
Submitted:
20 January 2023
Posted:
27 January 2023
You are already at the latest version
Abstract
Alzheimer’s Disease (AD) has no cure. Earlier, we showed that partial inhibition of mitochondrial complex I (MCI) with small molecule CP2 induces adaptive stress response activating multiple neuroprotective mechanisms. Chronic treatment reduced inflammation, improved synaptic and mitochondrial functions, and blocked neurodegeneration in symptomatic APP/PS1 mice, a translational model of AD. Here, using serial block-face scanning electron microscopy (SBFSEM) and three-dimensional (3D) EM reconstructions combined with Western blot analysis and next-generation RNA sequencing, we demonstrate that CP2 treatment also restores mitochondrial morphology and mitochondria-endoplasmic reticulum (ER) communication in the APP/PS1 mouse brain. Using 3D EM volume reconstructions, we show that mitochondria in AD dendrites exist primarily as mitochondria-on-a-string (MOAS). Compared to other morphological phenotypes, MOAS are extensively enveloped in the ER membranes forming multiple mitochondria-ER contact sites (MERCS) known to contribute to abnormal lipid and calcium homeostasis. CP2 treatment specifically reduced MOAS formation, consistent with improved energy homeostasis in the brain, with concomitant reduction in MERCS, ER stress, and improved lipid homeostasis. These data provide novel information on the role MOAS play in AD and additional support for further development of partial MCI inhibitors as disease modifying strategy for AD.
Keywords:
Alzheimer’s Disease (AD) mitochondria
; endoplasmic reticulum (ER)
; serial block-face scanning electron microscopy (SBFSEM)
; three-dimensional electron microscopy (3D EM)
; small molecule mitochondria targeted therapeutics
1. Introduction
Alzheimer’s Disease (AD) is a devastating neurodegenerative disorder without a cure. The accumulation of misfolded amyloid beta (Aβ) peptides and hyperphosphorylated microtubule-associated tau protein (pTau) represents two hallmarks of AD. However, incomplete understanding of the molecular mechanisms hampers the development of disease-modifying strategies. Limited success of clinical trials focused on Aβ and pTau-reducing therapies emphasizes the need to identify new targets and therapeutic approaches. AD is a multifactorial disease developed over time where neuroinflammation, altered energetics and metabolism together with genetic and environmental contributions play important roles [1]. One of the early molecular mechanisms affected in AD includes abnormal energy homeostasis and mitochondrial dysfunction [2,3]. Reduced brain glucose metabolism is seen in patients with mild cognitive impairment, a prodromal stage of AD, indicating that mitochondrial ability to produce energy to maintain synaptic activity essential for cognitive function may be compromised [4]. Indeed, reduced glucose uptake in the brain together with the lower synaptic density correlate best with the development of cognitive abnormalities than levels of Aβ or pTau [4,5]. The origin of mitochondrial dysfunction in AD is not clear. Multiple studies demonstrated that Aβ and pTau could directly co-localize with mitochondria and disrupt their function leading to increased production of reactive oxygen species (ROS), reduced ATP levels, and altered axonal trafficking that perturbs mitochondrial localization at the synapses and distal sites of axons [6,7]. Abnormal mitochondrial dynamics in AD affect biogenesis (a process associated with the production of new organelles), fission, fusion, and removal of damaged mitochondria via mitophagy, leading to the accumulation of dysfunctional organelles and compromised cellular energetics [8]. However, emerging data also support the reciprocal relationship where dysfunctional mitochondria accumulated with age affect Aβ and Tau homeostasis, raising a possibility that mitochondria could play an underlying and contributing roles in the AD development and progression [9,10,11,12]. Taken together, these findings support the notion that strategies to improve mitochondrial dynamics and function could be beneficial for AD.
Mitochondria produce energy in the form of ATP to support synaptic transmission and cognitive function, mitigate calcium (Ca2+) signaling, govern biosynthesis of macromolecules (e.g., lipids, heme, and iron-sulfur clusters), innate immunity, and apoptosis [13]. To maintain cellular and energy homeostasis, especially under stress conditions, mitochondria communicate with the nucleus and other cytoplasmic compartments by releasing various metabolites, ROS, peptides, and by changing mitochondrial membrane potential. Mitochondria directly interact with subcellular organelles, as in the case of the endoplasmic reticulum (ER), forming close contacts (below 25 nm) known as the mitochondria-ER contact sites (MERCS, also biochemically known as mitochondria-associated membranes, MAMs) [14]. MERCS were originally identified in 1950s using electron microscopy (EM) [15] and are recognized as platforms for the regulation of Ca2+ flux, lipid metabolism, autophagosome formation, ER stress, mitochondrial quality control, mitochondrial bioenergetics, and apoptosis [16]. Structural and molecular studies in AD reveal altered mitochondria-ER communication, a “hypergeneration” of MERCS, which was linked to increased formation of Aβ, pTau, abnormal phospholipid and calcium homeostasis, mitochondrial dysfunction, and neurodegeneration [16,17]. MERCS are involved in the production of ceramides, core constituents of sphingolipids important for the regulation of cell growth, differentiation, cell cycle arrest, cell survival, apoptosis, and inflammation [18]. Specifically, increased levels of ceramides Cer16, Cer18, Cer20, and Cer24 in blood of AD patients were directly linked to oxidative stress and Aβ pathology [19,20].
Along with the exaggerated formation of MERCS in AD, changes in mitochondrial morphology were reported in model systems and postmortem AD brains [7]. Multiple studies demonstrated extensive mitochondrial fragmentation in AD [21]. However, these findings were based almost exclusively on the application of two-dimensional (2D) transmission electron microscopy (TEM) or fluorescence microscopy that lack appropriate resolution. In recent years, the development of advanced imaging techniques, such as a serial block-face scanning electron microscopy (SBFSEM), allowed three-dimensional (3D) assessment of mitochondrial structures in the context of complex 3D environment of brain tissue [22]. 3D reconstructions of thin consecutive brain slices generated using SBFSEM in postmortem brain tissue of AD patients and multiple mouse models of AD allowed us to identify a novel mitochondrial phenotype named mitochondria-on-a-string (MOAS) [7,23,24]. MOAS consist of multiple teardrop-shaped organelles connected with a thin double membrane of various length and thickness that are difficult to detect using methods other than 3D EM since the total length of MOAS could exceed 5 μm [24]. The formation of MOAS was associated with energetic stress where fission arrest may maintain residual mitochondrial function ensuring neuronal survival [24]. No data exist on to what extent MOAS formation affects MERCS. Up until now, the complex structure of MERCS was also visualized using standard 2D TEM lacking the clarity of 3D architecture. Thus, mitochondria in AD undergo multiple morphological transformations emphasizing the complexity of their dynamics and interaction with other organelles and the need for advanced techniques to address these changes.
We recently identified a small molecule tricyclic pyrone compound (code name CP2) that penetrates the blood brain barrier and accumulates in mitochondria where it selectively and specifically reduces the activity of mitochondrial complex I (MCI) by ~30% [25,26]. Chronic CP2 administration in independent cohorts of transgenic AD mice starting in utero, and at pre- or symptomatic stages of the disease improved synaptic function, reduced levels of Aβ and pTau, decreased inflammation and oxidative stress, increased expression of Sirtuins and brain-derived neurotrophic factor (BDNF), restored long-term potentiation (LTP), ultimately blocking the ongoing neurodegeneration and cognitive dysfunction [25,26,27,28]. Targeted metabolomic profiling conducted in plasma of CP2-treated female mice harboring familial AD mutations in human amyloid precursor protein (APP) and presenilin 1 (PS1), the APP/PS1 mice, revealed a reduction in levels of ceramides Cer16, Cer18, and Cer24 implicated in excessive MERCS formation and ER stress [19], suggesting that treatment might also have improved mitochondria-ER communication. Cross-validation of the RNA-seq data generated in female APP/PS1 mice treated with CP2 for 14 months against the human dataset available via the Accelerating Medicine Partnership Program for AD (AMP-AD) confirmed translational value of the mouse model and demonstrated that CP2 treatment restored pathways essential for human AD, including inflammation, oxidative stress, and synaptic function [26]. The extensive evaluation of the molecular mechanisms induced by CP2 treatment suggests multifaceted adaptive stress response with the activation of AMP-activated protein kinase (AMPK), enhanced autophagy, mitochondrial biogenesis and turnover, and improved energy homeostasis in brain and periphery [26,28,29]. These findings support the notion that mild energetic stress could produce beneficial effect by engaging hormetic responses similar to the effect of exercise and caloric restriction [29]. The innovation of this therapeutic strategy includes the activation of multiple neuroprotective pathways without targeting them individually, which mimics polypharmacy approach that might be necessary for treatment of complex neurodegenerative diseases. Here, using advanced SBFSEM and 3D EM reconstruction, Western blot, and RNA-seq data analyses, we demonstrate that adaptive stress response to partial inhibition of MCI results in improved mitochondrial morphology, reduced MERCS, and alleviated ER stress in the hippocampus of APP/PS1 female mice, which additionally could contribute to the neuroprotection.
2. Materials and Methods
2.1. Reagents and CP2 synthesis
All reagents were purchased from Sigma-Aldrich unless indicated otherwise. Trump’s fixative solution consisting of 4% formaldehyde + 0.1% glutaraldehyde in 0.1 M phosphate buffer was made monthly in house and stored at 4°C. CP2 was synthesized by the Nanosyn, Inc biotech company as described previously [26] and purified using HPLC. Authentication was done using NMR spectra to ensure the lack of batch-to-batch variation in purity. CP2 was synthesized as free base.
2.2. Mice and chronic in vivo CP2 treatment
Brain tissue for all experiments described in this paper were used from the study reported here [26]. The following female mice were used: double transgenic APP/PS1 and their non-transgenic (NTG) littermates [30]. Genotypes were determined by PCR as described in [30]. All animals were kept on a 12 h–12 h light-dark cycle, with a regular feeding and cage-cleaning schedule. Mice were randomly selected to study groups based on their age and genotype. NTG and APP/PS1 female mice (n = 16 - 21 per group) were given either CP2 (25 mg/kg/day in 0.1% polyethylene glycol [PEG] dissolved in drinking water ad lib) or vehicle-containing water (0.1% PEG) starting at 9 months of age [26]. Mice were housed 5 per cage, water consumption and weight were monitored weekly. CP2 concentration was adjusted based on mouse weight/water consumption weekly. Independent groups of mice were continuously treated for 14 months until the age of 23 months. Seven to eight months after CP2 treatment, mice were subjected to the battery of tests, including in vivo FDG-PET and blood-based metabolomics, as described in [26]. After mice were sacrificed, brain tissue was collected for Western blot analysis, RNA sequencing, and electron microscopy examination.
2.3. Tissue dissection for electron microscopy examination
Non-sedated mice were sacrificed by cervical dislocation to avoid technical artifacts in mitochondrial morphology associated with perfusion fixation or application of anesthetics [31]. The whole brain was quickly removed (< 2 min), hemispheres were separated, and one of the hemispheres was used for dissection of the CA1 region for the EM examination. The rest of the brain was cut and frozen for Western blot analysis and RNA sequencing. Hippocampal CA1 region was dissected from vehicle and CP2-treated NTG and APP/PS1 mice, cut into 2 mm3 pieces, and immediately transferred to a glass bottom dish containing Trump’s fixative. Tissues were kept overnight at room temperature. The next day, tissues were placed into 0.1 M phosphate buffer and stored at 4°C till processing for SBFSEM.
2.4. Serial block-face scanning electron microscopy (SBFSEM) and 3D EM reconstruction
Serial images for 3D EM reconstructions were obtained using a Thermo Fisher VolumeScope 2 SEM, which combines an integrated microtome and high-resolution field emission scanning electron microscope (SEM) to image through a sample in the z-axis. Following 24 h fixation, tissues were prepared for SBFSEM using a protocol developed by the National Center for Microscopy and Imaging Research (La Jolla, CA) [32]: (1) samples were rinsed 4 × 3 min in 0.1 M cacodylate buffer + 2 mM CaCl2, (2) incubated in 2% osmium tetroxide in 0.15 M cacodylate buffer for 1.5 h rotating at RT, (3) incubated in 2% osmium tetroxide + 2% potassium ferrocyanide in 0.1 M cacodylate for 1.5 h rotating at RT, (4) rinsed in H2O 4 × 3 min, (5) incubated in 1% thiocarbohydrazide (TCH) 45 min at 50 °C, (6) rinsed in H2O 4 × 3 min, (7) incubated in fresh 2% osmium tetroxide in H2O 1.5 h rotating at RT, (8) rinsed in H2O 4 × 3 min, (9) incubated in 1% aqueous uranyl acetate overnight at 4 °C, (10) further incubation in uranyl acetate 1 h at 50 °C, (11) rinsed in H2O 4 × 3 min, (12) incubated in lead aspartate 1 h at 50 °C, (13) rinsed in H2O 4 × 3 min, (14) dehydrated through ethanol series (60, 70, 80, 95, 100, 100%) 10 min each, (15) two rinses in 100% acetone 10 min each, (16) resin 1:2, 1:1, 3:1 in acetone 0.5 h, 1 h, 2 h, overnight in 100% resin. Samples were embedded into Embed-812 hard resin (EMS, Hatfield, PA), and allowed to polymerize for a minimum of 24 h prior to trimming and mounting. Tissue was trimmed of all surrounding resin and adhered to 8 mm aluminum pins (Ted Pella Inc., Redding, CA) using EpoTek silver epoxy (EMS, Hatfield, PA). A square tower (0.5 mm) was trimmed from the tissue using a Diatome ultratrim knife (EMS, Hatfield, PA) and the entire pin was coated with gold palladium. Serial block-face images were acquired using a Thermo Fisher Volumescope 2 SEMTM (Thermo Fisher, Inc., Waltham, MA) under high-vac/low water vapor conditions with a starting energy of 1.5 keV. A sectioning depth of 50 nm provided a final voxel size of 8 nm x 8 nm x 50 nm. Segmentation and three-dimensional analysis were performed using Reconstruct [33] and Amira 6.4 software (Thermo Fisher, Inc., Waltham, MA).
2.5. Image segmentation and quantitative morphometric analysis using 3D EM
Dendrites, mitochondria, ER and MERCS were segmented manually tracing 10 – 40 consecutive serial sections (0.09 µm thick) to generate 3D voxel segmentations. After 3D reconstruction was completed using Reconstruct software, traces were modified by an absolute intensity and maximal thresholding approach and exported in JPEG format into Amira software. The Brush and Lasso tools in Amira segmentation mode were used to select each object. If borders of an object were incomplete, it was excluded. Multiple data sets were volume rendered simultaneously, and each cellular structure was assigned a different color during the segmentation process using 3D threshold-based selection.
Quantitative analysis of mitochondrial morphology was conducted using the label analysis and measure tool function in Amira project view to obtain parameters related to mitochondria shape (i.e., length, volume, aspect ratio, sphericity, Supplementary Figures 1,2). The length was estimated using the ruler icon viewing in the Amira Project view, based on the distance between any two most distant points in a 3D object. The volume was estimated using default measurements in Amira Native Measurements and measured in μm3. The aspect ratio (AR) was computed as [(major axis)/(minor axis)], which reflects the “length to width ratio”. The sphericity represents the adherence of the object to that of a sphere and was calculated using the equation:
where V refers to the volume of the object and SA refers to the surface area of the object, with value of 1 indicating perfect spheroid. SA for each object was estimated automatically and measured by Amira Native parameters based on a total area of all mitochondria faces.
The following classification for mitochondrial types observed in dendrites were adapted from [34]: Type I: round shaped, no longer than 0.5 μm; Type II: medium size, one mitochondrial tubule of 0.5 to 5 μm in length but no longer than 5 μm; Type III: elongated, one mitochondrial tubule of 5 μm or longer; Type IV: mitochondria-on-a-string (MOAS), elongated interconnected organelles with more than one teardrop shaped mitochondria (~0.5 µm in diameter) connected by a thin double membrane extending up to 5 μm long (uniformly ~65 nm in diameter). The length and volume of MERCS were established using the measure tool in Amira project view. The maximum distance between any segment of the ER to a mitochondrion was set at 25 nm to be considered a juxtaposed area of mitochondria–ER interface. To measure the length of the contact, a freehand line was drawn from the beginning to the end of the mitochondrion-ER contact that was considered before. These values were then averaged to generate the length of contact sites for a given sample. To estimate mitochondrial coverage with MERCS, we calculated the percentage of mitochondrial perimeter covered by area of individual MERCS (Supplementary Figures 1, 2). Representative images are included with the paper, additional images available upon request directed to Dr. E. Trushina.
2.6. Mitochondrial fractionation
Fresh mouse brain tissue was washed with ice-cold phosphate buffer saline (PBS) to remove excess blood. The tissue was suspended in mitochondrial isolation buffer (MIBA) containing 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.2 M D-mannitol, 0.05 M sucrose, 0.5 mM sodium orthovanadate, 1 mM sodium fluoride and 1×complete protease and phosphatase inhibitor cocktail (Roche, USA), and homogenized with 10-20 strokes using a Teflon pestle (motor speed as 100). The crude nuclei (CN) fraction was pelleted from the lysate by centrifugation at 500 × g at 4 °C for 5 min. The remaining supernatant was centrifuged at 500 × g at 4°C for 5 min to remove the micronuclei. Then, the supernatant was centrifuged at 8000 × g for 10 min at 4 °C yielding heavy mitochondrial (MT pellet) and cytoplasmic (CY supernatant) fractions. The MT pellet was washed two times with ice-cold MIBA buffer before it was resuspended in the lyses buffer. Three mice from each experimental group were taken into the analysis.
2.7. Western Blot analysis
Levels of proteins were determined in cortico-hippocampal regions of the brain or enriched mitochondria fractions obtained as described above from vehicle and CP2-treated NTG and APP/PS1 mice (n = 2 - 3 mice per group) using Western blot analysis. Samples were homogenized and lysed using 1× RIPA buffer plus inhibitors. Total protein lysates (20 µg) were separated in equal volume on 4 – 20% Mini-PROTEAN TGX™ Precast Protein Gels (Bio-Rad, cat. # 4561096) and transferred to PVDF membrane (cat. #1620177). The following primary antibodies were used: Drp1 (1:1000, BD Biosciences), phospho-Drp1 S616 (1:1000, Cell Signaling, cat. #3455), OPA1 (1:1000, Fisher Scientific, cat. #BDB612607), Mfn1 (1:1000, EMD Millipore, cat. #ABC41-M), Mfn2 (1:1000, Sigma, cat. # M6444-200UL), Vdac1 (1:1000, Proteintech, cat. #55259-1-AP), Tfam (1:1000, Sigma-Aldrich, cat. #AV36993), Tfeb (1:500, Thermo Fisher, cat. #PA5-75572), LC3b (1:1000, Novus Biologicals, cat. #NB100-2220), Pink1 (1:1000, Novus Biologicals, cat. #BC100-494), Parkin (PRK8) (1:1000, Santa Cruz, cat. #sc-32282), β-Actin (1:5000, Sigma-Aldrich, cat. #A5316). The following secondary antibodies were used: Peroxidase (HRP) Anti-Rabbit IgG (H+L) Goat Secondary Antibody (1:5000 dilution, Jackson ImmunoResearch, cat. #111-035-003) and Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) (1:5000 dilution, Jackson ImmunoResearch, cat. #115-035-003). Band quantification was done using ChemiDoc Imaging System from Bio-Rad. Data analysis was done using Image J software.
2.8. Next-generation RNA sequencing
Detailed description of the next-generation RNA-seq data generated in the brain tissue from APP/PS1 and NTG mice treated with vehicle of CP2 for 14 months (n = 5 mice per group) has been published here [26]. Differential expression analysis was performed using EdgeR package. For specific pathway analysis, we used loose threshold of log2-fold-change ≥ 0.2 and p-values ≤ 0.05 as cutoff. Barcode plots were generated using limma package. RNA-seq data is available from Gene Expression Omnibus (GEO accession ID is GSE149248). Code (R script) used to generate final RNA-seq analysis is available in Supplementary Data 22, 23 published here [26].
2.9. Statistics
Statistical analyses were performed using the GraphPad Prism (Version 8, GraphPad Software, Inc., La Jolla, Ca). Statistical comparisons among three groups were done by one-way ANOVA and the one-sided unpaired Student’s t test, where appropriate. Significant differences between vehicle and CP2-treated groups within the same genotype and differences among NTG, APP/PS1, and APP/PS1 + CP2 mice were considered in the final analysis. Data are presented as mean ± S.E.M. for each group of mice. Sample sizes were determined by setting a minimum n number for in vitro biological replicates at 3, to allow for statistical testing. All replicates displayed in this paper are biological replicates, technical replicates (usually 3) were performed and used to generate the means for each biological replicate. We were blinded to both the genetic and treatment information, being unblinded after the analyses were completed.
3. Results
3.1. Evaluation of mitochondrial morphology and MERCS in brain tissue of APP/PS1 and NTG mice using SBFSEM and 3D EM reconstruction
NTG and APP/PS1 female mice were treated with CP2 or vehicle starting at 9 months of age, after the onset of cognitive decline and neurodegeneration, to test efficacy of this novel therapeutic strategy under conditions most relevant to patients (Figure 1a) [26]. At the end of the study, the CA1 hippocampal tissue was dissected for SBFSEM [31]. For 3D EM reconstruction, 20 - 40 serial sections were collected, stacked, aligned, and visualized using Amira (Figure 1b) or Reconstruct software (Figure 1c). This approach provides high quality samples and 3D visualization of mitochondrial and the ER morphology (Figure 1c). An advantage of 3D EM is the ability to observe and quantitate morphological complexity of mitochondrial networks in great detail [35]. Amira allows for segmentation, rendering, and color-coded visualization of 3D EM reconstructions to obtain quantitative data (Supplementary Figures 1,2) [34,35]. Particularly, 3D volume segmentation provides clear identification of MERCS by visualizing contacts between mitochondria and the ER in 3D space. We limited volume reconstruction to dendrites since they could be unambiguously identified based on the presence of dendritic spines (Figure 1c, 2).
Data were collected from NTG and APP/PS1 mice treated with vehicle or CP2 to determine the effect of treatment on mitochondria shape, length, and the interaction with the ER (Figure 1 and 2). The representative reconstructions of mitochondria and the ER (Figure 1b) in dendrites (Figure 1c and 2) demonstrate 3D architecture of mitochondria including shape and volume and the distance between mitochondria-ER membranes. Detailed description of measurements using Amira software are presented in Supplementary Figure 1 and Figure 2.
3.2. CP2 treatment restores mitochondrial morphology in symptomatic APP/PS1 mice
We have previously shown that APP/PS1 mice have increased MOAS formation in the brain associated with abnormal glucose uptake and utilization [24]. We also found that CP2 treatment restored brain energy homeostasis in APP/PS1 mice suggesting an improvement in mitochondrial dynamics and function [25,26]. Thus, we first examined the effect of CP2 treatment on mitochondrial morphology in APP/PS1 mice compared to NTG littermates (Figure 3). 3D EM reconstructions of mitochondria were done in randomly selected dendrites with 60 - 100 organelles taken into the analysis per group (Figure 3a). To provide quantitative assessment of mitochondrial morphology, mitochondria were classified into four distinct types based on their 3D aspect ratio (length vs width) and volume (Figure 3b). Type I represents round mitochondria with the diameter below 1 μm. Type II includes mitochondria of elongated shape with the length of 1 to 5 μm. Type III consists of elongated organelles longer than 5 μm, and Type IV represents MOAS (Figure 3b).
We found that in NTG mice, dendritic mitochondria in the hippocampus were predominantly of Type II (~70%), with small percent of Types I (~5%) and III (and ~15%) (Figure 3c). NTG mice did not have MOAS in the hippocampus. Consistent with our previous observations [24,26], vehicle-treated APP/PS1 mice had increased MOAS (up to 80% of total mitochondria) and Type I (30% in APP/PS1 mice compared to 5% in NTG mice) (Figure 3c). We did not observe Type III mitochondria in APP/PS1 mice while in NTG mice this type represented ~15% of mitochondria population. CP2 treatment restored mitochondrial phenotype in APP/PS1 mice to the observed in NTG mice (Figure 3a-d). The most noticeable was the reduction of MOAS by 80% (Figure 3d). We also found that CP2 treatment reduced the number of round-shaped Type I organelles and increased the number of Types II and III mitochondria in the brain tissue of CP2-treated APP/PS1 mice compared to vehicle-treated counterparts (Figure 3c, Supplementary Movie 1). These data confirmed that CP2 treatment not only improved brain energy homeostasis but also restored mitochondrial morphology reducing fragmentation and MOAS formation in hippocampus of APP/PS1 mice.
3.3. CP2 treatment reduces MERCS in APP/PS1 mice
Extensive MERCS formation in AD was shown to facilitate Aβ production, abnormal lipid homeostasis, and ER stress [36,37,38]. We previously reported that CP2 treatment reduced levels of Aβ and ceramides in APP/PS1 mice, indicating a potential improvement of mitochondria-ER communication and a reduction of ER and unfolded protein response (UPR) stress [26]. To evaluate the relationship between different types of mitochondrial morphology and the ER, we utilized SBFSEM, 3D EM reconstructions and the Amira software to visualize and quantify MERCS architecture in the hippocampus of NTG and APP/PS1 mice treated with vehicle or CP2 (Figure 4, Supplementary Movie 2). MERCS were defined as the ER segments that are within the 25 nm or less proximity to mitochondria (Figure 1b, 4a, Supplementary Figures 1,2). We estimated MERCS length, volume and the percentage of mitochondrial surface covered with the ER. The analysis was done for mitochondrial Types I-III pooled together where MOAS were analyzed separately. Examination of 3D EM reconstructions revealed profound differences in the extent of mitochondria-ER interactions. For Types I-III, a presence of the ER membranes along mitochondria was sporadic without formation of continuous networks (Figure 4a). However, MOAS were extensively covered with the ER membranes (Figure 4a). The extent of mitochondrial Types I-III perimeter covered with the ER was alike in NTG and APP/PS1 mice (~18%, Figure 4b) while MOAS were covered to ~30% (Figure 4c). MERCS length did not differ significantly for Types I - III in NTG and APP/PS1 mice (~80 nm, Figure 4d) while mean MERCS length for MOAS was ~200 nm (Figure 4e). The volume of MERCS associated with Types I-III mitochondria was significantly greater in APP/PS1 mice compared to NTG mice (Figure 4f) and was even greater for MOAS (Figure 4g). CP2 treatment reduced MERCS and their volume for all types of mitochondria in APP/PS1 mice (Figure 4b,c,f,g), and specifically reduced MERCS length associated with MOAS (Figure 4e). Taken together, these data further support translational value of the APP/PS1 mouse model demonstrating increased MERCS in the hippocampus consistent with the presence of ER stress in AD. Furthermore, we found that compared to all mitochondrial morphological phenotypes, the interaction with the ER was specifically prominent in MOAS. CP2 treatment not only restored mitochondrial morphology decreasing MOAS in APP/PS1 mice but also reduced MERCS for all types of mitochondria.
3.4. CP2 treatment augmented mitochondrial biogenesis and turnover and reduced ER stress in APP/PS1 mice
The significant reduction of MOAS after CP2 treatment in APP/PS1 mice could be associated with improved glucose uptake and utilization in the brain detected using the in vivo 18F-fluorodeoxyglucose-positron emission tomography (FDG-PET) [24,26]. However, it remains unclear whether the reduction of MOAS after CP2 treatment was facilitated by changes in mitochondrial turnover, including enhanced fission, fusion, biogenesis and mitophagy. To address this question, we first conducted Western blot analysis in the whole brain extracts from NTG and APP/PS1 mice treated with vehicle or CP2 (Figure 5a,c, Supplementary Figure 3). Brain tissues were from the same cohort of mice used for 3D EM reconstructions. We found that CP2 treatment increased levels of the mitochondrial transcription factor A (Tfam), a master regulator of mitochondrial biogenesis. We next determined whether CP2 treatment enhanced mechanisms responsible for the removal of damaged organelles, which primarily occurs via autophagic degradation known as mitophagy. Indeed, levels of proteins essential for autophagy and mitophagy, including transcription factor EB (Tfeb), Parkin, and LC3b were elevated after CP2 treatment in APP/PS1 mice (Figure 5a,c). Levels of Pink1 remained unchanged.
We next examined the effect of CP2 treatment on the expression of mitochondrial fission and fusion proteins essential for mitochondrial turnover. Our earlier studies revealed that the most effective way to determine changes in these proteins was to assay mitochondrial fractions isolated from the brain regions of interest [23,24]. Indeed, no changes in fission and fusion proteins were detected in the whole brain extracts (Supplementary Figure 4). However, consistent with our previous findings showing increased translocation of fission protein Drp1 to MOAS during fission arrest, we found an increase in the expression of Drp1 phosphorylated at S616 in APP/PS1 mice compared to NTG mice (Figure 5b,d, Supplementary Figure 5). CP2 treatment did not decrease levels of Drp1 S616 associated with mitochondria in APP/PS1 mice and showed a trend toward additional increase. At the same time, we found increased levels of fusion proteins Mfn1 and Mfn2 (Figure 5b,d). No changes were detected in levels of fusion protein OPA1. These data indicate that reduction in MOAS after CP2 treatment in APP/PS1 mice was associated with enhanced mitochondrial dynamics, including augmented fission, fusion, biogenesis and mitophagy.
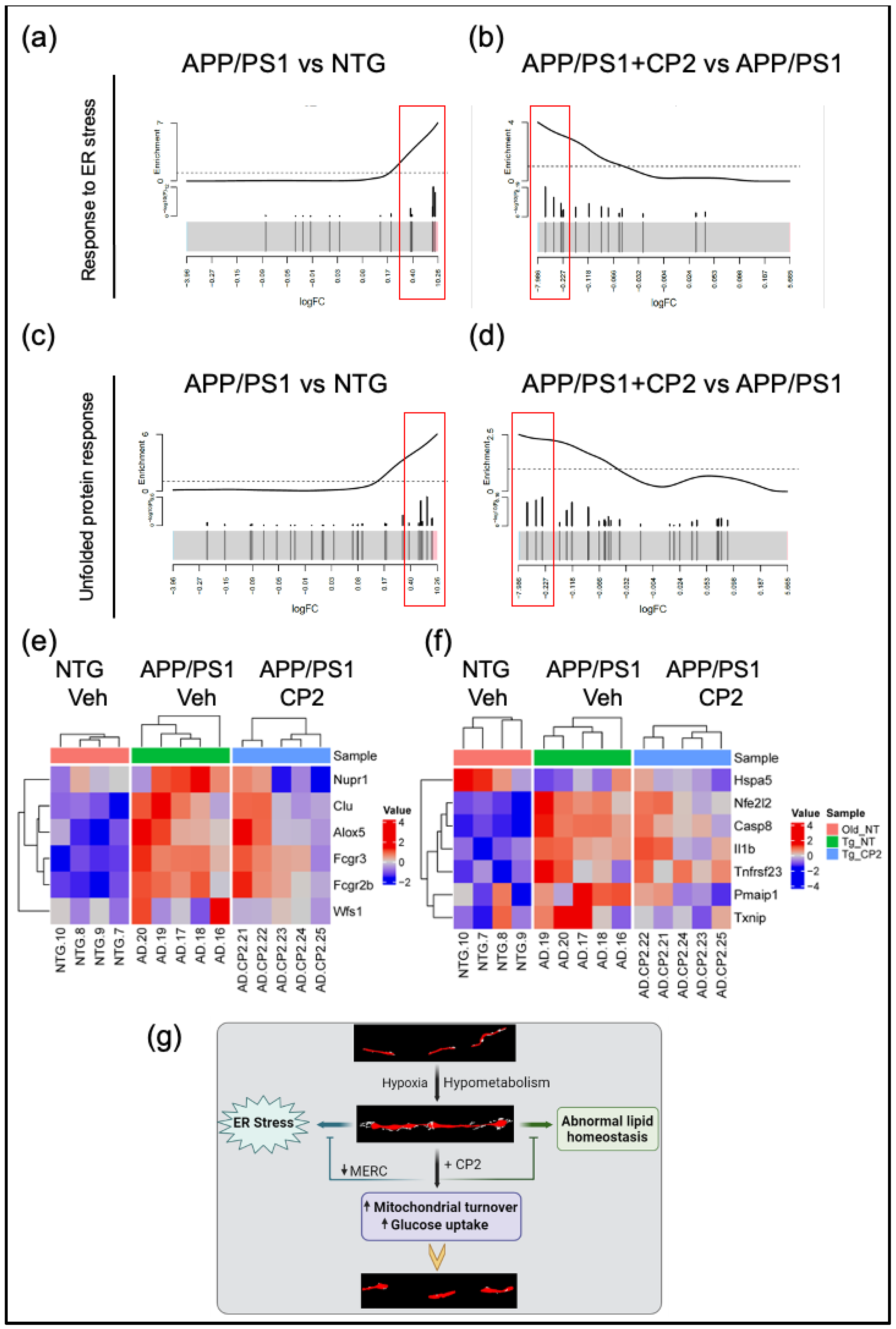
To further investigate the mechanism of CP2 action, we utilized the next-generation RNA sequencing (RNA-seq) data previously generated using brain tissue from vehicle- and CP2-treated NTG and APP/PS1 mice from the same cohorts that were used in the current study [26]. We identified 27 differentially expressed genes (DEGs) involved in the UPR, ER stress, mitochondria-ER tethering, mitochondrial dynamics, and autophagy (Figure 6, Supplementary Table 1). Key stress-response genes were upregulated in APP/PS1 mice compared to NTG littermates where chronic CP2 treatment restored their expression, indicating the relieve of cellular stress. Specifically, we found upregulation of genes involved in ER stress response mechanisms in APP/PS1 mice compared to NTG mice that were downregulated after CP2 treatment (Figure 6a,b,e). These genes included the Nuclear Transcriptional Regulator Protein 1 (Nupr1) involved in the regulation of autophagy-induced apoptosis through FOXO3 interaction [39]; Clusterin (Clu), which gene product functions as extracellular chaperone that prevents aggregation of proteins [40] and inhibits formation of amyloid fibrils [41]. Moreover, clusterin interacts with ubiquitin and SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complexes and promotes the ubiquitination and subsequent proteasomal degradation of target proteins [42]. Interestingly, mitochondrial clusterin suppresses BAX-dependent release of cytochrome c into the cytoplasm and inhibits apoptosis [43]. An intracellular form of clusterin suppresses stress-induced apoptosis by stabilizing mitochondrial membrane integrity through interaction with heat shock proteins HSPA5 [44]. Additional genes identified included arachidonate 5-lipoxygenase (Alox5), Fc Gamma Receptor IIIa (Fcgr3), Fc Gamma Receptor IIb (Fcgr2b), and Wolframin ER Transmembrane Glycoprotein (Wfs1) involved in mediating the inflammatory response, dendritic cell migration, apoptosis and intracellular calcium signaling [45,46,47,48]. Wfs1 gene product is directly involved in the regulation of cellular calcium homeostasis by modulating the ER calcium store [49].
Related to ER stress, another group of genes which expression was reversed by CP2 treatment in APP/PS1 mice included the UPR pathway (Figure 6c,d), which expression was downregulated in APP/PS1 mice (Figure 6f). Hspa5 protein localizes to the lumen of the ER where it is involved in the folding and assembly of the ER proteins serving as a master regulator of the ER homeostasis. During cellular stress, Hspa5 interacts with the transmembrane stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK), inositol-requiring kinase 1 (IRE1), and activating transcription factor 6 (ATF6) where it acts as a repressor of the UPR and also plays a role in cellular apoptosis and senescence [50]. Other genes are involved in anti-apoptotic, anti-inflammatory and antioxidant responses, including the master regulator of oxidative stress response Nuclear Factor Erythroid 2-Related Factor 2 (NFE2L2), Caspase 8 (Casp8), Interleukin 1 Beta (Il1b), Tumor Necrosis Factor Receptor Superfamily, Member 23 (Tnfrsf23), Phorbol-12-Myristate-13-Acetate-Induced Protein 1 (Pmaip1), and Thioredoxin Interacting Protein (Txnip). Importantly, thioredoxin-interacting protein has been recently identified as a link between redox state and metabolism where it plays an essential role in normal glucose homeostasis [51]. Furthermore, consistent with our observations of increased MERCS in APP/PS1 mice compared to NTG mice, we observed significant upregulation of RIPK1, RRBP1, SIGMAR1 and TGM2, which play important roles at MERCS [52,53,54,55]. CP2 treatment reversed trends in the expression of these genes (Supplementary Table 1). We also found that CP2 reversed changes in the expression of mitochondrial biogenesis regulator PGC-1α (Ppargc1a), mitochondrial dynamic protein MFN1 (Mfn1), and mitophagy related protein Pink1 (Pink1), consistent with the Western blot analysis (Figure 5, Supplementary Table 1). Collectively, these data suggest that CP2 treatment not only improves mitochondrial dynamics and reduces MOAS formation in APP/PS1 mice, but also alleviates MERCS dysregulation, ER and UPR stress.
4. Discussion
AD is a complex disorder with multiple pathways affected during the disease development. Some of these include inflammation, dysfunction of mitochondrial dynamics, function and bioenergetics, oxidative stress, altered protein and Ca2+ homeostasis, and ER stress. Since no biomarkers are available to clearly define at what stage of the disease what mechanisms become affected, the successful approach to treatment may require targeting multiple pathways at the same time. Such polypharmacy approach is very difficult given complicated etiology of AD. We identified MCI as a small molecule druggable target for AD [25,26,27,28,29]. Adaptive stress response associated with partial inhibition of MCI results in the activation of multiple mechanisms individually shown as neuroprotective [29]. Most important was the demonstration that treatment after the onset of cognitive decline and neurodegeneration was efficacious restoring key disease mechanisms, including abnormal energy homeostasis, synaptic dysfunction, inflammation, lipid homeostasis and accumulation of Aβ and pTau [26,28]. Using conventional 2D TEM, we found that CP2 treatment improved mitochondrial morphology [26]. Since MERCS are essential regulators of mitochondrial dynamics, we followed up with the in-depth evaluation of the effect of CP2 treatment on mitochondria-ER communication using advanced imaging technique with high resolution. Application of SBFSEM and 3D EM reconstructions allowed the visualization of changes in mitochondrial morphology and MERCS revealing novel insights into AD mechanisms.
One of the novel observations generated in this study was the demonstration that MOAS are increasingly associated with the ER forming extensive MERCS. Till now, this phenomenon has not been described in the context of AD. Our data suggest that MOAS, a mitochondrial phenotype formed in response to energetic stress in AD, have extensive interactions with the ER, greater than any other type of mitochondria. Currently, the precise mechanism of MOAS formation is not well understood, and it is unclear why MOAS are increasingly enveloped in the ER membranes. However, given the important role the ER plays in mitochondrial dynamics, fission in particular, such interaction may be instrumental for MOAS formation promoting fission arrest as well as MOAS elimination [56]. Indeed, data presented here demonstrate that CP2 treatment improves energy homeostasis in the brain leading to significant reduction in MOAS. Interestingly, the residual MOAS found in the brain of APP/PS1 mice after CP2 treatment had significantly decreased interaction with the ER, which may indicate that these structures were captured during their transition to different mitochondrial morphological types. It remains to be determined whether decreased interaction with the ER releases fission arrest associated with MOAS formation, activating Drp1-related fission, leading to MOAS elimination.
Along with the reduction of MOAS, CP2 treatment reduced the number of small mitochondria, which is commonly associated with mitochondrial dysfunction in AD [21]. The majority of mitochondria in CP2-treated APP/PS1 mouse brain were uniformly elongated, indicating increased fusion. Indeed, Western blot analysis and RNA-seq data indicate that treatment enhanced mitochondrial turnover restoring organelles and removing damaged ones. The increased organellar fusion, which is associated with improved function, is also consistent with reduced oxidative stress, enhanced bioenergetics, and improved ATP production in the brain of CP2-treated APP/PS1 mice detected in vivo using 31P Nuclear Magnetic Resonance (31P NMR) Spectroscopy and in neurons in vitro [26]. These data suggest a connection between abnormal glucose uptake and utilization in AD that results in increased MOAS and MERCS formation, ER stress and abnormal lipid homeostasis, consistent with the identification of these mechanisms as the underlying in AD etiology (Figure 6g).
Our findings further demonstrate that CP2 treatment may not only be beneficial for AD but also could improve health span delaying the onset of AD in aging individuals. We found that the mitochondrial MERCS coverage was reduced by CP2 treatment in APP/PS1 mice to the level below the observed in NTG aging mice. Since MERCS play the regulatory role in senescence and aging [57], this may indicate the potential rejuvenating effect of treatment not only in APP/PS1 but also in NTG mice. This is consistent with our observations that CP2 treatment reduced levels of senescent cells in adipose tissue of CP2-trearted APP/PS1 and NTG 24-month-old mice [26]. These data are also consistent with the findings showing the MCI as a small-molecule-sensitive modifier of lifespan where treatment of short-lived killifish with very low doses of MCI inhibitor rotenone reversed aging-related changes in gene expression and extended lifespan [58].
Other important outcome of this study includes the validation of translational value of APP/PS1 mice that recapitulate changes in mitochondrial morphology and MERCS hypergeneration found in AD patients [24,59]. Many promising therapeutic strategies shown efficacious in mouse models of AD failed clinical trials, emphasizing the need for better preclinical models. APP/PS1 mice recapitulate many AD pathologies providing confidence in translational success of this novel treatment. APP/PS1 mice have increased levels of blood ceramides associated with abnormal mitochondria-ER communication similar to found in blood of AD patients [26]. CP2 treatment reduced levels of ceramides [26] and MERCS, indicating that therapeutic efficacy could be monitored using blood-based translational biomarkers. This reduction of MECRS in APP/PS1 mice by CP2 treatment may also restore Ca2+ signaling that also might have contributed to the increased neuronal survival and cognitive protection [26,28]. Transcriptomic data generated in the brain of APP/PS1 mice overlapped with changes in AD patients [26]. Here, we re-analyzed these data and demonstrated the presence of UPR and ER stress, which was reduced by CP2 treatment, further supporting translational value of this mouse model. The ER stress is commonly defined as a response to the build-up of unfolded or misfolded proteins within the ER, affecting the ER and cellular homeostasis [60]. Recent data also suggest the reciprocal relationship between abnormal mitochondria-ER communication and ER stress, which contributes to the onset and development of AD. Given the significance of the impact of ER stress on AD progression, multiple therapeutic approaches have been developed to mitigate ER stress with few being tested in clinical trials [60]. Our data indicate that, without direct targeting the UPR or ER stress mechanisms, CP2 treatment reduced both, further strengthening therapeutic potential of this strategy.
Restored MERCS and mitochondrial morphology, improved lipid and glucose homeostasis, reduced UPR and ER stress in APP/PS1 mice after CP2 treatment coincide with reduction of Aβ and pTau [26,28]. The hierarchy of the beneficial mechanisms activated by CP2 treatment remains to be established. Our current findings suggest that activation of AMPK may play an essential role in the beneficial mechanistic cascade [25,26,27,28,29]. AMPK is a master regulator of cellular energy homeostasis that controls multiple mechanisms essential for neuronal survival and function. These mechanisms include synaptic transmission [61], mitochondrial biogenesis and turnover [62,63], inflammation [64,65], and the dynamics of MERCS [66], among others. However, excessive AMPK activation could be detrimental in AD [67]. Thus, future studies are needed to address safety and efficacy of targeting MCI for AD treatment. In this work, we focused exclusively on dendrites. Thus, it remains to be determined whether similar beneficial effects on mitochondrial morphology and MERCS are elicited by CP2 treatment in axons and other brain cells. It will also be interesting to examine the effect of disease and CP2 treatment on synaptic mitochondria. In conclusion, using advanced imaging technique, we provide new evidence that partial inhibition of MCI with small molecule CP2 improves mitochondrial dynamics and mitochondria-ER communication in dendrites in the hippocampus of APP/PS1 mice, and reduces UPR and ER stress. These data support further development of this strategy for AD.
Supplementary Materials
The following supporting information can be downloaded at: at the website of this paper posted on Preprints.org, Figure S1. Three-dimensional segmentation using Amira software. Protocol for generating surfaces and representative ortho slices of mitochondria-ER and MERCS, creating video maker option for better visualization using Amira protocols. Figure S2. Three-dimensional segmentation in Amira for MERCS identification. (a) Segmentation and 3D rendering of ER (yellow), mitochondria (b, green), and Mito-ER segmentation (c) to generate MERCS surfaces (d, pink). (e) Measuring MERCS morphology using label analysis measurements to determine volume, length, and MERCS coverage per mitochondria perimeter. Figure S3. Original uncropped Western blot images for Figure 5a. Cortico-hippocampal region from 3 mice per group was taken for Western blot analysis (NTG, n = 3; APP/PS1, n = 3; APP/PS1+CP2, n = 3) using whole brain lysate. Figure S4. (a) Expression of key fission/fusion proteins in whole brain extracts from NTG and APP/PS1 mice treated with vehicle or CP2. (b) Densitometry analysis of protein expression from (a) normalized to actin. Differences between individual groups were analyzed by one-sided unpaired Student’s t-test. Data are presented as mean ± SEM from 2 - 3 mice. * P < 0.05; ** P < 0.005. Figure S5. Original uncropped Western blot images for Figure 5b. Cortico-hippocampal regions from 3 mice per group were taken for Western blot analysis using mitochondrial factions. Movie S1. Representative 3D reconstructions from SBFSEM of dendrites (yellow), mitochondria (red) and ER (blue) in hippocampi from (a) NTG, (b) APP/PS1, (c) A/P+CP2 mouse. Movie S2. Representative 3D reconstructions highlighting MERCS (blue) in (a) NTG, (b) APP/PS1, and (c) APP/PS1 + CP2 mice. Table S1. Transcriptomic changes in brain tissue from vehicle and CP2-treated NTG and APP/PS1 mice determined using RNA-seq.
Author Contributions
Conceptualization, Eugenia Trushina; Investigation, Jessica Panes, Thi Kim Oanh Nguyen, Huanyao Gao, Trace Christensen, Andrea Stojakovic and Sergey Trushin; Supervision, Jorge Fuentealba; Writing – review & editing, Jeffrey Salisbury. ET designed the study, wrote the manuscript; ET and JF secured funding; AS treated mice, collected tissue; JP and TAC generated and analyzed 3D EM data; TKON and ST conducted WB analysis in brain tissue and isolated mitochondria; HG analyzed RNA seq data; JLS, JF and ET supervised study execution and data analysis. All authors contributed to data interpretation, manuscript editing and submission. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grants from the National Institutes of Health [grant numbers RF1AG55549, R01NS107265, RO1AG062135, AG59093, AG072899, UG3/UH3NS 113776, all to ET], Fondecyt 1200908 (to JF), and the ANID scholarship (21141247 to JP). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH and other funding organizations. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
All experiments with mice were approved by the Mayo Clinic Institutional Animal Care and Use Committee in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. IACUC Protocol A00001186-16-R18.
Informed Consent Statement
Not applicable
Data Availability Statement
RNA-seq data is available from Gene Expression Omnibus (GEO accession ID is GSE149248). Code (R script) used to generate final RNA-seq analysis is available in Supplementary Data 22, 23 published here [26].
Acknowledgments
We thank Ms. Shelly Gochnauer for help with manuscript submission.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat Neurosci 2020, 23, 311-322. [CrossRef]
- Mukherjee, S.; Heath, L.; Preuss, C.; Jayadev, S.; Garden, G.A.; Greenwood, A.K.; Sieberts, S.K.; De Jager, P.L.; Ertekin-Taner, N.; Carter, G.W.; et al. Molecular estimation of neurodegeneration pseudotime in older brains. Nat Commun 2020, 11, 5781. [CrossRef]
- Ke, J.; Tian, Q.; Xu, Q.; Fu, Z.; Fu, Q. Mitochondrial dysfunction: A potential target for Alzheimer’s disease intervention and treatment. Drug Discov Today 2021, 26, 1991-2002. [CrossRef]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol 2018, 17, 241-250. [CrossRef]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging 2009, 36, 811-822. [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol Cell Neurosci 2019, 98, 109-120. [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial Dysfunction in Alzheimer’s Disease and Progress in Mitochondria-Targeted Therapeutics. Current Behavioral Neuroscience Reports 2019, 6, 88–102. [CrossRef]
- Torres, A.K.; Jara, C.; Park-Kang, H.S.; Polanco, C.M.; Tapia, D.; Alarcon, F.; de la Pena, A.; Llanquinao, J.; Vargas-Mardones, G.; Indo, J.A.; et al. Synaptic Mitochondria: An Early Target of Amyloid-beta and Tau in Alzheimer’s Disease. J Alzheimers Dis 2021, 84, 1391-1414. [CrossRef]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp Neurol 2020, 330, 113321. [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J Alzheimers Dis 2018, 62, 1403-1416. [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener 2020, 15, 30. [CrossRef]
- Swerdlow, R.H. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid Redox Signal 2012, 16, 1434-1455. [CrossRef]
- Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab 2015, 22, 204-206. [CrossRef]
- Luan, Y.; Luan, Y.; Yuan, R.X.; Feng, Q.; Chen, X.; Yang, Y. Structure and Function of Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) and Their Role in Cardiovascular Diseases. Oxid Med Cell Longev 2021, 2021, 4578809. [CrossRef]
- Bernhard, W.; Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J Biophys Biochem Cytol 1956, 2, 73-78. [CrossRef]
- Barazzuol, L.; Giamogante, F.; Cali, T. Mitochondria Associated Membranes (MAMs): Architecture and physiopathological role. Cell Calcium 2021, 94, 102343. [CrossRef]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. Embo j 2012, 31, 4106-4123. [CrossRef]
- Zelnik, I.D.; Ventura, A.E.; Kim, J.L.; Silva, L.C.; Futerman, A.H. The role of ceramide in regulating endoplasmic reticulum function. Biochim Biophys Acta Mol Cell Biol Lipids 2020, 1865, 158489. [CrossRef]
- Filippov, V.; Song, M.A.; Zhang, K.; Vinters, H.V.; Tung, S.; Kirsch, W.M.; Yang, J.; Duerksen-Hughes, P.J. Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J Alzheimers Dis 2012, 29, 537-547. [CrossRef]
- Custodia, A.; Romaus-Sanjurjo, D.; Aramburu-Nunez, M.; Alvarez-Rafael, D.; Vazquez-Vazquez, L.; Camino-Castineiras, J.; Leira, Y.; Pias-Peleteiro, J.M.; Aldrey, J.M.; Sobrino, T.; et al. Ceramide/Sphingosine 1-Phosphate Axis as a Key Target for Diagnosis and Treatment in Alzheimer’s Disease and Other Neurodegenerative Diseases. Int J Mol Sci 2022, 23. [CrossRef]
- Reddy, P.H.; Reddy, T.P.; Manczak, M.; Calkins, M.J.; Shirendeb, U.; Mao, P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain research reviews 2011, 67, 103-118. [CrossRef]
- Trushina, E. A shape shifting organelle: unusual mitochondrial phenotype determined with three-dimensional electron microscopy reconstruction. Neural Regen Res 2016, 11, 900-901. [CrossRef]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer’s Disease. PLoS One 2012, 7, e32737. [CrossRef]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci Rep 2016, 6, 18725. [CrossRef]
- Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.; Yamazaki, Y.; et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s Disease. EBioMedicine 2015, 2, 294-305. [CrossRef]
- Stojakovic, A.; Trushin, S.; Sheu, A.; Khalili, L.; Chang, S.Y.; Li, X.; Christensen, T.; Salisbury, J.L.; Geroux, R.E.; Gateno, B.; et al. Partial inhibition of mitochondrial complex I ameliorates Alzheimer’s disease pathology and cognition in APP/PS1 female mice. Commun Biol 2021, 4, 61. [CrossRef]
- Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.T.; Yamazaki, Y.; et al. Corrigendum to "Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease" [EBioMedicine 2 (2015) 294-305]. EBioMedicine 2019, 42, 532. [CrossRef]
- Stojakovic, A.; Chang, S.Y.; Nesbitt, J.; Pichurin, N.P.; Ostroot, M.A.; Aikawa, T.; Kanekiyo, T.; Trushina, E. Partial Inhibition of Mitochondrial Complex I Reduces Tau Pathology and Improves Energy Homeostasis and Synaptic Function in 3xTg-AD Mice. J Alzheimers Dis 2020. [CrossRef]
- Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial complex I as a therapeutic target for Alzheimer’s disease. Acta Pharm Sin B 2022, 12, 483-495. [CrossRef]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 1998, 4, 97-100.
- Antentor Hinton Jr.; Prasanna Katti; Trace A. Christensen; Margaret Mungai; Jianqiang Shao; Liang Zhang; Sergey Trushin; Ahmad Alghanem; Adam Jaspersen; Rachel E. Geroux; et al. A comprehensive approach to artifact-free sample preparation and the assessment of mitochondrial morphology in tissue and cultured cells. 2021. [CrossRef]
- Deerinck;, T.J.; Bushong;, E.A.; Ellisman;, M.; Thor, A. Preparation of Biological Tissues for Serial Block Face Scanning Electron Microscopy (SBEM) v2. protocols.io, 2022. [CrossRef]
- Fiala, J.C. Reconstruct: a free editor for serial section microscopy. J Microsc 2005, 218, 52-61. [CrossRef]
- Garza-Lopez, E.; Vue, Z.; Katti, P.; Neikirk, K.; Biete, M.; Lam, J.; Beasley, H.K.; Marshall, A.G.; Rodman, T.A.; Christensen, T.A.; et al. Protocols for Generating Surfaces and Measuring 3D Organelle Morphology Using Amira. Cells 2021, 11. [CrossRef]
- Faitg, J.; Lacefield, C.; Davey, T.; White, K.; Laws, R.; Kosmidis, S.; Reeve, A.K.; Kandel, E.R.; Vincent, A.E.; Picard, M. 3D neuronal mitochondrial morphology in axons, dendrites, and somata of the aging mouse hippocampus. Cell Rep 2021, 36, 109509. [CrossRef]
- Schreiner, B.; Hedskog, L.; Wiehager, B.; Ankarcrona, M. Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J Alzheimers Dis 2015, 43, 369-374. [CrossRef]
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death & Disease 2018, 9, 335. [CrossRef]
- Yu, W.; Jin, H.; Huang, Y. Mitochondria-associated membranes (MAMs): a potential therapeutic target for treating Alzheimer’s disease. Clinical science (London, England : 1979) 2021, 135, 109-126. [CrossRef]
- Kong, D.K.; Georgescu, S.P.; Cano, C.; Aronovitz, M.J.; Iovanna, J.L.; Patten, R.D.; Kyriakis, J.M.; Goruppi, S. Deficiency of the transcriptional regulator p8 results in increased autophagy and apoptosis, and causes impaired heart function. Mol Biol Cell 2010, 21, 1335-1349. [CrossRef]
- Poon, S.; Easterbrook-Smith, S.B.; Rybchyn, M.S.; Carver, J.A.; Wilson, M.R. Clusterin is an ATP-independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 2000, 39, 15953-15960. [CrossRef]
- Hatters, D.M.; Wilson, M.R.; Easterbrook-Smith, S.B.; Howlett, G.J. Suppression of apolipoprotein C-II amyloid formation by the extracellular chaperone, clusterin. Eur J Biochem 2002, 269, 2789-2794. [CrossRef]
- Zoubeidi, A.; Ettinger, S.; Beraldi, E.; Hadaschik, B.; Zardan, A.; Klomp, L.W.; Nelson, C.C.; Rennie, P.S.; Gleave, M.E. Clusterin facilitates COMMD1 and I-kappaB degradation to enhance NF-kappaB activity in prostate cancer cells. Mol Cancer Res 2010, 8, 119-130. [CrossRef]
- Zhang, H.; Kim, J.K.; Edwards, C.A.; Xu, Z.; Taichman, R.; Wang, C.Y. Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol 2005, 7, 909-915. [CrossRef]
- Li, N.; Zoubeidi, A.; Beraldi, E.; Gleave, M.E. GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene 2013, 32, 1933-1942. [CrossRef]
- Smyrniotis, C.J.; Barbour, S.R.; Xia, Z.; Hixon, M.S.; Holman, T.R. ATP allosterically activates the human 5-lipoxygenase molecular mechanism of arachidonic acid and 5(S)-hydroperoxy-6(E),8(Z),11(Z),14(Z)-eicosatetraenoic acid. Biochemistry 2014, 53, 4407-4419. [CrossRef]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J Clin Invest 2011, 121, 569-581. [CrossRef]
- Lee, J.; Zhang, T.; Hwang, I.; Kim, A.; Nitschke, L.; Kim, M.; Scott, J.M.; Kamimura, Y.; Lanier, L.L.; Kim, S. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 2015, 42, 431-442. [CrossRef]
- Li, X.; Baskin, J.G.; Mangan, E.K.; Su, K.; Gibson, A.W.; Ji, C.; Edberg, J.C.; Kimberly, R.P. The unique cytoplasmic domain of human FcgammaRIIIA regulates receptor-mediated function. J Immunol 2012, 189, 4284-4294. [CrossRef]
- Takei, D.; Ishihara, H.; Yamaguchi, S.; Yamada, T.; Tamura, A.; Katagiri, H.; Maruyama, Y.; Oka, Y. WFS1 protein modulates the free Ca(2+) concentration in the endoplasmic reticulum. FEBS Lett 2006, 580, 5635-5640. [CrossRef]
- Fu, X.; Liu, J.; Liu, D.; Zhou, Y.; Guo, Y.; Wang, Z.; Yang, S.; He, W.; Chen, P.; Wang, X.; et al. Glucose-regulated protein 78 modulates cell growth, epithelial-mesenchymal transition, and oxidative stress in the hyperplastic prostate. Cell Death Dis 2022, 13, 78. [CrossRef]
- Chutkow, W.A.; Patwari, P.; Yoshioka, J.; Lee, R.T. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem 2008, 283, 2397-2406. [CrossRef]
- Zhao, H.H.; Han, Q.X.; Ding, X.N.; Yan, J.Y.; Li, Q.; Zhang, D.; Zhu, H.Y. Critical hubs of renal ischemia-reperfusion injury: endoplasmic reticulum-mitochondria tethering complexes. Chin Med J (Engl) 2020, 133, 2599-2609. [CrossRef]
- Bogar, F.; Fulop, L.; Penke, B. Novel Therapeutic Target for Prevention of Neurodegenerative Diseases: Modulation of Neuroinflammation with Sig-1R Ligands. Biomolecules 2022, 12. [CrossRef]
- Ismael, S.; Wajidunnisa; Sakata, K.; McDonald, M.P.; Liao, F.F.; Ishrat, T. ER stress associated TXNIP-NLRP3 inflammasome activation in hippocampus of human Alzheimer’s disease. Neurochem Int 2021, 148, 105104. [CrossRef]
- Kamboh, M.I.; Demirci, F.Y.; Wang, X.; Minster, R.L.; Carrasquillo, M.M.; Pankratz, V.S.; Younkin, S.G.; Saykin, A.J.; Alzheimer’s Disease Neuroimaging, I.; Jun, G.; et al. Genome-wide association study of Alzheimer’s disease. Transl Psychiatry 2012, 2, e117. [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 2020, 21, 204-224. [CrossRef]
- Ziegler, D.V.; Martin, N.; Bernard, D. Cellular senescence links mitochondria-ER contacts and aging. Commun Biol 2021, 4, 1323. [CrossRef]
- Baumgart, M.; Priebe, S.; Groth, M.; Hartmann, N.; Menzel, U.; Pandolfini, L.; Koch, P.; Felder, M.; Ristow, M.; Englert, C.; et al. Longitudinal RNA-Seq Analysis of Vertebrate Aging Identifies Mitochondrial Complex I as a Small-Molecule-Sensitive Modifier of Lifespan. Cell Syst 2016, 2, 122-132. [CrossRef]
- Morgado-Caceres, P.; Liabeuf, G.; Calle, X.; Briones, L.; Riquelme, J.A.; Bravo-Sagua, R.; Parra, V. The aging of ER-mitochondria communication: A journey from undifferentiated to aged cells. Front Cell Dev Biol 2022, 10, 946678. [CrossRef]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER stress and UPR in Alzheimer’s disease: mechanisms, pathogenesis, treatments. Cell Death Dis 2022, 13, 706. [CrossRef]
- Marinangeli, C.; Didier, S.; Ahmed, T.; Caillerez, R.; Domise, M.; Laloux, C.; Begard, S.; Carrier, S.; Colin, M.; Marchetti, P.; et al. AMP-Activated Protein Kinase Is Essential for the Maintenance of Energy Levels during Synaptic Activation. iScience 2018, 9, 1-13. [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 2018, 19, 121-135. [CrossRef]
- Vingtdeux, V.; Chandakkar, P.; Zhao, H.; d’Abramo, C.; Davies, P.; Marambaud, P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J 2011, 25, 219-231. [CrossRef]
- Saito, M.; Saito, M.; Das, B.C. Involvement of AMP-activated protein kinase in neuroinflammation and neurodegeneration in the adult and developing brain. Int J Dev Neurosci 2019, 77, 48-59. [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci Signal 2019, 12. [CrossRef]
- Hu, Y.; Chen, H.; Zhang, L.; Lin, X.; Li, X.; Zhuang, H.; Fan, H.; Meng, T.; He, Z.; Huang, H.; et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 2021, 17, 1142-1156. [CrossRef]
- Assefa, B.T.; Tafere, G.G.; Wondafrash, D.Z.; Gidey, M.T. The Bewildering Effect of AMPK Activators in Alzheimer’s Disease: Review of the Current Evidence. Biomed Res Int 2020, 2020, 9895121. [CrossRef]
Figure 1.
Treatment paradigm and workflow for 3D EM reconstruction of mitochondria and MERCS in brain tissue. (a) CP2 structure and a timeline of chronic CP2 administration to NTG and APP/PS1 female mice. (b) Serial block-face scanning electron microscopy (SBFSEM) with subsequent 3D EM reconstruction was done in the CA1 hippocampal region using Reconstruct and Amira software. Mitochondria (red) and associated ER (white arrows) were traced in a dendrite through consecutive brain slices generated using SBFSEM (left panels). Resulting 3D EM reconstructions of mitochondria (red) and the ER (brown) allow to identify MERCS with high accuracy (white arrows). Scale bar, 0.5 µm. (c) Representative 3D EM reconstruction of mitochondria (red) interacting with the ER (gold) in a dendrite of a WT mouse. Reconstruction was done using 28 0.09 μm thick TEM serial sections that were stacked, aligned, and reconstructed using Reconstruct software. Scale bar, 1 µm.
Figure 1.
Treatment paradigm and workflow for 3D EM reconstruction of mitochondria and MERCS in brain tissue. (a) CP2 structure and a timeline of chronic CP2 administration to NTG and APP/PS1 female mice. (b) Serial block-face scanning electron microscopy (SBFSEM) with subsequent 3D EM reconstruction was done in the CA1 hippocampal region using Reconstruct and Amira software. Mitochondria (red) and associated ER (white arrows) were traced in a dendrite through consecutive brain slices generated using SBFSEM (left panels). Resulting 3D EM reconstructions of mitochondria (red) and the ER (brown) allow to identify MERCS with high accuracy (white arrows). Scale bar, 0.5 µm. (c) Representative 3D EM reconstruction of mitochondria (red) interacting with the ER (gold) in a dendrite of a WT mouse. Reconstruction was done using 28 0.09 μm thick TEM serial sections that were stacked, aligned, and reconstructed using Reconstruct software. Scale bar, 1 µm.
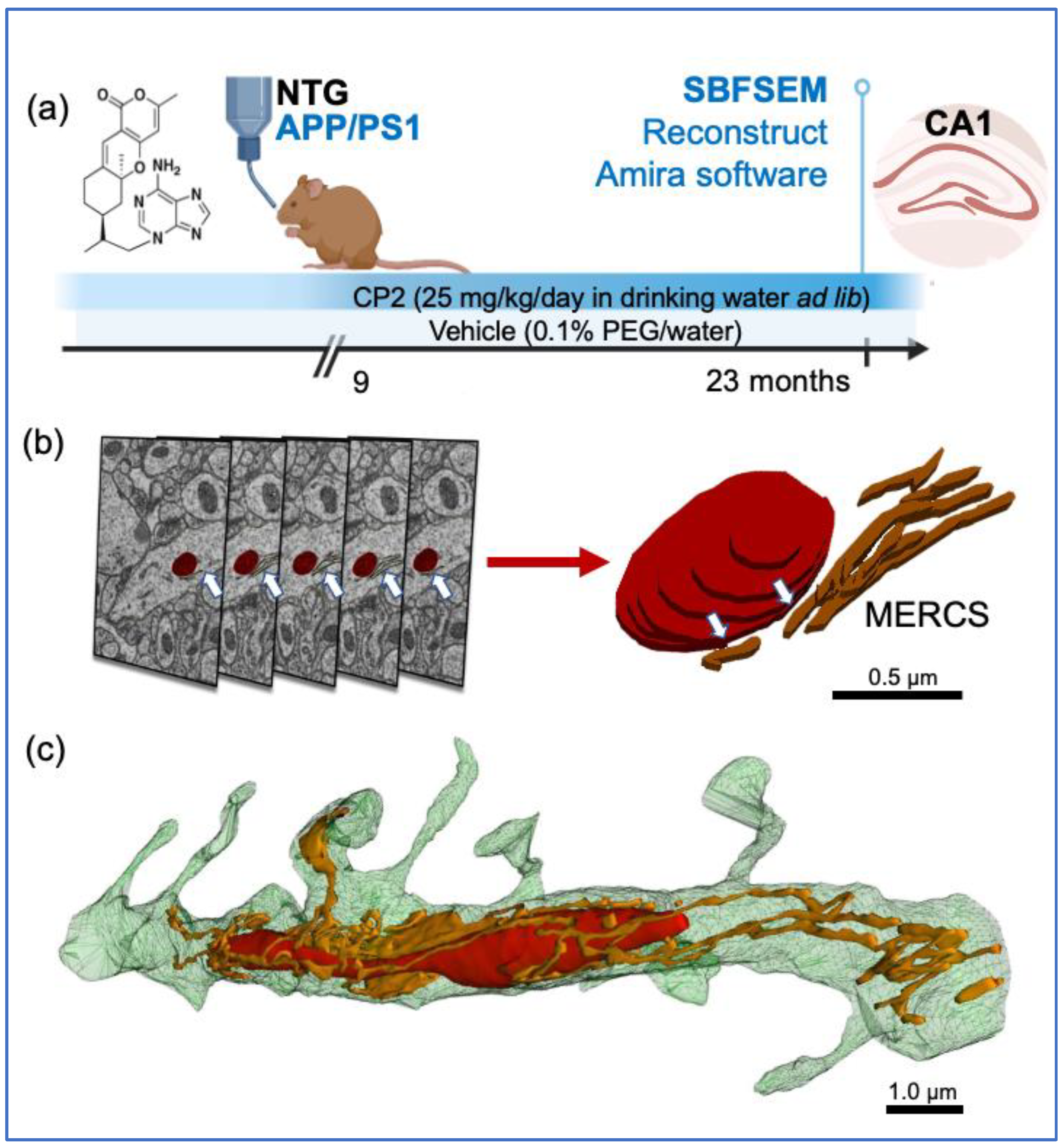
Figure 2.
Application of SBFSEM and 3D EM reconstructions to establish changes in mitochondrial morphology and MERCS in response to CP2 treatment. Representative 3D EM reconstructions of dendrites (yellow) and mitochondria (red) in hippocampal CA1 region of NTG (a), APP/PS1 (c) and APP/PS1+CP2 (e) mice. Scale bars, 5 μm. Representative 3D EM reconstructions of dendrites (yellow), mitochondria (red), and the ER (white) in hippocampal CA1 region of NTG (b), APP/PS1 (d) and APP/PS1+CP2 (f) mice. Scale bars, 2 μm. The Reconstruct software was applied to micrographs from 20 - 40 serial sections 0.09 μm thick generated using SBFSEM.
Figure 2.
Application of SBFSEM and 3D EM reconstructions to establish changes in mitochondrial morphology and MERCS in response to CP2 treatment. Representative 3D EM reconstructions of dendrites (yellow) and mitochondria (red) in hippocampal CA1 region of NTG (a), APP/PS1 (c) and APP/PS1+CP2 (e) mice. Scale bars, 5 μm. Representative 3D EM reconstructions of dendrites (yellow), mitochondria (red), and the ER (white) in hippocampal CA1 region of NTG (b), APP/PS1 (d) and APP/PS1+CP2 (f) mice. Scale bars, 2 μm. The Reconstruct software was applied to micrographs from 20 - 40 serial sections 0.09 μm thick generated using SBFSEM.
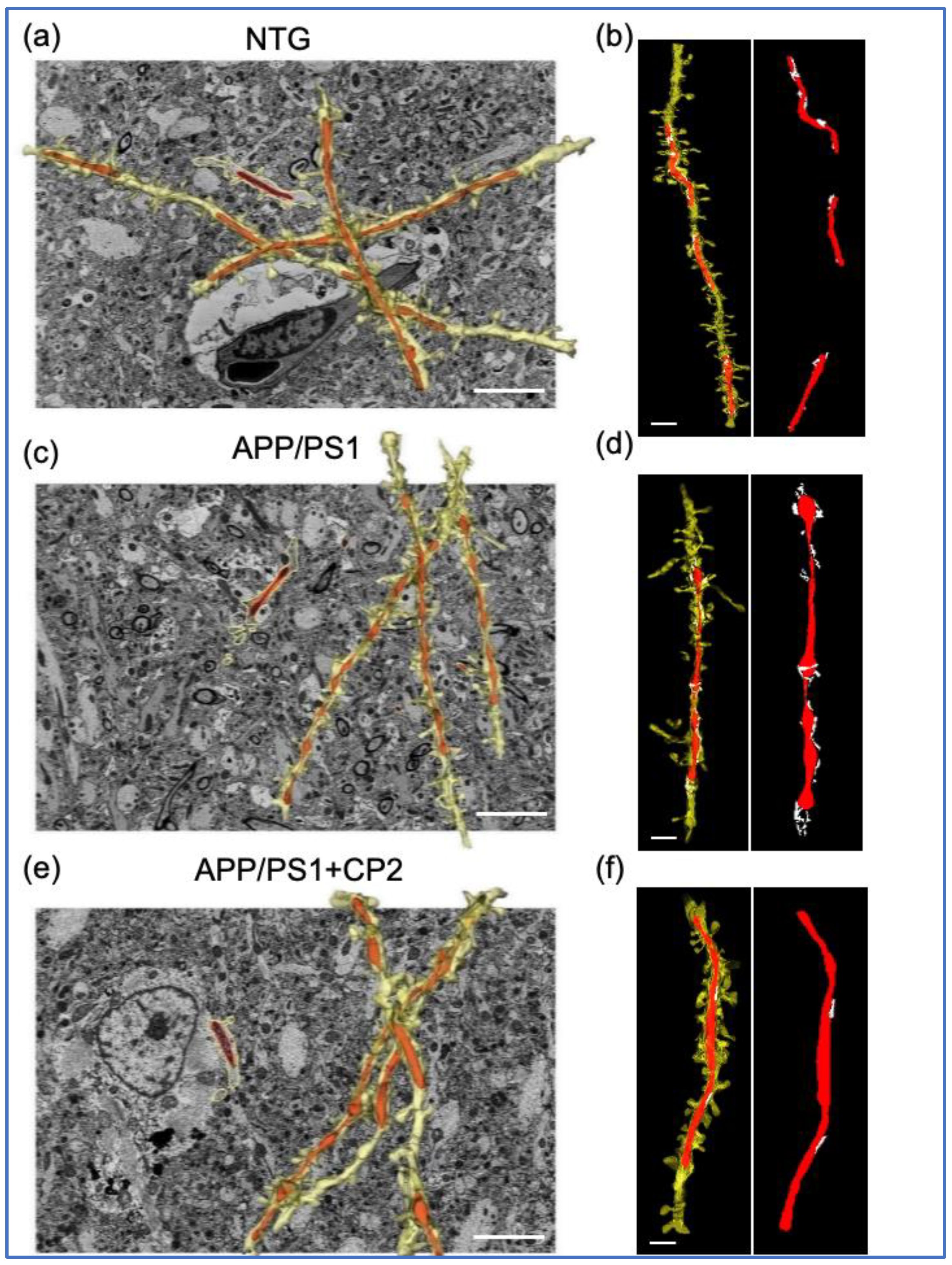
Figure 3.
Chronic CP2 treatment improves mitochondrial morphology in APP/PS1 (A/P) mouse brain. (a) Visualization of mitochondrial morphology in the hippocampi of NTG, APP/PS1 and APP/PS1+CP2 mice using SBFSEM and 3D EM reconstructions with the Amira software. Mitochondria are in red; the ER membranes are in green, yellow, or blue. White arrows indicate representative MERCS. Scale bars, 5 μm. (b) Four morphological profiles were defined using mitochondrial aspect ratio and volume estimated using 3D EM reconstructions and the Amira software. (c) Quantification of mitochondria Types I - IV in mouse brains. Morphometric analysis was conducted in a blind fashion using randomly selected 3D EM mitochondria reconstructions from NTG, APP/PS1 and APP/PS1+CP2 mice. Ten random sections with ~ 62 - 100 mitochondria were taken into the analysis for each group. (d) CP2 treatment reduces MOAS in APP/PS1 mice. Morphometric analysis was conducted as in (d). Unpaired Student’s t-test was used for all statistical analyses. Data are presented as mean ± SEM. *** P < 0.001.
Figure 3.
Chronic CP2 treatment improves mitochondrial morphology in APP/PS1 (A/P) mouse brain. (a) Visualization of mitochondrial morphology in the hippocampi of NTG, APP/PS1 and APP/PS1+CP2 mice using SBFSEM and 3D EM reconstructions with the Amira software. Mitochondria are in red; the ER membranes are in green, yellow, or blue. White arrows indicate representative MERCS. Scale bars, 5 μm. (b) Four morphological profiles were defined using mitochondrial aspect ratio and volume estimated using 3D EM reconstructions and the Amira software. (c) Quantification of mitochondria Types I - IV in mouse brains. Morphometric analysis was conducted in a blind fashion using randomly selected 3D EM mitochondria reconstructions from NTG, APP/PS1 and APP/PS1+CP2 mice. Ten random sections with ~ 62 - 100 mitochondria were taken into the analysis for each group. (d) CP2 treatment reduces MOAS in APP/PS1 mice. Morphometric analysis was conducted as in (d). Unpaired Student’s t-test was used for all statistical analyses. Data are presented as mean ± SEM. *** P < 0.001.
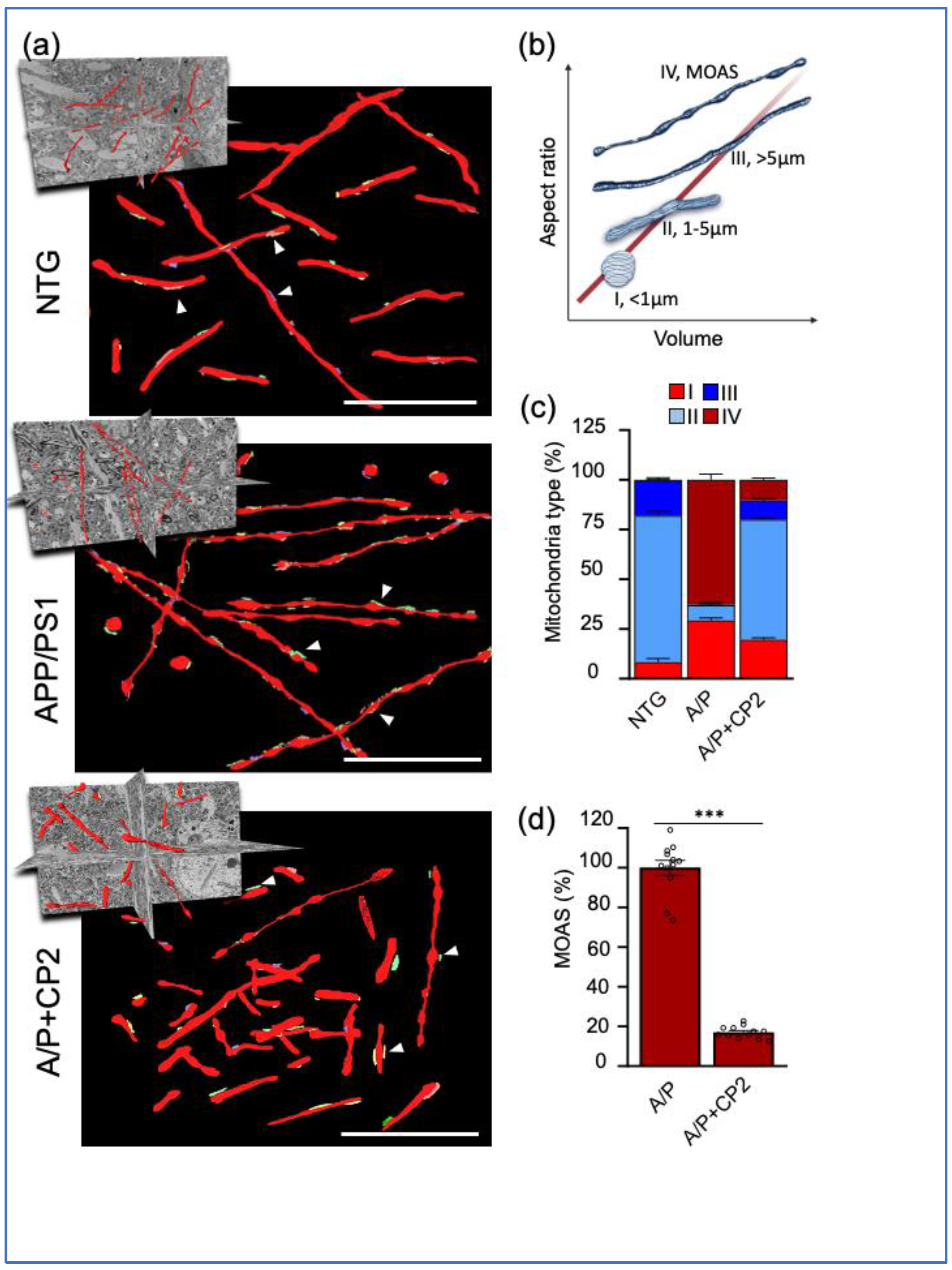
Figure 4.
Chronic CP2 treatment reduces MERCS in APP/PS1 (A/P) mice. (a) Representative 3D EM reconstructions of MERCS in NTG, APP/PS1 and APP/PS1+CP2 mice for randomly selected mitochondrial Types I - IV. Mitochondria are colored in red, blue, purple, green, and teal; the ER is yellow. Reconstruction was done using SBFSEM images from the CA1 hippocampal region using the Amira software. MOAS from the hippocampus of an APP/PS1 mouse have exaggerated MERCS along the whole organelle. CP2 treatment decreases MERCS in the residual MOAS (a, left panel). Scale bars, 0.5 μm. (b) Morphometric analyses show CP2 treatment significantly reduces MERCS coverage (b,c), MERCS length (d,e), and volume (f,g) for all Types of mitochondria and MOAS. Ten random sections with ~ 62 - 100 mitochondria were taken into the analysis for each group. One-way ANOVA and one-sided unpaired Student’s t-test were used for all statistical analyses. Data are presented as mean ± SEM. ** P < 0.005; *** P < 0.001.
Figure 4.
Chronic CP2 treatment reduces MERCS in APP/PS1 (A/P) mice. (a) Representative 3D EM reconstructions of MERCS in NTG, APP/PS1 and APP/PS1+CP2 mice for randomly selected mitochondrial Types I - IV. Mitochondria are colored in red, blue, purple, green, and teal; the ER is yellow. Reconstruction was done using SBFSEM images from the CA1 hippocampal region using the Amira software. MOAS from the hippocampus of an APP/PS1 mouse have exaggerated MERCS along the whole organelle. CP2 treatment decreases MERCS in the residual MOAS (a, left panel). Scale bars, 0.5 μm. (b) Morphometric analyses show CP2 treatment significantly reduces MERCS coverage (b,c), MERCS length (d,e), and volume (f,g) for all Types of mitochondria and MOAS. Ten random sections with ~ 62 - 100 mitochondria were taken into the analysis for each group. One-way ANOVA and one-sided unpaired Student’s t-test were used for all statistical analyses. Data are presented as mean ± SEM. ** P < 0.005; *** P < 0.001.
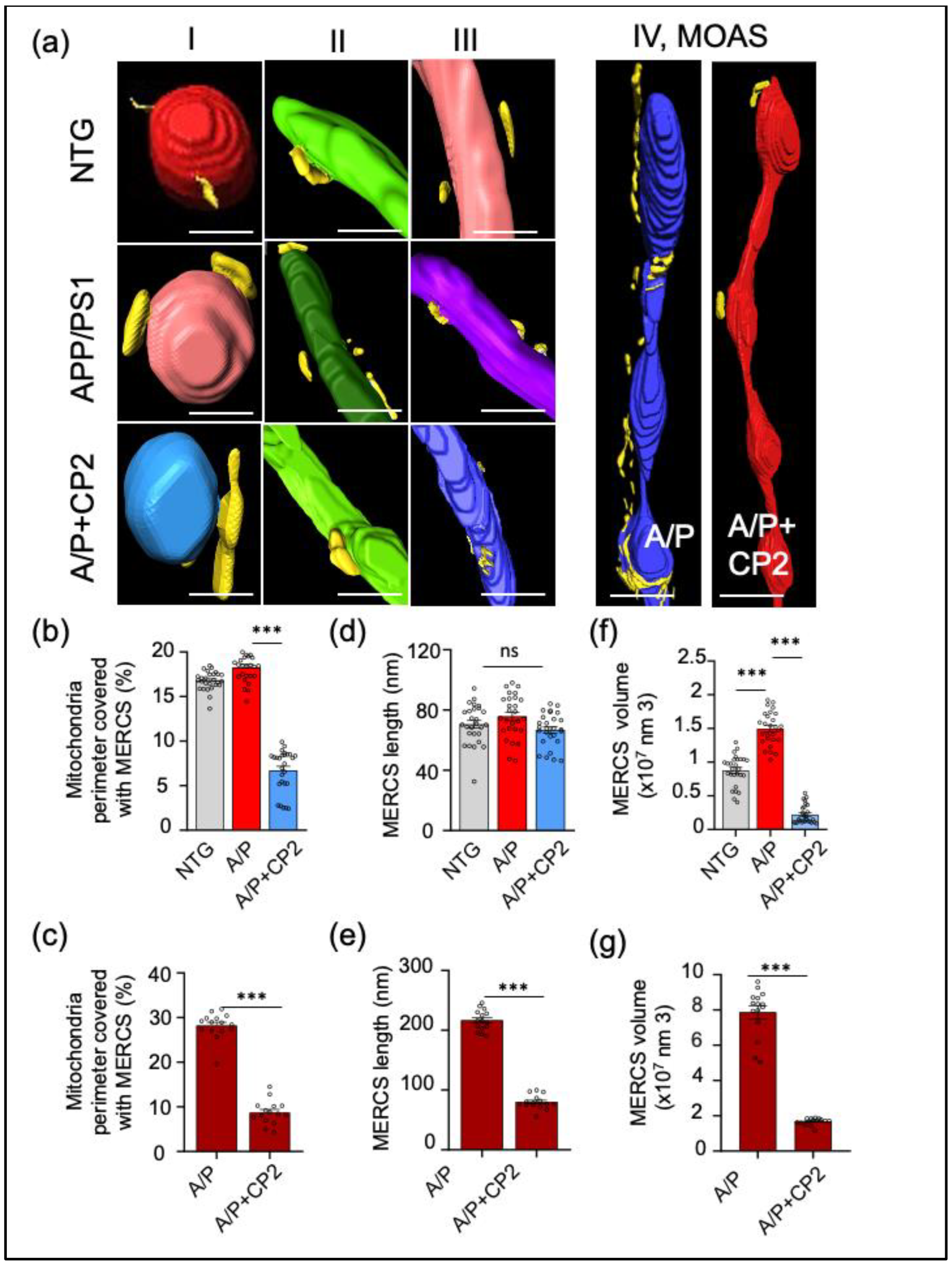
Figure 5.
CP2 treatment enhances mitochondrial biogenesis and turnover in APP/PS1 (A/P) mice. Western blot analysis of protein expression in the whole hippocampal brain tissue lysates (a) or mitochondrial fractions (b) isolated from hippocampal tissue of NTG, APP/PS1, and APP/PS1+CP2 mice. (c,d) Quantification of Western blots from (a) and (b), respectively. Differences between individual groups were analyzed using one-sided unpaired Student’s t-test. n = 3 mice per group. Data are presented as mean ± SEM. * P < 0.05, ** P < 0.005.
Figure 5.
CP2 treatment enhances mitochondrial biogenesis and turnover in APP/PS1 (A/P) mice. Western blot analysis of protein expression in the whole hippocampal brain tissue lysates (a) or mitochondrial fractions (b) isolated from hippocampal tissue of NTG, APP/PS1, and APP/PS1+CP2 mice. (c,d) Quantification of Western blots from (a) and (b), respectively. Differences between individual groups were analyzed using one-sided unpaired Student’s t-test. n = 3 mice per group. Data are presented as mean ± SEM. * P < 0.05, ** P < 0.005.
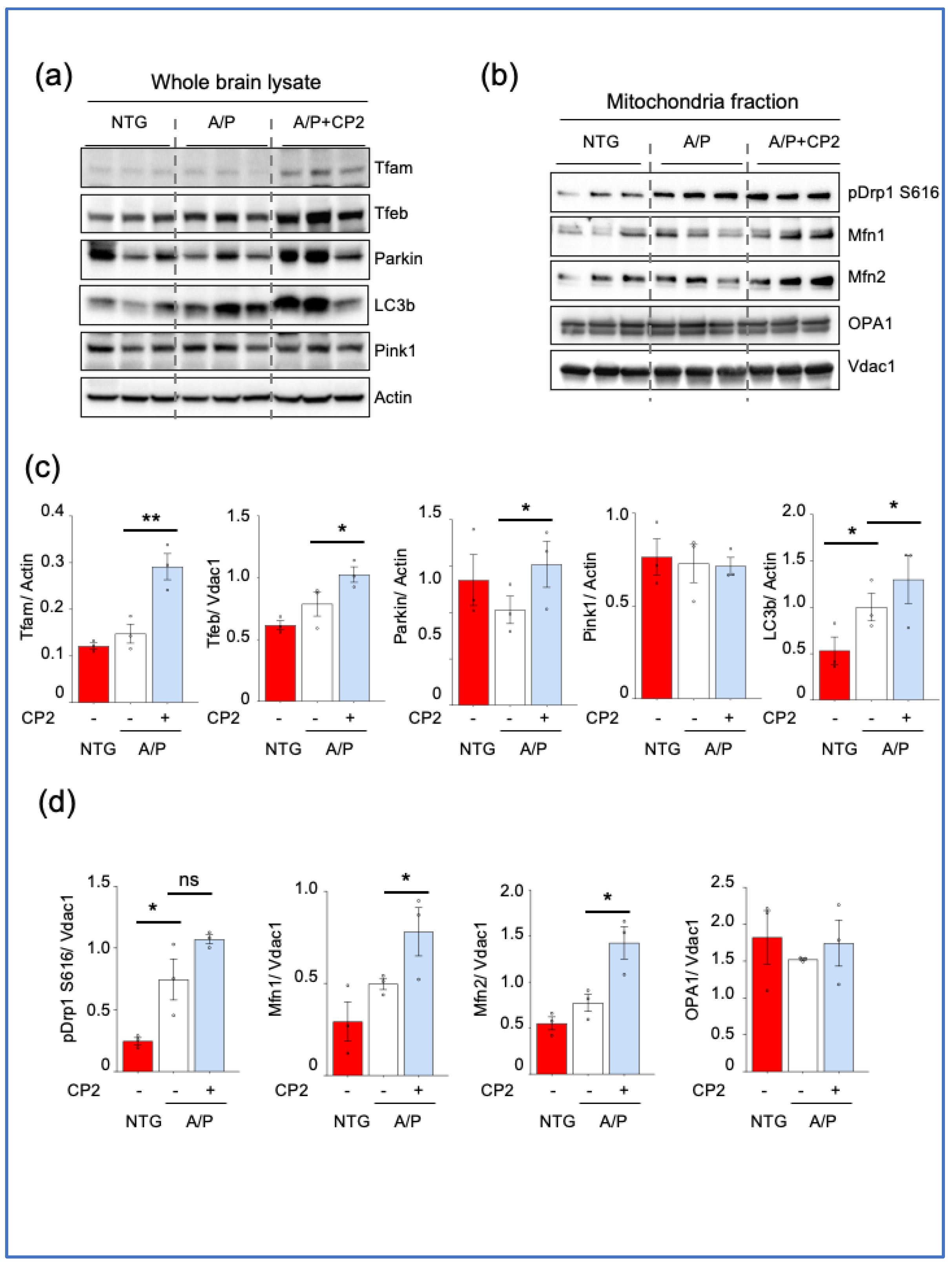
Figure 6.
CP2 treatment reduces UPR and ER stress in APP/PS1 mice. Barcode plots (a-d) and heatmaps (e,f) show upregulated genes involved in ER stress (GO:1905897) (a,b,e) and UPR (WikiPathway:WP4925) (c,d,f) pathways in APP/PS1 mice that were down-regulated after CP2 treatment. Plots were generated by enrichment analysis using RNA-seq data from APP/PS1 and NTG models. (a-d) Barcode plots of ER stress pathway in APP/PS1 vs NTG (a) and APP/PS1+CP2 vs APP/PS1 (b), and UPR pathway of APP/PS1 vs NTG (c) and APP/PS1+CP2 vs APP/PS1 (d). Genes that belong to specific pathway were marked by vertical lines in log2-fold-change scale, and enrichment was weighted by -log10 (p-values) from differential expression analysis between APP/PS1 and NTG or between APP/PS1+CP2 and APP/PS1. (e,f) Heatmaps of significantly increased genes in ER stress pathway (e) and UPR pathway (f) that were reversed by CP2 treatment. Genes are labeled on the right; samples are labeled at the bottom and grouped by genotype/treatment groups. Expression for each gene was z-scored; color indicates z-score. (g) Schematic diagram of the relationship between mitochondrial morphology and early dysfunctions involved in AD, and the reversal by CP2 treatment.
Figure 6.
CP2 treatment reduces UPR and ER stress in APP/PS1 mice. Barcode plots (a-d) and heatmaps (e,f) show upregulated genes involved in ER stress (GO:1905897) (a,b,e) and UPR (WikiPathway:WP4925) (c,d,f) pathways in APP/PS1 mice that were down-regulated after CP2 treatment. Plots were generated by enrichment analysis using RNA-seq data from APP/PS1 and NTG models. (a-d) Barcode plots of ER stress pathway in APP/PS1 vs NTG (a) and APP/PS1+CP2 vs APP/PS1 (b), and UPR pathway of APP/PS1 vs NTG (c) and APP/PS1+CP2 vs APP/PS1 (d). Genes that belong to specific pathway were marked by vertical lines in log2-fold-change scale, and enrichment was weighted by -log10 (p-values) from differential expression analysis between APP/PS1 and NTG or between APP/PS1+CP2 and APP/PS1. (e,f) Heatmaps of significantly increased genes in ER stress pathway (e) and UPR pathway (f) that were reversed by CP2 treatment. Genes are labeled on the right; samples are labeled at the bottom and grouped by genotype/treatment groups. Expression for each gene was z-scored; color indicates z-score. (g) Schematic diagram of the relationship between mitochondrial morphology and early dysfunctions involved in AD, and the reversal by CP2 treatment.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



