
Submitted:
16 January 2023
Posted:
17 January 2023
You are already at the latest version
Abstract
AIM: The study aims to identify if mutations in the SLC4A11 gene are present in Filipino families affected with Congenital Hereditary Endothelial Dystrophy (CHED). METHODS: This is a family cohort study that investigated the genetic profile of a selected family in Northern Luzon, Philippines, whose members were diagnosed with Congenital Hereditary Endothelial Dystrophy (CHED). A patient who was diagnosed with CHED prior to this study, served as the proband for this family. A detailed family history and a complete ophthalmologic examination were done to each of the family members. A total of six affected members and three unaffected members were included in this study. DNA was isolated from peripheral blood samples of family members, and polymerase chain reaction (PCR) was used to amplify the gene’s entire coding region, (19 exons and 2 putative promoter regions), and finally, the amplified regions were analyzed using DNA sequencing. RESULTS: Consanguinity was not present in the family. Corneal haze was reported to be present since birth or shortly thereafter in all affected patients. Slit-lamp examination showed edematous corneas. Molecular studies of the SLC4A11 gene revealed four novel homozygous point mutations variably present in six affected members as well as three unaffected members. One unaffected family member (I-1) had a novel sense mutation absent in other family members. All affected siblings showed little phenotypic variability. CONCLUSION: This is the first report that gives us a genetic profile of a Northern Luzon family with members affected by CHED. This study supports earlier findings that the mutations in the SLC4A11 gene are not consistently the same among different ethnic groups worldwide, probably due to the disease’s genetic heterogeneity. Our study documented five novel mutations adding to the growing list of mutations probably responsible for acquiring the CHED phenotype. It is possible that there are more novel mutations waiting to be discovered in this hereditary disease. Screening for these specific mutations in other families may prove useful for genetic counseling, prenatal diagnosis and the future development of gene therapy.
Keywords:
Congenital Hereditary Endothelial Dystrophy
; Mutation
; SLC4A11 gene
; Cornea
1. Introduction
Congenital hereditary endothelial dystrophy (CHED) is characterized by bilateral, diffuse corneal clouding that impairs visual acuity. The condition is classically known to occur in two genetic forms: autosomal dominant (CHED1) and autosomal recessive (CHED2), however this classification has been recently modified [1]. The severity of the disorder varies, the latter being more severe and usually more common and many patients require penetrating keratoplasty to restore vision. CHED1 and CHED2 have been mapped to chromosome 20 at two distinct loci. The primary abnormality is attributed to dysfunctional endothelial cells, resulting in thickening and opacification of the cornea [2].
The exact prevalence, sex distribution, and incidence of CHED are unknown at the moment while some authors reported an incidence of 3 per 100,000 births [3]. Most cases have been identified in children of consanguineous parents from Saudi Arabia, India, Pakistan, Myanmar (Burma) and Ireland [4].
CHED2 or now simply called CHED is reportedly caused by mutations in the SLC4A11 gene that is mapped to chromosome 20p13. This gene is made up of 20 exons, of which 19 of those exons, codes for an 891 amino acid membrane protein that functions as a sodium-coupled borate cotransporter 1 highly expressed in several organs and tissues including the eye (e.g., corneal endothelium).
More than 60 different homozygous or compound heterozygous mutations in this gene have been reported to cause either CHED or Harboyan syndrome (characterized by CHED in association with sensorineural hearing loss) with little evidence to support a genetic difference between these phenotypes. These individual mutations either affect the protein structure or its function, which ultimately leads to a defective corneal endothelium [5].
Mutational studies of SLC4A11 have been done which involves mainly patients from India [6] as well as in Korea [7] and in China [8]. At present, there are no published data available in the Philippines. This study determined the presence of mutations in the SLC4A11 gene, characterized the mutation, and assessed the role of these mutations in the causation of CHED2 in these particular Northern Luzon patients.
2. Methodology
Prior to the initiation of the study, the study protocol was reviewed and approved by the Institutional Ethics Review Committee (IERC) of St. Luke’s Medical Center, Philippines. Informed consent was obtained from all study subjects before any procedures were done.
A patient who has been diagnosed with CHED and who has been treated in our institution was selected as a proband for a family to be included in the study. The sample population included the proband’s nuclear family and extended family. The study group included all subjects diagnosed clinically with CHED during screening. A separate study group included all unaffected subjects in the family being screened. Detailed family histories were collected and a pedigree chart was constructed to represent the subjects. Consanguinity was also noted based on marriage histories if present.
The study included the proband and his/her family members within one generation higher and lower. Family members who are not directly related to the proband by blood and those unaffected by the disease who are below 18 years old were excluded.
3. Clinical Studies
The clinical examination primarily involved portable ophthalmic equipment since the family resided in a remote region of the Philippines (Mayoyao, Ifugao). These included routine slit-lamp biomicroscopy and a tonopen to take the intraocular pressure. Ancillary diagnostics such as specular microscopy, ultrasonic pachymetry, and ultrasonography for posterior segment evaluation were offered to the family members who decided to visit our hospital. The diagnosis of CHED was made clinically on the basis of the following characteristics: presence of mosaic corneal haze with corneal edema or an increased central corneal thickness (>0.7 mm in all cases), normal horizontal corneal diameter (10–11 mm), and no evidence of congenital glaucoma (e.g., no buphthalmos, Haab’s striae, or optic disc cupping). Affected family members formed the study group and a separate study group included all family members unaffected by the disease.
4. SLC4A11 Gene Mutation Screening Analysis
4.1. Sample Collection
Eight milliliters of peripheral blood were collected from enrolled patients. Blood was then drawn in ethylenediamine tetraacetic acid (EDTA) tubes and processed for buffy coat separation. Each tube was centrifuged at 4000 rpm for 10 mins at room temperature (250C). After centrifugation, the buffy coat layer was collected and placed in 1.5 ml microcentrifuge tubes. Each tube was centrifuged at 3000 rpm for 15 minutes at room temperature. Excess layer (plasma or RBC) was removed and the buffy coat was processed for extraction of genomic DNA.
4.2. DNA Extraction
Collected buffy coat was processed for DNA extraction according to QiaAmp DNA MiniKit ®. Briefly, 200 µl of the buffy coat was lysed using a lysis solution (Buffer AL) and proteinase K was added to degrade proteins. Cells were incubated at 560C for 10 minutes or until complete lysis. To precipitate the isolated DNA, ethanol was added to each sample. Wash buffers (AW1 and AW2) were added separately to the spin columns to facilitate the removal of contaminants. To elute purified genomic DNA (gDNA), Buffer AE was added to the spin columns. The DNA quality and quantity of the extracted eluent were assessed using Nanodrop® v1000 spectrophotometer. A final working concentration of at least 50 ng/µl of gDNA was used for each sample.
4.3. Gene Amplification
DNA sequences of certain regions of the SLC4A11 gene were amplified as described by Paliwal et al [9] with some modifications, mainly in the annealing temperatures conditions for each exon (Table 1). In a 25 ul PCR reaction, 10 ng DNA, 1.5 mM MgCl2, 0.25 mM of each dNTP, 10pM of each primer, and 0.7 units of Taq polymerase, and 1X Q-solution (Qiagen GmBH, Hilden, Germany) were mixed. Amplified samples were visualized on 2% agarose gel to determine fragment length. PCR products were purified and processed for DNA sequencing.
4.4. DNA Sequencing and Mutational analysis
All PCR products were purified using NucleoSpin® Gel and PCR Clean-up. Purified PCR products were sent to 1st Base Sequencing (Singapore) in triplicates for DNA Sequencing Services Single Pass Reaction.
Nucleotide sequences for the coding regions were compared with the reference nucleotide sequence of SLC4A11 gene on human chromosome 20 (NG_017072.1). Generated DNA sequences were analyzed using BioEdit, 4Peaks, and Seqotron software for alignment with the reference sequence and checked for mutations. Individuals who did not show SLC4A11 coding region changes were screened for mutations in the putative promoter region as well using their respective primers.
5. Results
Clinical Examination and Demographics
One family was recruited during the visit to Northern Luzon. The age of all family members being screened ranged from 11 to 57 years (Table 2). Based on the pedigree (Figure 1) constructed for the affected family, the disease appears to be transmitted recessively. The disease was only present in generation II where 6 out of 9 siblings (66.67%) were affected. In the family studied, both parents were unaffected and the presence of consanguinity was not noted.

Table 1.
Summary of forward and reverse primers that amplified certain exon regions for nucleotide sequence analysis. The expected product size and annealing temperatures used for each primer pair used for amplification are also presented. Reference gene sequence NG_017072.1 was used for succeeding comparison and analysis as well as exon designation to maintain consistency with NCBI website.
Table 1.
Summary of forward and reverse primers that amplified certain exon regions for nucleotide sequence analysis. The expected product size and annealing temperatures used for each primer pair used for amplification are also presented. Reference gene sequence NG_017072.1 was used for succeeding comparison and analysis as well as exon designation to maintain consistency with NCBI website.
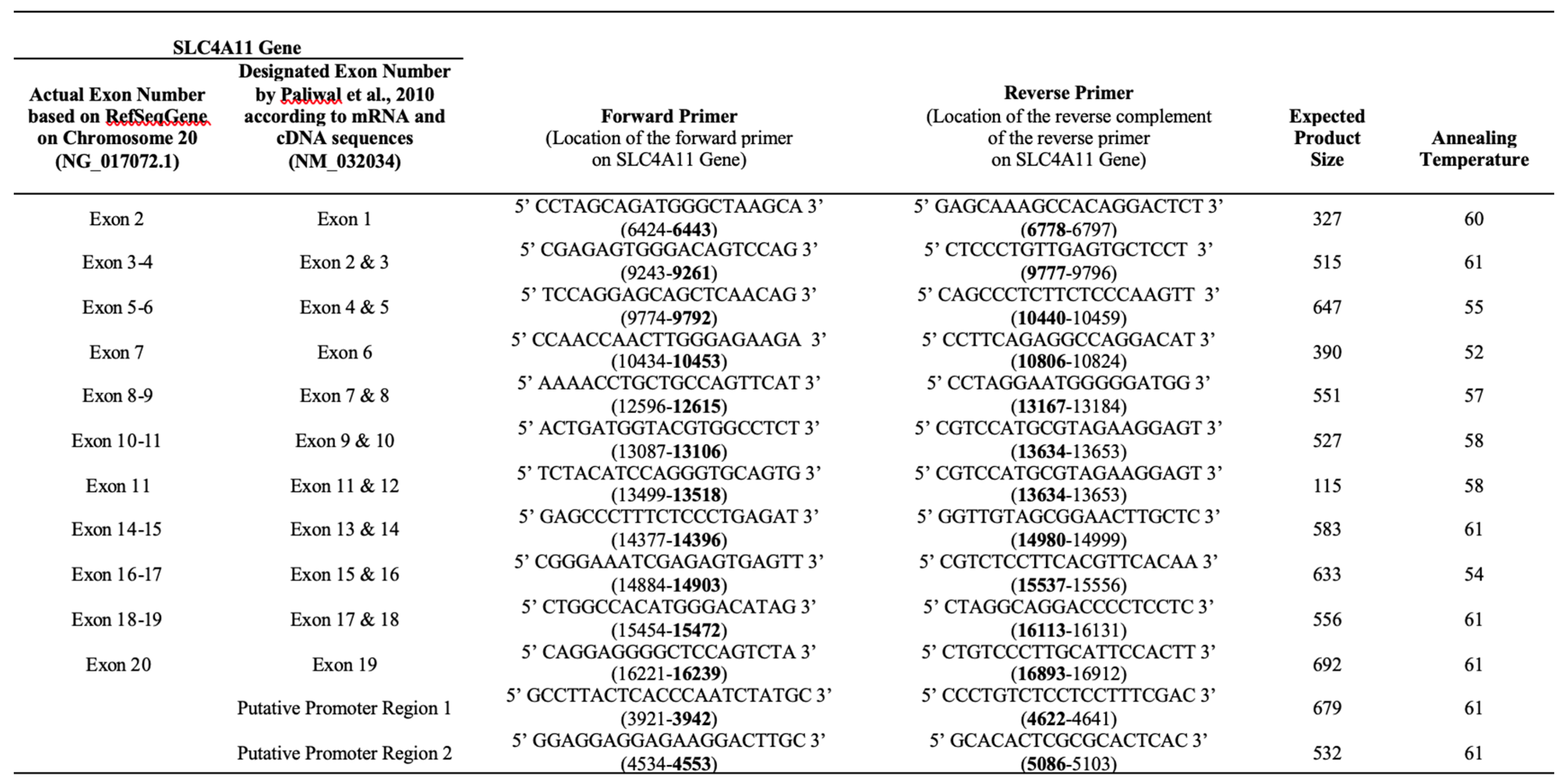
Table 2.
Summary of exon numbering and its corresponding locations in the SLC4A11 gene. This is based on the reference sequence gene NG_017072.1 (NCBI).
Table 2.
Summary of exon numbering and its corresponding locations in the SLC4A11 gene. This is based on the reference sequence gene NG_017072.1 (NCBI).
| Exon Number | Location | Exon Number | Location |
|---|---|---|---|
| Exon 1 | 5052-5147 | Exon 11 | 13397-13510 |
| Exon 2 | 6654-6698 | Exon 12 | 13595-13727 |
| Exon 3 | 9348-9500 | Exon 13 | 13982-14055 |
| Exon 4 | 9625-9674 | Exon 14 | 14466-14718 |
| Exon 5 | 9928-10159 | Exon 15 | 14790-14896 |
| Exon 6 | 10240-10321 | Exon 16 | 14979-15147 |
| Exon 7 | 10605-10728 | Exon 17 | 15231-15404 |
| Exon 8 | 12694-12912 | Exon 18 | 15535-15730 |
| Exon 9 | 13000-13093 | Exon 19 | 15814-15983 |
| Exon 10 | 13184-13309 | Exon 20 | 16386-16820 |
A total of 6 family members were clinically diagnosed with CHED with an age range of 11 to 37 years (Table 2). All of them presented with bilateral corneal opacity shortly after birth that gradually progressed towards early childhood. The average age of onset was noted to be 1 year after birth while significant visual symptoms were first appreciated at ages 6 to 7 years old. Corneal pachymetry revealed increased corneal thickness (>0.7 mm) in 2 patients prior to undergoing a corneal transplant. There was no pachymetry data for the other 4 patients, however slit-lamp examination revealed marked corneal edema bilaterally. The intraocular pressure was within the normal range (below 21 mmHg) for all patients. Best-corrected visual acuity (BCVA) among surgically untreated CHED patients ranged from 20/640 to 20/800 while the BCVA of surgically treated CHED patients ranged from 20/30 to 20/70 when excluding eyes with complications. Eyes with BCVA of CHED patients with a failed graft ranged from 20/800 to Hand movement. No other associated eye diseases were found during our examination. One patient (II-4) in family 1 reportedly had associated bilateral hearing loss though this was only subjective as there was no appropriate equipment available to properly document the associated symptoms.
Mutation Analysis
Certain challenges were met during PCR optimization as the published PCR conditions by Paliwal et al that was initially followed did not work in our laboratory. Furthermore, it was found that the primers used had not followed the conventional exon numbering being amplified in the whole SLC4A11 gene. The exon numbering that they had followed was based on the exons present in a messenger RNA (mRNA) transcript and the complementary DNA sequence (NM_032034). Nevertheless, the primers used were able to amplify sequences on both coding and non-coding regions of the SLC4A11 gene. The corrected numbering of exons as well as the amplified coding and non-coding regions were adapted based on the SLC4A11 reference gene sequence on chromosome 20 (NG_017072.1) available at the NCBI website as shown in Table 1. The exact locations of the 20 exons spanning the SLC4A11 gene are presented in Table 2.
One family was recruited during the visit to Northern Luzon. The age of all family members being screened ranged from 11 to 57 years (Table 2). Based on the pedigree (Figure 1) constructed for the affected family, the disease appears to be transmitted recessively. The disease was only present in generation II where 6 out of 9 siblings (66.67%) were affected. In the family studied, both parents were unaffected and the presence of consanguinity was not noted.
Direct DNA sequence analysis showed five novel point mutations scattered in both coding and non-coding regions of the SLC4A11 gene (Table 3): present in non-coding regions are 4291G>A (located before Exon 1), 6533G>C (located between Exon 1 and Exon 2), and 13165T>C (located between Exon 9 and 10); present in coding regions, specifically within Exon 6 are 10294G>C and 10307G>A (Figure 3). Both point mutations 4291G>A and 6533G>C are present in all the patients. The point mutation 13165T>C is absent in all patients except Patient I-1 and Patient II-8 who exhibited either presence or absence of nucleotide change suggesting heterozygosity.
Table 3.
Clinical details of screened family members and mutations found in the SLC4A11 gene.
|
Family 1 Generation No. |
Age/Sex |
CCT OD/OS |
Age at PK |
BCVAOD |
BCVAOS |
Complications |
Position of Nucleotide Change in SLC4A11 Gene | |||
|---|---|---|---|---|---|---|---|---|---|---|
| 4291G>A (Before Exon1) |
6533G>C (Between Exon1&2) |
10294G>C 10307G>A (in Exon6) |
13165T>C (Between Exon9&10) |
|||||||
| I-1 | 57/M | - | - | 20/30 | 20/30 | - | Present | Present | Absent | Present |
| I-2 | 57/F | - | - | 20/30 | 20/30 | - | Present | Present | Present | Absent |
| II-1* | 37/M | - | - | 20/640 | 20/800 | - | Present | Present | Present | Absent |
| II-2* | 35/M | - | - | 20/640 | 20/640 | - | Present | Present | Present | Absent |
| II-4* | 31/F | - | - | 20/800 | 20/800 | - | Present | Present | Present | Absent |
| II-6* | 27/F | >0.7 mm OU | 25 yrs. | 20/800 | 20/70 | Failed graft OD | Present | Present | Present | Absent |
| II-7* | 19/M | >0.7 mm OU | 17 yrs. | 20/30 | HM | Failed graft OS | Present | Present | Present | Absent |
| II-8 | 16/M | - | - | 20/20 | 20/20 | - | Present | Present | Present | Either (Probably heterozygous) |
| II-9* | 11/F | - | - | 20/800 | 20/800 | - | Present | Present | Present | Absent |
PK-Penetrating keratoplasty, P- Present, A- Absent, F- Female, M- male, “-”- No Data, CCT- Central corneal thickness, BCVA-Best corrected visual acuity, OD-Right eye, OS-Left Eye, HM-Hand movement, “*”-affected with CHED.
Interestingly, the two point mutations 10294G>C and 10307G>A, opposite to the 13165T>C mutation, are present in all patients except Patient I-1. Knowing that these mutations are located within Exon 6, mRNA transcripts of both normal and mutated forms were used to generate an amino acid sequence using ExPASy tool. The resulting 5’ to 3’ Frame was used as it showed the exact amino acid alignment with the reference amino acid sequence of the human sodium bicarbonate transporter-like protein 11 isoform 3 (NP_001167560.1). The 10307G>A point mutation did not result in a change in translated amino acid (Figure 4).
The 10294G>C point mutation is a missense mutation affecting the amino acid 193 to change from arginine to proline (aa193R>P). However, this missense mutation is predicted to be benign in nature using the Polyphen2 protein function prediction tool (Figure 5).
6. Discussion
The SLC4A11 gene has been extensively analyzed by numerous studies. This gene codes for BTR1 (Bicarbonate Transporter-Related protein-1) that functions as a sodium borate co-transporter (NaBC1) and plays a role in the activation of the mitogen-activated protein kinase pathway [9]. Various mutations scattered across the gene have been implicated in a few ophthalmologic conditions mainly involving the corneal endothelium. CHED, Harboyan syndrome and Fuch’s Endothelial Corneal Dystrophy (FECD) all share similar mutations all involving the same gene suggesting these conditions are part of a broader disease spectrum. This common disease feature, while underlying the importance of SLC4A11 protein for the correct development and differentiation of the corneal endothelium, may explain how the same gene can be involved in the pathogenesis of both congenital and late-onset disease [11].
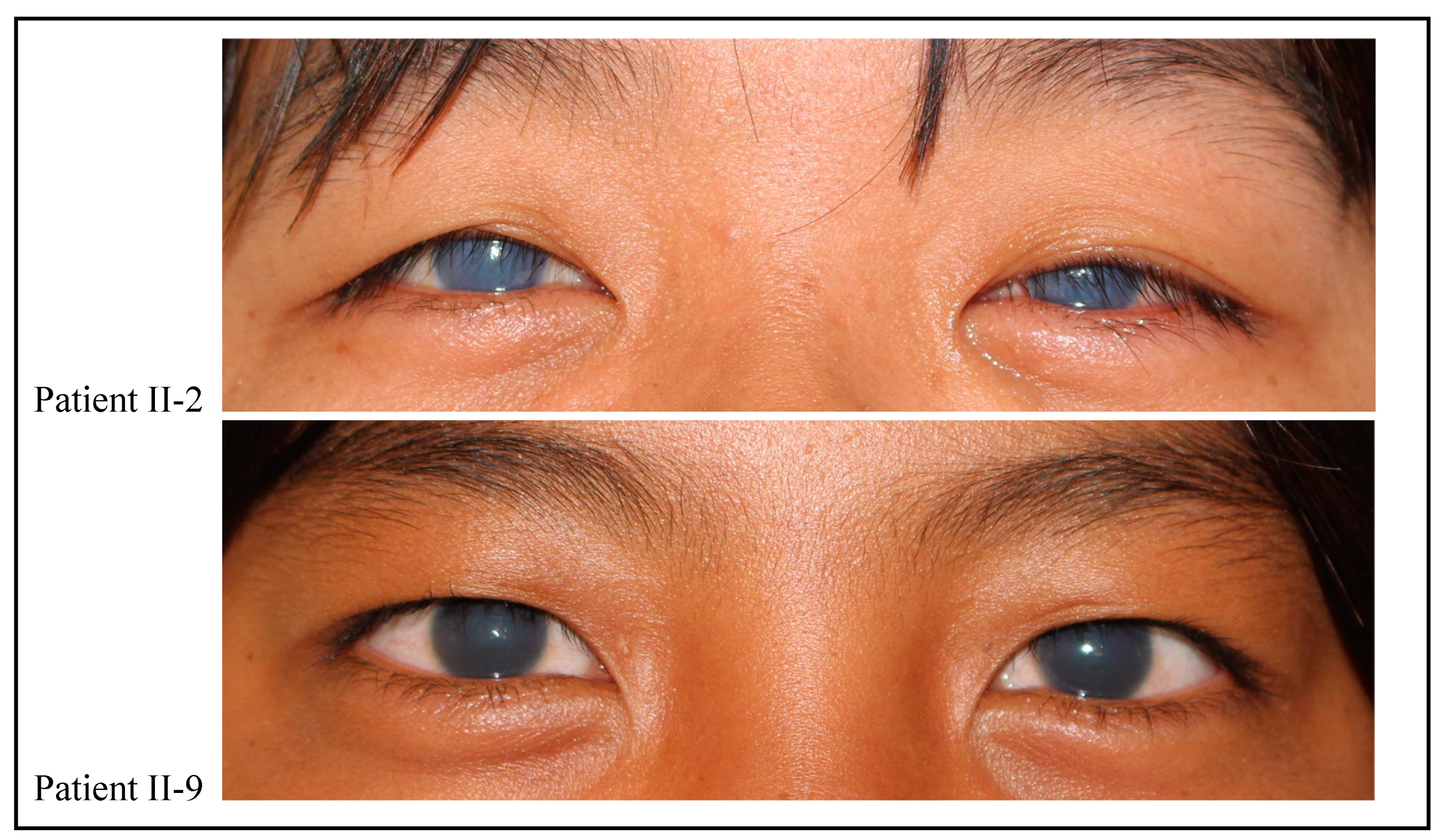
Moreover, the characteristic abnormal posterior non-banded zone of the Descemet’s membrane, which represents an abnormal function of the corneal endothelium in CHED and FECD, underlies the importance of the SLC4A11 protein for the proper development and differentiation of the corneal endothelium and may explain how the same gene can be involved in the pathogenesis of CHED [9]. Endothelial pathology would lead to a defective “corneal pump” causing water to accumulate in the corneal stroma thereby giving the clinical picture of CHED that is bilateral corneal edema (Figure 2).
The family that participated in study was located at Mayoyao, Ifugao in the Northern Luzon region. During the visit to the area, it was discovered that a lot of the locals actually had CHED. Though they were not included in our study, it is interesting to mention that CHED may have a geographic predilection to the area. One such possibility is that there may be a founder effect or mutation responsible for causing the disease to cluster among individuals within the area. One common ancestor may have passed down the CHED mutations to generations below. Despite our efforts in constructing a detailed family history, no consanguinity was reported upon interview. This may not have been the case since we only managed to extract data from 3 generations (Figure 2). Additionally, as mentioned in our results, one interesting feature in the family studied was the expressivity of the disease as if it were dominantly inherited.
This pilot study was conducted to analyze the genetic causes of CHED specifically among Filipinos. Our recommendations to further extend what we have reported are to find more epidemiological data since there is currently none and to do a histopathologic study and ultrastructural analysis on the corneas of such patients if possible. The mutations that may be present in this specific population are also yet to be analyzed for their specific gene products and their ultimate role whether it is pathogenic or not.
The only missense mutation found in this study, the 10294G>C that resulted to a change in the 193rd amino acid translation from arginine to proline, was only predicted to have a benign effect in a population. In addition, the presence of this point mutation together with the 10307G>A, both located at exon 6, varied in patients: specifically present in all patients except Patient I-1 despite the observed similar phenotype. The reverse is true for the point mutation 13165T>C which is absent in all patients, but present in Patient I-1. It is possible that there is a compensation between the variation on the presence of these point mutations resulting in similar observed phenotypes but could not be confirmed by this study yet as they are not within the coding regions for analysis using ExPASy and PolyPhen2. Still, its frequency in related patients suggest a unique marker for CHED population in a Philippine setting, including all other mutations found in this study.
Interestingly, both point mutations 4291G>A and 6533G>C are consistently present in all patients. These mutations, despite being not exactly within Exon 1 or the coding region sequence included in an mRNA sequence, are located near the Exon 1 and 2, and could possibly have an indirect influence to Exon 1 and/or Exon 2 at the transcription level process. We recommend isolating and sequencing mRNA transcript from the samples. As stated, there could be a pathologic process that is taking place at the level of transcription that could contribute to the disease phenotype. Analyzing the mRNA levels as well as its nucleotide sequence could provide an explanation why patients exhibit CHED phenotypes despite detecting most point mutations in non-coding regions. In this line of thought, a different protein at the transcription level might also be involved in the disease progression. If mRNA transcription was not found to be defective, then it is possible that post-translational processing could be involved rendering proteins defective.
To date, 74 mutations in 17 of the 19 coding exons of SLC4A11 have been identified, although 32 of the 136 (23.5%) pedigrees screened to date do not demonstrate coding region mutations in SLC4A11 (Aldave and Frausto, 2013). The majority of them have been missense mutations involving a base substitution. However, mutations possibly involving exon 1 have not been documented. Our first documented novel mutations flanking the said exon supports the genetic heterogeneity of the disease among races. The other exons have been previously known to harbor mutations responsible for the CHED phenotype. This study brings an addition to this growing list of point mutations not only in the coding regions but in non-coding regions as well.
Considering this was a preliminary report on the genetic profile of Filipino patients affected with CHED, we are limited by the fact that we managed to get samples from one family. Though our findings may very well be applicable to CHED patients in the Northern Luzon region, it is recommended to get samples from other unrelated families affected with CHED elsewhere to verify if these mutations are consistently present as a Filipino variant. The goal of ultimately screening all possible mutations responsible for the CHED phenotype in this specific racial group is to develop a panel to facilitate rapid prenatal diagnosis and aid families in genetic counseling. Lastly, the potential for the development of gene therapy for CHED remains on the horizon.
6. Conclusions
This is the first report that gives us a genetic profile of a Northern Luzon family with members affected with CHED. It is also evident that not all mutations are consistently present among different races probably due to the disease’s genetic heterogeneity. Our study documented five novel mutations adding to the growing list of mutations responsible for acquiring the CHED phenotype. It is possible that there are yet more novel mutations to be discovered causing the same disease. Screening for these specific mutations in other families may prove useful for genetic counseling, prenatal diagnosis and the future development of gene therapy.
Acknowledgments
The St Luke’s Medical Center Research and Biotechnology department has provided additional funding to support this project specifically in acquiring the reagents used for DNA sequencing.
Conflicts of Interest
Cabahug VLO, None; Llido JPS, None; Cabral LKD, None; Maynes TL, None; Pinuela CL, None; Tayengco-Tiu TL, None; Lim Bon Siong R, None; Enriquez MLD, None.
References
- Nischal, KK. Genetics of Congenital Corneal Opacification - Impact on Diagnosis and Treatment. Cornea 2015;34(Suppl): S24-S34. [CrossRef]
- Máire Callaghan, Collette K Hand, Susan M Kennedy, J Susan FitzSimon, Louis M T Collum, Nollaig A Parfrey. Homozygosity mapping and linkage analysis demonstrates that autosomal recessive congenital hereditary endothelial dystrophy (CHED) and autosomal dominant CHED are genetically distinct. Br J Ophthalmol 1999; 83:115–119. [CrossRef]
- Klintworth G. Congenital endothelial dystrophy type II. Orphanet2012. Available at: http://www.orpha.net/consor/cgi-bin/oc_exp.php?Ing=en&expert=293603. Accessed April 30, 2016.
- Yanoff M, Duker JS. Ophthalmology: Expert Consult: Online And Print. Elsevier Health Sciences; 2013.
- Siddiqui S, Zenteno JC, Rice A, Chacón-Camacho O, Naylor SG, Rivera-de la Parra D, Spokes DM, James N, Toomes C, Inglehearn CF, Ali M. Congenital Hereditary Endothelial Dystrophy Caused by SLC4A11 Mutations Progresses to Harboyan Syndrome. Cornea Volume 33, Number 3, March 2014 www.corneajrnl.com p. 247-251. [CrossRef]
- Boomiraj Hemadevi, MSc; Reiner A. Veitia, PhD; Muthiah Srinivasan, MD; et al 2008. Identification of Mutations in the SLC4A11 Gene in Patients With Recessive Congenital Hereditary Endothelial Dystrophy. Ophthalmic Molecular Genetics, 126(5): 700-708. [CrossRef]
- Jae-hyung Kim, Jung Min Ko & Hungwon Tchah. 2014. Fuchs Endothelial Corneal Dystrophy in a Heterozygous Carrier of Congenital Hereditary Endothelial Dystrophy Type 2 with a Novel Mutation in SLC4A11. Ophthalmic Genetics 36 (3). [CrossRef]
- Wang X, Zhu Y, Shen N, Peng J, Wang C, Liu H, Lu Y. Mutation analysis of a Chinese family with oculocutaneous albinism. Oncotarget, 2016, Vol. 7, (No. 51), pp: 84981-84988. [CrossRef]
- Preeti Paliwal, Arundhati Sharma, Radhika Tandon, Namrata Sharma, Jeewan S. Titiyal, Seema Sen, Tapas C. Nag, Rasik B. Vajpayee. Congenital hereditary endothelial dystrophy - mutation analysis of SLC4A11 and genotype-phenotype correlation in a North Indian patient cohort. Molecular Vision 2010; 16:2955-2963.
- Nejat Mahdieh, Bahareh Rabbani. An Overview of Mutation Detection Methods in Genetic Disorders. Iran J Pediatr Aug 2013; Vol 23 (No. 4), Pp: 375-388.
- Eranga N. Vithana, Patricio E. Morgan, Vedam Ramprasad, Donald T.H. Tan, Victor H.K Yong, Divya Venkataraman, Anandalakshmi Venkatraman, Gary H.F. Yam, Soumittra Nagasamy, Ricky W.K. Law, Rama Rajagopal, Chi P. Pang, Govindsamy Kumaramanickevel, Joseph R. Casey, Tin Aung. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Human Molecular Genetics, 2008, Vol. 17, No. 5 656–666. [CrossRef]
- Kodaganur SG, Kapoor S, Veerxappa AM, Tontanahal SJ, Sarda A, Yathish S, Prakash DR, Kumar A. Mutation analysis of the SLC4A11 gene in Indian families with congenital hereditary endothelial dystrophy 2 and a review of the literature. Molecular Vision 2013; 19:1694-1706.
- Kumar A, Bhattacharjee S, Prakash DR, Sadanand CS. Genetic analysis of two Indian families affected with congenital hereditary endothelial dystrophy: two novel mutations in SLC4A11. Molecular Vision 2007; 13:39-46.
- Ehlers N, Módis L, Møller-Pedersen T. A morphological and functional study of congenital hereditary endothelial dystrophy. Acta Ophthalmol Scand 1998; 76:314-8. [CrossRef]
Figure 1.
Pedigree showing the three generations of the family being investigated. Affected individuals are shown in black solid shapes Proband is marked with an arrowhead.
Figure 1.
Pedigree showing the three generations of the family being investigated. Affected individuals are shown in black solid shapes Proband is marked with an arrowhead.
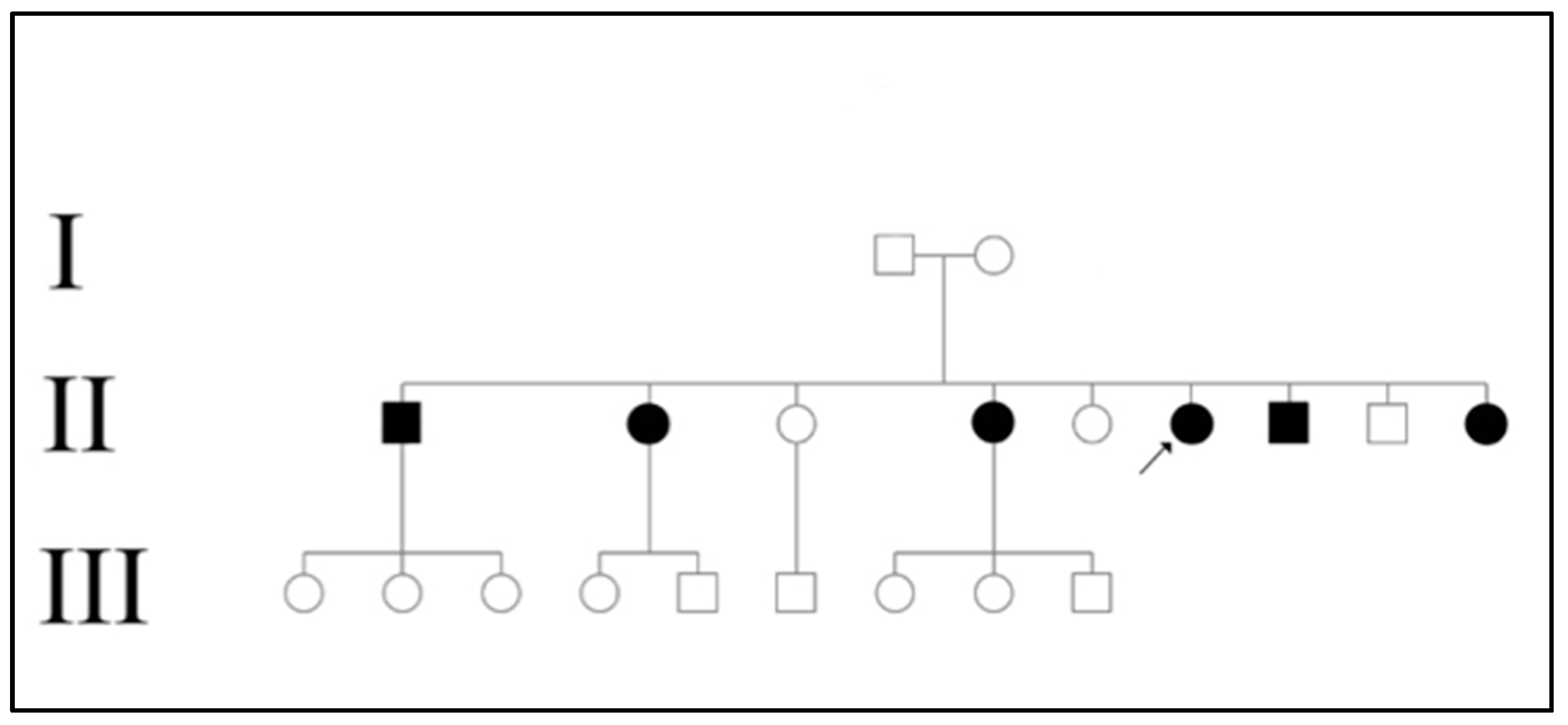
Figure 3.
Representative electropherograms of amplified DNA sequences analyzed in comparison with representative SLC4A11 reference sequence. Five point mutations have been observed, mostly located outside the exon regions except for 10294G>C and 10307G>A which are both located in Exon 6. Electropherograms were generated using the consensus of triplicate sequences, viewed and analyzed using BioEdit, Seqotron, and 4Peaks.
Figure 3.
Representative electropherograms of amplified DNA sequences analyzed in comparison with representative SLC4A11 reference sequence. Five point mutations have been observed, mostly located outside the exon regions except for 10294G>C and 10307G>A which are both located in Exon 6. Electropherograms were generated using the consensus of triplicate sequences, viewed and analyzed using BioEdit, Seqotron, and 4Peaks.
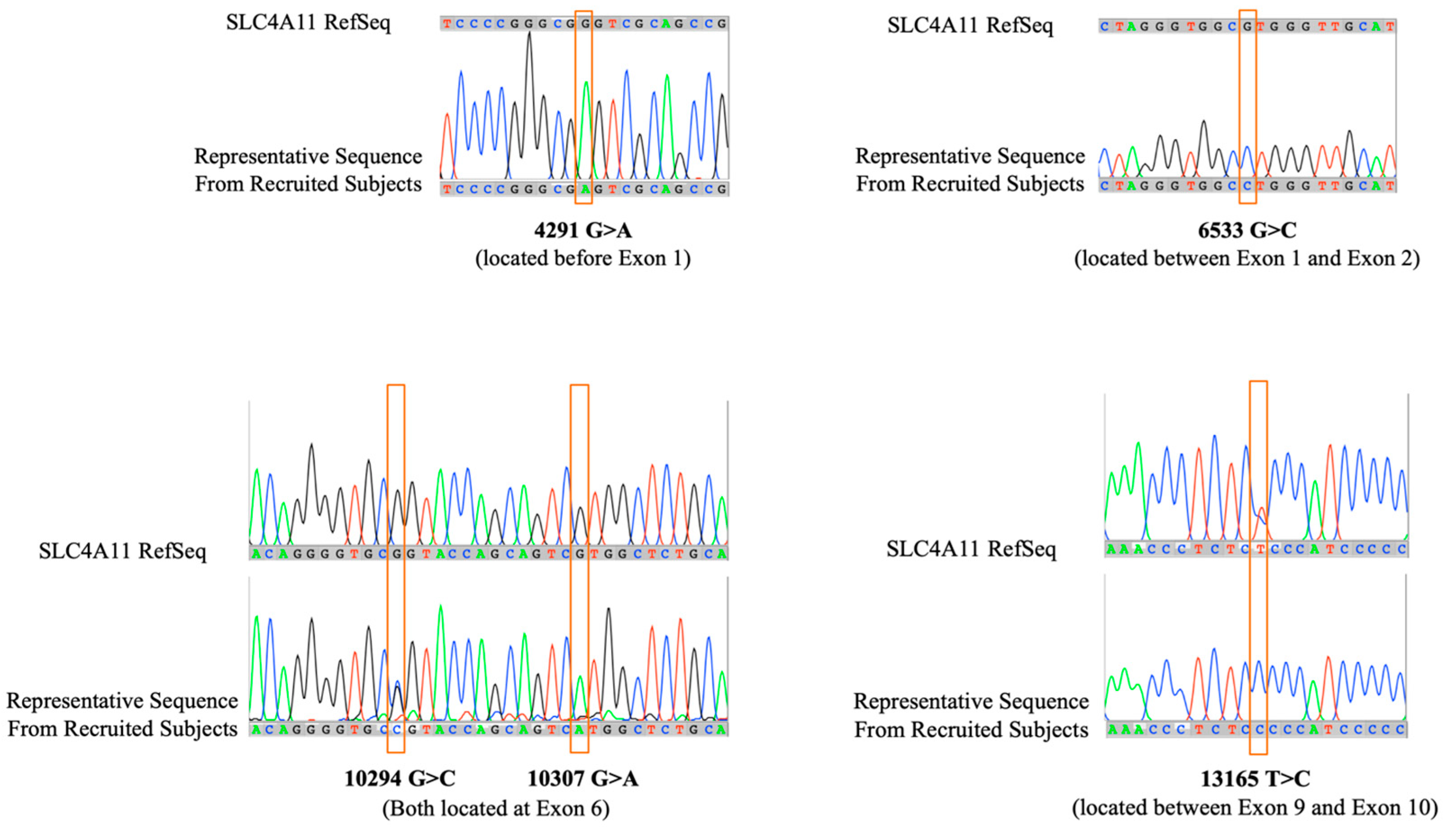
Figure 4.
Amino acid sequence alignment of reference and from patient samples. Two point mutations, 10294 G>C and 10307 G>A, at Exon 6 had different effects on translated amino acid sequence. The 10294G>C is a missense mutation, resulting in a change of translated amino acid at location 193 to change from arginine (R) to proline (P) while the point mutation at 10307G>A did not result in an amino acid change. Amino acid sequence was generated via ExPASy using the reconstructed DNA sequences as input. Seqotron was used for multiple sequence alignment.
Figure 4.
Amino acid sequence alignment of reference and from patient samples. Two point mutations, 10294 G>C and 10307 G>A, at Exon 6 had different effects on translated amino acid sequence. The 10294G>C is a missense mutation, resulting in a change of translated amino acid at location 193 to change from arginine (R) to proline (P) while the point mutation at 10307G>A did not result in an amino acid change. Amino acid sequence was generated via ExPASy using the reconstructed DNA sequences as input. Seqotron was used for multiple sequence alignment.

Figure 5.
Results of prediction/confidence on the effect of the change in the translated amino acid 193 from arginine to proline due missense mutation 10294G>C. The missense mutation was predicted to be “BENIGN” on both HumDiv and HumVar. According to the PolyPhen2 website (http://genetics.bwh.harvard.edu/pph2/dokuwiki/overview) “HumDiv was compiled from all damaging alleles with known effects on the molecular function causing human Mendelian diseases, present in the UniProtKB database, together with differences between human proteins and their closely related mammalian homologs, assumed to be non-damaging; HumVar consisted of all human disease-causing mutations from UniProtKB, together with common human nsSNPs (MAF>1%) without annotated involvement in disease, which were treated as non-damaging”.
Figure 5.
Results of prediction/confidence on the effect of the change in the translated amino acid 193 from arginine to proline due missense mutation 10294G>C. The missense mutation was predicted to be “BENIGN” on both HumDiv and HumVar. According to the PolyPhen2 website (http://genetics.bwh.harvard.edu/pph2/dokuwiki/overview) “HumDiv was compiled from all damaging alleles with known effects on the molecular function causing human Mendelian diseases, present in the UniProtKB database, together with differences between human proteins and their closely related mammalian homologs, assumed to be non-damaging; HumVar consisted of all human disease-causing mutations from UniProtKB, together with common human nsSNPs (MAF>1%) without annotated involvement in disease, which were treated as non-damaging”.
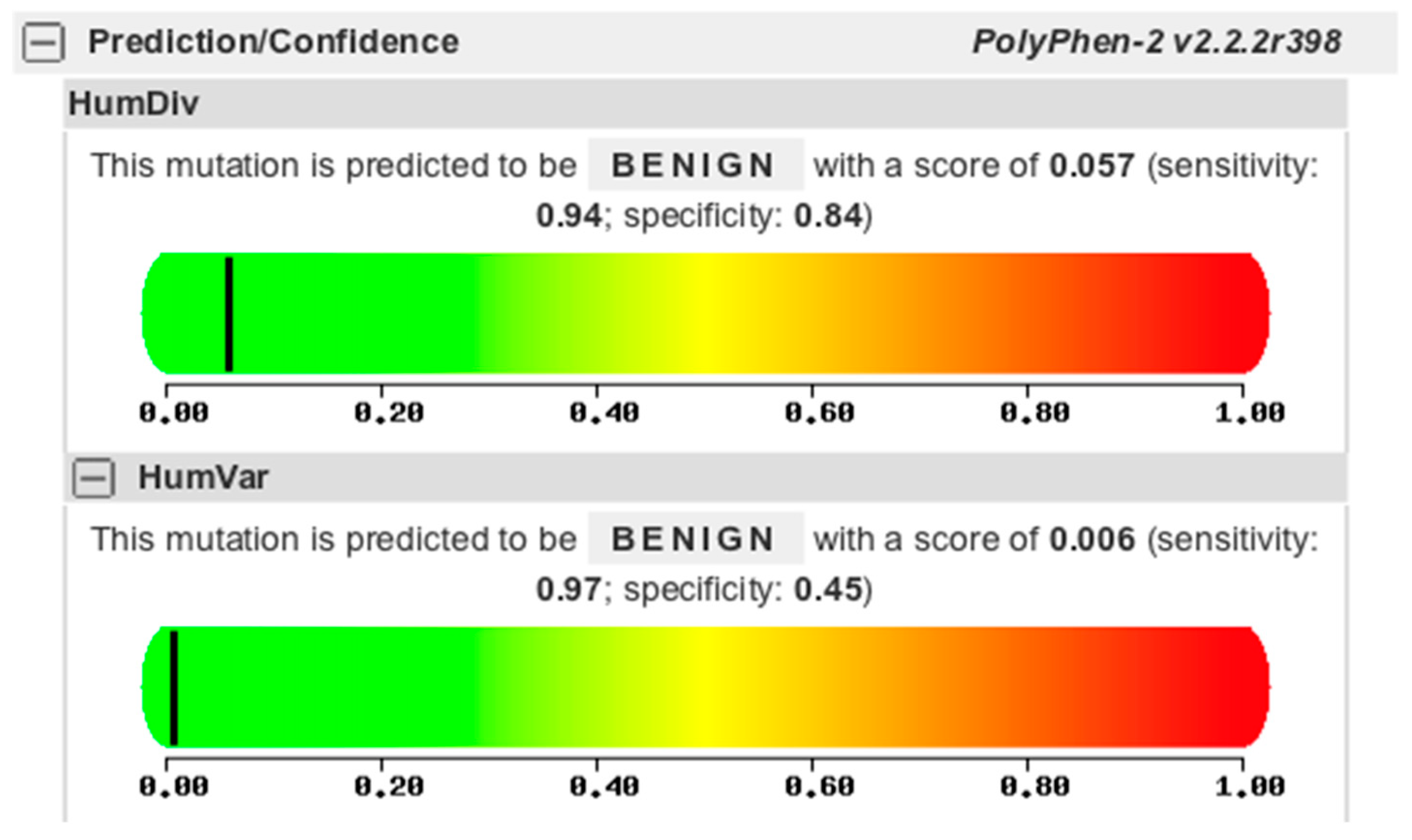
Figure 2.
Representative photographs of the eyes of patient II-2 (above) and patient II-9 (below) showing (described eye).
Figure 2.
Representative photographs of the eyes of patient II-2 (above) and patient II-9 (below) showing (described eye).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



