
Submitted:
20 January 2023
Posted:
26 January 2023
You are already at the latest version
Abstract
As exemplified by the global response to the SARS-CoV-2 pandemic, whole genome sequencing played an important role in monitoring the evolution of novel viral variants and provided guidance on potential antiviral treatments. The recent rapid and extensive introduction and spread of highly pathogenic avian influenza virus in Europe, North America and elsewhere raises the need for similarly rapid sequencing to aid in appropriate response and mitigation activities. To facilitate this objective, we investigated a next generation sequencing platform that uses a portable nanopore sequencing device to generate and present data in real time. This platform offers the potential to extend in-house sequencing capacities to laboratories that may otherwise lack resources to adopt sequencing technologies requiring large benchtop instruments. We evaluated this platform for routine use in a diagnostic laboratory. In this study we evaluated different primer sets for the whole genome amplification of influenza A virus and evaluated five different library preparation approaches for sequencing on the nanopore platform using the MinION flow-cell. A limited amplification procedure and a rapid procedure were found to be best among the approaches taken.
Keywords:
avian influenza
; highly pathogenic avian influenza
; next generation sequencing
; whole genome sequencing
; nanopore technology
; methods comparison
; clinical validation
1. Introduction
The emergence and global spread of SARS-CoV-2 has led to widespread use of whole genome sequencing to guide therapeutic countermeasures for individuals as well as for population-scale epidemiological monitoring. With over four million genomes sequenced, patterns of introduction, dissemination, and evolution of variants have been well described [1], but one area where the state of the art might improve is the ability to sequence in real-time or near real-time [2]. In recent years, highly pathogenic avian influenza (HPAI) viruses have been introduced and have circulated in wild birds and poultry in Asia, Europe, and North America [3,4,5,6]. Whole genome sequencing, in conjunction with epidemiological information, has often been used to elucidate routes of introduction and patterns of spread in HPAI outbreaks [7,8]. To date, however, there are no global programs for the large-scale sequencing and analysis of HPAI viral sequences analogous to that for SARS-CoV-2. For example, since 2021, in spite of nearly 2,400 HPAI H5Nx outbreaks in poultry and over 2,700 similar events in wild birds in Europe [9], plus another 673 detections in poultry [10], and 4,362 detections in wild birds in the United States [11], as of Dec. 8, 2022, only 3,116 HPAI H5Nx genomes were deposited in the GenBank and Global Initiative on Sharing All Influenza Data (GISAID) databases.
Avian influenza viruses are members of the family Orthomyxoviridae [12]. The genome contains eight single-stranded RNA segments ranging in size from 890 to over 2,280 nucleotides long. A conserved motif of 12 nucleotides is present at the 5’- and 3’-end of each RNA segment, and these conserved residues have been used as targets for cDNA synthesis and sequencing [13].
Reducing costs and turn-around times are among the key challenges that laboratories face when adopting next generation sequencing (NGS) technology [14,15]. Illumina sequencing systems, such as the MiSeq, are characterized by relatively short reads (2x300 bp) and long sequencing times (36 h), but feature high sample throughput, high data density, and high sequence accuracy [14]. They additionally require investment to purchase and maintain a benchtop-scale sequencing instrument and involve complex workflows. In contrast, Oxford Nanopore Technology (ONT) MinION utilizes a low-cost, portable nanopore sequencing device to yield longer read lengths and data availability in real time, but is limited by higher error rates and lower sensitivity [16]. Other NGS platforms exist, but Illumina and ONT were used to generate 90% (65% and 25%, respectively) of the 4.8 million SARS-CoV-2 genomes deposited in GISAID as of Nov 5, 2021 [16].
By producing readable sequencing data within 30 minutes, the MinION platform offers the ability to decrease time to pathogen identification when speed is of the essence [17]. Additionally, the smaller capacity of the MinION may be well-suited to the number of avian influenza surveillance samples that need to be sequenced at any one time by an individual diagnostic laboratory. Several studies have described the whole genome amplification and nanopore sequencing of avian influenza viruses from both wild bird and domestic samples [18,19,20]. Moreover, strategies to achieve >99% accuracy (often by increasing the depth of coverage, i.e., increasing the number of reads over each sequence location) in both SARS-CoV-2 and avian influenza virus sequencing have been published [19,20,21]. Although a variety of approaches and library preparation methods have been described for the sequencing of avian influenza viruses on the MinION platform (e.g. native barcoding kit (SQK-LSK109 [19], PCR barcoding kit (SQK-PBK004) [22], rapid barcoding kit (SQK-RBK004) [20]), the performance differences between these methods have not yet been evaluated.
In this study, we evaluated the performance of RT-PCR primers for the conversion of avian influenza virus RNA to cDNA, and the utility of different library preparation kits for the sequencing of the whole avian influenza genomes on the MinION platform.
2. Materials and Methods
2.1 Viruses used in this study
Three highly pathogenic H5N1 avian influenza virus-containing cloacal or tracheal swab samples collected from wild birds were chosen because not only do these samples represent the typical sample received in the laboratory for wild bird surveillance testing, but they also span the Cycle Threshold (Ct) range typically seen in positive samples (Table 1).
Viral RNA was extracted using the MagMAX-96 AI/ND Viral RNA Isolation Kit (ThermoFisher) using a KingFisher magnetic particle processor and 90-µl elution volume.
2.2 RT-PCR
Reverse-transcription – Polymerase Chain Reaction (RT-PCR) was performed using primer sets listed in Table 2. Briefly, 5 μl of extracted RNA was amplified in a 25-μl reaction with Invitrogen SuperScript III RT-PCR mix (ThermoFisher). Where referenced, the RT-PCR cycling conditions are as cited. For primer Set 2, the reaction was incubated as per Zhou and Wentworth [13].
cDNA was purified by diluting the reaction to 50 μl with nuclease-free water and adding 50 μl of AMPure beads (1:1 vol:vol) (Beckman-Coulter). Following incubation at room temperature for 5 minutes, the tubes were placed on a magnetic stand for 2 minutes. The supernatant was discarded, and the pellets were washed with 500 μl of 80% ethanol twice, allowed to dry briefly, and eluted in 20 μl of nuclease-free water. 1 μl of the recovered cDNA was quantitated using the Qubit dsDNA HS (High Sensitivity) Assay Kit (ThermoFisher).
2.3 Library kit comparison
The primer sets in Table 2 are designed to be used with different library preparation kits as some kits have their own specific requirements for purposes such as attachment or PCR amplification of barcodes (Supplementary Table S1). In Method A, libraries were constructed with cDNAs amplified with Set 1 primers using the native bar-coding kit SQK-LSK109. Method S used Set 2 primer amplified samples and the PCR barcoding kit (SQK-PBK004). Method E constructed libraries with Primer Set 3 amplified cDNA and the Rapid barcoding kit (SQK-RBK004). Method K used cDNAs amplified with Set 4 primers to prepare libraries with Rapid PCR Barcoding kit (SQK-RPB004). One additional experiment was performed with Set 4 amplified material using a newly described procedure for the whole genome sequencing of avian influenza viruses with the SQK-LSK109 kit (Method N, [23]). The samples were bar-coded, and library constructed as per manufacturer’s instructions except for Set 1, where equal volumes of New England Biolabs FFPE DNA repair mix were added to the end prep reaction.
2.4 MinION Runs
R9.4.1 version MinION flow cells were used in this study. Flow cells were loaded as per manufacturer’s instructions on an MK1c portable standalone sequencing unit. Runs were 48 h but were terminated early when little data continued to be generated, or when the Q score was below 10, depending on which came first.
2.5 Sequence Analysis
Base calls were exported as FASTQ files by the MK1c during the run, and the barcodes were deconvoluted. The FASTQ files were imported into CLC Genomic Workbench (Qiagen) where the sequences were mapped to a standard set of avian influenza reference sequences as described by Crossley et al. [19]. Mapped consensus sequences corresponding to each segment were compared to GenBank using BLAST and closer homologs were identified. The sequences of the closer homologs were then used as new reference sequences for a new round of mapping. The process was repeated recursively until no closer GenBank entries were found. The final consensus sequences for each segment were then exported in FASTA format. The coding region for each segment was examined using MacVector (GeneCode); and if the expected coding region was interrupted, the MinION reads over the corresponding region were manually examined for possible sequencing or assembly errors, as per Delahaye and Nicolas [24], and the sequence manually edited. Sequence accuracy was evaluated between the various methods using T-Coffee multiple sequence alignment against the sequencing of the same RNA samples on a benchtop instrument (Illumina MiSeq).
3. Results
3.1. cDNA generation by RT-PCR
The amount of cDNA generated was calculated from the Qubit quantitation value. Even though all primer sets essentially contain the same 12 or 13 nucleotide sequence homologous to the influenza RNA segments, the amount of products generated was variable (Figure 1). For example, the RNA from sample 245626 had a 38-fold difference among the four primer sets.
3.2. Library Construction Kits Evaluated
The major steps involved with each of the kits used in the methods evaluated in this study are summarized in Supplementary Table S1. SQK-LSK109 (Method A) is the kit often used for the bar-coding of samples if additional amplification is not required. The other kits examined all involve some degree of PCR amplification, with Method S taking the longest time to complete. Of note is the number of AMPure bead purification steps required in the different protocols, which ranged from two (Methods E, K, and S) to four (Methods A and N). The total time required for a single sample (not including RT-PCR step) until the sample is ready for loading onto the flowcell ranged from 18 minutes (Method E) to 228 min (Method S) (Figure 2).
3.3. Read comparisons
A total of 24.4 million reads were generated among the kits evaluated, with average read lengths between 300 bp (Method E) to 1.07 kb (Method N). Together, 20.5 million reads mapped to the HPAI H5N1 genome. The number of reads generated using each kit varied widely, and ranged from 1.7 million (Method N) to 9.4 million (Method S) (Table 4). Four of the methods had an average of 87-88% reads mapping to the H5N1 genome with Method E being an outlier with only 68% mapping to H5N1.

Table 3.
Overall reads generated by the ONT kits evaluated. The total number of reads generated between the three RNA samples, the number of reads mapped to the H5N1 genomes, and the percentage of reads mapped to H5N1 are presented.
Table 3.
Overall reads generated by the ONT kits evaluated. The total number of reads generated between the three RNA samples, the number of reads mapped to the H5N1 genomes, and the percentage of reads mapped to H5N1 are presented.
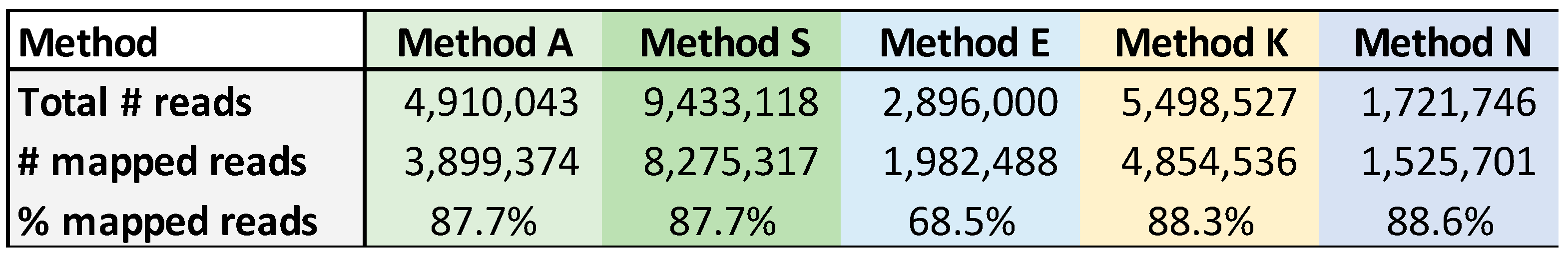
The total number of mapped reads was not a good measure of the kits’ performance, as some kits overamplified the ends of the viral RNA segments (e.g., Method S and segments PB2, PB1 and PA in Supplementary Figure S1b) or failed to span the length of some of the segments (Supplementary Figure S1a–c, Methods S and E). Using RNA 246038 as an example, between 10,758 (Method E) and about 3.6 million reads (Method S) were mapped to HPAI H5N1 by the five different methods, but only Methods A, K, and N were able to cover the entire genome (Table 4).
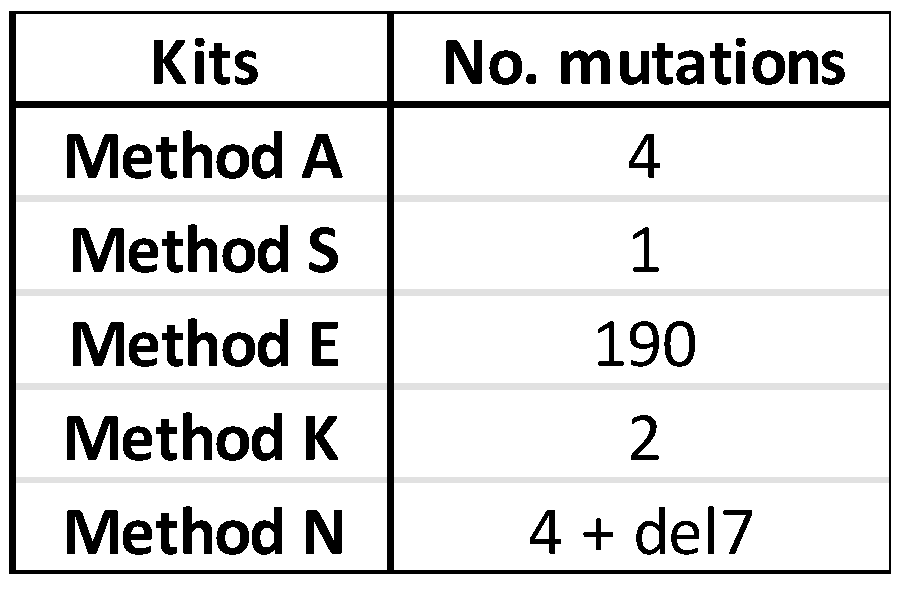
Several sequence variations were found in the consensus sequence generated from each kit. The number of variations for the three RNA samples are tabulated in Supplementary Table S2a–c, and the total number of sequence variations is given in Table 5.
The assembled consensus sequences from each kit were used in a multiple sequence aligment against the reference sequences generated on the MiSeq (Supplementary Figures S2a–h, S3a–h, and S4a–h). Only one sequence variation was shared across the kits; the distribution of all other variations did not follow a discernable pattern. The one common variation was in the PB2 sequence of Sample 245626, where C19 is changed to A19 in the sequence assembled from all four methods with complete PB2 sequences. The change from CTA to ATA changes the amino acid encoded from leucine to isoleucine. The seven nucleotide deletion in Method N is in the PB2 segment where only 695 reads in total were mapped to this gene (Table 4, Supplementary Figure S3a). The deletion results in a frame shift and a premature termination after residue 731 rather than 760 (data not shown). Method E produced a large number of sequence mutations, the variations of which could be seen across all three RNA samples but especially in those with lower Ct values (Supplementary Figures S2a–h, S3a–h, and S4a–h).
4. Discussion
The sequencing of avian influenza viruses (AIV) was instrumental in defining the multiple introductions of highly pathogenic avian influenza viruses into North America in 2021-2022 [5,6]. Moreover, detailed genetic studies are useful in defining the relationships between outbreaks, identifying independent introductions, farm to farm spread, and superspreader events (e.g., [7,8]). In a typical year, the Centers for Disease Control and Prevention (CDC) sequences approximately seven thousand influenza viruses from patient samples [25]; this is fewer than the number of SARS-CoV-2 viruses that are being sequenced [1]. Wider availability of rapid and preferably low-cost whole-genome sequencing methods for agents of emerging infectious diseases, including avian influenza viruses [26], could facilitate additional sequencing efforts. The nanopore technology-based sequencing systems described in this manuscript met both criteria [27].
Although some publications have described the sequencing of influenza viruses on the MinION platform, few have described the selection and optimization that led to the final publication [19,20,28]. In this study, we used three wild-bird swab samples that were positive for the HPAI H5N1 virus to examine the role of oligonucleotide primers for whole genome amplification and the use of different library preparation kits from ONT for their sequencing on the MinION platform. The RNA samples were chosen as they represented a typical range of samples that have been successfully sequenced on the Illumina MiSeq platform. The kits were chosen as their use has previously been published (Methods A, K, N, and S), or they had potential to offer useful features such as minimal handling steps and rapid time to first results (Method E).
The original Zhou and Wentworth primers (Set 1) generated much more cDNA than those using Sets 2 and 3 [13]. Set 4, which is a modification of the Mitchell et al. B primers [28], generated substantially more cDNA on each sample and particularly with the sample containing the lowest viral titer (Sample 245626). It is likely that secondary structural differences between the non-influenza A sequences on the primers might play a role in the efficiency of reverse transcriptase priming and cDNA synthesis. For example, primers in both Set 2 and Set 4 contain the same 3’ sequence as in the Zhou and Wentworth Set 1 as well as the same 5’ sequence (Table 2), which were added as anchor sequences for PCR amplification and barcoding with ONT’s SQK-PBK004 kit but differ in the sequence added as a spacer between the two domains. Based on our results, Set 4 primers were more appropriate for AIV whole genome amplification, but additional analysis of secondary structures and thermodynamics of these and other primers could yield further optimization.
The five procedures (called “Methods” for the sake of simplicity in this paper) differ in terms of complexity and hands-on time (Supplementary Table S1). The differences are expected to be further heightened when more samples than the three used in this study are processed together in a multiplexed sequencing run.
The use of only three samples vastly underutilized the capacity of the MinION flowcell, so the total number of reads generated and the degree of coverage in this study is not expected to be typical during a production run in the laboratory (Table 3). However, even with only three samples, it was unexpected that some methods (E and S) did not result in the assembly of the complete genome from any of the samples, including from the sample with the higher viral titer (Sample 246038, Table 4). Care is warranted in interpreting the results because the effects seen with the ONT kits are compounded by the primer sets used in the RT-PCR step. Not all primer sets are compatible with every kit because some kits require specific sequences in the primers for purposes such as barcoding; thus, a direct comparison utilizing a common primer set could not be determined in the present study. The following are a few caveated observations. Method E, which has the least degree of PCR amplification, resulted in many sequence variations (Supplementary Table S2a–c). This method also had the lowest number of reads mapped with every sample tested and on every viral segment. Method S generated uneven read coverage, failing to completely sequence the genome from Sample 245626, which had the lowest viral titer. Method N is based on a recent protocol, and the overall procedure was similar to that in Method A as both methods use ONT’s SQK-LSK109 library construction kit. Method N was designed for the simultaneous sequencing of Influenza A and B viruses from human samples, but we replaced the specified primers with Set 4, as that gave us superior amplification of avian influenza viruses. Method N generated complete genomes from all three RNA samples even though several segments (e.g., PB2) had very few mapped reads. In fact, across the five kits, Method N had the greatest number of viral segments with less than 10,000 reads per segment. The low number of reads in Method N, when much of the procedure is shared with Method A, indicates that optimization of the Method N procedure might result in improved generation of reads.
The principle of ONT’s nanopore sequencing is based on software interpretation of the changes in electrical voltage as a DNA molecule is passed through a synthetic protein pore in a membrane. This strategy is known to have issues of sequence accuracy of homopolymeric regions [24]. Indeed, several consensus sequences, particularly for the PB1 and PA segments, were found to have interrupted open reading frames using automated sequence assembly workflows (Table 4 and data not shown). When the reads in the region near the premature termination codon were examined, a frameshift caused by missing a nucleotide in a run of homopolymers was frequently found. For the MinION platform, 60-fold or higher read coverage has been found to result in a better than 99.95% sequencing accuracy [29,30]. In our study, Method K resulted in the identical sequence in two samples (245467 and 246038) when compared to the sequence generated from the same samples on the MiSeq (Supplementary Figures S2a–h and S4a–h). Two sequence differences were noted in Sample 245626, and this corresponds to an accuracy of 99.985% (Supplementary Figure S3a–h). A similar degree of sequence accuracy (>99%) between MinION and MiSeq was found by Wang et al. [31].
Both Method A and Method K were able to generate the complete genomes from all three test samples. Method A follows the classic process of RT-PCR followed by native bar-coding using ONT’s SQK-LSK109 kit. This process has been previously described [16]. We found four sequence deviations from the MiSeq reference sequence using Method A. In addition to the one common sequence variation in PB2 as discussed above, three other departures were found in the PA gene for Sample 245626. None of these variations were found in the genomes assembled with the other kits. Method K (ONT SQK-RBP004) utilizes an abbreviated PCR step to amplify and barcode samples. The procedure only has two bead purification steps and only one of which is kit specific. Manual bead purification is not only labor intensive, but products are often lost during purification; so minimizing bead purification steps meant that Method K was the second fastest among the kits evaluated. This may have contributed to more evenly mapped read coverage over each segment than the other kits (Supplementary Figures S2–S4a–h).
5. Conclusions
We evaluated four primer sets for the whole genome amplification of avian influenza viruses and tested several library preparation kits for the multiplexed sequencing of the samples using a nanopore next generation sequencing platform. Primer Set 4 and Methods A & K had the best performance as indicated by the generation of more cDNA, increased number of reads, more even genome coverage, and fewer sequencing errors, with Method K requiring less hands on time. Further studies on the optimization of these and other kits could help to improve the ability to rapidly characterize the genomes of avian influenza viruses for the study of virus phylogeny and to inform avian influenza outbreak response.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Supplementary Figure S1: Graphic representation of the read coverage of each influenza RNA segments by the methods tested; Supplementary Figure S2a–h. Sequence variations generated by the different methods tested for Sample 245467; Supplementary Figure S3a–h. Sequence variations generated by the different methods tested for Sample 245627; Supplementary Figure S4a–h. Sequence variations generated by the different methods tested of Sample 246038; Supplementary Table S1: Summary of key steps in the methods evaluated; Supplementary Table S2: The number of sequence variations in the final consensus sequence in each RNA segment by method.
Author Contributions
Conceptualization, Hon Ip; Data curation, Hon Ip and Sarah Uhm; Formal analysis, Hon Ip, Sarah Uhm, Mary Lea Killian and Mia Torchetti; Funding acquisition, Hon Ip and Mia Torchetti; Investigation, Hon Ip, Sarah Uhm, Mary Lea Killian and Mia Torchetti; Methodology, Hon Ip, Sarah Uhm, Mary Lea Killian and Mia Torchetti; Project administration, Hon Ip and Mia Torchetti; Resources, Hon Ip, Mary Lea Killian and Mia Torchetti; Supervision, Hon Ip and Mia Torchetti; Validation, Hon Ip, Sarah Uhm and Mia Torchetti; Writing – original draft, Hon Ip; Writing – review & editing, Hon Ip, Sarah Uhm, Mary Lea Killian and Mia Torchetti. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a U.S. Department of Agriculture Farm Bill grant and U.S. Geological Survey Ecosystems Grant GR.22.NC00.FV325.
Data Availability Statement
Data associated with this project are available at Ip, H.S., Uhm, S., Killian, M.L., and Torchetti, M., 2022, Raw MinION FASTQ datafiles corresponding to the paper “An evaluation of avian influenza virus whole genome sequencing approaches using nanopore technology”: U.S. Geological Survey data release, https://doi.org/10.5066/P93VXVGO.
Acknowledgments
We thank Katy Griffin for her expert assistance and members of the National Veterinary Services Laboratories for their excellent technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
Disclaimer
Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government. The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the U.S. Department of Agriculture.
References
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Worp, N.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Haagmans, B.; Fouchier, R.A.M.; Koopmans, M. The next Phase of SARS-CoV-2 Surveillance: Real-Time Molecular Epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Youk, S.; Lee, D. Role of Wild Birds in the Spread of Clade 2.3.4.4e H5N6 Highly Pathogenic Avian Influenza Virus into South Korea and Japan. Infect. Genet. Evol. 2022, 101, 105281. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, J.H.; Fouchier, R.A.M.; Lewis, N. Highly Pathogenic Avian Influenza Viruses at the Wild–Domestic Bird Interface in Europe: Future Directions for Research and Surveillance. Viruses 2021, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Caliendo, V.; Lewis, N.S.; Pohlmann, A.; Baillie, S.R.; Banyard, A.C.; Beer, M.; Brown, I.H.; Fouchier, R.A.M.; Hansen, R.D.E.; Lameris, T.K.; et al. Transatlantic Spread of Highly Pathogenic Avian Influenza H5N1 by Wild Birds from Europe to North America in 2021. Sci. Rep. 2022, 12, 11729. [Google Scholar] [CrossRef] [PubMed]
- Bevins, S.N.; Shriner, S.A.; Cumbee, J.C.; Dilione, K.E.; Douglass, K.E.; Ellis, J.W.; Killian, M.L.; Torchetti, M.K.; Lenoch, J.B. Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021. Emerg. Infect. Dis. 2022, 28, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Denzin, N.; Bölling, M.; Pohlmann, A.; King, J.; Globig, A.; Conraths, F.J. Investigation into a Superspreading Event of the German 2020–2021 Avian Influenza Epidemic. Pathogens 2022, 11, 309. [Google Scholar] [CrossRef]
- Engelsma, M.; Heutink, R.; Harders, F.; Germeraad, E.A.; Beerens, N. Multiple Introductions of Reassorted Highly Pathogenic Avian Influenza H5Nx Viruses Clade 2.3.4.4b Causing Outbreaks in Wild Birds and Poultry in The Netherlands, 2020-2021. Microbiol. Spectr. 2022, 10, e02499–21. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza; Adlhoch, C. ; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; et al. Avian Influenza Overview March – June 2022. EFSA J. 2022, 20. [Google Scholar] [CrossRef]
- US Department of Agriculture 2022 Confirmations of Highly Pathogenic Avian Influenza in Commercial and Backyard Flocks. https://www.aphis.usda.gov/aphis/ourfocus/animalhealth/animal-disease-information/avian/avian-influenza/hpai-2022/2022-hpai-commercial-backyard-flocks. Accessed 12/9/22.
- US Department of Agriculture 2022 Detections of Highly Pathogenic Avian Influenza in Wild Birds. https://www.aphis.usda.gov/aphis/ourfocus/animalhealth/animal-disease-information/avian/avian-influenza/hpai-2022/2022-hpai-wild-birds. Accessed 12/9/22.
- International Committee on Taxomomy of Viruses: ICTV Current ICTV Taxonomy Release: Orthomyxoviridae. 2021. https://ictv.global/taxonomy/taxondetails?taxnode_id=202103956. Accessed 12/9/22.
- Zhou, B.; Wentworth, D.E. Influenza A Virus Molecular Virology Techniques. In Influenza Virus; Kawaoka, Y., Neumann, G., Eds.; Humana Press: Totowa, NJ, 2012; Vol. 865, pp. 175–192. ISBN 978-1-61779-620-3. [Google Scholar]
- Lefterova, M.I.; Suarez, C.J.; Banaei, N.; Pinsky, B.A. Next-Generation Sequencing for Infectious Disease Diagnosis and Management. J. Mol. Diagn. 2015, 17, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Li, Z.; Li, R.; Tan, P.; Zhang, R.; Li, J. mNGS in Clinical Microbiology Laboratories: On the Road to Maturity. Crit. Rev. Microbiol. 2019, 45, 668–685. [Google Scholar] [CrossRef] [PubMed]
- Tshiabuila, D.; Giandhari, J.; Pillay, S.; Ramphal, U.; Ramphal, Y.; Maharaj, A.; Anyaneji, U.J.; Naidoo, Y.; Tegally, H.; San, E.J.; et al. Comparison of SARS-CoV-2 Sequencing Using the ONT GridION and the Illumina MiSeq. BMC Genomics 2022, 23, 319. [Google Scholar] [CrossRef] [PubMed]
- Quick, John Real-Time Gene Sequencing Can Help Control — and May Someday Prevent — Pandemics. Stat News. 2020. https://www.statnews.com/2020/09/11/real-time-gene-sequencing-can-help-control-and-may-someday-prevent-pandemics/. Accessed 12/9/22.
- Charre, C.; Ginevra, C.; Sabatier, M.; Regue, H.; Destras, G.; Brun, S.; Burfin, G.; Scholtes, C.; Morfin, F.; Valette, M.; et al. Evaluation of NGS-Based Approaches for SARS-CoV-2 Whole Genome Characterisation. Virus Evol. 2020, 6, veaa075. [Google Scholar] [CrossRef] [PubMed]
- Crossley, B.M.; Rejmanek, D.; Baroch, J.; Stanton, J.B.; Young, K.T.; Killian, M.L.; Torchetti, M.K.; Hietala, S.K. Nanopore Sequencing as a Rapid Tool for Identification and Pathotyping of Avian Influenza A Viruses. J. Vet. Diagn. Investig. Off. Publ. Am. Assoc. Vet. Lab. Diagn. Inc 2021, 33, 253–260. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Harder, T.; Globig, A.; Stacker, L.; Günther, A.; Grund, C.; Beer, M.; Pohlmann, A. Highly Pathogenic Avian Influenza Virus Incursions of Subtype H5N8, H5N5, H5N1, H5N4, and H5N3 in Germany during 2020-21. Virus Evol. 2022, 8, veac035. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical Validity of Nanopore Sequencing for Rapid SARS-CoV-2 Genome Analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.C.-Y.; Chan, W.-M.; Ip, J.D.; Seng, C.W.-M.; Leung, K.-H.; Poon, R.W.-S.; Ng, A.C.-K.; Wu, W.-L.; Zhao, H.; Chan, K.-H.; et al. Nanopore Sequencing Reveals Novel Targets for Detection and Surveillance of Human and Avian Influenza A Viruses. J. Clin. Microbiol. 2020, 58, e02127-19. [Google Scholar] [CrossRef] [PubMed]
- El Gazzar, M. ; Chaves Protocol for Whole Genome Sequencing of Influenza Virus. https://community.nanoporetech.com/docs/prepare/library_prep_protocols/ligation-sequencing-influenza-whole-genome/v/inf_9166_v109_reva_24aug2022. Accessed 12/9/22.
- Delahaye, C.; Nicolas, J. Sequencing DNA with Nanopores: Troubles and Biases. PLOS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef] [PubMed]
- Center for Disease Control and Prevention Influenza Virus Genome Sequencing and Genetic Characterization. https://www.cdc.gov/flu/about/professionals/genetic-characterization.htm. Accessed 12/9/22.
- Gardy, J.L.; Loman, N.J. Towards a Genomics-Informed, Real-Time, Global Pathogen Surveillance System. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.M.; Martin, I.W.; Moschetti, W.E.; Kershaw, C.M.; Tsongalis, G.J. Third-Generation Sequencing in the Clinical Laboratory: Exploring the Advantages and Challenges of Nanopore Sequencing. J. Clin. Microbiol. 2019, 58, e01315–19. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.K.; Cronk, B.D.; Voorhees, I.E.H.; Rothenheber, D.; Anderson, R.R.; Chan, T.H.; Wasik, B.R.; Dubovi, E.J.; Parrish, C.R.; Goodman, L.B. Method Comparison of Targeted Influenza A Virus Typing and Whole-Genome Sequencing from Respiratory Specimens of Companion Animals. J. Vet. Diagn. Invest. 2021, 33, 191–201. [Google Scholar] [CrossRef]
- Galli, C.; Ebranati, E.; Pellegrinelli, L.; Airoldi, M.; Veo, C.; Della Ventura, C.; Seiti, A.; Binda, S.; Galli, M.; Zehender, G.; et al. From Clinical Specimen to Whole Genome Sequencing of A(H3N2) Influenza Viruses: A Fast and Reliable High-Throughput Protocol. Vaccines 2022, 10, 1359. [Google Scholar] [CrossRef] [PubMed]
- Whitford, W.; Hawkins, V.; Moodley, K.S.; Grant, M.J.; Lehnert, K.; Snell, R.G.; Jacobsen, J.C. Proof of Concept for Multiplex Amplicon Sequencing for Mutation Identification Using the MinION Nanopore Sequencer. Sci. Rep. 2022, 12, 8572. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Moore, N.E.; Deng, Y.-M.; Eccles, D.A.; Hall, R.J. MinION Nanopore Sequencing of an Influenza Genome. Front. Microbiol. 2015, 6, 766. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Net cDNA generated with four primer sets. The amount, in nanograms, of cDNA recovered following bead purification of the SuperScript III RT-PCR reaction on the three HPAI H5N1 swab samples is shown on the Y axis. Primer sets used (from Table 2) are listed on the X axis and the RNA samples used (from Table 1) are listed in the figure legend.
Figure 1.
Net cDNA generated with four primer sets. The amount, in nanograms, of cDNA recovered following bead purification of the SuperScript III RT-PCR reaction on the three HPAI H5N1 swab samples is shown on the Y axis. Primer sets used (from Table 2) are listed on the X axis and the RNA samples used (from Table 1) are listed in the figure legend.
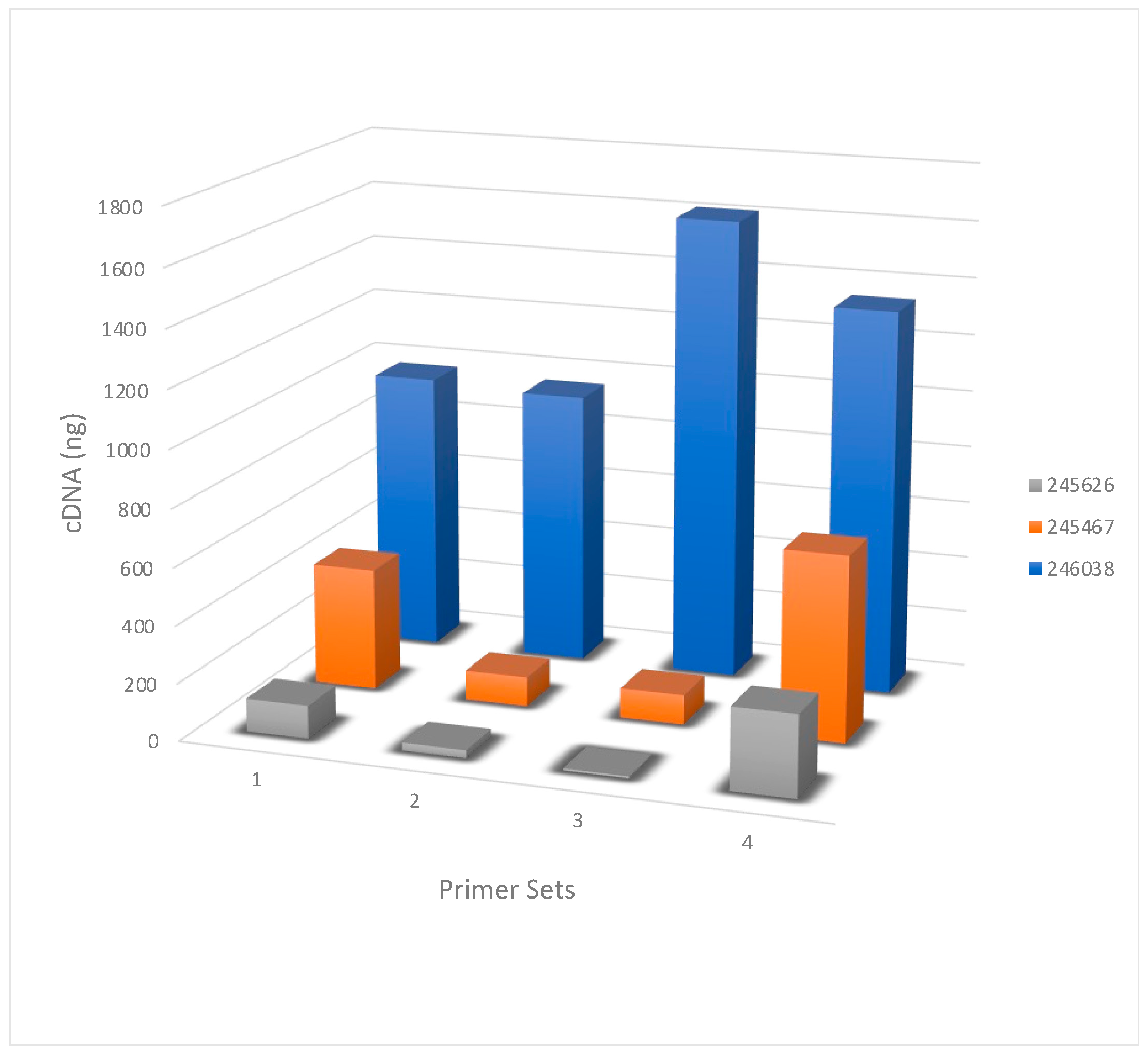
Figure 2.
Treemap representation of the time required for the methods evaluated.
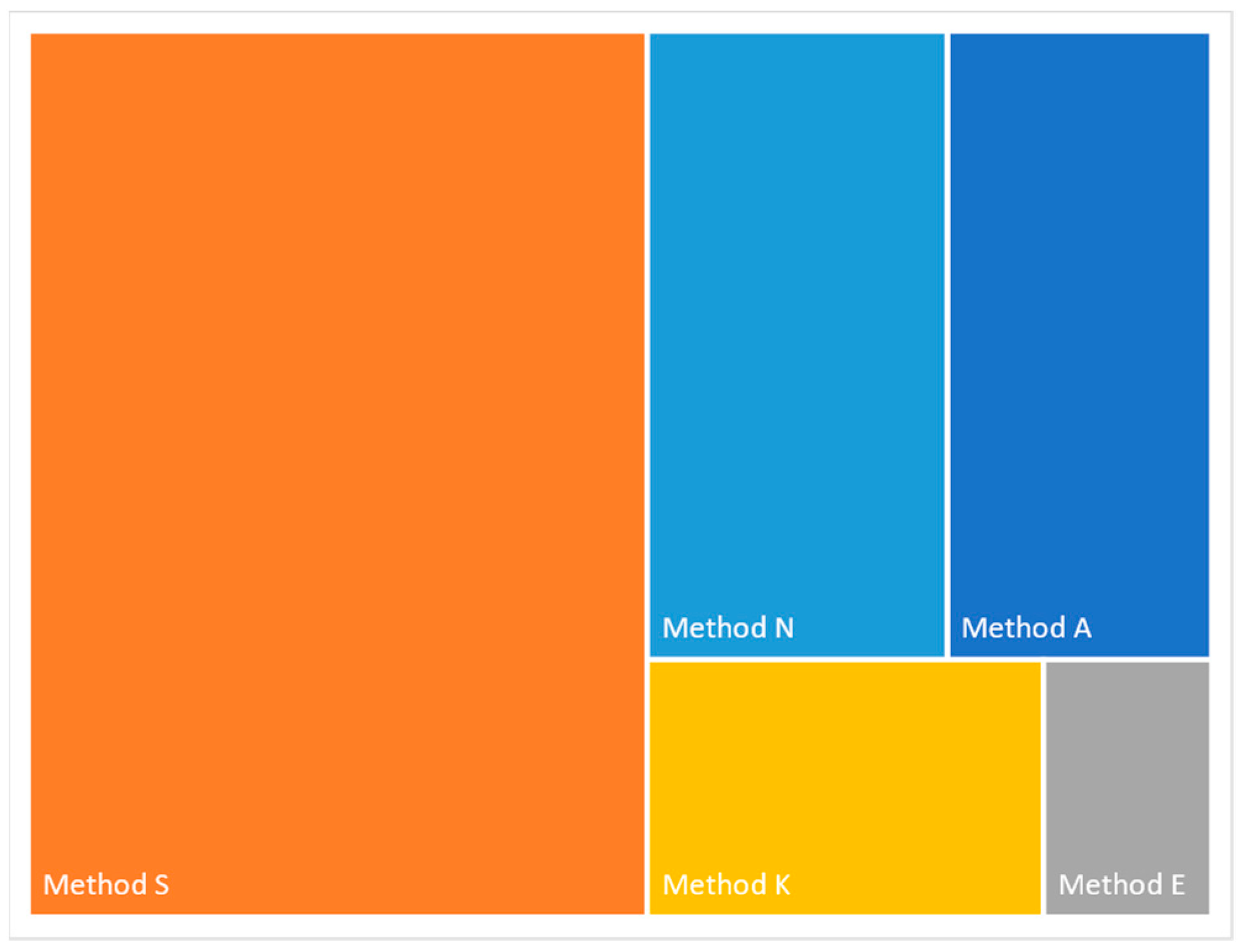
Table 1.
Highly pathogenic H5N1 avian influenza virus containing samples used in this study.

Table 2.
Primer sets for avian influenza virus whole genome amplification1.

1 Underlined sequences correspond to conserved sequences in influenza A viruses.
Table 4.
Summary of reads generated by each method with RNA sample 246038 that mapped to HPAI H5N1. Avian influenza virus (AIV) RNA segments, the number of reads mapped to each of the viral RNA segments, and the total number of mapped viral reads generated for each method are listed. Full length; the automatically assembled FASTA files were scored “Full” or “Partial” depending if the full length coding region was reconstructed or not. Genome Assembled; each kit was scored as “Complete” or “Incomplete” depending on whether the entire protein coding region for all eight segments was obtained or not.
Table 4.
Summary of reads generated by each method with RNA sample 246038 that mapped to HPAI H5N1. Avian influenza virus (AIV) RNA segments, the number of reads mapped to each of the viral RNA segments, and the total number of mapped viral reads generated for each method are listed. Full length; the automatically assembled FASTA files were scored “Full” or “Partial” depending if the full length coding region was reconstructed or not. Genome Assembled; each kit was scored as “Complete” or “Incomplete” depending on whether the entire protein coding region for all eight segments was obtained or not.
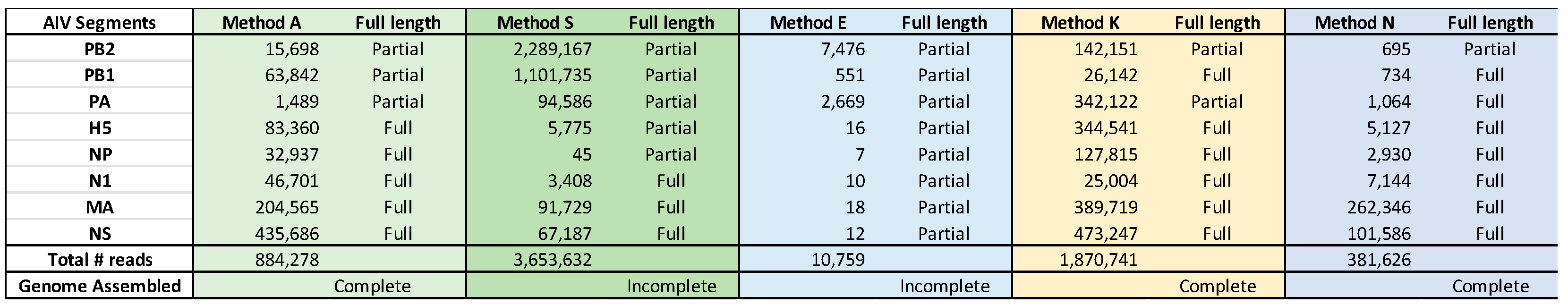
Table 5.
The total number of sequence variations across the virus genome per kit. The single nucleotide variations generated by each kit when evaluated against the reference sequence generated on the Illumina MiSeq are listed; del7; a deletion of seven nucleotides.
Table 5.
The total number of sequence variations across the virus genome per kit. The single nucleotide variations generated by each kit when evaluated against the reference sequence generated on the Illumina MiSeq are listed; del7; a deletion of seven nucleotides.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



