Submitted:
19 February 2023
Posted:
20 February 2023
You are already at the latest version
Abstract
Native reactive electrophile species (RES) are long-recognized regulators of pathophysiology; yet, knowledge surrounding how RES regulate context-specific biology remains limited. The latest technological advances in profiling and precision decoding of RES sensing and signaling have begun to bring about improved understanding of localized RES regulatory paradigms. However, studies in purified systems—prerequisites for gaining structure/function insights—prove challenging. We here introduce emerging chemical biology tools available to probe RES signaling, and the new knowledge that these tools have brought to the field. We next discuss existing structural data of RES-sensor proteins complexed with electrophilic metabolites or small molecule drugs (limited to < 300 Da), including challenges faced in acquiring homogenous RES-bound proteins. We further offer considerations that could promote enhanced understanding of RES regulation derived from three-dimensional structures of RES-modified proteins.
Keywords:
electrophile signaling
; reactive metabolites
; crystal structures of protein-electrophile complexes
1. Introduction
Reactive electrophile species (RES) act as cellular signals through non-enzymatic covalent modifications of sensor proteins and play functional roles in myriad pathophysiological processes, including aging, metabolic and cardiovascular diseases, inflammation and cancer (Figure 1A).[1] Different classes of RES are produced by various cellular physiological processes.[1,2] For instance, the tricarboxylic acid cycle (TCA) generates electrophilic metabolites directly (e.g. fumarate) or indirectly (e.g. itaconate, from cis-aconitate); peroxidation of membrane polyunsaturated fatty acids results in lipid-derived electrophiles (LDEs) formation.[1,3,4] While TCA cycle metabolites and LDEs were first characterized several decades ago, recent years have witnessed renewed interests towards understanding their subcellular functions; in particular, how they impinge on local proteome functions and signaling pathways. Beyond natural electrophiles, the inventory of small molecule covalent drugs and inhibitors housing electrophilic motifs is rapidly expanding.[5,6,7,8] Interestingly, the mode of action of some approved electrophilic drugs mimics that of natural electrophiles.[9] On the other hand, understanding of native electrophile signaling pathways has proven beneficial towards precision drug design and development.[7,10]
Despite such fundamental and translational importance of electrophile regulation, we know very little thus far about how these diffusible and reactive small molecules interact with individual protein targets and help shape biological decision making. Over the past ~25 years, ingenious work leveraging advanced chemoproteomics tools has enabled indirect profiling of electrophile-sensor proteins in both cells and animals.[2,11,12] However, these methods are unable to dissect spatiotemporal regulatory nuances of electrophile sensing and signaling, and are intrinsically limited in their ability to differentiate effects of on-target electrophilic modification(s) from off-target consequences, or cell toxicity/stress-related phenotypes. These major limitations are due to: (i) bulk administration from outside cells/animals (i.e., bolus dosing) of RES—highly toxic molecules that are often rapidly metabolized into additional reactive molecules in vivo; (ii) the use of lysates or homogenized tissue samples at the point of indirect target identification using proxy-electrophile probes—conditions that do not recapitulate nuances of the compartmentalized cellular environment (e.g., redox balance, physiological concentrations); (iii) indirect loss-of-signal readouts using non-biologically relevant electrophiles such as iodoacetamide; and (iv) low coverage of the cysteome (< 5 %).[13,14,15]
Recent innovations from our laboratory that leverage a “localized precision electrophile delivery concept” and associated REX technologies have begun to enable identification of individual protein/electrophile pairs and decoding the functional consequences of a single protein-specific electrophile sensing and signaling event in living systems.[16,17] Although different in their applications, all REX technologies are based on the same core system consisting of two main components: (i) in vivo locale-specific expression of HaloTag (either fused or not, to a putative electrophile-sensor protein of interest, and (ii) a cell/animal-permeable biocompatible and bioinert small-molecule photocaged electrophile probe that covalently reacts with HaloTag. Following wash-out of the excess unbound probe, light exposure releases a brief burst of the designated electrophile in the vicinity of HaloTag, at a preordained time. We refer interested readers to technological reviews and protocols focused on REX technologies.[16,17,18,19,20,21,22] These methods, equipped with rigorous technical and biological controls, lead us to several key findings, including: (i) low occupancy, i.e. substoichiometric modification of a RES on a given protein, is often sufficient to drive functional signaling outcomes; (ii) many electrophile-sensor proteins identified by REX technologies display enhanced RES adduction rates, i.e. are kinetically-privileged sensor (KPS) proteins, compared to others identified using the bolus RES treatment underpinning all state-of-the-art profiling methods; (iii) KPS proteins identified and validated by REX technologies have often not been previously profiled by existing chemoproteomics methods (fundamental underlying differences leading to this outcome have been comprehensively reviewed elsewhere); (iv) nuanced regulatory pathways involving RES are often masked by using bolus dosing conditions; and (v) applying REX technologies in zebrafish (D. rerio) and worms (C. elegans) reveals that mechanisms of electrophile sensing and signaling are largely conserved throughout evolution.[1,9,13,16,17,19,20,21,23,24,25,26,27,28,29,30,31,32]
Despite the myriad profiling tools [spearheaded by the innovative activity-based protein profiling (ABPP) method and its derivatives] and emerging function-guided proximity mapping-based REX technologies (T-REX, Z-REX, G-REX, and more recently, Localis-REX), several fundamental questions remain unaddressed, particularly with regard to structure/function relationship principles underlying electrophile-sensor protein regulation (Figure 2).[16,17,19,20,21] For instance, what makes a protein a KPS, given that a two units change in thiol pKa can only increase the reaction rate with peroxide by no more than 20-fold and that full cysteine thiol deprotonation only enhances the Michael addition reaction rate with the electrophile acrolein by a maximum of 10-fold, i.e. how is the more functionally relevant kinetic selectivity, such as transition state stabilization, achieved?[1] How does modification of a single site affect the sensor protein’s activity/function, or how does it modulate the function of a binding partner, the interactome or downstream signal transduction ? Insights into such fundamental mechanisms, especially from structure/function perspectives, and generalizable principles—should there be any—of electrophile sensing and signaling remain hugely limited, particularly for pleiotropic native electrophiles such as LDEs and TCA cycle metabolites, and related low molecular weight (< 300 Da) covalent drugs.
Three-dimensional structural information on proteins bound to small molecule ligands remain invaluable for potentially predicting molecular mechanisms governing ligand-gated protein activity, function or changes in interactions with macromolecular binding partners. Such structural information also heavily aids drug design and development, mode of action prediction and structure-activity relationship (SAR) efforts towards improving binding affinity. Despite increasing interests in electrophilic drugs and native electrophilic metabolites, the number of available protein structures bound to a small molecule electrophile (molecular weight < 300 Da) remains alarmingly low. An extensive search of the RCSB Protein Data Bank (PDB) and the literature yielded only two structures of proteins covalently bound to a small electrophilic drug (limited to < 300 Da) and two structures of electrophile-sensor proteins bound to an endogenous reactive electrophilic metabolite (limited to < 200 Da) (Table 1).[33,34,35,36,37]
Here, we review these available data and interrogate their functional relevance. We also offer some perspectives toward what we believe are key considerations in acquiring improved structural information of homogeneous protein states complexed to reactive and pleiotropic small molecule electrophilic ligands. Beyond the low molecular weight limit above, our search was also centered on endogenous LDEs, electrophilic metabolites and small molecule drugs (or their close derivatives) of fewer than 12 carbons bearing an α,β-unsaturated carbonyl motif and bound to their sensor/target protein (Figure 1B). Structures of enzymes catalyzing transformations of electrophilic metabolites such as fumarate reductases are omitted from the discussion. Where available and relevant, we have further discussed structures of proteins non-covalently bound to either synthetic drugs or endogenous electrophiles.
2. Structural basis of and putative insights into dimethyl fumarate mode of action
Dimethyl fumarate (DMF, Tecfidera) is an anti-inflammatory electrophilic drug structurally related to the endogenous metabolite fumarate (Figure 1B). DMF was initially approved for treatment of systemic psoriasis in 1994 and is used in the treatment of relapsing remitting multiple sclerosis (RRMS) since 2013.[38,39,40] Recently, two other fumarate analogs were approved for the treatment of RRMS: Vumerity in 2019 and monomethyl fumarate (MMF, Bafiertam) in 2020.[41,42] The exact mode of action of DMF and its derivatives had remained elusive for a long time. Although at its point of approval DMF was proposed to mainly target the Kelch-like ECH-associated protein 1 (Keap1)/nuclear factor erythroid 2-related factor 2 (Nrf2)-mediated antioxidant response (AR) pathway, this theory was questioned after a study revealed that DMF-mediated immune modulation is not ablated in Nrf2-knockout mice.[43] Additional target proteins and pathways identified by bolus dosing-based profiling methods also failed in functional genetic validation experiments.[44] The application of precision REX technologies in zebrafish, combined with functional and genetic validations in primary macrophages and other relevant models, has recently uncovered that immune cell-specific Keap1/WD repeat-containing protein 1 (Wdr1)-dependent mitochondrial-targeted apoptosis is necessary and sufficient to explain DMF-induced cell death, specifically in the innate immune system.[9] Besides functional studies in living systems, DMF’s mode of action has been investigated from a structure/function perspective. We identified crystal structures of three protein targets in complex with DMF, MMF and/or the native metabolite fumarate that we evaluate here.
Andersen et al. report the only structure available to date of DMF covalently complexed with the C-terminal kinase domain (CTKD) of ribosomal protein S6 kinase alpha-3 (RSK2), a mitogen-activated protein kinase (MAPK)-cascade effector found directly downstream of extracellular signal-regulated kinase (ERK).[45] Following incubation of RSK2 CTKD (8 mg/mL, corresponding to ~200 µM) with ~25-fold excess DMF (5 mM) [30 minutes at room temperature in 10 mM Tris-HCl (pH 8.0) and 5 mM tris(2-carboxyethyl)phosphine (TCEP)], co-crystallization experiments yielded a 1.90 Å resolution structure showing DMF covalently bound to Cys599. DMF is favorably positioned for Michael addition to Cys599 through contacts with several neighboring residues, including hydrophobic interactions with Ile633 and Leu710, only in the non-activated kinase state. This covalently bound ligand at Cys599 is thought to create steric hindrance preventing obligatory conformational change of the kinase activation loop required for phosphorylation-mediated kinase activation (Figure 3A).[34]
Notably, apparent IC50 of purified WT-RSK2 CTKD (IC50 ~250 µM) and C599V-RSK2 CTKD (IC50 ~400 µM) only become significantly different after 48 hours of DMF treatment, but remain similar at shorter time points (1 hour and 24 hours). These IC50 values are determined using a time resolved fluorophore-conjugated antibody-based activity assay following incubation of ~100 nM recombinantly-purified WT-RSK2 CTKD with increasing amounts of DMF (albeit at undisclosed concentrations) and subsequent ERK2-mediated RSK2 CTKD activation. Notably, excess reactive electrophile is not removed before incubation with ERK2 (at 0.1 µM) and may therefore negatively affect ERK2-mediated RSK2 CTKD activation. Interestingly, the combined mutations of the four other cysteine residues found in RSK2 CTKD (Cys436, Cys439, Cys560 and Cys579) initially lead to a decreased inhibitory capacity of DMF (IC50 ~700 µM), compared to WT-RSK2 CTKD, analyzed after 1 hour DMF treatment. However, the measured IC50 of this tetra-cysteine mutant significantly decreases to 350 µM after 48 hours. The authors thus postulate that the key labeling site (Cys599) is slowly modified over time and that the other cysteine residues play a role in the initial inhibitory effects observed of DMF. It is further speculated that the inhibitory effect due to the presence of the other four cysteine residues in the kinase domain is lost over time, as IC50 slightly increases from 1 hour to 48 hours for C599V-RSK2 CTKD, although this change is not analyzed as statistically significant. Such uncertainties underlying multi-site conjugation potential of reactive small electrophiles like DMF are not uncommon. Indeed, establishing a functional model of site-selective labeling at the most kinetically-privileged electrophile-sensor site that impacts protein function (over labeling at non-functional sites yielding thermodynamically more stable adducts) remains a thorny problem.
Since IC50 varies with time for covalent inhibitors, a more meaningful parameter reflecting covalent inhibitor efficacy in purified systems is the second-order rate of covalent inactivation, i.e., kinact/Ki, which is unfortunately not evaluated in this study.[10] Second-order rate constants for DMF addition with thiols vary from 0.79 M-1 s-1 for glutathione to 2.19 M-1 s-1 for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and 22.8 M-1 s-1 for Keap1, the latter two being DMF target candidates.[46] Estimating the second-order rate constants of DMF reaction with RSK2 CTKD WT, C599V and tetra-cysteine mutant would be a more relevant and informative way of comparing the relative functional contributions of different cysteines in DMF-mediated RSK2 inhibition.
On the other hand, transient expression of full length C599V-RSK2 in HEK293 cells ablates enzyme inhibition upon whole cell DMF treatment (140 µM, 1 hour) followed by epidermal growth factor (EGF) stimulation, as estimated by quantification of RSK2 autophosphorylation in western blot analysis of cell lysates. By contrast, ~50 % loss in kinase activity is observed when WT-RSK2 is expressed in an otherwise identical setup. The tetra-cysteine mutant previously used in in vitro assays is not assessed in these cell-based assays. Notably, determining the inhibitory dosage effective for purified proteins versus intact cells is less straightforward for RES since, beyond the extent of permeability and intracellular distribution (parameters that do not apply in purified systems), RES such as DMF or 4-hydroxynonenal (4-HNE) are vulnerable to metabolic conversion and degradation.[1,13,47] Interestingly, although the ATP-binding pocket residue Cys436 is reported in the same crystal structure as an additional (albeit less well-defined) modification site for DMF on RSK2 CTKD, its mutation to valine does not functionally impair DMF-mediated RSK2 inhibition in cells. Excess DMF used during crystal preparation may explain additional modification of this non-functionally relevant site. Indeed, this observation underscores the fundamental challenges often encountered in procuring homogenous single-site-modified states of proteins adducted with reactive small molecules. On the other hand, upregulation of ERK commonly associated with RRMS, presumably leads to hyperactivation of RSK2 and masking of Cys599 by the kinase activation loop.[48] Thus, the accessibility of RSK2 Cys599 to DMF in disease-relevant states remains unsettled.
As a postulated LDE- and fumarate-sensor protein, the essential glycolytic enzyme GAPDH has long been suspected to be a DMF target.[49,50] Using tandem mass spectrometry, Kornberg et al. identified a covalent modification of GADPH by DMF/MMF in DMF-treated mice and RRMS patient-derived samples.[51] Additional work in mouse and human immune cells led to the conclusion that DMF inhibits GAPDH through formation of a covalent bond with its catalytic cysteine, thereby leading to downregulation of aerobic glycolysis in activated immune cells and reduction of inflammatory responses.[51] A 2.29 Å crystal structure reports the covalent modification of human GAPDH catalytic site Cys152 by MMF.[35] The authors rationalize that inhibition of GAPDH occurs as a result of MMF covalent modification, preventing both the catalytic conversion of glyceraldehyde 3-phosphate to glycerate 1,3-bisphosphate within the catalytic site, and the binding of the co-factor NAD+ through steric hindrance (Figure 3B). Notably the GAPDH-MMF complex was obtained by incubation of the recombinant enzyme with ~18-fold excess DMF [3 hours at 37 °C in 250 mM HEPES-NaOH (pH 7.7), 1 mM EDTA, 0.1 mM neocuproine]. Although DMF is known to be rapidly converted to MMF by esterases after oral administration, DMF has a half-life of over 12 hours in phosphate buffer at pH 7.4.[52,53] The reason why only the MMF adduct is observed in the crystal structure is unclear.
The third reported structure of a DMF target protein complex is interestingly a non-covalent complex between DMF (or fumarate) and the C-terminal Kelch domain of murine Keap1. Keap1 is an established cellular LDE-sensor.[54] Under oxidative/electrophilic stress conditions, Keap1 is modified by electrophilic species and releases the transcription factor Nrf2 for nuclear translocation and activation of cytoprotective genes.[55] Incubation of purified Kelch domain of murine Keap1 (10.5 mg/mL, ~300 µM) with ~1.7-fold excess DMF or fumarate (500 µM) [overnight at 4 °C in 20 mM Tris-HCl (pH 8.3), 150 mM NaCl and 20 mM dithiothreitol (DTT)] followed by co-crystallization studies yielded protein structures at 1.54 Å and 1.75 Å resolution of the respective ligand-bound complexes.[56] Each structure reveals three non-covalent binding sites for DMF/fumarate, including two located at the Nrf2-binding interface, the target of numerous inhibitors currently in development.[57] Incubation of recombinant Keap1-Kelch domain with 1.8-fold excess DMF for 30 minutes inhibits binding of a fluorescein isothiocyanate (FITC)-tagged Nrf2 peptide by ~40 %, although there is no evidence that this effect is mediated by non-covalent binding of DMF to Keap1. Indeed, Keap1 is known to form covalent bonds with a number of electrophilic species through its multiple cysteine residues outside of its Kelch domain.[58] Recent studies in fact indicate that functional triple-cysteine-mutant of human Keap1 (Cys151S/Cys273W/Cys288E, all found outside the Kelch domain) renders the system refractory to 4-HNE-induced innate immune cell loss in zebrafish models, whereas wild type human Keap1 is covalently adducted by 4-HNE, leading to reduced innate immune cell count.[9] Of note, the same immune cell loss phenotype is observed upon DMF treatment in these studies. Collectively, these data highlight the criticality of functionally correlating RES-binding sites (whether this information be obtained from structural or biochemical data, or both) with target-specific biological output (through functional biological or phenotypic assays) wherever possible. In this specific case, the large excess (20 mM) of reducing agent used during the incubation of DMF/fumarate with the protein could have significantly depleted the pool of reactive electrophilic molecules available to form covalent bonds with the protein. The prolonged reaction time (overnight), followed by the crystallization period (7 days), also increase the likelihood of initially-formed Michael adducts being removed via retro-Michael addition reactions with residual DTT. Furthermore, such prolonged crystallization periods may result in Keap1 cysteines being air-oxidized and no longer available for covalent reaction with DMF. Taken together, the aforementioned issues could have contributed to the exclusive observation of only the non-covalent complex between Keap1 and the reactive Michael-acceptor ligand.
3. Structures of proteins covalently bound to native electrophiles resembling canonical substrates
Both itaconate and 4-HNE are well-studied native electrophiles, each known to interact with a number of context-dependent cellular target proteins.[1,59] The crystal structures of: (i) itaconate covalently bound to Mycobacterium tuberculosis isocitrate lyase isoform 1 (Mtb ICL-1); and (ii) murine adipocyte fatty acid binding protein (AFABP) complexed with 4-HNE, collectively offer some crucial structural insights into how each of these reactive metabolites may interact with its respective protein target, potentially explaining how these interactions result in their inhibitory behaviors.[33,36]
Itaconate is an anti-microbial metabolite generated from cis-aconitate in mammalian macrophages.[60,61] This small molecule electrophile is a known inhibitor of M. tuberculosis ICLs, enzymes that are essential for pathogen virulence and survival, as they catalyze the (retro)conversion of isocitrate (or methylisocitrate) to glyoxylate (or pyruvate) and succinate, a structural analog of itaconate, in the presence of magnesium ions (Figure 4A, top).[62,63] The reported 1.55 Å crystal structure, obtained after incubation of recombinant ICL-1 with 2.5-fold excess itaconate [4 hours on ice in 50 mM HEPES (pH 7.5), 500 mM NaCl and 5 mM MgCl2] reveals complexation via a covalent bond between the enzyme catalytic site Cys191 and C2 of itaconate (Figure 4A, bottom).[36] The two carboxylate moieties of itaconate additionally form critical hydrogen bonds with a number of other residues present in the catalytic pocket as well as water molecules. Importantly, the C5-carboxylate is stabilized by chelation to the magnesium ion. Consistent with this metal-ligand interaction, follow-up biochemical experiments using recombinant ICL-1 demonstrate that itaconate binding and covalent adduct formation is strictly magnesium-dependent. Interestingly, the rate of itaconate adduction significantly increases in the presence of glyoxylate. The authors rationalize that glyoxylate helps maintain itaconate in a conformation favorable for Michael addition to Cys191. Replacement of carboxylates by methylesters and/or derivatization of C2 leads to decreased inhibitory capacity of the derivatives, presumably indicating that both the negative charge and minimizing steric hindrance are key factors that favor ICL-1 covalent interaction with itaconate. However, since the relative inhibitory efficacies across different itaconate analogs are only assessed by IC50 values (6- to > 20-fold increase, as a result of above-mentioned derivatizations), we cannot discern to what extent the rates of covalent adduction (kinact) are also affected in each case. The authors propose derivatization of itaconate as a potential means to provide new perspectives for the design of itaconate-like ICL-1-selective inhibitors.
Modification of murine AFABP by 4-HNE is another example where relative resemblance of the native electrophilic metabolite to the canonical protein substrate, i.e. fatty acids, likely plays some role in covalent addition of the electrophile to the target protein. 4-HNE is a native LDE involved in several cellular processes, including the antioxidant response through covalent modification of Keap1 and upregulation of the Nrf2/AR pathway discussed above.[29] AFABP was first identified as a potential target of carbonyl-bearing electrophiles by tandem mass spectrometry analysis of protein extracts from obese mouse adipose tissue subjected to biotin-hydrazide treatment.[64] Carbonylated AFABP was detected by western blot analysis using streptavidin-HRP and an anti-4-HNE antibody. The corresponding signals were not present in similar samples derived from AFABP-null mice. These results lead to the proposal that AFABP is a 4-HNE target protein in murine adipose tissue.[33,64] Subsequent co-crystallization experiments yielded a 1.81 Å resolution crystal structure of AFABP covalently complexed with 4-HNE, following incubation of the recombinant protein at an unknown concentration with 500 µM 4-HNE [70 minutes at room temperature in 10 mM potassium phosphate (pH 7.4) and 150 mM NaCl]. The structure hints that 4-HNE may irreversibly inhibit AFABP by blocking the fatty acid binding pocket through covalent bond formation with Cys117 and several non-covalent hydrophobic interactions within the substrate binding site. The residues forming these hydrophobic contacts build similar interactions with the hydrocarbon chain of oleic acid, a known fatty acid substrate of AFABP. Additionally, complexation with 4-HNE leads to a 180° flip of the amide bond between Ala36 and Lys37 compared to the apo form of AFABP, an effect also observed in the presence of oleic acid in the substrate binding site. As 4-HNE adopts a similar conformation tooleic acid in the substrate binding site, and despite obvious chemical and structural differences between the two small molecules (9 versus 18 carbons, α,β-unsaturated aldehyde versus carboxylate head), the authors propose that AFABP may be potentially well-suited to act as an electrophile scavenger. Indeed, fatty acid binding proteins such as AFABP have been postulated to behave as reactive aldehyde scavengers, i.e. antioxidant proteins.[64,65] The authors postulate that as the most abundant protein in adipocytes, such antioxidant activity would only marginally impair fatty acid transport.[66] Impairing AFABP-mediated lipid transport results in a convoluted phenotype that encompasses inflammation and decreased insulin resistance.[67,68] Hellberg et al. rationalize these effects by proposing that AFABP knockout or small molecule inhibition could lead to increased cellular 4-HNE levels, and therefore additional reactions with other electrophile-sensor proteins eliciting cellular responses ultimately leading to additional biological effects, including decreased insulin resistance. Further investigations are however needed to identify protein players involved in this putative process.
4. Capturing structural data on kinetically-privileged electrophile-sensor proteins
Identification of physiologically relevant electrophile-sensor proteins remains challenging. This limitation is in large part due to continued reliance on bolus dosing-based approaches where cells/organisms are bathed in (often excess quantities of) highly reactive and toxic electrophiles, followed by either direct or indirect capture of modified proteins, post-cell lysis. These uncontrolled treatment approaches fail to recapitulate near-physiological conditions, where availability of electrophilic metabolites is limited, depends on specific space, time, and biological context, and where subtle alterations in electrophilic chemotypes can occur due to cellular metabolic processing, reactions with other intracellular small molecules, etc. As a result, top protein hits obtained from bolus treatment approaches often fail functional validation experiments, namely: (i) the observed electrophile-induced phenotype is not ablated by mutation of the electrophile-sensing site identified from bolus dosing; (ii) the observed electrophile-induced phenotype is not ablated (altered) by knocking out (down) the identified protein hit. Indeed, under bolus dosing conditions, many nucleophilic residues on numerous cellular proteins are modified. Parameters such as nucleophilicity of protein sites, nature of the surrounding residues, subcellular pH and redox states, etc., certainly play roles in tuning a given cysteine’s reactivity or kinetic privilege toward RES sensing. However, prediction of functional electrophile-sensing sites within a given target protein remains formidable, especially in the cellular context.[1]
REX technologies are able to deliver an electrophilic metabolite in near-physiological conditions, i.e. a limited amount of electrophile in a specific locale under temporal control. In the precision localized electrophile delivery set-up used in G-REX and Localis-REX, we allow the natural competition between the electrophile’s native diffusion rate and the rate of the electrophile’s irreversible reaction with local proteins/residues (and, to some extent, localized metabolic processing/degradation of the electrophile).[19,28] As a result, the best electrophile-sensor proteins within the locale compete for the electrophile transiently made available in limited quantities. Under these conditions, the KPS proteins in a given biological context can be quantitatively ranked based on ligand occupancy.[17,19,28] Substoichiometric ligand occupancy (10-50 %) are typically observed for numerous KPS proteins that we have validated thus far. [16,17,27,28,30,32] In many cases, such low occupancy modifications are sufficient to drive gain-of-function or dominant negative loss-of-function phenotypes, similarly to what is observed for enzyme-mediated post-translational modifications (PTMs). [10,16,17,27,28,30,32] Notably, functional electrophile-sensing sites are often identified outside of substrate-binding pockets, such as in loops [e.g., RAC-gamma serine/threonine-protein kinase (Akt3)], non-catalytically active sites [e.g., ubiquitin-conjugating enzyme E2 variant 1 and 2 (Ube2v1/2)] or in unstructured regions [e.g., Keap1, cyclin-dependent kinase 9 (Cdk9)].[19,27,28,29] Aside from the current limitations of REX methods that we have described elsewhere, these tools are designed specifically for probing electrophile-sensors and signal propagators in intact living models.[17] As such, they do not offer structure/function insights into electrophile-modified state of the proteins.
Overall, the existing crystal structures of RES-bound protein complexes discussed above provide a collective lens into how pleiotropic electrophilic drugs or native metabolites may help shape target selectivity and/or alter protein function/activity. Notably, all these structures were obtained from co-crystallization experiments, following incubation with supra-physiological amounts of electrophilic compounds. In three out of four of these available structures, the electrophile-binding site is the substrate binding pocket of the target protein, with ~100 % ligand occupancy in at least one case (RSK2/DMF) as estimated by mass spectrometry analysis of recombinant RSK2 CTKD pre-incubated with DMF. Given that functional electrophile signaling is often substoichiometrically operative, akin to canonical PTMs in cell signaling, fundamental structure/function understanding of substoichiometric electrophile sensing and signaling may not necessarily be derivable from these data. There is thus a crucial need for three-dimensional structural data of KPS proteins to which electrophilic drugs or reactive metabolites are functionally bound with substoichiometric ligand occupancy, and on which we can begin to build structure/function knowhow that remains missing to date. Beyond the reactive enals and ene-dicarbonyls such as respectively HNE and DMF, as discussed above, native reactive electrophilic molecules, such as methylglyoxal or 15-deoxy-Δ(12,14)-prostaglandin J₂ (15d-PGJ₂)—nature’s small molecules playing functional roles in various disease-relevant biological processes—constitute additional classes of native reactive electrophiles, where gaining a comprehensive picture of how these electrophiles form functional covalent complexes with their target proteins, could prove helpful.[69,70,71] A couple of crystal structures of 15d-PGJ₂-protein complexes have been reported, although we did not include them here as our discussion was restricted to native reactive electrophiles of 200 Da or less.[72,73,74] On the other hand, and to the best of our knowledge, no structural data exist for the non-Michael-acceptor-based electrophile methylglyoxal-bound proteins.
However, site selective RES modification of proteins in purified systems requires extensive and careful optimization of reaction conditions. In the cellular context, the presence of RES scavengers (e.g., glutathione) as well as the enzyme-assisted metabolic vulnerability of RES, render native RES to be short-lived and mitigate non-specific reactions with non-kinetically-privileged electrophile-sensor sites/proteins.[1,13] By contrast, when proteins in purified systems are subjected to RES in the absence of the above-mentioned ‘self-maintenance or intrinsic regulation’ commonly manifested in intact cells and living organisms, Michael acceptor-based RES often indiscriminately label several nucleophilic sites (typically Cys, Lys, His). As a result, selective labeling of kinetically-privileged functional sites is best achieved by setting the quantity of electrophilic ligand used in the labeling reaction to be substoichiometric (or at least in 1:1 molar ratio) to that of the protein under study.[28,30,32] The reaction time should also be limited to a few seconds/minutes.[28,30,32] Leveraging these conditions, we have successfully developed an in-gel fluorescence approach to derive the apparent bimolecular rate of reactive ligand labeling to multiple KPS proteins.[28,30,32] Careful investigations of a range of concentrations of both the reactive electrophile and KPS protein are recommended, typically in the low micromolar range or lower, using the wild type protein in direct comparison with a functional mutant where the kinetically-privileged sensing residue is mutated to a relevant non-nucleophilic residue. Such titration series using wild type against sensing-defective-but-otherwise-functional mutant provide a reliable approach to identify conditions that mitigate side reactions of the reactive electrophilic ligand under study with less kinetically-privileged nucleophilic sites. We propose that similar strategy can be applied to obtain homogenous proteins covalently bound to a given electrophile ligand at the desired functional sensing site for subsequent structural studies. It is worth noting that for KPS proteins with particularly low ligand occupancy, unreacted electrophile molecules need to be removed from the solution (e.g., by protein precipitation and subsequent refolding, dialysis or using desalting columns) as soon as the reaction is judged complete.
When designing these experiments, special considerations should be given to the choice of reducing agents and their concentrations used during RES-labeling reaction with KPS. Reducing agents are crucial to maintain protein-cysteines in a reduced state, but can lead to unwanted and uncontrolled reduction of/reaction with the RES under study, especially when used in large excess. DTT, β-mercaptoethanol (BME) and TCEP are among the most widely used reducing agents in biochemical and structural investigations. However, as thiol-based molecules, DTT and BME are prone to react with electrophilic compounds such as DMF or 4-HNE, through Michael addition reaction, and may outcompete protein cysteines. As alluded to above, DTT or BME may also lead to reversal of protein cysteine-RES adducts through retro-Michael addition reactions. On the other hand, as a phosphine-based and bulkier reducing agent, TCEP is less likely to react with α,β-unsaturated Michael acceptors, compared to DTT or BME, although the use of TCEP in large excess is best avoided. Quantitative information on rates of adduction of these common reducing agents on native RES or related electrophilic drugs is unfortunately limited. Second-order rate constants at pH 7.4 for (phospha-)Michael addition reaction between acrylamide and TCEP or BME, are approximated to be 0.07 M-1 s-1 and 0.004 M-1 s-1, respectively.[75,76] Reaction of TCEP with β-substituted electrophiles such as HNE and DMF, is however chemically less favored both from steric and electronic grounds. For instance, in our own hands, low concentration of TCEP (up to ~3 mM) is tolerable to numerous HNEylated proteins we have studied thus far, whereas this is not often the case for BME and DTT. It is also worth highlighting here the short half-lives of DTT and BME in solution (10 hours at pH 7.5 and 20 °C).[77] TCEP (in solution in a neutral pH range) is more stable, with only ~10 % oxidation in solution after a week, and should be preferred when setting-up crystallization screens that may remain in contact with air for several weeks/months.[78] A possible alternative, although logistically more complex, is to perform the crystallization screens under inert atmosphere (e.g. inside a glovebox). Some thought/attention should also be given to possible deleterious effects of crystallization buffer components, such as amine- or alcohol-based reagents, as the probability that these nucleophiles inadvertently react with, and impair the studies of, RES increases with time and dosage employed.
Furthermore, in procuring a homogenous ligand-bound state of the KPS protein for three-dimensional structural data acquisition, removal of non-ligand-bound protein molecules may prove necessary. In this case, enrichment of electrophile-bound protein may be achieved by introducing a biorthogonal chemical handle on the electrophilic ligand under study. Judicious choice of a minimally invasive chemical handle is essential to minimize potential bias and to preserve the structure of the electrophile as close as possible to its native state. Finally, where technically possible, the use of solution-based protein NMR spectroscopy or cryo-electron microscopy are likely more suited to study RES-modified proteins compared to protein crystallography. The prolonged crystallization period often required in the latter could result in inadvertent oxidation (due to depletion of available reducing agents), degradation, and/or further reaction of these reactive, and oftentimes labile, small molecules complexed with proteins under study. NMR spectroscopy that allows measurements of protein/ligand dynamics, can additionally offer an enhanced functional understanding of the ligand-bound state.
5. Summary and Outlook
Leveraging the innovative bolus-electrophile-dosing-based and activity-based target profiling methods, and emerging precision REX technologies, our broader understanding of the regulatory roles of electrophilic metabolites and related low molecular weight covalent drugs continues to expand rapidly. Encouragingly, applications of precision REX tools—that to date remain the only means to spatiotemporally map electrophile-sensor proteins and study their ‘on-target’ consequences—in multiple living models have unequivocally shown that precision electrophile regulation operates with low ligand occupancy, similarly to canonical enzyme-mediated PTMs. These results have fundamentally underlined the nuanced role of substoichiometric non-enzyme-assisted electrophilic PTMs in cell decision making.[13,24,79] They have further proven helpful in precision covalent inhibitor design.[10] While advances as such are being made at the live cell/animal-based levels with high spatiotemporal resolution, structure/function basis of substoichiometric electrophile-sensor protein regulation remains a critically missing knowledge gap. However, because of the practical challenges in dealing with reactive small molecules alluded to above, successfully achieving the homogenous protein state with the reactive ligand bound exclusively at the designated functional RES-sensing site clearly necessitates tailored conditions and dedicated reaction optimization efforts in purified protein systems. Nonetheless, we believe that these endeavors will prove worthwhile, and hope that our perspective shines light on the need, relevance, and importance, of being able to establish such structure/function knowhow.
Author Contributions
Compilation of manuscript draft, figures, and references: C.R; Proof-editing: Y.A. All authors agree to the final version of the manuscript.
Acknowledgements
The authors acknowledge SNSF Project funding n°184729.
Conflicts of Interest
Small-molecule inhibitors derived from applications of REX technologies have been filed for patent application.
References
- S. Parvez, M. J. C. Long, J. R. Poganik, Y. Aye, ‘Redox Signaling by Reactive Electrophiles and Oxidants’, Chem. Rev. 2018, 118, 8798–8888. [CrossRef]
- F. J. Schopfer, C. Cipollina, B. A. Freeman, ‘Formation and Signaling Actions of Electrophilic Lipids’, Chem. Rev. 2011, 111, 5997–6021. [CrossRef]
- V. Lampropoulou, A. Sergushichev, M. Bambouskova, S. Nair, E. E. Vincent, E. Loginicheva, L. Cervantes-Barragan, X. Ma, S. C.-C. Huang, T. Griss, C. J. Weinheimer, S. Khader, G. J. Randolph, E. J. Pearce, R. G. Jones, A. Diwan, M. S. Diamond, M. N. Artyomov, ‘Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation’, Cell Metabolism 2016, 24, 158–166. [CrossRef]
- M. Delmastro-Greenwood, B. A. Freeman, S. G. Wendell, ‘Redox-Dependent Anti-Inflammatory Signaling Actions of Unsaturated Fatty Acids’, Annual Review of Physiology 2014, 76, 79–105. [CrossRef]
- J. Singh, R. C. Petter, T. A. Baillie, A. Whitty, ‘The resurgence of covalent drugs’, Nature Reviews Drug Discovery 2011, 10, 307–317. [CrossRef]
- H.-P. Shih, X. Zhang, A. M. Aronov, ‘Drug discovery effectiveness from the standpoint of therapeutic mechanisms and indications’, Nature Reviews Drug Discovery 2018, 17, 19–33. [CrossRef]
- M. J. C. Long, Y. Aye, ‘Privileged Electrophile Sensors: A Resource for Covalent Drug Development’, Cell Chemical Biology 2017, 24, 787–800. [CrossRef]
- M. J. C. Long, A. Kulkarni, Y. Aye, ‘Can Precision Electrophile Signaling Make a Meaningful and Lasting Impression in Drug Design?’, ChemBioChem 2022, 23, e202100051. [CrossRef]
- J. R. Poganik, K.-T. Huang, S. Parvez, Y. Zhao, S. Raja, M. J. C. Long, Y. Aye, ‘Wdr1 and cofilin are necessary mediators of immune-cell-specific apoptosis triggered by Tecfidera’, Nat Commun 2021, 12, 5736. [CrossRef]
- X. Liu, M. J. C. Long, B. D. Hopkins, C. Luo, L. Wang, Y. Aye, ‘Precision Targeting of pten-Null Triple-Negative Breast Tumors Guided by Electrophilic Metabolite Sensing’, ACS Cent. Sci. 2020, 6, 892–902. [CrossRef]
- M. J. Niphakis, B. F. Cravatt, ‘Enzyme Inhibitor Discovery by Activity-Based Protein Profiling’, Annual Review of Biochemistry 2014, 83, 341–377. [CrossRef]
- A. J. Maurais, E. Weerapana, ‘Reactive-cysteine profiling for drug discovery’, Current Opinion in Chemical Biology 2019, 50, 29–36. [CrossRef]
- X. Liu, M. J. C. Long, Y. Aye, ‘Proteomics and Beyond: Cell Decision-Making Shaped by Reactive Electrophiles’, Trends in Biochemical Sciences 2019, 44, 75–89. [CrossRef]
- M. J. C. Long, Y. Aye, ‘Privileged Electrophile Sensors: A Resource for Covalent Drug Development’, Cell Chemical Biology 2017, 24, 787–800. [CrossRef]
- F. Yang, G. Jia, J. Guo, Y. Liu, C. Wang, ‘Quantitative Chemoproteomic Profiling with Data-Independent Acquisition-Based Mass Spectrometry’, J. Am. Chem. Soc. 2022, 144, 901–911. [CrossRef]
- S. Parvez, M. J. C. Long, H.-Y. Lin, Y. Zhao, J. A. Haegele, V. N. Pham, D. K. Lee, Y. Aye, ‘T-REX on-demand redox targeting in live cells’, Nature Protocols 2016, 11, 2328–2356. [CrossRef]
- M. J. C. Long, C. Rogg, Y. Aye, ‘An Oculus to Profile and Probe Target Engagement In Vivo: How T-REX Was Born and Its Evolution into G-REX’, Acc. Chem. Res. 2021, 54, 618–631. [CrossRef]
- M. J. C. Long, D. A. Urul, S. Chawla, H.-Y. Lin, Y. Zhao, J. A. Haegele, Y. Wang, Y. Aye, ‘Precision Electrophile Tagging in Caenorhabditis elegans’, Biochemistry 2018, 57, 216–220. [CrossRef]
- Y. Zhao, P. A. Miranda Herrera, D. Chang, R. Hamelin, M. J. C. Long, Y. Aye, ‘Function-guided proximity mapping unveils electrophilic-metabolite sensing by proteins not present in their canonical locales’, Proceedings of the National Academy of Sciences 2022, 119, e2120687119. [CrossRef]
- A. Van Hall-Beauvais, J. R. Poganik, K.-T. Huang, S. Parvez, Y. Zhao, H.-Y. Lin, X. Liu, M. J. C. Long, Y. Aye, ‘Z-REX uncovers a bifurcation in function of Keap1 paralogs’, eLife 2022, 11, e83373. [CrossRef]
- K.-T. Huang, J. R. Poganik, S. Parvez, S. Raja, B. Miller, M. J. C. Long, J. R. Fetcho, Y. Aye (in press), ‘Z-REX: Shepherding Reactive Electrophiles to Specific Proteins Expressed either Tissue-Specifically or Ubiquitously, and Recording the Resultant Functional Electrophile-Induced Redox Responses in Larval Fish’, Nat Protoc 2023. [CrossRef]
- J. R. Poganik, M. J. C. Long, Y. Aye, ‘Interrogating Precision Electrophile Signaling’, Trends in Biochemical Sciences 2019, 44, 380–381. [CrossRef]
- M. J. C. Long, Y. Aye, ‘The Die Is Cast: Precision Electrophilic Modifications Contribute to Cellular Decision Making’, Chem. Res. Toxicol. 2016, 29, 1575–1582. [CrossRef]
- J. R. Poganik, M. J. C. Long, Y. Aye, ‘Getting the Message? Native Reactive Electrophiles Pass Two Out of Three Thresholds to be Bona Fide Signaling Mediators’, BioEssays 2018, 40, 1700240. [CrossRef]
- M. J. C. Long, H.-Y. Lin, S. Parvez, Y. Zhao, J. R. Poganik, P. Huang, Y. Aye, ‘β-TrCP1 Is a Vacillatory Regulator of Wnt Signaling’, Cell Chemical Biology 2017, 24, 944-957.e7. [CrossRef]
- M. J. C. Long, D. A. Urul, S. Chawla, H.-Y. Lin, Y. Zhao, J. A. Haegele, Y. Wang, Y. Aye, ‘Precision Electrophile Tagging in Caenorhabditis elegans’, Biochemistry 2018, 57, 216–220. [CrossRef]
- M. J. C. Long, S. Parvez, Y. Zhao, S. L. Surya, Y. Wang, S. Zhang, Y. Aye, ‘Akt3 is a privileged first responder in isozyme-specific electrophile response’, Nat Chem Biol 2017, 13, 333–338. [CrossRef]
- Y. Zhao, M. J. C. Long, Y. Wang, S. Zhang, Y. Aye, ‘Ube2V2 Is a Rosetta Stone Bridging Redox and Ubiquitin Codes, Coordinating DNA Damage Responses’, ACS Cent. Sci. 2018, 4, 246–259. [CrossRef]
- S. Parvez, Y. Fu, J. Li, M. J. C. Long, H.-Y. Lin, D. K. Lee, G. S. Hu, Y. Aye, ‘Substoichiometric Hydroxynonenylation of a Single Protein Recapitulates Whole-Cell-Stimulated Antioxidant Response’, J. Am. Chem. Soc. 2015, 137, 10–13. [CrossRef]
- S. L. Surya, M. J. C. Long, D. A. Urul, Y. Zhao, E. J. Mercer, I. M. EIsaid, T. Evans, Y. Aye, ‘Cardiovascular Small Heat Shock Protein HSPB7 Is a Kinetically Privileged Reactive Electrophilic Species (RES) Sensor’, ACS Chem. Biol. 2018, 13, 1824–1831. [CrossRef]
- J. R. Poganik, M. J. C. Long, M. T. Disare, X. Liu, S.-H. Chang, T. Hla, Y. Aye, ‘Post-transcriptional regulation of Nrf2-mRNA by the mRNA-binding proteins HuR and AUF1’, The FASEB Journal 2019, 33, 14636–14652. [CrossRef]
- J. R. Poganik, A. K. Van Hall-Beauvais, M. J. C. Long, M. T. Disare, Y. Zhao, Y. Aye, ‘The mRNA-Binding Protein HuR Is a Kinetically-Privileged Electrophile Sensor’, Helvetica Chimica Acta 2020, 103, e2000041. [CrossRef]
- K. Hellberg, P. A. Grimsrud, A. C. Kruse, L. J. Banaszak, D. H. Ohlendorf, D. A. Bernlohr, ‘X-ray crystallographic analysis of adipocyte fatty acid binding protein (aP2) modified with 4-hydroxy-2-nonenal’, Protein Science 2010, 19, 1480–1489. [CrossRef]
- J. L. Andersen, B. Gesser, E. D. Funder, C. J. F. Nielsen, H. Gotfred-Rasmussen, M. K. Rasmussen, R. Toth, K. V. Gothelf, J. S. C. Arthur, L. Iversen, P. Nissen, ‘Dimethyl fumarate is an allosteric covalent inhibitor of the p90 ribosomal S6 kinases’, Nat Commun 2018, 9, 4344. [CrossRef]
- J. B. Park, H. Park, J. Son, S.-J. Ha, H.-S. Cho, ‘Structural Study of Monomethyl Fumarate-Bound Human GAPDH’, Molecules and Cells 2019, 42, 597–603. [CrossRef]
- B. X. C. Kwai, A. J. Collins, M. J. Middleditch, J. Sperry, G. Bashiri, I. K. H. Leung, ‘Itaconate is a covalent inhibitor of the Mycobacterium tuberculosis isocitrate lyase’, RSC Med. Chem. 2021, 12, 57–61. [CrossRef]
- X.-N. Guan, T. Zhang, T. Yang, Z. Dong, S. Yang, L. Lan, J. Gan, C.-G. Yang, ‘Covalent sortase A inhibitor ML346 prevents Staphylococcus aureus infection of Galleria mellonella’, RSC Med. Chem. 2022, 13, 138–149. [CrossRef]
- ‘FDA Approves Tecfidera - a New Treatment for Multiple Sclerosis’, can be found under https://www.drugs.com/newdrugs/fda-approves-tecfidera-new-multiple-sclerosis-3731.html, n.d.
- D. M. Balak, ‘Fumaric acid esters in the management of psoriasis’, PTT 2015, 5, 9–23. [CrossRef]
- H. A. Blair, ‘Dimethyl Fumarate: A Review in Relapsing-Remitting MS’, Drugs 2019, 79, 1965–1976. [CrossRef]
- ‘FDA Approves Vumerity (diroximel fumarate) for Multiple Sclerosis’, can be found under https://www.drugs.com/newdrugs/fda-approves-vumerity-diroximel-fumarate-multiple-sclerosis-5096.html, n.d.
- ‘FDA Approves Bafiertam (monomethyl fumarate) for Multiple Sclerosis’, can be found under https://www.drugs.com/newdrugs/fda-approves-bafiertam-monomethyl-fumarate-multiple-sclerosis-5252.html, n.d.
- U. Schulze-Topphoff, M. Varrin-Doyer, K. Pekarek, C. M. Spencer, A. Shetty, S. A. Sagan, B. A. C. Cree, R. A. Sobel, B. T. Wipke, L. Steinman, R. H. Scannevin, S. S. Zamvil, ‘Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2’, Proceedings of the National Academy of Sciences 2016, 113, 4777–4782. [CrossRef]
- J. R. Poganik, Y. Aye, ‘Electrophile Signaling and Emerging Immuno- and Neuro-modulatory Electrophilic Pharmaceuticals’, Front. Aging Neurosci. 2020, 12, DOI 10.3389/fnagi.2020.00001. [CrossRef]
- B. Gesser, M. K. Rasmussen, L. Raaby, C. Rosada, C. Johansen, R. B. Kjellerup, K. Kragballe, L. Iversen, ‘Dimethylfumarate inhibits MIF-induced proliferation of keratinocytes by inhibiting MSK1 and RSK1 activation and by inducing nuclear p-c-Jun (S63) and p-p53 (S15) expression’, Inflamm. Res. 2011, 60, 643–653. [CrossRef]
- M. Sauerland, R. Mertes, C. Morozzi, A. L. Eggler, L. F. Gamon, M. J. Davies, ‘Kinetic assessment of Michael addition reactions of alpha, beta-unsaturated carbonyl compounds to amino acid and protein thiols’, Free Radical Biology and Medicine 2021, 169, 1–11. [CrossRef]
- M. M. Blewett, J. Xie, B. W. Zaro, K. M. Backus, A. Altman, J. R. Teijaro, B. F. Cravatt, ‘Chemical proteomic map of dimethyl fumarate–sensitive cysteines in primary human T cells’, Science Signaling 2016, 9, rs10–rs10. [CrossRef]
- E. Kotelnikova, N. A. Kiani, D. Messinis, I. Pertsovskaya, V. Pliaka, M. Bernardo-Faura, M. Rinas, G. Vila, I. Zubizarreta, I. Pulido-Valdeolivas, T. Sakellaropoulos, W. Faigle, G. Silberberg, M. Masso, P. Stridh, J. Behrens, T. Olsson, R. Martin, F. Paul, L. G. Alexopoulos, J. Saez-Rodriguez, J. Tegner, P. Villoslada, ‘MAPK pathway and B cells overactivation in multiple sclerosis revealed by phosphoproteomics and genomic analysis’, Proceedings of the National Academy of Sciences 2019, 116, 9671–9676. [CrossRef]
- K. Uchida, E. R. Stadtman, ‘Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction.’, Journal of Biological Chemistry 1993, 268, 6388–6393. [CrossRef]
- M. Blatnik, N. Frizzell, S. R. Thorpe, J. W. Baynes, ‘Inactivation of Glyceraldehyde-3-Phosphate Dehydrogenase by Fumarate in Diabetes: Formation of S-(2-Succinyl)Cysteine, a Novel Chemical Modification of Protein and Possible Biomarker of Mitochondrial Stress’, Diabetes 2008, 57, 41–49. [CrossRef]
- M. D. Kornberg, P. Bhargava, P. M. Kim, V. Putluri, A. M. Snowman, N. Putluri, P. A. Calabresi, S. H. Snyder, ‘Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity’, Science 2018, 360, 449–453. [CrossRef]
- D. Werdenberg, R. Joshi, S. Wolffram, H. p. Merkle, P. Langguth, ‘Presystemic metabolism and intestinal absorption of antipsoriatic fumaric acid esters’, Biopharmaceutics & Drug Disposition 2003, 24, 259–273. [CrossRef]
- N. H. Litjens, E. van Strijen, C. van Gulpen, H. Mattie, J. T. van Dissel, H. B. Thio, P. H. Nibbering, ‘In vitro pharmacokinetics of anti-psoriatic fumaric acid esters’, BMC Pharmacology 2004, 4, 22. [CrossRef]
- H.-Y. Lin, J. A. Haegele, M. T. Disare, Q. Lin, Y. Aye, ‘A Generalizable Platform for Interrogating Target- and Signal-Specific Consequences of Electrophilic Modifications in Redox-Dependent Cell Signaling’, J. Am. Chem. Soc. 2015, 137, 6232–6244. [CrossRef]
- M. J. C. Long, Y. Aye, ‘Keap 1: The new Janus word on the block’, Bioorganic & Medicinal Chemistry Letters 2022, 71, 128766. [CrossRef]
- S. Unni, P. Deshmukh, G. Krishnappa, P. Kommu, B. Padmanabhan, ‘Structural insights into the multiple binding modes of Dimethyl Fumarate (DMF) and its analogs to the Kelch domain of Keap1’, The FEBS Journal 2021, 288, 1599–1613. [CrossRef]
- E. Crisman, P. Duarte, E. Dauden, A. Cuadrado, M. I. Rodríguez-Franco, M. G. López, R. León, ‘KEAP1-NRF2 protein–protein interaction inhibitors: Design, pharmacological properties and therapeutic potential’, Medicinal Research Reviews 2022, n/a. [CrossRef]
- L. Baird, M. Yamamoto, ‘The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway’, Molecular and Cellular Biology 2020, 40, e00099-20. [CrossRef]
- J. Lin, J. Ren, D. S. Gao, Y. Dai, L. Yu, ‘The Emerging Application of Itaconate: Promising Molecular Targets and Therapeutic Opportunities’, Frontiers in Chemistry 2021, 9. [CrossRef]
- T. Cordes, A. Michelucci, K. Hiller, ‘Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite’, Annual Review of Nutrition 2015, 35, 451–473. [CrossRef]
- L. A. J. O’Neill, M. N. Artyomov, ‘Itaconate: the poster child of metabolic reprogramming in macrophage function’, Nat Rev Immunol 2019, 19, 273–281. [CrossRef]
- J. D. McKinney, K. H. zu Bentrup, E. J. Muñoz-Elías, A. Miczak, B. Chen, W.-T. Chan, D. Swenson, J. C. Sacchettini, W. R. Jacobs, D. G. Russell, ‘Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase’, Nature 2000, 406, 735–738. [CrossRef]
- E. J. Muñoz-Elías, A. M. Upton, J. Cherian, J. D. McKinney, ‘Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence’, Molecular Microbiology 2006, 60, 1109–1122. [CrossRef]
- P. A. Grimsrud, M. J. Picklo, T. J. Griffin, D. A. Bernlohr, ‘Carbonylation of Adipose Proteins in Obesity and Insulin Resistance: Identification of Adipocyte Fatty Acid-binding Protein as a Cellular Target of 4-Hydroxynonenal*’, Molecular & Cellular Proteomics 2007, 6, 624–637. [CrossRef]
- A. Bennaars-Eiden, L. Higgins, A. V. Hertzel, R. J. Kapphahn, D. A. Ferrington, D. A. Bernlohr, ‘Covalent Modification of Epithelial Fatty Acid-binding Protein by 4-Hydroxynonenal in Vitro and in Vivo: EVIDENCE FOR A ROLE IN ANTIOXIDANT BIOLOGY*’, Journal of Biological Chemistry 2002, 277, 50693–50702. [CrossRef]
- V. Matarese, M. K. Buelt, L. L. Chinander, D. A. Bernlohr, in Methods in Enzymology, Academic Press, 1990, pp. 363–369.
- G. S. Hotamisligil, R. S. Johnson, R. J. Distel, R. Ellis, V. E. Papaioannou, B. M. Spiegelman, ‘Uncoupling of Obesity from Insulin Resistance Through a Targeted Mutation in aP2, the Adipocyte Fatty Acid Binding Protein’, Science 1996, 274, 1377–1379. [CrossRef]
- A. V. Hertzel, L. A. Smith, A. H. Berg, G. W. Cline, G. I. Shulman, P. E. Scherer, D. A. Bernlohr, ‘Lipid metabolism and adipokine levels in fatty acid-binding protein null and transgenic mice’, American Journal of Physiology-Endocrinology and Metabolism 2006, 290, E814–E823. [CrossRef]
- T. Shibata, ‘15-Deoxy-Δ12,14-prostaglandin J2 as an electrophilic mediator’, Bioscience, Biotechnology, and Biochemistry 2015, 79, 1044–1049. [CrossRef]
- T. Yagami, Y. Yamamoto, H. Koma, ‘Physiological and Pathological Roles of 15-Deoxy-Δ12,14-Prostaglandin J2 in the Central Nervous System and Neurological Diseases’, Mol Neurobiol 2018, 55, 2227–2248. [CrossRef]
- S. W. T. Lai, E. D. J. Lopez Gonzalez, T. Zoukari, P. Ki, S. C. Shuck, ‘Methylglyoxal and Its Adducts: Induction, Repair, and Association with Disease’, Chem. Res. Toxicol. 2022, 35, 1720–1746. [CrossRef]
- G. Abis, R. L. Charles, J. Kopec, W. W. Yue, R. A. Atkinson, T. T. T. Bui, S. Lynham, S. Popova, Y.-B. Sun, F. Fraternali, P. Eaton, M. R. Conte, ‘15-deoxy-Δ12,14-Prostaglandin J2 inhibits human soluble epoxide hydrolase by a dual orthosteric and allosteric mechanism’, Commun Biol 2019, 2, 1–14. [CrossRef]
- T. Waku, T. Shiraki, T. Oyama, K. Morikawa, ‘Atomic structure of mutant PPARγ LBD complexed with 15d-PGJ2: Novel modulation mechanism of PPARγ/RXRα function by covalently bound ligands’, FEBS Letters 2009, 583, 320–324. [CrossRef]
- T. Waku, T. Shiraki, T. Oyama, Y. Fujimoto, K. Maebara, N. Kamiya, H. Jingami, K. Morikawa, ‘Structural Insight into PPARγ Activation Through Covalent Modification with Endogenous Fatty Acids’, Journal of Molecular Biology 2009, 385, 188–199. [CrossRef]
- Y.-J. C. Lee, B. Wu, J. E. Raymond, Y. Zeng, X. Fang, K. L. Wooley, W. R. Liu, ‘A Genetically Encoded Acrylamide Functionality’, ACS Chem Biol 2013, 8, 1664–1670. [CrossRef]
- Y.-J. Lee, Y. Kurra, W. R. Liu, ‘Phospha-Michael Addition as a New Click Reaction for Protein Functionalization’, Chembiochem 2016, 17, 456–461. [CrossRef]
- R. Stevens, L. Stevens, N. C. Price, ‘The stabilities of various thiol compounds used in protein purifications’, Biochemical Education 1983, 11, 70–70. [CrossRef]
- E. B. Getz, M. Xiao, T. Chakrabarty, R. Cooke, P. R. Selvin, ‘A Comparison between the Sulfhydryl Reductants Tris(2-carboxyethyl)phosphine and Dithiothreitol for Use in Protein Biochemistry’, Analytical Biochemistry 1999, 273, 73–80. [CrossRef]
- M. J. C. Long, X. Liu, Y. Aye, ‘Genie in a bottle: controlled release helps tame natural polypharmacology?’, Current Opinion in Chemical Biology 2019, 51, 48–56. [CrossRef]
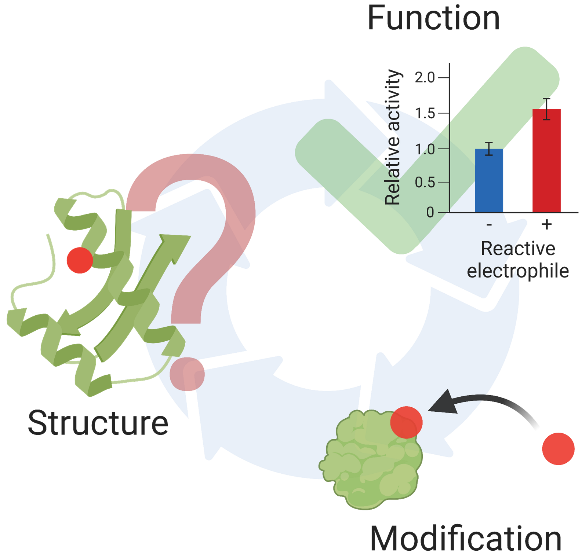
Figure 1.
(A) RES endogenously generated via different cellular processes such as membrane lipid peroxidation or the TCA cycle are “sensed” by kinetically-privileged sensor (KPS) proteins. Such non-enzyme-assisted post-translational electrophile modifications of KPS proteins often result in signaling events, in a process termed “electrophile signaling”. (B) Chemical structures of representative electrophilic metabolites and approved drugs discussed herein, with year of FDA approval shown in parentheses.
Figure 1.
(A) RES endogenously generated via different cellular processes such as membrane lipid peroxidation or the TCA cycle are “sensed” by kinetically-privileged sensor (KPS) proteins. Such non-enzyme-assisted post-translational electrophile modifications of KPS proteins often result in signaling events, in a process termed “electrophile signaling”. (B) Chemical structures of representative electrophilic metabolites and approved drugs discussed herein, with year of FDA approval shown in parentheses.
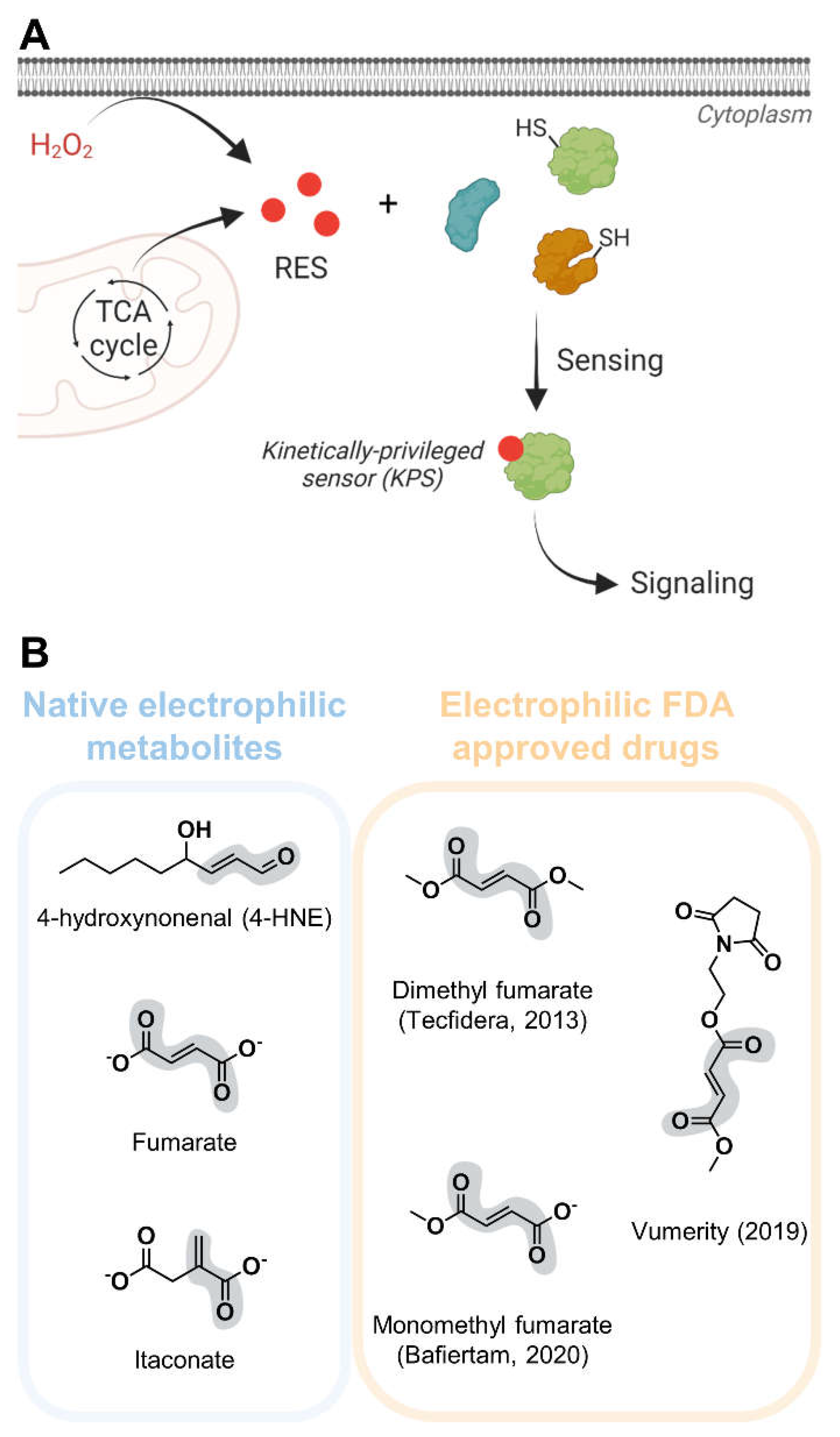
Figure 2.
Electrophile regulation of KPS proteins is often studied in purified systems or intact living models (e.g., cultured cells or whole animals). Studies in animal models with programmable spatiotemporal control are uniquely enabled by precision REX technologies. Accumulating cell-based data and in vivo investigations have shown that RES modification of a given KPS can affect, for instance, its subcellular localization and/or its function/activity, or that of binding partners, ultimately affecting downstream pathways.[9,10,19,27,28] By contrast, structural information on electrophile-bound proteins, especially KPS proteins, in purified systems is still sorely missing, despite its fundamental relevance in the process toward painting a complete picture of precision electrophile signaling regulation.
Figure 2.
Electrophile regulation of KPS proteins is often studied in purified systems or intact living models (e.g., cultured cells or whole animals). Studies in animal models with programmable spatiotemporal control are uniquely enabled by precision REX technologies. Accumulating cell-based data and in vivo investigations have shown that RES modification of a given KPS can affect, for instance, its subcellular localization and/or its function/activity, or that of binding partners, ultimately affecting downstream pathways.[9,10,19,27,28] By contrast, structural information on electrophile-bound proteins, especially KPS proteins, in purified systems is still sorely missing, despite its fundamental relevance in the process toward painting a complete picture of precision electrophile signaling regulation.
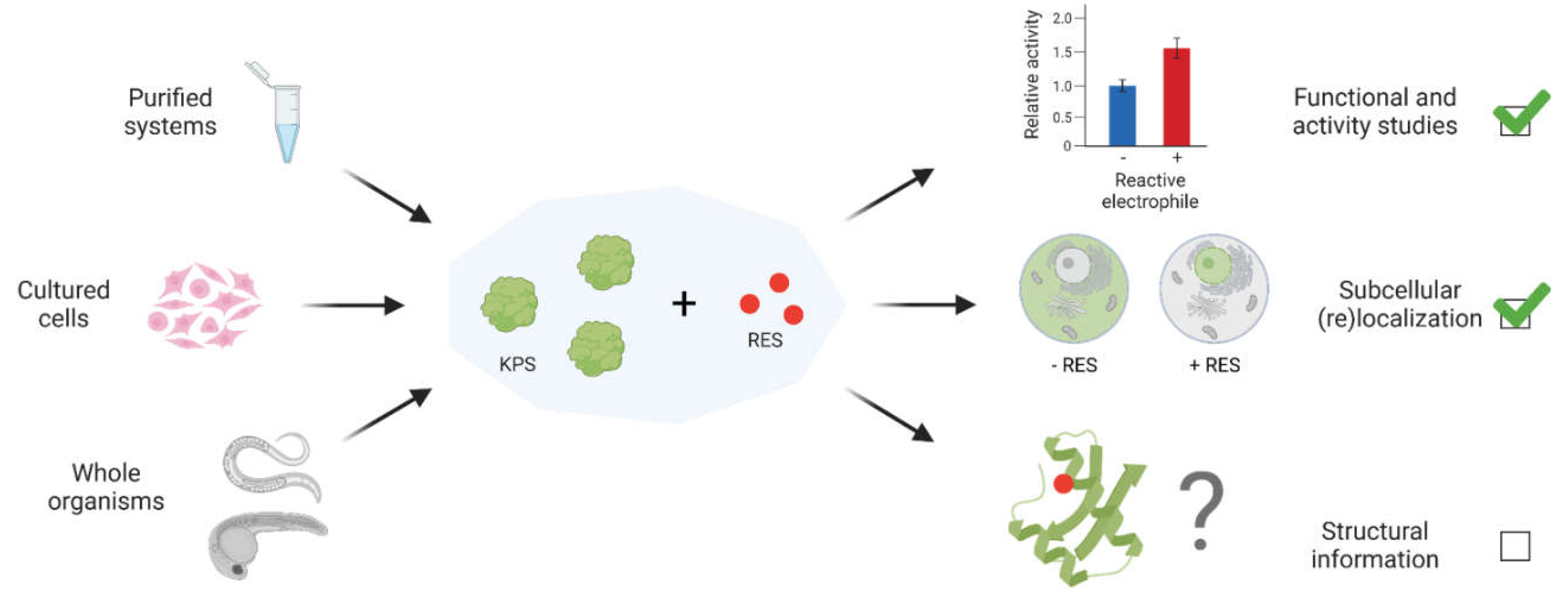
Figure 3.
(A) Crystal structure of RSK2 CTKD domain in its inactive state (light gray, PDB 5O1S) covalently modified by DMF (pink) at Cys436 and Cys599. Movement of the kinase activation loop (orange) into its active conformation (green, from the phosphorylated kinase p70S6K1, PDB 3A62) is postulated to be hampered by steric hindrance resulting from covalent adduction of DMF at Cys599. (B) Crystal structure of tetrameric human GAPDH covalently modified by MMF (pink) at Cys152 in each monomer (colored ribbons, PDB 6IQ6) superimposed with the crystal structure of NAD+-bound human GAPDH (light gray ribbons, NAD+ in dark grey, PDB 4WNC) shows steric clash between MMF and the co-factor in the substrate binding pocket. The crystal structure revealed two binding modes of MMF: in addition to the formation of a covalent bond with Cys152, MMF forms a hydrogen bond with either His179 (shown here) or Asn316.
Figure 3.
(A) Crystal structure of RSK2 CTKD domain in its inactive state (light gray, PDB 5O1S) covalently modified by DMF (pink) at Cys436 and Cys599. Movement of the kinase activation loop (orange) into its active conformation (green, from the phosphorylated kinase p70S6K1, PDB 3A62) is postulated to be hampered by steric hindrance resulting from covalent adduction of DMF at Cys599. (B) Crystal structure of tetrameric human GAPDH covalently modified by MMF (pink) at Cys152 in each monomer (colored ribbons, PDB 6IQ6) superimposed with the crystal structure of NAD+-bound human GAPDH (light gray ribbons, NAD+ in dark grey, PDB 4WNC) shows steric clash between MMF and the co-factor in the substrate binding pocket. The crystal structure revealed two binding modes of MMF: in addition to the formation of a covalent bond with Cys152, MMF forms a hydrogen bond with either His179 (shown here) or Asn316.
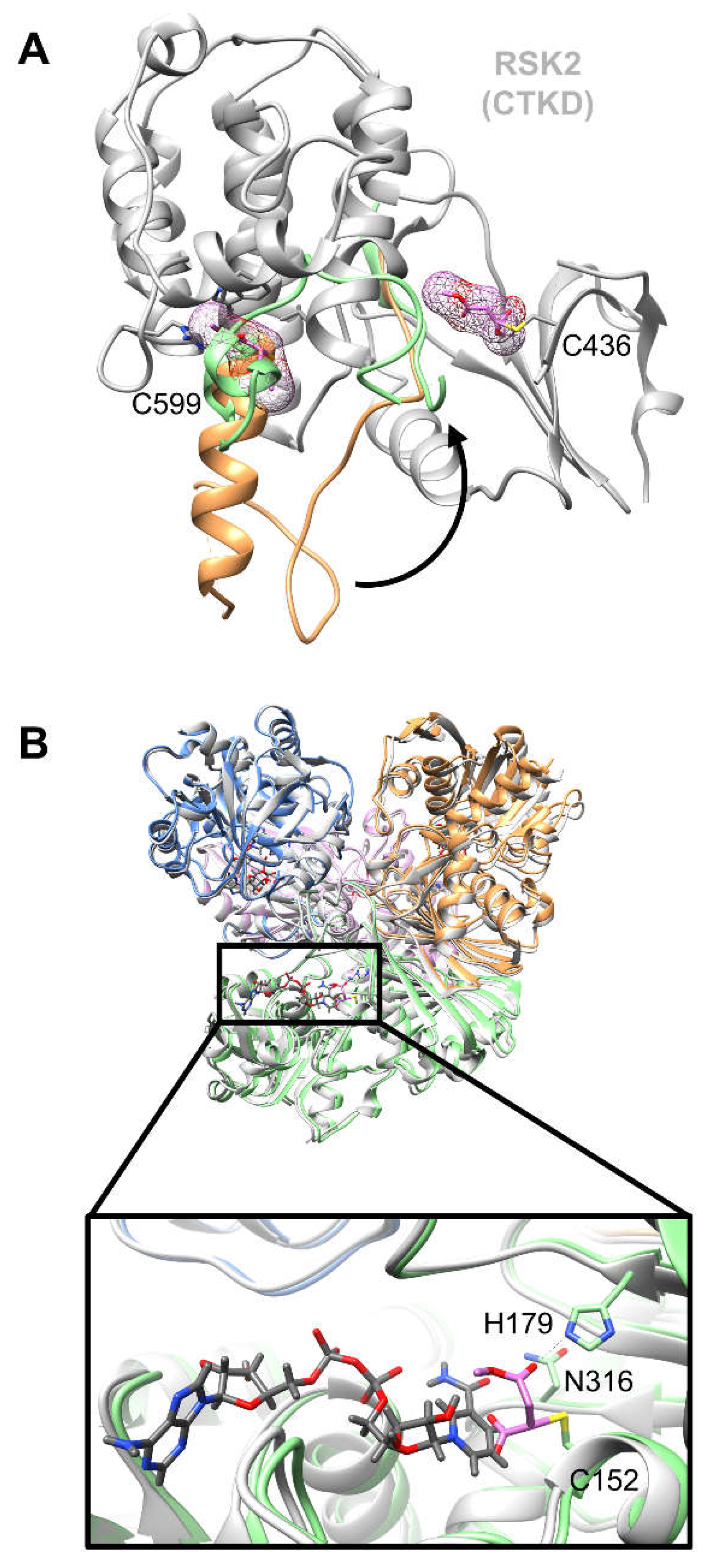
Figure 4.
(A) Itaconate is a known inhibitor of ICL-1-mediated conversion of (methyl)isocitrate to succinate and glyoxylate/pyruvate. The inhibition mechanism involves itaconate (pink) adduction of the catalytic site Cys191 within each monomer of tetrameric ICL-1 (colored ribbons, PDB 6XPP). Carboxylate groups within itaconate are stabilized in the substrate-binding pocket through columbic interactions with a magnesium ion (green sphere) and via hydrogen bonding with neighboring residues and water molecules (red spheres). (B) 4-HNE (pink) inhibits AFABP (green ribbons, PDB 3JS1) by forming a covalent bond with residue Cys117 and indirect hydrogen bonds with Arg126 and Tyr 128 supported by water molecules (red spheres); superimposing the structure of oleic acid-bound AFABP (oleic acid in dark grey, light gray ribbons, PDB 1LID) shows 4-HNE positions itself in the substrate-binding pocket, likely leading to blockage of AFABP-mediated fatty-acid transport due to steric hindrance. Note: in both structures, amino acids Asp71 to Lys81 are hidden for clarity.
Figure 4.
(A) Itaconate is a known inhibitor of ICL-1-mediated conversion of (methyl)isocitrate to succinate and glyoxylate/pyruvate. The inhibition mechanism involves itaconate (pink) adduction of the catalytic site Cys191 within each monomer of tetrameric ICL-1 (colored ribbons, PDB 6XPP). Carboxylate groups within itaconate are stabilized in the substrate-binding pocket through columbic interactions with a magnesium ion (green sphere) and via hydrogen bonding with neighboring residues and water molecules (red spheres). (B) 4-HNE (pink) inhibits AFABP (green ribbons, PDB 3JS1) by forming a covalent bond with residue Cys117 and indirect hydrogen bonds with Arg126 and Tyr 128 supported by water molecules (red spheres); superimposing the structure of oleic acid-bound AFABP (oleic acid in dark grey, light gray ribbons, PDB 1LID) shows 4-HNE positions itself in the substrate-binding pocket, likely leading to blockage of AFABP-mediated fatty-acid transport due to steric hindrance. Note: in both structures, amino acids Asp71 to Lys81 are hidden for clarity.
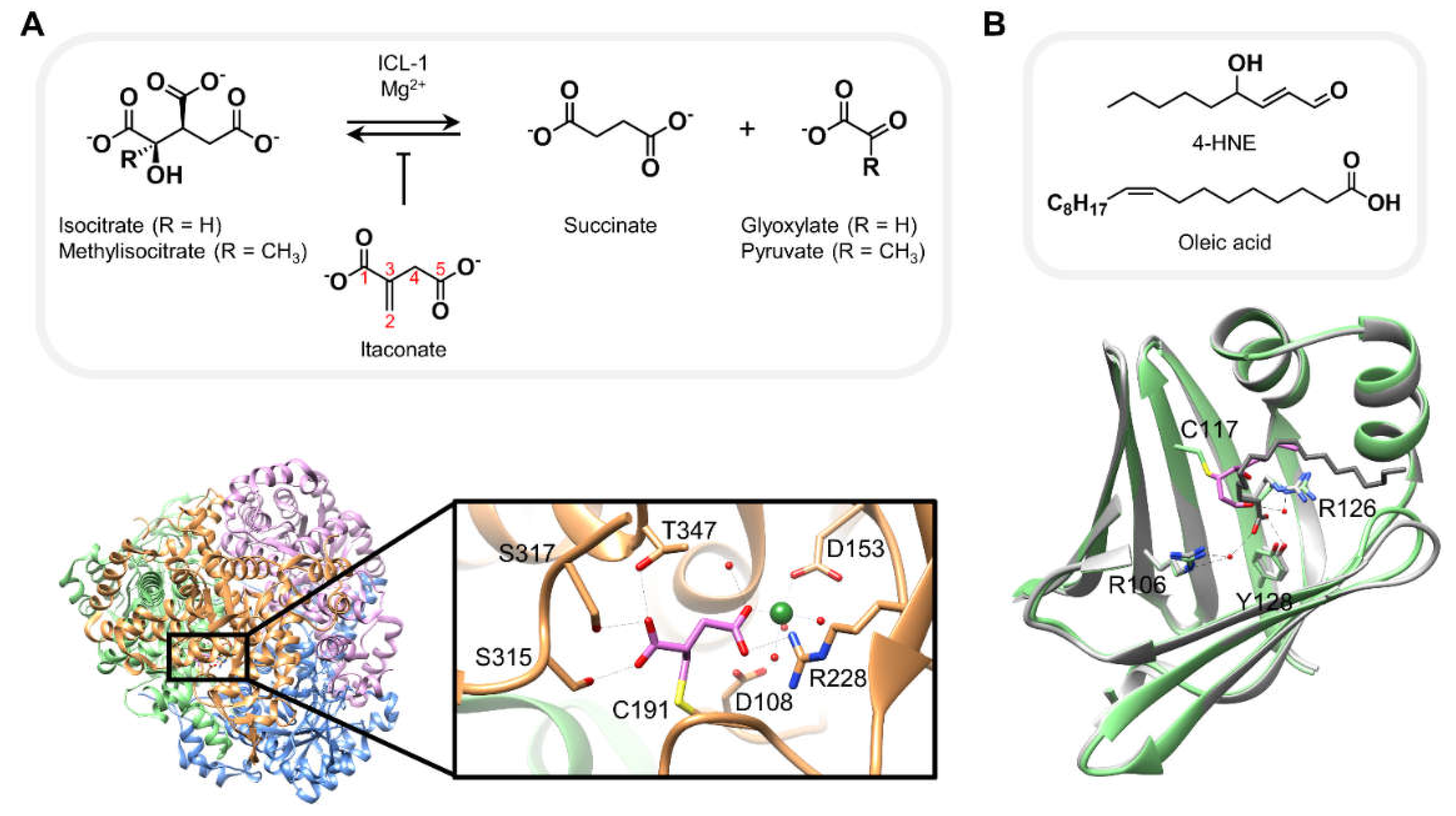

Table 1.
Available structures of proteins covalently complexed to small molecule electrophilic drugs and metabolites.
Table 1.
Available structures of proteins covalently complexed to small molecule electrophilic drugs and metabolites.
| PDB code | Electrophile | Protein | Binding site | Resolution (Å) |
|---|---|---|---|---|
| 6XPP | Itaconate (a) | ICL-1 | Cys191 (c) | 1.55 |
| 3JS1 | 4-hydroxynonenal (a) | AFABP | Cys117 (d) | 1.81 |
| 6IQ6 | Monomethyl fumarate (b) | GAPDH | Cys152 (c) | 2.29 |
| 5O1S | Dimethyl fumarate (b) | RSK2 | Cys599 (e), Cys436 (e) |
1.90 |
| (a) endogenous electrophile, (b) approved drug, (c) catalytic site, (d) endogenous ligand binding site, (e) allosteric site. | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



