulations were tested safe and alone had no effect on the cells. The coumarin-loaded BBN-conjugated nanocarriers showed an improved C6 cell uptake over PEG-PCL-Tat(49–57) (17 vs. 10%), and the researchers established their absorption into the tumor might occur through macropinocytosis by Tat(49–57) and clathrin-mediated endocytosis by BBN, making bombesin an important modulator of the intratumoral uptake. In vivo studies in orthotopic glioma-grafted rats confirmed in vitro findings and showed an enhanced coumarin uptake in cancerous tissue as well as reduced internalization into healthy tissues. In the case of CPT, the release of the drug from BBN-conjugated nanocarriers was greater as compared to pure PEG-PCL-Tat(49–57), but the difference was not significant. Nonetheless, the targeting transporters performed noticeably better in cytotoxicity studies in C6 glioma cells as well as prolonged the life of tumor-bearing rats. Their post-intranasal administration lifespan was extended from 17.8 days (for untreated rats) to 20.3 days, 30.8 days, and 42.2 days for pure CPT, CPT loaded PEG-PCL-Tat(49–57) and BBN/PEG-PCL-Tat(49–57) respectively, with 2/5 of the population surviving over 120 days in the case of GRPR targeting nanoformulations. What is also important is that the micelle encapsulation of the drug made it less toxic to other bodily tissues and organs. It was revealed that there was no weight loss post-intranasal administration, in contrast to a drastic drop in body weight in the case of pure CPT solution.
Fine-tuning of composition can relatively easily confer desired features to the assemblies, such as high drug-loading capacity, controlled release profile, prolonged circulation time, or slow bloodstream clearance. Because the hydrophobic compartments of liposomes and micelles can host poorly water-soluble compounds, their bioavailability and convenience of use can be significantly improved by carriers’ application. Ringhieri and colleagues used sterically stabilized micelles based on 1,2-stearoyl-sn-glycerol-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000) to encapsulate highly toxic anticancer drug with low water solubility Au(III)-dithiocarbamate complex (AuL12) [35]. Small micelles (~20 nm) were obtained, and the drug was successfully loaded into the micelles. Neither loading nor targeting moiety incorporation affected the structure size and the optimization of composition, namely, the incorporation of egg L-α-phosphatidylcholine (PC) or 1,2-dioleoyl-sn-glycerol-3-phosphocholine (DOPC), increased the loading capacity. The drug was shown to be stable in the liposomes for at least 72 h, and the slow release from the micelle revealed a high affinity of AuL12 towards the micelle core. Targeted micelles showed a marked decrease in cell viability when compared to non-targeted micelles or a free drug, indicating a beneficial influence of the targeting moiety. Hiding the drug within the liposomal or micellar structure has the potential to ameliorate the systemic toxicity of the drugs.
Monomethyl auristatin F (MMAF) is one of the highly efficient anticancer drugs, however, its application is limited due to its significant systemic toxicity. The success of Adcetris, a targeted MMAF–antibody conjugate, stimulated researchers to search for a way to deliver the drug specifically to the target site, thus maximizing the drug accumulation in malignant tissue. This, in turn, can ameliorate systemic toxicity and allow full exploitation of MMAF anticancer potential. Following the abovementioned concept, Kim et al. used polymeric micelles based on block copolymer of poly(ethylene glycol) and poly(1,4-phenylacetone dimethylene thioketal) (PEG-PPADT), tethered with BBN [65]. As expected, MMAF@BBN-PEG-PPADT showed higher anticancer activity than MMAF@mPEG-PPADT and free MMAF. The targeting peptide was shown to play an essential role in the enhancement of cellular uptake in vitro, implying promising anticancer efficiency and improved toxicity in vivo.
An emerging alternative to liposomes and micelles is represented by solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC). These constructs are based on the lipid-matrix core, composed of either solid crystalline lipids (SLN) or solid and liquid lipids with the less ordered crystalline arrangement (NLC), surrounded by a self-assembled layer of surfactants, stabilizing the whole construct. They offer improved drug encapsulation capacity and even better stability compared to typical micellar and liposomal systems [59,79]. GRPR-targeted SLNs loaded with DOX were reported by Wang and co-workers. Their system demonstrated an excellent in vitro cytotoxicity and in vivo antitumor effect both in MCF-7/MDR breast cancer cell lines and breast cancer animal models - the tumor inhibition rate of BBN-DOX/SLNs in mice was approx. 81%. However, drugs are not the only compounds that can be hidden inside the vesicular cavities. Du and Li have taken this system to the next level - they have complemented their DOX-loaded nanostructured lipid carrier with DNA, for which NLCs are a very effective delivery vehicle [17]. The formulations reached over 67% of transfection efficiency at 72 h, with targeted carriers outperforming the non-targeted counterparts in the lung cancer model both in vitro and in vivo. Such combination therapy may be a solution to multidrug resistance issues due to the synergistic anticancer effect of a drug and the genes delivered.
4.2. Gold nanomaterials
Among various delivery vehicles for therapeutic compounds, gold nanostructures have been one of the most widely studied because of the high degree of biocompatibility, ease of processing and functionalization as well as the possibility of size, shape, and surface optimization to suit stringent requirements of targeted therapies [80]. The surface chemistry of gold is relatively well understood and provides the opportunity for stable bonding with ligands ranging from synthetic polymers through proteins (such as antibodies or enzymes) to DNA fragments up to drugs and biosensing molecules. The aspect of interest for many research teams remains, however, the possibility of efficient conjugation with targeting moieties that could enhance cancer treatments' safety and efficacy through drug internalization mostly into the tumor cells. Moreover, when gold nanostructures are amassed in the tumor and then are treated with electromagnetic radiation from e.g., laser, they generate a substantial amount of heat and as a result damage the malignant cells without affecting healthy cells surrounding the tumor tissue [80]. This is possible only because of the surface plasmon resonance that these structures show, which can be exploited as an additional therapeutic modality called photothermal therapy. Last but not least, it is also worth highlighting that gold nanostructures are able to boost the absorption of ionizing radiation energy leading to radiosensitization. They promote the creation of secondary electrons that spur the formation of reactive oxygen species [80]. The latter prompts supplementary impairment of the DNA, cell membrane, and mitochondria of the cancer cells and further augments cancer cells eradication. All of these features taken together make AuNPs a particularly interesting material for further exploration.
Hosta-Rigau and colleagues synthesized and functionalized gold nanoparticles (AuNPs) with an RAF peptide analog (Ac-Cys-Ahx-RAF) serving as the anticancer moiety and a BBN analog (Ac-Cys-Ahx-BBN) with the role of targeting the overexpressed GRPR in HeLa cervical cancer cells [69]. They showed that conjugates of RAF derivative and AuNPs significantly decrease the cell viability, which confirms retaining the RAF peptide anticancer activity upon adding the Ac-Cys-Ahx spacer and conjugation. Interestingly, a significant reduction in cell viability was found both for AuNP-conjugates with the BBN derivative as compared to the control (which is explained by the successful receptor-mediated internalization of the conjugates) and for the AuNP-free counterpart, which is somewhat surprising, given the known effects of GRPR activation in cancer cells; authors do not comment on that. However, the cell viability for the mixed system - Ac-Cys-Ahx-Lys-BBN / Ac-Cys-Ahx-Lys-RAF@AuNPs was comparable to Ac-Cys-Ahx-Lys-RAF@AuNPs, and a notable drop in cell viability was observed (53% and 59% respectively in reference to untreated samples) for GRPR-positive cancer cells. Furthermore, virtually no toxicity of the system was shown for poorly GRPR-expressing neuroblastoma SHSY-5Y cells. Authors conclude that conjugation of the peptides may lead to changes in peptides conformation, altering their activity.
Different AuNP-based nanocarriers were synthesized and examined in treating both prostate and colon cancers by Chilug et al. They used engineered bombesin and neurotensin-like peptides and checked how these ligands bind to both types of tumors [25]. One of the targeting moieties was [68Ga]Ga-DOTA-PEG(4)-BBN(7-14), which, together with its AuNPs counterpart, was subjected to in vitro and in vivo tests. All the newly synthesized constructs showed promising imaging efficiency for colon and prostate malignancies. This is due to the fact that the conjugation of AuNPs with the modified BBN peptide resulted in roughly a threefold increase of the uptake by the colon tumor (9.82% ID / g vs 3.31% ID / g) observed in nude mice and almost its 3.5 times higher uptake by prostate cancer cells (3.78% ID / g vs 13.01% ID / g). Follow-up studies were pointed out to be focused on the application of the constructs in positron emission tomography/computed tomography (PET/CT) imaging.
Another attractive feature that can be introduced with targeted nanotherapeutics is the controlled release of the drugs if those are conjugated to the carrier with suitable cleavable links. Silva et al. postulated that the Au-S and S-S bonds in their BBN-AuNP-based conjugates coated with the thiolated radioisotope-chelating diethylenetriaminepentaacetic acid (DTPA) derivative ((2-[bis[2-[carboxymethyl-[2-oxo-2-(2-sulfanylethyl- amino)ethyl]amino]ethyl]amino]acetic acid - DTDTPA) can be selectively cleaved by glutathione (GSH), hence endow the system with the controlled release feature [24]. Interestingly, systems triggered with GSH can be even more selective since its intracellular concentrations are substantially greater (500-5000 times) than extracellular ones. The γ-counting and neutron activation experiments revealed that BBN-AuNP-DTDTPA radiolabeled with 99mTc release the radioisotope into the GRPR-positive cells in vivo, where a significant amount of GSH is present, while the PC-3 cell culture study with histidine (a powerful chelator for the radiolabeled precursor) did not result in the cleavage of 99mTc from the particles in spite of the BBN presence, indicating its high stability. The in vivo tests also revealed good blood clearance and excretion rate, with most formulations eliminated with urine. Some of the remaining Au and detached DTDTPA-[99mTc]Tc(CO)3 did accumulate in the liver and kidneys (10 and 30 % ID / g, respectively, at the 4 h mark) of the CD1 mice. However, this is expected from intravenous administration of the constructs. Therefore, GSH-cleavable BBN-AuNP-DTDTPA (Figure 10) still seems to be a promising platform for GRPR-targeting therapies.
Jimenez-Mancilla et al. addressed an additional not less important aspect of targeted imaging and therapy – drug internalization into the cancer cell nuclei [31]. As targeting ligand, they used a hybrid peptide consisting of the [Lys3]BBN sequence, short spacer, and additional Tat(49-57) sequence, which promotes cellular penetration and nuclear localization of the construct. The obtained Au-based nanosystem was radiolabeled with two radioisotopes, for which DOTA and HYNIC were used, respectively. 177Lu is depositing irradiation energy of β particles across hundreds of cancer cells due to low linear energy transfer (LET), while 99mTc targets cellular nuclei adjacent to the carrier due to high LET of Auger electrons [81]. Radiolabeled AuNPs linked with Tat(49-57)-[Lys3]bombesin (177Lu/99mTc-AuNP-Tat-BBN) were examined for their biodistribution in PC-3 xenograft-bearing athymic mice upon intratumoral injection. It was found that Tat(49-57)-[Lys3]bombesin functionalized carrier significantly increases tumor retention of radioactivity compared to its non-targeted counterpart and free radiolabeled targeting peptide. According to the authors, this significant improvement could be attributed to the successful targeting of GRPR, which was also confirmed in receptor blocking in the in vivo experiments. Subsequently, researchers also examined the potential of using the 177Lu/99mTc-AuNP-Tat-BBN particles as a thermal ablation system after internalization into the PC-3 cells and found it very effective not only in preventing cell proliferation but also in decreasing cell viability (down to about 1.3%), which together with the NIR fluorescence emitted from the conjugates make them applicable for both targeted radiotherapy and plasmonic photothermal therapy.
AuNPs can also serve as a core in more complex constructs where the metal nanoparticle is caped with various functional moieties. Such hybrid AuNPs, coated with water-soluble chitosan (WSCS) and [Lys1-Lys3(DOTA)]bombesin (DOTA-BBN), were prepared by Tangthong et al. to target GRPR-overexpressing PC-3 and LNCaP prostate cancer cells selectively [76]. Upon 48 h incubation, the examined AuNPs-WSCS-DOTA-BBN particles showed half maximal inhibitory concentration (IC50) values of 65.89 and 60.73 μg/mL, respectively, against PC-3 and LNCaP cells (compared to IC50 for AuNPs-WSCS being over 100 μg/mL) without any drug loaded; they also remained stable in biological media and proved safe for healthy HAEC aortic endothelial cells. Follow-up studies by the same team explored AuNPs reduced in situ within WSCS anchored with gallic acid (GA) and conjugated with DOTA-BBN [47]. The AuNPs-WCS-GA-DOTA-BBN exhibited an increased internalization into PC-3 cells when compared with the GA-free (previous study) or BBN-free conjugates as well as 2.4-fold lower IC50 values (5.94 μg/mL vs 12.05 μg/mL) after 48 h in comparison to AuNPs-WCS-GA. These findings confirm the targeting effect of the BBN analog incorporated into the system. GA also showed to have good anticancer properties, and although more potent when administered alone, considering its negative impact on healthy cells, such a targeted formulation seems very promising for more selective and thus more efficient carcinoma management, especially if the loading concentration was increased above ~15% used in the described study.
Most Au-based biomedical constructs are based on spherical particles, but it is certainly not the only available architecture of gold nanostructures; Heidari and colleagues exploited gold nanorods (GNRs) [39]. They have designed and successfully synthesized GNRs conjugated with a universal binding sequence derived- BBN analog through Nanothinks acid linkers and subsequently coated it with PEG for increased biocompatibility. The study on T47D cells showed that at the 24 h mark 12% or 50% of GNR-PEG or GNR-BBN-PEG, respectively, was absorbed into the cells, which is a significant improvement and a potential indication of the receptor-mediated nanoformulation internalization. Additionally, 24 h after near-infrared (NIR) laser irradiation of cancer cells incubated with GNR-BBN-PEG, nearly all of them were eradicated, which was not observed for any of the control samples (including cells incubated with GNR-PEG and treated photothermally). Biodistribution analysis in BALB/c mice revealed a doubling of uptake into the tumor tissue and a doubling of the photothermal treatment efficiency for GNR-BBN-PEG intravenously injected mice compared to subjects injected with the BBN-free control. What is important, 15 days after GNR-BBN-PEG and photothermal treatment, 8/10 mice exhibited complete tumor resorption and remained healthy 60 days post-treatment with no carcinoma regrowth, while the entirety of the control group medicated with GNR-PEG was dead by day 45. These results show a substantial improvement in the efficacy and safety of the treatment through the BBN-mediated targeting of GRPRs.
Gold nanorods were also the object of interest in the study carried out by Xu and co-workers [77]. They proposed a novel synergetic strategy combining GRPR-targeted delivery with photothermal effect derived from GNRs and the thermodynamic therapy, based on heat-labile cytotoxic free radical donor (2,2′-azobis[2-(2-imidazoline-2-yl)propane]dihydrochloride - AIPH). Upon heating produced by NIR laser irradiation of GNRs, AIPH generated oxygen-independent radicals in situ, eradicating the cancer cells while sparing healthy tissues exposed only to intact AIPH. Loading the compound to the gold nanostructures was achieved with the mesoporous silica shell deposited on the surface of the GNRs. Studies on the in vitro prostate cancer model (PC-3 cell line) showed highly specific uptake of BBN-modified, mesoporous silica-coated gold nanorods containing AIPH (MGN-A-BBN) and significant apoptosis of the cancer cells upon laser irradiation, resulting from heat and release of free radicals. In the mice model of subcutaneous prostate cancer xenografts, MGN-A-BBN with laser treatment almost completely eradicated the tumors after 21 days of treatment, while the mouse body weight and key blood biochemical parameters remained within a biosafety range - it proves the high efficacy and satisfactory biocompatibility of the MGN-A-BBN in vivo.
All of the presented studies provide an interesting insight into the use of AuNPs of different architectures and modifications for cancer treatment and undoubtedly reveal the importance of BBN in directed drug delivery into carcinomas. Some formulations would, however, require further examinations to establish whether the targeting moiety provides a sufficient anchor for the carrier to eliminate or at least mitigate the nonspecific binding into organs unaffected by the tumor and hence provide a less invasive treatment.
4.3. PLGA nanoparticles
Polymers are widely known for their versatility and impressive variety; therefore, unsurprisingly, they have interested scientists working on drug delivery systems. A broad range of polymers has already been tested and proved to be biocompatible and biodegradable [82,83], and many can be tuned to have stimuli-sensitive properties [84]. Most importantly, they can be combined with anticancer substances and targeting units for effective and safe tumor therapies.
Among biomaterials studied, poly(lactic-co-glycolic) acid nanoparticles seem to be of the greatest interest for the preparation of nanoparticles (NPs) for therapeutics. Kulhari et al. synthesized biodegradable PLGA carriers by a nano-precipitation method and successfully grafted them with BBN through amide bonding (Figure 11) [13]. The PLGA-BBN conjugate exhibited improved stability and aggregation resistance in relation to pure PLGA nanoparticles. Furthermore, the construct durability was examined through BBN release studies and revealed a slow peptide discharge of 6.8% within 1 h (under acceleration conditions), followed by around 12.8% being lost within 24 h, suggesting a satisfactory level of stabilization through covalent bonding. The research continued with experiments on DTX-loaded, BBN-conjugated PLGA particles prepared by the same method, with sodium cholate as the surfactant and BBN conjugated to the surface of already loaded particles. Formulation allowed for a steady drug release over the period of 120 h in contrast to pure drug suspension or Taxotere®, which showed the burst release of the drug within 10 h. Simultaneously, as in the case of most drug-loaded formulations, the initial 24 h was when the greatest amount (~21%) of DTX was released.
Furthermore, a bigger amount (36.2%) of the drug was released into the medium with lower pH (sodium acetate buffer, pH 5) as opposed to PBS with pH 7.4 (31.6%), which appears advantageous for targeted delivery into the tumor, since the pH of cancer environment is known to be acidic [85]. In vitro analysis of the DTX-PLGA-BBN cytotoxicity in GRPR-positive breast cancer cells (MDA-MB-231) indicated a significant improvement in efficacy over the free drug, Taxotere and DTX-PLGA with IC50 values being respectively 35.53 ng/mL for the targeted nanocarriers, 142.24 ng/μl for the non-targeted DTX-loaded NPs and more than 375 ng/μl for DTX and Taxotere®. The above data was very promising for concentrations as low as 3.125 ng/mL, but if the dose is increased, no advantage in toxicity was observed for the BBN-conjugated NPs.
Further studies on DTX-PLGA-BBN nanoparticles were focused specifically on human prostate cancer [75]. Using the same nanoconstructs, scientists investigated DU-145 and PC-3 cells and found IC50 values of 241.89, 126.04, and 77.41 ng/mL for DU-145 cells, respectively for DTX, DTX-PLGA and DTX-PLGA-BBN. In PC-3 cells, the values given for the samples in the same order were 122.95, 74.44 and 45.02 ng/mL, which indicated a clear and substantial increase in cytotoxicity for the examined nanoformulations. Intravenous administration of Taxotere®, DTX-PLGA, or DTX-PLGA-BBN to BALB/c mice (at a dose of 10 mg/kg body weight) revealed slower drug clearance and larger plasma half-life of the NP formulations, but no significant differences between DTX-PLGA or DTX-PLGA-BBN, confirming a similar stability level from the previous studies [14]. Biodistribution analysis indicated a rapid and rather high absorption of the nanoconstructs into the reticuloendothelial organs, with DTX-PLGA and DTX-PLGA-BBN accumulated mostly in the spleen and lungs, as compared to high drug concentrations observed in the kidneys for Taxotere®. Although at the 12 h mark, the DTX concentrations per gram of tissue were mostly below 1%, these results certainly do not seem promising in terms of targeted delivery, and the only advantage indicated by the authors seems to be the DTX absence in the brain during the course of the study.
The use of other polymers, although widely studied for drug delivery, has not been examined to a great degree for GRPR targeting. This might be because liposomes and micelles based on polymers offer better properties for loading, conjugation, or drug release and can be used for numerous therapeutics regardless of their hydrophobicity/hydrophilicity. All the abovementioned studies present some potential for improving tumor-specific drug delivery but unfortunately, none provided sufficiently satisfactory results. In Section 5.3, we showcase other interesting polymeric formulations with theranostic applications. The carrier's architecture was designed to allow particle tracking using various diagnostic modalities and improve drug delivery into cancer cells.
4.4. Other materials and structures
In light of the need for new therapeutic strategies, an out-of-the-box search yields sophisticated formulations brought together by a cross-domain approach. Zhang et al. have developed genetically engineered elastin-like peptides (ELP), with GRPR binding motif included in the N-termini of the peptides (ELP-GRP), expressed by Escherichia coli strain transfected with plasmid vectors prepared with molecular biology techniques [63]. Their peptides were able to form monodisperse temperature-responsive micellar structures with a size around 40-55 nm, capable of encapsulating a hydrophobic fluorescent label, 1-anilinonaphthalene-8-sulfonic acid (1,8-ANS). Confocal microscopy revealed that these structures were efficiently internalized in vitro in GRPR-positive PC-3 and DU-145 cells, but not in GRPR-negative 293T cells; high uptake in prostate cancer cells was mediated by receptor-ligand interaction, as proven by increased Ca2+ release upon incubation of cells with receptor-activating ELP-GRP micelles.
Subsequently, Zhang and colleagues took their ELP nanostructures to the next level and constructed innovative hybrid ELP/liposomal nanocarriers, combining liposomal formulation's advantages with the ELP-GRP targeting potential to form a docetaxel delivery vehicle with high specific binding and improved cytotoxicity [74]. As a result, 40-200nm monodisperse nanoparticles with neutral zeta potential showed high entrapment of DTX and GRP motif on the surface, enhanced specific binding of their hybrid carriers to prostate cancer cells, and not to GRPR-free cells (Figure 12). Moreover, targeting improved the cytotoxicity of the DTX-loaded nanostructures.
Interestingly, the authors have also elaborated on an important factor frequently overlooked, namely the influence of surface charges described by the zeta potential on the fate of the nanoparticles in vivo. In general, the surface charge is desired due to the fact that it stabilizes the nanoparticles in the solution. However, one should keep in mind that particles with negative potential (below -10 mV) would trigger the plasma proteins binding and opsonization, leading to an uptake in the reticuloendothelial system (RES) and fast clearance from the bloodstream. On the other side of the spectrum, particles with high surface potential (more than 10 mV) were found to aggregate in serum. These are the neutral particles (with a surface potential between -10 mV and 10 mV) that possess the most desired in vivo characteristics (least RES interactions, minimized nonspecific uptake, and the longest time of blood circulation), despite the fact that this is not the most desirable characteristics from the colloidal stability point of view.
Synergistic strategies for cancer treatment can also be an important contribution to current therapeutic needs. In research published by Dash and co-workers, a multifunctional system based on the combined action of a magnet, laser, and drug in magnetic reduced graphene oxide was demonstrated [58]. The magnetic nanocomposite (mrGOG) was obtained by conjugation of reduced graphene oxide with magnetic iron oxide nanoparticles, PEG moieties, and bombesin-like GRPR-binding peptide (Figure 13). Following functionalization, the construct was also loaded with DOX using π-π stacking between the drug and graphene rings (mrGOG/DOX). Dual targeting of the construct based on GRPR-binding peptide supported by magnetic targeting led to the successful delivery of DOX to cells, where the drug was successfully released in the endocytic compartments with acidic pH. In combination with NIR laser irradiation, nanoformulation significantly reduced the cell viability of U87 glioblastoma cells in vitro. In vivo studies confirmed these results: mrGOG, combined with magnetic targeting and laser irradiation, significantly slowed cell proliferation and tumor growth in mice bearing tumors with U87 xenograft.
5. Molecular imaging
Table 2 summarizes the contemporary (c.a. 2009 to date) advances in the research concerning molecular imaging applications of nanocarriers functionalized with GRPR- targeting moieties.
5.1. Gold nanomaterials
Gold nanomaterials are of huge importance when it comes to imaging. They have excellent optical properties of light absorbance and light scattering; not only do they work in a visible-light spectrum, but they are also able to operate in the near-infrared range [80]. Furthermore, depending on the extent to which the gold nanoparticles are modified, the electron charge density on the surface of an individual nanoparticle can differ, and this phenomenon is related to the already mentioned feature of AuNPs – surface plasmon resonance (SPR) [90]. This phenomenon leads to the creation of an electromagnetic field at the surface of the gold nanoparticle (Figure 14) and to surface-enhanced optical properties. Because of the high atomic number of gold, Au nanostructures can also find their application as imaging contrast agents. In fact, numerous studies [91,92] demonstrated that these materials can work as contrast agents in photoacoustic imaging [93], or computed tomography (CT), to name but a few. However, upon further functionalization, they may serve as excellent biocompatible carriers in multifunctional imaging nanosystems.
Photoacoustic imaging (PAI) relies on the formation of a sound wave following the absorption of light, hence offering high ultrasonic resolution and strong optical absorption. It was shown previously that gold nanorods, due to very strong light absorption compared to organic dyes, provide potent photoacoustic contrast and boost the diagnostic potential of PAI in vivo [93]. For this reason, Heidari and co-workers investigated bombesin-derivative-functionalized GNRs as potential nanotracers in PAI [40]. Their research proved that their nanomaterial was stable in human blood serum and able to selectively bind with GRPR-positive cells both in vitro in the T47D breast cancer cell line and in vivo in breast cancer tumor xenografts in BALB/c mice. Moreover, they showed that the uptake augmented in time, reaching the maximum signal intensity after 8 hours after intravenous injection.
Chanda and co-workers also focused on GNRs and their great photophysical properties but extended their research to targeting both breast and prostate tumors [88]. Their goal was to design and develop bombesin-conjugated nanoparticles in order to overcome contemporary issues with breast/prostate tumor imaging. To do so, they elaborated on the mechanism of the internalization of targeted nanostructures. The nanorods were of 40×13 nm size and functionalized with various BBN derivative ratios. At the highest level of decoration, the nanomaterial was shown to be chemically stable in various media. Targeted GNRs were effectively taken up by GRPR-positive cells (T47D and PC-3) by means of receptor-mediated endocytosis, but not phagocytosis. This has been proven by performing dark field light-scattering, fluorescence imaging, and transmission electron microscopy (TEM) measurements. When the fluorescence and dark-field scattering images overlapped, it could have been clearly seen that the GNR-BBN carriers are located in the cytosol. This fashion was not observed for GRPR-negative NIH-3T3 cells, where significantly fewer GNRs were internalized, phagocytosis played a major role, and nanostructures were trapped in endosomes.
Later, the same group of researchers investigated the performance of gold nanoparticles modified with different levels of bombesin substitution [68]. Their starch-encapsulated AuNPs were found to be the most stable, with the greatest amount of bombesin attached to the surface. Additionally, when the PC-3 cell binding affinity was tested, it turned out that when more bombesin molecules were attached to AuNPs, a 3.3-fold decrease in the IC50 value (2.45 μg/mL vs 8.10 μg/mL) was observed. In order to assess the in vivo performance, radioactive 198Au was utilized, and the SCID mice were injected with bombesin-altered nanoplatforms. Typical biodistribution was shown, with around 0.5% ID/g accumulation in prostate cancer tumors and promising diminished amassment in the liver despite a slightly bigger particle size than those reported in the earlier literature reports. Nanoparticles were also shown to significantly increase contrast in tumor imaging in CT after intraperitoneal administration of the nanocarrier. The signal was multiple times more intense than required to serve as a prostate cancer CT agent, and it could have been seen to deteriorate only after 6 hours after injection. Surprisingly enough, a very weak signal could still be detected after 48 hours, suggesting some enhanced retention in the tumor.
The great potential of gold nanoparticles as a contrast agent in X-ray imaging was investigated by Salouti and Saghatchi [41]. They found that their PEG-coated AuNPs functionalized with pan-bombesin had very good optical stability when introduced into blood serum, and by performing appropriate binding studies, they have evidenced that their conjugate could selectively target breast cancer tumor tissue (T47D cell line). Furthermore, researchers found out that this formulation could be successfully used in radiology, as the breast tumor in the BALB/c mice could be easily distinguished after 6 hours, and the clear signal was retained up to 8 hours post-injection. Afterward, no obvious accumulation trend is seen, and thus, it suggests that the construct clears from the tumor quite quickly - the signal in the X-ray images becomes somewhat faded. Furthermore, no amassment was observed when it comes to non-targeted NPs, proving the binding specificity of nanovectors.
Pretze and colleagues have exploited ultra-small (3±2 nm) GRPR-targeted AuNPs as biocompatible carriers for fluorescent probes in confocal fluorescence microscopy [86]. Their particles were functionalized with bombesin and the NIR dye - SIDAG. The performance of the particles modified with bombesin showed a good uptake by PC-3 cells when compared to the free targeting ligand. The results of in vitro experiments on prostate cancer cells revealed that the construct provided a good fluorescent signal, and the particles could be nicely visualized after 24 h of incubation. Subsequent in vivo studies on healthy mice, 21 h post-injection showed that the carrier accumulated mainly in the liver and spleen, however when a three-fold higher dose was tested on mice with PC-3 prostate cancer xenografts, it was found that SIDAG-BBN-AuNPs amassed in the tumor mass between 6 and 24 hours and allowed tumor visualization. Moreover, a preliminary in vitro study also proved utility of the system as an efficient contrast agent for CT with good signal-to-noise-ratio, even when the concentration of AuNPs was reduced by a factor of 13 compared to fluorescent imaging.
Following the success of the fluorescent system, Pretze et al. further developed the abovementioned nanosystem to visualize prostate cancer via PET scanning [87]. They again used very small AuNPs and conjugated them with a NIR fluorophore and targeting ligands, although this time, greater emphasis was placed on the functionalization with the chelator (NODAGA). Radiolabeling with 64Cu was achieved with high radiochemical yield and great stability in vitro. The authors provided an interesting explanation regarding the successful targeted delivery of the construct: since elemental copper binds to the DNA, it causes more cell damage than the nanocarrier-bound Cu, which in turn is retained in cell plasma and does not reach the nucleus. Cell viability experiments have shown higher cell toxicity of free Cu compared to Cu-labelled AuNPs, hence the conclusion about the successful targeting of the construct. To confirm in vivo whether the cancer-detecting units allowed visualization of the tumors, nanoplatform was administered intravenously into SCID mice. Distinguishable signals were obtained even 3 hours after injection of the moieties for PET/CT imaging and the prostate tumor could be easily recognized. However, fluorescence imaging did not yield the expected results as the signal was too intense at first, and only after 24 hours after injection could other organs be discerned. The biodistribution studies also confirmed a typical AuNPs demeanor where the moieties accumulated mostly in the liver and spleen.
Imaging of prostate tumors was also the goal of Mendoza-Sánchez and colleagues who wanted to exploit single-photon emission computed tomography (SPECT-CT) [19]. To do so, they synthesized a multifunctional construct based on AuNPs functionalized with HYNIC for 99mTc chelating. Selective binding to PC-3 prostate cancer cells was achieved by H3N+-Lys3-bombesin tethered to the surface gold atoms. Obtained nanovector, of ca. 20 nm size, was proven to be highly stable in human serum, and its radiochemical purity exceeded 95%. It was shown that targeted NPs could accumulate in the tumor 1.7 times better than unfunctionalized ones (36.93% vs 21.58%) in vivo. This trend was confirmed by microSPECT/CT imaging on athymic nude mice prostate with tumor xenografts. Malignant growth could easily be observed as the contrast between the carcinoma, and the surrounding tissues were considerably enhanced after 1 hour post-injection (6.39±0.83% injected activity). Engrossingly, the uptake by the tumor could be noticed as early as 30 minutes after the material administration. When the test was performed again 24 hours after injection - no signal was found in microSPECT/CT - full clearance was demonstrated in less than a day. Nevertheless, not differently than in many other GRPR-targeting projects, the typical accumulation region during this study turned out to be the pancreas (39.83±2.76% injected activity at 1 hour) as there are approximately 7 times more GRPR sites to bind than in prostate tumor.
Still, further elaboration on this construct was shown by Ocampo-García and colleagues – by altering and therefore boosting the current design, they developed the stable kit for 99mTc labeling of a multifunctional receptor-specific system based on gold nanoparticles [18]. The kit comprised two vials: one with a lyophilized chelating moiety (HYNIC derivative) and the second with a 20 nm AuNPs-biomolecule solution; studies demonstrated the 6-month stability of the abovementioned kit. Moreover, the bombesin-functionalized nanosystem obtained from the kit ([99mTc]Tc-AuNP–Lys3-bombesin) showed 1.8 times higher uptake in prostate cancer cells than non-targeted NPs. This fashion was confirmed by conducting in vivo studies, using micro-SPECT/CT imaging; the prostate cancer considerably took up AuNP-based nanosystem. Therefore, the newly synthesized kit was claimed to have well-fitting properties in order to be used as a target-specific agent for molecular imaging of tumors expressing GRPR.
5.2. Iron oxide nanoparticles
Iron oxide nanoparticles (IONPs) are one of the two most important inorganic nanomaterials, next to gold nanoparticles, owing to their outstanding magnetic properties [95]. GRPR-targeted cancer imaging efficiency of iron oxide nanoparticles is being investigated to a slightly lower extent than gold nanoparticles but considering their clinical success [96] they still pose a great importance in contemporary cancer management, especially in bioimaging of malignant tumors. The most commonly used IONPs are SPIONs – superparamagnetic iron oxide nanoparticles. Their most recognizable property is reversible magnetization [97], making them a promising material. When the external magnetic field is applied, SPIONs can be easily guided to the tumor location, and when it is removed, no magnetization is seen. Moreover, SPIONs may supply the negative MRI contrast in T2-weighted magnetic resonance imaging (MRI) [98]. This is particularly important in the intracellular magnetic labeling of tumor cells. When the iron-based nanoparticle is conjugated with the targeting ligand, the specific binding of the probe to the malignant tissue can be visualized in a more efficient manner.
There were many reports on research conducted on the performance of iron oxide nanoparticles in bioimaging. However, the yield of successful operations of them was mostly examined on prostate, breast, and pancreas cancers. Jafari created bombesin-modified SPIONs coated with carboxymethyl dextran for breast cancer magnetic resonance imaging [42]. Their construct was stable in human blood serum up to 24 hours of incubation and not toxic to breast cancer cells (T47D) up to 30 µg/mL. They have demonstrated that their carrier could selectively bind to the GRPR-expressing cells but not to cells lacking the receptor. Furthermore, in vivo MRI experiments in tumor-bearing BALB/c mice revealed favorable properties of their construct regarding the enhancement of the contrast of the tumor imaging with constantly excelling the MRI signal until 13 hours after nanocarrier administration and excreting the manufactured contrast agent after 30 hours. After that, the T2 relaxation time returned to its preinjection level. As a result, the novelty can be used as an attractive material for the T2 MRI contrast agent.
Negative contrast in T2–weighted MR imaging inspired Montet et al. to develop a highly out-of-the-box idea for imaging pancreatic ductal adenocarcinoma [66]. Their inversed approach was quite unique: they used superparamagnetic cross-linked iron oxide (CLIO) nanoparticles functionalized with bombesin to target healthy pancreatic tissue rich in GRPR (in contrast to malignant tissue). Their nanosystem selectively decreased the T2 parameter in the normal pancreas, thus giving rise to the enhancement of the signal shown by the pancreatic tumor when compared to the pre-contrast image. Fluorescent microscopy of cryosections of the organs of interest, performed following MRI, confirmed these finding, therefore proving the material to possess the desired properties for medical tumor imaging.
The performance of iron oxides was also tested by Martin and her colleagues in the management of prostate cancer [43]. They have prepared dextran-coated SPIONs, functionalized with fluorescent rhodamine derivative as well as clickable azides - they have demonstrated click cycloaddition as means of the successful functionalization of the nanocarrier with BBN peptide. Furthermore, cell-binding studies proved that the PC-3 cells can take up newly constructed carriers significantly more efficiently than unfunctionalized SPIONs. As for the fluorescence investigations, only after 2 hours of incubation of the prostate cancer cell line, it could be observed that an intense signal could be detected from PC-3 cytoplasm, reaching over 300% as intense signal as the one obtained from non-targeted NPs. Thus, their nanotransporter can serve as an excellent magnetofluorescent bioimaging probe for prostate carcinoma. Li et al. in turn, exploited SPIONs coated with DSPE-PEG, which provided the means for further functionalization with NIR fluorophore (Cy5) and bombesin analog [56]. The obtained nanomicellar system (Figure 15) was tested for its dual-modality imaging properties in breast cancer diagnosis. Not surprisingly, a strong signal was detected by fluorescence microscopy after the MDA-MB-231 cells were incubated with the nanomaterial in vitro, indicating that the newly synthesized material demonstrated high breast cancer targeting capabilities.
Furthermore, when this GRPR-targeted nanocarrier was injected into the BALB/c nude mice with MDA-MB-231 breast cancer xenografts, high-quality magnetic resonance and NIR fluorescence (NIRF) images of tumors were obtained with excellent contrast. These results prove that the construct is a suitable multimodal imaging probe since the non-targeted samples did not yield any significant improvements in the signal received from either MRI or NIRF imaging.
An unusual method was undertaken by Lee and co-workers where they combined iron oxide nanoparticles with nanostructured glycol chitosan (GC) to create a hybrid nanomaterial (IO-BC-NAHis-GC) for applications in magnetofluorescent imaging of prostate cancer tissue [67]. The polymeric nanostructure of a size range between 35 and 97 nm was obtained by self-assembly and further coupled with a Cy5.5 fluorescent dye. The loading of IONPs was achieved due to hydrophobic interactions between the oleic acid shell of IONPs and the hydrophobic core of the nanostructured GC, and the size range of the final product was 45-90 nm. In vitro studies evinced that those hybrid NPs are prone to bind to GRPR-positive cells. Although MRI was not part of this study, all the conducted imaging studies strongly support the statement that this hybrid material holds great potential in the field of prostate cancer fluorescent and magnetic resonance imaging.
Multimodal imaging properties of SPIONs can also be exploited by adding the radioactive component to the construct. Following this concept, Hajiramezanali and co-workers synthesized chitosan-derivative-coated SPIONs, conjugated with bombesin and isotope chelating DOTA moiety [50]. Radiolabeling of iron oxide nanoparticles with 68Ga rendered the formulation suitable for both MR and PET imaging. The examined product ([68Ga]Ga-DOTA–BN–TMC–MNPs) turned out to have very good activity obtained from gallium radiolabeling with decent biological stability both in vitro and in vivo. Furthermore, it was found that DOTA–BN–TMC–MNPs could be used successfully as a T2 MRI contrast agent for detecting breast cancer in vivo since the concentration of iron at the tumor site was high enough to be discerned. Additionally, PET studies indicated excellent visibility of breast cancer xenografts due to the significant uptake of NPs at that site.
Lahooti and colleagues chose an analogical approach, which proposed ultra-small SPIONs capped with 3-triethoxysilylpropyl succinic anhydride (TEPSA) [33]. In their research, they conjugated the NODAGA chelator and bombesin peptide to PEGylated USPIONs to selectively target breast cancer cells (MDA-MB-231 and MCF-7) and deliver 68Ga-derived activity. The sizes of the obtained nanoparticles did not exceed 24 nm, depending on the compounds coupled with NPs (transmission electron microscopy ~7 nm; dynamic light scattering ~22 nm). This material showed a great affinity for GRPR-positive cells in vitro and increased tumor uptake in vivo upon intravenous injection and hence, leading to improved tumor contrast in MRI images. In fact, the conjugation of the ligands to USPIONs affected the relaxivity – over time after the injection of the nanoformulation, the T2 parameter value in the breast tumor diminished by around 25%. Furthermore, PET/CT imaging 2 hours after injection showed tumor uptake, however, it was observed that, most likely due to the very small size or the protein corona formed over the nanoparticle, it was quickly cleared out by the urinary system.
5.3. Quantum Dots
Quantum Dots (QDs) undoubtedly are instrumental in the imaging of various tumors in contemporary medicine. Relatively high interest in the use of QDs in this area is provoked by the fact that their properties, such as long-lasting photostability, high signal brightness, or the ability of parallel excitation at several wavelengths, are highly favorable in visualizing tumors. So far, in the GRPR-positive malignancies targeting, the biggest number of research is focused on prostate cancer imaging.
One of the compounds suitable for QDs synthesis is CdTe. Hu et al. employed this material to develop ZnS-coated, bombesin-modified QDs (Figure 16) which could be used to visualize the prostate tumor via NIRF imaging [29]. Additionally, they have equipped obtained nanostructures with PET imaging-suitable signal groups containing 18F. In vivo and ex vivo, NIRF studies revealed that the accumulation at the target site proceeds relatively slowly, and the high tumor-to-background ratio (Tu/Mu) was obtained only after 5 hours post the intravenous injection, however, Tu/Mu parameter was still much greater than for the non-targeted quantum dots. When it comes to the assignation of the boundaries between the malignant and healthy tissue by PET imaging, the method turned out to be successful and required only an hour to show the greatest level of radioactivity in the prostate tumor xenograft.
On the contrary, Cai and co-workers investigated orthotopic prostate cancer [16]. They used it to examine the imaging potential of bombesin-functionalized, 64Cu-radiolabeled CuS QDs. By virtue of the smart choice of the particle’s composition, authors obtained simple, chelator-free positron-emitting probes suitable for PET/CT imaging. GRPR-targeting bombesin peptide allowed the selective accumulation of the probe in PC-3-KD1 cancer cells in vitro, as shown in the receptor saturation study. However, the greatest value of this research is focused on the abovementioned model of prostate cancer for examining nanoparticles in vivo performance – subcutaneous xenografts fail in accurately mimicking the tumor microenvironment and vasculature; therefore, orthotopic models provide more relevant experimental conditions. The team synthesized BBN-PEG-[64Cu]CuS NPs that have shown high accumulation at the tumor site even 1 hour after injection in orthotopic prostate cancer model of Nu/Nu mice and allowed high tumor-to-healthy tissue contrast, resulting in the acquisition of exceptionally clear images.
Another interesting take on QDs was shown by Young and Rozengurt – they checked the performance of quantum dots functionalized with targeting ligands using a biotin-streptavidin system [70]. They tethered biotinylated bombesin to the commercially available streptavidin-modified QDs, emitting the fluorescence at 655 nm, and examined them using fluorescence microscopy on bombesin receptor positive Swiss 3T3 cells, bombesin receptor negative Rat-1 cells as well as Rat-1 cells stably transfected with bombesin receptor. The investigations revealed that the conjugation of bombesin peptide to the Qdot 655-streptavidin promotes selective binding of the construct and enables high-quality imaging. Noteworthy, researchers also showed, using another short oligopeptide ligand (biotinylated angiotensin II - ANGII), that conjugation does not influence the intensity of QDs signal, and this nanoprobe is superior to low molecular weight fluorescent dye (Cy3) in terms of photostability, proving QDs to be an efficient tool in molecular imaging.
5.4. Other materials
- Besides the beforementioned groups of particles, scientists explore a variety of other nanosystems to target GRPR in bioimaging applications. Liposomes, graphene derivatives, and biobased structures are among the nanomaterials which are gaining more and more attention from the research community (Figure 17). What is important is that some of those are dedicated not only to managing breast and prostate malignancies but also, for example, head and neck cancer. It clearly shows how broad the spectrum of material science applications in nanomedicine can be.
To begin with, De Barros and colleagues prepared and characterized the liposome containing [99mTc]Tc-BBN(7–14) peptide so as to identify its ability to identify the GRPR-positive Ehrlich tumor [71]. In contrast to most of the research published in the field of cell membrane receptor targeting, in their formulation, BBN ligand was encapsulated inside the liposome in order to protect it from the circulating endopeptidases. Results proved that despite the unusual design, the carrier can be used successfully in diagnostic applications, as shown by the high uptake of radioactivity by the tumor. Scintigraphy images showed a preferential tumor-to-muscle ratio, supporting the tropism of the liposomes towards the malignancy in Swiss mice Ehrlich tumor model. The same research team has also published their results on the performance of the aforementioned liposome in targeting breast adenocarcinoma [73]. Their nanocarrier showed sufficient stability in plasma and relatively long circulation in blood. Not surprisingly, although liposomes displayed renal clearance and reticuloendothelial system uptake, there was a moderate uptake after 1 h in the tumor with no reduction after 4 h, which indicated a successful delivery of radioactivity-carrying peptide to the tumor site with the liposomal carrier. On the other hand, free nonprotected peptide showed a two-fold lower uptake compared to the liposomal formulation, and the ratio of the tumor-to-other tissue was significantly reduced as well. Similarly to the previous report, scintigraphy images and biodistribution studies evidenced satisfying discrimination between healthy and malignant tissue in the bioimaging of breast cancer.
Another very innovative idea was described by Steinmetz et al., who utilized a naturally occurring biobased nanoparticle type, a viral nanoparticle based on cowpea mosaic virus (CPMV), so as to target and visualize prostate cancer [44]. Their construct maintained its structure and integrity upon successful conjugation with a NIR fluorophore (Alexa Fluor 647) and targeting ligand (pan-bombesin analog). Obtained virus-based vehicle (ca. 30 nm) was able to specifically bind to and penetrate the PC-3 cells, contrary to its non-targeted counterpart - both in vitro and in vivo. CPMV nanocarrier could amass in prostate cancer of a 5 mm size, which implies that its use can be efficient in detecting really small malignancies within the body. Furthermore, a clear fluorescence signal coming from the cancer tissue could have been seen only after 2 h post-injection, and the tissue surrounding the tumor displayed a negligible fluorescence intensity, proving that the use of targeted CPMV has great potential. Fluorescent imaging was also within the interests of Li et al., however, they used nano-graphene oxide (NGO) as a carrier for further functionalization- the newly synthesized construct was equipped with a Sta-BBN antagonistic ligand and fluorescent Alexa Fluor 750 (NGO-BBN-AF750) [37]. The thorough studies revealed a high binding affinity and specificity towards oral squamous cell carcinoma (OSCC) HSC-3 cell line and not to GRPR-negative control, human oral kertinocytes HOK cell line. Having performed immunofluorescence studies, the authors obtained high-quality images and thus concluded that NGO-BBN-AF750 is suitable for imaging this kind of tumor.
Although iron oxide is the most popular material for MRI contrast nanoparticles, Cui et al. used a different compound- gadolinium oxide [89]. The primary difference between those two materials is the type of relaxation- while iron provides good contrast for T2-weighed visualization, gadolinium is useful in T1-weighed image acquisition. Nanoparticles obtained by Cui et al. were shown to function as an immensely efficient T1-weighed MRI contrast agent with magnetic properties comparable to the ones of the commercially available gadolinium-based equivalent (Magnevist). Upon functionalization with BBN and carboxyfluorescein (FAM), nanoprobes were providing strong green fluorescence when incubated with PC-3 prostate cancer cells in vitro, while this was not the case for the nanoparticles conjugated only with the FAM. This proved the specificity of binding to the PC-3 cell line in vitro. The desirable behavior of gadolinium oxide-based NPs was also corroborated by in vivo MRI and fluorescent imaging studies. While the strongest signal in magnetic resonance was to be seen 2 h post-injection for bombesin-altered specimens, the non-targeted nanoparticles did not demonstrate any considerable result. This was also verified by fluorescent imaging, where the construct showed a very intensive signal and, thus, confirmed the tendency described above.
Tang and colleagues proposed another multimodal imaging probe to detect and visualize prostate cancer - they used lanthanide oleate complexes as precursors and formed upconversion nanoparticles (UCNPs) based on yttrium, ytterbium, gadolinium, and thulium, capable of absorbing two or more low energy photons and converting them into one emitted photon with higher energy [45]. They modified them with BBN analog (Figure 18) and performed a very broad spectrum of tests, including in vitro and in vivo imaging to examine the targeting abilities of the UCNP-BBN probe. In the in vitro experiments, they used a PC-3 cell line and a GRPR-negative control group. Their findings confirmed the anticipated behavior of UCNPs-BBN, which are selectively bound to prostate cancer cells.
- Moreover, upconversion luminescence (UCL), MRI and CT examinations disclosed that the implementation of the newly synthesized construct greatly enhanced the tumor in vivo visualization in the Nude mice animal model. Interestingly, UCL turned out to be of great importance as the signal could have been observed only 3 minutes post-injection and was still detectable after 4 hours. For magnetic resonance and computed tomography, the signal amelioration was discerned after 4 hours of the administration of UCNPs-BBN, the former method being slightly more efficient as there was a 25% enhancement of MR signal (for CT: 20% signal intensity improvement).
6. Multifunctional particles for theranostic interventions
As already mentioned, theranostic interventions rely on the combination of therapeutic and diagnostic utilities in one nanosystem, as summarized in Table 3, based on the contemporary (c.a. 2009 to date) advances in this field.
Currently, the easiest (but not the only) way to achieve such an effect is to employ radiation derived from radioactive nuclides. Radionanomedicine [81] has become a brand-new field of medical science and is continuously gaining more attention, despite sophisticated requirements associated with appropriate facilities assuring radiation safety. Radionanomedicine takes advantage of already established fundamentals of nuclear medicine and simultaneously exploits the achievements of nanomedicine to deliver maximized performance for contemporary cancer management demands. In theranostic formulations, radioisotopes can play different roles, but practically, in GRPR targeting research one will find mostly two approaches. γ-emitting nuclides, such as 111In or 67Ga, provide means for nanocarrier tracking by SPECT-CT. On the other hand, there are isotopes such as 188Re or, commonly used, 177Lu, which can emit relevant doses of both β- (can cause significant radiobiological effects and lead to cell death) and γ radiation, which makes it the perfect radionuclide for theragnosis.
Usually, the incorporation of radionuclides requires the application of proper complexing moiety. One of the most frequently used chelators is DOTA and its derivatives. Silva et al. designed and synthesized AuNPs tethered with thiolated DOTA (TDOTA) to coordinate Gd3+ and 67Ga3+, potentially useful for MRI and SPECT, respectively [23]. For prostate cancer cells targeting, thioctic acid terminated bombesin (TA-BBN) was ligated to the AuNPs surface. Experiments with PC-3 cell line proved a significant influence of targeting ligands on the construct internalization in vitro during up to 3 hours of incubation. Moreover, upon exposition to 2 Gy of external gamma irradiation clear radiosensitization effect was found, as PC-3 cells incubated with targeted nanoparticles exhibited a significant decrease in cell viability. Biodistribution studies of construct, which was additionally radiolabeled with 67Ga, have shown excellent retention (nearly 80%) of the formulation in BALB/c mice PC-3 xenograft even up to 24 h upon direct intratumoral administration. Authors conclude their research with a remark about the possibility of customizing their Au-based system using other radioisotopes such as 68Ga, 90Y, 177Lu, or 165Er with known anticancer properties, which would improve the system efficacy.
Among Au-systems tested for cancer detection and treatment, some have been developed based on PAMAM dendrimers, with gold particles embedded in the dendritic cavities. For example, Mendoza-Nava et al. effectively prepared [177Lu]Lu-DOTA-PAMAM dendrimers with Au nanoparticles synthesized in situ. Subsequently, they have functionalized their hybrid nanosystem with breast cancer-targeting bombesin, but also with folate, to improve the targeting efficacy ([177Lu]Lu-DenAuNP-folate-bombesin) [48,51]. The in vitro studies performed on T47D breast cancer cell line, rich in both GRPR and folate receptor (FR), clearly showed specificity of the conjugates towards the receptors and presented better cellular uptake for the dendrimeric system as compared to free [177Lu]Lu-folate-bombesin, highlighting the beneficial effect of the carrier. Moreover, upon laser irradiation, DenAuNP-folate-bombesin led to a significant decrease in cell viability (down to 16%), thus exposing an efficient plasmonic photothermal effect. In vivo, in athymic mice breast cancer xenograft model, [177Lu]Lu-DenAuNP-folate-bombesin presented satisfactory radioactivity retention up to 96 h after intratumoral injection and allowed high-quality optical imaging. A similar approach was reported later by Wang and colleagues who also encapsulated AuNPs in the dendrimeric cavity of [177Lu]Lu-PAMAM-bombesin-folate conjugates and used them in the context of lung cancer [49]. Likewise, the presence of gold allowed a great enhancement of the bioimaging results, but also provided an opportunity for photothermal treatment in combination with the radiotoxicity of 177Lu. The researchers found selective absorption into the HEL-299 lung tumor cells overexpressing FRs and GRPRs, a greater carrier uptake than free peptides, and good retention even after 96 h post intratumoral injection.
Owing to their remarkable loading capacity, dendrimers were also the object of interest of Gibbens-Bandala and colleagues, who used them as carriers for a potent anticancer drug, hydrophobic paclitaxel (PTX) [53]. They exploited [177Lu]Lu-DOTA-PAMAM-bombesin for the theranostic delivery of PTX (Figure 19) to GRPR-positive T47D breast cancer cells. Combined radio- and chemotherapy resulted in satisfactory tumor eradication in vivo in athymic mice xenograft model- at the 120 h mark tumor size decreased by 15.6%. Furthermore, upon intratumoral administration, the construct is selectively bound to the receptor-bearing cells, with as much as 36.25% of the formulation retained at the tumor site and only small amounts accumulated in the pancreas and liver. Moreover, the carrier performed very well in terms of micro-SPECT/CT imaging, providing clear images of malignant tissues.
To further explore theranostic PTX delivery, Gibbens-Bandala and colleagues also evaluated PLGA nanocarriers, equipped with BBN and DOTA [52]. Even simple nanoformulation of PTX outperformed free drug in in vitro cytotoxicity assessment, yet its targeted and radioactive counterpart, [177Lu]Lu-BBN-PLGA(PTX), led to the highest observed viability decrease. Authors postulate that PTX delivered to the cells together with radiation led to radiosensitization, and thus the synergistic effect of radio- and chemotherapy can be seen. Biodistribution studies in athymic mice with subcutaneous xenograft of MDA-MB-231 breast cancer cells have shown very promising tumor uptake despite notable liver accumulation. In the pulmonary model of the same malignancy, clear SPECT images of lesions were acquired 72 h post-injection. Most importantly, however, eight days after administration of 3 MBq of [177Lu]Lu-BBN-PLGA(PTX), a more than 10-fold decrease in the tumor volume compared to the control (0.136 cm3 vs 1.83 cm3) was found. All the results taken together confirm the theranostic potential of the proposed PTX formulation.
Targeted delivery of the antineoplastic drug together with radionuclides can be a very promising strategy. It was also exploited by the Accardo team, who, as already mentioned, intensely worked on liposomal formulations. They examined several supramolecular aggregates based on their custom-designed amphiphilic peptide derivative containing both (7-14) BBN targeting moiety and DTPA chelator [34]. Their liposomes were loaded with DOX for prostate cancer treatment and radiolabeled with 111In for diagnostic nuclear medicine applications. Authors have found that they are able to bind to the GRPR receptors of PC-3 prostate cancer cells efficiently and selectively. In the former case, the aggregate retention in the carcinoma after 48 h is higher than for the control, but the improvement is rather minor (2.4% ID/g vs 1.6% ID/g), while the latter formulation provided a substantial carcinoma growth prevention (35%) compared with BBN-free liposomes, pointing to the importance of the targeting effect of the peptide. Furthermore, the DOTA-based liposomes were detectable under a clinical gamma camera, making them potentially applicable for theranostic purposes if the cytotoxicity to cancer cells was to be improved.
Chang et al. established a very different BBN-conjugated liposome system with DSPE-PEG as the base, 188Re radionuclide and DOX [99]. The effects of the [188Re]Re-DOX-liposome-BBN intravenous injection were clearly traceable with micro-SPECT/CT imaging which demonstrated successful targeting of the GRPR in AR42J pancreatic cancer cells and significantly reduced tumor growth rate, hence increasing the mice lifespan by as much as 86.96% compared to individuals treated with [188Re]Re-liposome-BBN or liposome-DOX-BBN whose lifespans were extended by 75% and 3.61% respectively. This is a promising result, particularly because, similarly to other nanocarriers that were not administered directly into the malignancy, some accumulation was also found in the spleen and liver.
Although radioisotopes are very useful in theranostic formulations, they are not the only solution. Last but not least, hybrid nanomaterials containing IONPs may provide convenient means for diagnostic imaging – as already mentioned, they produce efficient contrast in T2-weighted MRI. Bleul et al. reported Pluronic® L-121 delivery vehicles loaded with camptothecin (CPT) and magnetic IONPs with BBN targeting ligands covalently bound to the surface (Figure 20) [20]. The resulting formulation exhibited anticancer properties greatly increased compared to the free drug - MTT viability assay has reduced cell viability to around 20% compared to 40-45% for the free drug. Moreover, the applied polymer allowed for a sustained release of the drug [100]. In addition, the IONPs-containing carriers have shown very promising values of transverse relaxivity: despite the fact that loading with the CPT lowered this parameter in comparison to drug-free nanosystem, the value was still higher than commercially available contrast agent Feridex® (682 s-1 mM-1 vs 394 s-1 mM-1 vs. 111.5 s-1 mM-1, respectively). Unfortunately, the study was not followed-up with an in vivo protocol to assess the bioaccumulation in other organs or tissues and the tumor-targeting effect of these formulations.
7. Summary and conclusions
Cancer continues to be a major challenge for scientists and medical professionals around the world, therefore further development in the field of cancer detection and management remains a constant need. Oncological GRPR targeting represents a promising direction of research, from which the modern cancer management battlefield can benefit a lot. Keeping those facts in mind, this review article gives a general overview of gastrin-releasing peptide receptors, their expression, and biology in malignancies. Further, we have outlined the trends in GRPR targeting research, particularly those concerning the most frequently applied types of nanoparticles and most commonly addressed conditions. Furthermore, we have discussed the types of targeting ligands able to bind with GRPR and the possible rationale behind the current state of the art. It suggests that the future in this field might belong to engineered peptides which confer more than just a binding motif. Moreover, we have elaborated on strategies enabling combining the targeting ligand with the nanoparticle carrier, both covalently and noncovalently.
Last but not least, we have reviewed many research articles reporting cutting-edge methodologies for developing advanced nanoformulations. Described strategies were designed for the treatment, diagnosis as well as simultaneous treatment and diagnosis of various cancers. In both in vitro and in vivo preclinical animal models, most of the presented nanosystems have shown promising performance, i.e., successful targeting of GRPR-expressing cells and delivery of a variety of cargoes. However, one cannot also overlook a very important issue, typical for nanosystems in general, not only GRPR-targeting nanoplatforms, which is accumulation in the liver and spleen in vivo. The extent to which the carrier is retained in those organs differs among the systems, however, is omnipresent once the formulation is administered intravenously. Strategies such as PEGylation significantly ameliorate this issue but do not eliminate it completely. This fact requires further studies and should be kept in mind while developing new nanostrategies for therapeutic and diagnostic targeting of gastrin-releasing peptide receptor or any other molecular targets.
Author Contributions
Conceptualization, B.P.R. and A.P.C.; methodology, B.P.R.; writing—original draft preparation A.P.C., B.P.R., N.G., M.B.; writing—review and editing, B.P.R., A.P.C., P.U., U.K.; visualization; B.P.R., M.B.; supervision A.P.C., P.U., U.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Science Centre, Cracow, Poland (Grant no. 2019/33/B/ST5/02125), and National Centre of Research and Development, Warsaw, Poland, in the framework of the project InterChemMed (POWR.03.02.00–00–I029/16), co–funded by the European Social Fund.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Roser, M.; Ritchie, H. Cancer. Available online: https://ourworldindata.org/cancer (accessed on 14.12.2022).
- Arachchige, M.C.M.; Reshetnyak, Y.K.; Andreev, O.A. Advanced targeted nanomedicine. J. Biotechnol. 2015, 202, 88–97. [Google Scholar] [CrossRef]
- Pooja, D.; Gunukula, A.; Gupta, N.; Adams, D.J.; Kulhari, H. Bombesin receptors as potential targets for anticancer drug delivery and imaging. Int. J. Biochem. Cell Biol. 2019, 114, 105567. [Google Scholar] [CrossRef] [PubMed]
- Jalilian, A.R.; Ocampo-García, B.; Pasanphan, W.; Sakr, T.M.; Melendez-Alafort, L.; Grasselli, M.; Lugao, A.B.; Yousefnia, H.; Dispenza, C.; Janib, S.M.; et al. IAEA Contribution to Nanosized Targeted Radiopharmaceuticals for Drug Delivery. Pharmaceutics 2022, 14(5), 1060. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T. Gastrin-Releasing Peptide. In Encyclopedia of Gastroenterology; Johnson, L.R., Alpers, D.H., Barrett., K.E., Carethers., J.M., Feldman, M., Gores, G.J., Grand, R.J., Kagnoff, M.F., Liddle, R.A., Lu, S., Madara, J.L., II, C.M.M., Margulis, A.R., Williams, J.A., Wilmore, D.W., Wood, J.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 179–184. [Google Scholar]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R. V. International union of pharmacology. LXVIII. Mammalian bombesin receptors: Nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Akeson, M.; Sainz, E.; Mantey, S.A.; Jensen, R.T.; Battey, J.F. Identification of four amino acids in the gastrin-releasing peptide receptor that are required for high affinity agonist binding. J. Biol. Chem. 1997, 272, 17405–17409. [Google Scholar] [CrossRef] [PubMed]
- Tokita, K.; Katsuno, T.; Hocart, S.J.; Coy, D.H.; Llinares, M.; Martinez, J.; Jensen, R.T. Molecular basis for selectivity of high affinity peptide antagonists for the gastrin-releasing peptide receptor. J. Biol. Chem. 2001, 276, 36652–36663. [Google Scholar] [CrossRef] [PubMed]
- UniProt: P30550 GRPR_HUMAN. Available online: https://www.uniprot.org/uniprot/P30550 (accessed on 14.12.2022).
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, 1–16. [Google Scholar] [CrossRef]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef]
- Bombesin acetate salt hydrate. Available online: https://www.sigmaaldrich.com/PL/pl/product/sigma/b4272 (accessed on 14.12.2022).
- Kulhari, H.; Kulhari, D.P.; Singh, M.K.; Sistla, R. Colloidal stability and physicochemical characterization of bombesin conjugated biodegradable nanoparticles. Colloids Surfaces A Physicochem. Eng. Asp. 2014, 443, 459–466. [Google Scholar] [CrossRef]
- Kulhari, H.; Pooja, D.; Shrivastava, S.; V.G.M, N.; Sistla, R. Peptide conjugated polymeric nanoparticles as a carrier for targeted delivery of docetaxel. Colloids Surfaces B Biointerfaces 2014, 117, 166–173. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Pooja, D.; Kulhari, H.; Gudem, S.; Ravuri, H.G.; Bhargava, S.; Ramakrishna, S. Bombesin conjugated solid lipid nanoparticles for improved delivery of epigallocatechin gallate for breast cancer treatment. Chem. Phys. Lipids 2019, 224, 104770. [Google Scholar] [CrossRef]
- Cai, H.; Xie, F.; Mulgaonkar, A.; Chen, L.; Sun, X.; Hsieh, J.T.; Peng, F.; Tian, R.; Li, L.; Wu, C.; et al. Bombesin functionalized 64Cu-copper sulfide nanoparticles for targeted imaging of orthotopic prostate cancer. Nanomedicine 2018, 13, 1695–1705. [Google Scholar] [CrossRef]
- Du, J.; Li, L. Which one performs better for targeted lung cancer combination therapy: pre- or post-bombesin-decorated nanostructured lipid carriers? Drug Deliv. 2016, 23, 1799–1809. [Google Scholar] [CrossRef]
- Ocampo-García, B.; Ferro-Flores, G.; Morales-Avila, E.; Ramírez, F.D.M. Kit for preparation of multimeric receptor-specific 99mTc- radiopharmaceuticals based on gold nanoparticles. Nucl. Med. Commun. 2011, 32, 1095–1104. [Google Scholar] [CrossRef]
- Mendoza-Sánchez, A.N.; Ferro-Flores, G.; Ocampo-García, B.E.; Morales-Avila, E.; Ramírez, F.D.M.; De León-Rodríguez, L.M.; Santos-Cuevas, C.L.; Medina, L.A.; Rojas-Calderón, E.L.; Camacho-López, M.A. Lys3-bombesin conjugated to 99mTc-labelled gold nanoparticles for in vivo gastrin releasing peptide-receptor imaging. J. Biomed. Nanotechnol. 2010, 6, 375–384. [Google Scholar] [CrossRef]
- Bleul, R.; Thiermann, R.; Marten, G.U.; House, M.J.; Pierre, T.G.S.; Häfeli, U.O.; Maskos, M. Continuously manufactured magnetic polymersomes-a versatile tool (not only) for targeted cancer therapy. Nanoscale 2013, 5, 11385–11393. [Google Scholar] [CrossRef]
- [Lys3]-Bombesin. Available online: https://www.sigmaaldrich.com/PL/pl/product/sigma/b1647 (accessed on 14.12.2022).
- Suresh, D.; Zambre, A.; Chanda, N.; Hoffman, T.J.; Smith, C.J.; Robertson, J.D.; Kannan, R. Bombesin peptide conjugated gold nanocages internalize via clathrin mediated endocytosis. Bioconjug. Chem. 2014, 25, 1565–1579. [Google Scholar] [CrossRef]
- Silva, F.; Paulo, A.; Pallier, A.; Même, S.; Tóth, É.; Gano, L.; Marques, F.; Geraldes, C.F.G.C.; Castro, M.M.C.A.; Cardoso, A.M.; et al. Dual imaging gold nanoplatforms for targeted radiotheranostics. Materials (Basel). 2020, 13, 1–17. [Google Scholar] [CrossRef]
- Silva, F.; Gano, L.; Cabral Campello, M.P.; Marques, R.; Prudêncio, I.; Zambre, A.; Upendran, A.; Paulo, A.; Kannan, R. In vitro / in vivo “peeling” of multilayered aminocarboxylate gold nanoparticles evidenced by a kinetically stable 99mTc-label. Dalt. Trans. 2017, 46, 14572–14583. [Google Scholar] [CrossRef] [PubMed]
- Chilug, L.E.; Niculae, D.; Leonte, R.A.; Nan, A.; Turcu, R.; Mustaciosu, C.; Serban, R.M.; Lavric, V.; Manda, G. Preclinical evaluation of NHS-activated gold nanoparticles functionalized with bombesin or neurotensin-like peptides for targeting colon and prostate tumours. Molecules 2020, 25, 3363. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Galli, F.; Mansi, R.; Del Pozzo, L.; Aurilio, M.; Morisco, A.; Ringhieri, P.; Signore, A.; Morelli, G.; Aloj, L. Pre-clinical evaluation of eight DOTA coupled gastrin-releasing peptide receptor (GRP-R) ligands for in vivo targeting of receptor-expressing tumors. EJNMMI Res. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Matusiak, M.; Rurarz, B.P.; Kadłubowski, S.; Wolszczak, M.; Karczmarczyk, U.; Maurin, M.; Kolesińska, B.; Ulański, P. Synthesis and properties of targeted radioisotope carriers based on poly(acrylic acid) nanogels. Pharmaceutics 2021, 13. [Google Scholar] [CrossRef]
- Santos-Cuevas, C.L.; Ferro-Flores, G.; Arteaga de Murphy, C.; Ramírez, F. de M.; Luna-Gutiérrez, M.A.; Pedraza-López, M.; García-Becerra, R.; Ordaz-Rosado, D. Design, preparation, in vitro and in vivo evaluation of 99mTc-N2S2-Tat(49-57)-bombesin: A target-specific hybrid radiopharmaceutical. Int. J. Pharm. 2009, 375, 75–83. [Google Scholar] [CrossRef]
- Hu, K.; Wang, H.; Tang, G.; Huang, T.; Tang, X.; Liang, X.; Yao, S.; Nie, D. In vivo cancer dual-targeting and dual-modality imaging with functionalized quantum dots. J. Nucl. Med. 2015, 56, 1278–1284. [Google Scholar] [CrossRef]
- Trujillo-Benítez, D.; Ferro-Flores, G.; Morales-Avila, E.; Jiménez-Mancilla, N.; Ancira-Cortez, A.; Ocampo-García, B.; Santos-Cuevas, C.; Escudero-Castellanos, A.; Luna-Gutiérrez, M.; Azorín-Vega, E. Synthesis and biochemical evaluation of samarium-153 oxide nanoparticles functionalized with iPSMA-bombesin heterodimeric peptide. J. Biomed. Nanotechnol. 2020, 16, 689–701. [Google Scholar] [CrossRef]
- Jimenez-Mancilla, N.; Ferro-Flores, G.; Ocampo-Garcia, B.; Luna-Gutierrez, M.; De Maria Ramirez, F.; Pedraza-Lopez, M.; Torres-Garcia, E. Multifunctional targeted radiotherapy system for induced tumours expressing gastrin-releasing peptide receptors. Curr. Nanosci. 2012, 8, 193–201. [Google Scholar] [CrossRef]
- Jiménez-Mancilla, N.; Ferro-Flores, G.; Santos-Cuevas, C.; Ocampo-García, B.; Luna-Gutiérrez, M.; Azorín-Vega, E.; Isaac-Olivé, K.; Camacho-López, M.; Torres-García, E. Multifunctional targeted therapy system based on 99mTc/ 177Lu-labeled gold nanoparticles-Tat(49-57)-Lys3-bombesin internalized in nuclei of prostate cancer cells. J. Label. Compd. Radiopharm. 2013, 56, 663–671. [Google Scholar] [CrossRef]
- Lahooti, A.; Shanehsazzadeh, S.; Laurent, S. Preliminary studies of 68Ga-NODA-USPION-BBN as a dual-modality contrast agent for use in positron emission tomography/magnetic resonance imaging. Nanotechnology 2020, 31, 015102. [Google Scholar] [CrossRef]
- Accardo, A.; Mansi, R.; Salzano, G.; Morisco, A.; Aurilio, M.; Parisi, A.; Maione, F.; Cicala, C.; Ziaco, B.; Tesauro, D.; et al. Bombesin peptide antagonist for target-selective delivery of liposomal doxorubicin on cancer cells. J. Drug Target. 2013, 21, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Ringhieri, P.; Iannitti, R.; Nardon, C.; Palumbo, R.; Fregona, D.; Morelli, G.; Accardo, A. Target selective micelles for bombesin receptors incorporating Au(III)-dithiocarbamato complexes. Int. J. Pharm. 2014, 473, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Mannucci, S.; Nicolato, E.; Vurro, F.; Diaferia, C.; Bontempi, P.; Marzola, P.; Morelli, G. Easy formulation of liposomal doxorubicin modified with a bombesin peptide analogue for selective targeting of GRP receptors overexpressed by cancer cells. Drug Deliv. Transl. Res. 2019, 9, 215–226. [Google Scholar] [CrossRef]
- Li, R.; Gao, R.; Wang, Y.; Liu, Z.; Xu, H.; Duan, A.; Zhang, F.; Ma, L. Gastrin releasing peptide receptor targeted nano-graphene oxide for near-infrared fluorescence imaging of oral squamous cell carcinoma. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Tagliavini, V.; Honisch, C.; Serratì, S.; Azzariti, A.; Bonchio, M.; Ruzza, P.; Carraro, M. Enhancing the biological activity of polyoxometalate-peptide nano-fibrils by spacer design. RSC Adv. 2021, 11, 4952–4957. [Google Scholar] [CrossRef]
- Heidari, Z.; Salouti, M.; Sariri, R. Breast cancer photothermal therapy based on gold nanorods targeted by covalently-coupled bombesin peptide. Nanotechnology 2015, 26, 195101. [Google Scholar] [CrossRef]
- Heidari, Z.; Sariri, R.; Salouti, M. Gold nanorods-bombesin conjugate as a potential targeted imaging agent for detection of breast cancer. J. Photochem. Photobiol. B Biol. 2014, 130, 40–46. [Google Scholar] [CrossRef]
- Salouti, M.; Saghatchi, F. BBN conjugated GNPs: a new targeting contrast agent for imaging of breast cancer in radiology. IET Nanobiotechnology 2017, 11, 604–611. [Google Scholar] [CrossRef]
- Jafari, A.; Salouti, M.; Shayesteh, S.F.; Heidari, Z.; Rajabi, A.B.; Boustani, K.; Nahardani, A. Synthesis and characterization of Bombesin-superparamagnetic iron oxide nanoparticles as a targeted contrast agent for imaging of breast cancer using MRI. Nanotechnology 2015, 26, 075101. [Google Scholar] [CrossRef]
- Martin, A.L.; Hickey, J.L.; Ablack, A.L.; Lewis, J.D.; Luyt, L.G.; Gillies, E.R. Synthesis of bombesin-functionalized iron oxide nanoparticles and their specific uptake in prostate cancer cells. J. Nanoparticle Res. 2010, 12, 1599–1608. [Google Scholar] [CrossRef]
- Steinmetz, N.F.; Ablack, A.L.; Hickey, J.L.; Ablack, J.; Manocha, B.; Mymryk, J.S.; Luyt, L.G.; Lewis, J.D. Intravital imaging of human prostate cancer using viral nanoparticles targeted to gastrin-releasing peptide receptors. Small 2011, 7, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.H.; Wang, J.; Yang, C.X.; Dong, L.X.; Kong, D.; Yan, X.P. Ultrasonic assisted preparation of lanthanide-oleate complexes for the synthesis of multifunctional monodisperse upconversion nanoparticles for multimodal imaging. Nanoscale 2014, 6, 8037–8044. [Google Scholar] [CrossRef] [PubMed]
- Mansour, N.; Paquette, M.; Ait-Mohand, S.; Dumulon-Perreault, V.; Guérin, B. Evaluation of a novel GRPR antagonist for prostate cancer PET imaging: [64Cu]-DOTHA2-PEG-RM26. Nucl. Med. Biol. 2018, 56, 31–38. [Google Scholar] [CrossRef]
- Tangthong, T.; Piroonpan, T.; Thipe, V.C.; Khoobchandani, M.; Katti, K.; Katti, K. V.; Pasanphan, W. Bombesin peptide conjugated water-soluble chitosan gallate—a new nanopharmaceutical architecture for the rapid one-pot synthesis of prostate tumor targeted gold nanoparticles. Int. J. Nanomedicine 2021, 16, 6957–6981. [Google Scholar] [CrossRef]
- Mendoza-Nava, H.; Ferro-Flores, G.; Ramírez, F.D.M.; Ocampo-García, B.; Santos-Cuevas, C.; Aranda-Lara, L.; Azorín-Vega, E.; Morales-Avila, E.; Isaac-Olivé, K. 177Lu-dendrimer conjugated to folate and bombesin with gold nanoparticles in the dendritic cavity: a potential theranostic radiopharmaceutical. J. Nanomater. 2016, 2016, ID 1039258. [Google Scholar] [CrossRef]
- Wang, Z.; Ye, M.; Ma, D.; Shen, J.; Fang, F. Engineering of 177Lu-labeled gold encapsulated into dendrimeric nanomaterials for the treatment of lung cancer. J. Biomater. Sci. Polym. Ed. 2022 2022, 33, 197–211. [Google Scholar] [CrossRef]
- Hajiramezanali, M.; Atyabi, F.; Mosayebnia, M.; Akhlaghi, M.; Geramifar, P.; Jalilian, A.R.; Mazidi, S.M.; Yousefnia, H.; Shahhosseini, S.; Beiki, D. (68)Ga-radiolabeled bombesin-conjugated to trimethyl chitosan-coated superparamagnetic nanoparticles for molecular imaging: preparation, characterization and biological evaluation. Int. J. Nanomedicine 2019, 14, 2591–2605. [Google Scholar] [CrossRef]
- Mendoza-Nava, H.; Ferro-Flores, G.; Ramírez, F. de M.; Ocampo-García, B.; Santos-Cuevas, C.; Azorín-Vega, E.; Jiménez-Mancilla, N.; Luna-Gutiérrez, M.; Isaac-Olivé, K. Fluorescent, plasmonic, and radiotherapeutic properties of the 177Lu–dendrimer-AuNP–folate–bombesin nanoprobe located inside cancer cells. Mol. Imaging 2017, 16, 153601211770476. [Google Scholar] [CrossRef] [PubMed]
- Gibbens-Bandala, B.; Morales-Avila, E.; Ferro-Flores, G.; Santos-Cuevas, C.; Meléndez-Alafort, L.; Trujillo-Nolasco, M.; Ocampo-García, B. 177Lu-Bombesin-PLGA (paclitaxel): A targeted controlled-release nanomedicine for bimodal therapy of breast cancer. Mater. Sci. Eng. C 2019, 105, 110043. [Google Scholar] [CrossRef]
- Gibbens-Bandala, B.; Morales-Avila, E.; Ferro-Flores, G.; Santos-Cuevas, C.; Luna-Gutiérrez, M.; Ramírez-Nava, G.; Ocampo-García, B. Synthesis and evaluation of 177Lu-DOTA-DN(PTX)-BN for selective and concomitant radio and drug-therapeutic effect on breast cancer cells. Polymers (Basel). 2019, 11, 1572. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Xu, R.; Wu, X.; Gillespie, D.; Jensen, R.; Lu, Z.R. Targeted systemic delivery of a therapeutic siRNA with a multifunctional carrier controls tumor proliferation in mice. Mol. Pharm. 2009, 6, 738–746. [Google Scholar] [CrossRef]
- Akbar, M.J.; Ferreira, P.C.L.; Giorgetti, M.; Stokes, L.; Morris, C.J. Bombesin receptor-targeted liposomes for enhanced delivery to lung cancer cells. Beilstein J. Nanotechnol. 2019, 10, 2553–2562. [Google Scholar] [CrossRef]
- Li, L.; Wu, C.; Pan, L.; Li, X.; Kuang, A.; Cai, H.; Tian, R. Bombesin-functionalized superparamagnetic iron oxide nanoparticles for dual-modality MR/NIRFI in mouse models of breast cancer. Int. J. Nanomedicine 2019, Volume 14, 6721–6732. [Google Scholar] [CrossRef]
- Hussein, W.M.; Cheong, Y.S.; Liu, C.; Liu, G.; Begum, A.A.; Attallah, M.A.; Moyle, P.M.; Torchilin, V.P.; Smith, R.; Toth, I. Peptide-based targeted polymeric nanoparticles for siRNA delivery. Nanotechnology 2019, 30, 415604. [Google Scholar] [CrossRef] [PubMed]
- Dash, B.S.; Lu, Y.-J.; Chen, H.-A.; Chuang, C.-C.; Chen, J.-P. Magnetic and GRPR-targeted reduced graphene oxide/doxorubicin nanocomposite for dual-targeted chemo-photothermal cancer therapy. Mater. Sci. Eng. C 2021, 128, 112311. [Google Scholar] [CrossRef]
- Wang, C.; Sun, X.; Wang, K.; Wang, Y.; Yang, F.; Wang, H. Breast cancer targeted chemotherapy based on doxorubicin-loaded bombesin peptide modified nanocarriers. Drug Deliv. 2016, 23, 2697–2702. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Mansi, R.; Morisco, A.; Mangiapia, G.; Paduano, L.; Tesauro, D.; Radulescu, A.; Aurilio, M.; Aloj, L.; Arra, C.; et al. Peptide modified nanocarriers for selective targeting of bombesin receptors. Mol. Biosyst. 2010, 6, 878–887. [Google Scholar] [CrossRef]
- Kanazawa, T.; Taki, H.; Okada, H. Nose-to-brain drug delivery system with ligand/cell-penetrating peptide-modified polymeric nano-micelles for intracerebral gliomas. Eur. J. Pharm. Biopharm. 2020, 152, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Neef, T.; Mittelholzer, C.; Garcia Garayoa, E.; Bläuenstein, P.; Schibli, R.; Aebi, U.; Burkhard, P. The biodistribution of self-assembling protein nanoparticles shows they are promising vaccine platforms. J. Nanobiotechnology 2013, 11, 1–10. [Google Scholar] [CrossRef]
- Zhang, W.; Garg, S.; Eldi, P.; Zhou, F.H. huan; Johnson, I.R.D.; Brooks, D.A.; Lam, F.; Rychkov, G.; Hayball, J.; Albrecht, H. Targeting prostate cancer cells with genetically engineered polypeptide-based micelles displaying gastrin-releasing peptide. Int. J. Pharm. 2016, 513, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Achilli, E.; Flores, C.Y.; Temprana, C.F.; Alonso, S. del V.; Radrizzani, M.; Grasselli, M. Enhanced gold nanoparticle-tumor cell recognition by albumin multilayer coating. OpenNano 2021, 1–12. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.S.; Min, K.H.; Kim, Y.-H.; Chen, X.; Kim, J.; Kim, J.S.; Min, K.H.; Kim, Y.-H.; Chen, X. Bombesin-tethered reactive oxygen species (ROS)-responsive nanoparticles for monomethyl auristatin F (MMAF) delivery. Bioeng. 2021, 8, 43. [Google Scholar] [CrossRef]
- Montet, X.; Weissleder, R.; Josephson, L. Imaging pancreatic cancer with a peptide-nanoparticle conjugate targeted to normal pancreas. Bioconjug. Chem. 2006, 17, 905–91. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Jeong, H.J.; Cheong, S.J.; Kim, E.M.; Kim, D.W.; Lim, S.T.; Sohn, M.H. Prostate cancer-targeted imaging using magnetofluorescent polymeric nanoparticles functionalized with bombesin. Pharm. Res. 2010, 27, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Chanda, N.; Kattumuri, V.; Shukla, R.; Zambre, A.; Katti, K.; Upendran, A.; Kulkarni, R.R.; Kan, P.; Fent, G.M.; Casteel, S.W.; et al. Bombesin functionalized gold nanoparticles show in vitro and in vivo cancer receptor specificity. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8760–8765. [Google Scholar] [CrossRef] [PubMed]
- Hosta-Rigau, L.; Olmedo, I.; Arbiol, J.; Cruz, L.J.; Kogan, M.J.; Albericio, F. Multifunctionalized gold nanoparticles with peptides targeted to gastrin-releasing peptide receptor of a tumor cell line. Bioconjug. Chem. 2010, 21, 1070–1078. [Google Scholar] [CrossRef]
- Young, S.H.; Rozengurt, E. Qdot Nanocrystal Conjugates conjugated to bombesin or ANG II label the cognate G protein-coupled receptor in living cells. Am. J. Physiol. - Cell Physiol. 2006, 290, 728–732. [Google Scholar] [CrossRef] [PubMed]
- De Barros, A.L.B.; Mota, L.D.G.; Soares, D.C.F.; Coelho, M.M.A.; Oliveira, M.C.; Cardoso, V.N. Tumor bombesin analog loaded long-circulating and pH-sensitive liposomes as tool for tumor identification. Bioorg. Med. Chem. Lett. 2011, 21, 7373–7375. [Google Scholar] [CrossRef]
- De Barros, A.L.B.; Mota, L.D.G.; Soares, D.C.F.; De Souza, C.M.; Cassali, G.D.; Oliveira, M.C.; Cardoso, V.N. Long-circulating, pH-sensitive liposomes versus long-circulating, non-pH-sensitive liposomes as a delivery system for tumor identification. J. Biomed. Nanotechnol. 2013, 9, 1636–1643. [Google Scholar] [CrossRef]
- De Barros, A.L.B.; Das Graças Mota, L.; Coelho, M.M.A.; Corrêa, N.C.R.; De Góes, A.M.; Oliveira, M.C.; Cardoso, V.N. Bombesin encapsulated in long-circulating pH-sensitive liposomes as a radiotracer for breast tumor identification. J. Biomed. Nanotechnol. 2015, 11, 342–350. [Google Scholar] [CrossRef]
- Zhang, W.; Song, Y.; Eldi, P.; Guo, X.; Hayball, J.D.; Garg, S.; Albrecht, H. Targeting prostate cancer cells with hybrid elastin-like polypeptide/liposome nanoparticles. Int. J. Nanomedicine 2018, 13, 293–305. [Google Scholar] [CrossRef]
- Kulhari, H.; Pooja, D.; Singh, M.K.; Kuncha, M.; Adams, D.J.; Sistla, R. Bombesin-conjugated nanoparticles improve the cytotoxic efficacy of docetaxel against gastrin-releasing but androgen-independent prostate cancer. Nanomedicine 2015, 10, 2847–2859. [Google Scholar] [CrossRef]
- Tangthong, T.; Piroonpan, T.; Thipe, V.C.; Khoobchandani, M.; Katti, K.; Katti, K. V.; Pasanphan, W. Water-soluble chitosan conjugated DOTA-bombesin peptide capped gold nanoparticles as a targeted therapeutic agent for prostate cancer. Nanotechnol. Sci. Appl. 2021, 14, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Sheng, G.; Lu, L.; Wang, C.; Zhang, Y.; Feng, L.; Meng, L.; Min, P.; Zhang, L.; Wang, Y.; et al. GRPr-mediated photothermal and thermodynamic dual-therapy for prostate cancer with synergistic anti-apoptosis mechanism. Nanoscale 2021, 13, 4249–4261. [Google Scholar] [CrossRef] [PubMed]
- Engelberth, S.A.; Hempel, N.; Bergkvist, M. Development of nanoscale approaches for ovarian cancer therapeutics and diagnostics. Crit. Rev. Oncog. 2014, 19, 281–316. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, I.; Yasir, M.; Verma, M.; Singh, A.P. Nanostructured lipid carriers: a groundbreaking approach for transdermal drug delivery. Adv. Pharm. Bull. 2020, 10, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Chatterjee, D.K.; Lee, M.H.; Krishnan, S. Gold nanoparticles in breast cancer treatment: promise and potential pitfalls. Cancer Lett. 2014, 347, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S. Radionanomedicine. Combined Nuclear and Nanomedicine, 1st ed.; Lee, D.S., Ed.; Springer International Publishing AG: Cham, Switzerland, 2018. [Google Scholar]
- Vroman, I.; Tighzert, L. Biodegradable polymers. Materials (Basel). 2009, 2, 307–344. [Google Scholar] [CrossRef]
- Chandra, R.; Rustgi, R. Biodegradable polymers. Prog. Polym. Sci. 1998, 23, 1273–1335. [Google Scholar] [CrossRef]
- Efthimiadou, E.K.; Theodosiou, M.; Toniolo, G.; Abu-Thabit, N.Y. 15 - Stimuli-responsive biopolymer nanocarriers for drug delivery applications. In Woodhead Publishing Series in Biomaterials; Makhlouf, A.S.H., Abu-Thabit, N.Y., Eds.; Woodhead Publishing: Sawston, Great Britain, 2018; pp. 405–432. [Google Scholar]
- Lee, S.; Shanti, A. Effect of exogenous pH on cell growth of breast cancer cells. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Pretze, M.; Hien, A.; Rädle, M.; Schirrmacher, R.; Wängler, C.; Wängler, B. Gastrin-releasing peptide receptor- and prostate-specific membrane antigen-specific ultrasmall gold nanoparticles for characterization and diagnosis of prostate carcinoma via fluorescence imaging. Bioconjug. Chem. 2018, 29, 1525–1533. [Google Scholar] [CrossRef]
- Pretze, M.; van der Meulen, N.P.; Wängler, C.; Schibli, R.; Wängler, B. Targeted 64Cu-labeled gold nanoparticles for dual imaging with positron emission tomography and optical imaging. J. Label. Compd. Radiopharm. 2019, 62, 471–482. [Google Scholar] [CrossRef]
- Chanda, N.; Shukla, R.; Katti, K. V.; Kannan, R. Gastrin releasing protein receptor specific gold nanorods: breast and prostate tumor avid nanovectors for molecular imaging. Nano Lett. 2009, 9, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Lu, X.; Yan, C.; Liu, X.; Hou, M.; Xia, Q.; Xu, Y.; Liu, R. Gastrin-releasing peptide receptor-targeted gadolinium oxide-based multifunctional nanoparticles for dual magnetic resonance/fluorescent molecular imaging of prostate cancer. Int. J. Nanomedicine 2017, 12, 6787–6797. [Google Scholar] [CrossRef] [PubMed]
- Sztandera, K.; Gorzkiewicz, M.; Klajnert-Maculewicz, B. Gold Nanoparticles in cancer treatment. Mol. Pharm. 2019, 16, 1–23. [Google Scholar] [CrossRef]
- Mahan, M.M.; Doiron, A.L. Gold Nanoparticles as X-Ray, CT, and Multimodal imaging contrast agents: formulation, targeting, and methodology. J. Nanomater. 2018, 2018, ID 5837276. [Google Scholar] [CrossRef]
- Cheheltani, R.; Ezzibdeh, R.M.; Chhour, P.; Pulaparthi, K.; Kim, J.; Jurcova, M.; Hsu, J.C.; Blundell, C.; Litt, H.I.; Ferrari, V.A.; et al. Tunable, biodegradable gold nanoparticles as contrast agents for computed tomography and photoacoustic imaging. Biomaterials 2016, 102, 87–97. [Google Scholar] [CrossRef]
- Jokerst, J. V.; Cole, A.J.; Van De Sompel, D.; Gambhir, S.S. Gold nanorods for ovarian cancer detection with photoacoustic imaging and resection guidance via Raman imaging in living mice. ACS Nano 2012, 6, 10366–10377. [Google Scholar] [CrossRef]
- Fuller, M.A.; Köper, I. Biomedical applications of polyelectrolyte coated spherical gold nanoparticles. Nano Converg. 2019, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Gotman, I.; Psakhie, S.G.; Lozhkomoev, A.S.; Gutmanas, E.Y. Iron oxide and gold nanoparticles in cancer therapy. AIP Conf. Proc. 2016, 1760, 20020. [Google Scholar] [CrossRef]
- Dadfar, S.M.; Roemhild, K.; Drude, N.I.; von Stillfried, S.; Knüchel, R.; Kiessling, F.; Lammers, T. Iron oxide nanoparticles: diagnostic, therapeutic and theranostic applications. Adv. Drug Deliv. Rev. 2019, 138, 302–325. [Google Scholar] [CrossRef]
- Wei, H.; Hu, Y.; Wang, J.; Gao, X.; Qian, X.; Tang, M. Superparamagnetic iron oxide nanoparticles: cytotoxicity, metabolism, and cellular behavior in biomedicine applications. Int. J. Nanomedicine 2021, 16, 6097–6113. [Google Scholar] [CrossRef]
- Yen, S.K.; Padmanabhan, P.; Selvan, S.T. Multifunctional iron oxide nanoparticles for diagnostics, therapy and macromolecule delivery. Theranostics 2013, 3, 986–1003. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-J.; Yu, C.-Y.; Hsu, C.-W.; Lee, W.-C.; Chen, S.-J.; Chang, C.-H.; Lee, T.-W. Molecular imaging and therapeutic efficacy of 188Re-(DXR)-liposome-BBN in AR42J pancreatic tumor-bearing mice. Oncol. Rep. 2012, 28, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Bleul, R.; Thiermann, R.; Saatchi, K.; Häfeli, U.O.; Maskos, M. Multifunctional nanocarriers for biomedical applications. Colloid. Nanocrystals Biomed. Appl. VIII 2013, 8595, 85951N. [Google Scholar] [CrossRef]
Figure 1.
Structure of GRPR with seven transmembrane domains [9].
Figure 1.
Structure of GRPR with seven transmembrane domains [9].
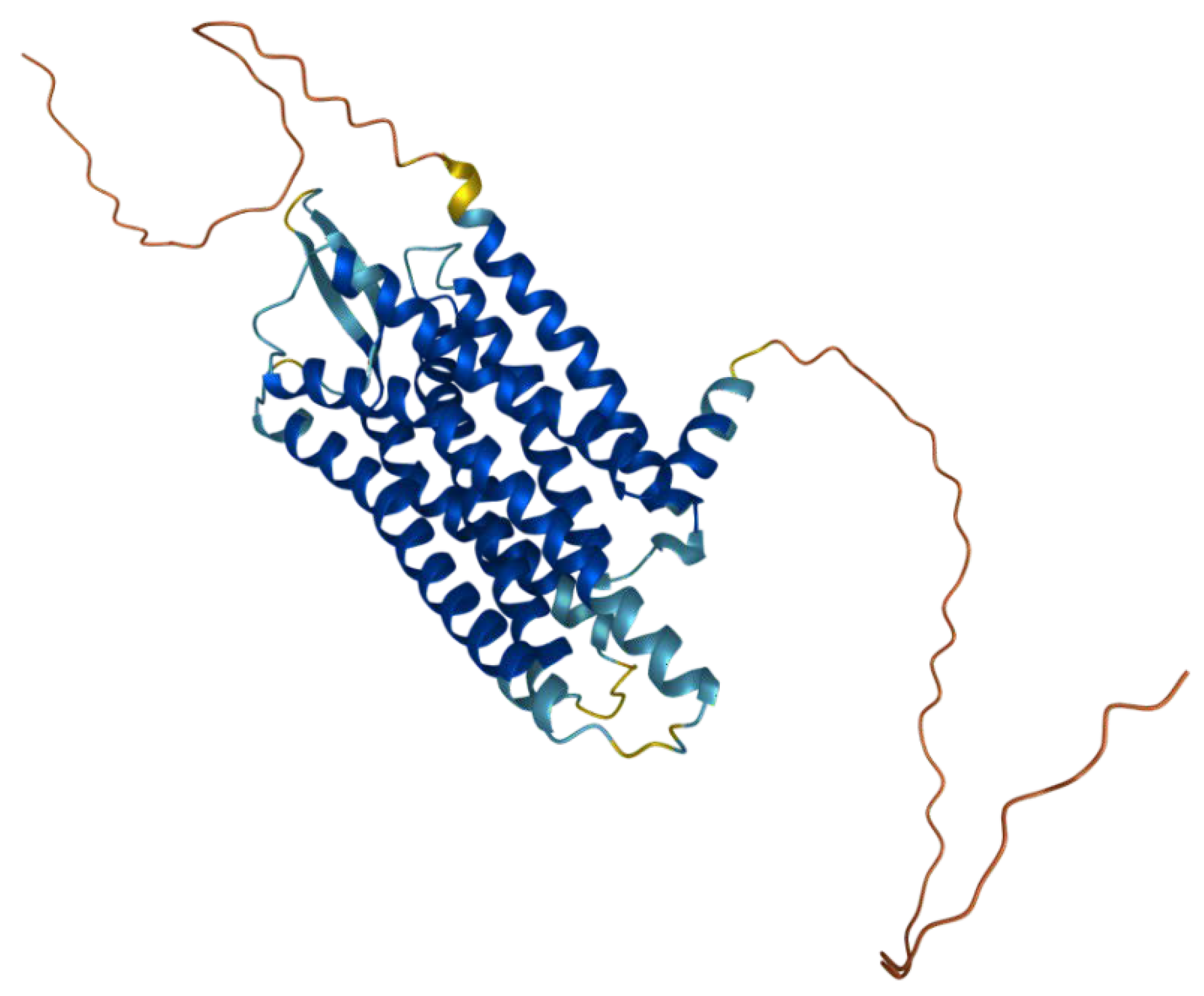
Figure 2.
Overview of GRPR signaling. Created with BioRender.com.
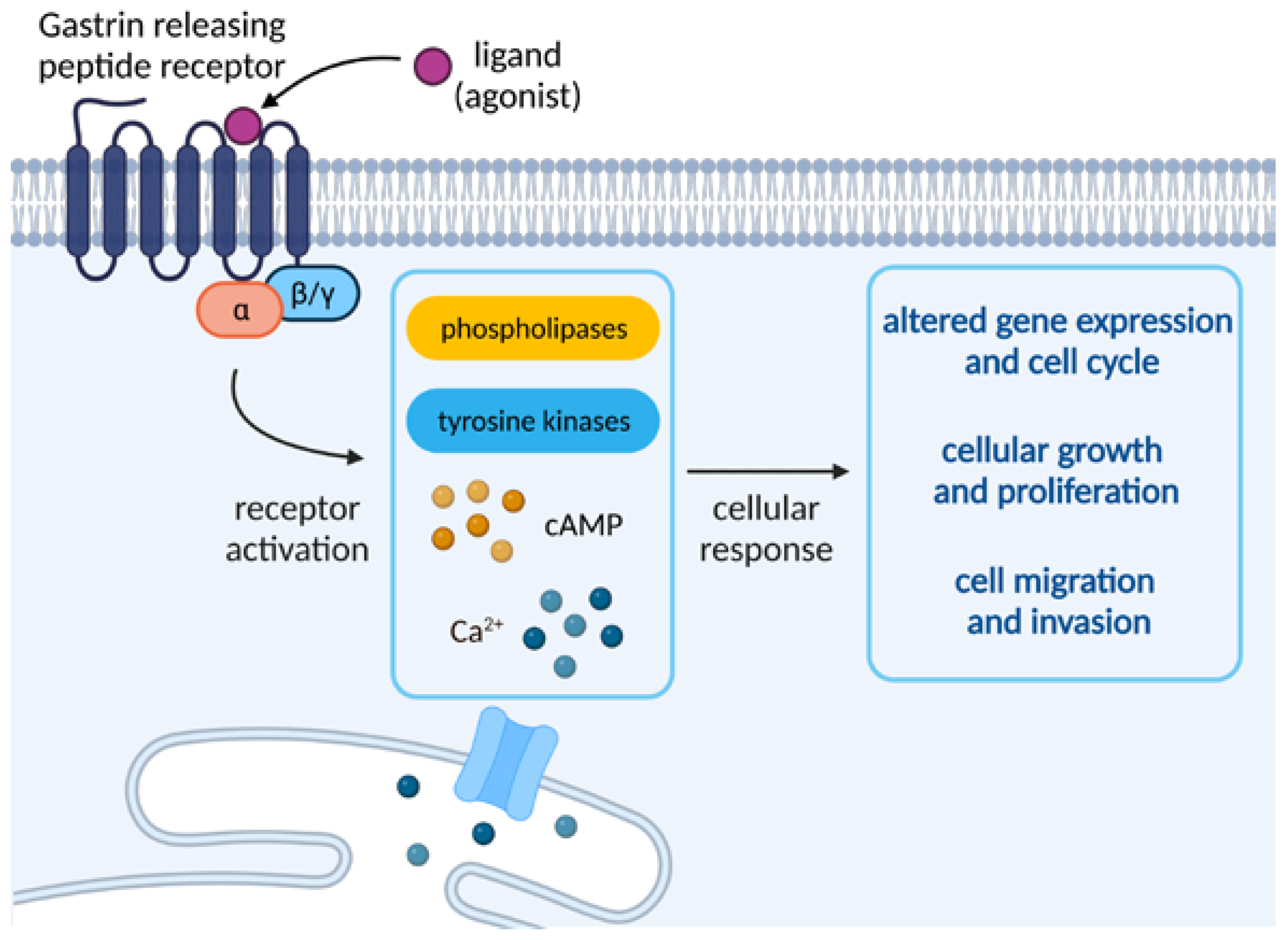
Figure 3.
Targeted nanosystems may perform different roles, but fundamentally three major aspects: carrier nanoparticle, targeting ligand and means of bringing them together, must be fine-tuned to yield successful formulation. Created with BioRender.com.
Figure 3.
Targeted nanosystems may perform different roles, but fundamentally three major aspects: carrier nanoparticle, targeting ligand and means of bringing them together, must be fine-tuned to yield successful formulation. Created with BioRender.com.
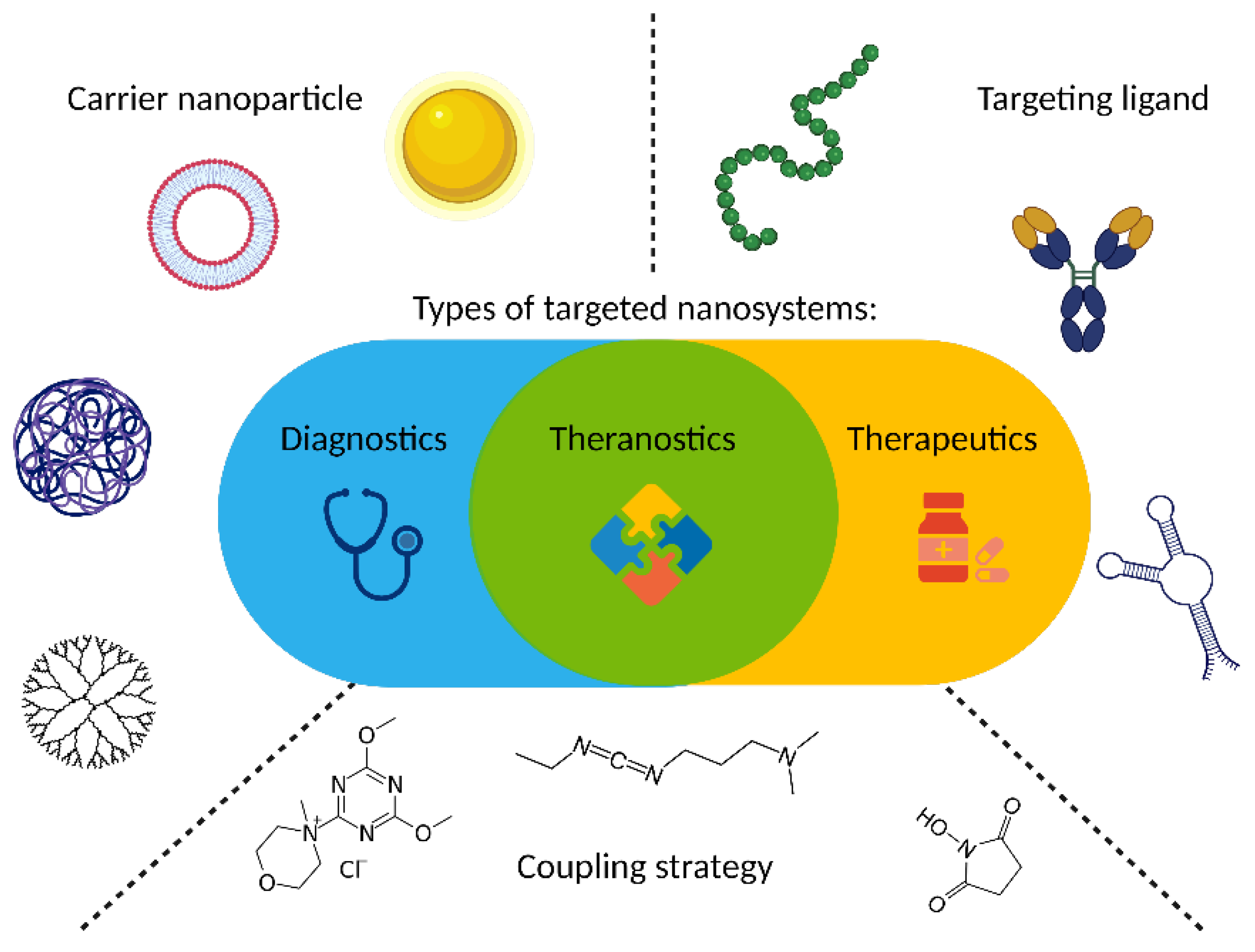
Figure 4.
Structures of commercially available bombesin peptides: (a) BBN (Pyr-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2); (b) [Lys3]BBN (Pyr-Gln-Lys-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2). Sketched with MarvinSketch 22.13 (ChemAxon Ltd., Hungary).
Figure 4.
Structures of commercially available bombesin peptides: (a) BBN (Pyr-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2); (b) [Lys3]BBN (Pyr-Gln-Lys-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2). Sketched with MarvinSketch 22.13 (ChemAxon Ltd., Hungary).
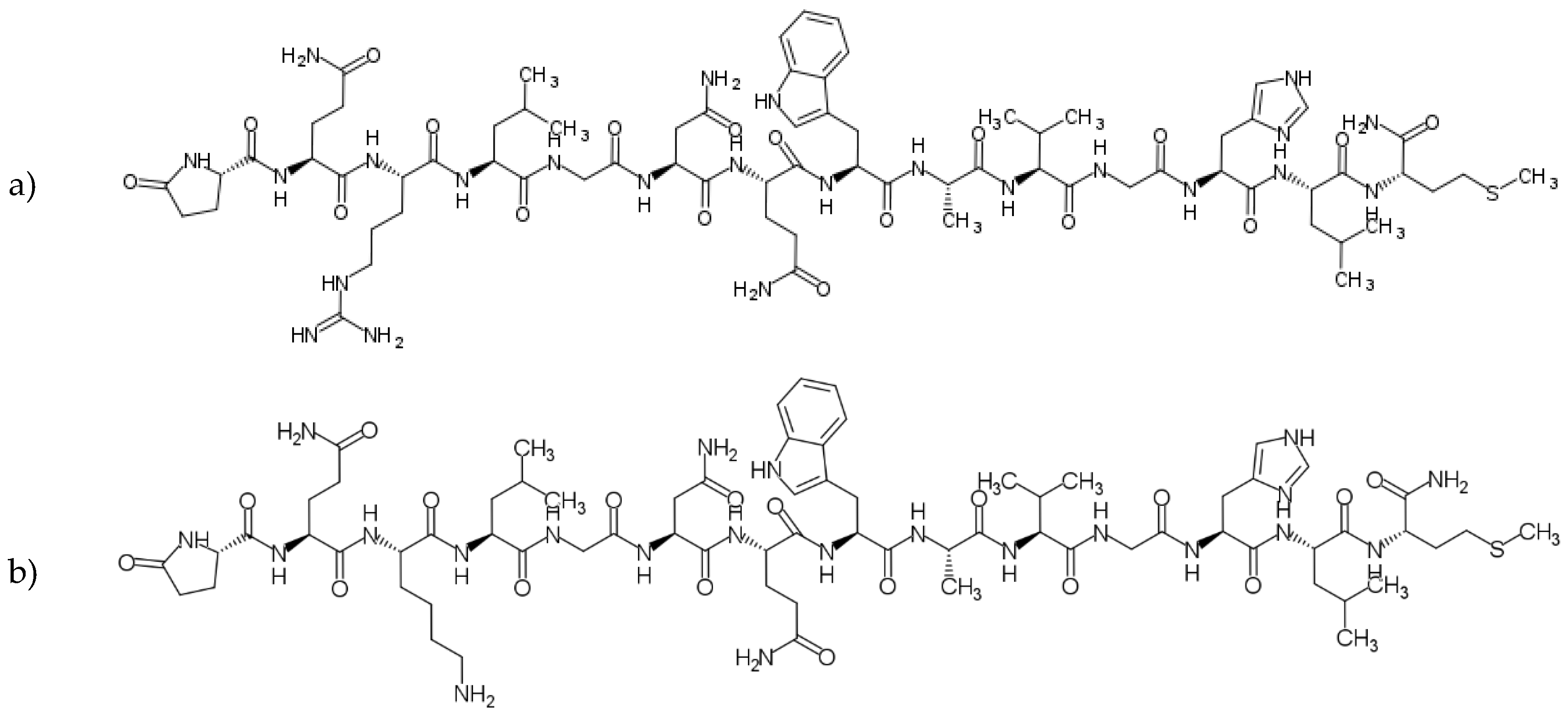
Figure 5.
Scheme of the EDC/NHS chemistry of ligand incorporation on the example of carboxylic group tethered nanoparticle. Created with BioRender.com. Chemical structures sketched with MarvinSketch 22.13 (ChemAxon Ltd., Hungary)
Figure 5.
Scheme of the EDC/NHS chemistry of ligand incorporation on the example of carboxylic group tethered nanoparticle. Created with BioRender.com. Chemical structures sketched with MarvinSketch 22.13 (ChemAxon Ltd., Hungary)
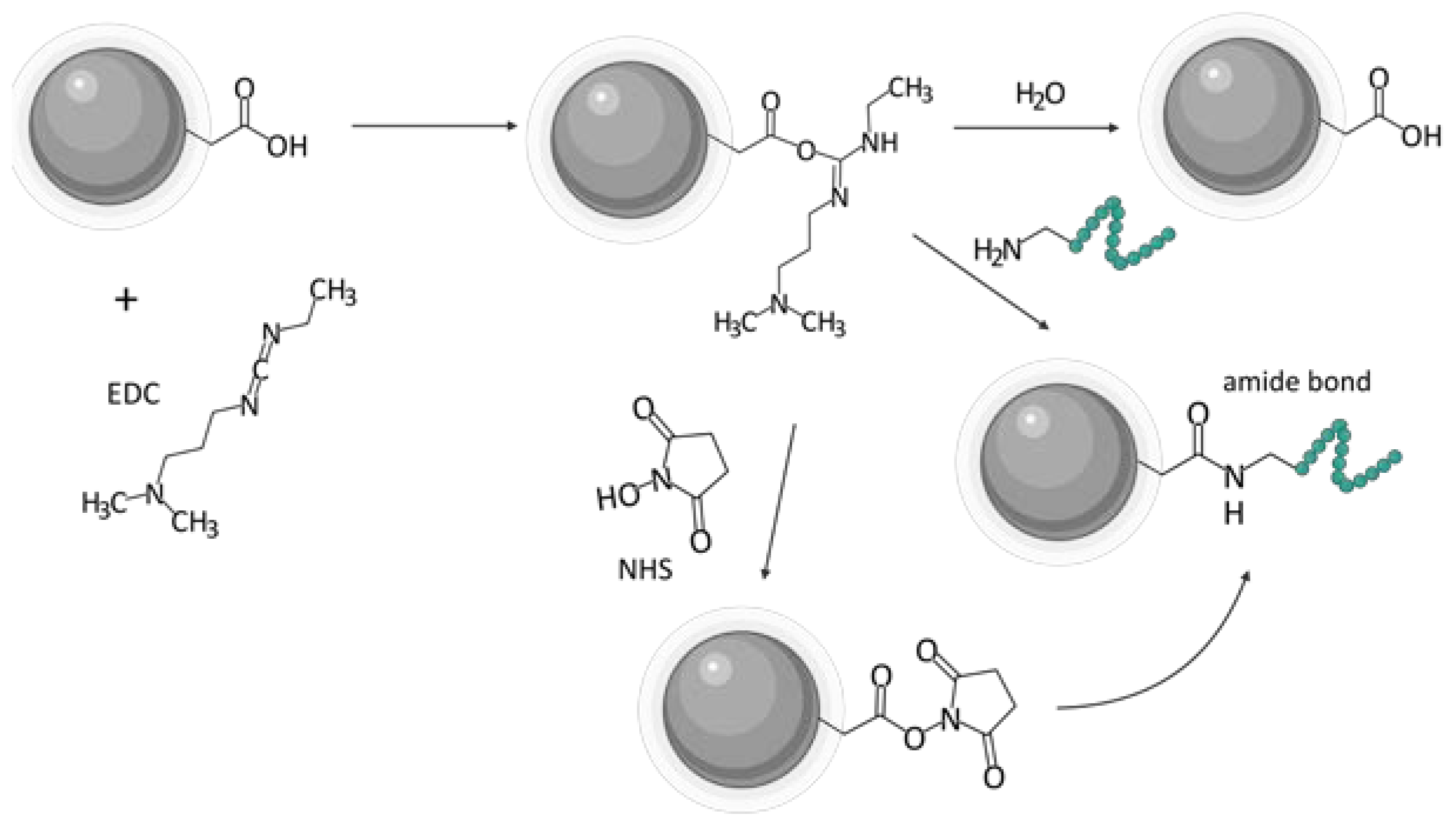
Figure 6.
GRPR binding motif can be incorporated in the structure of the building blocks of nanostructure prior to its assembly (adapted from [62]).
Figure 6.
GRPR binding motif can be incorporated in the structure of the building blocks of nanostructure prior to its assembly (adapted from [62]).
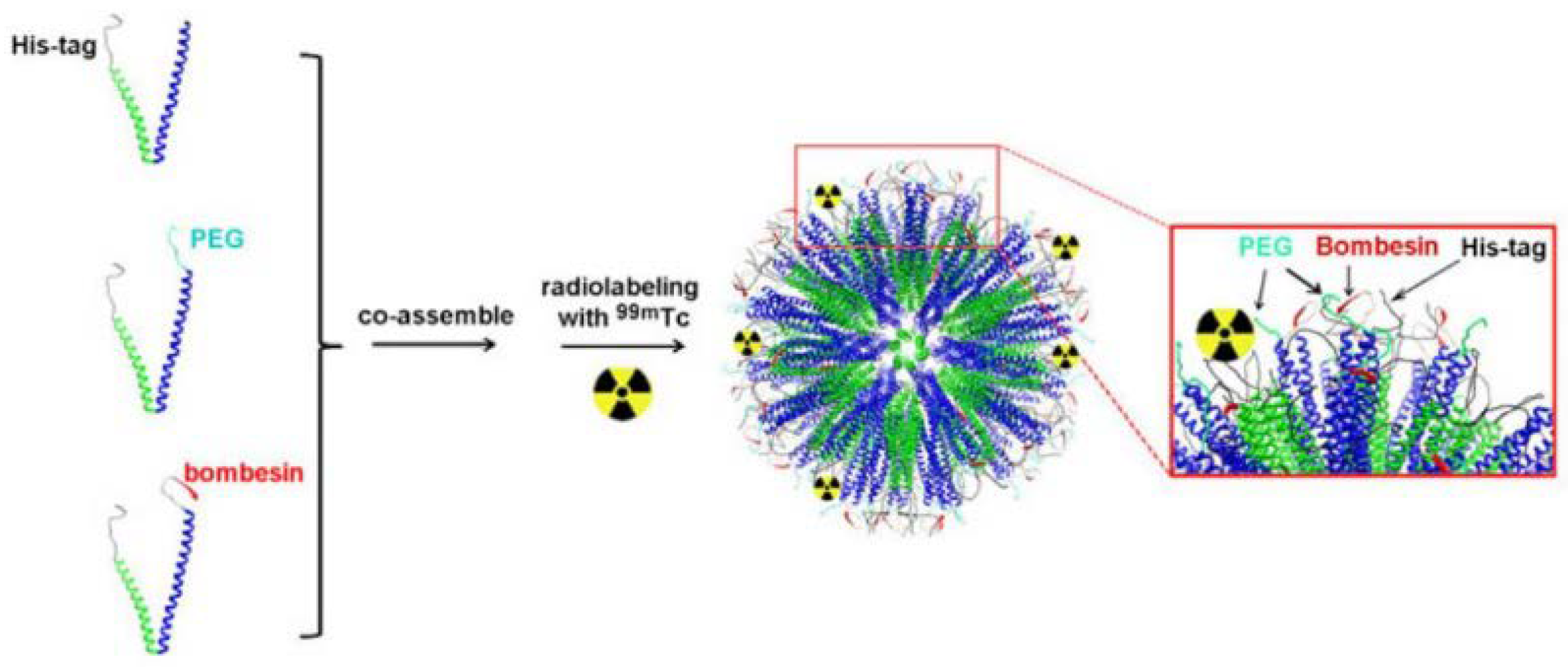
Figure 7.
Structures of bombesin derivatives: (a) modified with thioctic acid for binding with gold nanoparticles [68]; (b) AF750-6Ahx-Sta-BBN for biding with nanographene oxide via hydrogen and π–π interactions [37]. Sketched with MarvinSketch 22.13 (ChemAxon Ltd., Hungary).
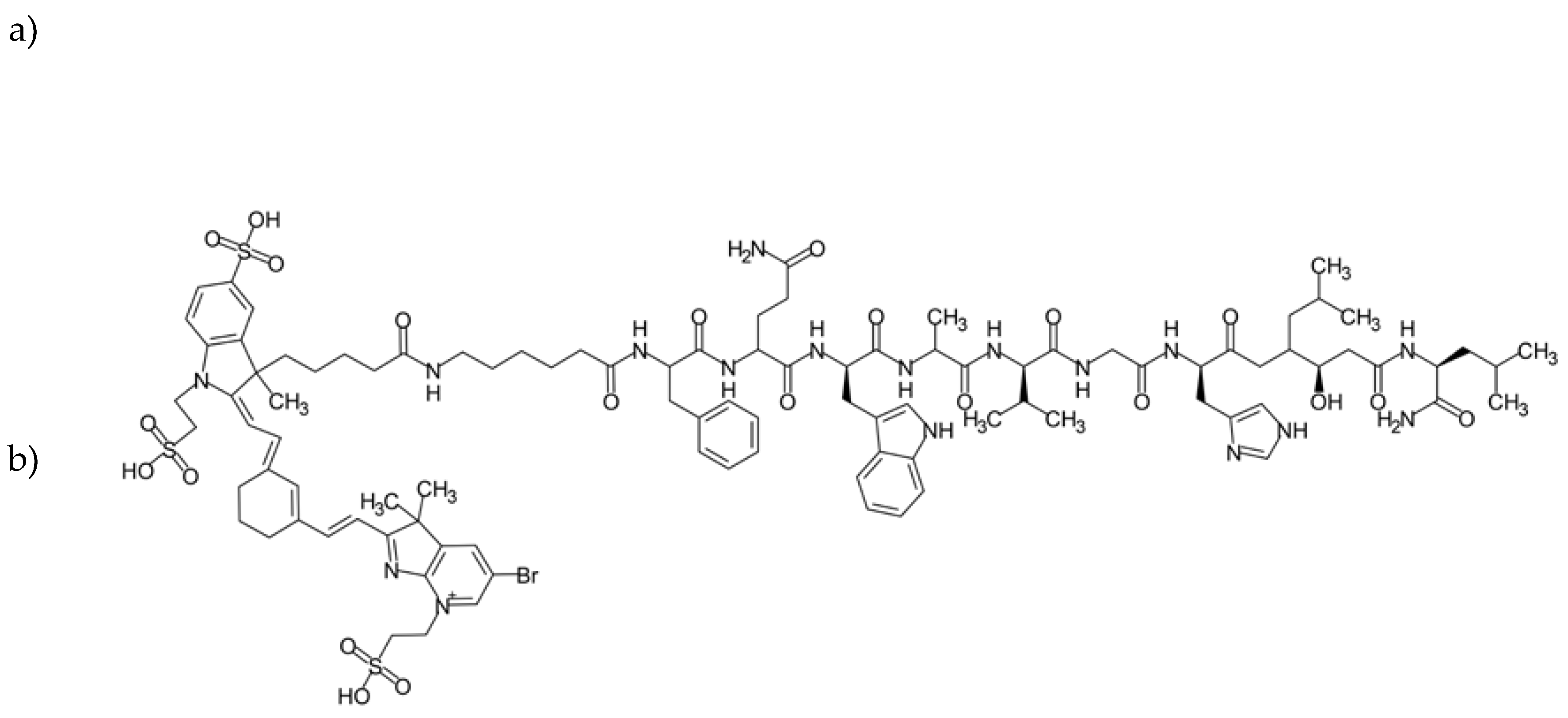
Figure 8.
Technetium-99m labeled BBN(7–14) ligand can be incorporated in the liposome by encapsulating it inside the vesicle (from [71]).
Figure 8.
Technetium-99m labeled BBN(7–14) ligand can be incorporated in the liposome by encapsulating it inside the vesicle (from [71]).
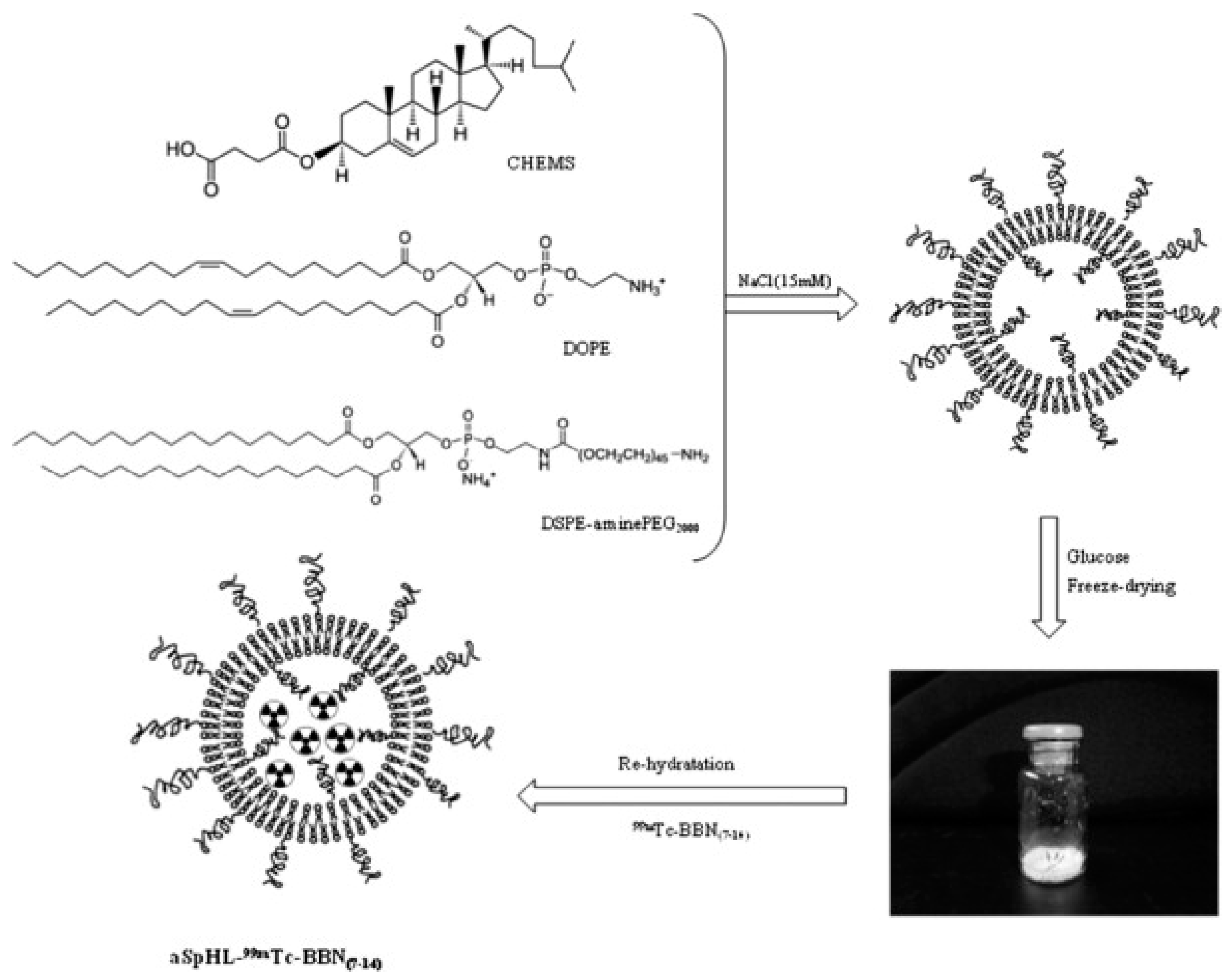
Figure 9.
Structures of (a) Doxil (PEGylated liposomal doxorubicin) (inspired by [78]) and (b) Doxil conjugated with BBN employing DSPE-PEG2000-maleimide (inspired by [36]). Created with BioRender.com.
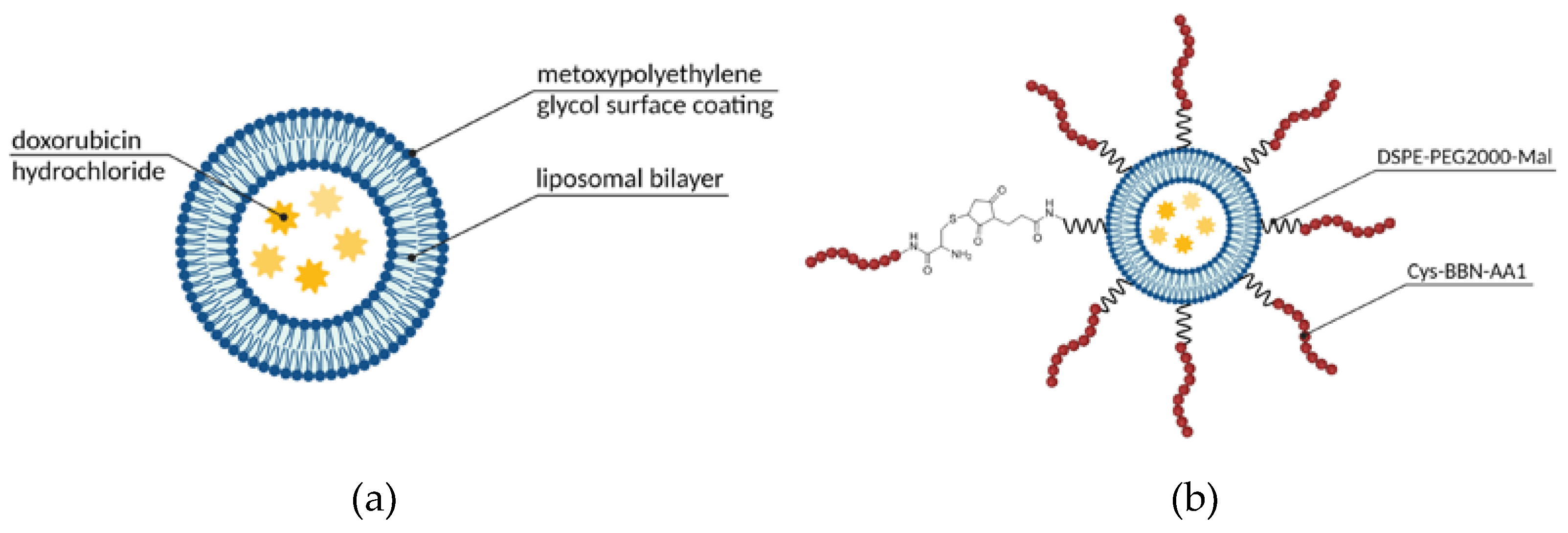
Figure 10.
Structure of 99mTc-labeled gold nanoparticles conjugated with BBN and DTDTPA (adapted from [24]).
Figure 10.
Structure of 99mTc-labeled gold nanoparticles conjugated with BBN and DTDTPA (adapted from [24]).
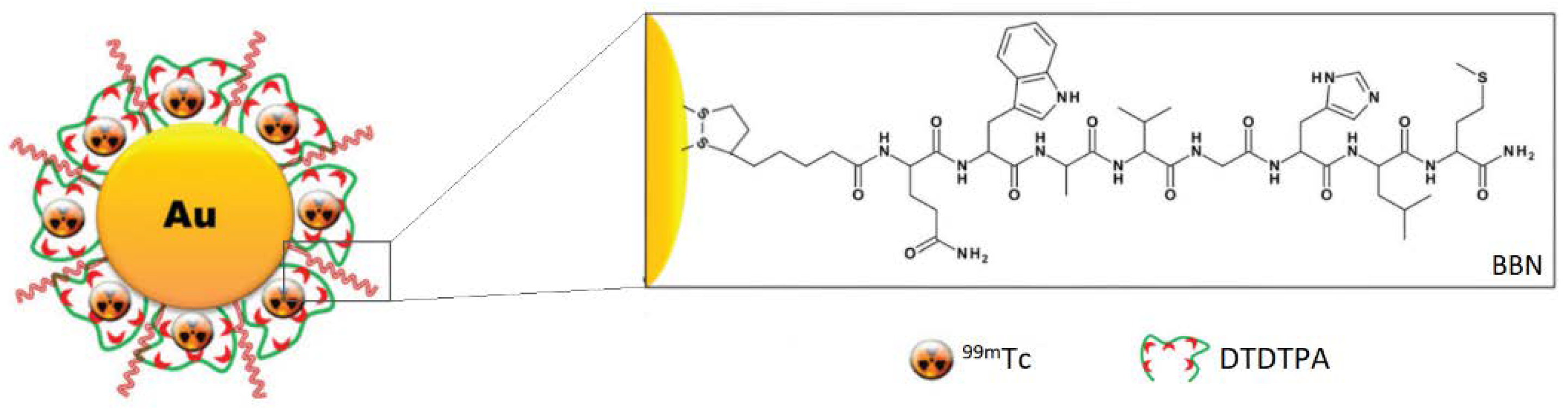
Figure 11.
An exemplary synthetic procedure for the preparation of PLGA nanoparticles (nanoprecipitation), surface conjugation of bombesin (BBN) to the nanoparticle surface as well as the stabilization and aggregation mechanisms for pure and conjugated nanoconstructs (adapted from [13]).
Figure 11.
An exemplary synthetic procedure for the preparation of PLGA nanoparticles (nanoprecipitation), surface conjugation of bombesin (BBN) to the nanoparticle surface as well as the stabilization and aggregation mechanisms for pure and conjugated nanoconstructs (adapted from [13]).
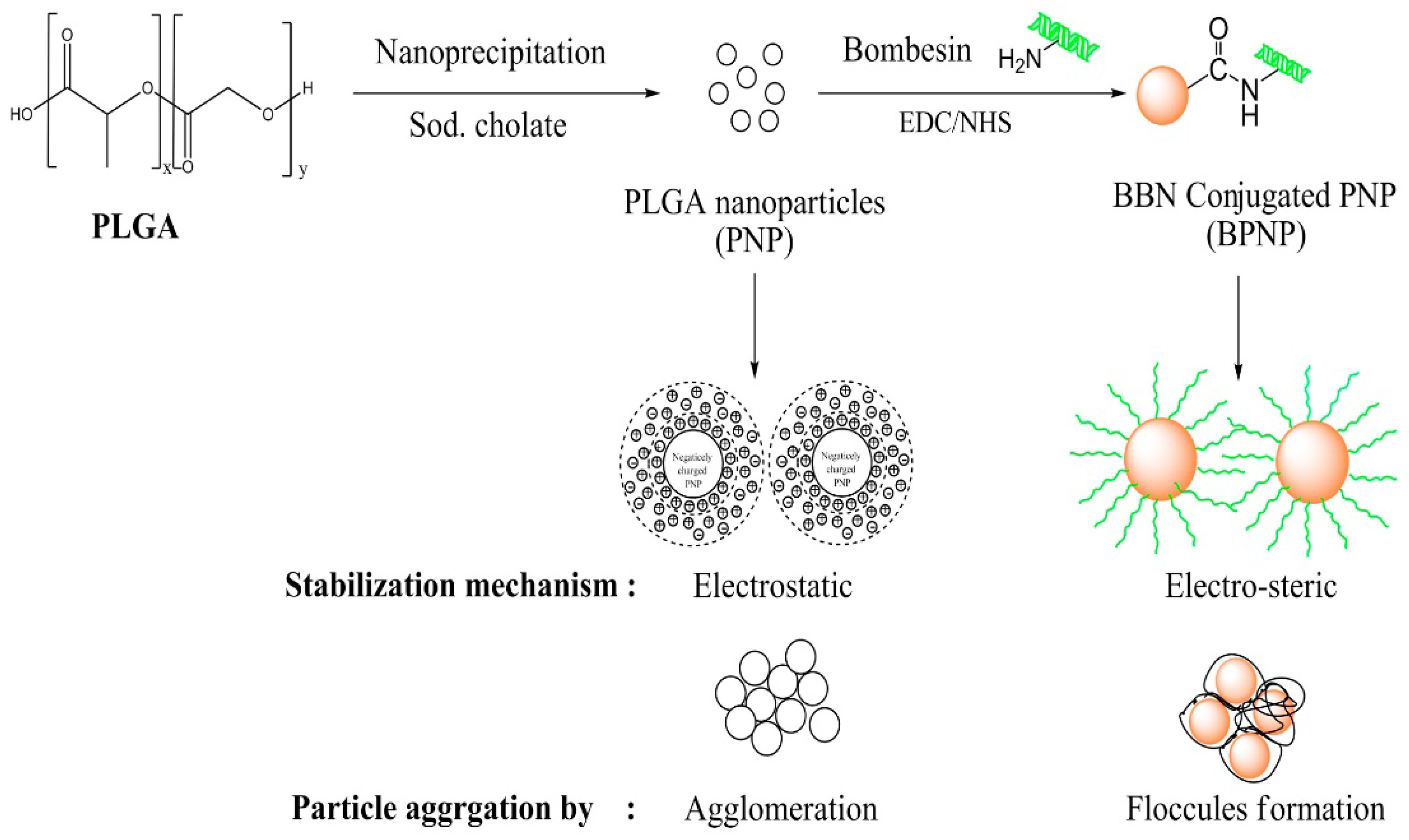
Figure 12.
Scheme of the process of specific targeting of prostate cancer cells with hybrid elastin-like polypeptide (ELP)/liposome nanoparticles (adapted from [74]).
Figure 12.
Scheme of the process of specific targeting of prostate cancer cells with hybrid elastin-like polypeptide (ELP)/liposome nanoparticles (adapted from [74]).
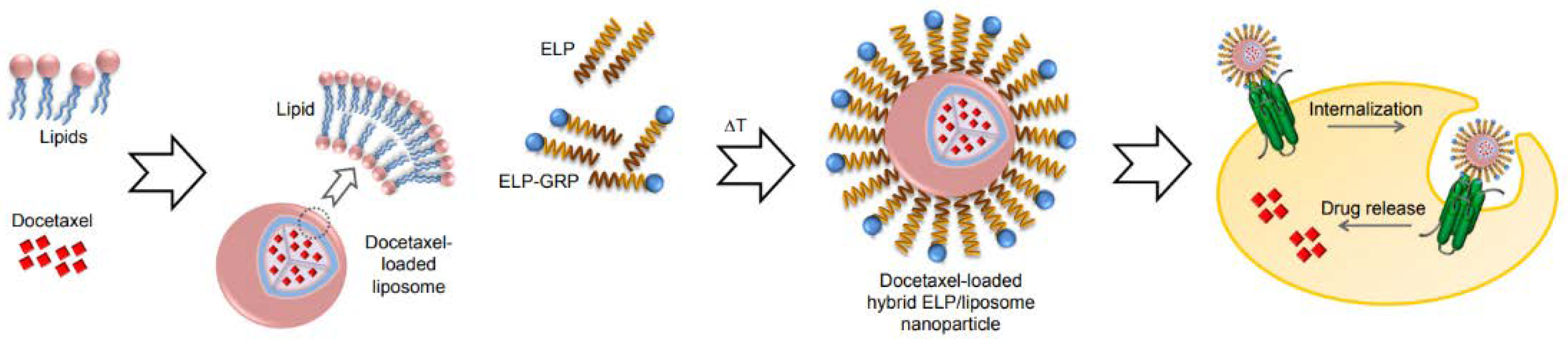
Figure 13.
The process of preparing BBN-conjugated, DOX-loaded magnetic reduced graphene oxide NPs (mrGOG/DOX) (from [58]).
Figure 13.
The process of preparing BBN-conjugated, DOX-loaded magnetic reduced graphene oxide NPs (mrGOG/DOX) (from [58]).
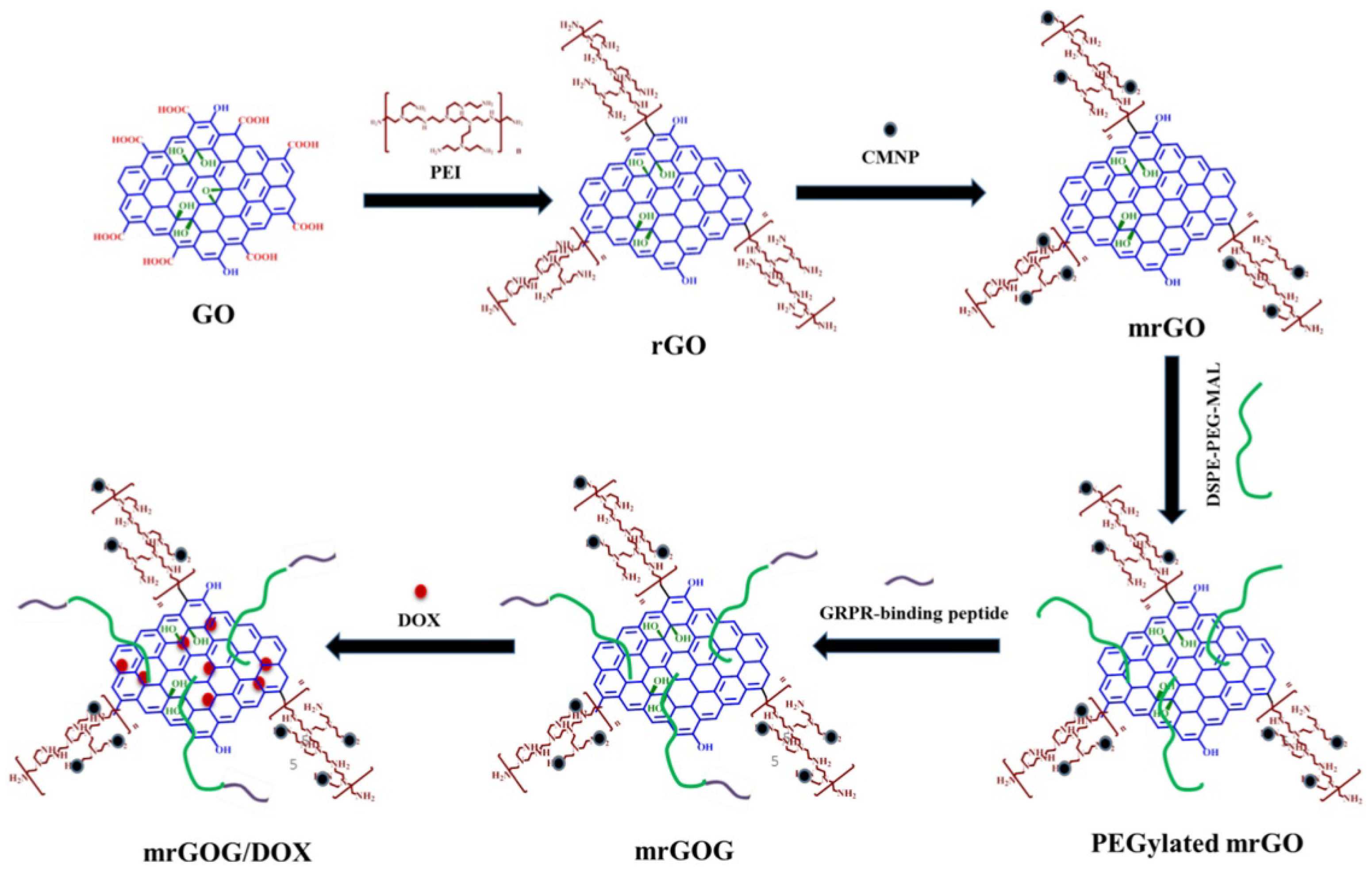
Figure 14.
Schematic representation of the surface plasmon resonance (SPR) effect in (a) gold nanoparticles (adapted from [94]) and (b) gold nanorods (adapted from [80]).
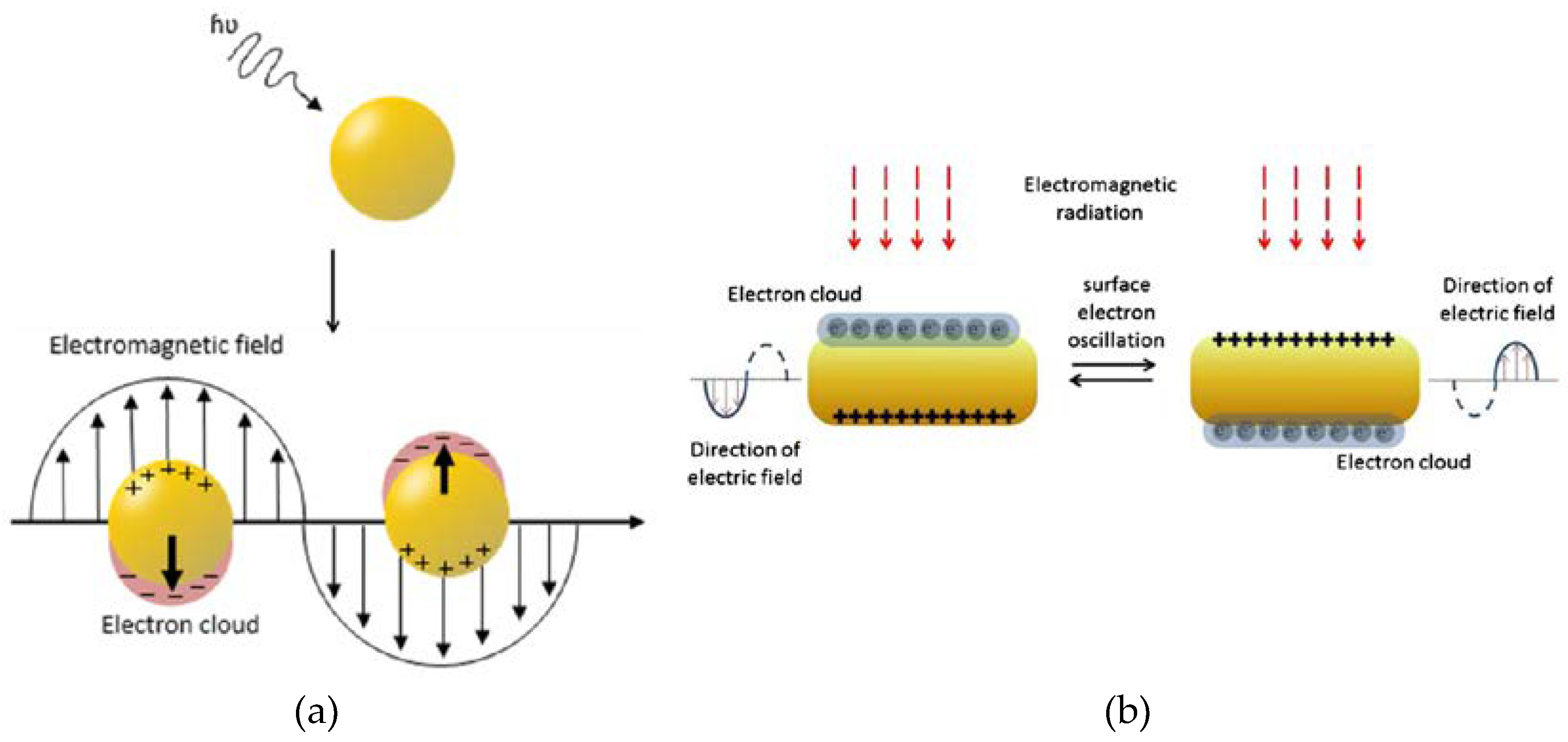
Figure 15.
The steps of the synthesis of PEGylated, superparamagnetic iron oxide (SPIO) nanoparticles coupled with Lys3-BBN and Cy5 fluorophore (adapted from [56]).
Figure 15.
The steps of the synthesis of PEGylated, superparamagnetic iron oxide (SPIO) nanoparticles coupled with Lys3-BBN and Cy5 fluorophore (adapted from [56]).
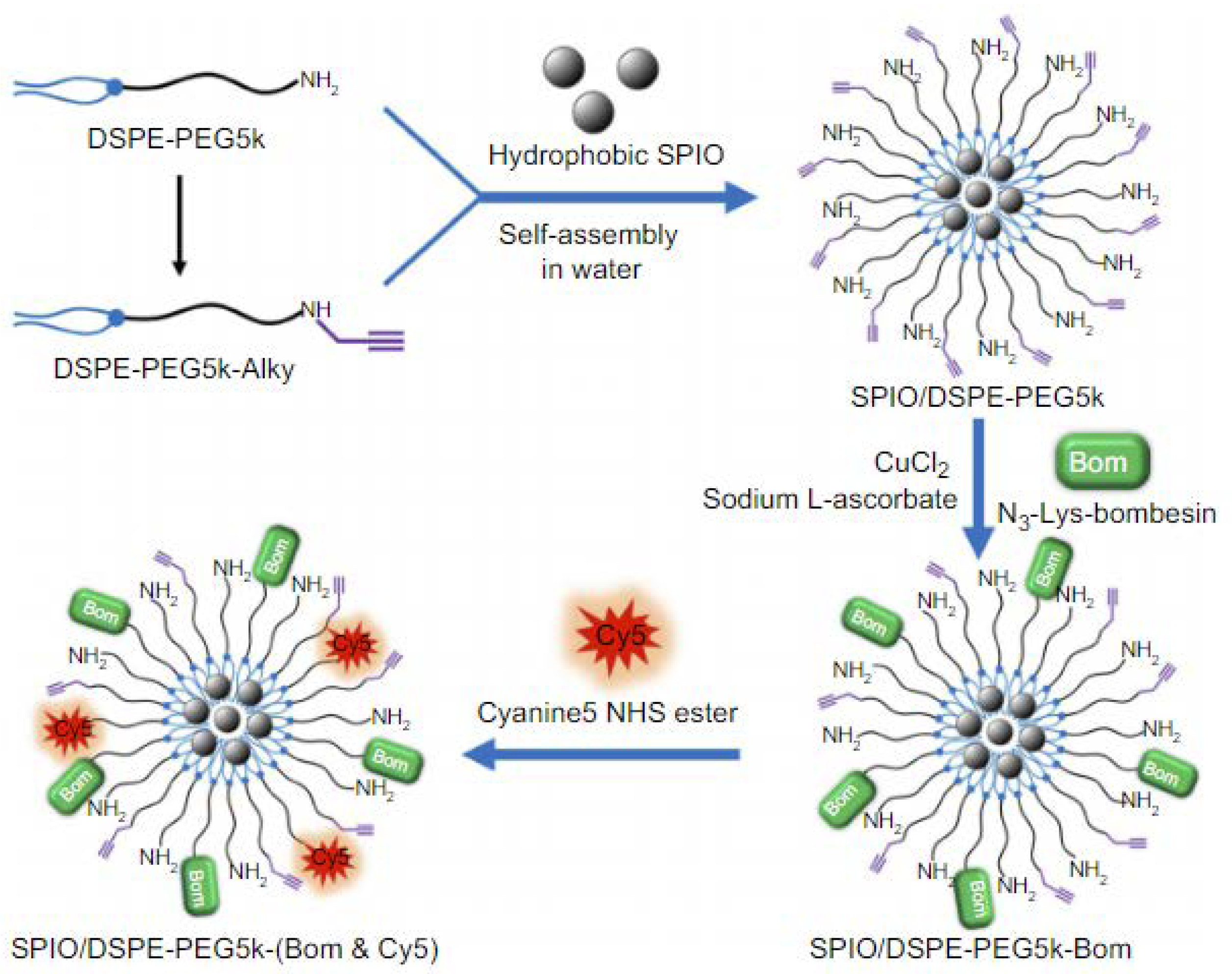
Figure 16.
Representation of QD functionalized for in vivo targeting and imaging including (a) the structure of a multiplex modifying multifunctional QD probe, (b) the structure of single modifying multifunctional QD probe and (c) the general idea of enhancing synergistic binding of heterodimeric multifunctional QD probe (adapted from [29]).
Figure 16.
Representation of QD functionalized for in vivo targeting and imaging including (a) the structure of a multiplex modifying multifunctional QD probe, (b) the structure of single modifying multifunctional QD probe and (c) the general idea of enhancing synergistic binding of heterodimeric multifunctional QD probe (adapted from [29]).
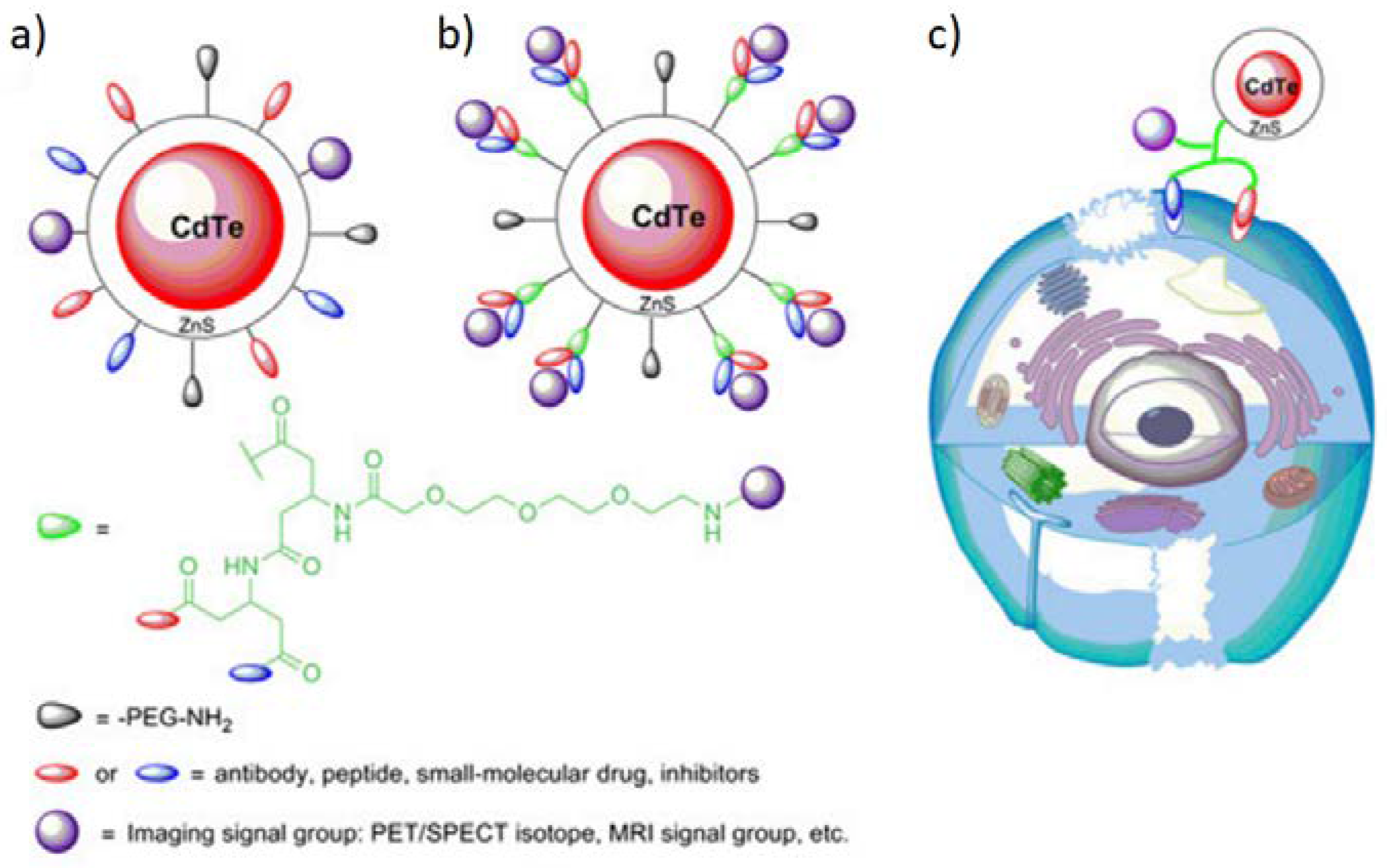
Figure 17.
Other representative nanoformulations used for tumor imaging including (a) long-circulating, pH-sensitive, BBN-labeled liposomes with 99mTc radiotracer (inspired by [71]), (b) dye-labeled, PEGylated cowpea mosaic virus (CPMV) conjugated with bombesin (inspired by [44]), (c) PEGylated, Gd2O3-based functional nanoparticles coupled with bombesin and 5(6)-carboxyfluorescein (inspired by [89]) and (d) lanthanide-oleate-based upconversion nanoparticles (UCNPs) functionalized with poly(acrylic acid) (PAA) and coupled with a BBN analog (inspired by [45]). Created with BioRender.com.
Figure 17.
Other representative nanoformulations used for tumor imaging including (a) long-circulating, pH-sensitive, BBN-labeled liposomes with 99mTc radiotracer (inspired by [71]), (b) dye-labeled, PEGylated cowpea mosaic virus (CPMV) conjugated with bombesin (inspired by [44]), (c) PEGylated, Gd2O3-based functional nanoparticles coupled with bombesin and 5(6)-carboxyfluorescein (inspired by [89]) and (d) lanthanide-oleate-based upconversion nanoparticles (UCNPs) functionalized with poly(acrylic acid) (PAA) and coupled with a BBN analog (inspired by [45]). Created with BioRender.com.
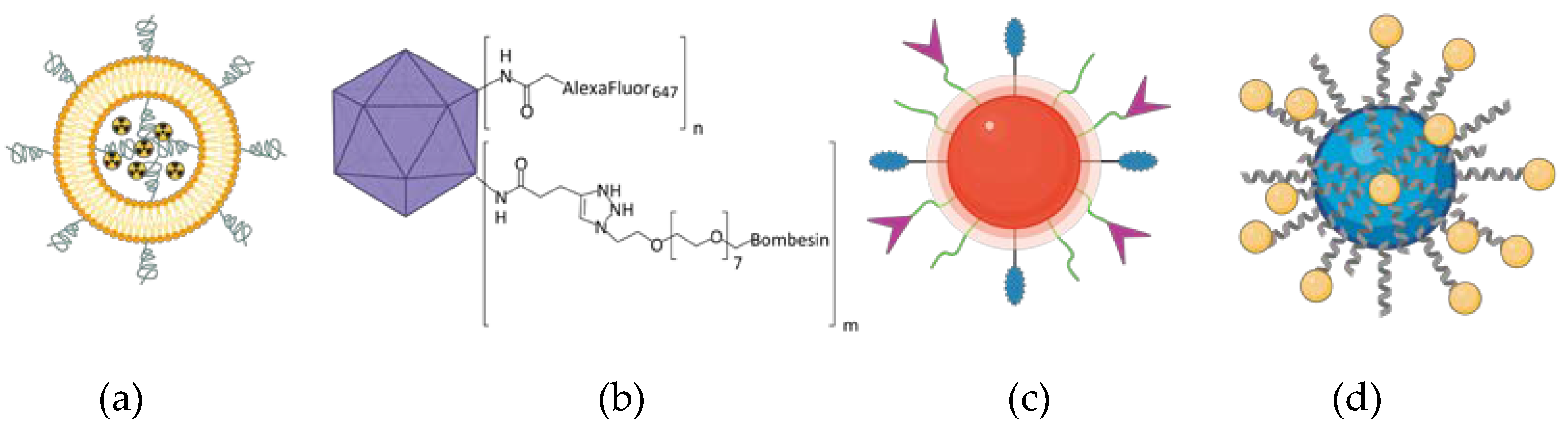
Figure 18.
The synthesis of PAA-coated, BBN-labelled upconversion nanoparticles (UCNPs) using lanthanide oleate as the precursor (inspired by [45]). Created with BioRender.com.
Figure 18.
The synthesis of PAA-coated, BBN-labelled upconversion nanoparticles (UCNPs) using lanthanide oleate as the precursor (inspired by [45]). Created with BioRender.com.
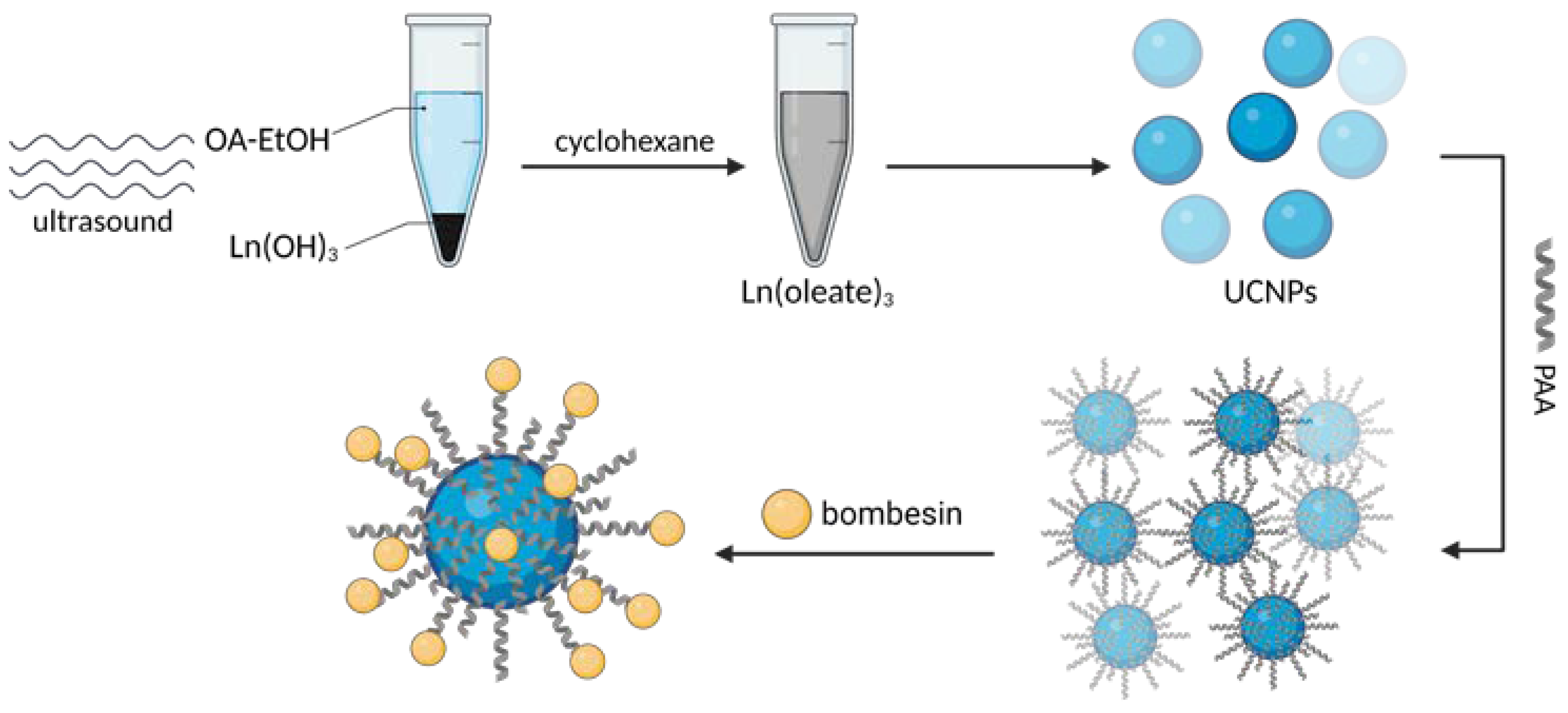
Figure 19.
The synthetic procedures involved in the preparation of paclitaxel(PTX)-loaded PAMAM dendrimers conjugated with BBN and DOTA-177Lu (from [53]).
Figure 19.
The synthetic procedures involved in the preparation of paclitaxel(PTX)-loaded PAMAM dendrimers conjugated with BBN and DOTA-177Lu (from [53]).
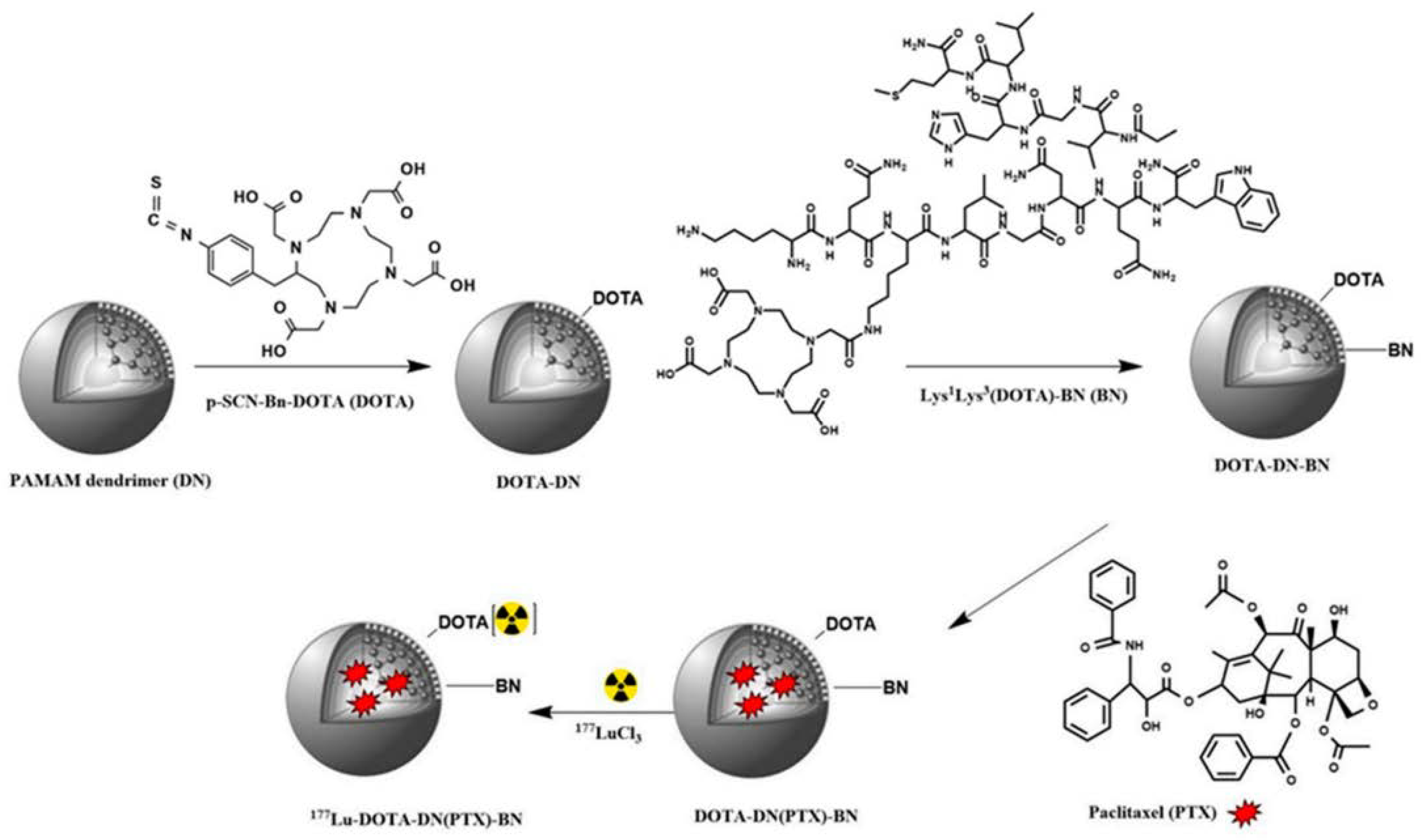
Figure 20.
Scheme of loaded magnetic polymersomes prepared from a PEO–PPO–PEO copolymer where the hydrophobic cargo (be it a magnetic nanoparticle or drug molecule) was implemented into the PPO liposome section (from [20]).
Figure 20.
Scheme of loaded magnetic polymersomes prepared from a PEO–PPO–PEO copolymer where the hydrophobic cargo (be it a magnetic nanoparticle or drug molecule) was implemented into the PPO liposome section (from [20]).
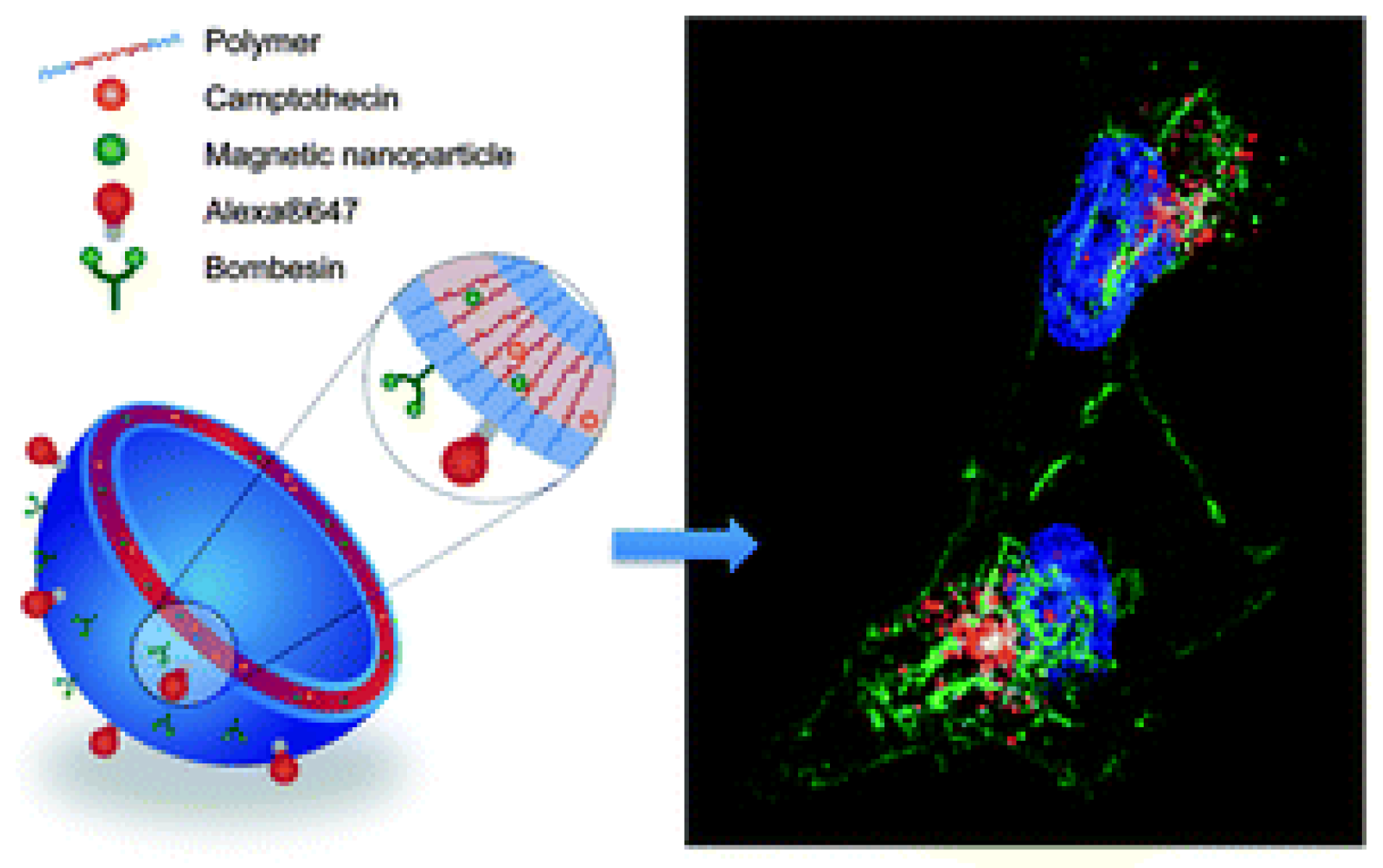

Table 1.
Contemporary GRPR-targeted nanosystems for cancer therapy
| Nanocarrier | Target condition | Drug/therapeutic means | Reference |
|---|---|---|---|
| phospholipid micelles | prostate cancer | Au(III)-dithiocarbamate complex (AuL12) | [35] |
| polymeric micelles | glioma | Coumarin-6 Camptothecin (CPT) |
[61] |
| prostate cancer | Monomethyl auristatin F (MMAF) | [65] | |
| elastin-like micelles | prostate cancer | - | [63] |
| Docetaxel (DTX) | [74] | ||
| nanostructured lipid carriers |
lung cancer | Doxorubicin (DOX) | [17] |
| solid lipid nanoparticles | breast cancer | Doxorubicin (DOX) | [59] |
| PLGA nanoparticles | breast cancer | Docetaxel (DTX) | [14] |
| prostate cancer | Docetaxel (DTX) | [75] | |
| gold nanoparticles | cervical cancer | RAF peptide analog (Ac-Cys-Ahx-RAF) | [69] |
| prostate cancer | 99mTc | [24] | |
| 99mTc, 177Lu | [31] | ||
| - | [76] | ||
| Gallic acid (GA) | [47] | ||
| prostate cancer, colon cancer |
68Ga | [25] | |
| gold nanorods | breast cancer | Photothermal therapy – use of NIR laser irradiation |
[39] |
| prostate cancer | Heat-labile cytotoxic free radical donor - AIPH | [77] | |
| graphene oxide | glioblastoma | Magnetic targeting, doxorubicin (DOX), NIR laser irradiation | [58] |
Table 2.
Contemporary GRPR-targeted nanosystems for cancer imaging
| Nanocarrier | Target condition | Imaging modality | Reference |
|---|---|---|---|
| gold nanoparticles | breast cancer | X-ray imaging | [41] |
| prostate cancer | CT imaging | [68] | |
| fluorescence imaging, CT imaging | [86] | ||
| PET imaging, CT imaging | [87] | ||
| microSPECT imaging, CT imaging | [19] | ||
| microSPECT imaging, CT imaging | [18] | ||
| gold nanorods | breast cancer | PA imaging | [40] |
| breast cancer, prostate cancer |
- | [88] | |
| iron oxide nanoparticles | breast cancer | MR imaging, NIRF imaging | [56] |
| MR imaging , PET imaging | [50] | ||
| PET imaging, CT imaging, MR imaging | [33] | ||
| MR imaging | [42] | ||
| pancreatic cancer | MR imaging | [66] | |
| prostate cancer | fluorescence imaging, MR imaging | [43] | |
| [67] | |||
| gadolinium oxide nanoparticles |
prostate cancer | fluorescence imaging, MR imaging | [89] |
| quantum dots | - | fluorescence imaging | [70] |
| prostate cancer | PET imaging, NIRF imaging | [29] | |
| PET imaging, CT imaging | [16] | ||
| liposomes | breast cancer | scintigraphy | [73] |
| Ehrlich tumor | [71] | ||
| upconversion nanoparticles |
prostate cancer | MR imaging, CT imaging, UC luminescence | [45] |
| graphene oxide | oral squamous cell carcinoma | fluorescence imaging | [37] |
| cowpea mosaic virus | prostate cancer | fluorescence imaging | [44] |
Table 3.
Contemporary GRPR-targeted nanosystems for cancer theranostics
| Nanocarrier | Target | Imaging modality | Drug/therapeutic means | Reference |
|---|---|---|---|---|
| gold nanoparticles | prostate cancer | MR imaging (Gd) SPECT imaging (67Ga) |
radiosensitization | [23] |
| hybrid gold nanoparticle - PAMAM dendrimer | breast cancer | optical imaging | 177Lu radiotherapy, plasmonic photothermal therapy | [51] |
| lung cancer | optical imaging | 177Lu radiotherapy, plasmonic photothermal therapy | [49] | |
| hybrid polymeric vesicles - iron oxide nanoparticles | prostate cancer | fluorescence imaging; potential for MRI | camptothecin delivery | [20] |
| liposomes | pancreatic cancer | microSPECT/CT imaging (188Re) | doxorubicin delivery | [99] |
| prostate cancer | gamma imaging (111In) | doxorubicin delivery | [34] | |
| PAMAM dendrimer | breast cancer | microSPECT imaging/CT imaging (177Lu) |
177Lu radiotherapy, paclitaxel delivery |
[53] |
| PLGA nanoparticles | breast cancer | microSPECT imaging/CT imaging (177Lu) |
177Lu radiotherapy, paclitaxel delivery |
[52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



