
Submitted:
22 January 2023
Posted:
26 January 2023
You are already at the latest version
Abstract
Transcription factors are diverse intracellular proteins facilitating cellular responses to inducing factors via gene expression. Regulatory signalling cascades from the membrane proteins (sensors) to transcription factors (effectors) are paramount to accurate phenotypic display against external factors. This review examines several transcription factors germane to Cryptococcus (C.) neoformans adaptation and survival for human infection. These opportunistic pathogenic single-cell yeasts (fungi) possess several gene duplications and peculiar membrane proteins due to adaptive phenotypes and morphological plasticity. Consequently, hundreds of responsible pleiotropic genes have been studied to understand how these genes are induced, regulated, and coordinated. However, these findings are sparsely converged and interlinked, making it cumbersome to relate one gene to the other or group them by their functions. We reviewed several wide-ranging transcriptional analysis works associated with C. neoformans into comparable phenotypic traits that necessitate adaptation, survival, and human infection. We present a robust work that addresses several transcription factors and their inducing factors. Lastly, we converge, link, and group several of these factors according to their multifunctional expression pattern. We also provide adequate information on certain genes critical to this fungus, which could be explored pharmaceutically in drug targeting for more effective antifungal management.
Keywords:
Cryptococcus
; transcription factor
; thermotolerance
; virulence factors
; antifungal
; oxidative and osmotic stress
; capsule
; melanin
; monokaryotic fruiting
; mating and filamentation
1. Introduction
C. neoformans is an environmental opportunistic and obligate aerobe, capable of colonising various human systemic organs [1]. It is an encapsulated pathogenic fungus with a high degree of ecological adaptation. It is found in soil enriched with bird droppings and around trees. The probability of finding cryptococcal cells around woody and shrub plants such as Eucalyptus [2], Carob [3], Olive [4], Pine [3], Platanus, and Prunus [3] trees is evidenced. The soil environment is a complete and complex ecosystem that allows for the survival of all living organisms through competition and adaptation. As such, C. neoformans is among the saprophytes exposed to the good, bad, and ugly environmental conditions that invariably cumulate and translate into the phenotypic adaptation for human infection. Besides the harsh environmental conditions, C. neoformans are constantly in predatory association with the water soil amoeba, enabling the cryptococcal cells to develop peculiar features to survive these menace [5,6]. These persistence associations reasonably equipped cryptococcal cells to survive the predatory phagocytic amoeba and improve the cellular virulence factors and self-defence phenotypic traits [7,8,9]. In that case, surviving the mammalian residential monocytes, macrophages, and dendritic cells is a pathogenic feature of invading C. neoformans. To further bolster this possibility, C. neoformans pre-adapted in Dictyostelium discoideum became enlarged/titanised (giant cell) and melanised faster than the wild type (wt). In addition, acapsular/avirulent C. neoformans became virulent in the myosin-mutant soil amoeba [10]. This pattern of infection re-emphasises why the immunocompromised individual is predisposed to cryptococcosis. By spore-inhalation, the C. neoformans pathogen becomes invasive (cryptococcemia) from the lungs to other parts of the body tissues and organs, particularly the CNS, brain, bones, skin, prostate, and heart [11].
C. neoformans isolated from the soil contaminated with the pigeon dropping/plant niche are regarded as environmental isolates, while those from infected humans are known as clinical/blood isolates. However, among the serotypes A, D, and AD of C. neoformans, serotype A is the most common environmental and clinical/veterinary type with genotype similarity among the isolated strains such as VNI [12,13]. Early investigation showed that environmental isolates might be less virulent in the animal model compared to the clinical isolate [14]. However, a recent study has shown that the pathogenicity of the VNI strain, whether an environmental or a clinical isolate, was the same in the animal model [11].
Several virulence factors associated with the pathogenesis of C. neoformans have been studied and characterised. To enhance the strategical launching of each factor, the fungus compartmentalises the factors and releases the virulence factors according to the inducing cue. Cryptococcal cells produce capsules, which release the immunomodulatory capsular polysaccharides – glucuronoxylomannan (GXM) and galactoglucuronoxylomannan (GalGXM) that majorly alter the phagocytosis of this fungus by macrophages. In addition, the presence of a cell wall, which is made with chitin, can release melanin. The melanin may facilitate cell wall titanisation/thickening and the membrane clustering of other virulent factors and enzymes. The desire to produce melanin drives the neurotaxis of C. neoformans to the catecholamine-enriched tissue, such as the basal ganglia of the brain [15]. These functional virulence factors sequentially orchestrate the expression and secretion of extracellular hydrolytic enzymes, the sequestration of host metabolic pathways, the coordination of robust antioxidant mechanisms, the attenuation of host NO-induction and PAMPs-PRRs (pathogen-associated molecular patterns-pattern recognition receptors) killing system, the resistance of C. neoformans to Ca2+ and CO2 influx, pH imbalance, nutrient, and oxygen deprivation. The constant modification of the dynamic components of the cryptococcal cell wall and membrane enables this pathogen to survive the environmental niche and host internal cellular homeostasis response [16].
Having highlighted some of the strategies launched by the cryptococcal cells for environmental adaptation, tissue invasion, and survival, it is paramount to harmonise how the transcription factors and intracellular proteins control each of these microbial phenotypic displays and, ultimately the release of various virulence factors. In this review, numerous works on the regulatory transcription factors of C. neoformans are critically examined and juxtaposed with fungal adaptation and survival during infection in the human host systems.
2. Regulatory Transcription Factors in Cryptococcal Cells
Various transcription factors are upregulated/downregulated when an amoeba or a macrophage engulfs cryptococcal cells. This happens because the infected cells launch a compulsory intracellular nutrient limitation and induce stress as a mode of pathogen attack. More than 525 inherent transcription factors are differentially upregulated in C. neoformans phagocytosed by macrophages [17,18], which is one of the reasons some basidiospores are constantly released non-lytically by vesicle secretion to promote cellular and tissue invasion, especially in the immunocompromised patients. These transcription factors cover different cellular systems, including translation, signal transduction, antioxidant induction, and carbon metabolism within the cryptococcal cells to facilitate cell dormancy, reproduction, infection, and escape the anti-cryptococcal-induced immune response [19,20,21]. In addition, the number of modulatory transcription factors expressed in C. neoformans when encountering stress from amoeba is usually more than that expressed in the macrophages purposely because of the concomitant effects of the environmental factors and predatory amoeba (with varying microbiota) as against the macrophages. Derengowski et al. reported more than double expressed genes in macrophages as expressed in Amoeba [8]. Several of these transcription factors are intrinsically linked to one another and the corresponding phenotypic expression [22,23,24]. Mutation (disruption/deletion) of such genes in cryptococcal cells produces defects that make them avirulent, easily phagocytosed, physiologically deformed, sterile, and easily killed. Paradoxically, mutation may sometimes enhance the survival, adaptation, and pathogenesis of cryptococcal infections (for details on the phenotypic display of different cryptococcal cell mutants, check Supplementary 1, and for details of cellular events that induced/repressed different membrane transporters and permeases in C. neoformans, check Supplementary 2).
Transcription factors are largely conserved and structurally similar in function among the cryptococcal species/strains; however, serotype-specific differences occur due to different environmental susceptibilities, evolution, and geolocation of each strain. This evolutionary divergence has brought about a discrepancy in the function of homologue transcription factors. Hicks et al. discovered that the Pka1 catalytic subunit of Pka is involved in the cAMP pathway major kinase function in serotype A and enhanced mating, haploid fruiting, capsule, and melanin formation, but Pka2 is engaged for the same purpose in serotype D [25]. The mating-type specific Pak (p21-activated protein kinase) homologues Ste20α in MATα and Ste20a in MATa are allelic genes found within the congenic mating-type loci controlling the mating and pheromone formation in C. neoformans. While the Ste transcription factors contribute to the mating and filamentation of MATα in serotype A, the congenic MATa is sterile with no pheromone and less virulence than MATα [26]. Further analysis showed that serotype A MATa mating-type locus lacked several mating-associated genes such as Ste12α, Ste20α, Ste11α, and Mfα2. This MATa serotype A was later confirmed to contain the Ste20a gene, which is similar to serotype D (95% homology) but diverged from the Ste20α of serotypes A and D (67% homology) [26].
Significant virulence reduction was reported in MATα serotype D Δste12α mutant but not in serotype A mutant. Also, this Ste12α controlled other phenotypic expressions, such as capsule and melanin formation in serotype D, which further underpinned the molecular linkage between mating type and virulence in serotype D [27,28]. Furthermore, a relatively low mating frequency and hyphal formation in serotype D were observed when the MATα Δste12α mutant strain was convergently crossed with the MATa strain [27]. This indicates the possibility of Ste12 homologues in different mating-type strains. Furthermore, the strain with Ste12α reconstituted locus exhibited the wt phenotypic expressions such as capsular size, mating, phospholipase activity, and haploid fruiting in serotype D [27]. Though serotype A Δste12α showed a modest defect in mating but completely defective in haploid fruiting/sporulation like serotype D; however, unlike serotype D, this homologue gene seems to be redundant with no effect on the virulence of serotype A [29].
Naturally, serotype D (like the JEC21 strain) is resistant to fludioxonil (FDX) like S. cerevisiae because Hog1 phosphorylation that translocates this MAPK protein into the nucleus is not initiated in the presence of this fungicide; hence the Hog1 MAPK signalling remains inactive. However, serotype A (such as the H99 strain) is sensitive to FDX because the Hog1-specific nuclear phosphatases activate Hog1 via the Hog1 osmosensing pathway. The Hog1 protein, an apparent functional divergent mitogen-activated protein kinase encoded by the Hog1 gene via the high osmolarity glycerol (HOG) pathway in response to high external osmotic stress, is a nuclear-localised dephosphorylation-activated protein regulated by the response regulator kinase Ssk1p. Optionally, it was proposed that Hog1-specific phosphatase could also be induced independent of Ssk1 but orchestrated by the response regulator phosphorelay histidine protein kinases, Tco1 and Tco2, in the yeast [30]. Generally, in its repressed state (phosphorylation), Hog1 prevents yeast virulence and mating by minimising the production of melanin, capsule, and mating pheromones. Contrarily, in its activated state (dephosphorylation), Hog1 mobilises the induction of stress response genes and the production of proteins against internally generated toxic molecules. Therefore, unless the Hog1 gene is repressed or deleted in serotype A, it remains sensitive [31,32]. It was further concluded that FDX resistance is a dominant phenotype because serotype AD obtained by hybrid crossing of serotype A (FDX sensitive) and serotype D (FDX resistant) showed FDX resistance [31].
Similarly, deletion of the Cna1 gene predisposes serotype B to a complete growth defect at 30oC. At 37oC, serotypes A and B, irrespective of mating strains, completely failed to grow and are rendered avirulent when Cna1 is deleted. However, only Δcna1 mutant of serotype A remained sensitive to osmotic stress induced by NaCl and LiCl [33] as well as FDX in a Hog1-independent manner [31]. In addition, there is a functional divergence of Snf1 between serotype A strain H99 and serotype D strain JEC21. Generally, Snf1p facilitates virulence production, alternative carbon utilisation, stress tolerance, and thermotolerance. A high temperature is needed for H99 Snf1 to function against stress response and the assimilation of alternative carbon sources, unlike strain JEC21 [34]. Likewise, disruption of Snf1 in JEC21 predisposes the strain to amphotericin B (AmpB) but not in the H99 strain [20,34]. Check Supplementary 4 for the meaning of the transcription factor, kinases, protein, and gene acronyms used in this review.
3. Regulatory Transcription Factors in Cryptococcal Cells Control Phenotypic Expression for Morphotypes, Adaptation, Survival, and Virulence
3.1. Cell Wall Chitin-Chitosan Components
C. neoformans overlays its cell wall matrix with chitin (a repeating unit of -1, 4 – N-acetyl-D-glucosamine). Chitin generally strengthens the cell walls and enables a rigid cellular shape [16,35]. With this, C. neoformans maintains a constantly balanced cellular turgor pressure against its cell wall and, together with capsules, helps to prevent desiccation injury when the soil is dry.
Though the presence of chitin and other unique cell wall components predispose the cryptococcal cells to the host PAMPs-PRRs (pathogen-associated molecular patterns-pattern recognition receptors) recognition to induce anti-cryptococcal defence strategies [36] but with the capsule formation and vesicular secretion of capsular components, C. neoformans can shield the chitin and circumvent the PAMPs effect [37,38]. Not only this, but Cryptococcus also has intrinsic chitin deacetylase encoded by seemingly redundant Cda1, Cda2, and Cda3 functional genes (the fourth, Fdp1, remained undetermined), which can convert cell wall chitin produced by chitin synthase (encoded by Chs3), in the presence of its regulator (encoded by Csr2), to chitosan (a non-rigid soluble polymer) [39,40]. Interestingly, C. neoformans seems to prefer the Cda1 gene, which is highly upregulated in mammalian lungs and primarily selected for fungal proliferation [16].
Eight genes encode chitin synthase, viz Chs1 to Chs8, but only three chitin synthase regulators, Csr1 – Csr3, are functionally active. Unexplainably, as many as these encoding genes are in cryptococcal cells, a compensatory expression for each mutant rarely occurs except for Δchs3, where a slight increase in Chs5 and Chs7 was detected [39]. Furthermore, the Chs3-Csr2-Cda1 complex is particularly important for chitosan production for the cryptococcal cells to grow, disseminate, and invade the host at a physiological temperature [41,42]. Under vegetative growth, the expression of Chs7 is entirely repressed, while Chs6 is extremely and minimally detected in contrast to Chs1 and Chs4 expression. However, Chs2, Chs3, Chs5, and Chs8 are highly induced during vegetative growth [39]. Surprisingly, none of these chitin-associated genes or regulators is essential for cryptococcal viability at 30oC. However, Δchs3 and Δcsr2 mutants are particularly hypersensitive to higher temperatures and cell wall stressors such as Calcofluor White (CFW), Congo red, SDS, and caffeine (for details on the phenotypic responses of different mutants of Cryptococcus against various quantitative external factors, check Supplementary 3).
Characteristically, all chitin mutants displayed similar phenotypic features as the Δcsr1 and Δcsr3, except the Δchs3 and Δcsr2 mutants. These two mutants could produce melanin around their cell wall but failed to retain this pigmentation ("leaky mutants"), unlike other chitin mutants [39]. Furthermore, cytokinesis in these mutants is impaired and characterised by incomplete budding. The CFW staining revealed intense septal and uneven cell surface staining. The aggregated mutants are proportionally larger than the wt, and the Δcsr2 mutant is even larger than the Δchs2 mutant. The two mutants also failed to produce chitosan by 48 hours of incubation but instead accumulated a higher level of chitin than any other mutants and the wt [39]. This cryptococcal strategy of protecting its cell wall seems evolutionary to prevent predatory and host-released chitinase, maintain cell wall integrity and capsule width, ensure melanin production and attachment, and budding during vegetative growth [39,40,43].
3.2. Cell Morphotypes and Aneuploidy
Reports have shown that small-size (<2 μm) basidiospores produced during sexual reproduction in Cryptococcus are deeply embedded in the alveoli making it difficult to be dislodged from the mammalian lung through airway ciliary movement [44]. These spores are highly pathogenic in an immunocompromised murine model and resistant to high temperature, oxidative stress, sunlight, and desiccation [45,46,47]. Conversely, the filamentous (hyphae or pseudohyphae) morphotype of Cryptococcus is not as highly pathogenic as the spores but is literally adopted to survive environmental amoeba [48]. Conditionally, pseudohyphae growth is usually initiated under a nitrogen-limiting condition (that is seldomly observed in murine and human infection but regularly formed when co-culture with amoeba), and it is phenotypically longer in the diploid unisexual than haploid strain; however, this seems unconnected to cell ploidy. Lee et al. showed that Amt1p and Amt2p are required for pseudohyphal growth under nitrogen catabolite repressor (NCR), NH4+, but these hyphae lacked clamp connection that could have formed basidiospores (monokaryotic fruiting) [49]. Irrespective of the serotype, either Δamt1 or Δamt2 but not Δamt1Δamt2 mutant can form pseudohyphae, which can fully develop back into the yeasts in yeast extract peptone dextrose (YPD) media (yeast ↔ pseudohyphae). Still, the colonies are wrinkled at the edges when cultured in invasive agar growth [49]. Formation of filaments in Cryptococcus seems to be microevolution depending on the host interaction but is unlikely to be significant in virulence [50,51,52]; however, this has been shown to improve agar adherence and invasion [49]. Thus, the reversible yeast-pseudohyphal switch may probably be responsible for the infection latency observed in humans.
Titanisation (cell giant) is another yeast-like morphotype, which has been linked to the accumulation of chitin within the cell wall to withstand several host-derived stresses [53,54,55]. Giant cells are conspicuously found in alveoli of the mammalian pulmonary system during infection, growing from 15 – 100 μm in size with polyploid genomes that can degenerate into haploid and aneuploid progenies; the bases of genetic variability, pathogen dormancy, and reactivation [56,57,58]. The formation of aneuploid spores during the sexual or non-sexual reproduction of MATa and MATα is the foundation of the infectious propagule of Cryptococcus [59]. Aneuploidy is a common phenomenon in the environmental strains to withstand external stress as well as the clinical isolates to initiate adaptation, survival, and infection [60,61].
However, aneuploidy is a rare event in C. neoformans. Out of the 75 basidiospores examined after crossing Δcbk1:Nat mutant with Δcna1:Neo mutant with both genes being far from each other on the serotype A genomic contigs; the MATa and MATα alleles segregated independently with 37.3% wt (NatS::NeoS), 30.7% cbk1 (NatR::NeoS), 30.7% cna1 (NatS::NeoR), and 1.35% cbk1cna1 double mutant (NatR::cna1:NeoR) (Nat - nourseothricin acetyltransferase; Neo – neomycin; S – sensitive; R – resistant) [62]. Further analysis showed that this aneuploidy progeny with cbk1:NatR::cna1:NeoR double mutant resistant strain displayed Δcbk1 phenotypic morphotype as a dominant allele because of the occurrence of heterozygous CNA1 locus (Cna1/Δcna1:Neo). This indicates a parallel function of Ram and Cna1 pathways to regulate cellular morphology at physiological temperature.
The catalytic subunit of Cna1p has been shown to relocate to ER-associated puncta and budding neck at 37oC. This thermal stress temperature induces a preponderant mRNA accumulation in the ER that attracts processing bodies (PBs) and stress granules (SGs). At the ER-associated puncta, a greater proportion of Cna1p localisation has been found with PBs decapping enzyme encoded by Dcp1 and SGs poly(A)-binding protein constituent encoded by Pub1 [63]. This co-localisation is to ease the thermal stress, which causes mRNA accumulation. The Ram-associated genes such as Cbk1, Kic1, Mob2, Sog2, and Tao3 are also crucial for normal cell polarisation, karyokinesis, and cytokinesis. The ram mutants displayed hyperpolarisation and incomplete cytokinesis leading to cell aggregation that forms hyper-elongated pseudohyphal, though karyokinesis leading to septate between the dividing cells appeared normal in these mutants [62]. This phenomenon is well displayed by each ram gene mutant (Δram), with actin being localised at the bud tips, unlike the wt where actins are well distributed throughout the cell for proper cell segregation. The mislocalisation of actin in the ram mutants has been traced to the absence of Cbk1 and Kic1 gene products at the punctate and septa structures of actively dividing cells (cytokinesis) [62,64]. The septal localisation of the Kic1 has been speculated to be controlled by other Ram genes, such as Tao3 [62]. The point mutation in the Mob2 gene usually found in C. gattii may be responsible for the pseudohyphal strain with incomplete mating when crossed with the wt, and this displayed normal mating hyphae and basidia formation but no sporulation [49]. Natural single nucleotide difference in Mob2 of C. gattii genes may have produced differentiated 37oC thermosensitive pseudohyphal strains and progenies, which failed to form yeast in YPD, and are more sensitive to Cna1 pathway inhibitor (FK-506) than the C. neoformans pseudohyphae [49].
Further investigation revealed that Ram protein kinases could interact to facilitate MAPK activity leading to complete and successful cytokinesis. By expressing C. neoformans Cbk1, Kic1, and Mob2 genes in S. cerevisiae, the yeast two-hybrid growth assays revealed a strong protein-protein interaction between Cbk1::Mob2, Cbk1::Kic1, and Kic1::Kic1 but a weak interaction between Mob2::Kic1 [62]. Despite this hybrid expression of the Ram kinases, none of the cryptococcal Cbk1, Kic1, and Mob2 genes could complement the corresponding cytokinesis defect in S. cerevisiae ram mutants; neither was the homologues of S. pombe complemented ram defective S. cerevisiae mutants as well [62,65]. Therefore, proteins from generally related organisms may structurally resemble but are functionally divergent sometimes, regulating different components of the pathways.
3.3. Capsule: Capsular Polysaccharides and Glycoproteins
The polysaccharide capsules afford Cryptococcus species to avoid and survive the attack of phagocytic cells like soil amoeba and mammalian macrophages, dendritic cells, and neutrophils [66]. The phenotypic switch in C. neoformans induced by immediate environmental stresses such as a change in pH, high CO2 level, iron and nutrient limitation, and antifungal has been shown to affect the resultant biophysical and biochemical natures of capsular GXM. This eventually allows the cryptococcal cells to survive different environmental conditions and brings about structural heterogeneity and antigenic variation [67,68,69]. Subtly, this invariably enables the cryptococcal cells to evade the physiological immune attack launched against this pathogen. This could be very deleterious and become aggravated, especially when the cryptococcal infection is being treated in immunocompromised patients. Coupled with the antifungal effect, there may be selective pressure and indirectly cause a phenotypic switch within the infected strain that can cross the blood-brain barrier (BBB), resulting in intracranial pressure. This is the cause of high morbidity and mortality in patients with meningoencephalitis [37,70].
Capsular components are immunomodulatory extracellular complex polysaccharides produced as a radial whitish cloud around the cryptococcal cells (Figure 1) and constantly shed as systemic antigens in the infected patients, which can be assessed from urine, serum, and cerebrospinal fluid (CSF). This enables the cells to suppress the host immune response, withstand the phagocytosis in the systemic milieus, and promote intracellular survival in the macrophages [1,71,72]. In vivo capsule production is usually induced by low iron content, physiological temperature, and CO2 [67,73]. The presence of capsular components such as glucuronoxylomannans (GXM), galactoxylomannans (GalXM), glucuronoxylomanogalactan (GXMGal), and a minor glycoprotein component known as mannoproteins (Mp) have been found in the systemic circulation, causing the shedding of L-selectin adhesion and integrin proteins from the surface of leukocytes such as neutrophils [74,75]. This shedding prevents polymorphonuclear leukocyte (PMN)-endothelial interaction and extravasation of the granulocytes into the inflammatory tissue.
Furthermore, these capsular components are chemotactic, downregulating the surface expression of the TNFα receptor from the PMN. Though the expression of cell-adhesion molecules such as CD11b and CD15 integrins are stimulated in the presence of capsular components yet, tissue inflammation and granulocyte infiltration are inhibited [75]. In addition to reducing pro-inflammatory cytokines, GXM also promotes the uncoupling activity of NF-κB to regulate growth, apoptosis, and inflammation in macrophages [76]. More dangerously, macrophage proliferation can be altered in a capsule-independent manner to promote cell cycle disruption and chromosomal aberration, known as aneuploidy in the macrophage. This type of systemic alteration, which is hitherto commonly found in bacterial and viral infections, has been suggested as a way of promoting tumorigenesis in fungi infection [76].
This effect is very similar to chemoattractants, such as formylmethionylleucylphenylalanine (FMLP) and IL-8, which promote pro-inflammation when located in the extravascular space or inhibit inflammation when situated within the intravascular space. Together with IL-8, GXM stimulates the endogenous production of IL-10 and IL-1β from the monocytes, which down-regulates pro-inflammatory cytokines to block PMN infiltration into the site of tissue inflammation caused by C. neoformans infection [72,77]. Substantially, the systemic concentration of cryptococcal GXM in untreated infected patients ranges from 250 – 500 μg/mL [72]. This is high enough to downplay the critical roles of pro-inflammatory cytokines from the peripheral blood monocytes.
Furthermore, the immunocontrol ability of cryptococcal cells, which resides in the expression of capsular polysaccharides during infection, differs by serotypes. For example, the dynamic structural complexity of the cryptococcal GXM reduces from serotypes C, B, A, to D [78]; however, the capsular diameter reduces from serotypes D, A, C, to B. This feature conferred a higher immunomodulatory effect on serotype B GXM than other serotype capsular components. To support this, Fonseca et al. reported that serotype B GXM, which has the least molecular mass, was the most potent activator of immunocellular responses such as NO production from macrophages and expression of toll-like receptors following the activation and nucleus translocation of NF-κB. On the contrary, this induction was independent of the resultant polysaccharide negative charges of the GXM [79].
It has been shown that while C. neoformans var. neoformans (serotypes A and D) culture filtrate antigen (CneF), which is majorly capsular GXM, induced chemotaxis and chemokinesis responses of hPMN (human polymorphonuclear leukocyte), the var. gattii (serotypes B and C) failed to stimulate but inhibit the hPMN responses to the chemoattractants [80]. This, therefore, means that PMN infiltration may be higher in the var. neoformans (serotypes A and D) immunocompromised infected patients than the var. gattii (serotypes B and C) immunocompetent infected patients. This implies that while the cryptococcal antigenemia may be almost completely cleared from the immunocompromised infected patients due to higher leukocyte mobilisation bolstered with antifungals, the clearance level in the immunocompetent may be inefficient due to lower leukocyte infiltration, despite the body resistance. This may also explain, in part, why var. neoformans serotypes A and D prefer immunocompromised patients to immunocompetent individuals. It is reasonable to think that the more the cryptococcal secretomes stay in the body, the higher the tendency of antibody identification and attack, which may happen in immunocompetent patients, unlike immunocompromised patients where the fungi secretomes are quickly cleared due to higher leukocyte mobilisation.
Hog1 plays an insignificant role in capsule formation in serotype D, unlike serotype A, where a mutation in the Hog1 gene enhances capsule formation [32]. Multiple mutated genes (Δgpa1, Δcac1, and Δpka1) in the Δhog1 background mutant completely hindered capsule production, just like when each gene was mutated alone or doubly mutated with Hog1 [32]. This shows that Hog1 expression must have prevented certain cAMP/Pka upstream cascade events that facilitated capsule production. To this effect, Δssk1, Δpbs2, and Δhog1 mutants have been characterised by hypercapsulation; however, Δtco mutant only showed wt capsule production [30]. In fact, the four major capsule-associated genes (Cap10, Cap59, Cap60, and Cap64) are significantly upregulated in Δhog1 and Δssk1 mutants more than the wt but not in the Δskn7 mutant [81]. Because of the difficulty in deleting Ypd1, it is uncertain whether the transcription factor from this gene plays a role in capsule formation. Notwithstanding, Lee et al. observed an increased capsule volume of Δhog1Δypd1 mutant similar to Δhog1, which is more significant than the wt [82].
D'Souza et al. discovered that while disruption of the Pka1 gene led to sterility and failure to produce capsule and melanin in serotype A, mutation of Pkr1 failed to overshadow the hypercapsulation and hypervirulent of the wt strain in an animal model [83]. This means that if cAMP is a potent inhibitor of Pkr1, then repressing/inducing the Pkr1 gene, even supplying cAMP exogenously, will not correct the Δpka1 defects. However, Pkr1 exhibited an epistatic effect on the Pka1 with a similar phenotypic appearance as the Δpka1 mutant. Furthermore, this sterile and avirulent double mutated Δpkr1Δpka1 strain showed that the catalytic subunit of Pka encoded by the Pka1 gene is a downstream functional/catalytic protein of the regulatory subunit of Pka encoded by the Pkr1 gene [83].
Mutation of Gpa1 is characterised by a phenotypic defect in mating and reduced production of melanin and capsule due to impaired function of the Gα-protein. However, the phenotypic features of attenuated Gα-protein-deficient avirulent sterile strain were restored by exogenous cAMP supply [84]. This means that cAMP is a direct secondary messenger signalling factor to the Gpa1. Unlike acapsular and avirulent Δgpa1 mutants, Δpkr1Δgpa1 double mutant and Δpkr1 mutant significantly produced larger capsule sizes with higher cell volume than the wt in the iron-limiting capsule-inducing media. This is due to the elevated and constitutive activation of Pka, with capsule size being larger in the doubly mutated strain, Δpkr1Δgpa1, than Δpkr1 mutant [83,84]. Such pkr1 mutation may be one of the unusual normal and natural transcriptional regulations in response to temperature to activate other more relevant factors. Indeed, some clinical isolates, characterised by enlarged cell volume, hypercapsulation, and melanisation, have been reported to infect immunocompetent individuals. These isolates were inexplicably larger in the infected tissues than in the in vitro culture [85,86]. Furthermore, mutation of the pkr1 gene showed increased virulence, lethality, tissue burden, and survival of this mutant in animal studies compared to the isogenic/wt strains [83]. Analysis from other G-protein encoding genes showed that Δgpb1, Δgpa2, Δgpa3, Δgpa2Δgpa3, Δgpg1, Δgpg2, and Δgpg1 Δgpg2 mutants displayed normal melanin and capsule formation and that all displayed normal/wt virulence but Δgpa2Δgpa3 mutant exhibited attenuated virulence due to longer surviving period of the inoculated mice [87].
Serotype A MATα Δste12α mutant consistently produced less capsule when compared to the wt or reconstituted strain [29] because Ste12α protein is a Hog1 repressing factor that activates cAMP/Pka pathways for capsule and melanin production [32]. Ordinarily, the expression rate of virulence-associated genes such as Cap59, Cap60, Cap64, and Lac1 – associated with melanin production are time-dependent, and their expression becomes conspicuous as glucose depletes. Cap60 gene, for example, increased the capsule size by 2-fold compared to the wt at 6-hour incubation in the low-iron media (LIM) [88]. However, with serial analysis of gene expression (SAGE), the tagging of other capsule genes, Cap10, Cap59, and Cap64, was not as significant as Cap60 irrespective of the iron level [88].
The stationary phase of C. neoformans will usually have a larger capsule [68] and higher melanin production [89] compared to the exponential phase when the glucose level is very high. In serotype D, Δste12α mutants are characterised with a low level of Cap and Lac1 gene expression compared to the wt over the course of growth using a promoter-coupling reporter gene [27]. With galactose as an expression inducer, overexpression of Ste12α promotes Cap and Lac1 gene expression in the Δste12α mutant better than the glucose. Unbelievably, by juxtaposing the relative effect of Ste12α when deleted or overexpressed, Chang et al. could not observe any significant difference in the capsule and melanin production when assessed on the agar culture [27]. This means that serotype D MATαΔste12α mutant may have encountered other environmental cues that facilitate alternative subways to subtly overshadow the phenotypic defects of deleting Ste12α when the mutant is cultured in agar media. In another way, deleting Ste12α may not be substantial enough to shut down the significant number of cooperative genes that facilitate virulence in C. neoformans, considering different environmental factors.
The Cas transcripts are homologs of Cap64 confirmed to be involved in the positional linkages of xylose and O-acetylation in the mannan backbone. Among the putative homologues identified, Cas3, Cas31, Cas32, Cas33, Cas34, and Cas35, none was shown to be actively involved in the CO2-mediated capsule formation like Cap64, but by speculation, they may be involved in capsule assembly and configuration, [90]. Though high CO2 promotes capsule formation, no capsule was formed in the Δcac1 mutant despite active Cas and Can2 gene expression/overexpression because capsule production is CO2-activated Cac1-dependent [91].
3.4. Melanin
The production of melanin is one of the critical environmental adaptive features of C. neoformans, and this is usually regulated by glucose and nitrogen catabolite repressions. Generally, low glucose induces melanin production in all cryptococcal strains, but nitrogen repression is strain-specific [89,92]. Through environmental sensing, C. neoformans can synthesise this heterologous and hydrophobic chromogenic polyphenol molecule within 4 – 48 hours under the influence of complexly regulated transcription factors using different exogenous molecules such as diphenolic/indole compounds [93], pigeon excreta [94], bacterial-presenting homogentisate [95], bacterial-presenting dopamine [96], catecholamine precursors (L-DOPA, dopamine, methyl-DOPA, epinephrine, norepinephrine, homovanillate, 5-hydroxylindolacetate, serotonin, catechol) [97,98], 1,8-dihydroxynaphthalene (DHN) [99], aminophenol and diaminobenzene compounds [92], and γ-glutaminyl-3,4-dihydroxybenzene (GDHB) [100]. In addition, other exogenous substrates have been well-characterised in different types of fungi [101]. These sources determined the eventual colour, chemical, and physical characteristics of melanin [102,103]; however, among the various reported colours – red, yellow, green, and purple; brown and black (eumelanin) are the major pigmentations [104].
This cell wall-associated biosynthetic process mostly involves the enzymatic conversion of exogenous catecholamine-intermediates into eumelanin [102]. Though melanin and capsular components are closely associated at the cell surface, evidence shows that the two components are independently regulated and differently shuttled to the cell wall. While melanin production is a cell surface synthesis, the components of the capsules are produced in the cytoplasm and transported in the vesicles for cell wall fusion; nevertheless, the heterologous nature of melanin particles can impose physical changes on the capsular size [102]. Notwithstanding, C. neoformans use molecular sorting mechanisms such as the endosomal sorting complex required for transport (ESCRT) to transport polysaccharides and conjugated molecules.
The Snf7p has been identified as a critical operator of ESCRT. In conjunction with other tagging chaperones and vacuolar proteins such as Vps20/25, Snf7p forms multivesicular bodies transported to the specific organelle, such as the plasma membrane, where the vesicular components can be directly fused or released extracellularly (exosome). Furthermore, ESCRTs have been implicated in other signalling events, such as the Rim101 pathway during the pH-sensing by the fungi [105]. The Vps20/25-Snf7 complex forms a bridge connecting the endosomal ESCRTs I, II, and III to the Rim20/13/101.
As a sequel to this, Godinho et al. mutated Snf7 and discovered impaired molecular trafficking of capsular and melanin components, and as such, the mutant displayed impaired polysaccharide secretion, capsule, and melanin formation, especially at physiological temperature with a complete loss of virulence in an intranasal model of murine cryptococcosis [106]. In addition, the depigmentation of the var. gattii appeared more intense in this mutant than the var. neoformans as temperature progressed from 30 to 37oC in either Niger or L-DOPA agar [106]. Furthermore, this mutant showed defective growth at pH > 7.5 and in the presence of LiCl2 solution with concomitant reduction of Rim101 expression; however, there is no significant effect on the phospholipase B (PLB) and urease activities [106].
C. neoformans produce melanin to protect and elongate propagules life span [107] and to survive antifungal/antimicrobial effects [108,109], oxidants [110], UV-light, engulfment/phagocytosis (camouflage) [98,111], heat (42 – 47oC)/cold (-20oC) [112], chelators/heavy metals [113,114], acid hydrolysis [102,115], and fungal cell wall denaturants [116]; however, melanised C. neoformans are susceptible to melanin-binding fungicidal like trifluoperazine [117]. Very importantly, melanin can sequestrate microbicidal peptides and reactive oxygen species generated by the macrophages against the internalised/phagocytosed C. neoformans, thereby promoting survival and pathogenesis [118].
Interestingly, C. neoformans strategically refused to cluster the Cu/Zn-Sod (encoded by the Sod1 gene) together with melanin produced from laccase activity (encoded by Lac1 and Lac2 genes). The two components are actively involved in oxidative stress response and are induced by exogenous copper, iron, and calcium but are repressed by glucose, nitrogen, and high temperature [73,119]. While Sod1p is restricted to the membrane lipid raft (enriched with sphingolipid and ergosterol), laccase is shuttled to the cell wall, where melanin is produced [120]. Therefore, melanin performs the first-line defence against the cell wall oxidants, while Sod1p is positioned as a second-line defence against the cell membrane oxidants.
Under the glucose-repressed Lac1/Lac2 transcription regulatory factors, C. neoformans expresses an iron-containing laccase enzyme similar to the copper-containing phenol/diphenol oxidase [121,122]. This enzyme is an analogue of tyrosinase in higher animals, which converts various exogenous phenolic substrates to pigmented compound, melanin, via auto-polymerization of quinine-like molecules [89,121]. The Lac1 gene seems more vital than the Lac2 transcript in laccase production [123]. The Lac1 gene is expressed as laccase, while the second laccase (75% homology) is produced from the adjacent Lac2 gene [123]. Four major transcription factors, Bzp4, Usv101, Mbs1, and Hob1, have been identified to be involved in the induction and regulation of the Lac1 gene [124]. Low glucose level facilitates cytoplasm-nucleus trans-localisation of Bzp4 and Usv101; and, together with the residential nuclear transcription factor, Mbs1, initiate the transcription of the Lac1 gene [124]. Conversely, Hog1 provides a repressive regulation of Bzp4 and Lac1 to coordinate melanin production. The Lac1 expression was particularly shown to be upregulated in Δssk1, Δhog1, and Δskn7 mutants [81]. In the early investigation, Snf5 and Mbf1 have been implicated in regulating laccase transcription, poor growth in non-glucose media, bilateral sterility, and mating defect [125] (for details on the phenotypic display of different cryptococcal cell mutants, check Supplementary 1). Also, because melanin production is anchored by the cell wall chitin and governed by the cytoplasmic ionic homeostasis, Chs3, Ccc2, and Atx1 have been further identified as important transcripts for melanisation [125].
Two oppositely regulated signalling pathways govern melanin production – cAMP/Pka and Hog1 [32,84]. Low glucose concentration activates cAMP/Pka-dependent pathways while repressing the Hog1 signalling pathways. This crosstalk significantly induced the Lac1 gene for melanin production. In serotype A, deletion of the Hog1 gene effectively restored melanin production in hitherto non-melanised Δgpa1, Δcac1, or Δpka1 mutant within 2 days in 0.1% glucose media. However, Hog1 seemed not to have a significant effect on the melanin production in serotype D because mutation of the Hog1 gene failed to restore melanin production in Δpka2 mutant [32]. Because of the close association of Hog1 with the cAMP/Pka signalling cascade, it can be deduced that Hog1 gene expression may have negatively hindered Pka downstream activity in melanin production. From the two-component system of the Hog1 pathway, deletion of Tco genes did not contribute significantly to melanisation; however, Δtco1 or Δtco1Δtoc2 mutant showed enhanced melanisation at 37oC even in up to 2% glucose media, and this melanin content was considerably higher than the Δhog1 mutant [30]. This shows that Tco1 kinase is a key repressor of Lac1 expression, just like Ssk1, Pbs2, Hog1, and Skn7 proteins but complementing the Δtco1 mutant (Δtco1::Tco1) reduced the melanin to the wt level.
The Ypd1 is also a component of the Hog1 pathway, which regulates melanin and capsule formation. At 0.1% glucose, Δypd1Δhog1 mutant produced a highly significant level of melanin just like Δhog1, Δssk1 and Δskn7 mutants at 30oC, and at 37oC, only the Δypd1Δhog1 and Δskn7 retained the melanin [82]. Similarly, Bahn et al. reported comparable melanin formation in the wt, Δhog1, Δpbs2, Δssk1, and Δskn7 mutants at 37oC [30]. In 1.0% glucose medium, Δssk1, Δhog1, and Δskn7 mutants melanised as wt, but all mutants, including the wt, failed to retain this melanin at 37oC. Surprisingly, the deletion of Ypd1 represses melanin formation in the Δhog1 background mutant irrespective of the temperature [82]. Contrary to this, Δhog1, Δpbs2, Δssk1, and Δskn7 mutants still retained pigmentation in 1.0% glucose media at 37oC while melanin formation had already been lost in the wt and reconstituted Δssk1. Yet, in 2% glucose media, pigmentations are still found in these same set of mutants with significant melanisation in the Δskn7 than the rest of the mutants [30]. Because of the regulatory effect of Hog1p on the expression of Mbs1, then Song et al. showed significant melanin and capsule defect in Δcac1 compared to Δmbs1 mutants at 37oC, notwithstanding Δmbs1 mutant still showed a reduced virulence with low degree tissue fungal burden and titanisation compared to the wt or complemented Δmbs1 mutant (Δmbs1::Mbs1) [126].
While it is reasonable to summate that increasing cAMP production would elevate melanin synthesis, D'Souza et al. showed that high levels of cAMP led to overexpression of Pka1 and repression of melanin production as compared to the wt [83]. However, capsule production appeared highly induced [83]. This means that capsule production may be more critical to virulence than melanin. Though the same upstream regulatory factors may control melanin and capsule productions, different transcription factors are involved in the terminal responses. Evidence exists that melanin is produced in the infected tissues but not as much as produced in vitro; nonetheless, the fact that larger capsules have been produced in the infected tissues than culture media means that capsules are more involved in the C. neoformans in vivo virulence and pathogenesis than melanin. Notwithstanding, an appreciable level of melanin is necessary for effective virulence, tissue invasion, antifungal resistance, and macrophage survival within the infected tissues [127,128].
Ironically, the presence of melanin in the infected tissues has been proposed with uncertainty. Liu et al. discovered that laccase-dependent catecholamine oxidative products such as pyrrole-2,3,5-tricarboxylic and pyrrole-2,3-dicarboxylic acids rather than melanin might be produced in the mouse brain during infection to induce oxidative cytotoxic effects within the infected tissue [129]. The accrued evidence came from the work of Ito et al. that dopamine-o-quinone is one of the reactive catecholamine oxidative intermediates, which are formed during the synthesis of melanin, and this intermediate strongly attacks the sulfhydryl groups of protein cysteine [130] and can be further oxidised to pyrrole acids in the presence of alkaline peroxide [131,132]. Therefore, this contrary discovery means that if melanin is actually produced during pathogenesis, then it may be stringently controlled to induce moderate pigmentation against oxidative damage. At the same time, most laccase products are diverted into making heterologous quinone-like derivatives, which are cytotoxic catecholamine oxidative intermediate products.
Mutation of the Pka1 gene in C. neoformans usually leads to sterility and avirulent because the strain failed to produce melanin and capsules; however, overexpression of Ste12α in the Δpka1 mutant restored the mating but was unable to restore the virulence of Δpka1 mutant [83]. Contrarily, the mutation of the Pkr1 gene, encoding the regulatory Pka subunit protein, failed to significantly affect the melanin production in serotype A MATα, unlike the mutation of Gpa1. Interestingly, it was shown that the Δpkr1Δgpa1 double mutant produced a comparable level of melanin to the wt better than the Δgpa1 mutant [83]. This indicates that melanin and capsule production is under the multifactorial transcription factors downstream of the Pka protein. Furthermore, mutation of the Crg1 gene has been shown to enhance melanin production in the MATα strain, which depends on the Ste12α expression. This mutation directly increased the virulence in the animal study in a Cpk1-independent manner [133].
Luberto et al. exposed the vital role of the Ipc1/Aur1 gene encoding inositol-phosphorylceramide synthase 1, which catalyses the formation of membrane-associated complex sphingolipids called inositol-phosphorylceramide (IPC) and diacylglycerol (DAG) from phytoceramides and phosphatidylinositol (PI) in fungi. The DAG is an essential second messenger of mitogen and protein kinase c (Pkc) activation. Under Pgal7-regulated inducible expression, it was shown that while the glucose-repressing condition reduced melanin production by 60%, the galactose-inducing condition increased melanin production by 80% [134]. Furthermore, this repressive condition impaired the virulence, survival rate, in vivo growth, replication, and cellular diffusion of infectious strain H99 in immunocompromised animal and macrophage-like cell line models [134]. Again, because of the increased melanin production in the hog1 deleted background mutant, Ko et al. discovered that upregulation of Ipc1 in the Δssk1 and Δhog1 mutants might synergistically enhance melanin production in C. neoformans [81].
3.5. Heterokaryotic Mating/Conjugation, Filamentation, and Sporulation/Haploid Fruiting
Like melanin and capsule formation in serotype D, Hog1 is less involved in mating in serotype D. Mutation in the Hog1 gene enhances mating filamentation and cell fusion in serotype A. Mating formations are strongly induced because mutation of the Hog1 gene re-activates yeast Ste4 analogue Gpb1 MAPK pathway to produce more pheromones from the induction of Mfα1 gene hence mutation of Hog1 gene will not improve mating defect in Δgpb1 mutant unlike Δras1, Δgpa1, Δcac1, and Δpka1 mutants [32]. Notwithstanding, the Δras1 mutants displayed defective mating, reduced in vivo viability and virulence with growth defect at 37oC [135]. The appearance of prototrophic progeny from unilateral mating of the two complementing auxotrophic mutants showed that Ras1 is required for cell fusion and maintenance of heterokaryotic/diploid cell formation [135]. The replacement of the H99 wt Ras1 gene background with a site-specific mutated ras1Q67L tagged as the dominant gene evidently induced excessive haploid filamentation after 4 weeks of incubation at 25oC [135]. So, besides controlling cell fusion and dikaryotic cell formation, Ras1 also promotes haploid fruiting filamentation under nutrient starvation.
The second Ras protein, Ras2p, is expressed at a low level, and its absence caused no defect in filamentation, cell differentiation, and virulence factor production [136]. Furthermore, the double mutant Δras1Δras2 strain exhibited poorer temperature-dependent growth than either of the single mutant, and the overexpression of Ras2 entirely suppressed the mating defect but partially suppressed the actin polarisation defect in the budding of Δras1 mutant at higher temperature [136]. The observation that defects in mating, filamentation, basidia, and basidiospores formation in Δras1 mutant can be partially suppressed by exogenous cAMP or fully suppressed by the overexpression of Gpb1 (a pheromone-sensing MAPK signalling element) under the control of serotype D Pgal7 in glucose/galactose enriched media showed that Ras protein functions upstream of pheromone-response MAPK signalling pathway and this is the reason Ras1 activation will not suppress the mating defects in Δgpa1 and Δgpb1 mutants. Ras1 coordinates the downstream activation of Gpb1 and Ste12α to regulate haploid fruiting, but then, constitutive activation of Ras1p will not induce filamentation/haploid fruiting during starvation in Δgpb1 and Δste12α mutants except if each of these genes is re-introduced [135]. The fact that cAMP and Gpb1 failed to suppress the growth defect at 37oC in Δras1 mutant showed that Ras1 regulates the vegetative growth of C. neoformans at 37oC in a separate pathway independent of cAMP, osmotic-rescue sorbitol, and MAPK pheromone sensor [135].
Reconstituting Δras1 mutant (Δras1::Ras1) restored the virulence and induced the haploid fruiting (cell differentiation), filamentation, agar invasion, and sporulation in nitrogen-limited media. Selectively, exogenous cAMP that restored mating in the Δgpa1 mutant only partially suppressed mating in the Δras1 mutant and restored agar adherence as well in this mutant (agar adherence by cAMP-dependent pathway). However, because the dominant ras1Q67L allele induced a mixture of yeast and hyphae filament at 25oC, only a mutant of this dominant allele could adhere, invade, and retain the invasion in the presence/absence of cAMP (cAMP-independent pathway) [135]. This shows that cAMP, the primary target of Gpa1, is an auxiliary downstream effector regulator of Ras1 function because Δras1 mutant apparently showed no significant defect in melanin and capsule production at 30oC in an iron-limiting media when compared to the wt. After all, evidence shows a parallel expression pattern of ras-dependent and cAMP-dependent genes in a DNA microarray analysis [135,137].
C. neoformans produced G proteins in the form of 3Gα (Gpa), 1Gβ (Gpb), and 2Gγ (Gpg), which are needed for pheromone production, mating, and virulence. There are two putative mating-specific-pheromone-receptor encoding genes in C. neoformans – Cpr1 and Cpr2, which are homologues of yeast Ste3α/a. During the pheromone-sensing event, pheromone-activated receptor Cpr1α interacts with Gpa2 or Gpa3, and the Gpa2 interacts with the heterotrimer G protein (Gpb1-Gpg1/Gpg2) and Rgs1 domain of Crg1. However, coupling Gpa3 to Crg1, which does occur during pheromone induction, may not be via Gpb1 [87]. Though all the G proteins interact with Ste3, Gpb1, and Crg1 in vitro, only Gpa2 has a strong in vivo interaction. Crg1, on the other hand, can act as a pheromone desensitiser via Rgs protein to disengage Gpa2 or Gpa3, thereby inhibiting mating/cell fusion. The Gpa2 GAP activity is greatly enhanced by Crg1 for conserved active involvement in pheromone response and mating. No observable pheromone response when the MATα wt, Δgpa2, Δgpa3 or Δgpa2Δgpa3 mutant was crossed with MATa Δcrg1 mutant; however, shmoos cells with conjugation tubes were observed in the MATa Δcrg1 mutant crossed with MATα Δgpa3 or Δgpa2Δgpa3 mutant. This shows that the latter mutants produced pheromones, and the significantly high pheromone production by the MATα Δgpa2Δgpa3 is similar to MATα Δcrg1 mutant that stimulated a large number of shmoos cells [87]. Further analysis showed the importance of Gpa3 in pheromone response (conjugation tube formation), while Gpa2 is important in pheromone production, reaffirming the coupling of Gpa2 to Gpb1. Unlike the MATα strain, the phenotypic changes accompanying the deletion of Gpa in the MATa strain are less conspicuous; however, this may affect the conjugation tube stimulated from the MATα Δcrg1 when crossed with MATa Δgpa2, Δgpa3, Δgpa2Δgpa3, and the wt. Nevertheless, in general, the conjugation tube induced in the Δcrg1 mutant, whether MATα or MATa, is rationally higher when crossed with either MATα Δgpa2Δgpa3 or MATa Δgpa2Δgpa3 mutant.
The significant regulatory contribution of Gpa2 and Gpa3 to mating, dikaryotic filamentation, basidia, and basidiospores increased considerably compared to the Gpa1. Therefore, mating mutant, either MATα or MATa, with the deletion of Gpa1, significantly increased mating with the wt MATa or MATα, respectively. However, unilateral mating seemed to be attenuated with MATα/a of Δgpa2Δgpa3 x MATa/α of the wt respectively but completely abrogated in bilateral mating MATα Δgpa2Δgpa3 x MATa Δgpa2Δgpa3 [87]. On the other hand, the unilateral mating of MATα Δcrg1Δgpa2 or Δcrg1Δgpa3 with the wt MATa is also defective, unlike the unilateral mating of MATα Δcrg1 with the wt. There is an in vitro interaction of Gpg1 and Gpg2 with Gpb1 to exercise pheromone response. No pheromone response was detected in the MATα Δgpg1, Δgpg2, and Δgpg1Δgpg2 mutants when crossed with MATa Δcrg1 mutant; however, the formation of conjugation tubes appeared more promising on the MATα Δcrg1 mutant when crossed with MATa Δgpg1 and Δgpg2 [87]. In addition, unilateral mating of MATα/a of Δgpg1 was attenuated while MATα/a of Δgpg2 and MATα Δgpg1Δgpg2 were utterly sterile, just like Δgpb1.
In addition, the pheromone-responsive Cpk1 MAPK inherently enhances mating in the MATα Δcrg1 mutant [133] because Crg1 is a negative regulator of mating and when studied in a confrontational assay with MATa Δssk1, Δpbs1, and Δhog1 mutants produced more pheromones than the wt, but with Δskn7 mutant, no filament was induced [30]. Similarly, bilateral mating between the MATα and MATa mutants of Δssk1, Δpbs2, Δhog1, and Δskn7 was significantly enhanced in V8 media at pH 5.0 incubated at 25oC in the dark and even when co-cultured with Δcac1 mutant [30]. This shows that transcription factors of the Hog1 pathway are potent repressors of mating pheromones.
Confrontational assays are best done in the dark because light induces some mating repressing genes such as Crk1. C. neoformans senses light via the putative membrane photoreceptor, Cwc1p (a blue light chromophore-binding protein) that conjugates with another transcription factor, Cwc2p, to form an oligomeric protein that migrates to the nucleus to induce various light-dependent transcription factors that majorly suppressed pheromone production and filamentation. The Crk1, for example, is a protein kinase that is induced in the light to suppress Mat2, Znf2, and Sxi1α that are involved in bisexual mating; however, its effect in monokaryotic fruiting is ambiguous because unisexual mating remained attenuated in the Δcrk1 mutant and even when overexpressed [138].
Though the G-protein β-subunit (Gpb1) and MAP kinase (Cpk1) are not mating-strain specific factors [139,140], unlike Sxi1α, Ste11α/a, Ste12α/a, Ste20α/a, and Mfα/a, which are mating-type specific [26,141] (check Supplementary 1 for mating-type specific factors) but mutation of Gpb1 completely abrogated mating while its overexpression induced conjugation tubes and cell fusion through the regulation of pheromones secretion via the upstream regulation of Cpk1 MAPK cascade event irrespective of the mating-type. This regulation is, however, independent of the Ste12α factor [140] but highly dependent on Ste20α expression [142]. In fact, overexpression of the G protein β-subunit Gpb1 could not rescue the mating defect of Δste20α [142,143,144].
Furthermore, Ste50p is a vital adaptor protein for Ste11 autophosphorylation to regulate pheromone-dependent Cpk1 MAPK sexual reproduction in any serotype of Cryptococcus and other yeasts. However, unlike other yeasts, Ste50 expression is not involved in any stress response, titan cell formation, and virulence factors as controlled by Hog, Ras, and cAMP/Pka cascade pathways in C. neoformans [145]. Therefore, it is not surprising to observe cell fusion and pheromone-inducing mating defects in Δste50 mutant due to the repression of the Mfα1 pheromone gene. Not only this but even the mating enhancing Δcrg1 mutant also showed severe impairment in pheromone production and filamentation when Ste50 (the same for Aca1) is deleted in this background mutant [145]. This means that neither MATα Δste50Δcrg1 nor MATa Δcrg1 mutant will form any conjugation tube when confronted.
In the same way, none of MATa Δste50Δcrg1 and MATα Δcrg1 mutants will form any conjugation tube. Therefore, in the pheromone-response MAPK pathway, Ste50 appears to be a downstream functional protein to Crg1, while Aca1 acts as an upstream regulatory protein (check mitogen activation pathways in Supplementary 2). In addition, the deletion of Ste50 fails to affect pheromone-independent enhanced melanin production from Δcrg1 mutants.
The Gpb1 is another pheromone-sensing transcription factor regulating mating and haploid/monokaryotic fruiting via the Cpk1 MAPK signalling cascade. The Gpa1 senses the nutrients to regulate mating, filamentation, and virulence via the cAMP signalling cascade. No wonder the pre-suppose phenotypic defects of Δgpa1 mutant are suppressed by cAMP cascade events [84]; however, virulence expression (melanin and capsule production) is independent of the Gpb1 factor [140]. Contrarily, the Gα-Gpa1-cAMP regulatory cascade cannot suppress the mating defect incurred from gpb1 mutation. This indicates the separate role of Gpa1 and Gpb1 in C. neoformans. Furthermore, overexpression of the Gβ-subunit, which associates with the γ-subunit of Gpb1, enhances conjugation tube formation in MATα and MATa mating strains [139]. This confirmed that the Gβγ-complex subunit of the Gpb1 protein constitutes an important activation factor in mating, but this may probably be antagonised by the Gα-subunit of the Gpb1.
Lengeler et al. overexpressed Gpb1 and Ste12α in a serotype A MATa and discovered that Ste12α induced filament formation while Gpb1 failed to induce filamentation [26]. This shows that serotype A MATa might probably be a sterile mating strain. Nevertheless, the existence of diploid/aneuploid hybrid serotype AD might have demonstrated that fertile serotype A MATα with MATa mating type may naturally exist, albeit with a very low mating efficiency. This is more evidenced because analysis showed that the hybrid AD is heterologous with MATα and MATa loci derived from parent serotypes D and A, respectively [146]. Naturally, mating still occurs in serotype D MATα and MATa mating types; however, most isolated serotype A, whether clinical/environmental, are MATα. Being the most prevalent aetiology of cryptococcosis, virulent serotype A has been proposed as an asexual propagule because the serotype A MATα can mate with serotype D MATa to produce serotype AD; however, the rare serotype A MATa cannot cross-fertile with serotypes A and D MATα mating-strain. In fact, Lengeler et al. could not recover any fertile serotype A MATa mating strain even from the self-fertilised serotype AD basidiospores [146].
In another scenario, overexpression of Ste12α could not rescue the mating defect in Δgpb1 mutant [29] in the same way overexpression of Gpb1 could not rescue the mating defect in Δste20α [142]. However, overexpression of MAP kinase Cpk1 or Ste11α restored the mating defect not only in the Δste20α but also in the Δpak1 mutants [142]. Hierarchically, the flow of MAP kinase activation proceeds from the inducing activity of Gpb1 and the terminal activity of Ste12, Gpb1→Ste20α→Ste11α→Cpk1→Mat2→Ste12 (check mitogen activation pathways in Supplementary 2). This explains why overexpression of Cpk1 or Ste11α can rescue the mating defect in Δste20α mutant because they are functional downstream PAK kinases to Ste20α; however, monokaryotic fruiting defect in MATα Δste20 and Δpak1 mutants cannot be rescued by the overexpression of either Cpk1 or Ste11α MAP kinase but Ste12α [142]. This shows that Ste12 is a multifunctional transcription factor robust enough to circumvent the mating roles of Ste20 and Pak1 and relay the information from the pheromone-sensing factor Gpb1 to the MAP kinase Cpk1 to promote mating and monokaryotic fruiting.
Further works showed that “Ste” transcription factors were majorly involved in MAP kinase signalling events to regulate haploid fruiting and filamentation on the filament agar but not primarily engaged in mating and virulence [28,29]. Contrarily, Chang et al. showed that serotype D Δste12α mutant was marked with a lower virulence as compared to the wt; however, overexpression of this gene enhances hyphal projection with fertile basidiospores in serotype D MATα Δste12α when co-cultured with MATa on a V-8 juice agar [27]. This mating expression displayed by the Δste12α progeny is similar to the wt serotype D MATα X MATa mating progeny but with significantly reduced viable basidia [27]. Nevertheless, the deletion of Ste12α seemed not to impede hyphal formation and mating in the serotype D strain, but haploid fruiting is usually impaired, just as found in serotype A.
cAMP orchestrates the activation of Pka1 to regulate mating and filamentation. Generally, Δgpa1 and Δpka1 mutants are sterile and failed to produce basidia/basidiospores when co-cultured with serotype D MATa strain; however, exogenous cAMP restored the mating defect in the Δgpa1 just like how the virulence would have been restored. Although the Pka is a downstream functional protein to cAMP, exogenous cAMP could not restore mating defect in Δpka1 [83]. Notwithstanding, because Pka targets Ste12α expression, among others, to induce filamentation, then overexpression of Ste12α was shown to restore mating and dikaryotic/haploid filamentation with preponderant basidiospores in serotype A MATα Δpka1 mutant co-cultured with serotype D MATa. Emphatically, the interplay of Pka1 and Ste12α to initiate filament differentiation is well characterised in the nitrogen-limiting filamentation media. Compared to the wt, the overexpression of Ste12α in Δpka1 mutant produced elongated filaments in the nitrogen-limiting filamentation liquid culture that failed to form basidia/basidiospores; however, overexpression of Ste12α in the wt had less elongation but formed basidia [83]. Thus, this filament differentiation may, in addition to the Pka1, require further environmental cues to produce basidia. On this note, the acquisition of iron from the iron-rich medium may pose ionic stress in C. neoformans, which induces several genes encoding calcium-calmodulin-calcineurin signalling components to facilitate cell wall integrity, growth at 37oC, mating, haploid fruiting, and virulence [88,147].
The regulatory (R) and catalytic (C) subunits of Pka are sequential and functional proteins downstream of Gα-Gpa1-cAMP-Pka(R)-Pka(C) cascade events to promote differentiation and virulence in C. neoformans. Mutation of Pkr1 showed no effect on the mating in the wt strain but restored mating in otherwise non-mating serotype A MATα Δgpa1 mutant when co-culture with the serotype D MATa mating strain; however, Δpkr1Δpka1 double mutant failed to restore mating [83]. This means that Pka(C) is the last functional protein in this cascade event through which other regulatory factors will be activated for mating and virulence production, including the regulatory feedback inhibition of cAMP production. Literarily, exogenous glucose increases cAMP production, which inhibits Pkr1 expression to activate Pka. However, at the threshold level, Pka inhibits further production of cAMP to keep the intracellular level of this second messenger. Thus, mutated pka1 strains accumulated cAMP production in the glucose-rich media, but the Δpkr1 mutant negated the overproduction of intracellular cAMP to prove that the Pka autoregulates cAMP production, possibly by activating the phosphodiesterase, Pde1 [83], to hydrolyse cAMP to AMP [148].
The Tco transcription factors are expressed as multiple regulatory sensor kinases such as Tco1, Tco2, Tco3, Tco4, Tco5, and Tco7. These sensor kinases control the response regulators such as Ssk1 (an upstream signalling component modulating the Pbs2-Hog1 MAPK-dependent phenotypes) and Skn7 (a Pbs2-Hog1 MAPK-independent regulator) [149]. Among the Tco kinases, Tco1 and Tco2 are the most significant kinases. Surprisingly, there is increasing evidence of the regulatory effect of Tco kinases in mating. The majority of the Δtco mutants displayed normal mating like the wt. However, mating was found completely defective in the bilateral mutant crossing of Δtco1 (Δtco1 MATα x Δtco1 MATa), and a reduced mating was also observed with a unilateral mutant crossing of Δtco1 (Δtco1 MATα x MATa) [30]. Further investigation showed a relatively efficient dikaryotic cell fusion in bilateral mutant crossing, though with heterologous sizes and uneven filamentation compared to the wt or unilateral mutant crossing [30]. This means that Tco1 is highly essential for cell viability and dikaryotic stability but not necessarily for cell fusion. Mating is also enhanced in the Δhog1, Δpbs2, and Δssk1 mutants, making them negative modulators of mating.
C. neoformans prefer mating in an ambient environment with about 0.036% CO2 because higher CO2 (4 – 10%) naturally inhibits mating in cryptococcal cells by preventing the expression of Mfα1; however, deletion of the Can2 gene rescues this mating inhibition [150]. Similar to Can2 gene deletion, mating is highly promoted in Δhog1 and Δcrg1 mutants in the ambient environment but less efficiently, and deletion of the Hog1 gene partially restores mating in the presence of high CO2 while Δcrg1 mutant failed to engage in bilateral mating in the presence of high CO2 [32,150].
C. neoformans deploy carbonic anhydrase (encoded by Can genes) to harness the CO2 as HCO3— and H+ in the presence of water molecule, and the HCO3— are used in regulating other transcription factors such as Cac1 or metabolic reaction involving carboxylation such as lipogenesis [150]. Similar to serotype A MATα x MATa, high CO2 disrupted the bilateral mating of MATα Δcan1 x MATa Δcan1 and unilateral mating of MATα Δcan2 x MATa but not in the bilateral mating of MATα Δcan2 x MATa Δcan2 [150]. Despite the good state of the dikaryotic cells in the MATα Δcan2 x MATa Δcan2 crossing filaments, the basidia and basidiospore formations are highly impaired, which means that Can2 is highly important for sporulation. Being the primary encoding transcript, accumulation of HCO3— by Can2 expression caused by the high level of CO2 was proposed as the basis of mating inhibition but not the H+. So, the absence of Can2 drastically reduces the cytoplasmic HCO3— to the level only generated nonenzymatically, probably via aquaporin/water channel (encoded by Aqp1/Aqy gene) hydration of the diffused CO2, and this can support vegetative growth and promote mating in the bilateral crossing. Further analysis showed that high CO2 arrests the cell fusion in the wt while a normal cell fusion was displayed in the Δcan2 but with impaired sporulation [150]. Just as high CO2 rescues growth and mating defects in Δcan2 and Δcan1Δcan2 mutants, the same way exogenous cAMP (≈1.0 mM) rescued the mating defects in Δgpa1, Δaca1, and Δcac1 mutants; and again, as accumulated HCO3— disrupted cell fusion in the wt or basidia formation in the Δcan2 mutant, the same way the excessive supply of exogenous cAMP (10 mM) will arrest the cell fusion [150]. So, it is not unusual to predict the inevitable involvement of a Can2-mediated HCO3— dependent Cac1p activation for basidia, basidiospore, and mature sporulation in C. neoformans in the same way it was discovered in C. albicans [151].
On the contrary, the effect of CO2-sensing linking to pheromone production is less pronounced in serotype D mating compared to serotype A. Pheromone and mating defective serotype D mutants such as Δste11, Δste7, and Δcpk1 highly exhibited defective unilateral mating and pheromone production in low and high CO2 levels [150]. Thus, mating could be pheromone-dependent like MATα-MATa bisexual development and pheromone-independent like MATα unisexual development. Mat2 is a high-mobility group (HMG)-box homologue of Prf1, recognising various PREs to promote bisexual reproduction of the fungi. It is a negative regulator of unisexual development, and its deletion favours hyphal formation and monokaryotic filamentation under the regulation of Znf2 and Cna1/Cnb1 [152]. This condition is favoured by high copper, calcium, and temperature at 37oC but not magnesium, zinc, and iron. The presence of FK506 (Cna1/Cnb1 inhibitor) and bathocuproine disulfonate (BCS) (copper chelator) will prevent hyphal formation.
Transposable genomes and transgene repeats do cause genomic instability and mutation during meiosis; hence C. neoformans employs RNAi silencing pathways using a quelling model to defend the genome against these transposons. Among the conserved regulatory genes for this purpose are Rdp1, Ago1, Dcr1, Znf3, Qip1, Cpr2, and Fzc28. All these transcription factors favour sex-induced pheromone-dependent and mitosis-induced silencing except Cpr2 and Fzc28, which are not involved in yeast-hyphae formation during mitosis-induced silencing [153]. Transposon interacts with the DNA template during replication as a tandem to induce RNAi, which begins with the deployment of Qde3 to resolve the secondary structure of the tandem, promote the transcription, and enable the formation of ds-genome by the Qde1. The duplex genome, made up of passenger and guide strands, is shortened to 21 – 25 nucleotides siRNA by the redundant dicer activity of RNase III encoded by the Dcl1 and Dcl2 genes [154]. As a result, the passenger strand is nicked and degraded by the slicer and Qip activities of Qde2.
In contrast, the guide strand, by complementary pairing, identifies other homologous mRNA and targets them for Qde2-dependent sequence-specific degradation [155]. This degradation is orchestrated by the binding of Ago1p to the siRNA duplex to activate the RNA-induced silencing complex (RISC) [156]. By so doing, C. neoformans maintain genomic integrity during sexual development and yeast-hyphae formation (vegetative growth) at the post-transcriptional level and exclude the interfering short-sequence RNAs or other interfering transposons.
3.6. Cell Wall and Membrane Integrity: Sensitivity to Temperature, Radiation/Light, Salinity, pH, Antifungal, Genotoxicants, Reactive Radicals (Oxidative and Nitrosative Stress), and Quorum-Sensing Molecules
The cell wall components are majorly glucan fibrils, chitin, and melanin. The unique biochemical process and synthesis at the cell wall have made it a potential site for drug targets against cryptococcal cells. Cell wall integrity is highly important to environmental and common stress survival, adaptation, and development. It has been earlier confirmed that 1 M of NaCl was strong enough to disrupt cell wall integrity and impose stress on the cell membrane, thereby reducing the capsule size [157] (for details on the phenotypic responses of different mutants of Cryptococcus against various quantitative osmolytes and oxidants, check Supplementary 3). The cell wall is the first point of contact to various environmental effects (Figure 1), and it is enriched with various sensory proteins to relay the message for the recruitment of transcription factors. These factors, with other messengers, induce gene expression for nutrient assimilation, environmental adaptation, and survival. The Hog1 gene is crucial to cellular response against various environmental cues such as temperature, radiation, oxidative stress, and salinity. This gene island encodes p38-like MAP kinases known as Hog1 MAP kinases in the yeast [158].
Different serotypes shared the same conserved structural functions of the Hog1 gene yet displayed distinctive roles in response to temperature. Serotype A Δhog1 mutant was sensitive to peroxide and temperature at 40oC but not at 37oC or 30oC; however, no peroxide and temperature sensitivity growth defect in serotype D Δhog1 mutant [32]. The Δhog1 mutants of the two serotypes were sensitive to UV radiation between 480 – 720 J/m2, and more than 1.0 M chloride salts are needed to significantly alter their growth [32] (for details on the phenotypic responses of different mutants of Cryptococcus against various quantitative external factors, check Supplementary 3).
In addition to the Hog1 pathway, C. neoformans synergistically harnesses arrays of other signalling pathways such as Ras/Cdc, cAMP/Pka, Cam/Cna, Pkc/Mpk, Rho/Mpk, and Rim against various environmental and common stress agents (reviewed in [149]). There is evidence of interconnections among these signalling pathways. For example, methylglyoxal (MG) is a ubiquitous reduced form of pyruvate in fast metabolic cells, imposing stress against C. neoformans. Mutants lacking Hog1 or any cAMP/Pka pathway components are variously sensitive to MG, but Δras1 mutants showed no observable changes against this stress-inducing intermediate metabolite. Further analysis showed that tco2 was downregulated in mutants lacking Hog1 and cAMP/Pka pathways; however, this double-hybrid histidine kinase transcript was slightly upregulated in Δras1 mutants – a condition that conferred a wt resistance to Δras1 mutants against MG effect [137]. In another scenario, C. neoformans may deploy one or two strategic pathways for a particular resistance while other pathways are routed against other stress to avoid futile cycles, metabolic redundancy, and excessive energy dispensary. For example, genotoxic and oxidative resistance is jointly controlled by the Ras and Hog pathways, thermotolerance is controlled by the Ras and Can pathways, osmotic resistance is jointly controlled by the cAMP/Pka and Hog pathways, common and heavy metal stress resistance is controlled by Ras pathway, cell wall stress resistance and integrity are jointly controlled by the Pkc and Ras pathways, and antifungal resistance is jointly controlled by cAMP/Pka, Hog, and Ras pathways in a differentially repressed/induced mode to elevate/depress ergosterol biosynthesis – a condition that respectively facilitates resistance to azole/polyene antifungal agents [137]. Special attention has been drawn to the independent regulation of cell wall integrity by the Aca1 and Ras1 pathways and how the double deletion Δaca1Δras1 mutants usually displayed higher sensitivity to cell wall disruptors compared to Δaca1 or Δras1 [137]. The Δaca1 mutants usually display phenotypic appearances similar to Δras1 mutants but distant from other cAMP family components, Δgpa1, Δcac1, Δpka1, and Δpka2.
Surprisingly, this fungus and other related human pathogenic fungi have developed a plethora of transcription factors associated with each pathway to ensure independent or complementary control to resist the imposing cytotoxic agents. Notwithstanding, mutants of different transcription factors deficient may respond similarly to the same stress agents. This means the involved signalling pathways must have shared some common routes at the upstream/downstream regulatory points. On the other hand, the lack of one transcription factor in a mutant can be complemented by other factors. For example, MG had no effect on the Δras1 mutant because the presence of the cAMP/Pka pathway stimulates the expression of tco2 for Hog1 pathway induction; however, Δaca1, Δgpa1, Δcac1, and Δhog1 mutants are hypersensitive to MG while Δpka1 and Δpka1Δpka2 are slightly sensitive. This is further corroborated by the susceptibility of Δaca1Δras1 and Δcac1Δras1 mutants to MG [137] (check Supplementary 3 for phenotypic responses of different mutants).
Among the well-studied transcription factors that regulate temperature are Ras proteins (Ras1p and Ras2p). In every stressful condition, especially under nitrogen starvation, C. neoformans prefers to express Ras1 while silencing or minimising the Ras2 expression. Most of the Ras-dependent genes of C. neoformans are unique and bear no orthologues with other yeast, just like the cAMP-dependent genes, but among the evolutionarily conserved few genes that are involved in the regulation of Rho-GTPase and activation of Cdc42 via Cdc24 are Pxl1, Rdi1, and Bem3 [137]. Some of these genes have been implicated in the full virulence and gene repair in C. neoformans (for details of cellular events that induced/repressed different transcription factors for full virulence and gene repair in C. neoformans, check Supplementary 2). Through the guanine nucleotide exchange effector Cdc24, Ras1 is activated to mediate thermotolerance and infection in the infected hosts. In terms of the oxidative and genotoxic stress responses, Δcdc24 and Δras1 mutants responded similarly, which supported the downstream roles of Cdc24p to Ras1p [137,159]. The Δras2 mutants, on the other hand, displayed the wt features against most of the stresses, which shows a minor role against stress; however, careful observation showed a slight sensitivity to amphotericin B (AmpB), fludioxonil (FDX), and tert-butyl hydroperoxide (t-BOOH) agents [137] (check Supplementary 3 for phenotypic responses of different mutants to antifungals).
The avirulent nature of Δras1 mutants is basically due to defective growth at 37oC because, together with Δras2 mutant, there are no observable differences in virulence expression compared to the wt [135]. The growth of Δras1 mutants decreased sharply at a temperature higher than 30oC. Still, the mutants were not quickly killed even at 39oC for 24 hours and transferring this mutant into YPD culture at non-permissive temperature survived with efficient growth [135]. Unfortunately, the temperature-dependent growth defect imposed by the deletion of Ras1 (Δras1) appeared irreversible even in the presence of 1 M sorbitol or 1 – 50 mM cAMP unless the mutant is complemented with the wt Ras1 gene (Δras1::Ras1) [135]. Like Δcan1 mutants, Δras1 mutants are also sensitive to immunosuppressive agents FK-506 and CsA at temperatures >30oC because both transcription factors modulate thermotolerance. The Δcna1 mutants are sensitive to NaCl and LiCl, but Δras1 mutants showed a wt phenotype with NaCl and LiCl [135] (check Supplementary 3 for phenotypic responses of different mutants to different salt concentrations). Again, this shows osmotolerance control of Cna1 and Ras1 in different and distinctive pathways. Waugh et al. overexpressed the Ras2 in the ras1 background mutant under the Gdp1 promoter and Ura5 selectable marker and discovered that both transcription factors overlapped in their functions [136]. Furthermore, overexpressed Ras2 can partially restore some of the phenotypic defects associated with Δras1 mutants; hence both may be redundantly functional.
Contrarily, Rho1 wt expression under Gal1 promoter in the C. neoformans Δade2Δura5 auxotrophic mutant showed that the overexpression of this Ras-related superfamily small G proteins had no significant impact on the growth of this mutant at 30 and 37oC in glucose- or galactose-enriched media. Again, this overexpression showed no effect on pneumocandin Bo inhibition and the activity level of cell wall glucan synthase [160]. Attempt to create Δrho1 mutants failed because Rho1 is highly required for growth; therefore, the rho1 gene replacement mutant was created with deregulated rho1E41I allele and discovered that the glucan synthase activity remained unaltered while the mutant failed to grow at 37oC, but this condition could be rescued with 1 M sorbitol at 25oC, and such mutant could survive 37oC thereafter [160]. Furthermore, because Pkc1 is a downstream functional protein of Rho1, any alteration with this transcription factor may nullify the thermosensitivity of the rho1E41I auxotrophic mutant [161].
Similarly, rho1 mutants like rho1G15V survived quite well at 25 and 30oC, even at 37oC, but were hypersensitive to 39oC, which could not be rescued with sorbitol except by complementing the mutated allele. The rho1Q64L, however, showed no significant growth defect at all tested temperatures [162]. These two mutants, including Δrho10, which is nevertheless hypersensitive to 37oC, constitutively activate Mpk1 independent of the heat shock/stress at 24 or 39oC contrary to 39oC-induced Mpk1 activation by Rho1p in the wt, Δrho11, Δrho10Δrho11, and Δrho10::Rho10 [162]. This shows that uncoordinated constitutive hyperphosphorylation of Mpk1 in Δrho10 and point mutated mutants (rho1G15V and rho1Q64L) at 24 and 39oC predisposed these mutants to growth defects observed at temperature >37oC, which means that the Rho transcriptional factors play an essential role in the Pkc1 pathway for cell wall integrity against stress. Moreover, Rho10 and Rho11 may have opposite roles regarding Mpk1 phosphorylation in the Pkc1 signalling pathway. In terms of virulence, all the mutants produced a wt melanin level, but the cell body size and the capsule diameter of the point mutated rho1 mutants are significantly lower than the wt [162].
The Pkc1 encodes protein kinase c with pleiotropic effects on other transcription factors to regulate defence mechanisms against oxidative and nitrosative stress, osmotic imbalance, high temperature, cell wall disruptors, and anti-virulence agents. In addition, it maintains chitin and chitosan localisation within the cell wall and melanin production and attachment. Among the popular putative and probably membrane glycoprotein sensors are Slg1p, Wsc2p, and Mid2p, which initiate the activation of Pkc1 via Ipc1p, which eventually phosphorylates Bck1, Mkk2, and Mpk1 for subsequent activation of different Pkc1-dependent pathways (reviewed in [149]). This activation has been strongly correlated with the presence of oxidative and nitrosative stress agents, but the downstream Bck1, Mkk2, and Mpk1 appeared dispensable because their mutants displayed wt resistance against oxidative and nitrosative agents [163] except against SDS (a membrane destabiliser) and Congo red (cell wall/membrane stressor) [164]. The growth of Δbck1 and Δmkk2 mutants at 30 and 37oC was significantly considerable compared to the wt except at 39oC, where a significant defect was observed. This defect could be restored with 1 M sorbitol [164]. Being a protein kinase, Pkc1 phosphorylates Bck1p (MAPKKK) that activates Mkk2p (MAPKK) in a cascade event, Pkc1→Bck1p→Mkk1p/Mkk2p→Mpk1/Slt2p; however, the activation of Bck1p could also be from Rho1 or Rho11 [149]. This phosphorylation has been observed to be thwarted in Δmkk2 and Δbck1 mutants at a permissive and non-permissive temperature but not in the Δpkc1 mutants [162], which means that activation of Bck1p and Mkk2p is highly important in the Mpk1p phosphorylation against thermal stress; however, Pkc1 seems dispensable [162].
By interpretation, other transcription factors, such as activated Rho1p and Rho11p, are promoted by guanine nucleotide exchange factors (GDP↔GTP) and GEFs (such as Rom1p and Rom2p/Rom20p/Rom21p) may be activating the Bck1p in the Δpck1 mutants in thermal stress response. Surprisingly, repression of Pkc1 by dephosphorylation appeared to favour capsule formation, but capsules are not adequately attached to the cell wall because of the impaired cell wall integrity caused by the Pkc1 dephosphorylation [163]. Therefore, apart from the integral function of the Rho1-Pkc1 cascade pathway to regulate cell wall biogenesis, Mpt5p and Ssd1p are potential alternative transcriptional factors for initiating cell wall biogenesis [164]. Further investigation showed the possible involvement of three GEFs homologues (Rom2, Rom20, and Rom21) and four GTPase-activating proteins, GAPs (Lrg1, Bag7, Bem3, and Rga1), in the regulation of C. neoformans Rho1, Rho10, and Rho11 regulatory pathway for the Pkc1-mediated cell wall biogenesis [164].
In addition, small heat shock proteins (Hsp) are induced due to temperature changes to facilitate thermotolerance (check Supplementary 2 for detailed transcription factors induced/repressed by temperature). Reports showed that Δpka1 mutants are more heat-tolerant than the wt and Δpkr1 mutants. On this point, it was demonstrated that the Δpka1, Δpka2, and Δova1 mutants subjected to heat shock at 50oC for as high as 30 min still survived and remained viable better than the Δpkr1 and the wt when cultured at 30oC in YPD [165]. The Hsp10, Hsp12, Hsp60, Hsp70, Hsp90, Hsp122, Sks2, and Gre2, are particularly under the influence of cAMP, and their expression seems induced in Δpka1 mutants irrespective of the stress or nutrient availability; however, the Pkp1 gene encoding dehydrogenase kinase is negatively controlled by Pka1 expression (as induced by Gpa1 or Aca1 in the cAMP pathway). This scenario was well examined, and discovered that Cac1 is not involved in the repression of Pkp1 [137]. Therefore, an unaltered expression of Pkp1 in the Δcac1 mutants may be the reason Δpkp1 mutants displayed hypocapsulation (less capsulation) similar to Δcac1 mutants while the Δgre2, Δhsp12, Δhsp122, and Δhsp12Δhsp122 are characterised with a wt capsule production. Nevertheless, Δpkp1 mutants deviated from Δcac1 in terms of melanin production. All the mutants of these cAMP-dependent genes displayed wt melanin production in the L-DOPA (3,4-dihydroxyphenylalanine) media fortified with 0.1 – 0.3% glucose irrespective of the temperature, but Δcac1 mutants are hypomelanised [137]. Regarding osmotic stress, cell wall/membrane stress, oxidative stress, genotoxic stress, MG, azoles, and FDX, each of the mutants displayed similar phenotypes as mutants from the cAMP pathway except Δaca1 [137]. This means that though oxidative and thermotolerance genes seem induced in Δpka1 mutants, this condition could not contribute to or improve the attenuated virulence in this mutant.
Together, the basal production of these hypoxic-responsive genes prepares the fungus against temperature, antifungals, and hypoxia. Therefore, the repression of Pkp1 may probably increase the availability of reducing equivalent needed for reductive pathways such as ergosterol biosynthesis, usually induced by the hypoxic condition. However, despite the involvement of cAMP in capsule and melanin production, none of these genes participates in the virulence of C. neoformans [137]. Notwithstanding, each of the mutants (Δgre2, Δhsp12, Δhsp122, Δhsp12Δhsp122, and Δpkp1) are slight to moderately sensitive to heavy-metal toxicity and that Δhsp12, Δhsp122, and Δhsp12Δhsp122 are more sensitivity to AmpB than their corresponding wts, unlike the Δgre2 and Δpkp1 that showed a wt sensitivity level to AmpB under normoxic or osmotic condition. This observation indicated that Hsp12 and Hsp122 expression might be redundant against AmpB resistant [137]. Besides the cAMP influence on these transcription factors, the expression of Hsp12 and Hsp122 seemed to be Hog1-dependent. Complete repression of these transcripts was observed in Δhog1 and Δssk1 mutants, unlike the Δskn7 mutant that showed comparable expression to wt. On the other hand, the basal expression of Gre2 is slightly repressed in the Δhog1 and Δssk1 mutants, but the Δskn7 mutant displayed a wt expression of Gre2 [137].
The Cnn1 gene encodes 34-amino acid helix-turn loosely conserved protein containing 16 tandem copies of tetratricopeptide-repeat (TPR). These polypeptides associate together to form complex regulatory proteins by protein-protein interaction to coordinate different cellular processes. These processes include cell cycle/division, DNA replication, RNA transcriptional repression and splicing, protein trafficking and kinesin-mediated intracellular cargo, signal transduction, stress response, peroxisome and mitochondrion biogenesis, protein kinase R (PKR, a dsRNA-activated kinase) inhibition, and neurogenesis [166,167,168,169,170]. The Cnn1 is a homologue of the S. cerevisiae Clf1 gene [171] and an orthologue of the Crn gene that controls Drosophila melanogaster neurogenesis, normal cell proliferation, and embryo development [172].
In yeast, this protein engages in 5′-pre-mRNA splicing, cell cycle control and progression, and DNA replication (by modulating the activity of Orc2p while initiating DNA replication). Furthermore, as a U2-snRNP component, it associates with Syf1, Syf2, Mod2, and U1-Prp40 to displace the branchpoint binding protein (BBP) known as splicing factor 1 (SF1) and forms a pre-spliceosome complex, which attracts the U4/U6.5 tri-snRNP. An active spliceosome is formed when the incoming U4/U6.5 ribonucleosome displaces the U1-Prp40 to orchestrate the mRNA splicing and cell cycle progression [173]. The absence of Clf1p together with any other TPR-containing proteins such as Prp1p/Zer1p, Prp4p, Prp6p, Prp9p, Prp11p, Prp13p, Cdc16p, Cdc23, Cdc28p, and Nuc2+p in the yeast usually promotes ts mutants, defective poly(A)-RNA nuclear export, G2 cell cycle arrest and S phase delay transition, and G2→M growth transition arrest [171,174].
Intriguingly, a genetic lesion resulting in the deletion of Lys217 residue from Cnn1p of B-4551 serotype A MATα strain of C. neoformans isolated from the chronic granulomatous lesion in the nasal cavity of a cat (feline) is the basis for the temperature sensitivity of this natural strain [175,176]. This strain can only grow to the optimum temperature of 35oC in vitro, characterised by short hyphae but normal melanisation and capsulation; however, the strain failed to cause systemic infection [176]. Notwithstanding, reconstitution of this strain with the wt Cnn1 gene restored the growth at 37oC with systemic infection [175]. The eukaryotic expression of Cnn1 or its homologues is iron-dependent [88], performing different structural and functional cellular activities but related to achieving a common goal in the cell. The Cnn1 gene seems to be specific to C. neoformans because complementing the ΔCnn1 mutant with S. cerevisiae Clf1 restored the thermosensitivity; however, C. neoformans Cnn1 transcript could not rescue the Δclf1 S. cerevisiae mutant [175].
Ras-signalling cascade plays a significant role in cryptococcal thermotolerance. This GTP-activated protein (Ras1 and Ras2) effectively interacts with other effector proteins, such as Aca1, to synthesise cAMP and MAPK signalling cascade components. The Δras1 mutants are viable at 25oC with normal melanin and capsule productions even at 30oC; however, this mutant failed to grow at 37oC, and the in vivo virulence is drastically reduced [135]. Moreover, though Ras1 signal is a Gpa1cAMP-dependent (nutrient-regulated sensor) and Gpb1-MAPK (pheromone-responsive) signalling event yet, exogenous cAMP and expression of MAPK could not restore the thermotolerance defect in Δras1 mutant [135] but overexpression of Ras2 partially suppressed the high-temperature growth defect in Δras1 mutant [136]. Therefore, together with the Cna1 signalling cascade, the thermotolerance ability of C. neoformans is controlled by the Ras1 cascade event but with distinct regulatory pathways. No wonder the Cna1 inhibitors, cyclosporine A (CsA) and tacrolimus (FK-506/Fujimycin/Prograf/FR900506), which are popularly used as an immunosuppressant in organ transplant patients, inhibited the growth of Δras1 mutant even at 30oC [135]. The poor physiological thermotolerance of this mutant and the blocking of the calcineurin pathway complement the lethality of this drug. This calcineurin is a Ca2+-calmodulin-dependent (Cam1) serine/threonine protein phosphatase that activates the nuclear translocation of Crz1p via dephosphorylation to activate other transcription factors against environmental stress and induces virulence expression. Besides, Cna1 expression is induced in high temperature and other stress indicators to activate P-bodies and stress granules RNP transcription factors such as Puf4p, Lhp1p, Pbp1p, Pab1p, and Gwo1p (check Supplementary 2 for events that induce P-bodies and stress granules RNP transcription factor).
Cam1-Cnb1 complex activates the Cna1p to cause the activation of Crz1p by dephosphorylation for nuclear translocation to coordinate cell wall integrity via the expression of Chs5 – 7. Park et al. discovered that deletion of Cna1 or Crz1 repressed Chs5 and Chs6 at 37oC [177]. The Chs7 expression remains unchanged in Δcna1 compared to the wt but significantly reduced in Δcrz1 mutant [177]. Similarly, transcription factors like Puf4p, Pbp1p, Tif3p, Vts1p, and Gwo1p in the P-bodies and probably Lhp1p, Gcd2p, and Anb1p in the stress granules are also activated by dephosphorylation via active Cna1p and all act within the cytoplasm to orchestrate virulence, thermotolerance, and sexual reproduction [177]. Thus, evidence exists that Lhp1p, and yet unknown transcription factors may be an indirect target of Cna1p or are probably activated by PΒ/GS-unrecruited Cna1p; conversely, Puf4p and Pbp1p are directly activated by the PΒ/GS-translocated Cna1p [177]. It is important to note that Lhp1p and Puf4p are located differently but are activated in parallel to Crz1p by Cna1 for additive function in thermotolerance and virulence expression. Strategically, Pbp1p with Crz1p activation seems to perform opposite roles against heat stress because the thermosensitivity of Δcrz1Δpuf4 or Δcrz1Δlhp1 mutants is usually higher than the individual mutants while Δcrz1Δpbp1 mutants lie generally in between the Δcrz1 and Δpbp1 mutants [177]. To bolster the parallel regulation and additive phenotypic effects of Crz1p and these mRNA-binding proteins, every double mutant of Δcrz1Δpuf4, Δcrz1Δlhp1, and Δcrz1Δpbp1 consistently exhibits higher attenuated virulence than their corresponding individual mutants (check Supplementary 1 for detail). Regarding mating and hyphal formation, sexual reproduction is more severely defective in Δcna1 than the Δpbp1 mutants but with marked reduced Mfα1 expression in the Δpbp1 than the Δcna1 mutant during bilateral mating assay [177]. This shows that the phosphatase activity of Cna1p also controls the phosphate level of Pbp1p to regulate pheromone production, sexual reproduction, hyphal formation, and elongation.
The Mga2p is another temperature-regulated transcription activator protein and an orthologue of the components of fatty acid biosynthesis. With WSC domain orthologue Slg1, Mga2p constitutes the cell surface proteins essential for heat-stress perturbation signal control via the Pkc-Mpk MAPK pathway. Apart from these two transcription factors, Pps1, Thr4, Glt1, Dur3, Lys2, Rim15, Pma1, Chs6, Chl1, Clc1, Mdr1, Rds1, and Smg1 are highly involved in the thermotolerance of C. neoformans [178] (check Supplementary 2). These putative proteins are clustered as WSC domain proteins, chitin synthase, trehalose-associated enzymes, glycan-forming enzymes, proteases, amino acid permease and oxidase, multidrug-resistant proteins, pentose phosphate pathway enzymes, DNA helicase, oxidoreductases, peroxidase, and catalase. Specifically, genes involved in ribosomal, amino acids (such as isoleucine or valine) and pyrimidine biosynthesis are repressed at 37oC than 25oC [178], which re-iterates that cell development is not favoured at higher temperatures but rather C. neoformans divert resources to adaptation and survival. Surprisingly, deletion of Ilv2 and Ura5, especially in serotype A, still produced high-temperature growth defects [179], which means stress-accumulated intermediates must have developed at higher temperature in these mutants leading to non-viable cell growth.
Drugs such as CsA, FK-506, and Rapamycin (RPM) are potential antifungals; however, due to their immunosuppressive effect on the calcineurin signal transduction and T-cell activation, non-immunosuppressant analogues are recommended. These drugs have an affinity for Frr1p (a homologue of Fkbp12 prolyl isomerase encoded by the Frr1 gene in C. neoformans), and the drug-Frr1p complex targets Tor1-like kinase to interrupt necessary MAPK needed for cell cycle (G1→S phase) and signal transductions (such as calcineurin signalling event). Therefore, any mutation in either Frr1 and Tor1 will confer resistance against immunosuppressants without affecting the fungi growth, prototrophy, mating, sporulation, cell differentiation, and virulence [180]. Reports have shown that C. neoformans is resistant to FK-506 and its non-immunosuppressive analogue (L-685,818) at 24oC but sensitive at 37oC; even the FCZ-resistant strains are also susceptible at this temperature [181]. The CsA, FK-506, and L-685,818 are active against pulmonary cryptococcosis but not against cryptococcal meningitis. These drugs are relatively bigger to cross the BBB, and their immunosuppression outweighs the in vivo antifungal action at 37oC. This, therefore, calls for further investigation into the use of non-immunosuppressive agents as a potential antifungal.
C. neoformans displays multiple copies of gene-encoding peptidyl-prolyl cis-trans isomerases (PPIases), also known as rotamases/foldases, including cyclophilins A (encoded by Cpa1 and Cpa2), parvulin (encoded by Ess1), and FK-506 binding proteins (Fkbps). These enzymes catalyse the isomerisation of cis-trans peptide bonds preceding prolyl residue and bring about protein folding and conformational change [182,183]. Deletion of Cpa1 alone or with Cpa2 produced ts mutants with attenuated virulence, unlike the Δcpa2 mutants [184]. The double mutant, Δcpa1Δcpa2, is resistant to the CsA effect just like a site-specific mutation on the Cna1p conferred resistance against CsA [33]. This shows a link between the Cna1p and Cpa1p-Cpa2p in the same way Cna1p is connected to Tor1p. Neither Δcpa1 nor Δcpa2 mutant displayed any defect in unilateral mating and formation of filament, basidia, and basidiospores; however, these phenotypic defects are manifested in Δcpa1Δcpa2 in a similar way to Δcna1 mutant but with a bilateral mating defect. Though Cpa1p and Cpa2p may differ in their structure and function, evidence shows synergistic functions may exist between them. From Wang et al., only the Δcpa1Δcpa2 mutant displayed defective capsule and melanin formation in a glucose-limiting medium, and only the Δcap1 and Δcpa2 mutants displayed hypersensitivity to CsA stress with mating defect but not Δcpa1Δcpa2 mutant [184].
The Ess1 gene, on the other hand, was found to be dispensable for cell growth, mating pheromone response, haploid fruiting, and capsule formation; however, Ess1 expression is highly important for melanin and urease activity and for this reason, Δess1 mutants displayed impaired virulence in murine model cryptococcosis [185]. It is important to note further that Δess1 mutants appeared to have delayed growth in standard medium without any PPIase inhibitor. It is very likely that each component of the PPIase compensates for one another in an overlapping function though there may not be a full replacement. Since the viability of these mutants above 30oC is Cna1-dependent, in vitro assays must be maintained below 30oC in these mutants due to ineffective Cna1p. Both Δcpa1 and Δcpa2 mutants are found with Fkbp12 expression, which is insufficient to confer resistance against CsA, and again, Δess1 mutant may have produced Cpa1 and Cpa2 but also insufficient against CsA at 25 – 28oC [184,185]. The Δess1 mutant showed no response to the FK-520 analogue at 25oC (check Supplementary 3 for mutant responses to FK-520). Surprisingly, both works concluded that the reconstitution of the mutants, Δcpa1::Cpa1//Δcpa2::Cpa2 and Δess1::Ess1, appeared incomplete as the phenotypic traits were still less than the corresponding wts.
DNA topoisomerase I, encoded by the Top1 gene in fungi, is an enzyme in genome replication and transcription. This enzyme features a unique fungal pocket sequence region, which is not found in mammalian topoisomerase I. Under the influence of inducible promoter, Del Poeta et al. demonstrated that moderate overexpression of Top1 gene predisposed serotype A strain H99 to heat shock, γ-ray, and camptothecin (a topoisomerase inhibitor) as compared to the isogenic strain [186]; though capsule and melanin production remained unchanged. Previous work to generate S. cerevisiae Δtop1 mutant promoted the resistance of this strain against camptothecin; however, the wt strains were sensitive to this antifungal, possibly by accumulating preponderant levels of cleaved DNA duplexes, which topoisomerase failed to ligate because of camptothecin. This invariably leads to a cytotoxic effect [187,188]. Contrarily, Jiang et al. had shown the essentiality of the Top1 gene for the survival and full virulence of C. albicans wt compared to the Δtop1/Top1 heterozygote mutant [189]. Meanwhile, the impaired expression of this nucleus-located repairing enzyme in C. neoformans serotype A showed no effect on the toxicity of dicationic aromatic compounds (DACs) such as bis-benzimidazoles, carbazoles, furans, and pentamidine analogues, which are novel antifungal agents and that such strain was fully pathogenic. However, the reported initial stress response experienced by C. neoformans H99 strain in the presence of these antifungals may probably depend on the initial low expression level of the Top1 gene, which gradually increased over a period [186].
DNA topoisomerase has been proposed as a target enzyme for drug inhibition against various infectious agents, including cryptococcal cells [190]. C. neoformans possesses the second Top2 gene for DNA topoisomerase 2, but then there is no evidence of functional replacement/complementarity between the Top1 and Top2 genes [186]. Strategically, C. neoformans serotype A may likely engage in a housekeeping regulation of the production of topoisomerase, even under the induce-promoter system, to avoid initial stress from antifungals, radiation effect, and heat shock. This may also be tantamount to the expression of genes involved in producing heat-shock protein (Hsp70) and melanin (Lac1) to cope with heat shock and radiation, respectively.
The UV-light incidence on C. neoformans can inhibit mating or haploid fruiting; in the same way, it will induce DNA damage in the yeast via the production of cyclobutane pyrimidine dimers (CPDs) or photoconjugate products. Such photoconjugates include the formation of dipyrimidine from adjacent thymine or cytosine bases, 6 – 4 pyrimidine–pyrimidone or 6 – 4 pyrimidine–pyrimidinone, and Dewar pyrimidinone isomers of 6 – 4 photoproducts. In each case, failure of the DNA repair enzymes (repairsome) and photolyase to repair these damages can lead to a double-strand breakage due to base mismatch, thereby impairing the mating and haploid fruiting. This has been shown by Idnurm et al. that dark incubation favoured mating and hyphal formation more than light/illuminated incubation irrespective of the mating type, with evidence of hyper-hyphal formation in either bilateral or unilateral crossing involving Δbwc1 mutant in the V8 media at pH 7.0 [191].
Furthermore, light signalling/transducer homologues, including Ops1, Phy1, and Bwc1, were identified in C. neoformans. The Δops1, Δphy1, and Δops1Δphy1 mutants showed no effect on the mating and haploid fruiting in the presence of light, unlike the Δbwc1 mutant, which is sensitive to light [191]. However, mutation of these genes (single, double, or triple) has no significant effect on the virulence factors of C. neoformans. Bwc1 and Bwc2 are the two major photo-inducing genes in C. neoformans. The Bwc1p significantly controls the cell fusion and filament development when the Δbwc1 mutant is crossed with either ade2 or lys1 auxotrophic Bwc1 wt [191]. Similar to Neurospora crassa Wc-1 protein, blue light induces the non-zinc finger DNA binding protein Bwc1p to regulate cell fusion and repress hyphal development in C. neoformans serotype A or D [191]. This step inhibits further growth pending favourable conditions to preserve the integrity of the genome. Unlike Bwc1p, Bwc2p appeared to have the PAS and zinc-finger DNA binding domain, like N. crassa Wc-2. The Bwc1p perceives the photon as a light sensor and induces a conformational change, which the transcription factor Bwc2p can recognise to favour protein-protein interaction.
The ability of C. neoformans to replicate and proliferate within the micro-acidic phagolysosome of macrophages is partly controlled by the Ipc1 expression. The outcome from Luberto et al. showed that downregulation of the Ipc1 gene under the influence of glucose-repressing Pgal7::Ipc1 induction expression reduced the exponential proliferation of strain H99 at pH 4.0 in a macrophage-like cell line, but galactose-inducing condition stimulated the rate of replication even than the wt strain [134]. The implication is that glucose-enriched tissues like CNS, which have 50 – 60% of systemic glucose (800 μg/mL), may impact early stress on C. neoformans to prevent proliferation. Nevertheless, the effect of Top1 and many other transcription factors may come to play to oppose this stress and promote cell replication.
Strategically, C. neoformans demobilises the activation of the cAMP/Pka-Rim101 signalling pathway in the acidic medium (such as phagosomes) because this pathway is generally activated in the neutral-to-alkaline media. The acidic nature of the phagosomes usually excludes glucose to maintain low osmolarity. This condition favours the activation of cryptococcal cAMP/Pka sensors, which will not initiate the Rim101 cascade event but Gpa1. The induction of the Gpa1 gene and the concurrent release of DAG to activate Pkc favour the Lac1 and Cap genes for the formation of melanin and capsule, respectively. Together with chitin expression, this virulence enables the cryptococcal cells to withstand the acidic phagosomes and promote endosomal and lysosomal survival of C. neoformans within the phagolysosome. Consequentially, titanisation of the phagocytosed cryptococcal cells is initiated – because of the chitin formation, leading to phagosome rupturing. Here, the cell can then utilise the cytoplasmic glucose of the macrophage for metabolism and growth while initiating the time-dependent macrophage apoptosis. Indeed, C. neoformans is a battle-ready pathogen with numerous counter-attacking accessory genes.
C. neoformans prefers an acidic environment to an alkali medium. Gradual reduction of H+ in a culture medium induces Ena1 expression. From Supplementary 3, Δena1 and Δcna1 mutants are hypersensitive to alkali pH [192]. This shows that the two separate pathways regulated by ATP and Ca2+, respectively, are involved in the survival of C. neoformans under elevated pH. Though Ena1 expression has been speculated to be induced for long-term hyperosmotic adaptation under the influence of Hog1 expression; however, the results from Idnurm et al. showed that there is no evidence of increasing Ena1 expression under the influence of elevated salinity of monovalent salts of Li+, Na+, and K+ or divalent salts of Ca2+ or even 1 M sorbitol but somewhat under the alkali medium when Na+ or K+ is low [192]. Besides, the expression of Ena1 could be tightly regulated by Can1, Hog1, and Rim101 [192]. The Nha1 expression, on the other hand, was induced by osmotic stress in probably a Hog1-dependent pathway and plays a role in survival under high K+ osmotic stress and acidic conditions [193].
Plc1p is another important membrane-localised protein that releases phospholipase B from the glycosylphosphatidylinositol (GTI)-anchor. A Δplc1 mutant is excessively susceptible to azoles, 5-flucytosine (5-FC), and AmpB due to defective cell wall integrity in this mutant [194]. Similarly, U73122 ((1-[6-[((17β)-3-Methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione)) is a PLC and PLA2 synthetic inhibitor that prevents the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) to two intracellular second messengers, 1,2-diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG activates Ca2+-dependent Pkc that modulates other MAPKs to restore cell wall integrity, which is usually perturbed by high temperature and cell wall disruptors. In contrast, IP3 binds the ER to release storage Ca2+ for Ca2+- and calmodulin-regulated signalling pathways, which means that Plc1 activity contributes significantly to calmodulin-activation but to calcineurin-activation, its contribution is insignificant [195]. Back to the synthetic phospholipase inhibitor, incubating C. neoformans in the presence of U73122 at 2.5 μM prevented the growth for over 12 hours, and the effect was more devastating as the temperature increased; however, the analogue of this inhibitor (U73343) displayed no antifungal effect [194].
Further studies have shown again that Ipc1 expression may encourage sphingolipid interaction with the membrane acyl chains and ergosterol to promote a more compact, resilient, and less permeable bilipid layer surrounding the cryptococcal cells [134]. This excludes extracellular solutes and prevents ionic perturbation while maintaining intracellular homeostasis. In addition, repression of the Ipc1 gene delayed macrophage toxicity and disruption, extracellular predominance, and tissue invasion [1,134]. Similarly, iron absorption encouraged ergosterol biosynthesis to maintain membrane integrity. Deficiency in iron acquisition has been shown to influence fungi susceptibility to antifungal drugs [196]. Though Δcft1 and Δcft2 mutants displayed normal growth on YPD, but these mutants have been characterised with impaired ergosterol biosynthesis, and that Δcft1 is more sensitive to miconazole (MCZ) and AmpB than the Δcft2 (check Supplementary 1 for detail). Therefore, drug targeting against the functional iron permease, especially the Cft1p, may be an additional benefit to the current antifungal drugs.
Till now, the most widely used azole antifungal, such as fluconazole, itraconazole (ICZ), and voriconazole, target one or two enzymes in the ergosterol biosynthesis. These enzymes may be oxygen-dependent/independent. The motif behind this is that in all circumstances, cryptococcal cells modify the membrane components to survive every environmental impediment. In doing so, azole drugs can easily block the deployment of such key enzymes needed for membrane sterol production. Erg11 encodes oxygen-dependent lanosterol-14α-demethylase in the production of ergosterol. This enzyme is the target of azole drugs. The Mbs1 negatively regulates basal expression of Erg11 and, when deleted, increases the azole resistance; however, the resistance against polyene is drastically reduced as par Δert1, Δjjj1, Δhcm1 and Δecm22 [126,197] (mutant responses to polyene drugs are highlighted in Supplementary 3). Also, Hob1 is another negative regulator of Erg11, and its deletion remarkably increased the basal expression of Erg2 and Erg11. However, under sterol depletion, Δhob1 mutants are characterised by a remarkable reduction in the induction of Erg2, Erg3, Erg5, Erg11, and Erg25 involved in ergosterol biosynthesis [197]. It is, therefore, possible that C. neoformans partially represses Hob1 expression to elevate ergosterol accessory enzymes in the wts.
Unlike Hob1, Sre1 is a positive key regulator of ergosterol synthesis. It is probably possible that the FCZ resistance observed in Δhob1 mutant results from the background expression of Sre1 to rescue impaired ergosterol synthesis in Δhob1 mutants. Surprisingly, while Δhob1 mutants are hyper-resistant to FCZ but hypersensitive to AmpB, the Δsre1 mutants are hyper-resistant to AmpB but hypersensitive to FCZ (mutant responses to FCZ are highlighted in Supplementary 3). Realistically, the expression of Sre1 in Δhob1 mutant induces most of the oxygen-dependent enzymes for sterol biosynthesis, including Erg1, Erg2, Erg3, Erg5, Erg7, Erg11, and Erg25, in the presence of FCZ [197]. Notwithstanding, Sre1 expression can also regulate and control the expression of other oxygen-independent enzymes involved in the upstream catalysis of ergosterol intermediate formation, and this includes the expression of Erg6, Erg10, and Erg13 [198]. To this extent, Sre1 and Hob1 expressions may be playing complementary tête-à-tête roles against the oxidative, osmotic, genotoxic, ER, cell wall, and membrane stress by jointly regulating the ergosterol biosynthesis [197]. Consequentially, Δstp1 and Δscp1 mutants are unequivocally susceptible to FCZ even in the normoxic conditions (normal atmospheric oxygen concentration, 20 – 21% or physiological oxygen concentration, 2 – 3%) at 30oC; not only FCZ but also ICZ and voriconazole [198]. Convincingly, reconstituting this mutant with the wt Sre1 gene restored normal growth even under the inhibitory concentration of the antifungals. Paradoxically, oxygen availability is insufficient to prevent the growth defect displayed by the Δsre1 mutant in the azole-containing media. With this sterol biosynthesis and proper incorporation of melanin and chitin, cell wall integrity is confirmed to withstand antifungal and increasing temperatures.
Also, the cell wall integrity and antifungal are jointly controlled by the Mpk1 and Can expression. The Δmpk1 mutants are attenuated for virulence with poor cell wall integrity and are thermosensitive and susceptible to antifungals such as nikkomycin Z (which inhibits chitin synthase) and caspofungin (CpF, which inhibits β-1,3-glucan synthase) (other mutants are discussed in Supplementary 1) [199]. Surprisingly, disruption of calcineurin function activates the expression of a single-copy Fks1 homologue to produce β-1,3-glucan synthase, which is essential for cell viability in a low-iron medium [200]. Based on this, capsule induction is expected to increase; however, ΔcanA and ΔcanB mutants showed no significant difference in their capsule sizes whether cultured in a low or enriched iron media [88]. Thus, by inference, calcineurin function may not be essential for capsule formation.
Apart from Sre1 expression controlling ergosterol biosynthesis, disruption of the Hog1 components and Skn7 independently controls ergosterol synthesis to promote antifungal resistance. The Δhog1 and Δssk1 mutants showed upregulation of major Erg genes such as Erg4, Erg5, Erg6, Erg11, Erg20, Erg25, and Erg28 with a concomitant preponderance of cellular ergosterol content, while Erg8, Erg10, Erg13, Egr26, Erg27, Hmg2, and Idi1 are more upregulated in Δssk1 mutant; however, Δskn7 showed no upregulation of these genes [81]. Because Ko et al. discovered a significantly higher level of cellular sterol in Δssk2 (MAPKKK) and Δpbs2 (MAPKK) than in the wt and Δskn7 mutant, it is therefore concluded that constitutive phosphorylation of Hog1p represses ergosterol biosynthesis under normal conditions [81]. To this effect, all four mutants are hypersensitive to AmpB, but Δskn7 showed a wt resistance level. Contrarily, all four mutants, together with Δskn7, are KCZ- and FCZ-resistant, but to imidazole (ICZ) at low concentration, a wt-resistance was displayed (mutant responses to antifungals are highlighted in Supplementary 2). This outcome has presented a differential response of the Hog1 pathway to different antifungals. The resistance observed with the Δskn7 mutant was purely aberration and certainly not Erg-dependent resistance because there was no significant difference in the expression of Erg genes in this mutant compared to the wt. However, an observation from Wormley et al. showed that Skn7 might be responsible for the expression of thioredoxin reductase [201] – an enzyme released against oxidative stress.
To mollify this observation, Bahn et al. discovered that mutation of Skn7 showed no significant effect on the sensitivity to H2O2, UV-irradiation at 720 J/m2, high temperature, methylglyoxal (MG), and high salt solution (especially KCl); however, Δssk1, Δpbs2, and Δhog1 mutants are sensitive to all these factors except FDX [30]. Precisely, the in vitro flocculating Δskn7 mutants of C. neoformans var. grubii (serotype A) are highly susceptible to 0.025 mM t-BOOH, 1 M NaCl, and significantly less virulent due to oxidative arrest; nevertheless, the non-flocculating Δskn7 serotype D mutants displayed a wt similar stress, antifungal, and adaptations to t-BOOH, 1.5 M NaCl, AmpB, and 38oC [202]. The Skn7 function against oxidative stress is independent of Trx2 and Glr1 but induces Trr1 and Sod1 to maintain intracellular redox balance [201]. Because Δskn7 mutant can still deploy other oxidative and nitrosative stress regulatory/response groups of genes (OSR and NSR genes), its pathogenesis and survival in the macrophage-killing assay appeared unaltered [201]. Intraendothelial survival, virulence, and brain and lung colony recovery of Δsnk7 serotype D mutants appeared significantly reduced compared to the isogenic control, but the tissue adherence and fungaemia are similar to the wt [202].
The dependence of Hog1 activation on Ssk1p is very paramount in C. neoformans. Most times, Δssk1 mutants display similar phenotypic defects as Δhog1 and Δpbs2 mutants; however, Ssk1 plays a less significant role in osmosensing pathways because, in the Δssk1 mutant, Hog1p can still be phosphorylated to a lesser extent in the presence of 1 M NaCl but not in the presence of FDX or MG [30]; no wonder Δssk1 mutant displayed a wt phenotype against 1 M NaCl and 1.5 M KCl in YPD [82]. Paradoxically, Ssk1p is needed to keep Hog1p constitutively phosphorylated under normal conditions. However, in the absence of the second two-component system response regulator, Skn7, Hog1 exhibited a more progressive time-dependent dephosphorylation in 20 μg/mL FDX than the wt. This Hog1 dephosphorylation and activation in Δskn7 are, however, similar to the wt in the presence of 1 M NaCl and 20 mM MG [30]. The observation that Δhog1, Δssk1, and Δpbs2 mutants are hypersensitive to MG but hyper-resistant to FDX (while Δskn7 mutant showed opposite phenotypic features) showed that dephosphorylation of the Hog1p is differentially regulated towards different osmotic, oxidative, and antifungal stress. In addition, Δssk1 and Δpbs2 shared similar phenotypic defects with respect to H2O2, UV-radiation, and FDX (Supplementary 3 highlights responses of several mutants to various quantified factors).
The roles of the thioredoxin system (Trx1 and Trx2) in maintaining the cytosolic redox-equilibrium glutathione reductase activity in C. neoformans deserve high commendation. Functionally, the two proteins appear redundant and can replace each other to enhance growth, promote membrane integrity, antioxidative, antinitrosative, and influence the virulence of this pathogen. The induction of Trx1 in the presence of H2O2, Trx1 and Trx2 in the presence of t-BOOH, and Trx1 and Trx2 in the presence of NaNO2 confirmed the antioxidative and antinitrosative roles of thioredoxin proteins. The Δtrx1 and Δtrx1Δtrx2 mutants exhibited severe growth defects due to increased levels of oxidised glutathione compared to the total glutathione level, but the Δtrx2 mutant only showed comparable wt growth phenotypes [203]. Therefore, trx1 expression is highly important in macrophage survival and virulence. Likewise, the expression of thiol peroxidase Tsa1, Tsa3, and Tsa4 in the presence of H2O2, t-BOOH, and NO at 37oC also complements the roles of the thioredoxin system. Among these, Tsa1 appeared to play the most significant role against oxidative and nitrosative stress to bring about thermotolerance and virulence in the mouse model [204]. Unfortunately, while an exogenous ascorbate supply restored all the growth defects in Δtsa mutants, neither ascorbate nor dithiothreitol could easily restore the growth defects in the Δtrx mutants (check mutants in Supplementary 1 and 3 for other responses to different oxidants).
Glutathione peroxidase is another plausible cytoplasmic localised antioxidative enzyme encoded by Gpx1 and Gpx2. These redundant transcripts are differentially induced in response to stress agents but have no impact on virulence expression. Missall et al. discovered that only the Gpx2 is induced in response to H2O2 while the two transcription factors are induced in the presence of t-BOOH and cumene hydroperoxide (COOH) but are repressed with NO stress in the wt [205]. Interestingly, H2O2 can induce Gpx1 in the Δgpx2 mutant as potential transcriptional compensation, but t-BOOH failed to induce Gpx2 in the Δgpx1 mutant showing that the Δgpx1 mutant is particularly sensitive to t-BOOH without any compensation from the Gpx2 [205].
Another well-studied C. neoformans antioxidative enzyme is Mn-SOD, an important component of the mitochondrial antioxidant defence system encoded by the Sod2 gene. From Giles et al. analysis, Δsod2 mutants showed increased sensitivity to superoxide radicals generated by Antimycin A or ParaquatTM (N,N′-dimethyl-4,4′-bipyridinium dichloride or Methyl Viologen) with O2-dependent temperature-sensitive (ts) growth defect. However, these phenotypic defects were observed to be restored with increased exogenous supplementation of 20 mM ascorbate or 200 μM Mn-salen-type ligand (a Jacobsen's catalyst known as N,N'-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediaminomanganese(III) chloride) at 30oC but not 37oC [206]. Surprisingly, an equal concentration of inorganic MnCl2 failed to restore this ts phenotypic defect, and these restoring concentrations appeared to inhibit the wt growth at 37oC [206]. Contrarily, Narasipura et al. achieved progressive growth of the Δsod2 mutant in the presence of 5 mM and 10 mM of ascorbate at 37oC, and this growth was comparable to the wt at 37oC [207].
Regarding the electron transport chain in mitochondria, the two antioxidative enzymes, Ccp1p and Aox1p, are very important. For example, C. neoformans cultured in the yeast nitrogen base (YNB) at pH 4.0 and 30oC are moderately inhibited by 1 mM of H2O2 but highly exacerbated in the presence of 0.5 μg/mL of Antimycin A (an inhibitor of Ccp1p-mediated electron transport chain) or slightly inhibited in the presence of 2 mM of SHAM (an inhibitor of Aox1p-mediated electron transport chain). Additively, the combination of the two inhibitors prevents the growth of C. neoformans either in the presence or absence of H2O2 [208]. This shows the significant role of Ccp1p in the antioxidative effect against H2O2; nevertheless, the supportive role of the Aox1p-mediated pathway is highly commendable, especially in reducing the preponderant formation of ROS during redox reactions. Furthermore, the wt strain had shown early downregulation of Aox1 transcript, which remained unperturbed when exposed to exogenous H2O2 effect, unlike its functionally redundant Ccp1 transcript that was later upregulated [208]. On the other hand, the presence of 0.25 μg/mL of FCCP (4-trifluoromethoxycarbonylcyanide phenylhydrazone), a protonophore (proton translocator/ionophore) that dissipates the H+ gradient across the inner mitochondrial membrane, has no effect on C. neoformans but a 50% growth reduction when added with 1 mM H2O2. Contrarily, 0.5 μg/mL of FCCP is sufficient to moderately reduce the growth of C. neoformans, unlike S. cerevisiae [209] and C. albicans [210]; and when combined with 1 mM H2O2 drastically inhibited the growth [208]. This demonstrated mitochondria oxidative phosphorylation pathways against oxidative stress imposed by exogenous peroxide, perhaps by providing ATP for reparative and homeostasis cellular processes after a series of oxidative damages. One such is proteasomal ubiquitin-dependent protein catabolism, which actively involves Ubc8p against oxidative stress imposed by H2O2 [81].
Structurally, Mn-salen is a less complex structure than Mn-porphyrin and possesses a high-valent central Mn in a nitrogen and oxygen donor environment [211]. This condition may have improved cellular entry and redox participation of the Mn better than the inorganic salt or a more complex Mn-porphyrin. Due to its function in maintaining the steady-state cellular endogenous reactive oxidants, Δsod2 mutants in an Mn-salen medium are non-viable in aerobic conditions at 37oC for 24 hours because of the accumulated reactive oxidants [206]. This is not surprising because C. neoformans is a facultative aerobe; however, the Δsod2 mutants are viable under the anaerobic condition at 37oC and can as well survive ambient conditions when transferred to 25oC [206]. Deletion of the Sod2 gene has no significant effect on capsule production, melanin formation, and urease activity; however, surviving the oxidative attack by the macrophage is impossible in the absence of Sod2, and this is the reason Giles et al. could not recover the Δsod2 mutant from either the lung or brain in murine inhalation model of cryptococcosis (MIMC) [206]. Thus, this is one of the antioxidative enzymes not linked to virulence per se but to survival, adaptation, and infection in the host.
Contrarily, Narasipura et al. discovered that the killing of the Δsod2 mutants is necessarily not from the oxidative attack by the phagocytes but from the high oxygen environment [207]. This observation is true for the in vitro system but achieving such a high level of O2 in vivo may be difficult. Therefore, the major killing of the Δsod2 mutants may still be from the oxidative attack by the macrophage. There was a slight difference in the growth of Δsod2 mutants at 30oC in the Narasipura (at 95% air) and Giles (no percentage stated) works. Convergently, when Giles et al. incubated the mutant at 37oC under anaerobic conditions and transferred the mutant to a condition as low as 25oC, viable growth was observed, which is as good as the growth Narasipura et al. observed at 30oC. However, Δsod2 practically remained unviable at 30oC, making the mutant isolation difficult, unlike 25oC [206]. In the presence or absence of O2, Δsod2 and Δsod1Δsod2 mutants displayed similar ts growth defect at 37oC contrary to Δsod1 [207]. In addition to the oxidative attack by oxygen, Δsod2 and Δsod1Δsod2 mutants are equally hypersensitive to all common superoxide- and osmotic-inducing agents (check Supplementary 3 for other responses); however, the cytosolic sod1 mutants behaved alike except against ParaquatTM where a slight sensitivity was observed [207]. It was further discovered that Δsod2 and Δsod1Δsod2 viability reduced drastically in the stationary phase nutrient limitation study. The Δsod2 mutants are completely avirulent in the murine inhalation study, but the virulence is attenuated in the murine intravenous study. The Δsod1Δsod2 mutants, on the other hand, lost their virulence completely with murine inhalation/intravenous model of cryptococcosis with a complete fungi clearance in the lungs; however, traces of this mutant were recovered from the brain after 120 days post-infection. The analysis further showed that these mutants were the same as the inocula mutants lacking Sod1p and Sod2p [207].
Unlike thioredoxin, peroxidase, and dismutase systems, the catalase system seems to play a less significant role against oxidative stress and virulence factors. Among the four Cat genes identified in C. neoformans, Cat1 and Cat3 (encoding putative spore formation-specific catalase), Cat2 (encoding putative peroxisomal catalase), and Cat4 (encoding putative cytosolic catalase), none of the mutants (Δcat1, Δcat2, Δcat3, Δcat4, and Δcat1Δcat2Δcat3Δcat4) showed any phenotypical defect related to oxidative stress irrespective of the temperature, just as observed in S. cerevisiae [212]. Furthermore, it was shown that none of the Cat genes is required for survival, infection, and virulence expression; however, Cat4 seemed to be required for mating and spore viability.
Out of the seven different hybrids of sensor histidine kinases (Tco1, Tco2, Tco3, Tco4, Tco5, Tco6, and Tco7), Tco1 and Tco2 are the most studied two-component phosphorelay system expressing sensor kinases. In tandem with Ypd1p, it activates response regulator kinase Skn7p in the nucleus or Ssk1p in the cytoplasm [213]. The C. neoformans Ypd1 is an intermediate and dynamic phosphorelay histidine-containing phosphotransfer protein (HPt) and a structural homolog of yeast HPt protein. The Ssk1 kinase activates the Ssk2-Pbs2-Hog1 MAPK cascade event, and this subsequently induces a plethora of downstream target genes for various cellular activities, including cell wall/membrane stress response, sterol formation, virulence factors, and cell differentiation and filamentation [32,81]. Furthermore, the disruption of Tco and Hog1 genes differentially predisposes C. neoformans to antifungal, osmotic shock, and membrane disruptors.
5-FC induces the regulation of several genes under the control of Tco kinases and the Hog1 pathway. These genes are majorly involved in signal transduction, cell cycle, replication, translation, ribosomal maturation, and post-translational processing. Since 5-FC is a pyrimidine analogue, there is a possibility that when incorporated during replication/transcription, it will accumulate in either truncated or non-functional transcripts, which may invariably produce biologically toxic proteins and compromise the induction of genes majorly involved in the quality control of DNA, RNA, and protein production. The Δtco1 mutant is sensitive to 5-FC, but Δtco2 is more resistant to 5-FC even more than the Δssk1, Δssk2, Δpbs2, Δhog1, and Δskn7 mutants under the same conditions. Unequivocally, the Δtco2 resistance against 5-FC was not attributed to a natural alteration in the putative genes encoding cytosine deaminase (encoded by Fcy1), cytosine permease (encoded by Fcy2, Fcy3, and Fcy4), or UPRT (encoded by Fur1) but a metabolic alteration was suspected rather than the transcription factors [126].
The unique wt upregulation of Ste14 transcript encoding isoprenylcysteine carboxylmethyltransferase (ICMT) [214], which in addition to palmitoylation, prenylates the CAAX motif of Ras1p for proper membrane localisation to promote functional GTPase and thermotolerance trait deserves closer attention to unravel the 5-FC-resistance. So, for this connection, it is possible that active and membrane-located Ras1p promotes resistance to 5-FC in a similar way that Atf1p promotes 5-FC- resistance [91]. Previously, none of the hybrid sensor kinases was differentially involved in the resistance/sensitivity to AmpB, FCZ, ketoconazole, itraconazole, osmotic shock, UV irradiation, and high temperature (as high as 40oC) except the Δtco2 mutant that showed hypersensitivity to AmpB and 1.5 M KCl [30,81].
Though Ypd1p is an intermediate HPt, its roles in the viability of C. neoformans are inevitable. The initial attempt to delete this gene in a wt was unsuccessful but from the Δhog1 background mutant [82]. Changing its promoter to a copper regulatory promoter (Pctr4) showed that it regulates the growth in a Hog1-dependent manner. The Pctr4::Ypd1 strain grew under the influence of BCS but not CuSO4, meaning that CuSO4 is a repressor; however, the Pctr4::Ypd1 strain in Δhog1 background grew in both BCS- and CuSO4-containing media [82]. Characteristically, Δhog1Δypd1 and Δhog1 mutants displayed osmosensitivity to 1.5 M KCl but not 1.0 M NaCl solution, unlike Δskn7, which is hypersensitive to 1.0 M NaCl. Basically, Ypd1p controls the Hog1-dependent pathway to maintain membrane integrity against perturbation caused by osmolytes. This HPt protein can as well function independently of the Hog1 pathway to confer resistance to oxidative stress caused by diamide (thiol-specific oxidant) and H2O2. The Δhog1Δypd1 mutant conspicuously showed hyper-resistance against azole better than Δssk1, Δhog1, and Δskn7 mutants (check Supplementary 3 for other mutant responses). This may reflect the additive repression of Ssk1 and Skn7 expression in the Δhog1Δypd1 double mutant to promote the expression of the Erg11 gene.
Oxidative stress imposed by H2O2 induces specific stress regulatory genes, which outnumber the specific genes induced by osmotic and antifungal stress, probably because H2O2 produces reactive radicals (oxidants). This means that C. neoformans possesses a unique genome-wide regulatory expression profile induced by oxidative stress. This dynamic expression profile has been streamlined into the array of genes involved in signal transduction, membrane transport (ionic, solutes, and secondary metabolites), metabolism, transcription, post-translational modification, and ubiquitin-regulatory proteins with full dependence on the Hog1, Ssk1, and Skn7 expressions [81] (check Supplementary 2 additional information). Just as Hog1 and Ssk1 expressions control ergosterol biosynthesis, evidence showed that Ubc6-2 might also be involved in ergosterol synthesis because Δubc6-2 mutants are resistant to FCZ but sensitive to AmpB. However, these mutants failed to show any increased susceptibility to osmotic and oxidative stress except for a slight sensitivity to CdSO4 and FDX [81] (check Supplementary 3 for further information).
This seems contrary to the ubiquitin-proteasome system in S. cerevisiae, where Ubc7p/Qri8p selectively degraded unphosphorylated Ssk1p two-component system that activated Ssk2p MAPKKK during hyperosmotic stress [215]. Though the Δubi4 mutant of S. cerevisiae was hypersensitive to H2O2 with evidence of respiratory-induced basal production of Ubi4 in the wt [216], it was the Δubc8 mutant of C. neoformans that showed hypersensitive to H2O2 [81]. The production of Ubi4p has been further shown to be induced by starvation, CdSO4, genotoxic compounds, and heat shock [216]. This indicates that the roles of the ubiquitin-conjugating system are highly laudable in the antioxidative and antiosmotic stress strategies of C. neoformans as well. Among the downregulating genes in the wt exposed to oxidative- and osmotic-stress are translational and ribosomal structure-expressing genes, but these arrays of genes are unaffected in Δhog1 mutant [81].
Further analysis showed the implication of mbs1 expression in the resistance against 5-FC and potentially involved in thermotolerance. 5-FC induces mbs1 transcripts in the Δtco1 mutant, unlike Δtco2 and Δhog1 mutants, where the basal transcript was slightly higher than the 5-FC-induced transcript [126]. By interpretation, the interplay between Tco2p and Hog1p to regulate Mbs1 basal transcription promotes 5-FC resistance. Nevertheless, Ko et al. had projected redundant and distinct roles of Tco1 and Tco2 kinases in modulating the Hog1-dependent phenotypic expression [81]. There is a suspicion that mbs1 expression, proposed to be controlled by Tco2p and Hog1p, could be modulating ergosterol synthesis in C. neoformans because the Δmbs1 mutant was AmpB-sensitive but resistant to ketoconazole and FCZ through its controlling expression of Erg11 [81]. The basal transcriptional level of Erg11 and the cellular ergosterol level are quite higher in the Δmbs1 mutant, just as in the Δhog1 mutant, than in the wt [81]. Therefore, the low level of the membrane sterol may be the reason for polyene susceptibility, while the high activity level of lanosterol 14α-demethylase (encoded by Erg11) promotes azole resistance. The same reason predisposes Δmbs1 and Δhog1 mutants to membrane perturbation by SDS and osmotic stress caused by NaCl and KCl in the absence of glucose [126]. Therefore, in addition to antifungal resistance and potential thermotolerance, Mbs1p also ensures membrane stability against osmotic stress.
Because of the close association of Mbs1p with the nucleic acid to maintain DNA integrity and repair during cell growth, the Δmbs1 mutant displayed high susceptibility to genotoxic agents such as hydroxyurea (HU) just like Δhog1 mutant, unlike Δtco1 and Δtco2 mutants. However, the Δmbs1 mutant showed resistance against methylmethanesulfonate (MMS) and thiabendazole (TBZ) but a slight resistance against TBZ in the Δtco1 and Δtco2 mutants [126]. Since Tco1 and Tco2 are upstream of Hog1, their presence may slightly negate TBZ-resistance, so if Hog1 and Tco2 modulatory effect on the Mbs1 is repressed, then this condition will favour TBZ-resistance. However, house-keeping expression of Mbs1 (together with Rad9 and Ddc1 expression as discovered in the yeast) may still be needed to initiate a complete transcriptional response of Rnr2 (encoding ribonucleotide reductase) during DNA damage checkpoint control initiated by genotoxic agents such as MMS [217]. The connection among these three transcription factors is evidenced in the similar susceptibility of the Δtco2, Δtco1Δtco2, Δssk1, Δhog1, and Δtbs1 mutants to H2O2 and similar resistance to diamide; however, Δtco1, Δtco3, Δtco4, Δtco5, and Δtco7 mutants showed a wt-resistance to H2O2 but Δtco1 only slightly resistant to diamide [30,126]. Thus, the resistance to diamide might be because it is an exogenous oxidant, which failed to produce reactive radicals during the redox process, unlike the H2O2 that is endogenously produced with concomitant reactive radicals.
Remarkably, resistance against FDX and MG appeared to be controlled by the two-component system. Mutation of Tco1 improves MG resistance while Δtco2 and Δtco1Δtco2 mutants are highly sensitive, however, reconstituted Δtco2 mutants showed a high MG tolerance as high as Δtco1, Δtco3, Δtco4, and Δtco5 but Δtco7 and reconstituted ΔTco1 mutants showed a wt MG-tolerance [30]. MG, therefore, represses Tco genes, but the Tco2 gene is induced for concerted resistance effect. Just as found in the mutation of the Hog1 pathway, FDX-tolerance is highly improved in Δtco1, Δtco1Δtco2, and moderately improved in Δtco2 mutants but restoring the corresponding Tco gene into the Δtco1 and Δtco2 mutants drastically reduced the FDX-tolerance to the wt susceptibility level, which Δtco3, Δtco4, Δtco5, and Δtco7 mutants naturally displayed [30]. This observation further confirmed the biological importance of Tco1 and Tco2 sensor kinases on the Hog1 regulation compared to other hybrids of sensor histidine kinase. By interpretation, the Hog1 pathway must naturally be repressed perpetually for the yeast to withstand FDX. This means that though the Tco genes may be less involved against the oxidative and osmotic stresses, but their roles in FDX are inevitable as they are the upstream regulatory sensors that can modulate Hog1 MAPK activity and, in so doing, negate FDX resistance.
The evidence of consistent early or late dephosphorylation of Hog1p when exposed to NaCl, H2O2, MG, and FDX in Δtco1, Δtco2, and Δtco1Δtco2 mutants showed that Hog1 phosphorylation may still be initiated by some other subcellular activation systems sensitive to the same environmental cue as Tco sensor kinases or, yet some undiscovered MAPKKKK that may agitate Ssk2 kinase. Specifically, Hog1p is dephosphorylated early when Δtco1 mutants are exposed to 1 M NaCl, MG, and FDX and when Δtco2 mutants are exposed to MG and FDX. However, late dephosphorylation of the Hog1p is observed in Δtco2 mutants when exposed to 1 M NaCl and when Δtco1Δtco2 mutants are exposed to 1 M NaCl, MG, and FDX [30]. This may be the reason Δtco2 and Δtco1Δtco2 mutants showed comparable defects to Δssk1 and Δhog1 mutants when exposed to MG and H2O2 and that Tco1 and Tco2 independently conjoined against FDX stress.
By inference, Tco1 and Tco2 may be redundant sensors modulating the activation of Hog1p. However, late dephosphorylation of Hog1p in Δtco1, Δtco2, and Δtco1Δtco2 mutant could have brought about the wt resistance against NaCl and 15 mM MG. In the actual sense, however, the Δhog1 mutants are hypersensitive to 1 M NaCl but moderately sensitive to 15 mM MG (check Supplementary 3). The Tco2 and probably along with other sensors, are solely responsible sensors against MG levels >15 mM because the Δtco2 and Δtco1Δtco2 are the more sensitive mutants with 20 mM MG after Δhog1 and Δssk1 mutants [30].
Furthermore, Δmbs1 mutant showed the wt phenotypic characteristics to FDX, CdSO4, and t-BOOH, but Δssk1, Δssk2, Δpbs2, and Δhog1 mutants exhibited cadmium-tolerance by inducing cadmium-responsive genes higher than the Δskn7 mutant and the wt [81]. In addition, Bahn et al. observed that Δssk1, Δpbs2, and Δhog1 are completely resistant to FDX, but the Δskn7 mutant with its Hog1 MAPK activation showed moderately high tolerance to FDX compared to the wt [30] (other phenotypic responses of mutants are in Supplementary 3), which means Hog1 pathway negatively influences resistance to FDX and that the function of Skn7 kinase response is parallel and independent of Hog1 pathway. Further analysis showed that all genes involved in the posttranslational modification, protein turnover, lipid transport, and secondary metabolite biosynthesis and transport are upregulated in the wt strain exposed to FDX. However, genes involved in the transport and metabolism of carbohydrates, nucleotides, lipids, and other metabolites are downregulated [81]. Contrary to the Hog1 pathway components, all Δtco mutants except Δtco2 showed only a wt cadmium tolerance, similar to AmpB response [81]. This is a reminiscence that Hog1p and Tco2p control the regulation of Mbs1, and cadmium toxicity is motivated by their presence. There are extensive works on the phenotypic responses of C. neoformans mutants to various environmental stress, and these are summarised in Supplementary 3.
C. neoformans detoxify xenobiotics by expressing ABD-type multidrug homologues transporters such as Pdr5, Pdr5-2, Pdr5-3, Yor1, and Snq1. The result from Ko et al. showed a several-fold increase in Pdr5 in the Δhog1 mutant, which explained the resistance of this mutant against FDX [81]. FDX induces specific response genes, which might explain why deletion of specific drug efflux transporter showed no significant growth defect compared to the wt with respect to the general stress response. Specifically, the deletion of Pdr5 and Yor1 increases sensitivity to FDX and FCZ (check Supplementary 3). There is speculation that both genes may play a redundant role regarding drug efflux; after all, C. neoformans has been identified to contain several copies of ABC efflux pump-related genes [81]. The Afr1 is another ABC pump encoding transporter in C. neoformans that is upregulated in the wt when exposed to FCZ to facilitate resistance, virulence, and macrophage survival in an animal model without altering the thermotolerance and virulence-associated genes such as Cap10, Cap59, Cap60, Cap64, Lac1, Plb1, Ure1, Sod1, and Vph1 [218]. Therefore, multiple copies of these Pdr-like genes might have conferred resistance against various antifungal drugs. Serendipitously, Ko et al. discovered that the cAMP-Pka signalling event is co-induced in a Hog1-dependent manner against FDX stress; however, only Aca1 mutation yielded significant growth defect compared to the wt [81] (check Supplementary 3). This means that the deletion of Hog1 gene must have induced the expression of Aca1 gene component of cAMP-Pka cascade unit.
Cell-to-cell communication via the release of adhesin protein is important for cell aggregation and population density. In addition, the physiological state of the cell determines the rate of growth and cell density. Studies showed that quorum sensing-like molecules regulate not only the cell density-dependent growth in C. neoformans but also sexual reproduction. Quorum sensing-like molecules are biomolecules that generally arouse cell growth to higher density. Lee et al. discovered that inoculum of MATα serotype D Δtup1 mutant at 5 x 106 and MATa serotype D Δtup1 mutant at 5 x 105 at 25oC and 30oC, respectively, grew as much as the wt followed by the isolation of an 11-mer peptide that was proposed as a growth factor. This mer autoregulated the density-dependent growth of these mutants irrespective of the media used, and any inoculum of fivefold less than this specific density stated above will thwart Δtup1 growth [219]. This quorum sensing-like peptide 1(Qsp1) was later identified to match a putative cqs1 gene product.
Double deletion mutant Δtup1Δcqs1 culture was found with minimal production of Qsp1, which could barely support Δtup1 growth, but on the contrary, the culture filtrate from Δtup1 can support Δtup1Δcqs1 mutant better than the self-culture filtrate [219]. The lower expression rate of Tup1 in hypoxic and other environmental conditions during exponential growth phase shines a light on the master controlling effect of Tup1p as cells transition to the stationary growth phase where Cqs1 expression may not have a direct correlation with Tup1 expression. In addition, Cqs1p has been implicated in the pheromone-controlling effect of bisexual reproduction and the initiation of unisexual differentiation, but these modulatory effects can be attenuated in the absence of the second Qsp2 encoded by the cqs2 gene [220].
The extracellular release of Qsp1 and Qsp2 is catalysed by the cell-associated protease Pqp from their corresponding pro-Qsp. The mature Qsp is re-absorbed into the cytoplasm via an oligopeptide transporter, Opt, to orchestrate differential transcriptional responses such as virulence factors, sexual reproduction, cell wall biogenesis, and extracellular proteomes, which are coordinated for higher cell density. This Qsp is described as an autoregulatory peptide that gets matured extracellularly but functions in the cytoplasm [221]. Simultaneously, the production of Qsp1p is under the concerted regulation of Gat201, Gat204, and Liv3 to ensure the virulence of C. neoformans (check the mutants in Supplementary 1). Apart from the attenuated infection and lower tissue burden, Δqsp1 mutant displayed dried and wrinkled cell morphology between 25 – 30oC, and this was restored to smooth and mucoid colonies when confronted with the wt patch. Morphologically, the Δliv3 mutant appeared as Δqsp1 mutant, but it can neither be rescued by confrontation assay nor by synthetic Qsp1 supplementation; however, it can assume a wt appearance at 37oC. This indicates that Liv3p is probably a downstream functional protein to Qsp1p and that overexpression of liv3 bypassed all the phenotypic deformations associated with the lack of quorum peptide secretion [221].
Mutants such as Δarf1, Δpka2, Δndh1, Δdrp1, Δcbk1, Δkic1, Δtao3, Δpgi1, Δcap10, Δcap60, Cap64, Δpbx1, Δpqp1, and Δshe4 have been identified with dried, hyperpolarised, actin delocalisation/mislocalisation due to perturbation and wrinkled colony-forming mutants [62]. However, mutants deficient in serine protease, an aspartyl protease, metalloprotease, carboxypeptidase, Tco1, Tco2, Tco3, Tco4, Tco5, Tco7, Gpr1-7, and Cpr2 failed to produce dried and wrinkled colonies like Δqsp1 mutant [221]. Besides, deleting any of these morphologically related genes usually increases susceptibility to immunosuppressive drugs (check the mutant responses to immunosuppressive drugs in Supplementary 3). Therefore, mutants compromised for Cna1- and RAM-signalling pathways will likely be inviable at >30oC in the presence of immunosuppressive drugs. A proper investigation eventually showed that RAM genes are necessary for proliferation at 37oC but not required for viability at 37oC. In the same way, dormancy at 37oC is fungistatic, but the presence of immunosuppressive drugs killed the mutants at this temperature [62].
Further analysis showed that Qsp1 and Opt1 are nested to each other and probably share the same promoter associated with Gat201, Gat204, and Liv3; therefore, Δqsp1 and Δopt1 mutants displayed similar phenotypic features [221]. Logically, this hypercapsulated Δqsp1 mutant can be complemented with Δopt1 mutant for quorum-sensing and cell aggregation because Qsp1p is produced in the Δopt1 mutant, which means that only the Δqsp1 mutant can respond to exogenous Qsp1p, mutant expressing Qsp1p or be complemented with the wt patches. The Δqsp1 mutant is yet attenuated for infection in the pulmonary tissue but not in the CNS. Though this mutant is phenotypically hypomelanised yet, this may not justify its attenuation in the pulmonary tissue; after all, this mutant was hypercapsulated [221]. Thus, the attenuated infection may be due to the impacts of the deletion on various proteolytic enzymes, such as virulence-promoting aspartyl proteinase, whose secretion is significantly reduced in the Δqsp1 mutant.
Ko et al. compared the regulatory genes that are either upregulated or downregulated among two pathogenic fungi (C. neoformans and C. albicans) with two non-pathogenic fungi (S. cerevisiae and S. pombe) to delineate the evolutionary relationship in terms of the gene-relatedness but then there were no exact common genes among these fungi that were equally regulated against osmotic stress except Ald5, Ena1, Prm10, and Stl1, which were common but differentially regulated among them [81]. Further comparison showed that C. neoformans deployed more genes against oxidative stress among the fungi. Again, there are no common-stress regulatory genes (osmotic and oxidative stress) common to them as well [81].
Convergently, different types of stress can eventually result in ER stress with the accumulation of unfolded or misfolded proteins around the ER, which can trigger unfolded protein response (UPR) signalling pathways. This well-conserved eukaryotic ER stress-protective pathway has been characterised in C. neoformans. One major transcription factor identified is Ire1, an ER stress sensor kinase similar to Tco in the Hog1 pathway. The Ire1 receives stress information to regulate other downstream transcription factors such as Hxl1/Bzp1, which is structurally and conventionally conserved within cryptococcal species but unique and diverged from other yeast Hac1 and human Xbp1 [222,223]. The Hxl1p is a Hac1 and Xbp1-like bZIP transcription factor that is UPR-induced via Ire1-dependent unconventional splicing, and its absence is more devastating in C. neoformans than the Ire1 gene. The unconventional intron splicing of Hxl1 mRNA is the regulatory rate-limiting step toward UPR activation. It has been shown to be governed by Puf4, which regulates the splicing rate of Hxl1 but can also initiate the attenuation of the UPR signalling pathway, not through Kar4 but via the hxl1 mRNA decay process orchestrated by Ccr4p relative to thermotolerance adaptation [223,224]. The wt virulence observed in Δpuf4 mutant in murine cryptococcosis is attributed to late activation of Hxl1p to compensate for the early induction of UPR signalling and considerable growth of this mutant at physiological temperature, though a temperature of ≥ 37oC seems to attenuate the growth of this mutant [223]. On the other hand, Δccr4 mutant has been characterised by elevated constitutive activation of UPR-related transcription factors such as Ost2, Sss1, Kar2, Per1 and many others (check Supplementary 2 for relevant transcription factors to UPR signalling pathway). This constitutive activation, together with stabilised ER stress transcripts, may probably be the reason for thermotolerance and moderate resistance observed with Δccr4 mutant in the presence of tunicamycin (TCM) (check Supplementary 3) [224].
Among the functions associated with the Ire1-Hxl1-dependent UPR pathway are the response to ER stress, cell wall integrity, thermotolerance, drug resistance, and virulence [222]. Obviously, the avirulence Δire2 and Δhxl1 mutants are highly susceptible to physiological temperature, ER stress agents, and cell wall destabilising agents; however, because most of the defects in the ire1 mutant can be suppressed by the active form of Hxl1p from spliced mRNA, then Hxl1 is a downstream functional protein to Ire1p. Also, Ire1p activity can be Hxl1-dependent or -independent because both mutants showed differential responses to temperature, capsule synthesis, and diamide resistance. The Δire1 mutants are defective in capsule formation, but Δhxl1 mutants are not. Again, both mutants showed normal melanin production and resistance to H2O2 (check Supplementary 3). Non-radical forming diamide affects Δire1 mutants only, which predicted Ire1 gene expression to control capsule formation and diamide-resistance in an Hxl1-independent route [222].
Essentially, UPR regulates the expression of genes in response to the stress signal received from the ER, and any impairment in this tandem stress response makes the yeast inviable [179]. Among the UPR-dependent transcription factors, which are related to heat- and osmotic-induced ubiquitin- or proteasomal-tagged ER-mediated protein degradation, are Mga2, Kar2, Alg7, Pps1, and Sod2; however, Erv29, Ost1, Pmt1, Pmt2, Pmt4, and Wbp1, which are involved in glycosylation processes are Hxl1-dependent genes and are repressed in Δhxl1 mutant but not in Δire1 mutant. The Pmt4 and Chs2, which are involved in glycan formation, are significantly induced in Δire1 mutants [222]. This means that cell wall stressors, such as Congo red and Calcofluor white repress Ire1 expression to induce cell wall integrity and promote relevant transcription factors. Kar2 is an ER luminal Hsp70 molecular chaperone, which is needed for cell viability, and it is induced by heat shock or TCM treatment to promote and regulate UPR signalling event via Ire-Kar2 interaction [225,226,227]. In addition, it works together with protein disulphide isomerase (encoded by the Pdi gene) to bring about the folding of non-glycoproteins, while the same Pdip in the Calnexin cycle brings about the folding of glycoproteins [228]. Overexpression of kar2 partially suppressed the growth defect associated with Δire1 and Δhxl1 mutants in low TCM and DTT (dithiothreitol) concentrations; however, Kar2p appeared not to suppress the growth defect at higher concentrations of these ER stress-inducing agents. Apart from the Hxl1 transcription factor, Kar2p is a sub-level downstream effector protein to Ire1 sensor kinase [225].
Further investigation showed that the restoration effect of Kar2 overexpression in Δire1 mutant seems more significant in the presence of DDT (a disulphide bridge disruptor) and diamide (thiol oxidant that induces abnormal disulphide bridge) than TCM because the tendency of forming unfolded/misfolded proteins is higher in the former two agents [225]. Furthermore, the Δire1 mutants are more sensitive to diamide than Δhxl1 mutants; therefore, the restoration effect of Kar2 appeared better in the Δhxl1 mutant than the Δire1 mutant, just like in the presence of TCM. However, the opposite is the case in the presence of DTT, where Δire1 mutant appeared better restored. Also, because Δhxl1 mutants are more sensitive to temperature, the restoration is better in the Δire1 mutants, but no restoration of growth defects above 37oC. Though both mutants are hypersensitive to cell wall disruption agents; however, restoration appeared better with the Δire1 mutant than Δhxl1. Collectively, Kar2 can be induced in the presence of azole and phenylpyrrole antifungals to regulate susceptibility in an Erg-independent manner. Though not as much as the reconstituted mutants, the overexpression of Kar2 can partially restore growth defects in Δire1 and Δhxl1 mutants at 37oC as well as support the cell wall integrity, genotoxic and antifungal responses (exclusively in Ire1-dependent manner at a low concentration of each antifungal). However, Kar2 appeared not to have any significant effect on the Ire1-mediated capsule production [225].
3.7. Osmotic Shock
Every eukaryotic cell has designed various mechanisms to ensure osmoregulation within the cytoplasm. An imbalance between the solute concentration and water content within the cell results in osmotic shock/stress. C. neoformans has evolved a unique mechanism to address solute fluctuation in the cytoplasm. This function is majorly centred on the phosphorylation-dephosphorylation relay activities of Pbs MAPKK and phosphotyrosine phosphatase (Ptp)/phosphoserine or phosphothreonine phosphatase (Ptc) associated with Hog1 protein. The Hog1 activity is primarily under the negative feedback repression of distinctive but redundant Ptp1 and Ptp2 and the downstream Aft1 regulator [149]. The Ptp generally promotes Hog1 dephosphorylation, thereby reducing the hyperphosphorylation of this p38-MAPK transcription factor. This phosphatase also promotes thermotolerance, osmotic, and oxidative stress resistance in C. neoformans via the Atf1p downstream regulator. Further evidence showed that both Ptp1p and Ptp2p localised more to the nucleus than the cytoplasm to anchor the nuclear transient movement of Hog1p in a stressed cellular state [229].
Hog1 MAPK is primarily responsible for the osmotic-stress response, and this becomes more effective as the level of constitutive phosphorylation of the Hog1p increases [81]. The expression of glycerol-3-phosphatase encoded by Gpp1 and glycerol-3-phosphate dehydrogenase encoded by Gpd1 is significantly linked to osmotic-stress response, and this expression has been shown to be 2-fold reduced in Δhog1 and Δssk1 mutants [81]. With the evidence of overlapping gene regulation, the Hog1 and Ssk1 expressions significantly controlled C. neoformans genes under stressed and unstressed conditions more than the Skn7 – a response regulator kinase, like Ssk1, activated by Tco system [81]. Ample of these genes are environmental stress regulatory (ESR) genes, common stress regulatory (CSR) genes, and stress-specific regulatory (SSR) genes. Apart from this, Ssk1p and Hog1p can independently regulate other cellular responses.
According to Bahn et al., the Hog1p of serotype A is constantly in the phosphorylated state (Hog1-P) in a normal cellular state; however, phosphorylation of the Hog1 in serotype D is induced by osmotic stress [32]. With respect to Tco sensor kinases, Hog1p, in the absence of Tco1 expression, is generally under dephosphorylation, but activation of Hog1 by dephosphorylation is delayed in the Δtco1Δtco2 or Δtco2 mutants, which possibly means that regulation of Hog1p may partly rely on the distal Tco2 sensor kinase via the Ssk1-Ssk2 MAPKKK signalling [149]. Generally, stress response is an energy-consuming process due to fashionably coordinated MAPK. Therefore, less attention is given to other high-energy consuming metabolisms, which leads to their genes being downregulated during the osmotic-stress response by C. neoformans.
The transient translocation of Hog1-P into serotype A nucleus ensures that the transcription initiation for virulence genes and mating is perpetually repressed. At the same time, osmotic stress induces dephosphorylation of the Hog1-P to promote adaptation to the osmotic shock via a concomitant activation of Ena1 (encoding a putative P-type ATPase Na+ pump), Nha1 (encoding a Na+/H+ antiporter), and Aqp1 (encoding aquaporin) genes for osmoregulation and ionic balance. Osmotically, 1 M sorbitol could only and transiently induce Ena1 but no effect on Nha1; likewise, 1 M NaCl or KCl is enough to induce Ena1 and Nha1 with higher induction in the formal than the latter [193]. This shows that under the control of the Hog1 pathway, the two transporters are involved in the Na+/K+ efflux in C. neoformans to maintain ionic homeostasis, but only Ena1 is involved in osmotic shock response. Specifically, both Ena1 and Nha1 are essential for the less toxic K+ homeostasis, but only Ena1 is majorly involved in toxic Na+ and Li+ cation homeostasis. Also, none of them is involved in Ca2+ homeostasis [193]. Regarding the environmental influence, Ena1 is induced in an alkaline medium where the concentration of the H+ is low to control Na+ or K+ stress. Nha1 is otherwise induced in an acidic environment to control K+ stress, while Ena1 expression is dispensable under this condition. Surprisingly, Jung et al. actually discovered that though Nha1p seemed to be active in the acidic medium, its induction was not strongly induced in the acidic medium per se under K+ stress [193].
Furthermore, apart from maintaining the intracellular pH (H+ homeostasis across plasma membrane), these membrane-located cation transporters, though not really involved in cell wall integrity but are required to maintain membrane potential, integrity, and stability for an effective antifungal counterattack under a moderate to low level production of capsule and melanin. Like Δhog1 mutants, deletion of Ena1 and Nha1 cumulatively improved capsule and melanin production at 37oC and 30oC, respectively. From Jung et al. observation, capsule diameter/volume marginally increased from Δnha1, Δena1 to Δena1Δnha1 mutants, with the double mutants having as much as a 2.8-fold increase in the Lac1 transcripts after 2 hours of incubation in YNB medium at 30oC. Nevertheless, none of these cationic transporters played any significant role in cell differentiation and mating, unlike Hog1 MAPK [193].
In terms of infection, C. neoformans seems to shut down completely the expression of Nha1 but not Ena1 for virulence factors. The expression of Nha1 is dispensable for virulence probably because the activating condition of Nha1 is scarcely experienced in vivo, but Ena1 expression is highly needed partly because of the alkaline systemic condition and micro-alkaline environment created by the C. neoformans urease activity [192,193,230]. This observation seems very subtle with C. neoformans during infection to reduce the cumulative effect of the two redundant cationic transporters on the virulence expression while maintaining ionic homeostasis with Ena1p and other essential ionic transporters such as Cft1, Pho84, Vph1, Ccc1, and Ctr1. Thus, the avirulence features of Δena1 and Δena1Δnha1 mutants in an animal model of cryptococcosis seem to come from impaired tissue dissemination and persistence but not systemic clearance.
The Δhog1 mutants are known to resist azole because of the increased expression of the Erg genes but are sensitive to polyene. One could have expected the same for the Δena1 and Δnha1 mutants because both are under the regulation of the Hog1 pathway; however, the Δena1Δnha1 mutant appeared highly sensitive to azole and polyene antifungal drugs more than the Δhog1 mutants in a manner independent of ergosterol biosynthesis [193]. It is not surprising, therefore, to observe hypersensitivity to Hygromycin B in Δhog1 mutants but hyper-resistance in Δena1, Δnha1, and Δena1Δnha1 mutants because the presence of cationic transporter allows for the hyper-polarisation caused by Hygromycin B in the Δhog1 mutants [193].
To avoid the redundancy of transcription factors and the bioeconomical loss of metabolites, any environmental cue that induces osmotic stress may likely repress the formation of virulence factors in C. neoformans. No wonder the mutation of any Hog1 pathway-associated transcription factors promotes oxidative stress and reduces osmo-tolerance, yet such mutations enormously enhance mating and the formation of virulence factors. The skn7 mutation, however, has no significant contribution to capsule production but melanin [30]. Contrarily, Hog1-P is continuously retained in the nucleus of serotype D after osmotic stress to initiate adaptation [32]. This means that the phosphorylation-dephosphorylation relay activity of Hog1p is differentially regulated in different serotypes to achieve the same goal. Interestingly, the upstream activity of Pbs2-MAPKK to phosphorylate Hog1 protein is highly conserved and inevitable in the functioning of the Hog1 cascade event. Therefore, any mutation in the Pbs2 gene will produce the same phenotypic defect as the Δhog1 mutant strain.
The Δena1 mutants are highly sensitive to osmotic stress caused by KCl and NaCl, especially in the absence of glucose, but in the presence of glucose, Δena1 mutants showed a wt resistance to osmotic stress caused by KCl and oxidative stress caused by H2O2 [81]. This indicates that Ena1p may not be needed against oxidative stress under normal conditions. Similar to Δcna1, Δena1 mutants are susceptible to alkaline pH and are upregulated at high pH in the wt [192]. Hyperosmolarity also upregulates the expression of Hnm1 (encoding putative high-affinity choline/ethanolamine transporter/permease) to cope with common environmental stress [81]. The upregulation of Hnm1, as observed in S. cerevisiae, may lead to a high turnover rate of membrane phosphatidylcholine to glycerophosphocholine in the presence of choline [231]. This glycerophosphocholine is an osmoprotective accumulated methylamine, earlier identified in Madin-Darby Canine Kidney renal (MDCK) cell line in the presence of high salt content and urea [232]. Perhaps, C. neoformans may be using a similar mechanism to defend against the high salinity and the urea content of the bird droppings.
In the wt strain, osmotic stress downregulates the expression of genes involved in carbohydrate metabolism, as evidenced in the reduced transcript level of Gal2, Hxt5, Hxt13, and Hxt17 and every other gene involved in actin/cytoskeleton formation, signal transduction, intracellular trafficking/secretion, and vesicular transport but because of the proximity of ionic homeostasis to osmotic stress, Cfo1 and Fre2 are concurrently upregulated [81]. In addition, diverse transporters and permeases such as Dur3, Mep2/Amt2, Stl1, Aqy1, Aqp1, Pho84, and Qdr1 are all activated to allow for the movement of osmolytes in a mechanism to promote cellular homeostasis during osmotic stress [81]. Therefore, the expression of all these specific osmotic stress regulatory genes is automatically let down in Δhog1 and Δssk1 mutants exposed to osmotic stress caused by NaCl and KCl. Unfortunately, but strategically, C. neoformans failed to deploy Pdr-like ABC efflux pumps against general osmotic stress in the presence or absence of glucose, oxidative stress, UV, and metal-induced stress, except against high concentrations of FDX and FCZ.
Lastly, the Δtco1 and Δtco2 mutants showed a wt resistance to osmotic stress caused by 1.5 M KCl. While Δtco1 was resistant to 0.05% SDS, Δtco2 mutant was relatively susceptible [126]. Though there was no significant effect of H2O2, UV exposure, high temperature, MG, and 1 – 1.5 M KCl osmotic stress on the Δskn7 mutant yet, the mutant showed extreme sensitivity to as low as 1 M NaCl in YPD media despite a similar wt Hog1p activation in this mutant [30]. This re-emphasises the scrupulous involvement of Skn7 expression in Na+-tolerance. On the contrary, Ko et al. observed a wt growth of Δskn7 to KCl only in the glucose-deprived media but a complete hypersensitivity to KCl and NaCl in YPD and glucose-deprived media, respectively [81] (check Supplementary 3 mutant responses to osmolytes). In view of this, C. neoformans has shown an intricately networking yet highly regulating transcription factors in response to stresses.
6. Conclusions and Perspectives
Signalling cascades, wired by intracellular proteins, as induced by various environmental factors are the basis for expressing multiple transcription factors that facilitate responses to immediate inconveniences. This review identified different clusters of transcription factors and extensively discussed their biological functions and associated phenotypic deficiencies. Furthermore, we identified cascades of events that showed the interconnection of these transcription factors and how they coordinate and regulate phenotypic traits and cellular responses to nutrients, light, oxidative and nitrosative stressors, disruptive membrane dyes/detergent, antifungals, CO2, pH, temperature, genotoxic, and cytotoxic compounds. These phenotypic characteristics manifest in virulent expressions, such as capsule, melanin, secretory, and hydrolytic enzymes. Besides, with the recruitment of these transcription factors, pathogenic Cryptococcus species invariably develop resistance to temperature, pH, nutrients depletion, oxidative and osmotic stress, phagocytosis, irradiation, and antifungals.
Strategically, C. neoformans cautiously repress some genes for others to function, while some are co-expressed for survival, adaptation, and infection. Being dominantly haploid, the mating of different serotypes facilitates survival, adaptation, aneuploidy, and genetic variation among the basidiospores. Natural diploid isolate also exists after the unisexual activity of the haploids. This provides the basis for gene segregation, redundancy, and ploidy. All these are carefully controlled under the regulation of different transcription factors related to mating, filamentation, and monokaryotic fruiting.
Targeting these key transcription factors by gene disruption or deletion has enabled an understanding of how this pathogenic/opportunistic fungus switches the arrays of genes to augment the deficiencies or give in to the impeding factors in some instances where such deletion is critical to cellular activity. It seems much is known yet there are transcription factors whose functions are unknown and their relevance to human infection remains folded. With this extensive review work, the importance of numerous but specific transcription factors is further expatiated, grouped, and linked together based on their functional expression regarding survival, adaptation, and human infection. This article provides adequate information to understand the cellular framework of C. neoformans relative to other fungi and offers insight into attractive transcription factors suitable for a therapeutic approach in drug targeting for more effective management and control of cryptococcal infection, especially in immunocompromised and organ-recipient individuals. With this, the antifungal chemotherapy will go beyond the classical ergosterol and translational process blockers.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, File: Genes, Transcription Factors, Protein Kinases, Intracellular Proteins, and Other Abbreviations as Used in the Article.
Author Contributions
Conceptualisation, O.S. and O.M.; methodology, O.S.; software, O.S.; validation, O.S. and O.M.; formal analysis, O.S.; investigation, O.S.; resources, O.S. and O.M.; data curation, O.S.; writing—original draft preparation, O.S.; writing—review and editing, O.S. and O.M.; visualization, O.S. and O.M.; supervision, O.M.; project administration, O.M.; funding acquisition, O.M. All authors have read and agreed to the published version of the manuscript.
Funding
The authors declared that no funding was obtained for this article.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors appreciated the conducive research environment and administrative procedures put in place by the Research Office of University of the Free State.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Feldmesser, M.; Kress, Y.; Novikoff, P.; Casadevall, A. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect Immun 2000, 68, 4225–4237. [Google Scholar] [CrossRef] [PubMed]
- Elhariri, M.; Hamza, D.; Elhelw, R.; Refai, M. Eucalyptus Tree: A Potential Source of Cryptococcus neoformans in Egyptian Environment. Int J Microbiol 2016, 2016, 4080725. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, M.; D'Amicis, R.; Zani, A.; Montagna, M.T.; Caggiano, G.; De Giglio, O.; Balbino, S.; De Donno, A.; Serio, F.; Susever, S. Environmental distribution of Cryptococcus neoformans and C. gattii around the Mediterranean basin. FEMS yeast research 2016, 16. [Google Scholar] [CrossRef]
- Ergin, C.; Sengul, M.; Aksoy, L.; Dogen, A.; Sun, S.; Averette, A.F.; Cuomo, C.A.; Seyedmousavi, S.; Heitman, J.; Ilkit, M. Cryptococcus neoformans Recovered From Olive Trees (Olea europaea) in Turkey Reveal Allopatry With African and South American Lineages. Front Cell Infect Microbiol 2019, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, J.N.; Casadevall, A. The origin and maintenance of virulence for the human pathogenic fungus Cryptococcus neoformans. Microbes Infect 2003, 5, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A. Evolution of intracellular pathogens. Annu Rev Microbiol 2008, 62, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, J.N.; Shuman, H.A.; Casadevall, A. Cryptococcus neoformans interactions with amoebae suggest an explanation for its virulence and intracellular pathogenic strategy in macrophages. Proc Natl Acad Sci U S A 2001, 98, 15245–15250. [Google Scholar] [CrossRef]
- Derengowski Lda, S.; Paes, H.C.; Albuquerque, P.; Tavares, A.H.; Fernandes, L.; Silva-Pereira, I.; Casadevall, A. The transcriptional response of Cryptococcus neoformans to ingestion by Acanthamoeba castellanii and macrophages provides insights into the evolutionary adaptation to the mammalian host. Eukaryot Cell 2013, 12, 761–774. [Google Scholar] [CrossRef]
- Casadevall, A. Amoeba provide insight into the origin of virulence in pathogenic fungi. In Recent advances on model hosts; Springer: 2012; pp. 1-10. [CrossRef]
- Steenbergen, J.N.; Nosanchuk, J.D.; Malliaris, S.D.; Casadevall, A. Cryptococcus neoformans virulence is enhanced after growth in the genetically malleable host Dictyostelium discoideum. Infect Immun 2003, 71, 4862–4872. [Google Scholar] [CrossRef]
- Paul, C.; Emeka, N. Pathogenicity of Cryptococcus neoformans VNI (ST 32) recovered from environmental and clinical isolates in Nigeria. Comparative Clinical Pathology 2019, 28, 1013–1024. [Google Scholar] [CrossRef]
- Litvintseva, A.P.; Kestenbaum, L.; Vilgalys, R.; Mitchell, T.G. Comparative analysis of environmental and clinical populations of Cryptococcus neoformans. J Clin Microbiol 2005, 43, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Litvintseva, A.P.; Thakur, R.; Vilgalys, R.; Mitchell, T.G. Multilocus sequence typing reveals three genetic subpopulations of Cryptococcus neoformans var. grubii (serotype A), including a unique population in Botswana. Genetics 2006, 172, 2223–2238. [Google Scholar] [CrossRef] [PubMed]
- Litvintseva, A.P.; Mitchell, T.G. Most environmental isolates of Cryptococcus neoformans var. grubii (serotype A) are not lethal for mice. Infection and immunity 2009, 77, 3188–3195. [Google Scholar]
- Casadevall, A.; Rosas, A.L.; Nosanchuk, J.D. Melanin and virulence in Cryptococcus neoformans. Curr Opin Microbiol 2000, 3, 354–358. [Google Scholar] [CrossRef]
- Upadhya, R.; Baker, L.G.; Lam, W.C.; Specht, C.A.; Donlin, M.J.; Lodge, J.K. Cryptococcus neoformans Cda1 and Its Chitin Deacetylase Activity Are Required for Fungal Pathogenesis. mBio 2018, 9, e02087–02018. [Google Scholar] [CrossRef]
- Fan, W.; Kraus, P.R.; Boily, M.J.; Heitman, J. Cryptococcus neoformans gene expression during murine macrophage infection. Eukaryot Cell 2005, 4, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Goulart, L.; Rosa e Silva, L.K.; Chiapello, L.; Silveira, C.; Crestani, J.; Masih, D.; Vainstein, M.H. Cryptococcus neoformans and Cryptococcus gattii genes preferentially expressed during rat macrophage infection. Med Mycol 2010, 48, 932–941. [Google Scholar] [CrossRef]
- Himmelreich, U.; Allen, C.; Dowd, S.; Malik, R.; Shehan, B.P.; Mountford, C.; Sorrell, T.C. Identification of metabolites of importance in the pathogenesis of pulmonary cryptococcoma using nuclear magnetic resonance spectroscopy. Microbes Infect 2003, 5, 285–290. [Google Scholar] [CrossRef]
- Hu, G.; Cheng, P.Y.; Sham, A.; Perfect, J.R.; Kronstad, J.W. Metabolic adaptation in Cryptococcus neoformans during early murine pulmonary infection. Mol Microbiol 2008, 69, 1456–1475. [Google Scholar] [CrossRef]
- Kronstad, J.; Saikia, S.; Nielson, E.D.; Kretschmer, M.; Jung, W.; Hu, G.; Geddes, J.M.; Griffiths, E.J.; Choi, J.; Cadieux, B.; et al. Adaptation of Cryptococcus neoformans to mammalian hosts: integrated regulation of metabolism and virulence. Eukaryot Cell 2012, 11, 109–118. [Google Scholar] [CrossRef]
- Chun, C.D.; Brown, J.C.S.; Madhani, H.D. A major role for capsule-independent phagocytosis-inhibitory mechanisms in mammalian infection by Cryptococcus neoformans. Cell Host Microbe 2011, 9, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Chun, C.D.; Liu, O.W.; Madhani, H.D. A link between virulence and homeostatic responses to hypoxia during infection by the human fungal pathogen Cryptococcus neoformans. PLoS Pathog 2007, 3, e22. [Google Scholar] [CrossRef]
- Chun, C.D.; Madhani, H.D. Ctr2 links copper homeostasis to polysaccharide capsule formation and phagocytosis inhibition in the human fungal pathogen Cryptococcus neoformans. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.K.; D'Souza, C.A.; Cox, G.M.; Heitman, J. Cyclic AMP-dependent protein kinase catalytic subunits have divergent roles in virulence factor production in two varieties of the fungal pathogen Cryptococcus neoformans. Eukaryot Cell 2004, 3, 14–26. [Google Scholar] [CrossRef]
- Lengeler, K.B.; Wang, P.; Cox, G.M.; Perfect, J.R.; Heitman, J. Identification of the MATa mating-type locus of Cryptococcus neoformans reveals a serotype A MATa strain thought to have been extinct. Proc Natl Acad Sci U S A 2000, 97, 14455–14460. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Wickes, B.L.; Miller, G.F.; Penoyer, L.A.; Kwon-Chung, K.J. Cryptococcus neoformans STE12α regulates virulence but is not essential for mating. J Exp Med 2000, 191, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Wickes, B.L.; Edman, U.; Edman, J.C. The Cryptococcus neoformans STE12alpha gene: a putative Saccharomyces cerevisiae STE12 homologue that is mating type specific. Mol Microbiol 1997, 26, 951–960. [Google Scholar] [CrossRef]
- Yue, C.; Cavallo, L.M.; Alspaugh, J.A.; Wang, P.; Cox, G.M.; Perfect, J.R.; Heitman, J. The STE12α Homolog Is Required for Haploid Filamentation But Largely Dispensable for Mating and Virulence in Cryptococcus neoformans. Genetics 1999, 153, 1601–1615. [Google Scholar] [CrossRef]
- Bahn, Y.S.; Kojima, K.; Cox, G.M.; Heitman, J. A unique fungal two-component system regulates stress responses, drug sensitivity, sexual development, and virulence of Cryptococcus neoformans. Mol Biol Cell 2006, 17, 3122–3135. [Google Scholar] [CrossRef]
- Kojima, K.; Bahn, Y.S.; Heitman, J. Calcineurin, Mpk1 and Hog1 MAPK pathways independently control fludioxonil antifungal sensitivity in Cryptococcus neoformans. Microbiology (Reading) 2006, 152, 591–604. [Google Scholar] [CrossRef]
- Bahn, Y.S.; Kojima, K.; Cox, G.M.; Heitman, J. Specialisation of the HOG pathway and its impact on differentiation and virulence of Cryptococcus neoformans. Mol Biol Cell 2005, 16, 2285–2300. [Google Scholar] [CrossRef]
- Cruz, M.C.; Sia, R.A.L.; Olson, M.; Cox, G.M.; Heitman, J. Comparison of the Roles of Calcineurin in Physiology and Virulence in Serotype D and Serotype A Strains ofCryptococcus neoformans. Infection and Immunity 2000, 68, 982–985. [Google Scholar] [CrossRef]
- Yang, J.; Li, D.; Liu, X.; Pan, J.; Yan, B.; Zhu, X. Regulation of virulence factors, carbon utilisation and virulence by SNF1 in Cryptococcus neoformans JEC21 and divergent actions of SNF1 between cryptococcal strains. Fungal Genet Biol 2010, 47, 994–1000. [Google Scholar] [CrossRef]
- Free, S.J. Fungal cell wall organisation and biosynthesis. In Advances in genetics; Elsevier: 2013; Volume 81, pp. 33-82. [CrossRef]
- Lee, C.G.; Da Silva, C.A.; Dela Cruz, C.S.; Ahangari, F.; Ma, B.; Kang, M.J.; He, C.H.; Takyar, S.; Elias, J.A. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol 2011, 73, 479–501. [Google Scholar] [CrossRef]
- Fries, B.C.; Goldman, D.L.; Cherniak, R.; Ju, R.; Casadevall, A. Phenotypic Switching in Cryptococcus neoformans Results in Changes in Cellular Morphology and Glucuronoxylomannan Structure. Infection and immunity 1999, 67, 6076–6083. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nakayasu, E.S.; Oliveira, D.L.; Nimrichter, L.; Nosanchuk, J.D.; Almeida, I.C.; Casadevall, A. Extracellular vesicles produced by Cryptococcus neoformans contain protein components associated with virulence. Eukaryot Cell 2008, 7, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Banks, I.R.; Specht, C.A.; Donlin, M.J.; Gerik, K.J.; Levitz, S.M.; Lodge, J.K. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot Cell 2005, 4, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.G.; Specht, C.A.; Donlin, M.J.; Lodge, J.K. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell 2007, 6, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.G.; Specht, C.A.; Lodge, J.K. Cell wall chitosan is necessary for virulence in the opportunistic pathogen Cryptococcus neoformans. Eukaryot Cell 2011, 10, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, R.; Lam, W.C.; Maybruck, B.; Specht, C.A.; Levitz, S.M.; Lodge, J.K. Induction of Protective Immunity to Cryptococcal Infection in Mice by a Heat-Killed, Chitosan-Deficient Strain of Cryptococcus neoformans. mBio 2016, 7, e00547–00516. [Google Scholar] [CrossRef] [PubMed]
- Ohtakara, A.; Izume, M.; Mitsutomi, M. Action of Microbial Chitinases on Chitosan with Different Degrees of Deacetylation. Agricultural and Biological Chemistry 2014, 52, 3181–3182. [Google Scholar] [CrossRef]
- Botts, M.R.; Hull, C.M. Dueling in the lung: how Cryptococcus spores race the host for survival. Curr Opin Microbiol 2010, 13, 437–442. [Google Scholar] [CrossRef]
- Botts, M.R.; Giles, S.S.; Gates, M.A.; Kozel, T.R.; Hull, C.M. Isolation and characterisation of Cryptococcus neoformans spores reveal a critical role for capsule biosynthesis genes in spore biogenesis. Eukaryot Cell 2009, 8, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Sukroongreung, S.; Kitiniyom, K.; Nilakul, C.; Tantimavanich, S. Pathogenicity of basidiospores of Filobasidiella neoformans var. neoformans. Med Mycol 1998, 36, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, R.; Hsueh, Y.P.; Geunes-Boyer, S.; Wright, J.R.; Heitman, J. Spores as infectious propagules of Cryptococcus neoformans. Infect Immun 2009, 77, 4345–4355. [Google Scholar] [CrossRef]
- Neilson, J.B.; Ivey, M.H.; Bulmer, G.S. Cryptococcus neoformans: pseudohyphal forms surviving culture with Acanthamoeba polyphaga. Infect Immun 1978, 20, 262–266. [Google Scholar] [CrossRef]
- Lee, S.C.; Phadke, S.; Sun, S.; Heitman, J. Pseudohyphal growth of Cryptococcus neoformans is a reversible dimorphic transition in response to ammonium that requires Amt1 and Amt2 ammonium permeases. Eukaryot Cell 2012, 11, 1391–1398. [Google Scholar] [CrossRef]
- Wang, L.; Zhai, B.; Lin, X. The link between morphotype transition and virulence in Cryptococcus neoformans. PLoS Pathog 2012, 8, e1002765. [Google Scholar] [CrossRef]
- Lin, X.; Jackson, J.C.; Feretzaki, M.; Xue, C.; Heitman, J. Transcription factors Mat2 and Znf2 operate cellular circuits orchestrating opposite- and same-sex mating in Cryptococcus neoformans. PLoS Genet 2010, 6, e1000953. [Google Scholar] [CrossRef]
- Magditch, D.A.; Liu, T.B.; Xue, C.; Idnurm, A. DNA mutations mediate microevolution between host-adapted forms of the pathogenic fungus Cryptococcus neoformans. PLoS Pathog 2012, 8, e1002936. [Google Scholar] [CrossRef]
- Okagaki, L.H.; Nielsen, K. Titan cells confer protection from phagocytosis in Cryptococcus neoformans infections. Eukaryot Cell 2012, 11, 820–826. [Google Scholar] [CrossRef]
- Okagaki, L.H.; Wang, Y.; Ballou, E.R.; O'Meara, T.R.; Bahn, Y.S.; Alspaugh, J.A.; Xue, C.; Nielsen, K. Cryptococcal titan cell formation is regulated by G-protein signaling in response to multiple stimuli. Eukaryot Cell 2011, 10, 1306–1316. [Google Scholar] [CrossRef]
- Okagaki, L.H.; Strain, A.K.; Nielsen, J.N.; Charlier, C.; Baltes, N.J.; Chretien, F.; Heitman, J.; Dromer, F.; Nielsen, K. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog 2010, 6, e1000953. [Google Scholar] [CrossRef]
- Alspaugh, J.A. Virulence mechanisms and Cryptococcus neoformans pathogenesis. Fungal Genet Biol 2015, 78, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, O.; Garcia-Rodas, R.; Nosanchuk, J.D.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L.; Casadevall, A. Fungal cell gigantism during mammalian infection. PLoS Pathog 2010, 6, e1000945. [Google Scholar] [CrossRef]
- Zaragoza, O.; Nielsen, K. Titan cells in Cryptococcus neoformans: cells with a giant impact. Curr Opin Microbiol 2013, 16, 409–413. [Google Scholar] [CrossRef]
- Idnurm, A. A tetrad analysis of the basidiomycete fungus Cryptococcus neoformans. Genetics 2010, 185, 153–163. [Google Scholar] [CrossRef]
- Brandt, M.E.; Pfaller, M.A.; Hajjeh, R.A.; Graviss, E.A.; Rees, J.; Spitzer, E.D.; Pinner, R.W.; Mayer, L.W. Molecular subtypes and antifungal susceptibilities of serial Cryptococcus neoformans isolates in human immunodeficiency virus-associated Cryptococcosis. Cryptococcal Disease Active Surveillance Group. J Infect Dis 1996, 174, 812–820. [Google Scholar] [CrossRef]
- Fries, B.C.; Chen, F.; Currie, B.P.; Casadevall, A. Karyotype instability in Cryptococcus neoformans infection. J Clin Microbiol 1996, 34, 1531–1534. [Google Scholar] [CrossRef]
- Walton, F.J.; Heitman, J.; Idnurm, A. Conserved elements of the RAM signaling pathway establish cell polarity in the basidiomycete Cryptococcus neoformans in a divergent fashion from other fungi. Mol Biol Cell 2006, 17, 3768–3780. [Google Scholar] [CrossRef] [PubMed]
- Kozubowski, L.; Aboobakar, E.F.; Cardenas, M.E.; Heitman, J. Calcineurin colocalises with P-bodies and stress granules during thermal stress in Cryptococcus neoformans. Eukaryot Cell 2011, 10, 1396–1402. [Google Scholar] [CrossRef]
- Nelson, B.; Kurischko, C.; Horecka, J.; Mody, M.; Nair, P.; Pratt, L.; Zougman, A.; McBroom, L.D.; Hughes, T.R.; Boone, C.; et al. RAM: a conserved signaling network that regulates Ace2p transcriptional activity and polarised morphogenesis. Mol Biol Cell 2003, 14, 3782–3803. [Google Scholar] [CrossRef]
- Racki, W.J.; Becam, A.M.; Nasr, F.; Herbert, C.J. Cbk1p, a protein similar to the human myotonic dystrophy kinase, is essential for normal morphogenesis in Saccharomyces cerevisiae. EMBO J 2000, 19, 4524–4532. [Google Scholar] [CrossRef]
- Zaragoza, O.; Rodrigues, M.L.; De Jesus, M.; Frases, S.; Dadachova, E.; Casadevall, A. The capsule of the fungal pathogen Cryptococcus neoformans. Adv Appl Microbiol 2009, 68, 133–216. [Google Scholar] [CrossRef]
- Vartivarian, S.E.; Anaissie, E.J.; Cowart, R.E.; Sprigg, H.A.; Tingler, M.J.; Jacobson, E.S. Regulation of cryptococcal capsular polysaccharide by iron. J Infect Dis 1993, 167, 186–190. [Google Scholar] [CrossRef]
- Granger, D.L.; Perfect, J.R.; Durack, D.T. Virulence of Cryptococcus neoformans. Regulation of capsule synthesis by carbon dioxide. J Clin Invest 1985, 76, 508–516. [Google Scholar] [CrossRef]
- McFadden, D.C.; Fries, B.C.; Wang, F.; Casadevall, A. Capsule structural heterogeneity and antigenic variation in Cryptococcus neoformans. Eukaryot Cell 2007, 6, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Fries, B.C.; Cook, E.; Wang, X.; Casadevall, A. Effects of antifungal interventions on the outcome of experimental infections with phenotypic switch variants of Cryptococcus neoformans. Antimicrob Agents Chemother 2005, 49, 350–357. [Google Scholar] [CrossRef]
- Vecchiarelli, A.; Retini, C.; Pietrella, D.; Monari, C.; Tascini, C.; Beccari, T.; Kozel, T.R. Downregulation by cryptococcal polysaccharide of TNFα and IL-1β secretion from human monocytes. Infect Immun 1995, 63, 2919–2923. [Google Scholar] [CrossRef] [PubMed]
- Vecchiarelli, A.; Retini, C.; Monari, C.; Tascini, C.; Bistoni, F.; Kozel, T.R. Purified capsular polysaccharide of Cryptococcus neoformans induces interleukin-10 secretion by human monocytes. Infect Immun 1996, 64, 2846–2849. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, E.S.; Compton, G.M. Discordant regulation of phenoloxidase and capsular polysaccharide in Cryptococcus neoformans. J Med Vet Mycol 1996, 34, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Reiss, E.; Cherniak, R.; Eby, R.; Kaufman, L. Enzyme immunoassay detection of IgM to galactoxylomannan of Cryptococcus neoformans. Diagn Immunol 1984, 2, 109–115. [Google Scholar] [PubMed]
- Dong, Z.M.; Murphy, J.W. Cryptococcal polysaccharides induce L-selectin shedding and tumor necrosis factor receptor loss from the surface of human neutrophils. J Clin Invest 1996, 97, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Ben-Abdallah, M.; Sturny-Leclere, A.; Ave, P.; Louise, A.; Moyrand, F.; Weih, F.; Janbon, G.; Memet, S. Fungal-induced cell cycle impairment, chromosome instability and apoptosis via differential activation of NF-kappaB. PLoS Pathog 2012, 8, e1002555. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Hughes, T.M.; Nguyen, H.Q.; Trebasky, L.D.; Danilenko, D.M.; Medlock, E.S. Long-term impaired neutrophil migration in mice overexpressing human interleukin-8. J Clin Invest 1994, 94, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.K.; Bennett, J.E.; Glaudemans, C.P. Capsular polysaccharides of Cryptococcus neoformans. Rev Infect Dis 1984, 6, 619–624. [Google Scholar] [CrossRef]
- Fonseca, F.L.; Nohara, L.L.; Cordero, R.J.; Frases, S.; Casadevall, A.; Almeida, I.C.; Nimrichter, L.; Rodrigues, M.L. Immunomodulatory effects of serotype B glucuronoxylomannan from Cryptococcus gattii correlate with polysaccharide diameter. Infect Immun 2010, 78, 3861–3870. [Google Scholar] [CrossRef]
- Dong, Z.M.; Murphy, J.W. Effects of the two varieties of Cryptococcus neoformans cells and culture filtrate antigens on neutrophil locomotion. Infect Immun 1995, 63, 2632–2644. [Google Scholar] [CrossRef]
- Ko, Y.J.; Yu, Y.M.; Kim, G.B.; Lee, G.W.; Maeng, P.J.; Kim, S.; Floyd, A.; Heitman, J.; Bahn, Y.S. Remodeling of global transcription patterns of Cryptococcus neoformans genes mediated by the stress-activated HOG signaling pathways. Eukaryot Cell 2009, 8, 1197–1217. [Google Scholar] [CrossRef]
- Lee, J.W.; Ko, Y.J.; Kim, S.Y.; Bahn, Y.S. Multiple roles of Ypd1 phosphotransfer protein in viability, stress response, and virulence factor regulation in Cryptococcus neoformans. Eukaryot Cell 2011, 10, 998–1002. [Google Scholar] [CrossRef]
- D'Souza, C.A.; Alspaugh, J.A.; Yue, C.; Harashima, T.; Cox, G.M.; Perfect, J.R.; Heitman, J. Cyclic AMP-dependent protein kinase controls virulence of the fungal pathogen Cryptococcus neoformans. Mol Cell Biol 2001, 21, 3179–3191. [Google Scholar] [CrossRef] [PubMed]
- Alspaugh, J.A.; Perfect, J.R.; Heitman, J. Cryptococcus neoformans mating and virulence are regulated by the G-protein alpha subunit GPA1 and cAMP. Genes Dev 1997, 11, 3206–3217. [Google Scholar] [CrossRef]
- Cruickshank, J.G.; Cavill, R.; Jelbert, M. Cryptococcus neoformans of unusual morphology. Appl Microbiol 1973, 25, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Love, G.L.; Boyd, G.D.; Greer, D.L. Large Cryptococcus neoformans isolated from brain abscess. J Clin Microbiol 1985, 22, 1068–1070. [Google Scholar] [CrossRef]
- Li, L.; Shen, G.; Zhang, Z.G.; Wang, Y.L.; Thompson, J.K.; Wang, P. Canonical heterotrimeric G proteins regulating mating and virulence of Cryptococcus neoformans. Mol Biol Cell 2007, 18, 4201–4209. [Google Scholar] [CrossRef]
- Lian, T.; Simmer, M.I.; D'Souza, C.A.; Steen, B.R.; Zuyderduyn, S.D.; Jones, S.J.; Marra, M.A.; Kronstad, J.W. Iron-regulated transcription and capsule formation in the fungal pathogen Cryptococcus neoformans. Mol Microbiol 2005, 55, 1452–1472. [Google Scholar] [CrossRef]
- Polacheck, I.; Hearing, V.J.; Kwon-Chung, K.J. Biochemical studies of phenoloxidase and utilisation of catecholamines in Cryptococcus neoformans. J Bacteriol 1982, 150, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Moyrand, F.; Chang, Y.C.; Himmelreich, U.; Kwon-Chung, K.J.; Janbon, G. Cas3p belongs to a seven-member family of capsule structure designer proteins. Eukaryot Cell 2004, 3, 1513–1524. [Google Scholar] [CrossRef]
- Kim, M.S.; Ko, Y.J.; Maeng, S.; Floyd, A.; Heitman, J.; Bahn, Y.S. Comparative transcriptome analysis of the CO2 sensing pathway via differential expression of carbonic anhydrase in Cryptococcus neoformans. Genetics 2010, 185, 1207–1219. [Google Scholar] [CrossRef]
- Nurudeen, T.A.; Ahearn, D.G. Regulation of melanin production by Cryptococcus neoformans. J Clin Microbiol 1979, 10, 724–729. [Google Scholar] [CrossRef]
- Hamilton, A. Cryptococcus neoformans-the encapsulated menace. Mycologist 2002, 16, 125–126. [Google Scholar] [CrossRef]
- Nosanchuk, J.D.; Rudolph, J.; Rosas, A.L.; Casadevall, A. Evidence that Cryptococcus neoformans is melanized in pigeon excreta: implications for pathogenesis. Infect Immun 1999, 67, 5477–5479. [Google Scholar] [CrossRef] [PubMed]
- Frases, S.; Salazar, A.; Dadachova, E.; Casadevall, A. Cryptococcus neoformans can utilise the bacterial melanin precursor homogentisic acid for fungal melanogenesis. Appl Environ Microbiol 2007, 73, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Frases, S.; Chaskes, S.; Dadachova, E.; Casadevall, A. Induction by Klebsiella aerogenes of a melanin-like pigment in Cryptococcus neoformans. Appl Environ Microbiol 2006, 72, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Piattelli, M.; Fattorusso, E.; Magno, S.; Nicolaus, R.A. Ustilago Melanin, a Naturally Occurring Catechol Melanin. Tetrahedron Letters 1963, 4, 997–998. [Google Scholar] [CrossRef]
- Wang, Y.; Aisen, P.; Casadevall, A. Cryptococcus neoformans melanin and virulence: mechanism of action. Infect Immun 1995, 63, 3131–3136. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.E.; Wheeler, M.H.; Szaniszlo, P.J. Evidence for Pentaketide Melanin Biosynthesis in Dematiaceous Human Pathogenic Fungi. Mycologia 1987, 79, 320–322. [Google Scholar] [CrossRef]
- Stüssi, H.; Rast, D.M. The biosynthesis and possible function of γ-glutaminyl-4-hydroxybenzene in Agaricus bisporus. Phytochemistry 1981, 20, 2347–2352. [Google Scholar] [CrossRef]
- Leonowicz, A.; Cho, N.S.; Luterek, J.; Wilkolazka, A.; Wojtas-Wasilewska, M.; Matuszewska, A.; Hofrichter, M.; Wesenberg, D.; Rogalski, J. Fungal laccase: properties and activity on lignin. J Basic Microbiol 2001, 41, 185–227. [Google Scholar] [CrossRef]
- Garcia-Rivera, J.; Eisenman, H.C.; Nosanchuk, J.D.; Aisen, P.; Zaragoza, O.; Moadel, T.; Dadachova, E.; Casadevall, A. Comparative analysis of Cryptococcus neoformans acid-resistant particles generated from pigmented cells grown in different laccase substrates. Fungal Genet Biol 2005, 42, 989–998. [Google Scholar] [CrossRef]
- Chaskes, S.; Tyndall, R.L. Pigment production by Cryptococcus neoformans from para- and ortho-Diphenols: effect of the nitrogen source. J Clin Microbiol 1975, 1, 509–514. [Google Scholar] [CrossRef]
- Wheeler, M.H.; Bell, A.A. Melanins and their importance in pathogenic fungi. In Current topics in medical mycology; Springer: 1988; pp. 338-387. [CrossRef]
- Maeda, T. The signaling mechanism of ambient pH sensing and adaptation in yeast and fungi. FEBS J 2012, 279, 1407–1413. [Google Scholar] [CrossRef]
- Godinho, R.M.; Crestani, J.; Kmetzsch, L.; Araujo Gde, S.; Frases, S.; Staats, C.C.; Schrank, A.; Vainstein, M.H.; Rodrigues, M.L. The vacuolar-sorting protein Snf7 is required for export of virulence determinants in members of the Cryptococcus neoformans complex. Sci Rep 2014, 4, 6198. [Google Scholar] [CrossRef] [PubMed]
- Sussman, A.S. The Fungal Population: An Advanced Treatise; Elsevier Science: 2013; pp. 447-486.
- Garcia-Rivera, J.; Casadevall, A. Melanisation of Cryptococcus neoformans reduces its susceptibility to the antimicrobial effects of silver nitrate. Med Mycol 2001, 39, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Casadevall, A. Growth of Cryptococcus neoformans in presence of L-dopa decreases its susceptibility to amphotericin B. Antimicrob Agents Chemother 1994, 38, 2648–2650. [Google Scholar] [CrossRef]
- Wang, Y.; Casadevall, A. Susceptibility of melanised and nonmelanised Cryptococcus neoformans to nitrogen- and oxygen-derived oxidants. Infect Immun 1994, 62, 3004–3007. [Google Scholar] [CrossRef] [PubMed]
- Durrell, L.W. The Composition and Structure of Walls of Dark Fungus Spores. Mycopathol Mycol Appl 1964, 23, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Rosas, A.L.; Casadevall, A. Melanisation affects susceptibility of Cryptococcus neoformans to heat and cold. FEMS Microbiol Lett 1997, 153, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Morison, W.L. What is the function of melanin? Archives of dermatology 1985, 121, 1160–1163. [Google Scholar] [CrossRef]
- Hill, H.Z. The function of melanin or six blind people examine an elephant. Bioessays 1992, 14, 49–56. [Google Scholar] [CrossRef]
- Wang, Y.; Aisen, P.; Casadevall, A. Melanin, melanin "ghosts," and melanin composition in Cryptococcus neoformans. Infect Immun 1996, 64, 2420–2424. [Google Scholar] [CrossRef]
- Rosas, A.L.; Nosanchuk, J.D.; Gomez, B.L.; Edens, W.A.; Henson, J.M.; Casadevall, A. Isolation and serological analyses of fungal melanins. J Immunol Methods 2000, 244, 69–80. [Google Scholar] [CrossRef]
- Wang, Y.; Casadevall, A. Susceptibility of melanised and nonmelanised Cryptococcus neoformans to the melanin-binding compounds trifluoperazine and chloroquine. Antimicrob Agents Chemother 1996, 40, 541–545. [Google Scholar] [CrossRef]
- Doering, T.L.; Nosanchuk, J.D.; Roberts, W.K.; Casadevall, A. Melanin as a potential cryptococcal defence against microbicidal proteins. Medical Mycology 1999, 37, 175–181. [Google Scholar] [CrossRef]
- Zhu, X.; Gibbons, J.; Zhang, S.; Williamson, P.R. Copper-mediated reversal of defective laccase in a Deltavph1 avirulent mutant of Cryptococcus neoformans. Mol Microbiol 2003, 47, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Siafakas, A.R.; Wright, L.C.; Sorrell, T.C.; Djordjevic, J.T. Lipid rafts in Cryptococcus neoformans concentrate the virulence determinants phospholipase B1 and Cu/Zn superoxide dismutase. Eukaryot Cell 2006, 5, 488–498. [Google Scholar] [CrossRef]
- Williamson, P.R. Biochemical and molecular characterisation of the diphenol oxidase of Cryptococcus neoformans: identification as a laccase. J Bacteriol 1994, 176, 656–664. [Google Scholar] [CrossRef]
- Torres-Guererro, H.; Edman, J.C. Melanin-deficient mutants of Cryptococcus neoformans. J Med Vet Mycol 1994, 32, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Pukkila-Worley, R.; Gerrald, Q.D.; Kraus, P.R.; Boily, M.J.; Davis, M.J.; Giles, S.S.; Cox, G.M.; Heitman, J.; Alspaugh, J.A. Transcriptional network of multiple capsule and melanin genes governed by the Cryptococcus neoformans cyclic AMP cascade. Eukaryot Cell 2005, 4, 190–201. [Google Scholar] [CrossRef]
- Lee, D.; Jang, E.H.; Lee, M.; Kim, S.W.; Lee, Y.; Lee, K.T.; Bahn, Y.S. Unraveling Melanin Biosynthesis and Signaling Networks in Cryptococcus neoformans. mBio 2019, 10, e02267–02219. [Google Scholar] [CrossRef] [PubMed]
- Walton, F.J.; Idnurm, A.; Heitman, J. Novel gene functions required for melanisation of the human pathogen Cryptococcus neoformans. Mol Microbiol 2005, 57, 1381–1396. [Google Scholar] [CrossRef]
- Song, M.H.; Lee, J.W.; Kim, M.S.; Yoon, J.K.; White, T.C.; Floyd, A.; Heitman, J.; Strain, A.K.; Nielsen, J.N.; Nielsen, K.; et al. A flucytosine-responsive Mbp1/Swi4-like protein, Mbs1, plays pleiotropic roles in antifungal drug resistance, stress response, and virulence of Cryptococcus neoformans. Eukaryot Cell 2012, 11, 53–67. [Google Scholar] [CrossRef]
- Nosanchuk, J.D.; Rosas, A.L.; Lee, S.C.; Casadevall, A. Melanisation of Cryptococcus neoformans in human brain tissue. Lancet 2000, 355, 2049–2050. [Google Scholar] [CrossRef]
- Rosas, A.L.; Nosanchuk, J.D.; Feldmesser, M.; Cox, G.M.; McDade, H.C.; Casadevall, A. Synthesis of polymerised melanin by Cryptococcus neoformans in infected rodents. Infect Immun 2000, 68, 2845–2853. [Google Scholar] [CrossRef]
- Liu, L.; Wakamatsu, K.; Ito, S.; Williamson, P.R. Catecholamine oxidative products, but not melanin, are produced by Cryptococcus neoformans during neuropathogenesis in mice. Infect Immun 1999, 67, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Chemical degradation of melanins: application to identification of dopamine-melanin. Pigment Cell Res 1998, 11, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Pezzella, A.; d'Ischia, M.; Prota, G. New pyrrole acids by oxidative degradation of eumelanins with hydrogen peroxide. Further hints to the mechanism of pigment breakdown. Tetrahedron 1996, 52, 8775–8780. [Google Scholar] [CrossRef]
- Napolitano, A.; Pezzella, A.; Vincensi, M.R.; Prota, G. Oxidative degradation of melanins to pyrrole acids: A model study. Tetrahedron 1995, 51, 5913–5920. [Google Scholar] [CrossRef]
- Wang, P.; Cutler, J.; King, J.; Palmer, D. Mutation of the regulator of G protein signaling Crg1 increases virulence in Cryptococcus neoformans. Eukaryot Cell 2004, 3, 1028–1035. [Google Scholar] [CrossRef]
- Luberto, C.; Toffaletti, D.L.; Wills, E.A.; Tucker, S.C.; Casadevall, A.; Perfect, J.R.; Hannun, Y.A.; Del Poeta, M. Roles for inositol-phosphoryl ceramide synthase 1 (IPC1) in pathogenesis of C. neoformans. Genes Dev 2001, 15, 201–212. [Google Scholar] [CrossRef]
- Alspaugh, J.A.; Cavallo, L.M.; Perfect, J.R.; Heitman, J. RAS1 regulates filamentation, mating and growth at high temperature of Cryptococcus neoformans. Mol Microbiol 2000, 36, 352–365. [Google Scholar] [CrossRef]
- Waugh, M.S.; Nichols, C.B.; DeCesare, C.M.; Cox, G.M.; Heitman, J.; Alspaugh, J.A. Ras1 and Ras2 contribute shared and unique roles in physiology and virulence of Cryptococcus neoformans. Microbiology (Reading) 2002, 148, 191–201. [Google Scholar] [CrossRef]
- Maeng, S.; Ko, Y.J.; Kim, G.B.; Jung, K.W.; Floyd, A.; Heitman, J.; Bahn, Y.S. Comparative transcriptome analysis reveals novel roles of the Ras and cyclic AMP signaling pathways in environmental stress response and antifungal drug sensitivity in Cryptococcus neoformans. Eukaryot Cell 2010, 9, 360–378. [Google Scholar] [CrossRef]
- Liu, K.H.; Shen, W.C. Mating differentiation in Cryptococcus neoformans is negatively regulated by the Crk1 protein kinase. Fungal Genet Biol 2011, 48, 225–240. [Google Scholar] [CrossRef]
- Wang, P.; Heitman, J. Signal transduction cascades regulating mating, filamentation, and virulence in Cryptococcus neoformans. Curr Opin Microbiol 1999, 2, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Perfect, J.R.; Heitman, J. The G-protein beta subunit GPB1 is required for mating and haploid fruiting in Cryptococcus neoformans. Molecular and Cellular Biology 2000, 20, 352–362. [Google Scholar] [CrossRef]
- Clarke, D.L.; Woodlee, G.L.; McClelland, C.M.; Seymour, T.S.; Wickes, B.L. The Cryptococcus neoformans STE11alpha gene is similar to other fungal mitogen-activated protein kinase kinase kinase (MAPKKK) genes but is mating type specific. Mol Microbiol 2001, 40, 200–213. [Google Scholar] [CrossRef]
- Wang, P.; Nichols, C.B.; Lengeler, K.B.; Cardenas, M.E.; Cox, G.M.; Perfect, J.R.; Heitman, J. Mating-type-specific and nonspecific PAK kinases play shared and divergent roles in Cryptococcus neoformans. Eukaryot Cell 2002, 1, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Perfect, J.R.; Heitman, J. The G-protein β subunit GPB1 is required for mating and haploid fruiting in Cryptococcus neoformans. Molecular and Cellular Biology 2000, 20, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Leeuw, T.; Wu, C.; Schrag, J.D.; Whiteway, M.; Thomas, D.Y.; Leberer, E. Interaction of a G-protein beta-subunit with a conserved sequence in Ste20/PAK family protein kinases. Nature 1998, 391, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.W.; Kim, S.Y.; Okagaki, L.H.; Nielsen, K.; Bahn, Y.S. Ste50 adaptor protein governs sexual differentiation of Cryptococcus neoformans via the pheromone-response MAPK signaling pathway. Fungal Genet Biol 2011, 48, 154–165. [Google Scholar] [CrossRef]
- Lengeler, K.B.; Cox, G.M.; Heitman, J. Serotype AD strains of Cryptococcus neoformans are diploid or aneuploid and are heterozygous at the mating-type locus. Infect Immun 2001, 69, 115–122. [Google Scholar] [CrossRef]
- Kraus, P.R.; Heitman, J. Coping with stress: calmodulin and calcineurin in model and pathogenic fungi. Biochem Biophys Res Commun 2003, 311, 1151–1157. [Google Scholar] [CrossRef]
- Ma, P.; Wera, S.; Van Dijck, P.; Thevelein, J.M. The PDE1-encoded low-affinity phosphodiesterase in the yeast Saccharomyces cerevisiae has a specific function in controlling agonist-induced cAMP signaling. Mol Biol Cell 1999, 10, 91–104. [Google Scholar] [CrossRef]
- Maliehe, M.; Ntoi, M.A.; Lahiri, S.; Folorunso, O.S.; Ogundeji, A.O.; Pohl, C.H.; Sebolai, O.M. Environmental Factors That Contribute to the Maintenance of Cryptococcus neoformans Pathogenesis. Microorganisms 2020, 8, 180. [Google Scholar] [CrossRef]
- Bahn, Y.S.; Cox, G.M.; Perfect, J.R.; Heitman, J. Carbonic anhydrase and CO2 sensing during Cryptococcus neoformans growth, differentiation, and virulence. Curr Biol 2005, 15, 2013–2020. [Google Scholar] [CrossRef]
- Klengel, T.; Liang, W.J.; Chaloupka, J.; Ruoff, C.; Schroppel, K.; Naglik, J.R.; Eckert, S.E.; Mogensen, E.G.; Haynes, K.; Tuite, M.F.; et al. Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr Biol 2005, 15, 2021–2026. [Google Scholar] [CrossRef]
- Gyawali, R.; Zhao, Y.; Lin, J.; Fan, Y.; Xu, X.; Upadhyay, S.; Lin, X. Pheromone independent unisexual development in Cryptococcus neoformans. PLoS Genet 2017, 13, e1006772. [Google Scholar] [CrossRef]
- Feretzaki, M.; Billmyre, R.B.; Clancey, S.A.; Wang, X.; Heitman, J. Gene Network Polymorphism Illuminates Loss and Retention of Novel RNAi Silencing Components in the Cryptococcus Pathogenic Species Complex. PLoS Genet 2016, 12, e1005868. [Google Scholar] [CrossRef]
- Catalanotto, C.; Pallotta, M.; ReFalo, P.; Sachs, M.S.; Vayssie, L.; Macino, G.; Cogoni, C. Redundancy of the two dicer genes in transgene-induced posttranscriptional gene silencing in Neurospora crassa. Mol Cell Biol 2004, 24, 2536–2545. [Google Scholar] [CrossRef]
- Maiti, M.; Lee, H.C.; Liu, Y. QIP, a putative exonuclease, interacts with the Neurospora Argonaute protein and facilitates conversion of duplex siRNA into single strands. Genes Dev 2007, 21, 590–600. [Google Scholar] [CrossRef]
- Catalanotto, C.; Azzalin, G.; Macino, G.; Cogoni, C. Involvement of small RNAs and role of the qde genes in the gene silencing pathway in Neurospora. Genes Dev 2002, 16, 790–795. [Google Scholar] [CrossRef]
- Jacobson, E.S.; Tingler, M.J.; Quynn, P.L. Effect of Hypertonic Solutes Upon the Polysaccharide Capsule in Cryptococcus neoformans: Die Wirkung hypertonischer Lösungen auf die Polysaccharid-Kapsel von Cryptococcus neofomans. Mycoses 1989, 32, 14–23. [Google Scholar] [CrossRef]
- Bahn, Y.S. Master and commander in fungal pathogens: the two-component system and the HOG signaling pathway. Eukaryot Cell 2008, 7, 2017–2036. [Google Scholar] [CrossRef]
- Nichols, C.B.; Perfect, Z.H.; Alspaugh, J.A. A Ras1-Cdc24 signal transduction pathway mediates thermotolerance in the fungal pathogen Cryptococcus neoformans. Mol Microbiol 2007, 63, 1118–1130. [Google Scholar] [CrossRef]
- Chang, Y.C.; Penoyer, L.A. Properties of various Rho1 mutant alleles of Cryptococcus neoformans. J Bacteriol 2000, 182, 4987–4991. [Google Scholar] [CrossRef]
- Nonaka, H.; Tanaka, K.; Hirano, H.; Fujiwara, T.; Kohno, H.; Umikawa, M.; Mino, A.; Takai, Y. A downstream target of RHO1 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. The EMBO journal 1995, 14, 5931–5938. [Google Scholar] [CrossRef]
- Lam, W.C.; Gerik, K.J.; Lodge, J.K. Role of Cryptococcus neoformans Rho1 GTPases in the PKC1 signaling pathway in response to thermal stress. Eukaryot Cell 2013, 12, 118–131. [Google Scholar] [CrossRef]
- Gerik, K.J.; Bhimireddy, S.R.; Ryerse, J.S.; Specht, C.A.; Lodge, J.K. PKC1 is essential for protection against both oxidative and nitrosative stresses, cell integrity, and normal manifestation of virulence factors in the pathogenic fungus Cryptococcus neoformans. Eukaryot Cell 2008, 7, 1685–1698. [Google Scholar] [CrossRef]
- Gerik, K.J.; Donlin, M.J.; Soto, C.E.; Banks, A.M.; Banks, I.R.; Maligie, M.A.; Selitrennikoff, C.P.; Lodge, J.K. Cell wall integrity is dependent on the PKC1 signal transduction pathway in Cryptococcus neoformans. Mol Microbiol 2005, 58, 393–408. [Google Scholar] [CrossRef]
- Hu, G.; Steen, B.R.; Lian, T.; Sham, A.P.; Tam, N.; Tangen, K.L.; Kronstad, J.W. Transcriptional regulation by protein kinase A in Cryptococcus neoformans. PLoS Pathog 2007, 3, e42. [Google Scholar] [CrossRef]
- Goebl, M.; Yanagida, M. The TPR snap helix: a novel protein repeat motif from mitosis to transcription. Trends in biochemical sciences 1991, 16, 173–177. [Google Scholar] [CrossRef]
- Lamb, J.R.; Tugendreich, S.; Hieter, P. Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends in biochemical sciences 1995, 20, 257–259. [Google Scholar] [CrossRef]
- Sikorski, R.S.; Michaud, W.A.; Wootton, J.C.; Boguski, M.S.; Connelly, C.; Hieter, P. TPR proteins as essential components of the yeast cell cycle. 1991, 1991; pp. 663-673.
- Gindhart, J.G., Jr.; Goldstein, L.S. Tetratrico peptide repeats are present in the kinesin light chain. Trends in biochemical sciences 1996, 21, 52–53. [Google Scholar] [CrossRef]
- Kyrpides, N.C.; Woese, C.R. Tetratrico-peptide-repeat proteins in the archaeon Methanococcus jannaschii. Trends in biochemical sciences 1998, 23, 245–247. [Google Scholar] [CrossRef]
- Zhu, W.; Rainville, I.R.; Ding, M.; Bolus, M.; Heintz, N.H.; Pederson, D.S. Evidence that the pre-mRNA splicing factor Clf1p plays a role in DNA replication in Saccharomyces cerevisiae. Genetics 2002, 160, 1319–1333. [Google Scholar] [CrossRef]
- Zhang, K.; Smouse, D.; Perrimon, N. The crooked neck gene of Drosophila contains a motif found in a family of yeast cell cycle genes. Genes & Development 1991, 5, 1080–1091. [Google Scholar]
- Chung, S.; McLean, M.R.; Rymond, B.C. Yeast ortholog of the Drosophila crooked neck protein promotes spliceosome assembly through stable U4/U6.U5 snRNP addition. RNA (New York, N.Y.) 1999, 5, 1042–1054. [Google Scholar] [CrossRef]
- Russell, C.S.; Ben-Yehuda, S.; Dix, I.; Kupiec, M.; Beggs, J.D. Functional analyses of interacting factors involved in both pre-mRNA splicing and cell cycle progression in Saccharomyces cerevisiae. RNA (New York, N.Y.) 2000, 6, 1565–1572. [Google Scholar] [CrossRef]
- Chung, S.; Mondon, P.; Chang, Y.C.; Kwon-Chung, K.J. Cryptococcus neoformans with a mutation in the tetratricopeptide repeat-containing gene, CCN1, causes subcutaneous lesions but fails to cause systemic infection. Infect Immun 2003, 71, 1988–1994. [Google Scholar] [CrossRef]
- Bemis, D.A.; Krahwinkel, D.J.; Bowman, L.A.; Mondon, P.; Kwon-Chung, K.J. Temperature-sensitive strain of Cryptococcus neoformans producing hyphal elements in a feline nasal granuloma. J Clin Microbiol 2000, 38, 926–928. [Google Scholar] [CrossRef]
- Park, H.S.; Chow, E.W.; Fu, C.; Soderblom, E.J.; Moseley, M.A.; Heitman, J.; Cardenas, M.E. Calcineurin Targets Involved in Stress Survival and Fungal Virulence. PLoS Pathog 2016, 12, e1005873. [Google Scholar] [CrossRef]
- Kraus, P.R.; Boily, M.J.; Giles, S.S.; Stajich, J.E.; Allen, A.; Cox, G.M.; Dietrich, F.S.; Perfect, J.R.; Heitman, J. Identification of Cryptococcus neoformans temperature-regulated genes with a genomic-DNA microarray. Eukaryot Cell 2004, 3, 1249–1260. [Google Scholar] [CrossRef]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.C.; Cavallo, L.M.; Gorlach, J.M.; Cox, G.; Perfect, J.R.; Cardenas, M.E.; Heitman, J. Rapamycin antifungal action is mediated via conserved complexes with FKBP12 and TOR kinase homologs in Cryptococcus neoformans. Mol Cell Biol 1999, 19, 4101–4112. [Google Scholar] [CrossRef] [PubMed]
- Odom, A.; Del Poeta, M.; Perfect, J.; Heitman, J. The immunosuppressant FK506 and its nonimmunosuppressive analog L-685,818 are toxic to Cryptococcus neoformans by inhibition of a common target protein. Antimicrob Agents Chemother 1997, 41, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G. Peptidyl-prolyl cis/trans isomerases and their effectors. Angewandte Chemie International Edition in English 1994, 33, 1415–1436. [Google Scholar] [CrossRef]
- Fischer, G.; Tradler, T.; Zarnt, T. The mode of action of peptidyl prolyl cis/trans isomerases in vivo: binding vs. catalysis. FEBS Lett 1998, 426, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cardenas, M.E.; Cox, G.M.; Perfect, J.R.; Heitman, J. Two cyclophilin A homologs with shared and distinct functions important for growth and virulence of Cryptococcus neoformans. EMBO Rep 2001, 2, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Rossettini, A.; Chaturvedi, V.; Hanes, S.D. The Ess1 prolyl isomerase is dispensable for growth but required for virulence in Cryptococcus neoformans. Microbiology (Reading) 2005, 151, 1593–1605. [Google Scholar] [CrossRef]
- Del Poeta, M.; Toffaletti, D.L.; Rude, T.H.; Dykstra, C.C.; Heitman, J.; Perfect, J.R. Topoisomerase I Is Essential in Cryptococcus neoformans: Role in Pathobiology and as an Antifungal Target. Genetics 1999, 152, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Wang, J.C. Cloning of yeast TOP1, the gene encoding DNA topoisomerase I, and construction of mutants defective in both DNA topoisomerase I and DNA topoisomerase II. Proc Natl Acad Sci U S A 1985, 82, 7178–7182. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.; Redinbo, M.R.; Qiu, X.; Hol, W.G.; Champoux, J.J. A model for the mechanism of human topoisomerase I. Science 1998, 279, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Gerhold, D.; Kmiec, E.B.; Hauser, M.; Becker, J.M.; Koltin, Y. The topoisomerase I gene from Candida albicans. Microbiology (Reading) 1997, 143 ( Pt 2) Pt 2, 377–386. [Google Scholar] [CrossRef]
- Shen, L.L.; Baranowski, J.; Fostel, J.; Montgomery, D.A.; Lartey, P.A. DNA topoisomerases from pathogenic fungi: targets for the discovery of antifungal drugs. Antimicrob Agents Chemother 1992, 36, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Idnurm, A.; Heitman, J. Light controls growth and development via a conserved pathway in the fungal kingdom. PLoS Biol 2005, 3, e95. [Google Scholar] [CrossRef] [PubMed]
- Idnurm, A.; Walton, F.J.; Floyd, A.; Reedy, J.L.; Heitman, J. Identification of ENA1 as a virulence gene of the human pathogenic fungus Cryptococcus neoformans through signature-tagged insertional mutagenesis. Eukaryot Cell 2009, 8, 315–326. [Google Scholar] [CrossRef]
- Jung, K.W.; Strain, A.K.; Nielsen, K.; Jung, K.H.; Bahn, Y.S. Two cation transporters Ena1 and Nha1 cooperatively modulate ion homeostasis, antifungal drug resistance, and virulence of Cryptococcus neoformans via the HOG pathway. Fungal Genet Biol 2012, 49, 332–345. [Google Scholar] [CrossRef]
- Chayakulkeeree, M.; Sorrell, T.C.; Siafakas, A.R.; Wilson, C.F.; Pantarat, N.; Gerik, K.J.; Boadle, R.; Djordjevic, J.T. Role and mechanism of phosphatidylinositol-specific phospholipase C in survival and virulence of Cryptococcus neoformans. Mol Microbiol 2008, 69, 809–826. [Google Scholar] [CrossRef]
- Lev, S.; Desmarini, D.; Li, C.; Chayakulkeeree, M.; Traven, A.; Sorrell, T.C.; Djordjevic, J.T. Phospholipase C of Cryptococcus neoformans regulates homeostasis and virulence by providing inositol trisphosphate as a substrate for Arg1 kinase. Infect Immun 2013, 81, 1245–1255. [Google Scholar] [CrossRef]
- Prasad, T.; Chandra, A.; Mukhopadhyay, C.K.; Prasad, R. Unexpected link between iron and drug resistance of Candida spp.: iron depletion enhances membrane fluidity and drug diffusion, leading to drug-susceptible cells. Antimicrob Agents Chemother 2006, 50, 3597–3606. [Google Scholar] [CrossRef]
- Jung, K.W.; Yang, D.H.; Maeng, S.; Lee, K.T.; So, Y.S.; Hong, J.; Choi, J.; Byun, H.J.; Kim, H.; Bang, S.; et al. Systematic functional profiling of transcription factor networks in Cryptococcus neoformans. Nat Commun 2015, 6, 6757. [Google Scholar] [CrossRef]
- Chang, Y.C.; Bien, C.M.; Lee, H.; Espenshade, P.J.; Kwon-Chung, K.J. Sre1p, a regulator of oxygen sensing and sterol homeostasis, is required for virulence in Cryptococcus neoformans. Mol Microbiol 2007, 64, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Kraus, P.R.; Fox, D.S.; Cox, G.M.; Heitman, J. The Cryptococcus neoformans MAP kinase Mpk1 regulates cell integrity in response to antifungal drugs and loss of calcineurin function. Mol Microbiol 2003, 48, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.R.; Douglas, C.M.; Li, W.; Jue, C.K.; Pramanik, B.; Yuan, X.; Rude, T.H.; Toffaletti, D.L.; Perfect, J.R.; Kurtz, M. A glucan synthase FKS1 homolog in Cryptococcus neoformans is single copy and encodes an essential function. J Bacteriol 1999, 181, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Wormley, F.L., Jr.; Heinrich, G.; Miller, J.L.; Perfect, J.R.; Cox, G.M. Identification and characterisation of an SKN7 homologue in Cryptococcus neoformans. Infect Immun 2005, 73, 5022–5030. [Google Scholar] [CrossRef]
- Coenjaerts, F.E.; Hoepelman, A.I.; Scharringa, J.; Aarts, M.; Ellerbroek, P.M.; Bevaart, L.; Van Strijp, J.A.; Janbon, G. The Skn7 response regulator of Cryptococcus neoformans is involved in oxidative stress signalling and augments intracellular survival in endothelium. FEMS Yeast Res 2006, 6, 652–661. [Google Scholar] [CrossRef]
- Missall, T.A.; Lodge, J.K. Function of the thioredoxin proteins in Cryptococcus neoformans during stress or virulence and regulation by putative transcriptional modulators. Mol Microbiol 2005, 57, 847–858. [Google Scholar] [CrossRef]
- Missall, T.A.; Pusateri, M.E.; Lodge, J.K. Thiol peroxidase is critical for virulence and resistance to nitric oxide and peroxide in the fungal pathogen, Cryptococcus neoformans. Mol Microbiol 2004, 51, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Missall, T.A.; Cherry-Harris, J.F.; Lodge, J.K. Two glutathione peroxidases in the fungal pathogen Cryptococcus neoformans are expressed in the presence of specific substrates. Microbiology (Reading) 2005, 151, 2573–2581. [Google Scholar] [CrossRef]
- Giles, S.S.; Batinic-Haberle, I.; Perfect, J.R.; Cox, G.M. Cryptococcus neoformans mitochondrial superoxide dismutase: an essential link between antioxidant function and high-temperature growth. Eukaryot Cell 2005, 4, 46–54. [Google Scholar] [CrossRef]
- Narasipura, S.D.; Chaturvedi, V.; Chaturvedi, S. Characterisation of Cryptococcus neoformans variety gattii SOD2 reveals distinct roles of the two superoxide dismutases in fungal biology and virulence. Mol Microbiol 2005, 55, 1782–1800. [Google Scholar] [CrossRef]
- Upadhya, R.; Campbell, L.T.; Donlin, M.J.; Aurora, R.; Lodge, J.K. Global transcriptome profile of Cryptococcus neoformans during exposure to hydrogen peroxide induced oxidative stress. PLoS One 2013, 8, e55110. [Google Scholar] [CrossRef] [PubMed]
- Demasi, A.P.; Pereira, G.A.; Netto, L.E. Yeast oxidative stress response. Influences of cytosolic thioredoxin peroxidase I and of the mitochondrial functional state. FEBS J 2006, 273, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.H.; Seyfang, A. High-affinity myo-inositol transport in Candida albicans: substrate specificity and pharmacology. Microbiology (Reading) 2003, 149, 3371–3381. [Google Scholar] [CrossRef]
- Crabtree, R.H. The organometallic chemistry of the transition metals; John Wiley & Sons: 2009.
- Giles, S.S.; Stajich, J.E.; Nichols, C.; Gerrald, Q.D.; Alspaugh, J.A.; Dietrich, F.; Perfect, J.R. The Cryptococcus neoformans catalase gene family and its role in antioxidant defense. Eukaryot Cell 2006, 5, 1447–1459. [Google Scholar] [CrossRef]
- Lu, J.M.; Deschenes, R.J.; Fassler, J.S. Saccharomyces cerevisiae histidine phosphotransferase Ypd1p shuttles between the nucleus and cytoplasm for SLN1-dependent phosphorylation of Ssk1p and Skn7p. Eukaryot Cell 2003, 2, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.B.; Ferreyra, J.; Ballou, E.R.; Alspaugh, J.A. Subcellular localisation directs signaling specificity of the Cryptococcus neoformans Ras1 protein. Eukaryot Cell 2009, 8, 181–189. [Google Scholar] [CrossRef]
- Sato, N.; Kawahara, H.; Toh-e, A.; Maeda, T. Phosphorelay-regulated degradation of the yeast Ssk1p response regulator by the ubiquitin-proteasome system. Mol Cell Biol 2003, 23, 6662–6671. [Google Scholar] [CrossRef]
- Cheng, L.; Watt, R.; Piper, P.W. Polyubiquitin gene expression contributes to oxidative stress resistance in respiratory yeast (Saccharomyces cerevisiae). Mol Gen Genet 1994, 243, 358–362. [Google Scholar] [CrossRef]
- Walsh, L.; Schmuckli-Maurer, J.; Billinton, N.; Barker, M.G.; Heyer, W.D.; Walmsley, R.M. DNA-damage induction of RAD54 can be regulated independently of the RAD9- and DDC1-dependent checkpoints that regulate RNR2. Curr Genet 2002, 41, 232–240. [Google Scholar] [CrossRef]
- Sanguinetti, M.; Posteraro, B.; La Sorda, M.; Torelli, R.; Fiori, B.; Santangelo, R.; Delogu, G.; Fadda, G. Role of AFR1, an ABC transporter-encoding gene, in the in vivo response to fluconazole and virulence of Cryptococcus neoformans. Infect Immun 2006, 74, 1352–1359. [Google Scholar] [CrossRef]
- Lee, H.; Chang, Y.C.; Nardone, G.; Kwon-Chung, K.J. TUP1 disruption in Cryptococcus neoformans uncovers a peptide-mediated density-dependent growth phenomenon that mimics quorum sensing. Mol Microbiol 2007, 64, 591–601. [Google Scholar] [CrossRef]
- Tian, X.; He, G.J.; Hu, P.; Chen, L.; Tao, C.; Cui, Y.L.; Shen, L.; Ke, W.; Xu, H.; Zhao, Y.; et al. Cryptococcus neoformans sexual reproduction is controlled by a quorum sensing peptide. Nat Microbiol 2018, 3, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Homer, C.M.; Summers, D.K.; Goranov, A.I.; Clarke, S.C.; Wiesner, D.L.; Diedrich, J.K.; Moresco, J.J.; Toffaletti, D.; Upadhya, R.; Caradonna, I.; et al. Intracellular Action of a Secreted Peptide Required for Fungal Virulence. Cell Host Microbe 2016, 19, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.A.; Jung, K.W.; Chen, Y.L.; Heitman, J.; Bahn, Y.S.; Kang, H.A. Unique evolution of the UPR pathway with a novel bZIP transcription factor, Hxl1, for controlling pathogenicity of Cryptococcus neoformans. PLoS Pathog 2011, 7, e1002177. [Google Scholar] [CrossRef] [PubMed]
- Glazier, V.E.; Kaur, J.N.; Brown, N.T.; Rivera, A.A.; Panepinto, J.C. Puf4 regulates both splicing and decay of HXL1 mRNA encoding the unfolded protein response transcription factor in Cryptococcus neoformans. Eukaryot Cell 2015, 14, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Havel, V.E.; Wool, N.K.; Ayad, D.; Downey, K.M.; Wilson, C.F.; Larsen, P.; Djordjevic, J.T.; Panepinto, J.C. Ccr4 promotes resolution of the endoplasmic reticulum stress response during host temperature adaptation in Cryptococcus neoformans. Eukaryot Cell 2011, 10, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.W.; Kang, H.A.; Bahn, Y.S. Essential roles of the Kar2/BiP molecular chaperone downstream of the UPR pathway in Cryptococcus neoformans. PLoS One 2013, 8, e58956. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Kimata, Y.; Higashio, H.; Tsuru, A.; Kohno, K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun 2000, 279, 445–450. [Google Scholar] [CrossRef]
- Pincus, D.; Chevalier, M.W.; Aragon, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol 2010, 8, e1000415. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Vishwanath, P.; Cui, J.; Kelleher, D.J.; Gilmore, R.; Robbins, P.W.; Samuelson, J. The evolution of N-glycan-dependent endoplasmic reticulum quality control factors for glycoprotein folding and degradation. Proc Natl Acad Sci U S A 2007, 104, 11676–11681. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.T.; Byun, H.J.; Jung, K.W.; Hong, J.; Cheong, E.; Bahn, Y.S. Distinct and redundant roles of protein tyrosine phosphatases Ptp1 and Ptp2 in governing the differentiation and pathogenicity of Cryptococcus neoformans. Eukaryot Cell 2014, 13, 796–812. [Google Scholar] [CrossRef]
- Erickson, T.; Liu, L.; Gueyikian, A.; Zhu, X.; Gibbons, J.; Williamson, P.R. Multiple virulence factors of Cryptococcus neoformans are dependent on VPH1. Mol Microbiol 2001, 42, 1121–1131. [Google Scholar] [CrossRef]
- Kiewietdejonge, A.; Pitts, M.; Cabuhat, L.; Sherman, C.; Kladwang, W.; Miramontes, G.; Floresvillar, J.; Chan, J.; Ramirez, R.M. Hypersaline stress induces the turnover of phosphatidylcholine and results in the synthesis of the renal osmoprotectant glycerophosphocholine in Saccharomyces cerevisiae. FEMS Yeast Res 2006, 6, 205–217. [Google Scholar] [CrossRef]
- Zablocki, K.; Miller, S.P.; Garcia-Perez, A.; Burg, M.B. Accumulation of glycerophosphocholine (GPC) by renal cells: osmotic regulation of GPC:choline phosphodiesterase. Proc Natl Acad Sci U S A 1991, 88, 7820–7824. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Exogenous activities that promote the expression and regulation of transcription factors in C. neoformans for adaptation, survival, mating, and pathogenesis. Many of these external factors include common environmental factors and interactions, as listed at the center of the figure above. These factors interact with the fungi cell wall, interpret by the specialised cell membrane proteins, and relay the message as signals via transcription factors to induce specific and non-specific genes in an attempt to subvert and resist invading factors. Overwhelming situations could force the fungi into some phenotypic cell shapes, including titanisation (relatively bigger cell size compared to the wild type), pigmentation (melanisation), capsulation (source of antigenic factors), and shmoo-like cell formation (unusual cell morphotype).
Figure 1.
Exogenous activities that promote the expression and regulation of transcription factors in C. neoformans for adaptation, survival, mating, and pathogenesis. Many of these external factors include common environmental factors and interactions, as listed at the center of the figure above. These factors interact with the fungi cell wall, interpret by the specialised cell membrane proteins, and relay the message as signals via transcription factors to induce specific and non-specific genes in an attempt to subvert and resist invading factors. Overwhelming situations could force the fungi into some phenotypic cell shapes, including titanisation (relatively bigger cell size compared to the wild type), pigmentation (melanisation), capsulation (source of antigenic factors), and shmoo-like cell formation (unusual cell morphotype).
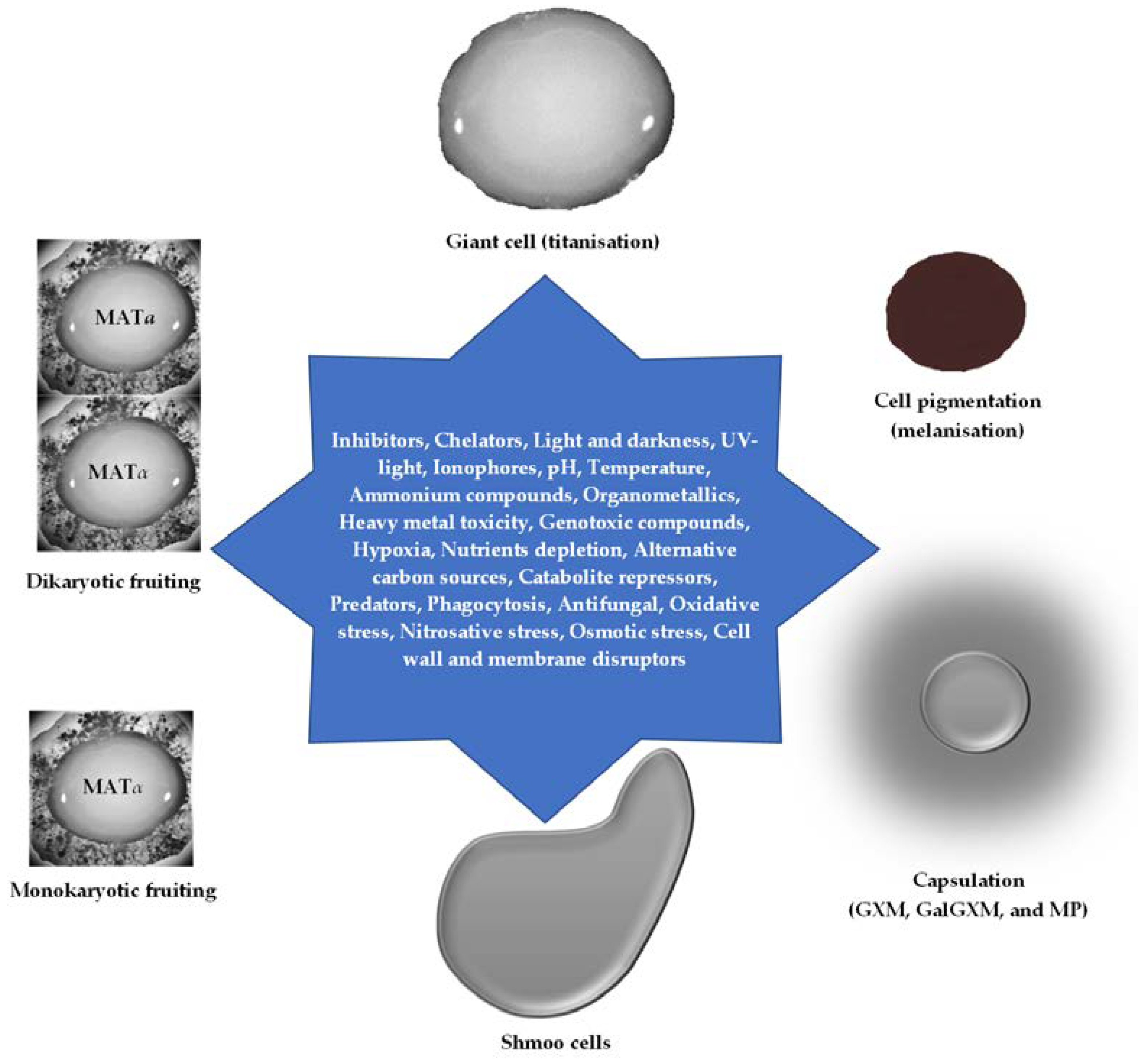
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



