
Submitted:
21 January 2023
Posted:
23 January 2023
You are already at the latest version
Abstract
Endothelial integrity plays a major role in homeostasis and is responsive to the numerous endogenous factors released. While its functional role in vascular tone is well described, its role in the pathophysiology of cardiovascular disease is of interest as a potential therapeutic target. We performed a systematic review to provide an overview of new therapeutic and diagnostic targets for the treatment of coronary artery disease related to endothelial dysfunction. Databases of PubMed, Ovid’s version of MEDLINE, and EMBASE were interrogated with appropriate search terms. 28 studies met inclusion criteria and were included in the final systematic review. We identified inflammation, pulmonary hypertension, diabetes mellitus and Fabry disease as pathophysiological mechanisms and explored the therapeutic options related to these conditions including medications such as Canakinumab. Endothelial dysfunction has a key role in several different pathophysiological processes which can be targeted for therapeutic options. Ongoing research should be targeted at making the transition to clinical practice. Further research is also needed on understanding the amelioration of endothelial dysfunction with the use of cardiovascular medications.
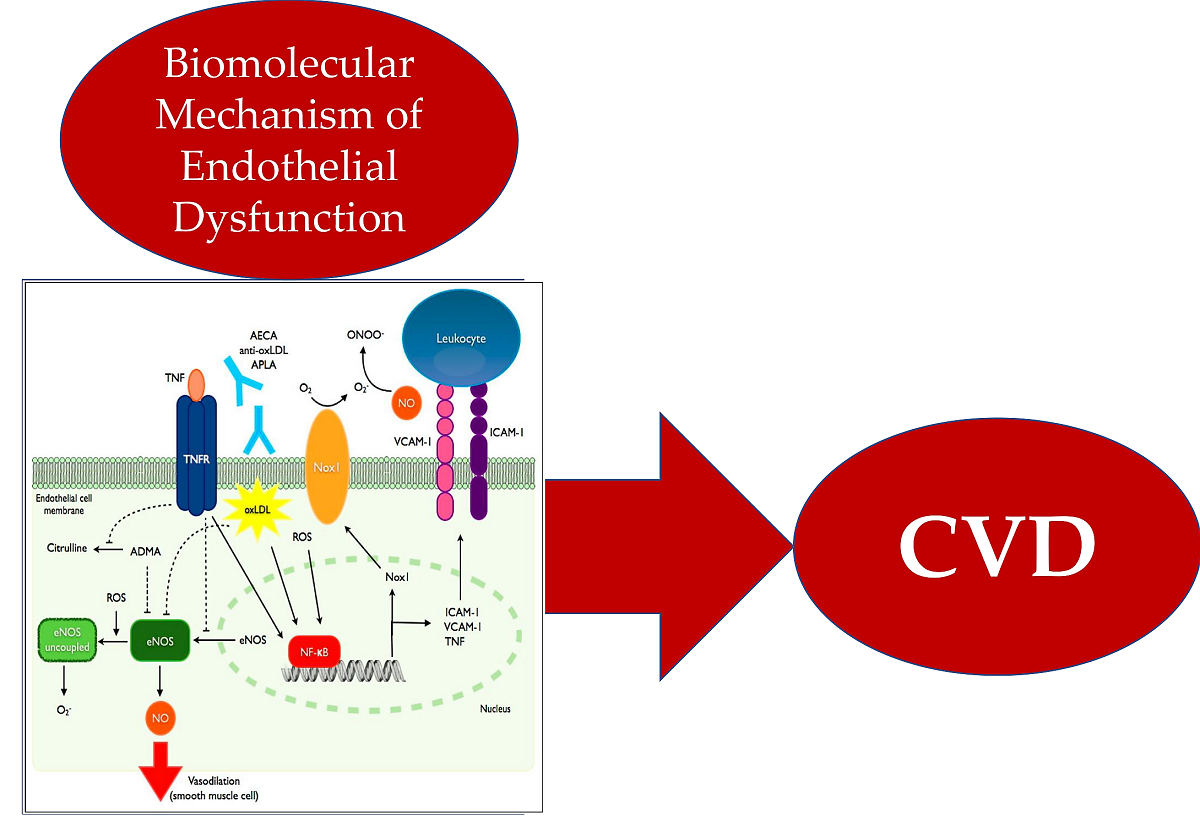
Keywords:
Endothelial dysfunction
; inflammation
; pulmonary hypertension
; diabetes
; car-diovascular disease
; endothelium-derived relaxing factor
1. Introduction
Although studies on the endothelium function have been performed for more than 30 years, to date, no detailed guidelines on the choice of novel therapeutic and diagnostic targets have been published to attenuate the onset of cardiovascular disease. This evidence collides with current clinical practice that demonstrates an important role exhibited by endogenous hyperglycemic hormones, which are advantageous for ensuring proper maintenance of vascular tone under pathological conditions [1,2,3]. In these circumstances the role of endothelium-derived relaxing factors is crucial and is mediated by the action of vasodilator prostaglandins, NO, and endothelium-dependent hyperpolarization (EDH) factors, as well as endothelium-derived contracting factors. Insulin levels influence synergistically coupling with endothelium-derived relaxing factors the correct functioning of the macro and microcirculation. However, when the correct responses mediated by at least one of the endothelium-derived relaxing is impaired, vasoconstriction, alongside a prothrombotic and proinflammatory state is favored [4,5]. Given the importance that the endothelial function exerts in humans, its assessment is the subject of particular attention in the clinical field because if properly studied it offers the possibility of being a reliable surrogate marker of cardiovascular events. Endothelial dysfunction, when detected by impaired brachial artery flow-mediated dilatation or digital reactive hyperemia index in peripheral arterial tonometry, may be the marker of future cardiovascular events in patients with coronary artery disease [6,7,8]. In patients revealing a 1-SD decrease in flow-mediated dilatation or index reactive hyperemia, a duplication of risk of cardiovascular events in patients with cardiovascular disease (CVD) is markedly superimposed [9,10,11]. These considerations support the determinant role that endothelial function exerts in peripheral vascular circulation that could predict future cardiovascular events, to the point of a potential predictor of future cardiovascular events [9].
To encourage a broader understanding of the hallmarks of disease interfering with endothelial function and to provide guidance for clinicians, we discuss the current evidence base on the role of inflammation, diabetes mellitus, and pulmonary hypertension that may drive impaired endothelial function. In addition, we provide an overview of new therapeutic and diagnostic targets for the treatment of coronary artery disease related to endothelial dysfunction. Graphical abstract
2. Methods
2.1. Search Strategy
In September 2022, PubMed, Ovid’s version of MEDLINE, and EMBASE the systematic review was investigated using the terms “Endothelial vasodilation (5.202 to the present)” “Endothelial vasoconstriction (1.102 to the present)” “nitric oxide inflammation (3, 133 to the present)” “ nitric oxide diabetes (1, 117 to the present) ” nitric oxide, “pulmonary hypertension (481 to the present) ”, “endothelium-derived relaxing factor inflammation (3, 136 to the present)”, “ endothelium-derived relaxing factor diabetes (1, 124 to the present) ” and “ endothelium-derived relaxing factor pulmonary hypertension (483 to the present) ”. The search was directed to the identification of data from basic research articles and randomized controlled trials (RCT). The review was registered with the OSF register of systematic reviews and followed the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) reporting guidelines. The project files are available online at (https://osf.io/tbm8y/)
2.2. Study Selection and Data Extraction
Searches retrieved 15, 778 relevant abstracts, and after deduplication 6 928 relevant citations were screened by 2 reviewers on their own (F.N, SSAS). Inconsistency was resolved by the first author who performed the conceptualization (F.N). The predefined inclusion criteria guided the review of titles and abstracts. The articles included were in English and were very impressive research articles based on the function of NO, EDRF, and hallmarks of disease that occurred in cases of endothelial dysfunction. Also of great relevance were the RCTs focused on novel therapeutic and diagnostic targets and observational studies on the role of inflammation, diabetes, and pulmonary. Relevant animal studies were included because they were of higher impact on the emerged role of endothelial dysfunction in supporting cardiovascular disease. The most relevant articles performed on the animal models were published in Arteriosclerosis, Thrombosis, and Vascular Biology journal. Case reports, conference presentations, editorials, and expert opinions were excluded. A total of 390 citations were evaluated of which 28 studies met inclusion criteria and were included in the final systematic review. Figure 1 PRISMA Flowchart. PRISMA 2020 checklist in Supplementary Material.
2.3. Endpoints and Effect Summary
The endpoints evaluated the effects of the emerging role of the hallmarks of disease that may support endothelial dysfunction focusing the analysis on studies that investigated the role of inflammation, diabetes, and pulmonary hypertension. She studies are reported in Table 1, Table 2 and Table 3 alongside the discussion of results.
3. Results
3.1. Presence of Inflammation in Endothelial Dysfunction
Inflammatory conditions are closely implicated in supporting the development of endothelial dysfunction, as has been demonstrated by the detection of IL-1β (interleukin-1β) which is a major proinflammatory cytokine [12,13,14]. Figure 2
The study by Honda et al [15] revealed a close correlation between endothelial dysfunction assessed by flow-mediated dilatation and the development of vascular inflammation. The authors evaluated the endothelial function and vascular inflammation by flow-mediated dilatation of the brachial artery and [(18)F]-fluorodeoxyglucose positron emission tomography/computed tomography of the carotid arteries, respectively, of 145 patients (95 men and 50 women; mean age 61,8±9.5 years) who underwent cardiovascular disease risk screening tests. Determination of the degree of vascular inflammation was achieved by measuring the normalized standardized uptake value in the blood, known as the target: background ratio (TBR). Absolute changes from baseline in % FMD after 6 months of antihypertensive treatment (Δ% FMD) were investigated and whether they correlated with those in TBR in 33 drug-innocent patients with essential hypertension. Statistical significance was demonstrated by multiple logistic regression analysis in which low-density lipoprotein-cholesterol (odds ratio, 1.630 for an increase of 26 mg/dL), TBR values (odds ratio, 1.759 for an increase of 0.2), male sex (odds ratio, 0.434), and age (odds ratio, 1.767 for a 10-year increase) were substantially independently associated with % of flow-mediated dilation in 145 patients. The report of Honda et al initially suggested the association of vascular inflammation with endothelial dysfunction in humans disclosing the evidence that the vascular inflammation in the carotid arteries analyzed was one of the independent correlations of decreased % of flow-mediated dilation [15].
Given the crucial role of inflammation in endothelial dysfunction, furthermore in the presence of atherosclerotic plaques, the investigation oriented towards the role of statins in influencing a robust anti-inflammatory effect of atherosclerotic plaques primarily during the early treatment period or continuously throughout use was considered a valid field of study. Kang et al [16] reported findings from a prospective three timepoint 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT) study. Investigators evaluated the carotid artery by means of an assessment of the anti-inflammatory effects of statin during the early treatment period considered as the initiation of therapy to 3 months and the late treatment period relating to the period of 3 months to 1 year and their correlation with lipid and inflammatory profile changes during a year of therapy. Briefly, 20 mg/day atorvastatin was administered in nine statin-naïve stable angina patients with inflammatory carotid plaques after undergoing initial 18F-FDG PET/CT scanning of carotid arteries and ascending thoracic aorta and then completed serial 18F-FDG PET/CT imaging at 3 and 12 months. The primary outcome was the inter-scan percent change in a target-to-background ratio within the index vessel was established. Importantly, at 3 months of atorvastatin treatment results revealed that mean serum low-density lipoprotein cholesterol level decreased by 36.4% to < 70 mg/dL (p = 0.001) and mean serum high-density lipoprotein cholesterol level increased to > 40 mg/dL (p = 0.041). Again, both these parameters were preserved with no further reduction at up to 1 year (p = 0.516 and 0.715, respectively) while mean serum high sensitivity C-reactive protein level only numerically decreased (p = 0.093). The index vessel of target-to-background ratio disclosed continuous plaque inflammation reduction over 1 year, by 4.4% (p = 0.015) from the initiation to the 3rd month and 6.2% (p = 0.009) from 3rd month to 1 year, respectively, without correlation with lipid profile changes. The second crucial finding was disclosed by the delta target-to-background ratio of the bilateral carotid arteries and ascending aorta which decreased from 3 months to 1 year. Evidence suggested that the three timepoint 18F-FDG PET/CT imaging was instrumental in proving that the anti-inflammatory effect of the statin continues throughout its use up to 1 year, even if it produces plasma LDL-C levels were stable below the 3-month target [16].
Four independent studies have studied the potential role exhibited by proinflammatory molecules in endothelial dysfunction that occurs in the course of diabetes and atherosclerosis [17,18,19,20]. The lack of evidence emerges from prospective human studies that have been aimed at understanding the mechanism between the initiating factor of the lectin pathway, i.e., mannose-binding lectin (MBL), and low-grade inflammation, endothelial dysfunction, or carotid intima-media thickness. Furthermore, MBL-associated proteases (MASPs) and MBL-associated proteins (MAps), which mediate downstream complement activation, have not been investigated in patients who experience the development of CVD [17,18]. Based on the previous observation that suggested the action of the lectin-complement pathway offered as molecules with a complex role in CVD, by Hertle et al [19] in the Cohort on Diabetes and Atherosclerosis Maastricht (CODAM) Study. The prospective cohort enrolled 574 individuals (age 60±7 years; 7-year follow-up), who were investigated to establish the longitudinal associations of plasma MBL, MASP-1, MASP-2, MASP-3, and MAp44 with biomarker scores that reflect low-grade inflammation and endothelial dysfunction, respectively, and with carotid intima-media thickness. Another priority of the authors was to determine their association with CVD manifestation in 73 patients adjusted analysis recorded values for low-grade inflammation that was lower in the middle tertile (TMiddle) of MBL, i.e., TMiddle was 0.19 SD (0.03 to 0.34) less than TLow and 0.15 SD (- 0.02 to 0.31) less than THigh. The value of carotid intima-media thickness reached 28 μm (-50 to -5) lower in patients with the highest MBL tertile (THigh) as compared to those with TMiddle and did not differ between TLow and TMiddle. Again, the evidence revealed no association between MBL and endothelial dysfunction or CVD. Furthermore, MASP-1 and MASP-2 did not show any association with any cardiovascular outcomes. In contrast, MASP-3 and MAp44 achieved, regardless of MBL levels, statistically significant values for endothelial dysfunction (for 1 SD higher MASP-3: β=0.10 SD [0.02 to 0.18]; for 1 SD higher MAp44 β=0.12 SD [0.04 to 0.20]), although data did not confirm the occurrence of low-grade inflammation, carotid intima-media thickness, or CVD. Evidence suggested three crucial findings. Firstly, although MBL may concur to low carotid intima-media thickness, however, the association of MBL with low-grade inflammation showed a non-linearity. Second, MASP-1 and MASP-2 estimates were not demonstrative for adverse cardiovascular outcomes. Third, the role of MASP-3 and MAp44 in endothelial dysfunction may be established and was potentially independent of lectin-pathway activation [18].
In a recent report, Hertle et al [20] defined the associations of the C1q pattern recognition factor and the C1-INH (C1-inhibitor) regulator with the occurrence of CVD events, carotid intima-media thickness, endothelial dysfunction, and low-grade inflammation. Baseline concentrations of C1q and C1-INH were determined in patients enrolled in the CODAM study (n=574; 61% men; age, 60±7 years). To evaluate the 7-year incidence of CVD in participants who did not experience CVD at baseline, the investigators performed a logistic regression analysis (n=342; 73 cases). The results reported a lower incidence of CVD that was documented in the middle tertile of C1q (Tlow versus Tmiddle: odds ratio, 2.38 [95% confidence interval, 1.14-4.95]; Thigh versus Tmiddle: odds ratio, 1.96 [95% confidence interval, 0.94 -4.07]). Furthermore, C1-INH values did not reach significance for the occurrence of CVD. The data analyzed, encompassing the 7-year follow-up period, suggested that C1q and C1-INH were not, or were inconsistently linked with carotid intima-media thickness or biomarker scores and pondered the role of endothelial dysfunction and low-grade inflammation. Once again, the results offered by the CODAM study leaned towards a nonlinear association between C1q and CVD incidence. This suggests the speculation that early activation of the classical complement pathway may have both protective and pathological effects on human CVD [20].
Interleukin (IL)-1β works as a cytokine with a pivotal key role in the development of CVD. An endogenous inhibitor identified as the IL-1 receptor antagonist (IL-1RA), has the function of counter-regulating IL-1β levels. This concern has been addressed in a systematic review and meta-analysis aimed to identify population-based studies on circulating IL-1RA and incident CVD [21,22,23].
Herder et [23] al assessed the association between IL-1RA and incident CVD and studied the association between IL-1RA and incident CVD along with the possible involvement of other inflammation-related biomarkers in the MONICA/KORA Augsburg case-cohort study (Multinational Monitoring of Trends and Determinants in Cardiovascular Disease/Cooperative Health Research in the Region of Augsburg). A total of 1855 CVD cases and 18 745 noncases with follow-up times between 5 and 16 years. 5 cohort studies on IL-1RA and incident CVD were included in the analysis. To reach this significant population of patients, investigators identified a relevant number of patients enrolled in the MONICA/KORA Augsburg case cohort study that completed the cohort for the meta-analysis. The pooled standardized hazard ratio (95% confidence interval) for the incidence of CVD was 1.11 (1.06-1.17) after adjustment for age, sex, lifestyle factors, metabolic, and anthropometric markers(P<0.0001). Again, no heterogeneity in effect sizes (I2=0%; P=0.88) was reported. Importantly, more comprehensive analyses in the MONICA/KORA trial revealed that the surfeit risk for CVD events was mitigated by ≥10% after additional adjustment for serum levels of high-sensitivity C-reactive protein, IL-6, myeloperoxidase, soluble E-selectin, or soluble intercellular adhesion molecule-1. The observed metanalytical results from 6 population-based cohorts proved that serum IL-1RA levels were substantially linked with the risk of CVD after adjustment for multiple confounders. This combination may at least partially explain a response to triggers that induce subclinical inflammation, oxidative stress, and endothelial activation [20].
The role of inflammation as a contributory factor in all phases of the atherothrombotic process and the detection of patients with elevated inflammatory biomarkers such as high sensitivity C-reactive protein (hsCRP) may indicate an increased vascular risk. The CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcomes Study) RCT investigated how direct inhibition of inflammation can reduce cardiovascular event rates [23]. Ridker et al [24] in a double-blind trial of canakinumab randomized 10,061 patients with previous myocardial infarction and a high-sensitivity C-reactive protein level of 2 mg or more per liter to receive canakinumab a therapeutic monoclonal antibody targeting interleukin-1β. The trial was designed to compare patients who received three doses of canakinumab (50 mg, 150 mg, and 300 mg, administered subcutaneously every 3 months) and a placebo cohort. Nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death constituted the primary efficacy endpoints. At 48 months, investigators recorded that the median decrease from baseline in the high-sensitivity C-reactive protein level was 26 percentage points higher in the recipients of the 50-mg dose of canakinumab, to 37 percentage points higher in patients who received the 150-mg, and 41 percentage points higher in those who were treated with 300-mg as compared to the placebo cohort of patients. Of note that the administration of Canakinumab did not support a reduction of lipid levels from baseline. Results at a median follow-up of 3.7 years, revealed that the incidence rate for the primary endpoint recorded 4.50 events per 100 person-years in the placebo group, 4.11 events per 100 person-years in the 50-mg group, 3.86 events per 100 person-years in the 150-mg group, and 3.90 events per 100 person-years in the 300-mg group. The hazard ratios of patients treated with canakinumab (compared to the recipients of placebo) was 0.93 (95% confidence interval [CI], 0.80 to 1.07; P=0.30) in the 50-mg cohort; 0.85 (95% CI, 0.74 to 0.98; P=0.021) in the 150-mg cohort; and 0.86 (95% CI, 0.75 to 0.99; P=0.031) in the 300-mg cohort. Recipients of the 150 mg dose, but not other dose-patient-groups, achieved the prespecified multiplicity-adjusted threshold for statistical significance for the primary endpoint and the secondary endpoint which additionally included hospitalization for unstable angina which led to urgent revascularization (hazard ratio vs. placebo, 0.83; 95% CI, 0.73 to 0.95; P=0.005). It should be that there was a higher incidence of fatal infections with the administration of Canakinumab when compared to the placebo. There was no significant difference in all-cause mortality (hazard ratio for all canakinumab doses vs. placebo, 0.94; 95% CI, 0.83 to 1.06; P=0.31). The evidence proved the efficacy of Canakinumab as anti-inflammatory therapy targeting the interleukin-1β innate immunity pathway. The drug administered at a dose of 150 mg every 3 months led to a significantly lower rate of recurrent cardiovascular events compared to the placebo, regardless of lowering of lipid levels.
Likewise, Choi et al [25] discussed the pathophysiology of endothelial dysfunction as drivers of early manifestation of atherosclerosis with particular regard to the role of the inflammatory response and vasa vasorum responsibility, which sustain a substantial role in the pathophysiology of plaque initiation, development, and complications. The authors investigated the hypothesis that segments with endothelial dysfunction revealed an accumulation of macrophages and changes at the level of vasa vasorum in patients with early CAD. Optical coherence tomography images were acquired from 40 patients who had mild coronary atherosclerosis and underwent coronary endothelial function assessment. The findings obtained included macrophages and microchannels that were examined in 76 coronary segments corresponding to those assessed for endothelial response to acetylcholine. A change of diameter was observed in the coronary artery that manifested a response to acetylcholine, however, this modification was more pronounced in segments disclosing macrophages (-17.7±14.7% versus -6.3±13.9%; P<0.01) and microchannels (-15.9±15.9% versus -6.4±13.5%; P<0.01) than those without. Increased trends of the prevalence of macrophages and microchannels combined with endothelial dysfunction as stratified by quartiles of coronary artery diameter change were reported (P<0.01 and P=0.02 for trend, respectively). A substantial discovery concerned those segments which revealed both the presence of macrophages and microchannels (n=12) had worse endothelial function than those with macrophages alone (n=15) and microchannels alone (n=15; -22.1±14.6% versus -10.9±15.6% and -10.9±15.6%; P=0.07 and P=0.06, respectively). Results indicated how epicardial endothelial dysfunction was linked with optical coherence tomography -identified macrophages and microchannels in mild coronary atherosclerosis suggesting the crucial role of inflammation and vasa vasorum proliferation in the early stage of coronary atherosclerosis. Table 1 shows the characteristics of the included studies relating to inflammation.
3.2. Pulmonary Hypertension Mediates Endothelial Dysfunction
The pulmonary endothelium performs multiple functions which include the control of blood fluidity, platelet aggregation, and vascular tone in the pulmonary circulation. It works as an important player in the regulation of immunology, inflammation, and angiogenesis and is an important metabolizing and endocrine organ. Pulmonary endothelial cells control vascular tone, and therefore local blood flow, synthesizing and releasing fundamental relaxing and contracting factors such as nitric oxide, arachidonic acid metabolites via the cyclooxygenase, lipoxygenase and cytochrome P450 pathways, various peptides (endothelin, urotensin, CNP, adrenomedullin, etc..), adenosine, purines, reactive oxygen species and so on. Furthermore, endothelial ectoenzymes are necessary steps in the generation of vasoactive hormones such as angiotensin II. Pulmonary endothelial dysfunction dependent on an imbalance in the synthesis and/or release of these various endothelial factors may explain the onset of cardiovascular pathologies such as pulmonary hypertension may develop and perpetuate [26,27]. Figure 3.
In the context of cardiovascular pathologies, pulmonary arterial hypertension (PAH) still constitutes a life-threatening disorder with serious concern for practitioners regarding clinical management. A better understanding of the underlying mechanisms is still needed. The literature today offers a satisfactory body of knowledge to better address this pathology. The role of cyclophilin in this specific context has been widely discussed [28,29,30,31,32].
The oxidative stress and inflammation work in a substantial manner to favour the development of pulmonary arterial hypertension (PAH). Oxidative stress determines a biochemical response leading to the secretion of Cyclophilin A (CypA) thus promoting inflammation and cardiovascular disease. In the pulmonary vascular environment, endothelial cell (EC) dysfunction is involved in driving the early pathogenesis of PAH [30].
Satoh et al [28] investigated the role of extracellular CypA in PAH and compared the effects of acetylated CypA (AcK-CypA), which was increased by oxidative stress, with those of CypA on EC dysfunction. High levels of expressed CypA in EC specifically were observed in transgenic mice to generate a PAH phenotype. The cohort was evaluated at 3 months accounting for several parameters which included increased right ventricular systolic pressure, α-smooth muscle actin expression in small arterioles, and CD45-positive cells in the lungs. Investigators assessed a mechanistic analysis with the use of cultured mouse pulmonary microvascular EC and human pulmonary microvascular EC revealing that extracellular CypA and AcK-CypA stimulated EC inflammatory signals. Results proved an enhancement in the production of VCAM1 (vascular cell adhesion molecule 1) and ICAM1 (intercellular adhesion molecule 1), degradation of IkB (nuclear factor-kappa B), and phosphorylation of p65. Increased EC apoptosis was induced by extracellular CypA and AcK-CypA and was determined by TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) staining, Apo-ONE assay, and caspase 3 cleavage. The vicious circle that was generated was due to oxidative stress stimulating the secretion of CypA and AcK-CypA, which further promoted the oxidative stress of EC. A difference was observed according to the effect of AcK-CypA, compared to CypA. The former induced greater increases in apoptosis, inflammation, and oxidative stress. Using MM284 acting as a specific inhibitor of extracellular CypA, the investigators observed attenuation of EC apoptosis induced by CypA and AcK-CypA. These findings revealed a substantial role of EC-derived CypA, superimposed to the action of AcK-CypA, that promoted the development of PAH. The presumptive mechanism involved an increased process of EC apoptosis, inflammation, and oxidative stress. The results revealed a substantial role of CE-derived CypA, more determinant than the action of AcK-CypA, which promoted the development of PAH. The putative mechanism involved an increase in the EC apoptosis process, inflammation, and oxidative stress. The elaborated evidence have been an open window for new applicable therapies [28].
Cyclophilin A (CyPA) is a molecule produced from vascular smooth muscle cells (VSMCs) by oxidative stress and promotes VSMC proliferation. The role of extracellular CyPA and its interaction with receptor Basigin (Bsg, encoded by Bsg) has been considered in the pathogenesis of PH. Satoh et al [29] observed that the pulmonary arteries of patients who experienced PH immunostaining disclosed strong expression of CyPA and Bsg. In addition, in a mice model, the pulmonary arteries of CyPA (±) and Bsg (±) CyPA mice exposed to hypoxia for 4 weeks recorded a substantially decreased right ventricular systolic pressure, pulmonary artery remodeling, and right ventricular hypertrophy compared with their littermate controls. Pulmonary VSMCs from Bsg (+/+) and Bsg (±) mice were harvested to study the role of vascular Bsg. Firstly, investigators noted that proliferation was markedly reduced in Bsg (±) compared with Bsg (+/+) VSMCs. Secondly, based on the mechanistic analysis they proved that Bsg (±) VSMCs recorded a contracted extracellular signal-regulated kinase 1/2 activation and less secretion of cytokines/chemokines and growth factors such as the platelet-derived growth factor-BB. Finally, in the clinical study, patients with PH revealed augmented plasma CyPA levels in accordance with the acuteness of pulmonary vascular resistance. Furthermore, the event-free curve disclosed that the occurrence of high plasma CyPA levels was strongly predictive of poor outcomes in patients with PH. The substantial role of extracellular CyPA and vascular Bsg in the pathogenesis of PH was carefully documented [29].
Recently Rosa et al [32] evaluated the importance of acetylation at sites K82 and K125 of two lysines that was potentially required for the release of CyPA from platelets. This process may support the potential for local delivery of CyPA thus exacerbating cardiovascular disease events. Investigators found the actual presence of CyPA in human and mouse platelets and thrombin stimulation resulted in CyPA release from platelets However, investigators revealed no reaction of acetylation neither in cell lysates nor in supernatants of both untreated and activated platelets. The absence of acetylation was corroborated after immunoprecipitation of CyPA from platelets. Evidence suggested that acetylation of CyPA was not a crucial protein alteration in platelets and that secretion of CyPA from platelets could occur also without acetylation [32].
Pulmonary arterial hypertension is a clinical pathological entity with manifestations not confined to the lungs [33-35]. Several studies have reported that many signaling pathways described in PAH also play a crucial role in other diseases in which vascular remodeling occurs, which is also a pathoanatomical characterization of CAD [36-38]. Evidence has documented that CAD occurs 4 times more frequently in PAH than in the world population, providing a plausible reason to assert a link between these 2 diseases. Both PAH and CAD patients develop sustained inflammation and deranged smooth muscle cell proliferation/apoptosis. Meloche et al in two landmark papers [39,40] worked on miR-223 function that reversed experimental pulmonary arterial hypertension and, in the PAH, coupled CAD mating phenotypes with a well-defined role in the increased expression of miR-223/DNA/Poly [ADP-ribose] polymerase 1/miR -204 in causing the damage. The mechanism is underpinned by activation of the miR-223/DNA/Poly [ADP-ribose] polymerase 1/miR -204 complex with subsequent overexpression of bromodomain protein 4 (BRD4) [39]. Firstly, the restoration of the expression of miR-223 in the lungs of rats with monocrotaline-induced PAH inverted the generated PAH thus providing beneficial effects on vascular remodeling, pulmonary resistance, right ventricle hypertrophy, and survival. In the occurrence of PAH miR-223 downregulation worked with a substantial role in numerous pathways implicated in the disease. The retrieval of its expression was effective in reversing PAH. Secondly, BRD4 expression in PAH was microRNA-204 dependent. The investigators through JQ1 or siBRD4 reduced the expression of BRD4 favoring a reduction of the expression of 3 major oncogenes, which are overexpressed in PAH: nuclear factor of activated T cells, B-cell lymphoma 2, and Survivin. Blockade of this oncogenic signature led to a decrease in PAH-PASMC proliferation and an increase in apoptosis linked to BRD4 expression. Therefore, the mechanisms leading to pharmacological inhibition of JQ1 or molecular (siRNA) of BRD4 caused both a reversal of this pathological phenotype and the restoration of the mitochondrial membrane potential to increase the respiratory reserve capacity of the cells [39,41]. Furthermore, the investigators demonstrated that in vivo BRD4 inhibition reversed PAH induced in the Sugen/hypoxia rat model [39]. The former study of Meloche et al implemented the evidence already reported by Archer et al [41] that worked on the expression and activity of mitochondrial superoxide dismutase-2 (SOD2) which is recognized as the major generator of H2O2 leading to a reduction of PAH. The latter may be caused by excessive proliferation and impaired apoptosis of pulmonary artery smooth muscle cells (PASMC) contributing to vascular obstruction in patients and fawn-hooded rats (FHR) with pulmonary arterial hypertension (PAH). The findings emerged were strongly suggestive of tissue-specific and epigenetic role of SOD2. Its deficiency commenced and supported a heritable form of PAH by impairing redox signaling and creating a proliferative, apoptosis-resistant PASMC. SOD augmentation reverted experimental PAH.
More recently Meloche et al. [40] hypothesized that since BRD4 also promotes triggers for calcification and remodeling processes, both important in CAD, BRD4 activation in PAH could play a substantial role in determining the development of CAD. Investigators reported that in patients with PAH distal pulmonary arteries, coronary arteries also recorded increased DNA damage, inflammation, and BRD4 overexpression. In vitro testing with the use of human coronary artery smooth muscle cells from PAH, CAD, and non-PAH-non-CAD patients, was achieved revealing that both PAH and CAD smooth muscle cells exhibited increased proliferation and suppressed apoptosis in a BRD4-dependent manner. In vivo tests demonstrated an improvement in PAH by the BRD4 inhibitor which was linked to a reduction in coronary remodeling with an increase in the expression level of interleukin-6. These results suggested that the increased expression of BRD4 in the coronary arteries of the patients with PAH could contribute to supporting a process of vascular remodeling and the development of comorbidities [40].
Several studies demonstrated that in rodents, hypoxia-induced mitogen factor (HIMF; also known as FIZZ1 or resistin-like molecule-β) was a potent effector for eliciting PH through triggering pulmonary vascular inflammation [42,43,44,45]. In particular Yamaji-Kegan et al [44], studying the initial phase of inflammation induced in a mouse model, reported that the systemic administration of HIMF could favor an increase in the apoptotic process in EC associated with higher levels in the expression of vascular inflammatory markers related to the interleukin (IL)-4 production Importantly HIMF, human homolog resistin (hRETN), and IL-4 activated pulmonary microvascular ECs (PMVECs) by increasing angiopoietin-2 expression and induced PMVEC apoptosis. The investigators further observed that the environment containing the hRETN-treated ECs revealed higher levels of endothelin-1 leading to marked increases in pulmonary vascular smooth muscle cell proliferation. Likewise, HIMF administration resulted in the development of PH characterized by evident pulmonary vascular remodeling with right heart failure in wild-type mice but not in IL-4 knockout mice.
In a subsequent study Johns et al [45] demonstrated that hypoxia-inducible factor-1 (HIF-1) was a crucial downstream signal mediator of HIMF during PH development. In the mouse model studied, the degree of HIMF-induced pulmonary vascular remodeling and PH development in wild-type (HIF-1α (+/+)) and heterozygous HIF-1α null (HIF-1α (+/-)) mice was compared. The results revealed that HIMF-induced PH was significantly reduced in HIF-1α (+/-) mice and was driven by activation of a dysregulated vascular endothelial growth factor-A-vascular endothelial growth factor receptor 2 pathway. The work of John et al [45] is of crucial importance because it offers a clear explanation of the interconnected mechanisms between HIMF, production of inflammatory mediators, cellular infiltration, and pulmonary vascular remodeling. In particular, HIMF and its human homolog, the molecule resistin-β, induced a marked increase in interleukin (IL)-6 levels in macrophages and lung-resident cells by mean of a mechanism related to the activity of HIF-1α and, at least to some extent, on nuclear factor κB. Therefore HIF-1α was found to play a key role as a substantial downstream transcription factor for HIMF-induced pulmonary vascular remodeling and PH development. These data suggested the critical role of HIMF in promoting increased expression of HIF-1, vascular endothelial growth factor-A, and interleukin-6, which are critical mediators of both hypoxic inflammation and PH pathophysiology sustained by remodeling vascular [45].
Newly Lin et al [46], in the wake of previous experience [45], studied the immunomodulatory effects of HIMF signaling in PH pathogenesis. In vivo experiments were achieved using a gene-modified mice that lacked HIMF (KO [knockout]) or overexpressed HIMF human homolog resistin (hResistin). In the first step the pro-PH role of HIMF was checked in HIMF-KO mice uncovered to chronic hypoxia or sugen/hypoxia. In the second step, based on the human in vitro and the in vivo murine model of EC, a mechanistic evaluation was performed. Activation of HIMF/hResistin that triggered the HMGB1 (high mobility group box 1) and RAGE (receptor for advanced glycation end products) pathway in pulmonary endothelial cells (ECs) of both hypoxic mouse lungs in vivo and in human pulmonary microvascular ECs in vitro was evaluated. The results recorded an induction of the autophagic response, defects in BMPR2 (bone morphogenetic protein receptor 2) expression and subsequent apoptosis-resistant proliferation in human pulmonary artery smooth muscle (vascular) cells closely related to HMGB1 levels. The described effects were corroborated by analyzes of ECs and smooth muscle cells isolated from the pulmonary arteries of patients with idiopathic PH. Importantly, both HIMF/HMGB1/RAGE-mediated autophagy and BMPR2 impairment were also recorded in pulmonary artery smooth muscle (vascular) cells of hypoxic mice. Both described effects could probably be supported by a damping phenomenon of FoxO1 (forkhead box O1) by HIMF. Experiments performed in transgenic mice overexpressing EC-specific hResistin corroborated the evidence that increased expression of EC-derived HMGB1 tempered hResistin-driven pulmonary vascular remodeling and PH. Given the results reported by Lin et al it appears evident that in HIMF-induced PH, HMGB1-RAGE signaling is crucial for arbitrating EC-smooth muscle cell crosstalk. Reported evidence from humanized mouse models further supports the clinical implications for the HIMF/HMGB1 signaling axis and suggests that hResistin and its downstream pathway may serve as targets for the evolution of new anti-PH therapies in humans [46].
The development of chronic thromboembolic pulmonary hypertension can be considered a distinct type of pulmonary hypertension which is due to the formation of organized thrombi of unknown origin in the pulmonary arteries causing a mechanical obstruction phenomenon. The pulmonary endothelium is composed of a monolayer of endothelial cells that constitutes the inner cellular lining of the blood vessels represented by arteries, veins and capillaries, and the lymphatic system, and therefore is in direct contact with the blood/lymph and the circulating cells. A disorder of the endothelial function supported by coagulation dysregulation can trigger the thrombotic process. Although the endothelium exerts decisive actions in the regulation of thrombosis, hemostasis, and fibrinolysis the pathophysiology of chronic thromboembolic pulmonary hypertension persists to be poorly understood [26,27,47].
In two reports, Yaoita et al [48,49] investigated the pathophysiological and molecular mechanisms of chronic thromboembolic pulmonary hypertension (CTEPH) and how it should be regarded as a fatal disease distinct from PAH. Although CTEPH is distinguished by considerable pulmonary artery obstruction related to a chronic thrombus formation, it is unclear whether CTEPH is associated with a prothrombotic condition. Patients without pulmonary hypertension (non-PH, n=15), patients with PAH (n=19), and patients with CTEPH (n=25) were investigated through measurement of conventional markers, GTP-bound levels of Rap1, RhoA, RalA, Rac1, and Ras in platelets. Moreover, the reactivity to ex vivo thrombin stimulation was assessed. Results showed that the ratios of the P-selectin positive platelets in the non-PH patients, patients with PAH, and patients with CTEPH were 1.40% (median and interquartile range, 0.83-1.82), 2.40% (1.80-3.39), and 2.63% (1.90-8.22), respectively (non-PH versus CTEPH, P<0.01). Secondly, the activated GPIIb/IIIa-positive platelets reached the rate of 6.01% (1.34-7.87), 11.39% (5.69-20.86), and 9.74% (7.83-24.01), respectively (non-PH versus CTEPH, P=0.01). GTP-bound RhoA attained the rate of 1.79% (0.94-2.83), 4.03% (2.01-5.14), and 2.01% (1.22-2.48), respectively (non-PH versus PAH, P=0.04), and GTP-bound RalA achieved the rate of 1.58% (1.08-2.11), 3.02% (2.03-3.54), and 2.64% (1.42-4.28), respectively (non-PH versus PAH, P=0.023; non-PH versus CTEPH, P=0.048). In contrast, Rap1, Rac1, or Ras disclosed no activation in any groups. The platelets of patients with CTEPH displayed hyperreactivity to ex vivo thrombin stimulation confronted to those of non-PH patients when assessed for the surface markers. Either D-dimer or fibrin degradation product level revealed no augmentation in patients with CTEPH. The success of this study provided evidence that the platelets of patients with CTEPH were extremely activated and exhibit hyperactivity to thrombin stimulation [48]. Another report by Yaoita et al [49] investigated the role of TAFI in the involvement of the pathogenesis of CTEPH by enrolling 68 consecutive patients undergoing right heart catheterization. Cohorts included patients with CTEPH (n=27), those with pulmonary arterial hypertension (n=22), and controls (non-pulmonary hypertension, n=19). Overall blood clot lysis assay revealed that the proportion of clot residual after 4 hours was markedly higher in CTEPH as compared to the cloth retrieved from patients with pulmonary arterial hypertension or controls (41.9 versus 26.5 and 24.6%, both P<0.01). Moreover, the determination of plasma levels of TAFI was significantly augmented in patients with CTEPH than in pulmonary arterial hypertension or control cohorts (19.4±4.2 versus 16.1±4.5 or 16.3±3.3 μg/mL, both P<0.05), which remained unchanged even after the achievement of percutaneous transluminal pulmonary angioplasty which led to hemodynamic improvement. Furthermore, the extension of the clot persisted after 4 hours and was notably improved with the administration of either CPI-2KR (an inhibitor of activated TAFI) or prostaglandin E1 (an inhibitor of activation of platelets). It should be highlighted that plasma levels of TAFI were substantially correlated with the persistent of clot formation after 4 hours. In addition, the range of clots that persist after 4 hours was improved with an activated TAFI inhibitor. Cohorts examined revealed that plasma levels of TAFI resulted elevated in patients with CTEPH and were related to clot lysis resistance [49]. Table 2 reports the characteristics of the included studies relating to pulmonary hypertension.
3.3. Diabetes Induce Injury in Endothelium
The effect of acute hyperglycemia on vascular function remains controversial. Several studies reported that the effect of acute hyperglycemia on endothelial and vascular smooth muscle functions across healthy and cardiometabolic diseased individuals is of relevance [1,50,51,52,53,54,55,56,57,58] Figure 4
A meta-analysis compared the standardized mean difference (SMD) in endothelial and vascular smooth muscle functions between acute hyperglycemia and normoglycemia [50]. Thirty-nine articles were evaluated which included a large cohort of healthy subjects (n=525) and patients with cardiometabolic disease (n=540). In 39 studies, decreased endothelial function was reported (n=1065; SMD, -1.25; 95% confidence interval, -1.52 to -0.98; P<0.01), while in 6 studies investigators recorded that vascular smooth muscle function was preserved (n=144; SMD, -0.07; 95% confidence interval, -0.30 to 0.16; P=0.55) during acute hyperglycemia versus normoglycemia. However, it is important to note significant heterogeneity was observed between studies of endothelial function (P < 0.01). A subgroup analysis of 30 studies revealed that although the endothelial function was impaired in the macrocirculation (n=884; SMD, -1.40; 95% confidence interval, -1.68 to -1.12; P<0.01) in 9 studies analysed, it did not show reductions in microcirculation (n = 181; SMD, -0.63; 95% confidence interval, -1.36 to 0.11; P=0, 09). The study of macrovascular endothelial function revealed an inverse association with age, blood pressure, and low-density lipoprotein cholesterol. In contrast, macrovascular endothelial function was positively associated with the post-occlusion interval of vascular assessment. The study suggested that macrovascular endothelial dysfunction occurred during acute hyperglycemia in both healthy and diseased subjects as opposed to microvascular dysfunction that was not revealed [50]. Recently, Lespagnol et al [51] evaluated the extent and the contributing risk factors of early endothelial dysfunction, and of the possible concomitant vascular smooth muscles (VSM) dysfunction, in subjects with type 1 diabetes using a meta-analysis. Of 58 total studies, 21 studies evaluated VSM dysfunction reporting either a standardized mean difference (SMD) (Cohen's d) impairment of endothelial function ( -0.61 (95% CI: -0.79, -0.44) that of SMD related to VSM dysfunction ( -0.32 (95% CI: -0.57, -0.07). Some points deserve consideration. First, different types of stimuli were analyzed, such as exercise, occlusion-reperfusion, pharmacological substances, and heat, which did not support any impairment of the vasodilatory capacity. Secondly, endothelial dysfunction appeared more striking within the macrovascular than in microvascular beds. The latter was particularly altered in cases of poor glycemic control [HbA1c > 67 mmol/mol (8.3%)]. The evidence from the study not only corroborated the early impairment of endothelial function, which persisted despite responses to physiological stimuli such as exercise but also highlighted VSM dysfunction in children and adults with type 1 diabetes. However, the endothelial dysfunction appeared to be more pronounced in large than small vessels, fueling the debate about their pertinent temporal aspect [51].
Loader et al [53] assessed the vascular function during acute hyperglycemia that was induced by commercial sugar-sweetened beverage (SSB) consumption. Investigators addressed the effect on underlying mechanisms of the nitric oxide pathway. The randomized, single-blind designed crossover study enrolled 12 healthy male participants who were scheduled to consume 600 mL (20 oz) of water or commercial SSB at 2 visits. Methods used to evaluate endothelial and vascular smooth muscle functions included laser contrast imaging coupled with iontophoresis for microcirculation while in macrocirculation evaluations were performed by brachial artery ultrasound with flow- and nitrate-mediated dilation. Impairment of microvascular and macrovascular endothelial function has been reported in subjects consuming SSB which was not manifested in those consuming water. In detail, a decrease in the vascular response to acetylcholine iontophoresis (208.3±24.3 versus 144.2±15.7%, P<0.01) and a reduced flow-mediated dilatation (0.019±0.002 versus 0.014 ±0.002%/s, P<0.01) were recorded. One substantial finding was that systemic vascular smooth muscle remained preserved. Similar changes in endothelial function were also observed in the presence of induced acute hyperglycemia in an in-vivo rat model. However, endothelial function was completely restored after treatment with antioxidants of the type N-acetylcysteine and apocynin. Furthermore, it is important to underline, as revealed by ex vivo experiments, that although the production of reactive oxygen species was increased during acute hyperglycemia, nevertheless a reduction in the bioavailability of nitric oxide in the endothelium was recorded, even if no change in the activation state of endothelial nitric oxide synthase was detectable. Evidence from the RCT suggested that SSB-mediated endothelial dysfunction was in part due to increased oxidative stress that was associated with a reduction in nitric oxide bioavailability [53].
In patients with diabetes mellitus, endothelial dysfunction is a process associated with insulin resistance, inflammatory activation, and increased cardiovascular risk. Substantial evidence has emerged demonstrating the role of proinflammatory wingless-type (Wnt) family member 5a signaling via c-jun N-terminal kinase (JNK) as a crucial regulator of metabolic dysfunction, attributing to its potential relevance for vascular function [54,55]. Walther et al [56] investigated differences within endothelial-dependent and endothelial-independent vasoreactivity both in macro- and microcirculation beds among patients with metabolic syndrome (MetS) with and without type 2 diabetes mellitus (T2D) compared with healthy counterparts. In addition, investigators determined the existence of relationships among the function of macro- and microvascular systems and abdominal adiposity, as well as inflammatory markers by studying 3 groups. The RCT randomized enrolled 53 MetS patients without T2D and 25 with T2D who underwent cross-sectional analyses. In addition, the study included 40-year-old, sex-matched healthy controls in which microvascular function (cutaneous blood flow measured by laser Doppler flowmetry in response to acetylcholine and sodium nitroprusside iontophoresis), and macrovascular reactivity (flow-mediated dilatation and nitrate-mediated dilatation) together with anthropometric, plasma glucose, insulin, and inflammatory markers measurements. The MetS cohort demonstrated depressed endothelial function of both micro and macrocirculatory beds compared to the control group. Patients with MetS combined with TED revealed exacerbated vascular smooth muscle dysfunction in micro- and macrocirculation compared with those with MetS but without T2D. Furthermore, upon examination of micro and macrocirculation indices, an inverse correlation between abdominal fat accumulation and inflammatory markers was reported. These findings prove that MetS was associated with endothelial-dependent and endothelial-independent dysfunction, affecting both the macro- and the microvascular systems. Subjects who experienced diabetes mellitus demonstrated the most severe smooth muscle dysfunction. Importantly, central abdominal fat accumulation and systemic inflammation were implicated in the pathogenesis of vascular dysfunctions in MetS when they occurred in RCT participants [56].
Breton Romero et al [57] looked for evidence demonstrating enhanced activation of Wnt5a-JNK signaling that could contribute to impaired endothelial function in patients with diabetes mellitus. In 85 age- and sex-matched subjects, which included patients with type 2 diabetes mellitus (n = 42), nondiabetic controls (n = 43), and Wnt5a-treated human aortic endothelial cells, flow-mediated dilatation of the brachial artery was measured. In addition, the function of freshly isolated endothelial cells was characterized by protein expression. Endothelial cells from patients with diabetes mellitus revealed 1.3-fold higher Wnt5a levels (P=0.01) along with 1.4-fold higher JNK activation (P<0.01), without reporting a substantial difference in total JNK levels. The investigators also observed increased JNK activation that was associated with lower flow-mediated dilatation, which was broadly consistent with endothelial dysfunction (r = 0.53, P = 0.02). In patients with diabetes mellitus, insulin administration resulted in the inhibition of Wnt5a and JNK signaling while the addition of A23187 mediated eNOS activation by enhancing nitric oxide production in endothelial cells. Instead, in the endothelial cells of the non-diabetic control group, treatment with rWnt5a forbade the activation of eNOS to reproduce the diabetic endothelial phenotype. In human aortic endothelial cells, Wnt5a-induced impairment of eNOS activation and nitric oxide generation was attenuated by inhibition of Wnt5a and JNK. Findings revealed that noncanonical Wnt5a signaling and JNK activity concurred with the vascular reluctance to insulin action and to induce endothelial dysfunction thus offering a novel therapeutic opportunity to protect the vasculature in patients with diabetes mellitus [57].
Cho et al [58] examined whether SFRP5 could restore WNT5A-induced endothelial dysfunction in vitro and ex vivo and whether the serum concentration of SFRP5 may be associated with atherosclerosis in humans. A Sprague-Dawley rat model was used to determine endothelium-dependent vasorelaxation in the isolated thoracic aorta. In addition, intracellular nitric oxide (NO) in human endothelial cells was measured. Moreover, the protein plenty of total and phosphorylated JNK (c-Jun N-terminal kinase), AKT (protein kinase B), and endothelial NO synthase were evaluated in human endothelial cells. At the same time, 282 human subjects with type 2 diabetes mellitus were studied by measuring circulating levels of SFRP5 and WNT5A and brachial-ankle pulse wave velocity. In the rat thoracic aorta, the results showed that the SFRP5 dose-dependently restored the impaired vasodilation that was sustained by Wnt5 via an endothelial NO synthase-dependent mechanism. Furthermore, in human endothelial cells, the authors demonstrated that SFRP5 treatment reversed the WNT5A-induced reduction in NO production via endothelial NO synthase. Evidence proved that WNT5A-induced changes in the phosphorylation of JNK, AKT, and endothelial NO synthase were ameliorated by SFRP5 administration. Again, in the human population with type 2 diabetes mellitus, serum SFRP5 concentration was positively correlated with brachial-ankle pulse wave velocity (r=0.146; P=0.024). Multivariate linear regression analysis also demonstrated that serum SFRP5 concentration was independently associated with brachial-ankle pulse wave velocity after adjustment for potential confounders [B (SE)=7.40 (3.35); P=0.028]. These findings suggested the potential compensatory role exerted by SFRP5 against atherosclerosis under a state of metabolic dysfunction [58].
We learned that in humans’ high glucose level concentrations acutely lead to endothelial cell oxidative stress and his process is suggested to trigger diabetes-related macro- and microvascular disorders. Although numerous clinical studies have established that acute hyperglycemia, that induced by mixed meals or oral glucose, decreases arterial vascular function in healthy humans; however, diet has an influence on autonomic production, blood pressure, and insulin and incretin secretion, which can independently alter vascular function and obscure the effect of acute hyperglycemia per se. Given this evidence, surprisingly, the literature lacks a body of studies evaluating the effect of acute hyperglycemia on both macro and microvascular function during the control of plasma insulin concentrations. Based on this evidence, Horton et al [52] designed an RCT to compare macrovascular and microvascular functional responses to euglycemia and hyperglycemia. In detail, in healthy young adults’ functional responses such as brachial artery flow-mediated dilatation, carotid-femoral pulse wave velocity, and post-ischemic brachial artery flow velocity cardiac and skeletal muscle perfusion by ultrasound with medium contrast agents after octreotide infusion in both protocols to prevent endogenous insulin release were investigated. Of note that acute hyperglycemia determined by intravenous glucose infusion ameliorated flow-mediated dilation of the brachial artery (P = 0.004), increased skeletal muscle microvascular blood volume and blood flow (P = 0.001), and heart muscle microvascular blood volume expansion (P = 0.014). In addition, the investigators reported no changes in measures of vascular function during euglycemia sustained by octreotide administration. The evidence demonstrated in the randomized study suggested a substantial difference between acute hyperglycemia induced by the meal and that induced by intravenous glucose infusion over 4 hours. The latter improved flow-mediated dilatation of the brachial artery, stimulated the microvascular function of cardiac and skeletal muscle, and did not compromise aortic stiffness. This study opposes different results compared to the previous ones reported on acute vascular dysfunction of the great arteries during oral ingestion of glucose or mixed meals which may be favored by marked differences in the analyzed populations and by the presence of meal-induced humoral or neural factors in addition to hyperglycemia itself [52]. Table 3 highlights the included studies relating to diabetes.
3.4. Endothelial Dysfunction and Fabry Disease
Evidence reported a specific role of Globotriaosylceramide (Gb3) in inducing KCa3.1 downregulation in patients who experienced Fabry disease (FD) [59,60]. Choi et al [61] studied KCa3.1 endocytosis and degradation caused by Gb3. The two cohorts used had KCa3 downregulation, mainly plasma membrane-localized KCa3.1, in both Gb3-treated mouse aortic endothelial cells (MAEC) and human umbilical vein endothelial cells. Gb3-induced KCa3.1 downregulation was avoided by lysosomal inhibitors but not by a proteasomal inhibitor. Agents causing endoplasmic reticulum stress were not responsible for causing the downregulation of KCa3.1. Gb3 had the effect of upregulating the protein levels of early endosome antigen 1 and lysosome-associated membrane protein 2 in MAECs. In an animal model of FD reproduced in aged α-galactosidase A (Gla) knockout mice, with downregulation of KCa3.1, early endosome antigen 1 and lysosome-associated membrane protein 2 expression was reported. In contrast, no fundamental difference was recorded in the exhibition of early endosome antigen 1 and lysosome-associated protein 2 between Gla-knockout juveniles and wild-type MAEC. The significant data observed demonstrated that in aged Gla-knockout MAECs, clathrin was translocated near the cell border and clathrin knockdown recovered KCa3.1 expression. This effect was enhanced by Rab5, an effector of early endosome antigen 1, which was upregulated, and the knockdown of Rab5 restored KCa3.1 expression, current, and endothelium-dependent relaxation. Evidence suggested an acceleration of the endocytosis and lysosomal degradation phenomena of endothelial KCa3.1 that induced by Gb3 and via a clathrin-dependent process, leading to endothelial dysfunction in FD [61].
4. Conclusions
Endothelial dysfunction has a key role in several different pathophysiological processes which can be targeted for therapeutic options. Ongoing research should be targeted at making the transition to clinical practice. There is significant overlap between the use of cardiovascular medications and its effects on the endothelium. Further research is also needed on understanding the amelioration of endothelial dysfunction with the use of cardiovascular medications which may explain some of the prognostic benefits that reduce both morbidity and mortality.
Author Contributions
Conceptualization, F.N.; methodology, F.N. and S.S.A.S.; software, S.S.A.S.; validation, F.N. and S.S.A.S.; formal analysis, F.N. and S.S.A.S.; investigation, F.N.; data curation, F.N. and S.S.A.S.; writing—original draft preparation, F.N.; writing—review and editing, F.N. and S.S.A.S.; visualization, F.N. and S.S.A.S.; supervision, F.N. and S.S.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations: in table and figures.
References
- Nappi F, Fiore A, Masiglat J, Cavuoti T, Romandini M, Nappi P, Avtaar Singh SS, Couetil JP. Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review. Biomedicines. 2022 Nov 10;10(11):2884. [CrossRef]
- Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol (Oxf). 2017; 219:22–96. [CrossRef]
- Shimokawa H. 2014 Williams Harvey lecture: importance of coronary vasomotion abnormalities-from bench to bedside. Eur Heart J. 2014; 35:3180–3193. [CrossRef]
- Godo S, Takahashi J, Yasuda S, Shimokawa H. Endothelium in Coronary Macrovascular and Microvascular Diseases. J Cardiovasc Pharmacol. 2021 Dec 3;78(Suppl 6): S19-S29. [CrossRef]
- Godo S, Shimokawa H. Endothelial Functions. Arterioscler Thromb Vasc Biol. 2017 Sep;37(9): e108-e114. [CrossRef]
- Matsuzawa Y, Kwon TG, Lennon RJ, Lerman LO, Lerman A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc. 2015;4: e002270. [CrossRef]
- Kitta Y, Obata JE, Nakamura T, Hirano M, Kodama Y, Fujioka D, Saito Y, Kawabata K, Sano K, Kobayashi T, Yano T, Nakamura K, Kugiyama K. Persistent impairment of endothelial vasomotor function has a negative impact on outcome in patients with coronary artery disease. J Am Coll Cardiol. 2009; 53:323–330. [CrossRef]
- Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Kuvin JT, Lerman A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J Am Coll Cardiol. 2004; 44:2137–2141. [CrossRef]
- Chaulin AM, Sergeev AK. The Role of Fine Particles (PM 2.5) in the Genesis of Atherosclerosis and Myocardial Damage: Emphasis on Clinical and Epidemiological Data, and Pathophysiological Mechanisms. Cardiol Res. 2022 Oct;13(5):268-282. [CrossRef]
- Tremblay JC, Pyke KE. Flow-mediated dilation stimulated by sustained increases in shear stress: a useful tool for assessing endothelial function in humans? Am J Physiol Heart Circ Physiol. 2018 Mar 1;314(3):H508-H520. [CrossRef]
- Matsuzawa Y, Kwon TG, Lennon RJ, Lerman LO, Lerman A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc. 2015;4: e002270. [CrossRef]
- Donato AJ, Machin DR, Lesniewski LA. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ Res. 2018 Sep 14;123(7):825-848. [CrossRef]
- Paneni F, Diaz Cañestro C, Libby P, Lüscher TF, Camici GG. The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol. 2017; 69:1952–1967. [CrossRef]
- Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. 2013 May 23;368(21):2004-13. [CrossRef]
- Honda A, Tahara N, Nitta Y, Tahara A, Igata S, Bekki M, Nakamura T, Sugiyama Y, Kaida H, Kurata S, Fujimoto K, Abe T, Enomoto M, Adachi H, Narula J, Yamagishi S, Fukumoto Y. Vascular inflammation evaluated by [18F]-fluorodeoxyglucose-positron emission tomography/ computed tomography is associated with endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2016; 36:1980–1988. [CrossRef]
- Kang MK, Kim CJ, Choo EH, Han EJ, Hwang BH, Kim JJ, Kim SH, O JH, Chang K. Anti-inflammatory effect of statin is continuously working throughout use: a prospective three time point 18F-FDG PET/CT imaging study. Int J Cardiovasc Imaging. 2019 Sep;35(9):1745-1753. [CrossRef]
- Frauenknecht V, Thiel S, Storm L, Meier N, Arnold M, Schmid JP, Saner H, Schroeder V. Plasma levels of mannan-binding lectin (MBL)-associated serine proteases (MASPs) and MBL-associated protein in cardio- and cerebrovascular diseases. Clin Exp Immunol. 2013 Jul;173(1):112-20. [CrossRef]
- Krogh SS, Holt CB, Steffensen R, Funck KL, Høyem P, Laugesen E, Poulsen PL, Thiel S, Hansen TK. Plasma levels of MASP-1, MASP-3 and MAp44 in patients with type 2 diabetes: influence of glycaemic control, body composition and polymorphisms in the MASP1 gene. Clin Exp Immunol. 2017 Jul;189(1):103-112. [CrossRef]
- Hertle E, Arts IC, van der Kallen CJ, Feskens EJ, Schalkwijk CG, Hoffmann-Petersen IT, Thiel S, Stehouwer CD, van Greevenbroek MM. Distinct longitudinal associations of MBL, MASP-1, MASP-2, MASP- 3, and MAp44 with endothelial dysfunction and intima-media thickness: the cohort on diabetes and atherosclerosis maastricht (CODAM) study. Arterioscler Thromb Vasc Biol. 2016 ; 36 :1278–1285. [CrossRef]
- Hertle E, Arts ICW, van der Kallen CJH, Feskens EJM, Schalkwijk CG, Stehouwer CDA, van Greevenbroek MMJ. Classical Pathway of Complement Activation: Longitudinal Associations of C1q and C1-INH With Cardiovascular Outcomes: The CODAM Study (Cohort on Diabetes and Atherosclerosis Maastricht)-Brief Report. Arterioscler Thromb Vasc Biol. 2018 May;38(5):1242-1244. [CrossRef]
- Emeny RT, Zierer A, Lacruz ME, Baumert J, Herder C, Gornitzka G, Koenig W, Thorand B, Ladwig KH; KORA Investigators. Job strain-associated inflammatory burden and long-term risk of coronary events: findings from the MONICA/KORA Augsburg case-cohort study. Psychosom Med. 2013 Apr;75(3):317-25. [CrossRef]
- Herder C, Kannenberg JM, Carstensen-Kirberg M, Huth C, Meisinger C, Koenig W, Peters A, Rathmann W, Roden M, Thorand B. Serum levels of interleukin-22, cardiometabolic risk factors and incident type 2 diabetes: KORA F4/FF4 study. Cardiovasc Diabetol. 2017 Jan 31;16(1):17. [CrossRef]
- Herder C, de Las Heras Gala T, Carstensen-Kirberg M, et al. Circulating levels of interleukin 1-receptor antagonist and risk of cardiovascular disease: meta-analysis of six population-based cohorts. Arterioscler Thromb Vasc Biol. 2017; 37:1222–1227. [CrossRef]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ; CANTOS Trial Group Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017 Sep 21;377(12):1119-1131. [CrossRef]
- Choi BJ, Matsuo Y, Aoki T, Kwon TG, Prasad A, Gulati R, Lennon RJ, Lerman LO, Lerman A. Coronary endothelial dysfunction is associated with inflammation and vasa vasorum proliferation in patients with early atherosclerosis. Arterioscler Thromb Vasc Biol. 2014; 34:2473–2477. [CrossRef]
- Zaric B, Obradovic M, Trpkovic A, Banach M, Mikhailidis DP, Isenovic ER. Endothelial Dysfunction in Dyslipidaemia: Molecular Mechanisms and Clinical Implications. Curr Med Chem. 2020;27(7):1021-1040. [CrossRef]
- Feletou M. The Endothelium: Part 1: Multiple Functions of the Endothelial Cells-Focus on Endothelium-Derived Vasoactive Mediators. San Rafael, CA: Morgan & Claypool Life Sciences Publishers; 2011.
- Satoh K, Matoba T, Suzuki J, O'Dell MR, Nigro P, Cui Z, Mohan A, Pan S, Li L, Jin ZG, Yan C, Abe J, Berk. Cyclophilin A mediates vascular remodeling by promoting inflammation and vascular smooth muscle cell proliferation. Circulation. 2008 Jun 17;117(24):3088-98. [CrossRef]
- Satoh K, Satoh T, Kikuchi N, Omura J, Kurosawa R, Suzuki K, Sugimura K, Aoki T, Nochioka K, Tatebe S, Miyamichi-Yamamoto S, Miura M, Shimizu T, et al. Basigin mediates pulmonary hypertension by promoting inflammation and vascular smooth muscle cell proliferation. Circ Res. 2014 Sep 26;115(8):738-50. [CrossRef]
- Xue C, Sowden M, Berk BC. Extracellular Cyclophilin A, Especially Acetylated, Causes Pulmonary Hypertension by Stimulating Endothelial Apoptosis, Redox Stress, and Inflammation. Arterioscler Thromb Vasc Biol. 2017 Jun;37(6):1138-1146. [CrossRef]
- Xue C, Senchanthisai S, Sowden M, Pang J, White RJ, Berk BC. Endothelial-to-Mesenchymal Transition and Inflammation Play Key Roles in Cyclophilin A-Induced Pulmonary Arterial Hypertension. Hypertension. 2020 Oct;76(4):1113-1123. [CrossRef]
- Rosa A, Butt E, Hopper CP, Loroch S, Bender M, Schulze H, Sickmann A, Vorlova S, Seizer P, Heinzmann D, Zernecke A Cyclophilin A Is Not Acetylated at Lysine-82 and Lysine-125 in Resting and Stimulated Platelets. Int J Mol Sci. 2022 Jan 27;23(3):1469. [CrossRef]
- Liu SF, Nambiar Veetil N, Li Q, Kucherenko MM, Knosalla C, Kuebler WM. Pulmonary hypertension: Linking inflammation and pulmonary arterial stiffening. Front Immunol. 2022 Oct 5; 13:959209. [CrossRef]
- Rogula S, Pomirski B, Czyżak N, Eyileten C, Postuła M, Szarpak Ł, Filipiak KJ, Kurzyna M, Jaguszewski M, Mazurek T, Grabowski M, Gąsecka A. Biomarker-based approach to determine etiology and severity of pulmonary hypertension: Focus on microRNA. Front Cardiovasc Med. 2022 Oct 6 ;9 :980718. [CrossRef]
- Kherbeck N, Tamby MC, Bussone G, Dib H, Perros F, Humbert M, Mouthon L. The role of inflammation and autoimmunity in the pathophysiology of pulmonary arterial hypertension. Clin Rev Allergy Immunol. 2013 Feb ;44(1) :31-8. [CrossRef]
- Dutta P, Gomez D, Gladwin MT. Do BRD (4)S of a Feather Flock Together? How an Inflammation-Driven Epigenetic Regulator May Link Pulmonary Hypertension and Coronary Artery Disease. Arterioscler Thromb Vasc Biol. 2017 Aug;37(8):1428-1430. [CrossRef]
- Borck PC, Guo LW, Plutzky J. BET Epigenetic Reader Proteins in Cardiovascular Transcriptional Programs. Circ Res. 2020 Apr 24;126(9):1190-1208. [CrossRef]
- Li MX, Jiang DQ, Wang Y, Chen QZ, Ma YJ, Yu SS, Wang Y. Signal Mechanisms of Vascular Remodeling in the Development of Pulmonary Arterial Hypertension. J Cardiovasc Pharmacol. 2016 Feb;67(2):182-90. [CrossRef]
- Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, Courboulin A, Breuils-Bonnet S, Tremblay E, Couture C, Michelakis ED, Provencher S, Bonnet S. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation. 2014 Feb 18;129(7):786-97. [CrossRef]
- Meloche J, Le Guen M, Potus F, Vinck J, Ranchoux B, Johnson I, Antigny F, Tremblay E, Breuils-Bonnet S, Perros F, Provencher S, Bonnet S. miR-223 reverses experimental pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2015 Sep 15 ;309(6):C363-72. [CrossRef]
- Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thébaud B, Husain AN, Cipriani N, Rehman. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010 Jun 22;121(24):2661-71. [CrossRef]
- Yamaji-Kegan K, Su Q, Angelini DJ, Myers AC, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. J Immunol. 2010 Nov 1;185(9):5539-48. [CrossRef]
- Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Skinner JT, Poloczek A, El-Haddad H, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMα) in chronic hypoxia- and antigen-mediated pulmonary vascular remodeling. Respir Res. 2013 Jan 4;14(1):1. [CrossRef]
- Yamaji-Kegan K, Takimoto E, Zhang A, Weiner NC, Meuchel LW, Berger AE, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (FIZZ1/RELMα) induces endothelial cell apoptosis and subsequent interleukin-4-dependent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014 Jun 15;306(12): L1090-103. [CrossRef]
- Johns RA, Takimoto E, Meuchel LW, Elsaigh E, Zhang A, Heller NM, Semenza GL, Yamaji-Kegan K. Hypoxia-inducible factor 1α is a critical downstream mediator for hypoxia-induced mitogenic factor (FIZZ1/RELMα)-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2016; 36:134–144. [CrossRef]
- Lin Q, Fan C, Gomez-Arroyo J, Van Raemdonck K, Meuchel LW, Skinner JT, Everett AD, Fang X, Macdonald AA, Yamaji-Kegan K, Johns RA. HIMF (Hypoxia-Induced Mitogenic Factor) Signaling Mediates the HMGB1 (High Mobility Group Box 1)-Dependent Endothelial and Smooth Muscle Cell Crosstalk in Pulmonary Hypertension. Arterioscler Thromb Vasc Biol. 2019 Dec;39(12):2505-2519. [CrossRef]
- Félétou M, Köhler R, Vanhoutte PM. Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep. 2010 Aug;12(4):267-75. [CrossRef]
- Yaoita N, Shirakawa R, Fukumoto Y, Sugimura K, Miyata S, MiuraY, Nochioka K, Miura M, Tatebe S, Aoki T, Yamamoto S, Satoh K, Kimura T, Shimokawa H, Horiuchi H. Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2014; 34:2486–2494. [CrossRef]
- Yaoita N, Satoh K, Satoh T, Sugimura K, Tatebe S, Yamamoto S, Aoki T, Miura M, Miyata S, Kawamura T, Horiuchi H, Fukumoto Y, Shimokawa H. Thrombin-activatable fibrinolysis inhibitor in chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2016; 36:1293– 1301. [CrossRef]
- Loader J, Montero D, Lorenzen C, Watts R, Méziat C, Reboul C, Stewart S, Walther G. Acute hyperglycemia impairs vascular function in healthy and cardiometabolic diseased subjects: systematic review and meta-analysis. Arterioscler Thromb Vasc Biol. 2015; 35:2060–2072. [CrossRef]
- Lespagnol E, Dauchet L, Pawlak-Chaouch M, Balestra C, Berthoin S, Feelisch M, Roustit M, Boissière J, Fontaine P, Heyman E. Early Endothelial Dysfunction in Type 1 Diabetes Is Accompanied by an Impairment of Vascular Smooth Muscle Function: A Meta-Analysis. Front Endocrinol (Lausanne). 2020 Apr 17; 11:203. [CrossRef]
- Horton WB, Jahn LA, Hartline LM, Aylor KW, Patrie JT, Barrett EJ. Acute hyperglycaemia enhances both vascular endothelial function and cardiac and skeletal muscle microvascular function in healthy humans. J Physiol. 2022 Feb;600(4):949-962. [CrossRef]
- Loader J, Meziat C, Watts R, Lorenzen C, Sigaudo-Roussel D, Stewart S, Reboul C, Meyer G, Walther G. Effects of sugar-sweetened beverage consumption on microvascular and macrovascular function in a healthy population. Arterioscler Thromb Vasc Biol. 2017; 37:1250–1260. [CrossRef]
- Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N, Farb MG, Rosenzweig J, Ruderman N, Vita JA, Hamburg NM. Protein kinase C-β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013 Jan 1;127(1):86-95. [CrossRef]
- Farb MG, Karki S, Park SY, Saggese SM, Carmine B, Hess DT, Apovian C, Fetterman JL, Bretón-Romero R, Hamburg NM, Fuster JJ, Zuriaga MA, Walsh K, Gokce N. WNT5A-JNK regulation of vascular insulin resistance in human obesity. Vasc Med. 2016 Dec;21(6):489-496. [CrossRef]
- Walther G, Obert P, Dutheil F, Chapier R, Lesourd B, Naughton G, Courteix D, Vinet A. Metabolic syndrome individuals with and without type 2 diabetes mellitus present generalized vascular dysfunction: crosssectional study. Arterioscler Thromb Vasc Biol. 2015; 35:1022–1029. [CrossRef]
- Bretón-Romero R, Feng B, Holbrook M, Farb MG, Fetterman JL, Linder EA, Berk BD, Masaki N, Weisbrod RM, Inagaki E, Gokce N, Fuster JJ, Walsh K, Hamburg NM. Endothelial dysfunction in human diabetes is mediated by Wnt5a-JNK signaling. Arterioscler Thromb Vasc Biol. 2016; 36:561–569. [CrossRef]
- Cho YK, Kang YM, Lee SE, Lee Y, Seol SM, Lee WJ, Park JY, Jung CH. Effect of SFRP5 (Secreted Frizzled-Related Protein 5) on the WNT5A (Wingless-Type Family Member 5A)-Induced Endothelial Dysfunction and Its Relevance With Arterial Stiffness in Human Subjects. Arterioscler Thromb Vasc Biol. 2018 Jun;38(6):1358-1367. [CrossRef]
- Park S, Kim JA, Joo KY, Choi S, Choi EN, Shin JA, Han KH, Jung SC, Suh SH. Globotriaosylceramide leads to K(Ca)3.1 channel dysfunction: a new insight into endothelial dysfunction in Fabry disease. Cardiovasc Res. 2011 Feb 1;89(2):290-9. [CrossRef]
- Satoh K. Globotriaosylceramide induces endothelial dysfunction in fabry disease Arterioscler Thromb Vasc Biol. 2014 Jan;34(1):2-4. [CrossRef]
- Choi S, Kim JA, Na HY, Cho SE, Park S, Jung SC, Suh SH. Globotriaosylceramide induces lysosomal degradation of endothelial. KCa3.1 in fabry disease. Arterioscler Thromb Vasc Biol. 2014; 34:81–89. [CrossRef]
Figure 1.
The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372: n71. doi: 10.1136/bmj. n71. For more information, visit: http://www.prisma-statement.org/.
Figure 1.
The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372: n71. doi: 10.1136/bmj. n71. For more information, visit: http://www.prisma-statement.org/.
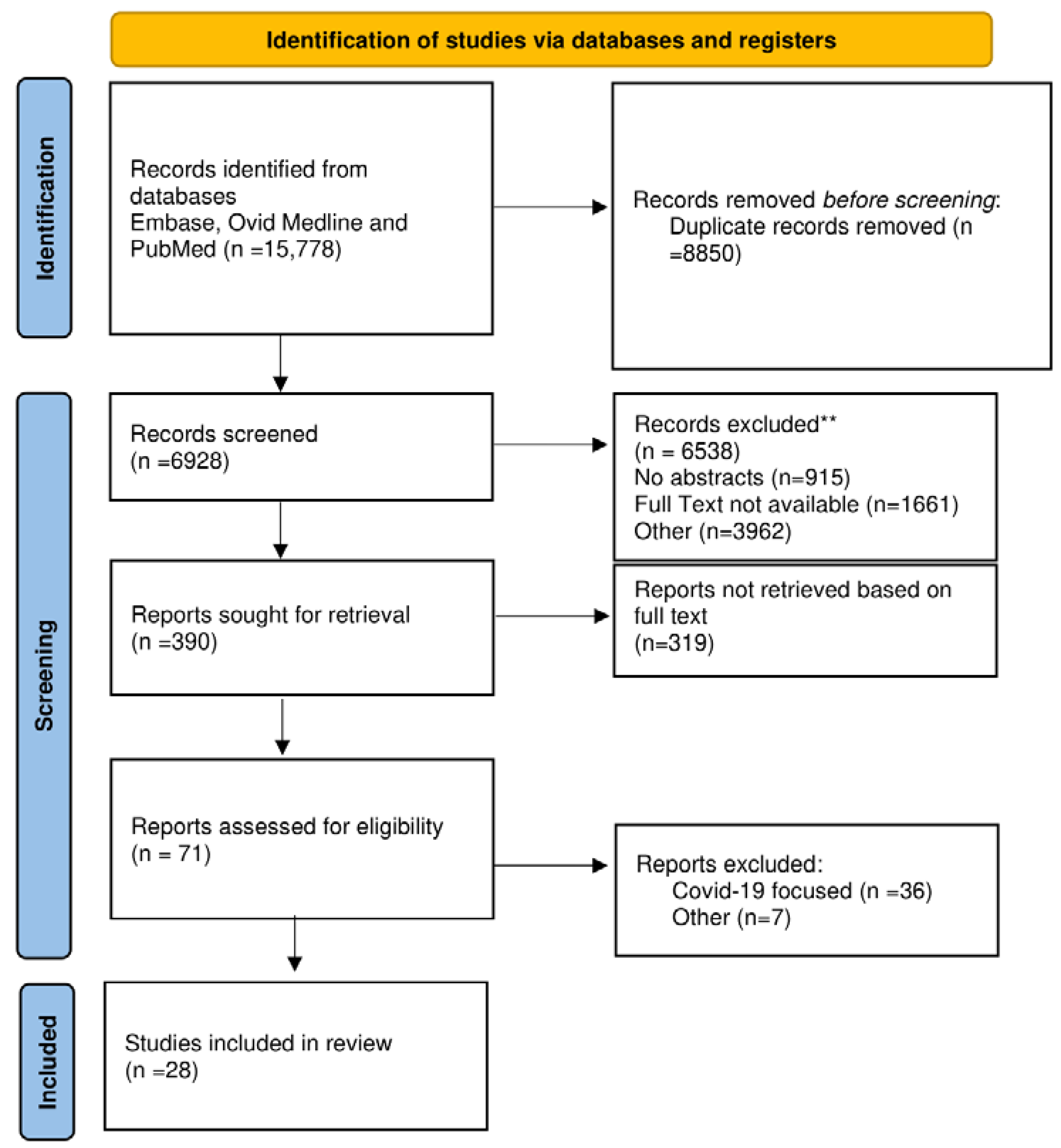
Figure 2.
Inflammatory pathways involved in atherosclerosis is depicted. A complex balance between pro-inflammatory and anti-inflammatory pathways emerged from preclinical and clinical trials. Inflammatory cells (macrophages and T-cells) and liver (CRP) regulate the balance that promotes endothelial dysfunction and the progression of atherosclerotic plaque through the production of molecules with a proinflammatory (red box) or anti-inflammatory (blue box) effect. Plaque rupture can be a consequence of the exacerbated inflammatory process. Abbreviations; CRP, C-reactive protein; MMPs, matrix metalloproteinases; IFN-γ, interferon-gamma; IL-1α, interleukin-1-alpha; IL-1β, interleukin-1-beta; IL-2, interleukin-2; IL-6, interleukin-6; IL-10, interleukin-10; IL-18, interleukin-18; Lp-PLA2, lipoprotein-associated phospholipase A2; TGF-β, transforming growth factor beta; Th-1, T-helper-1 lymphocyte; TNF-α, tumor necrosis factor alpha; T-reg, regulatory T lymphocyte.
Figure 2.
Inflammatory pathways involved in atherosclerosis is depicted. A complex balance between pro-inflammatory and anti-inflammatory pathways emerged from preclinical and clinical trials. Inflammatory cells (macrophages and T-cells) and liver (CRP) regulate the balance that promotes endothelial dysfunction and the progression of atherosclerotic plaque through the production of molecules with a proinflammatory (red box) or anti-inflammatory (blue box) effect. Plaque rupture can be a consequence of the exacerbated inflammatory process. Abbreviations; CRP, C-reactive protein; MMPs, matrix metalloproteinases; IFN-γ, interferon-gamma; IL-1α, interleukin-1-alpha; IL-1β, interleukin-1-beta; IL-2, interleukin-2; IL-6, interleukin-6; IL-10, interleukin-10; IL-18, interleukin-18; Lp-PLA2, lipoprotein-associated phospholipase A2; TGF-β, transforming growth factor beta; Th-1, T-helper-1 lymphocyte; TNF-α, tumor necrosis factor alpha; T-reg, regulatory T lymphocyte.
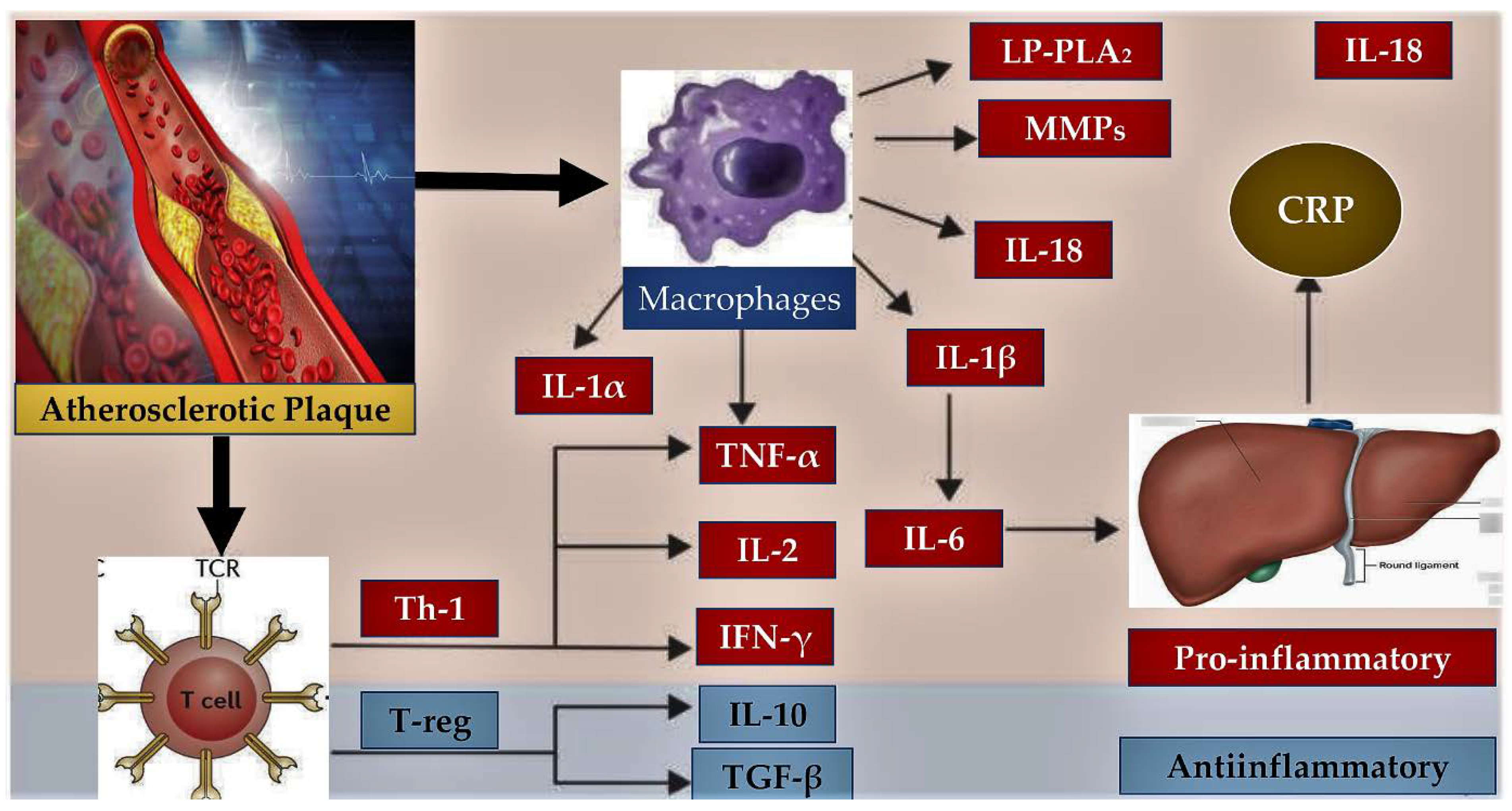
Figure 3.
Cyclophilin A and Basigin participles with a markedly role in cell proliferation of vascular smooth muscle cells (VSMCs). In red box and arrow mechanisms triggered in VSMCs by hypoxia that induces the production of reactive oxygen species (ROS), which promotes cyclophilin A (CyPA) secretion. Rho-kinase also promotes the secretion of CyPA. In purpure box and arrow is showed how extracellular CyPA recruits and stimulates inflammatory cells promoting secretion of inflammatory cytokines. In violet box is highlighted the role of CyPA which directly stimulates proliferation of VSMCs through Basigin, leading to additional secretion of cytokines/chemokines and growth factors. The interaction between extracellular CyPA and Basigin in VSMCs may contribute to VSMC proliferation and pulmonary vascular remodeling. VAMP indicates vesicle-associated membrane protein.
Figure 3.
Cyclophilin A and Basigin participles with a markedly role in cell proliferation of vascular smooth muscle cells (VSMCs). In red box and arrow mechanisms triggered in VSMCs by hypoxia that induces the production of reactive oxygen species (ROS), which promotes cyclophilin A (CyPA) secretion. Rho-kinase also promotes the secretion of CyPA. In purpure box and arrow is showed how extracellular CyPA recruits and stimulates inflammatory cells promoting secretion of inflammatory cytokines. In violet box is highlighted the role of CyPA which directly stimulates proliferation of VSMCs through Basigin, leading to additional secretion of cytokines/chemokines and growth factors. The interaction between extracellular CyPA and Basigin in VSMCs may contribute to VSMC proliferation and pulmonary vascular remodeling. VAMP indicates vesicle-associated membrane protein.
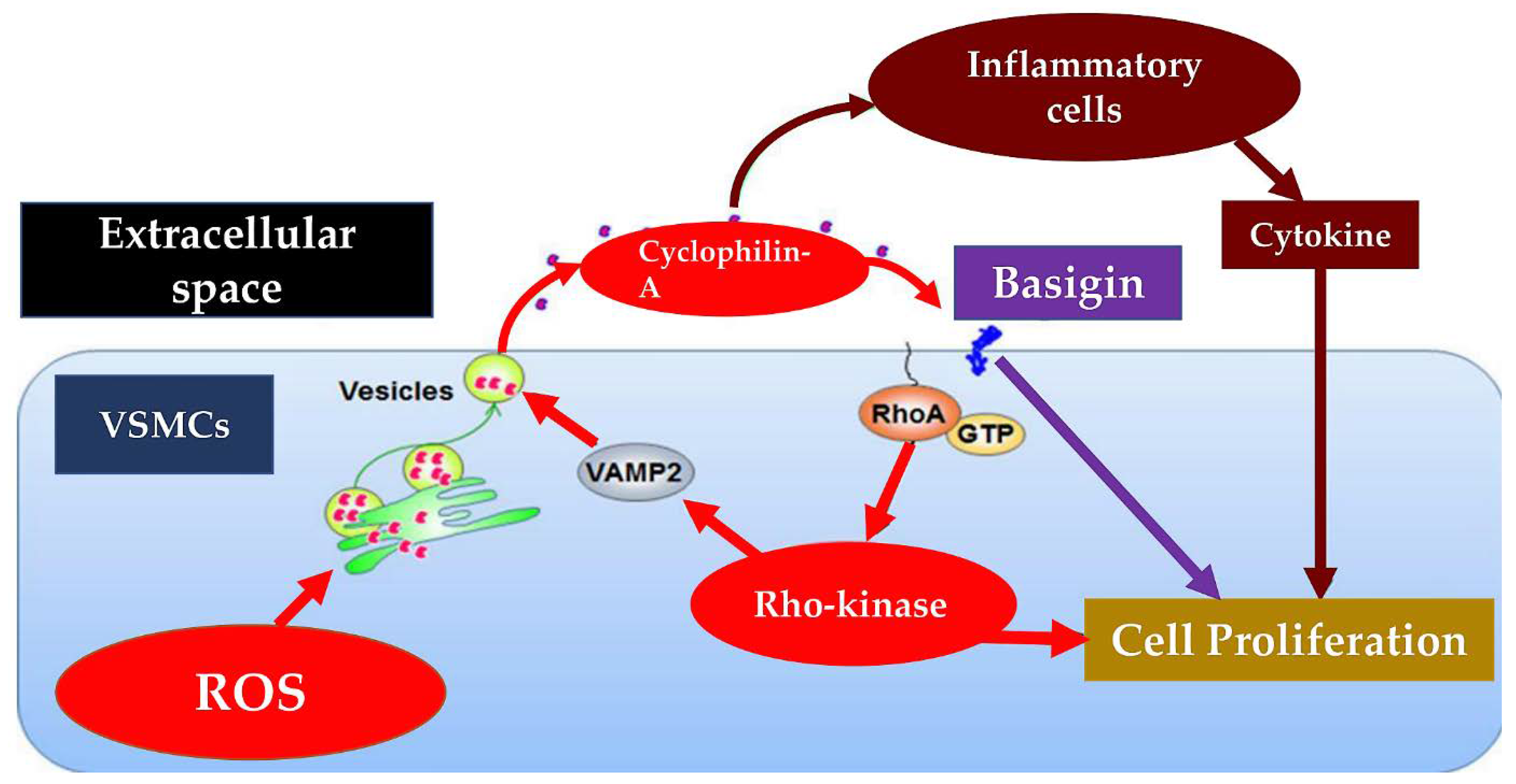
Figure 4.
Cellular and molecular basis for endothelial dysfunction in diabetes are reported.
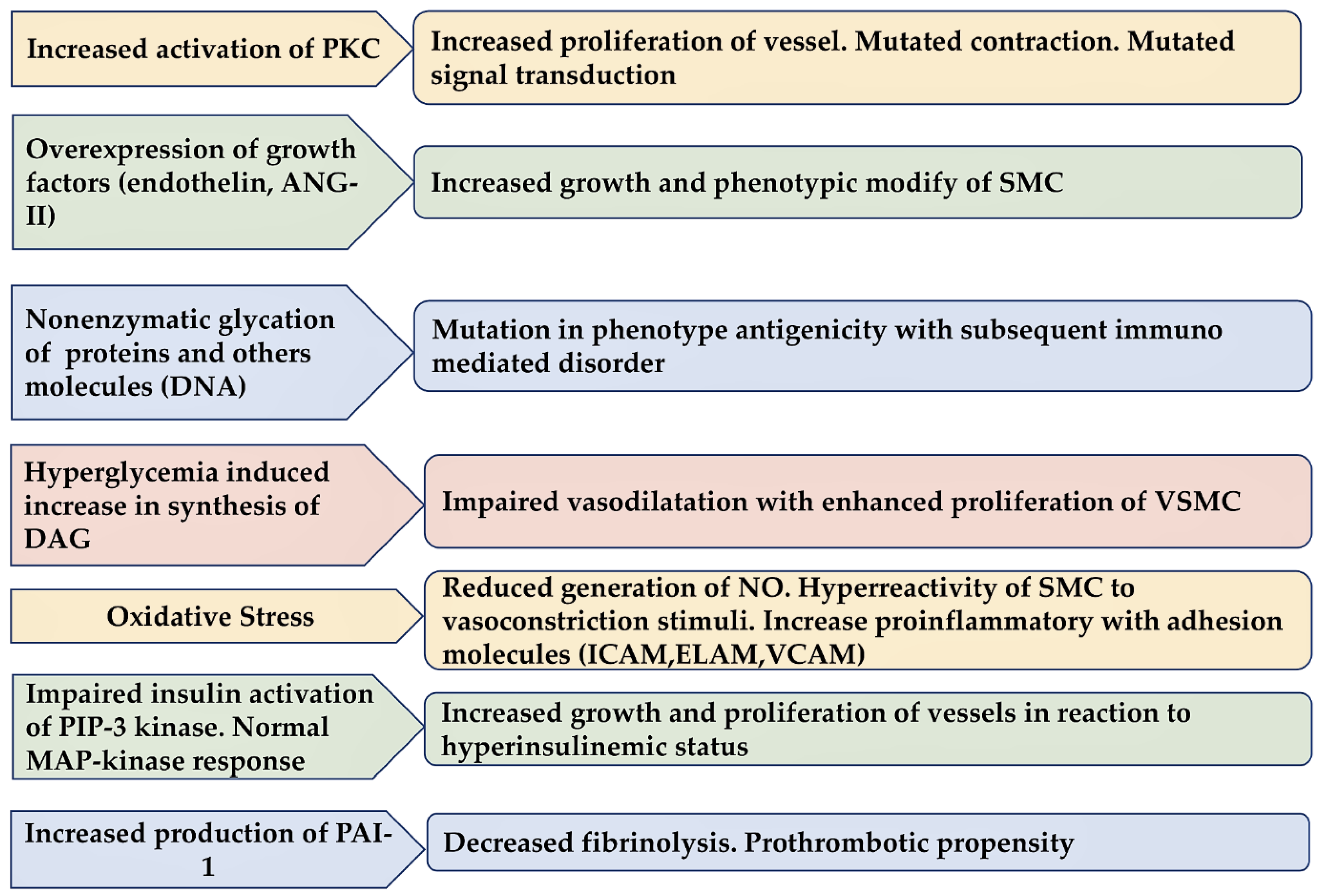

Table 1.
Characteristics of the Included Studies.
| First Author/Year Ref | Type of Study | Cohort | Aims | Finding |
| Honda et al (2016) Arterioscler Thromb Vasc Biol [15] |
Human Prospective Single Center (USA ) |
145 pts ((95 men and 50 women) | To evaluate the relationship between endothelial function and vascular inflammation by mean of flow-mediated dilation (FMD) and 18FDG-PET | Vascular inflammation in the carotid arteries evaluated was independently correlates to decreased %FMD suggesting the association of vascular inflammation with endothelial dysfunction |
| Kang et al (2019) Front Immunol [16] |
Prospective Single Center (Korea) |
Nine statin-naïve SA patients with inflammatory carotid plaques | To evaluate anti-inflammatory effects of statin initiation to 3 and 3 months to 1 year) by mean o 18F-FDG PET/CT | The anti-inflammatory effect of the statin continues to maintain an effect for up to 1 year. However, stable plasma LDL-C levels below the 3-month target were produced. |
| Krog et al (2017) Clin Exp Immunol [18] |
Human/Animal model Prospective Single Center (Denmark.) |
100 pts with type 2 diabetes Vs 100 sex- and age-matched controls Streptozotocin-induced diabetes mouse |
To study SNPs in the MASP1 gene and altered MASP-1, MASP-3 and MAp44 | Higher levels of MASP-1 levels among pts with type 2 diabetes and diabetic mice |
| Hertle et al (2016) Arterioscler Thromb Vasc Biol [19] |
Prospective Multicenter (Netherlands, Denmark) CODAM study (Cohort on Diabetes and Atherosclerosis Maastricht) |
574pts cIMT 73 pts CVD |
To study MBL-associated proteases (MASPs) and MBL-associated proteins (MAps) in complement activation and CVD | High MBL may contribute to low cIMT. MASP-1 and MASP-2 were not associated with adverse cardiovascular outcomes. MASP-3 and MAp44 crucial role in endothelial dysfunction |
| Hertle et al (2018) Arterioscler Thromb Vasc Biol [20] |
Prospective Multicenter (Netherlands Denmark) CODAM study (Cohort on Diabetes and Atherosclerosis Maastricht) |
574pts cIMT 73 pts CVD |
To determine the associations between factor C1q its regulator C1-INH and CVD | Nonlinear association between C1q and incident CVD |
| Herder et al (2017) Cardiovasc Diabetol. [22] |
Prospective Single Center (Germany) |
1107 pts KORA F4 study. |
Whether higher IL-22 levels are associated with lower diabetes incidence. | High serum levels of IL-22 were inversely associated with cardiometabolic risk factors. these associations did not translate into an increased risk for type 2 diabetes |
| Herder et al (2017) Arterioscler Thromb Vasc Biol [23] |
Meta-Analysis Single Center (Germany) |
5 cohort studies IL-1RA MONICA/KORA Augsburg case-cohort study 1855 pts CVD 18 745 noncases CVD |
To evaluate circulating IL-1RA and incident of CVD | Serum IL-1RA levels were associated with risk of CVD after adjustment for multiple confounders. IL-1RA lead to subclinical inflammation, oxidative stress, and endothelial activation. |
| Ridker et al (2017) NEJM J [24] |
RCT Multicenter Center CANTOS Trial |
10,061 patients canakinumab (50 mg, 150 mg, and 300 mg) 3,344 placebo group |
To compare therapeutic effect of monoclonal antibody targeting interleukin-1β | Canakinumab at a dose of 150 mg every 3 months was effective as anti-inflammatory against interleukin-1β laeding to a significantly lower rate of recurrent cardiovascular events than placebo, |
| Choi et al (2014) Arterioscler Thromb Vasc Biol [25] |
Multicenter Center (USA/Korea) |
40 ptswith mild coronary atherosclerosis | To study endothelial dysfunction in pts with early CAD presenting macrophages and vasa vasorum infiltrates. | Epicardial endothelial dysfunction was associated with optical coherence tomography -identified macrophages and microchannels in mild coronary atherosclerosis. |
Abbreviations; cIMT, carotid intima-media thickness; C, complement; C1-INH, C1-inhibiteur; CAD, CVD, cardiovascular disease; 18FDG-PET, [18F]-fluorodeoxyglucose-positron emission tomography; FMD, flow-mediated dilation; KORA, Kooperative Gesundheitsforschung in der Region Augsburg; MAp44; mannan-binding lectin-associated protein of 44 kDa; Map, MBL-associated protein; MASP, MBL-associated serin protease; MBL, mannose-binding lectin; SA, stable angina; pts, patients; SNPs, nucleotide polymorphisms.
Table 2.
Characteristics of the Included Studies.
| First Author/Year Ref | Type of Study | Cohort | Aims | Finding |
| Satoh et al (2008) Antioxid Redox Signal [28] |
Animal Model Single Center (Japan) |
CyPA knockout mice vs Wild-type mice vs VSMC-Tg mice |
To evaluate contribute of CyPA to vascular remodeling | CyPA regulate inflammatory cell accumulation, flow-mediated vascular remodeling and intima formation. |
| Satoh et al (2014) Circ Res [29] |
Animal Model Single Center (Japan) |
CyPA (±) mice Vs Bsg (±) mice |
To determine the role of CyPA/Bsg signaling in the development of PH. | Increased CyPA levels in patients with PH. Cell proliferation reduced in Bsg (±) compared with Bsg (+/+) VSMCs. |
| Xe et al.(2017) Arterioscler Thromb Vasc Biol [30] |
Human/Animal Model Single Center (USA) |
CyPA (±) mice Vs Human pulmonary EC |
To evaluated the role of extracellular CypA in PH. To compare effects of acetylated CypA (AcK-CypA) and CypA on EC dysfunction. | EC-derived CypA (especially AcK-CypA) favor PH due to apoptosis, inflammation, and oxidative stress |
| Rosa et al (2022) Int J Mol Sci. [32] |
Animal/Human Model Prospective Multicenter (GermanyUK,) |
CyPA (±) mice Vs Human pulmonary EC |
Whether K82 and K125 acetylation is required for release of CyPA from platelets | Acetylation of CyPA no major protein modification in platelets. CyPA acetylation is not required |
| Meloche et al (2014) Circulation [39] |
Prospective Single Center (Canada) |
Human PH EC Vs Healthy tissues/cells |
To study PAH-PASMCs increasing during activation of poly (ADP-ribose) polymerase-1 (PARP-1) | PH development related to DNA damage/PARP-1 signaling pathway |
| Meloche et al (2015) Am J Physiol Cell Physiol [40] |
Prospective Single Center (Canada) |
Human PH EC Vs Healthy tissues/cells |
Wether miR-223 downregulation triggers PARP-1 overexpression | Downregulation of miR-223 in PH |
| Archer et al (2010) Circulation [41] |
Animal/ Human Model Single center (USA) |
FHR (PH rat) Vs *Sprague-Dawley rat PH pts |
To study expression of SOD2 and its correlation with PH | Epigenetic SOD2 deficiency induce PH due to impairing redox signaling and creating a proliferative, apoptosis-resistant PASMC |
| Yamaji-Kegan et al (2014) Am J Physiol Lung Cell Mol Physiol [44] |
AnimalModel Single Center (USA) |
IL-4/STAT6 KO mice Vs wild-type (WT) mice |
To evaluate how HIMF lead to lung inflammation and vascular remodeling | IL-4 signaling exert substantial role in HIMF-induced lung inflammation and vascular remodeling. |
| Johns et al (2016) Arterioscler Thromb Vasc Biol [45] |
Animal Model Single Center (USA) |
HIF-1α(+/-) mice Vs wild-type (HIF-1α (+/+) vs Human resistin-like molecule-β |
To evaluate hypoxia-inducible factor-1 (HIF-1) is a critical downstream signal mediator of HIMF during PH development. | HIMF can induce HIF-1, vascular endothelial growth factor-A, and interleukin-6. Mediators for hypoxic inflammation and PH pathophysiology. |
| Lin et al (2018) Arterioscler Thromb Vasc Biol [46] |
Animal/Human Model Single Center (USA) |
HIMF KO mice Vs Human RELM-β |
To investigated the immunomodulatory properties of HIMF signaling in PH pathogenesis | In HIMF-induced PH, HMGB1-RAGE mediates EC-smooth muscle cell crosstalk |
Abbreviations; AcK-CypA, CypA; Basigin; Cyclopillin A; EC, Endothelial cell; FHR, Fawn-hooded rat; PARP-1, poly (ADP-ribose) polymerase-1; h-restin; homolog resistin; HIF-1, hypoxia-inducible factor-1; HIF-1α(+/-), HIF-1α heterozygous null; †HIMF, Hypoxia-induced mitogenic factor; ; HMGB1, high mobility group box 1; KO, knockout; PASMC, pulmonary arterial smooth muscle cell; PH, pulmonary hypertension; Pt, patient; RAGE, receptor for advanced glycation end products; RELM, Resistin-like molecule; SOD2, mitochondrial superoxide dismutase-2; VSMC, smooth muscle cell; VSMC-Tg, overexpress CyPA; WT, wild-type. * Sprague-Dawley rat selective for PASMCs. † HIMF or FIZZ1 (found in inflammatory zone-1) or RELM (resistin-like molecule-α).
Table 3.
Characteristics of the Included Studies.
| First Author/Year Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Loader et al (2015) Arterioscler Thromb Vasc Biol [50] |
Human Study level meta-analysis |
525 healthy pts 540 cardiometabolic pts |
To compare acute hyperglycemia in EF and VSMF | In healthy and diseased subjects macrovascular but not microvascular endothelial dysfunction during acute hyperglycemia were revealed |
| Lespagnol et al (2020) Front Endocrinol. [51] |
Human Study level meta-analysis |
21 study | To evaluate early EF and VSMF alteration in type 1 diabetes. | In children and adults VSM dysfunction with type 1 diabetes is demonstrated. Endothelial dysfunction s more pronounced in large than small vessels. |
| Horton et al. (2022) J Physiol [52] |
Human RCT |
Healthy young adults 6 males 7 females |
To compared macrovascular and microvascular functional responses to euglycemia and hyperglycaemia | Unlike meal-promoted acute hyperglycaemia 4 h of intravenous glucose-induced hyperglycaemia enhances brachial artery flow-mediated dilatation evokes cardiac and skeletal muscle microvascular function without impairing aortic stiffness. |
| Loader et al (2017) Arterioscler Thromb Vasc Biol [53] |
Human RCT |
Healthy young adults (12 males) 600 mL (20 oz.) of water Vs SSB |
To compare EF and VSMF. | SSB infusion mediates endothelial dysfunction with increasing oxidative stress and decreasing NO bioavailability after SSB infusion. |
| Tabit et al (2013) Circulation [54] |
Human Prospective comparative |
40 diabetics type 2 Vs 36 nondiabetic controls |
To study activity of PKCβ nuclear factor κB and reduced No | Altered eNOS activation, reduced insulin action and increased inflammatory activation. Increased PKCβ activity in endothelial insulin resistance. |
| Farb et al (2016) Vasc Med [55] |
Human Visceral adipose tissue arterioles |
43 obeses pts | To investigate the role of WNT5A-JNK leading to insulin-mediated vasodilator responses | Up-regulation WNT5A-JNK signaling and impaired endothelial eNOS activation |
| Walther et al (2015) Arterioscler Thromb Vasc Biol [56] |
Human RCT |
53 pts MetS without T2D Vs 25 pts T2D Vs 40 pts healthy |
To compare EF and VSMF. To measure plasma glucose, insulin and inflammatory markers | MetS was associated with endothelial-dependent and endothelial-independent dysfunction, affecting both the macro- and the microvascular systems. |
| Bretón-Romero et al (2016) Arterioscler Thromb Vasc Biol [57] |
Human Prospective comparative |
42 pts T2D Vs 43 pts healthy |
To evaluate whether increased activation of Wnt5a-JNK signaling contributes to impaired EF. To determine eNOS activation and NO production | Wnt5a-induced impairment of eNOS activation and NO that was reversed by Wnt5a and JNK inhibition. Noncanonical Wnt5a signaling and JNK activity contribute to vascular insulin resistance and endothelial dysfunction. |
| Cho et al (2018) Arterioscler Thromb Vasc Biol [58] |
Human/Animal model | Sprague-Dawley rat Vs Human EC |
To investigate whether SFRP5 could restore WNT5A-induced endothelial dysfunction in vitro and ex vivo. | Compensatory action of SFRP5 against atherosclerosis under conditions of metabolic dysfunction. SFRPS restored WNT5A-induced reduction of NO production via eNOS |
Abbreviations; EC, endothelial cell; EF, endothelial function; eNOS, endothelial nitric oxide synthase; MetS, metabolic syndrome; NO, nitric oxide; PKCβ, protein kinase C-β; RCT, randomized clinical trial; SFRP5, secreted frizzled-related protein 5; SSB, sweetened beverage; T2D, type 2 diabetes mellitus; VSM, vascular smooth muscles; VSMF, vascular smooth muscle function; WNT5A-JNK, Wingless-Related Integration Site 5A- Jun N-terminal kinase.;. rWnt5a, recombinant Wingless-Related Integration Site 5A.
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



