
Submitted:
18 January 2023
Posted:
19 January 2023
You are already at the latest version
Abstract
Prostate cancer (PC) is the most frequently diagnosed non-skin cancer in the world. Previous studies showed that genomic alterations represent the most common mechanism for molecular alterations that cause the development and progression of PC. Great efforts have been done to identify common protein-coding genetic variations; however, the impact of non-coding variations including regulatory genetic variants is not still well understood. To gain an understanding of the functional impact of genetic variants, particularly, regulatory variants in PC, we developed an integrative pipeline (AGV) that used whole genome/exome sequences, GWAS SNPs, chromosome conformation capture data, and ChIP-Seq signals to investigate the potential impact of genomic variants on the underlying target genes in PC. We identified 646 putative regulatory variants, of which 30 of them significantly altered the expression of at least one protein-coding gene. Our analysis of chromatin interactions data (Hi-C) revealed that the 30 putative regulatory variants may affect 131 coding and non-coding genes. Interestingly, our study showed the 131 protein-coding genes are involved in disease-related pathways including Reactome and MSigDB in which for most of them targeted treatment options are currently available. Together, our results provide a comprehensive map of genomic variants in PC and revealed their potential contribution to prostate cancer progression and development.
Keywords:
Prostate cancer
; Somatic point mutations
; Copy number variation
; Regulatory variant
; Hi-C
; Per-sonalized medicine
; Biomedical machine learning
1. Introduction
Prostate cancer is the second most common cancer and the fifth leading cause of cancer death among men, with almost 1.3 million new cases and 359,000 associated deaths in 2018 worldwide [1]. Genetic instability is one of the hallmarks of cancer cells. This is happened both by single point mutations or at the chromosomal abnormality. However, a few of them, called drivers, contribute to oncogenesis, whereas the majority are passenger mutations accumulated during cancer progression. Systematic identification of driver genes from large background noise is important. In this study, we identify putative genomic variants associated with the increased risk of cancer susceptibility from large background noises to provide an appropriate list of genes with potential impact on PC progression.
Identification of cancer-associated genomic variants has been focused on both protein-coding and non-coding genes. For example, Functional Analysis through Hidden Markov Models (FATHMM) [2] was used to prioritize genomic variants in the protein-coding genes. However, most of the genome is in non-coding regions including non-coding RNAs and non-annotated regions and the majority (> 90%) of genomic variants occur in these regions [3] Thus, determining the effect of genomic variants in non-coding regions is necessary. To this aim, there are computational tools that link genomic variants to different regulatory elements obtained from international projects such as ENCyclopedia of DNA Elements (ENCODE), Functional Annotation of the Mammalian Genome (FANTOM), Roadmap Epigenomics Project, and Genotype-Tissue Expression (GTEX). For instance, FunSeq2 [4] was designed to identify and prioritize non-coding somatic point mutations using various resources including ENCODE and other publications [5]. This pipeline firstly assigns a score to genomic variants based on overlapping these genomic variants with different genomic features including regulatory elements (enhancer marks H3K4me1 and H3K27ac, DNA methylation), network of genomic variants associated with genes, and recurrent elements across cancer samples (i.e., those variants identified on whole genome sequencing of at least two samples). Then, FunSeq2 assigns specific weight to features based on the 1-Shannon entropy. RegulomeDB is another tool [6] that was designed to prioritize disease-associated Single Nucleotide Polymorphisms (SNPs). This method employs a heuristic scoring system that assign a specific score for each SNPs based on the number of overlapping between SNPs and integrated regulatory database including TFBSs, chromatin states of different cell types and eQTL data. Chen et al. [7] also developed an enrichment analysis to test whether any risk-associated SNPs are located in the functional genomic regions including UCSC annotated coding regions (exon and snoRNA/miRNA) and regulatory regions, as well as binding regions for transcription factors (TFs), histone modifications (HMs), DNase I hypersensitivity (DHSs), and RNA Polymerase IIA (POLR2A) more than expected. RegulemeDB, HAploReg and Variant Effect Predictor (VEP) toolsets also map GWAS SNPs to regulatory elements to identify functional GWAS variants [8].
There is another category of methods using machine learning techniques to predict the potential impact of genomic variants. These methods are supervised methods, which have been trained using functional annotations to determine pathogenic variants. and then, new genomic variants can be classified using this information. For example, DeepSEA (deep learning based sequence analyzer) [9] uses a convolutional neutral network (CNN) based framework to predict the effect of chromatin factors (transcription factor binding, DNase I sensitivity, histone mark profile) in genomic sequence. In the prioritization part, DeepSEA predicts regulatory mutations using boosted logistic classifiers via eQTL data, non-coding trait-associated SNPs identified in GWAS studies from the US National Human Genome Research Institute’s GWAS Catalog. Chengliang et al. also presented iCAGES (integrated CAncer GEnome Score) [10], a statistical framework that prioritizes cancer driver mutations, genes, and targeted drugs. This method firstly integrates different prioritization tools (FunSeq2, SIFT, FATHMM, VEST, Mutation Taster, Phylop, PolyPhen2, GERP++, Mutation Assessor, LRT, SiPhy, and LRT) to determine specific score into protein-coding mutations, non-coding mutations, and structural variations. Then, in the second layer, iCAGES takes the relevant genes from the last step and gene list from Phenolyzer tool to provide the score to each gene based on a logistic regression model. Lastly, this method links identified genes to specific drugs and calculates specific score for each drug, based on its effectiveness. Shengcheng et al. also presented SURF (Score of Unified Regulatory Features) [11] that uses features from RegulomeDB and DeepSEA tools and then apply a random forest model to predict the effect of genomic variant (SNP) in promoters and enhancers regions.
The above-mentioned methods determine the overlapping of genomic variants in coding and non-coding regions, however, they are not able to identify the potential impact of the variants and how these variants affect gene expression. Integrative analyses have been used previously in cancer biomarkers discovery [12], however, none of these platforms integrate chromosome confirmation capture data to identify the impact of regulatory variants in PC. Here, we develop a new integrative pipeline, Associated Genomic Variants (AGV), that uses high-throughput chromosome confirmation capture data (Hi-C), RNA-Seq, ChIP-Seq, and a list of genetic variants to link the variants to target genes in prostate cancer. We applied AGV on genomic variants of 194 PC patients obtained from International Cancer Genome Consortium (ICGC) and PC-associated GWAS SNPs from GWAS Catalog and identified the candidate coding and non-coding variants and their associated target genes.
To do this, AGV first identifies PC-associated somatic point mutations hotspot regions and CNV regions (genomic regions that CNVs are overlapping – CNVRs) and coding and non-coding genes affected by these variants. AGV then uses H3K27ac ChIP-Seq marks to identify variants that happened in the enhancer regions. Using Hi-C interactions from normal and cancer cell lines, AGV generates a list of genetic variants with potential regulatory function. Lastly, we validated the PC-associated variant identified in this study using an independent whole genome sequencing data from the same PC cell line. An overview of AGV pipeline is provided in Figure 1.
The main novelties and contributions of our work are as follows:
- This is the first study that comprehensively considers GWAS SNPs, somatic point mutations and CNVs, while the previous methods only considered somatic mutations and GWAS SNPs to identify functional cancer-associated variants.
- In comparison to other studies [2] which mainly considered genomic variants in protein-coding genes, in this study, we performed the analysis on both coding and non-coding regions.
- We used an innovative strategy to identify hotspot somatic point mutation regions, which can also be used in the further studies for identifying hotspot regions in cancer. The proposed method is built upon window analysis for detection of hotspot somatic mutation regions, which is an effective strategy for identification of hotspot regions, whereas other methods such as FunSeq2 [2] and iCAGES [10] were not to report highly mutable regions.
2. Results
2.1. Making a comprehensive map of prostate cancer associated genomic variants
We first collected all prostate cancer associated GWAS SNPs from [13] considering GWAS SNPs with (P-value < 5E-8) (Supplementary Table 1a).We then used somatic point mutations from 194 ICGC PC samples (including 10,154,740 single point mutations) to identify hotspot regions. Since somatic point mutations (SPMs) are distributed in the whole genome randomly and the vast majority of them are passengers. We therefore consider somatic hotspot regions as the genomic regions with an enrichment of somatic point mutations in PC samples. Hotspot regions have been highly mentioned to be important in different cancer types [14,15]. Identification of somatic point mutations hotspot regions has three main steps including window analysis, selection of significant windows, and filtering process. Figure 2 describes the framework used for the identification of hotspot region in this study.
In the first step, window analysis is used to divide genome into fix-binned size windows and count the number of samples encompassing SPM within the window. In this study, window sizes 9, 21, 50 and 5000bp were tested to detect the optimal window size. We selected window length 21bp as the optimal window length. To select the optimal window length. we run different window lengths (9bp, 21bp, 50bp and 1000bp) and the result shows that there was no significant difference in terms of number of samples in each region between window 21bp and 9bp (Figure 3A).
Totally, 21,966 windows were detected containing at least one sample with SPM. We then used Poisson binomial distribution to determine the significance of observing k samples containing somatic mutations in a 21bp window. To determine the best fitted distribution for selection of statistically significant windows (P-value < 0.001) we used Skewness-kurtosis and CDF (Figure 3B) (see method for more details).
As a result, we identified 71 somatic mutation hotspot regions that were significantly associated with PC (Figure 4 and Supplementary Table 1b).
To have a comprehensive list of PC-associated variants, we also used copy number variants available for PC samples from ICGC datasets to identify PC-associated CNVRs (genomic regions that CNVs are overlapping). To do this, we used CNV maps for 11,564 CNVs (3,625 deletions and 7,939 duplications) of 194 patients from ICGC [16] and 2,392 publicly available healthy individuals from 1000 genome project [17] containing the genomic coordinates for 32,449 CNVs (22,318 deletions and 10,131 duplications. To identify PC associated CNVs, a genome-wide genetic association analysis needs to be performed between the CNV regions and the observed phenotypes. However, one of the major obstacles in a CNV-based genome-wide association study occurs when categorizing CNVs across all cases (individuals with the phenotype of interest) and controls (healthy individuals), because CNVs are inconsistent in sequence, size and genomic coordinates across individuals. To address this issue, one effective approach is to build CNVRs (genomic region that CNVs are overlapping - CNVRs) prior to identifying those CNVRs statistically associated with the phenotype of interest. In this study, we used PeakCNV [18] method which can determine CNVRs that are significantly associated with PC. It considers the dependency between CNVs to remove CNVRs which overlap or co-occur with true positive CNVRs. PeakCNV uses an artificial intelligence-based technique that firstly identify deleted and duplicated CNVRs that are significantly overrepresented among cancer samples. It then identifies cluster of CNVRs which have deleted/duplicated in the samples and are proximally closed to each other. PeakCNV then reports the best representative CNVR for each cluster as the candidate CNVRs. As a result, we identified 216 duplicated CNVRs and 75 deleted CNVRs that were significantly associated with PC (Supplementary Table 1c).
Totally, we made a list of 2,354 PC-associated genomic variants including 1,992 GWAS SNPs, 71 hotspot regions, and 291 CNVRs. We next investigate how these variants contribute to the progress of prostate cancer.
2.2. Linking PC-associated genomic variants to coding and non-coding genes
To determine genes relating to PC-associated genomic variants from the analysis in the last step, we overlapped the coordinate of genomic variants with the human reference genome (see method section for more details). Notably, we identified that a greater portion of genomic variants (70% hotspot regions, 87% GWAS SNPs, 99% of duplicated and 97% of deleted CNVRs) were associated to coding and non-coding genes (Figure 5 and Supplementary Table 2). Interestingly, we observed that a greater fraction of hotspot regions and CNVRs are in non-coding genes, while a greater portion of GWAS SNPs are in protein-coding genes. We also explored the distribution of genes associated to genomic variants in non-coding RNAs and found out that more than 50% of these non-coding genes are lncRNAs (Figure 5).
2.3. Identify variants with likely regulatory function
Of 2,354 PC-associated genomic variants identified in this study, 1026 of them are located in non-coding regions, in particular of interest, non-coding RNAs. Despite this, the majority of these non-coding variants has unknown function. Here, we hypothesize that some fraction of these variants might have regulatory function, called as regulatory variants. To identify regulatory variants, we first used Hi-C interactions and H3K27ac Chip-Seq signals to identify enhancer-promoter interactions. We used Hi-C interactions from two prostate cancer cell lines (PC3 and LNCaP) and one healthy cell line (PrEC). HiC-Pro [19] was used for mapping, trimming and valid interaction calling. MaxHiC [20] and MHiC [21] were used to identify statistically significant interactions (P-value < 0.001). As a result, 107,705, 235,181, and 82,334 significant Hi-C interactions were identified in PC3, LNCaP, and PrEC cell lines, respectively. The number of significant Hi-C interactions and their distance were higher in both prostate cancer cells compared to normal PrEC cell line (Figure 6 and Supplementary Table 3), which indicates that Hi-C interactions in normal cells were often subdivides into multiple smaller interactions in cancer cells.
H3K27ac signals were then used to identify enhancer marks. We considered those Hi-C interactions that one side of the interactions overlapped with H3K27ac signals as an enhancer mark and another side overlapped with promoter region of protein-coding genes, resulting in identification of enhancer-promoter interactions (EPIs). We identified 12,266, 3,653, 3,690 EPIs in LNCaP, PC3, and PrEC cell lines, respectively (Supplementary Table 4). Of these 1,130 and 3,593 EPIs were only observed in PC3 or LNCaP cell lines and not in the healthy cell line (PrEC). We then focus on these EPIs and intersect them with PC-associated genomic variants to identify regulatory variants with potential functional impact in PC. We only considered those variants that overlapped with the enhancer side of the interactions. As a result, 135 SNPs, 14 hotspot regions, 213 duplicated and deleted CNVRs were overlapped with EPIs in LNCaP cell line. We also identified 51 SNPs, 7 hotspot regions, 226 duplicated and deleted CNVRs that overlapped with EPIs in PC3 cell line (Supplementary Table 5). Of the particular of interest, we identified GWAS SNP rs10993994 that overlapped with EPI chr10:5130000-51535000;chr10:51580000-51585000 An study by Bicak et al. [22] on this GWAS SNP showed that it has a regulatory function for genes MSMB [22].
The 646 potential regulatory variants interacted with 13,858 protein-coding genes. Interestingly, 278 of these variants located in the body of non-coding RNAs, mostly lncRNAs. For example, lncRNA HOTAIR encompassed 2 regulatory variants. This lncRNA has been previously identified as an enhancer RNA that regulate protein-coding gene MDM2, validated by different integrative meta-analysis [23,24].
We then used whole genome sequencing (WGS) of prostate cancer cell lines (PC3 and LNCaP) to see if how many of the regulatory variants we used in this study replicated in a whole genome sequence of the same cancer cell line. As a result, 23 GWAS SNPs, 2 hotspot regions, 93 duplicated and deleted CNVRs that overlapped with EPIs in LNCaP cell line were also replicated in the WGS data. 2 GWAS SNPs, 1 hotspot regions, 67 duplicated and deleted CNVRs were also replicated in the PC3 cell line (see Supplementary Table 6 for more details).
For example, CNVR (chr8:127394134-127501076) overlaps with enhancer side of EPI, in which the other side overlaps with protein-coding genes TATDN1 and RNF139. More importantly, both H3K27ac and RNA-seq data showed much higher signal in the cancer cell line compared to the healthy cell line (Figure 7A), indicating the possible impact of this PC-associated duplicated CNVR on enhancing the expression of genes NDUFB9 and MTSS1 in prostate cancer. Interestingly, MTSS1 has been reported as the metastasis driver gene in a subset of human melanomas [25].
Deleted CNVR (chr2:73916673-73947014) is another example of the PC-associated regulatory variants identified in this study that was also observed in the whole genome sequence of the prostate cancer cell line. As Figure 7B shows there is a Hi-C interaction in PC3 cancer cell line which one side of the interaction overlapped with the potential enhancer region and another side overlapped with NAT8 and ALMS1P. The expression of these genes was significantly increased in the cancer cell line indicating that this CNVR may act as the potential genomic variant disrupting this enhancer-promoter interaction. Based on the literature search, ALMS1P is one of the causative genes identified for different disease, while its physiological function and pathological significance in different diseases are still unknown [26].
We next performed a pathway analysis on the genes associated with the identified regulatory regions. We used ShinyGO [27] to determine genes that were enriched in disease-related pathways. To this aim, we first used a whole list of pathway databases in ShinyGO to assess the relative biological importance of identified regulatory genes (see methods for more details). We then mapped the regulatory genes to curated gene sets/pathways to screen for involvement in known cancer and other molecular processes.
Our analysis showed that ~44% of interacting genes are associated with previously known curated gene sets/pathways (cutoff of P-value < 0.05). The most highlighted gene set is LASTOWSKA_NEUROBLASTOMA_COPY_NUMBER_DN database from msigdb [28] database which containing genes with copy number losses in primary neuroblastoma tumors. These deleted copy number variations are the major cause of gene transcription. We identified 17% of interacting genes were involved in this pathway (9.55-fold change, ). Furthermore, 15 genes were expressed in the CUX1-19635798-MULTIPLE HUMAN CANCER CELL TYPES-HUMAN transcription factor binding site profile database [29], which contains 2406 expressed genes with transcription factor binding evidence in the multiple human cancer cell types (Supplementary Table 7).
Intriguingly, we also identified two other cancer-associated pathways including WOO_LIVER_CANCER_RECURRENCE_DN [30] and VANTVEER BREAST CANCER ESR1 UP [31]. Some of the genes in these pathways include ALAS1, ACAA1, ACOX2 which are negatively correlated with recurrence free survival in patients with hepatitis B-related (HBV) hepatocellular carcinoma (HCC). Interestingly, it has been shown that chronic hepatitis B virus (HBV) infection is a leading cause of hepatocellular carcinoma (HCC) [32].
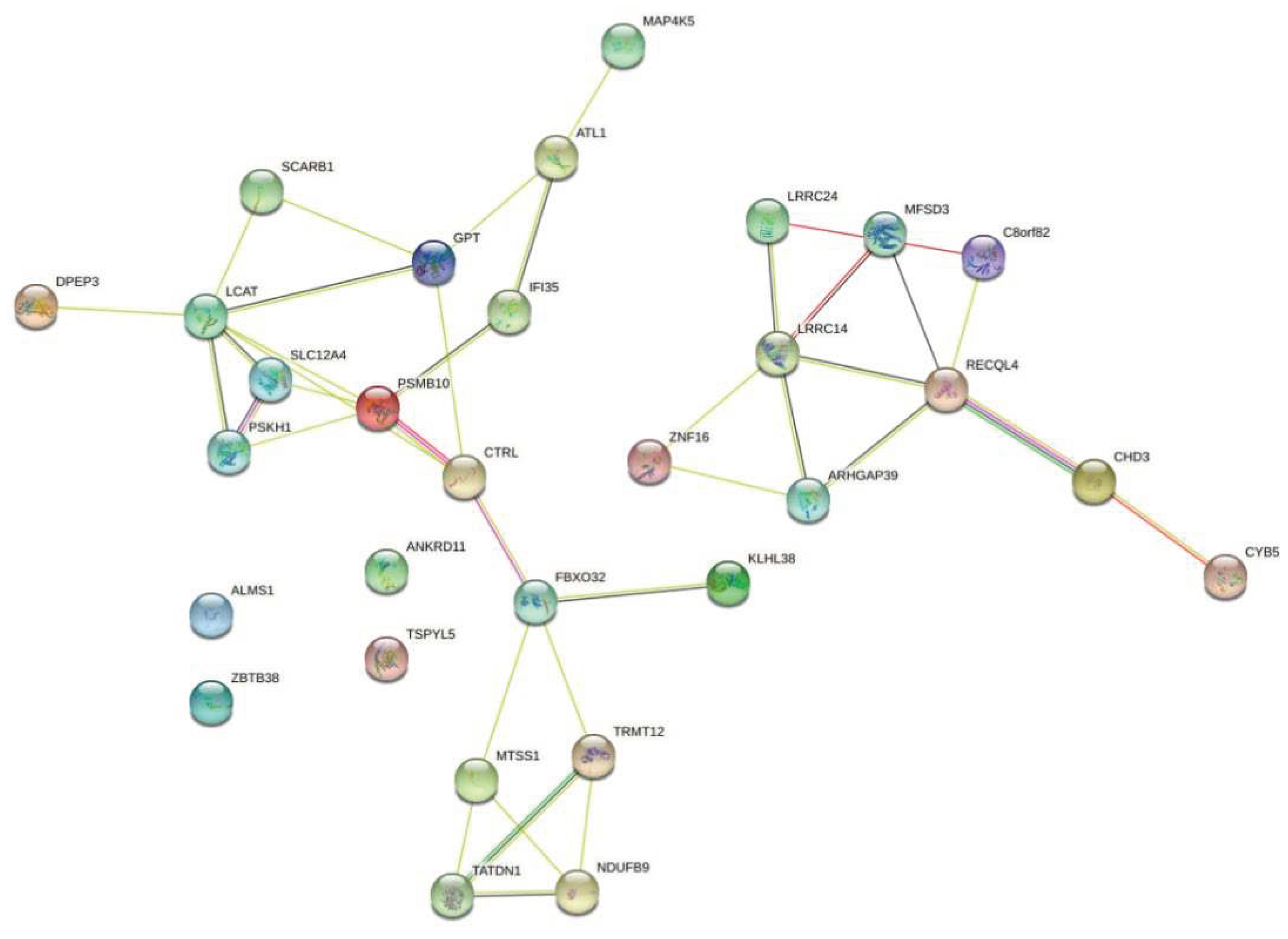
We then used STRING-db website [33] to retrieve the protein-protein interactions for the interacting protein-coding genes. This network gives the insight to the which proteins are associated with other proteins and it can be beneficial for disease therapy to develop new molecule drugs that control interactions between causal proteins interactions. Figure 8 illustrates protein networks for the top 30 enriched genes in prostate cancer related KEGG pathway. This analysis provides list of most significant target proteins with cutoff P-value < 0.05. For example, our analysis finds zinc finger protein 16 (ZNF16) which has been shown to have a potential role in DNA damage, and Cisplatin (used as an anticancer drug) to prevent from the overexpression of this protein [34]. Furthermore, RecQL4 has been reported as a novel molecular target for cancer therapy in 2021 [35], which has a prognostic role in metastatic tumor samples [36].
3. Conclusion
In this study, we have developed a new pipeline, AGV, to systematically detect putative regulatory variants including copy number variations, SNPs and hotspot somatic mutations for prostate cancer. AGV pipeline can be easily integrated into any other pipeline, thus it is useful for downstream analysis of any disease. AGV contains three main steps that can be run independently based on the user request. Firstly, it generates a list of hotspot somatic mutations, CNVRs and GWAS SNPs with their associated coding and non-coding genes. To determine hotspot somatic mutation regions, AGV employs a sliding window algorithm that split the human genome into fixed size windows and then compute the significant windows. AGV then uses an AI-based algorithm (PeakCNV) to generate a list of true positive CNVRs. The identified genomic variants will then be integrated with Hi-C data and H3k27ac signals to provide a list of potential functional EPIs. We identified 30 regulatory variants that potentially disrupt enhancer promoter interactions in PC-related cancer cell line. The regions that encompass these variants, interact with 131 genes, in which each gene can be targeted by multiple regulatory variants.
The development of innovative deep learning algorithms, which have proven to outperform traditional approaches in genomics, transcriptomics, and clinical biomarker identification [37,38,39], our strategy has the potential to be used in integration with these methods to provide a better understanding of the mechanisms underline cancers.
4. Materials and Methods
GWAS dataset
GWAS SNPs were downloaded from GWAS Catalog (https://www.ebi.ac. uk/gwas/docs/file-downloads) and GWASdb v2 (http://jjwanglab.org/gwasdb). We only considered those SNPs that were associated with Prostate cancer. All GWAS SNPs with were excluded from the analysis.
Somatic point mutations dataset
The genomic coordinates of Somatic point mutation (SPM) for prostate cancer were obtained from International Cancer Genome Consortium (ICGC) [40]. Totally, there were 10,154,740 SPMs from 1,037 PC patients across six projects (PRAD-US, PRAD-CA, PRAD-UK, EOPC-DE, PRAD-CN and PRAD-FR) from United States, United Kingdom, Canada, Germany, China and France.
Identification of somatic point mutation hotspots
To identify somatic point mutation hotspots, our pipeline firstly counts the mutation recurrence for fixed binned size regions (bin length=21bp). The user can set the window length based on the desired minimum recurrence frequency. The P-value of mutation recurrence is computed using a Poisson binomial distribution model to determine the significance of observing k samples containing somatic mutations in a 21bp window. Skewness-kurtosis graph and CDF plot were executed by “fitdistrplus” in R package. In the next step, the problematic hotspot regions, such as masked regions (regions with mappability score < 1 in the ENCODE 75mers alignability track in the UCSC genome browser) and Repetitive regions (RepeatMasker track and simpleRepeat tracks in the UCSC Genome browser) [41] were excluded. We also excluded chromosome Y in our analysis.
PeakCNV
To determine CNV regions (genomic region that CNVs are overlapping - CNVRs) that are associated to disease, we proposed an AI-based method calling PeakCNV, which is an extension of SNATCNV toolset [42].
PeakCNV selects CNVRs with the lowest confounding with true positive CNVRs. To this aim, PeakCNV has three main steps: including CNVR map building, clustering process and selection process. In the first step, it builds deletion and duplication CNVR maps for case and control, independently, then, it selects CNVRs that are significantly represented in cases over controls at nucleotide base. In the next step, it groups significant CNVRs into different clusters based on the similar association of CNVRs with the phenotype of interest. To this aim, we used DBSCAN clustering algorithm with two input features including, CNVR uniqueness (the number of case samples covered by a given CNVR after subtracting the common case samples between each pair of CNVRs) and the genomic distance between CNVRs. Lastly, it selects the most independent CNVRs from each cluster using a novel score IR-score. Independent CNVRs are those detected in the greatest number of cases and having a minimum co-occurrence with other CNVRs. PeakCNV runs with the default parameters (P-value <0.05).
Reference gene annotations
FANTOM5 [43], Ensemble [44] and GENCODE [45] gene annotation files were used to curate a comprehensive reference gene list. The FANTOM5 gene annotation file was used as the backbone of our reference gene list, but when the gene annotation was absent from FANTOM5 these were acquired from Ensembl and GENCODE. The final reference gene list contained 82,539 genes including 58,000, 24,501 and 38 genes from FANTOM5, Ensembl and GENCODE, respectively. The genomic coordinates for CNVRs, somatic point mutations, GWAS SNPs and gene annotations were in hg19 genome assembly.
Identification of genomic variants affecting coding and non-coding genes
This analysis is performed to indicate which genes are affected by the observed genomic variants in prostate cancer including GWAS SNP (1,992 SNPs), hotspot regions (71 regions), and CNVRs (duplication: 216 CNVRs, deletion: 75 CNVRs). Bedtools v2.30.0 [46] was used to identify the overlapping between the genomic coordinates of genomic variants and genes [46]. The risk SNPs, hotspot regions, and CNVRs were used for this analysis are provided in Supplementary Table 1. The list of genes affected by different types of genomic variants is also provided in Supplementary Table 2.
Preparation of Hi-C libraries
Hi-C data from Normal human prostate epithelia cells (PrEC) prostate cancer cell lines PC3 and LNCaP with GEO GSE73785 were downloaded using KARAJ toolset [47] from previously published data [48]. We used KARAJ [47] to download datasets and the supplementary files. Two replicates were available for each cell line. We used HiC-Pro v2.11 [49] with the default parameters for analyzing and aligning Hi-C data in 5kb fragment size. Hg19 genome building was uses form mapping. We then used MaxHiC [20] and MHiC identify statistically significant cis-interactions. Here, we only considered those significant cis-interactions with P-value < 0.01, read-count >= 10, and distance between two sides of interaction more 5k and less than 10M. We then used our genes list to annotate Hi-C interactions with coding and non-coding genes. At least 10% overlap between gene and Hi-C fragments been considered to annotate Hi-C fragments with genes. Two replicates of each Hi-C cell line were merged to enhance the statistical power (Supplementary Table 3).
Identification of H3K27ac ChIP-Seq peak regions
H3K27ac ChIP-Seq fastq files PC3, LNCaP and PrEC cell lines were downloaded from GEO GSE57498, GSE73785, GSE57498, respectively [48,50]. Bowtie2 [51] were then used to map the fastq file to hg19 human genome reference. Peaks were then called using Model-based Analysis of ChIP-Seq (MACS2) [52] with the (Supplementary Table 9).
Literature search strategy
Our literature searches were focused on human and mice English language papers available in the PubMed, Scopus, and Web of Science. We also used data and text mining techniques to extract additional related studies [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68]. A knowledge-based filtering system technique has been also used to categorize the texts from the literatures search [69,70,71,72,73,74]. The search terms included "Cancer", "Prostate cancer", "noncoding RNA", "enhancer", "CNV", "mutation", "copy number variations".
Whole genome sequencing data processing
- a.
- Mapping of fastq reads of prostate cell lines to reference genome
We obtained WGS data of LNCaP (ATCC CRL-1740) and PC3 (ATCC CRL-1435) from published work [75] with Karaj pipeline. The quality checking of fastq files were performed using FastQC v0.11.9 [76]. Trimmomatic v0.40 [77] was then used to filter poor quality reads and trim poor quality bases (phred score < 30) from our samples. BWA-MEM v0.7.17 (r1188) [78] was then used to map sequencing reads to the human reference genome (hg19) and a sorted BAM file was generated by SAMtools v1.12 [79].
- b.
- Variant calling
To call single nucleotide polymorphisms (SNP) and short indels from the bam files, SAMtools v1.12 mpileup and bcftools [80] were used to interrogate indexed BAM files of reads aligned to the reference genome and generate a VCF (Variant Call Format) file of SNP and short indel variants. Variant files (VCF) were next filtered using bcftools with the following parameters: QUAL <=30 && DP <=10; where QUAL denotes minimum variance confidence and DP total depth threshold. Control-FREEC v11.6 pipeline [81] were also used to call copy number variations from the sorted BAM files and generate duplicated and deleted variants.
Data visualization
To visualize the impact of regulatory variants in Hi-C interaction and gene expression, Washu Epigenome Browser [82] was used. In this analysis, the Hi-C interactions in conjunction with gene expression, ChIP-Seq and genomic variants data was used.
Pathway analysis
To validate the capability of AGV in identifying meaningful genes, we used ShinyGO v0.4 [27]. It contains 72,394 gene-sets for human genome including KEGG [83], MSigDB [84], GeneSetDB [85], REACTOME [86], etc. It has also access to STRING-db [87] for retrieving protein–protein interaction networks. we analyzed a set of 131 genes (Supplementary Table 6) which identified as the potential regulatory genes in our analysis. These genes list is mapped to the all collection of human gene-sets in ShinyGo for enrichment analysis. ShinyGo uses a hypergeometric distribution over-representation test to calculate the p value for gene set overlaps. We run ShinyGo with the default values () (Supplementary Table 8).
5. Conclusions
This section is not mandatory but can be added to the manuscript if the discussion is unusually long or complex.
6. Patents
This section is not mandatory but may be added if there are patents resulting from the work reported in this manuscript.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table 1: List of genomic variants for PC were used in this study. (a) GWAS SNPs. (b) hotspot somatic mutations. (C) CNVRs. Supplementary Table 2: List of genomic variants affecting genes. (a) SNPgenes, (b) somatic genes, (c) CNVgenes. Supplementary Table 3: List of statistically significance Hi-C interactions was used in this study. (a) LNCaP cell line. (b) PC3 cell line. (c) PrEC cell line. Supplementary Table 4: List of identified enhancer-promoter interactions from Hi-C interactions for PC. (a) LNCaP cell line. (b) PC3 cell line. SupplementartTable5: List of potential regulatory variants in (a)LNCaP cell line. (b) PC3 cell line. Supplementary Table 6: List of determined regulatory variants in (a) LNCaP cell line. (b) PC3 cell line. Supplementary Table 7: List of associated genes into regulatory variants. Supplementary Table 8: List of enriched genes with the pathway analysis. Supplementary Table 9: List of H3k27ac ChIP-seq peak regions was used in this study. (a) (a) LNCaP cell line. (b) PC3 cell line. (c) PrEC cell line. Supplementary Table 10: Definition of promoter regions that we used in our analysis.
Author Contributions
HAR and ML conceived the project. ML carried out all the analyses. ML generated all figures and supplementary tables with the supervision of HAR. HAR, ARA, AB, and ML revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the UNSW Scientia Program Fellowship and the Australian Research Council Discovery Early Career Researcher Award (DECRA) under grant DE220101210 to HAR. ML was funded by a Macquarie PhD Scholarship.
Institutional Review Board Statement
NA.
Informed Consent Statement
NA.
Data Availability Statement
All data are publicly available and cited in the paper. The source code is available at https://github.com/mahdieh1/ProstateCancer.
Acknowledgments
The authors wish to thank Ali Afrasiabi (BioMedical Machine Learning Lab (BML), The Graduate School of Biomedical Engineering, UNSW Sydney) for his constructive comments.
Conflicts of Interest
The authors declare no competing financial/non-financial interests.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.; Torre, L.; Jemal, A. Erratum: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2020, 70, 313. [Google Scholar]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef]
- Rojano, E.; Seoane, P.; Ranea, J.A.; Perkins, J.R. Regulatory variants: From detection to predicting impact. Brief. Bioinform. 2019, 20, 1639–1654. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, Z.; Lou, S.; Bedford, J.; Mu, X.J.; Yip, K.Y.; Khurana, E.; Gerstein, M. FunSeq2: A framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014, 15, 1–15. [Google Scholar] [CrossRef]
- Yip, K.Y.; Cheng, C.; Bhardwaj, N.; Brown, J.B.; Leng, J.; Kundaje, A.; Rozowsky, J.; Birney, E.; Bickel, P.; Snyder, M. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012, 13, 1–22. [Google Scholar] [CrossRef]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef]
- Chen, H.; Yu, H.; Wang, J.; Zhang, Z.; Gao, Z.; Chen, Z.; Lu, Y.; Liu, W.; Jiang, D.; Zheng, S.L. Systematic enrichment analysis of potentially functional regions for 103 prostate cancer risk-associated loci. Prostate 2015, 75, 1264–1276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tillmans, L.S.; Thibodeau, S.N.; Wang, L. Single-nucleotide polymorphisms sequencing identifies candidate functional variants at prostate cancer risk loci. Genes 2019, 10, 547. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Guo, Y.; Yang, H.; He, Z.; Liu, X.; Wang, K. iCAGES: Integrated CAncer GEnome Score for comprehensively prioritizing driver genes in personal cancer genomes. Genome Med. 2016, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Boyle, A.P. Predicting functional variants in enhancer and promoter elements using RegulomeDB. Hum. Mutat. 2019, 40, 1292–1298. [Google Scholar] [CrossRef]
- Parhami, P.; Fateh, M.; Rezvani, M. A comparison of deep neural network models for cluster cancer patients through somatic point mutations. J. Ambient Intell. Humaniz. Comput. 2023, 14, 10883–10898. [Google Scholar] [CrossRef]
- Dashti, H.; Dehzangi, I.; Bayati, M.; Breen, J.; Beheshti, A.; Lovell, N. Integrative analysis of mutated genes and mutational processes reveals novel mutational biomarkers in colorectal cancer. BMC Bioinform. 2022, 23, 1–24. [Google Scholar] [CrossRef]
- Heidari, R.; Akbariqomi, M.; Asgari, Y.; Ebrahimi, D. A systematic review of long non-coding RNAs with a potential role in Breast Cancer. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108375. [Google Scholar] [CrossRef]
- Ghareyazi, A.; Mohseni, A.; Dashti, H.; Beheshti, A.; Dehzangi, A.; Rabiee, H.R. Whole-genome analysis of de novo somatic point mutations reveals novel mutational biomarkers in pancreatic cancer. Cancers 2021, 13, 4376. [Google Scholar] [CrossRef] [PubMed]
- Bayati, M.; Rabiee, H.R.; Mehrbod, M.; Vafaee, F.; Ebrahimi, D.; Forrest, A.R. CANCERSIGN: A user-friendly and robust tool for identification and classification of mutational signatures and patterns in cancer genomes. Sci. Rep. 2020, 10, 1–11. [Google Scholar]
- Seim, I.; Jeffery, P.L.; Thomas, P.B.; Nelson, C.C.; Chopin, L.K. Whole-genome sequence of the metastatic PC3 and LNCaP human prostate cancer cell lines. G3 Genes Genomes Genet. 2017, 7, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Park, E.S.; Cheon, J.H.; Kim, J.H.; Lee, J.S.; Park, B.J.; Kim, W.; Park, S.C.; Chung, Y.J.; Kim, B.G.; et al. Gene expression–based recurrence prediction of hepatitis b virus–related human hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 2056–2064. [Google Scholar] [CrossRef]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, Z.; Zhou, W.; Chong, Z.; Meric-Bernstam, F.; Mills, G.B.; Chen, K. Hotspot mutations delineating diverse mutational signatures and biological utilities across cancer types. BMC Genom. 2016, 17, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Nesta, A.V.; Tafur, D.; Beck, C.R. Hotspots of human mutation. Trends Genet. 2021, 37, 717–729. [Google Scholar] [CrossRef]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B. International Cancer Genome Consortium Data Portal—A one-stop shop for cancer genomics data. Database 2011, 2011, bar026. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.P. A global reference for human genetic variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef] [PubMed]
- Labani, M.; Afrasiabi, A.; Beheshti, A.; Lovell, N.H.; Alinejad-Rokny, H. PeakCNV: A multi-feature ranking algorithm-based tool for genome-wide copy number variation-association study. Comput. Struct. Biotechnol. J. 2022, 20, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.-J.; Vert, J.-P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 1–11. [Google Scholar] [CrossRef]
- Alinejad-Rokny, H.; Ghavami Modegh, R.; Rabiee, H.R.; Ramezani Sarbandi, E.; Rezaie, N.; Tam, K.T.; Forrest, A.R. MaxHiC: A robust background correction model to identify biologically relevant chromatin interactions in Hi-C and capture Hi-C experiments. PLOS Comput. Biol. 2022, 18, e1010241. [Google Scholar] [CrossRef]
- Khakmardan, S.; Rezvani, M.; Pouyan, A.A.; Fateh, M.; Alinejad-Rokny, H. MHiC, an integrated user-friendly tool for the identification and visualization of significant interactions in Hi-C data. BMC Genom. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- Bicak, M.; Wang, X.; Gao, X.; Xu, X.; Väänänen, R.-M.; Taimen, P.; Lilja, H.; Pettersson, K.; Klein, R.J. Prostate cancer risk SNP rs10993994 is a trans-eQTL for SNHG11 mediated through MSMB. Hum. Mol. Genet. 2020, 29, 1581–1591. [Google Scholar] [CrossRef]
- Misawa, A.; Takayama, K.i.; Inoue, S. Long non-coding RNAs and prostate cancer. Cancer Sci. 2017, 108, 2107–2114. [Google Scholar] [CrossRef]
- Leite, K.R.; Franco, M.F.; Srougi, M.; Nesrallah, L.J.; Nesrallah, A.; Bevilacqua, R.G.; Darini, E.; Carvalho, C.M.; Meirelles, M.I.; Santana, I. Abnormal expression of MDM2 in prostate carcinoma. Mod. Pathol. 2001, 14, 428–436. [Google Scholar] [CrossRef]
- Mertz, K.D.; Pathria, G.; Wagner, C.; Saarikangas, J.; Sboner, A.; Romanov, J.; Gschaider, M.; Lenz, F.; Neumann, F.; Schreiner, W. MTSS1 is a metastasis driver in a subset of human melanomas. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Braune, K.; Volkmer, I.; Staege, M.S. Characterization of alstrom syndrome 1 (ALMS1) transcript variants in hodgkin lymphoma cells. PLoS ONE 2017, 12, e0170694. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Łastowska, M.; Viprey, V.; Santibanez-Koref, M.; Wappler, I.; Peters, H.; Cullinane, C.; Roberts, P.; Hall, A.G.; Tweddle, D.A.; Pearson, A.D.J.; et al. Identification of candidate genes involved in neuroblastoma progression by combining genomic and expression microarrays with survival data. Oncogene 2007, 26, 7432–7444. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Van’t Veer, L.J.; Dai, H.; Van De Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T. Gene expression profiling predicts clinical outcome of breast cancer. nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Arbuthnot, P.; Kew, M. Hepatitis B virus and hepatocellular carcinoma. Int. J. Exp. Pathol. 2001, 82, 77–100. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- George, C.L. Analyzing ZNF16: An Understudied Gene. The University of Texas at El Paso, 2020. [Google Scholar]
- Balajee, A.S. Human recql4 as a novel molecular target for cancer therapy. Cytogenet. Genome Res. 2021, 161, 305–327. [Google Scholar] [CrossRef]
- Su, Y.; Meador, J.A.; Calaf, G.M.; De-Santis, L.P.; Zhao, Y.; Bohr, V.A.; Balajee, A.S. Human RecQL4 helicase plays critical roles in prostate carcinogenesis. Cancer Res. 2010, 70, 9207–9217. [Google Scholar] [CrossRef]
- Nasab, R.Z.; Ghamsari, M.R.E.; Argha, A.; Macphillamy, C.; Beheshti, A.; Alizadehsani, R.; Lovell, N.H.; Lotfollahi, M.; Alinejad-Rokny, H. Deep Learning in Spatially Resolved Transcriptomics: A Comprehensive Technical View. arXiv 2022, arXiv:2210.04453. [Google Scholar]
- Razzak, I.; Naz, S.; Alinejad-Rokny, H.; Nguyen, T.N.; Khalifa, F. A Cascaded Mutliresolution Ensemble Deep Learning Framework for Large Scale Alzheimer’s Disease Detection using Brain MRIs. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Argha, A.; Celler, B.G.; Alinejad-Rokny, H.; Lovell, N.H. Blood Pressure Estimation From Korotkoff Sound Signals Using an End-to-End Deep-Learning-Based Algorithm. IEEE Trans. Instrum. Meas. 2022, 71, 1–10. [Google Scholar] [CrossRef]
- Consortium, I.C.G. International network of cancer genome projects. Nature 2010, 464, 993. [Google Scholar]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J. The UCSC genome browser database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Alinejad-Rokny, H.; Heng, J.I.; Forrest, A.R. Brain-enriched coding and long non-coding RNA genes are overrepresented in recurrent neurodevelopmental disorder CNVs. Cell Rep. 2020, 33, 108307. [Google Scholar] [CrossRef]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Labani, M.; Beheshti, A.; Lovell, N.H.; Alinejad-Rokny, H.; Afrasiabi, A. KARAJ: An Efficient Adaptive Multi-Processor Tool to Streamline Genomic and Transcriptomic Sequence Data Acquisition. Int. J. Mol. Sci. 2022, 23, 14418. [Google Scholar] [CrossRef]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.-J.; Vert, J.-P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Druliner, B.R.; Vera, D.; Johnson, R.; Ruan, X.; Apone, L.M.; Dimalanta, E.T.; Stewart, F.J.; Boardman, L.; Dennis, J.H. Comprehensive nucleosome mapping of the human genome in cancer progression. Oncotarget 2016, 7, 13429. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef]
- Rajaei, P.; Jahanian, K.H.; Beheshti, A.; Band, S.S.; Dehzangi, A.; Alinejad-Rokny, H. VIRMOTIF: A user-friendly tool for viral sequence analysis. Genes 2021, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Pho, K.H.; Akbarzadeh, H.; Parvin, H.; Nejatian, S.; Alinejad-Rokny, H. A multi-level consensus function clustering ensemble. Soft Comput. 2021, 25, 13147–13165. [Google Scholar] [CrossRef]
- Mahmoudi, M.R.; Akbarzadeh, H.; Parvin, H.; Nejatian, S.; Rezaie, V.; Alinejad-Rokny, H. Consensus function based on cluster-wise two level clustering. Artif. Intell. Rev. 2021, 54, 639–665. [Google Scholar] [CrossRef]
- Hosseinpoor, M.; Parvin, H.; Nejatian, S.; Rezaie, V.; Bagherifard, K.; Dehzangi, A.; Beheshti, A.; Alinejad-Rokny, H. Proposing a novel community detection approach to identify cointeracting genomic regions. Math. Biosci. Eng. 2020, 17, 2193–2217. [Google Scholar] [CrossRef]
- Bahrani, P.; Minaei-Bidgoli, B.; Parvin, H.; Mirzarezaee, M.; Keshavarz, A.; Alinejad-Rokny, H. User and item profile expansion for dealing with cold start problem. J. Intell. Fuzzy Syst. 2020, 38, 4471–4483. [Google Scholar] [CrossRef]
- Alinejad-Rokny, H. Proposing on Optimized Homolographic Motif Mining Strategy Based on Parallel Computing for Complex Biological Networks. J. Med. Imaging Health Inform. 2016, 6, 416–424. [Google Scholar] [CrossRef]
- Alinejad-Rokny, H.; Pourshaban, H.; Orimi, A.G.; Baboli, M.M. Network motifs detection strategies and using for bioinformatic networks. J. Bionanoscience 2014, 8, 353–359. [Google Scholar] [CrossRef]
- Ahmadinia, M.; Alinejad-Rokny, H.; Ahangarikiasari, H. Data aggregation in wireless sensor networks based on environmental similarity: A learning automata approach. J. Netw. 2014, 9, 2567. [Google Scholar] [CrossRef]
- Parvin, H.; Alinejad-Rokny, H.; Minaei-Bidgoli, B.; Parvin, S. A new classifier ensemble methodology based on subspace learning. J. Exp. Theor. Artif. Intell. 2013, 25, 227–250. [Google Scholar] [CrossRef]
- Parvin, H.; Alinejad-Rokny, H.; Parvin, S. A classifier ensemble of binary classifier ensembles. Int. J. Learn. Manag. Syst. 2013, 1, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Javanmard, R.; JeddiSaravi, K.; Alinejad-Rokny, H. Proposed a new method for rules extraction using artificial neural network and artificial immune system in cancer diagnosis. J. Bionanoscience 2013, 7, 665–672. [Google Scholar] [CrossRef]
- Parvin, H.; Alinejad-Rokny, H.; Seyedaghaee, N.; Parvin, S. A heuristic scalable classifier ensemble of binary classifier ensembles. J. Bioinform. Intell. Control 2012, 1, 163–170. [Google Scholar] [CrossRef]
- Hasanzadeh, E.; Poyan, M.; Alinejad-Rokny, H. Text clustering on latent semantic indexing with particle swarm optimization (PSO) algorithm. Int. J. Phys. Sci. 2012, 7, 16–120. [Google Scholar] [CrossRef]
- Esmaeili, L.; Minaei-Bidgoli, B.; Alinejad-Rokny, H.; Nasiri, M. Hybrid recommender system for joining virtual communities. Res. J. Appl. Sci. Eng. Technol. 2012, 4, 500–509. [Google Scholar]
- Parvin, H.; Alinejad, H.; Minaei-Bidgoli, B. Using Clustering for Generating Diversity in Classifier Ensemble. JDCTA 2011, 3, 51–57. [Google Scholar] [CrossRef]
- Parvin, H.; Alinejad-Rokny, H.; Asadi, M. An ensemble based approach for feature selection. J. Appl. Sci. Res. 2011, 9, 33–43. [Google Scholar]
- Alinejad-Rokny, H.; Pedram, M.M.; Shirgahi, H. Discovered motifs with using parallel Mprefixspan method. Sci. Res. Essays 2011, 6, 4220–4226. [Google Scholar]
- Alinejad-Rokny, H.; Sadroddiny, E.; Scaria, V. Machine learning and data mining techniques for medical complex data analysis. Neurocomputing 2018, 276, 10–1016. [Google Scholar] [CrossRef]
- Niu, H.; Khozouie, N.; Parvin, H.; Alinejad-Rokny, H.; Beheshti, A.; Mahmoudi, M.R. An ensemble of locally reliable cluster solutions. Appl. Sci. 2020, 10, 1891. [Google Scholar] [CrossRef]
- Niu, H.; Xu, W.; Akbarzadeh, H.; Parvin, H.; Beheshti, A.; Alinejad-Rokny, H. Deep feature learnt by conventional deep neural network. Comput. Electr. Eng. 2020, 84, 106656. [Google Scholar] [CrossRef]
- Parvin, H.; MirnabiBaboli, M.; Alinejad-Rokny, H. Proposing a classifier ensemble framework based on classifier selection and decision tree. Eng. Appl. Artif. Intell. 2015, 34–42. [Google Scholar] [CrossRef]
- Parvin, H.; Alinejad-Rokny, H.; Minaei-Bidgoli, B. Detection of cancer patients using an innovative method for learning at imbalanced datasets. In Proceedings of the International Conference on Rough Sets and Knowledge Technology; pp. 376–381.
- Andrew, S. A quality control tool for high throughput sequence data. Fast QC 2010, 390, 391. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Zhou, X.; Lowdon, R.F.; Li, D.; Lawson, H.A.; Madden, P.A.; Costello, J.F.; Wang, T. Exploring long-range genome interactions using the WashU Epigenome Browser. Nat. Methods 2013, 10, 375–376. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Araki, H.; Knapp, C.; Tsai, P.; Print, C. GeneSetDB: A comprehensive meta-database, statistical and visualisation framework for gene set analysis. FEBS Open Bio 2012, 2, 76–82. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, gkw937. [Google Scholar] [CrossRef]
Figure 1.
An overview of AGV. AGV pipeline first makes a list of associated genomic variants including GWAS SNPs, somatic point mutation and CNV regions. AGV then uses Hi-C and H3K27ac to determine variants with possible rules in disrupting promoter-enhancer interactions. Lastly, AGV reports a list of functional genomic variants with possible role in PC.
Figure 1.
An overview of AGV. AGV pipeline first makes a list of associated genomic variants including GWAS SNPs, somatic point mutation and CNV regions. AGV then uses Hi-C and H3K27ac to determine variants with possible rules in disrupting promoter-enhancer interactions. Lastly, AGV reports a list of functional genomic variants with possible role in PC.
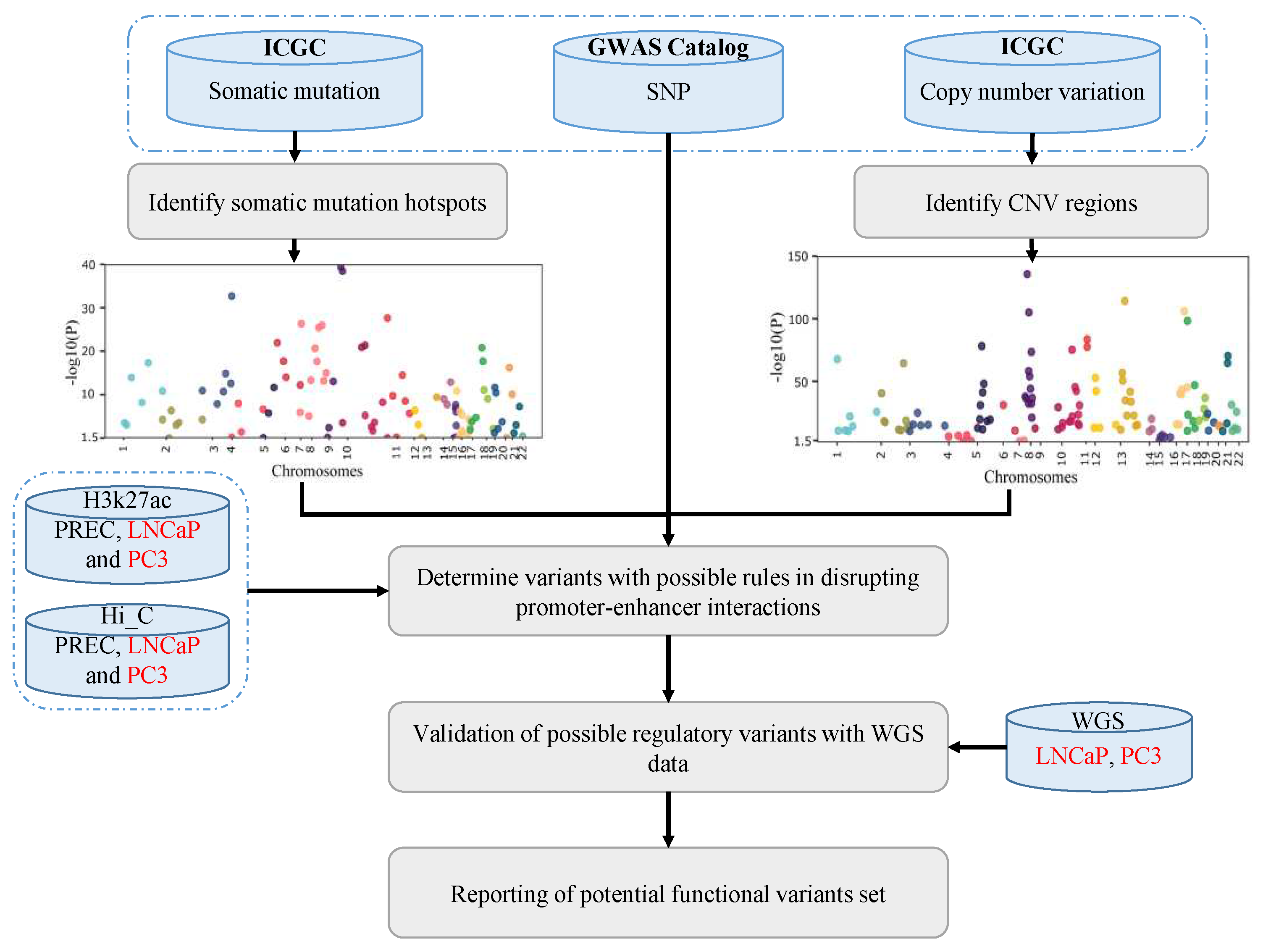
Figure 2.
The schematic workflow used in this study to identify somatic point mutation hotspot regions. This analysis consists of three main steps: (1) window analysis, (2) selection, and (3) filtering. In the first step, the tool divides the genome into 21bp bins and then counts the number of samples with at least one SPM that overlapped with the window. In the selection step, a Poisson binomial distribution was used to select significant bins (P-value < 0.05). Lastly, in the filtering step, the problematic hotspot regions and chromosome Y was excluded from the final list of hotspot regions.
Figure 2.
The schematic workflow used in this study to identify somatic point mutation hotspot regions. This analysis consists of three main steps: (1) window analysis, (2) selection, and (3) filtering. In the first step, the tool divides the genome into 21bp bins and then counts the number of samples with at least one SPM that overlapped with the window. In the selection step, a Poisson binomial distribution was used to select significant bins (P-value < 0.05). Lastly, in the filtering step, the problematic hotspot regions and chromosome Y was excluded from the final list of hotspot regions.
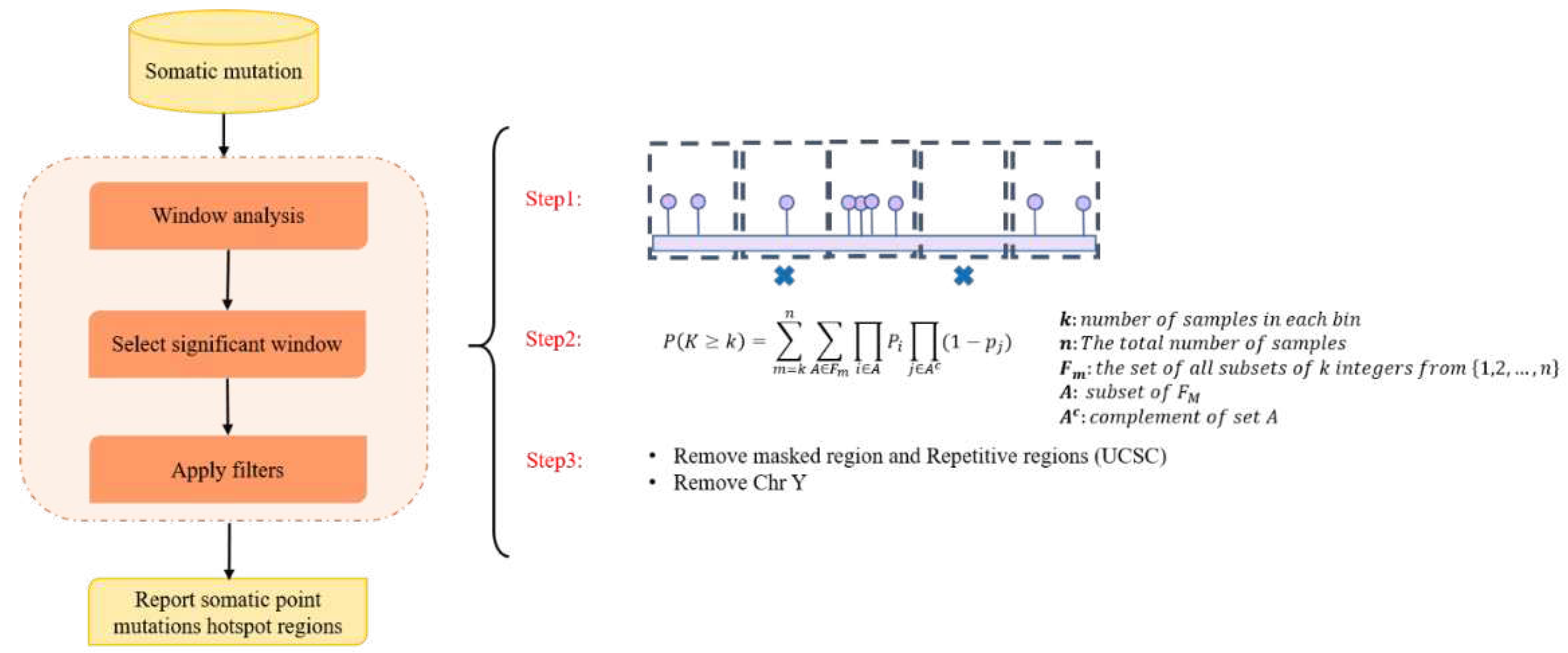
Figure 3.
A) Distribution of SPMs for different window sizes (500bp, 50, 21, and 9bp) on chromosome 22. B) Probability distribution for identified 21 bp bins by Cullen and Frey graph, and CDF plot.
Figure 3.
A) Distribution of SPMs for different window sizes (500bp, 50, 21, and 9bp) on chromosome 22. B) Probability distribution for identified 21 bp bins by Cullen and Frey graph, and CDF plot.
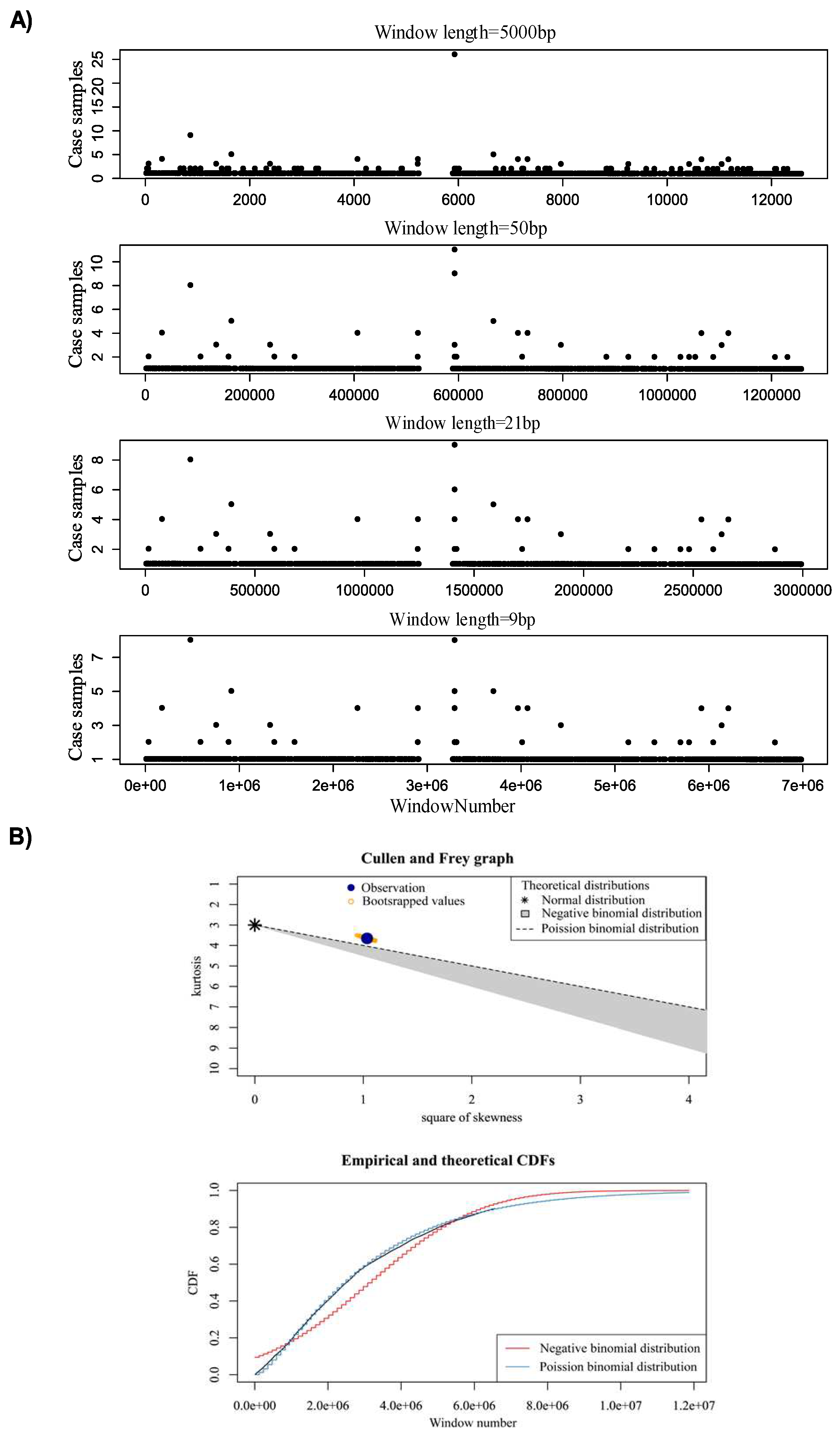
Figure 4.
A genome-wide overview of somatic mutation hotspots (red dots) and filtered regions (containing masked regions and repetitive regions (blue dots) and non-significant regions (black dots)) identified in this study. The figure illustrates the distribution of 21bp bin-size windows encompassing PC related somatic point mutations across the genome. x-axis shows the window number and y-axis shows the number of case samples covered by the window. For each window, our proposed method calculates the P-value of mutation recurrence using the Poisson binomial distribution Then, problematic regions including masked regions and repetitive regions were excluded and bins with P-value < 0.001 were selected.
Figure 4.
A genome-wide overview of somatic mutation hotspots (red dots) and filtered regions (containing masked regions and repetitive regions (blue dots) and non-significant regions (black dots)) identified in this study. The figure illustrates the distribution of 21bp bin-size windows encompassing PC related somatic point mutations across the genome. x-axis shows the window number and y-axis shows the number of case samples covered by the window. For each window, our proposed method calculates the P-value of mutation recurrence using the Poisson binomial distribution Then, problematic regions including masked regions and repetitive regions were excluded and bins with P-value < 0.001 were selected.
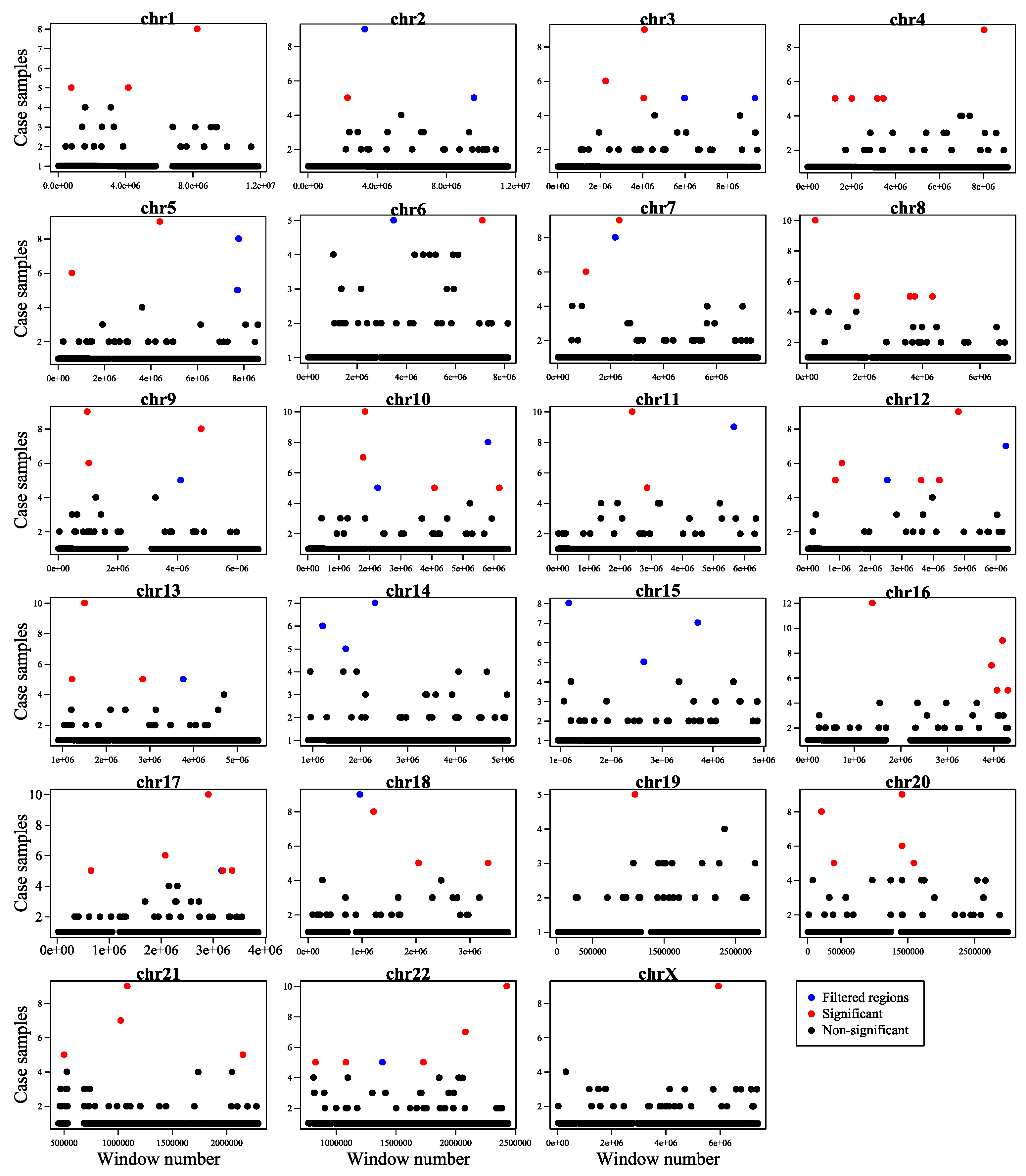
Figure 5.
Linking Genomic variants to coding and non-coding genes. A) Somatic point mutation hotspots; B) GWAS SNPs; C) Duplicated CNVRs; D) Deleted CNVRs. The left panel shows the percentage of genomic variants associated to the genes (Annotated regions) or there are not genes relating to the genomic variants (non-annotated regions). The right panel shows the fraction of these genomic variants are protein coding or non-coding genes. The greater fraction of hotspot regions and CNVRs are located in non-coding genes, while less than 30% of GWAS SNPs are located in non-coding genes. E-I show the percentage of linking different types of genomic variants including E) Somatic point mutation hotspots; F) GWAS SNPs; G) Duplicated CNVRs; H) Deleted CNVRs into noncoding RNA. y-axis represents the number of different types of RNA associating to genomic variants.
Figure 5.
Linking Genomic variants to coding and non-coding genes. A) Somatic point mutation hotspots; B) GWAS SNPs; C) Duplicated CNVRs; D) Deleted CNVRs. The left panel shows the percentage of genomic variants associated to the genes (Annotated regions) or there are not genes relating to the genomic variants (non-annotated regions). The right panel shows the fraction of these genomic variants are protein coding or non-coding genes. The greater fraction of hotspot regions and CNVRs are located in non-coding genes, while less than 30% of GWAS SNPs are located in non-coding genes. E-I show the percentage of linking different types of genomic variants including E) Somatic point mutation hotspots; F) GWAS SNPs; G) Duplicated CNVRs; H) Deleted CNVRs into noncoding RNA. y-axis represents the number of different types of RNA associating to genomic variants.

Figure 6.
The number and distance of statistically significant Hi-C interactions in cancer cell lines (PC3 and LNCaP) and healthy cell line (PrEC).
Figure 6.
The number and distance of statistically significant Hi-C interactions in cancer cell lines (PC3 and LNCaP) and healthy cell line (PrEC).
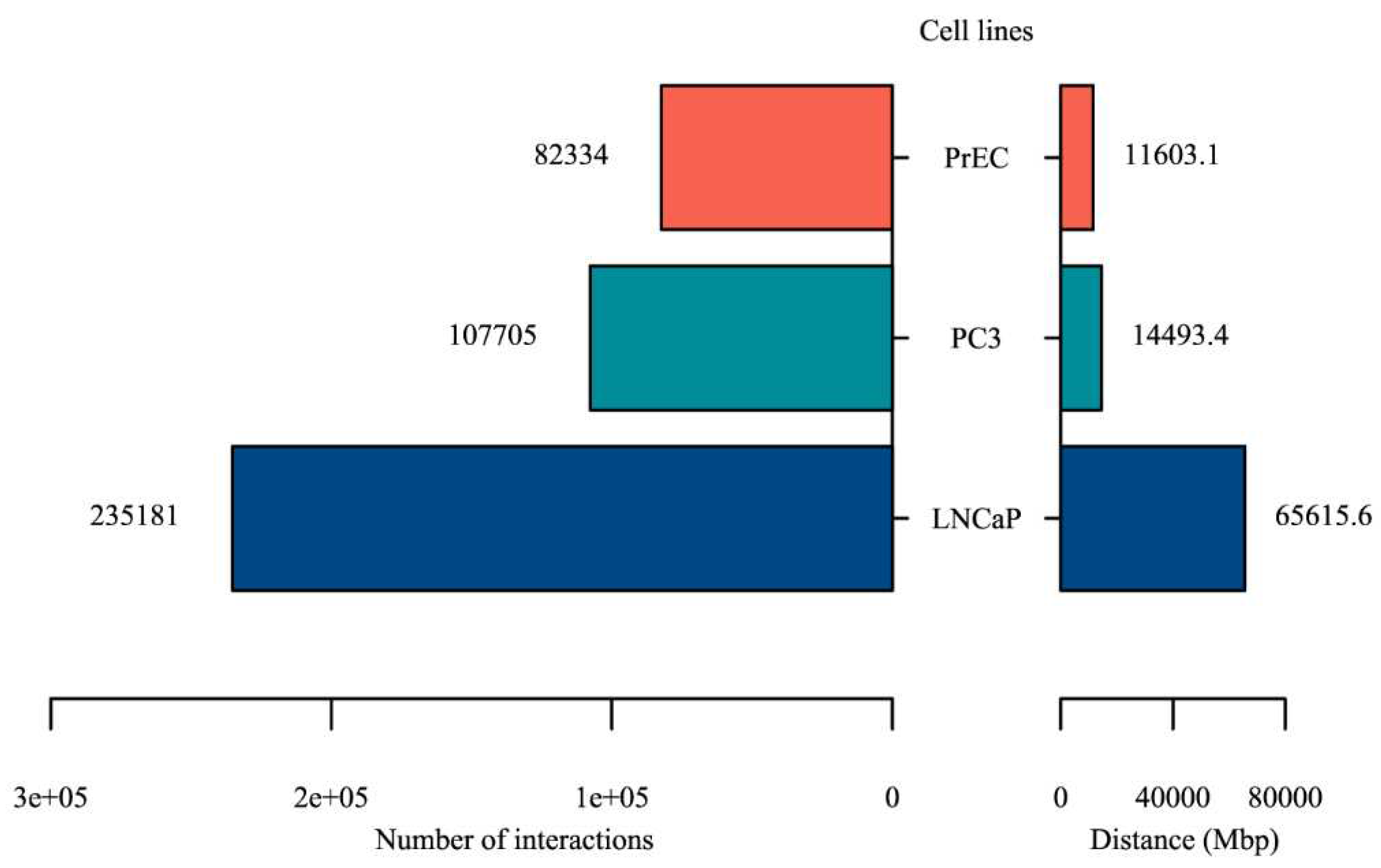
Figure 7.
A) Example of a regulatory variant in cancer cell line PC3. The figure demonstrates the RNA-Seq, H3K27ac signals, and Hi-C chromatin interactions map of normal (PREC) and prostate cancer (PC3) cells on Chromosome 8. The highlighted box shows one of the PC-associated CNVR identified in this study that was also observed in PC3 WGS. There is an EPI in cancer cell line (the EPI was not observed in the healthy cell line) that the enhancer side of the interaction overlapped with CNVR. Interestingly. The left side of this interaction is promoter regions of RNF139 and TADN1, and the right side (enhancer region) has also an active H3K27ac signal. The expression of NDUFB9 is much higher in cancer cell compared to healthy cell line. B) Example of regulatory variant in cancer cell line LNCaP. The figure demonstrates the RNA-seq, H3K27ac signals, and Hi-C chromatin interactions map of normal (PREC) and prostate cancer (LNCaP) cells on Chromosome 2. The highlighted box shows one of the PC-associated CNVRs identified in this study that was also observed in LNCaP WGS. There is an EPI in cancer cell line (the EPI was not observed in the healthy cell line) that the enhancer side of the interaction overlapped with CNVR. Interestingly. The left side of this interaction is promoter regions NAT8 and ALMS1P genes, and the right side (enhancer region) has also an active H3K27ac signal. WashU Epigenome Browser has been used to generate the figure.
Figure 7.
A) Example of a regulatory variant in cancer cell line PC3. The figure demonstrates the RNA-Seq, H3K27ac signals, and Hi-C chromatin interactions map of normal (PREC) and prostate cancer (PC3) cells on Chromosome 8. The highlighted box shows one of the PC-associated CNVR identified in this study that was also observed in PC3 WGS. There is an EPI in cancer cell line (the EPI was not observed in the healthy cell line) that the enhancer side of the interaction overlapped with CNVR. Interestingly. The left side of this interaction is promoter regions of RNF139 and TADN1, and the right side (enhancer region) has also an active H3K27ac signal. The expression of NDUFB9 is much higher in cancer cell compared to healthy cell line. B) Example of regulatory variant in cancer cell line LNCaP. The figure demonstrates the RNA-seq, H3K27ac signals, and Hi-C chromatin interactions map of normal (PREC) and prostate cancer (LNCaP) cells on Chromosome 2. The highlighted box shows one of the PC-associated CNVRs identified in this study that was also observed in LNCaP WGS. There is an EPI in cancer cell line (the EPI was not observed in the healthy cell line) that the enhancer side of the interaction overlapped with CNVR. Interestingly. The left side of this interaction is promoter regions NAT8 and ALMS1P genes, and the right side (enhancer region) has also an active H3K27ac signal. WashU Epigenome Browser has been used to generate the figure.
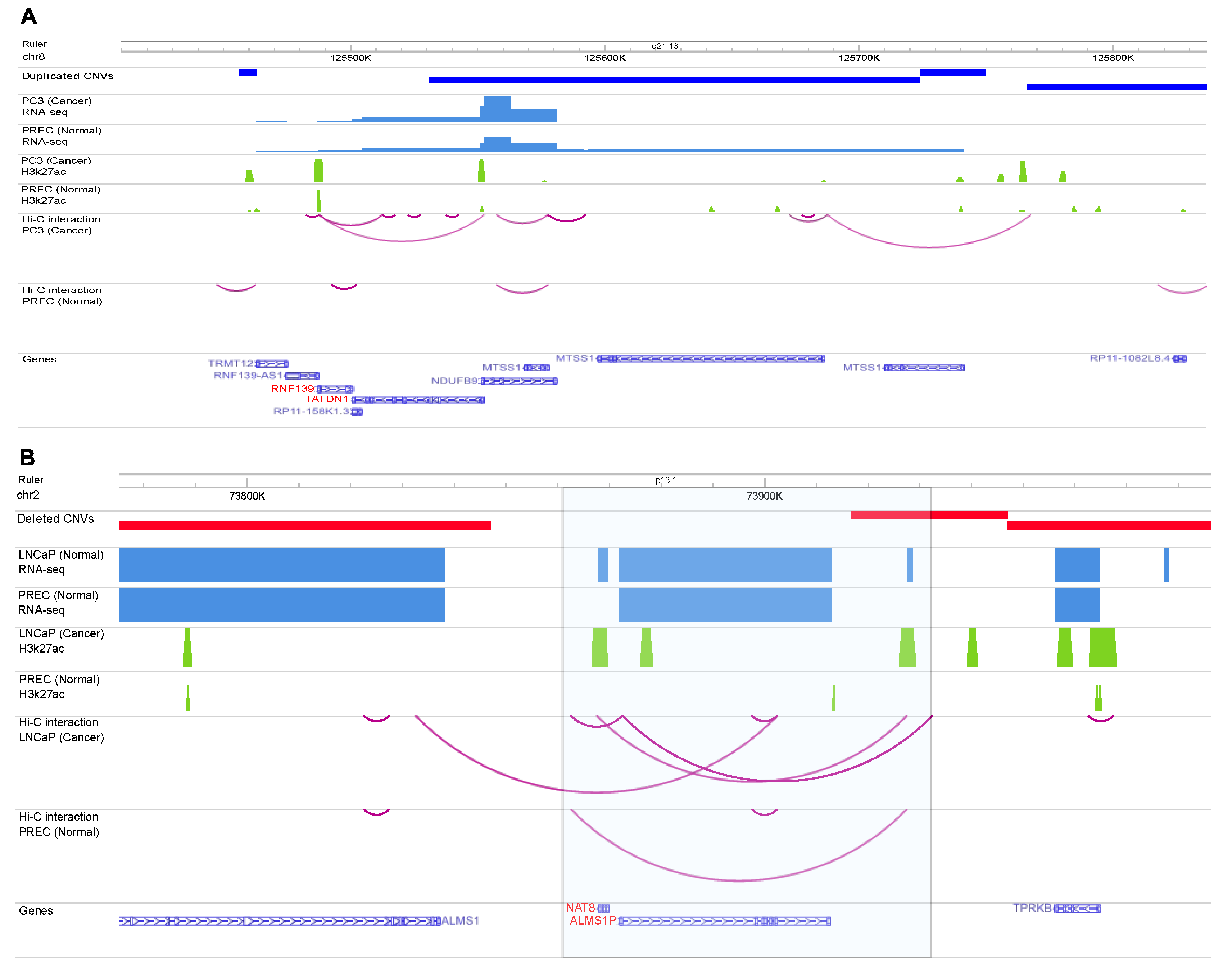
Figure 8.
Enriched proteins in the KEGG pathway for cancer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



