
Submitted:
09 February 2023
Posted:
10 February 2023
You are already at the latest version
Abstract
The placenta is a vital organ of pregnancy, regulating adaptation to pregnancy, gestational-parent/fetal exchange and ultimately fetal development and growth. Not surprisingly, in cases of placental dysfunction - where aspects of placental development or function become compromised - adverse pregnancy outcomes can result. One common placenta-mediated disorder of pregnancy is preeclampsia (PE), a hypertensive disorder of pregnancy with a highly heterogeneous clinical presentation. The wide array of clinical characteristics observed in pregnant individuals and neonates of a PE pregnancy are likely the result of distinct forms of placental pathology underlying the PE diagnosis, explaining why no one common intervention has proven effective in the prevention or treatment of PE. The historical paradigm of placental pathology in PE highlights an important role for utero-placental malperfusion, placental hypoxia and oxidative stress, and a critical role for placental mitochondrial dysfunction in the pathogenesis and progression of the disease. In the current review, the evidence of placental mitochondrial dysfunction in the context of PE will be summarized, highlighting how altered mitochondrial function may be a common feature across distinct PE subtypes. Further, advances in this field of study and therapeutic targeting of mitochondria as a promising intervention for PE will be discussed.
Keywords:
placenta
; mitochondria
; preeclampsia
; disease subclasses
; pregnancy
; hypertension
; reactive oxygen species
; therapies
1. Introduction
The human placenta serves as a metabolic and endocrine exchange barrier between the gestational parent and fetus, regulating adaptation to pregnancy and fetal growth and development [1,2]. Abnormalities in the development and function of this organ underlie the majority of obstetrical disorders faced today, many of which can be classified under the umbrella term of “placenta-mediated diseases”. One of the most common of these is preeclampsia (PE), a hypertensive disorder of pregnancy that affects ~ 3-5% of all pregnancies and to date has no effective therapeutic interventions [3]. A large body of research has focused on understanding the placental pathology(/ies) that lead to gestational parent hypertension and end organ damage. Much of this research focused on a paradigm of utero-placental malperfusion, placental hypoxia and oxidative stress resulting in shedding of placental debris, proinflammatory and anti-angiogenic mediators into the circulation of the gestational parent. This initiates a heightened inflammatory response and endothelial dysfunction that precipitate the clinical symptoms observed in the gestational parent [3,4].
The primary functions of the placenta are to regulate gestational parent-fetal exchange (gas exchange, nutrients/micronutrient and ion exchange, waste removal); hormone and growth factor production (required for uterine quiescence, gestational parent adaptation to pregnancy, fetal growth and development); and to act as a selective barrier to the fetus (fetal protection from xenobiotics, enzymatic inactivation of gestational parent hormones - i.e. cortisol) [5]. Each of these functions is highly energy dependent and consequently highly reliant on mitochondria function.
Mitochondrial dysfunction has been widely implicated as a central component to the establishment of placental and gestational parent disease in cases of PE [6]. Mitochondria not only harvest energy to produce adenosine triphosphate (ATP) but also play a vital role in signaling pathways that are essential for growth, development, and homeostasis of any organ. Mitochondrial dysfunction is a central mediator of disease pathophysiology in many organ systems, owing to the numerous processes that can be directly influenced by mitochondrial function, including biosynthetic and bioenergetic pathways, reduction-oxidation (redox) signalling, antioxidant defence, hypoxia adaptations, inflammatory signaling, the mitochondrial unfolded protein response, calcium signalling and apoptotic signaling [7]. Similarly, when placental mitochondrial functions become dysregulated, placental development and function may be compromised, resulting in detrimental health outcomes for both the developing fetus and the gestational parent [6,8,9,10]. In the current review, literature on the central role of mitochondria in the establishment of placental disease will be summarized, highlighting the possibility that placental mitochondrial dysfunction may be a central and common aspect of pathophysiology across distinct subclasses of PE. Further, recent advancements in mitochondria-targeting therapeutics to treat PE will be discussed.
2. Mitochondria in the Healthy Placenta
Nutrient and energy demand of the placenta change across pregnancy to match and optimize fetal development and growth patterns [11,12,13]. Further, the placenta requires a considerable amount of ATP for its own growth and expansion, for example, during developmental processes that include vascularization, cell differentiation and the formation of function-specific structures within the placenta [14]. Mitochondrial content, structure, and function play an important role in supporting the placenta’s energetic demands during these highly dynamic transitions. A brief overview is provided below and summarized in Table 1, but for a more comprehensive discussion on its role in healthy placental development and function can be found in the following reviews [6,15,16,17].
2.1. Placental Mitochondrial Content, Structure, and Function across Pregnancy
During most of the 1st trimester of human pregnancy, feto-placental development takes place in a low-oxygen environment [18,19,20]), accompanied by low placental mitochondrial content. At this gestational phase, mitochondria found within the chorionic villi structures are relatively large in size, with an oval or circular shape. At the onset of the 2nd trimester, when the gestational parent-fetal circulation is established, placental mitochondrial biogenesis is upregulated, leading to increased mitochondrial content that continues to rise until the end of pregnancy [21]. Mitochondrial structure during this trimester likewise demonstrates dynamic remodeling, with the presence of smaller mitochondria with irregular cristae, likely a function of rapid placental mitochondrial turnover in response to increased oxygen supply [22]. During this gestational phase, placental respiration and ATP production increases; however, the ratio of respiration rates to mitochondrial content demonstrates a slowing within the third trimester of pregnancy, perhaps to maintain respiration capacity for acute energetic insults [21].
In addition to meeting the specific temporal energy demands, the placental mitochondria must be capable of maintaining cellular redox homeostasis across pregnancy in the face of a changing oxygen landscape. Mitochondria are a major source of reactive oxygen and nitrogen species (ROS, RNS) generation. They also house several key antioxidant enzymes, required to maintain cellular redox balance [23]. When compared to pre-pregnancy state, an increase in the maternal blood levels of superoxide is required for healthy pregnancy; and this level remains stable throughout the gestation [24]. With the rapid increase in oxygen availability at the transition between first and second trimester, a burst of ROS production is also noted, with elevated ROS production maintained across the remainder of gestation. Importantly, placental ROS generation is considered an important second messenger system to promote development of the placental vasculature [25]. On the other hand, the expression and activity of mitochondrial superoxide dismutase (mnSOD2) and glutathione peroxidase (GPX) enzymes could be upregulated in response to increased oxidative stress as an adaptive mechanism under both physiological and pathological conditions [22,26]. In line with this, results from a mouse study showed that placental mitochondria are highly responsive to environmental changes, such as oxygen level, and can modulate its oxidative capacity and substrate preference accordingly [27].
Placental mitochondria are also involved in regulating cellular apoptosis. Upon cellular stimuli such as nutrient stress, mitochondrial membrane permeability may increase, leading to swelling and release of proapoptotic proteins to initiate the mitochondria mediated caspase 9 apoptotic cascade [28]. The mitochondrial-driven apoptotic cascade is a central biological process required for normal placental developmental processes, such as: gestational parent-fetal immune tolerance, trophoblast differentiation, syncytium formation, trophoblast invasion and remodelling of uterine spiral arteries [29,30]. Thus, aberrant regulation of apoptosis within the placenta at any point in pregnancy can disrupt placental development and function and contribute to the establishment of placental dysfunction and obstetrical disease.
2.2. Mitochondrial Content, Structure, and Function in Distinct Placental Cell Types
The human placenta demonstrates considerable anatomical complexity, with heterogenous structures and cellular composition - characteristics required to support the diverse functions of this critical rogan of pregnancy. Unsurprisingly, the distinct placental cell populations demonstrate unique oxygen availability and energetic needs, dramatically influencing the mitochondrial content, structure, and function in the distinct populations. As such, in addition to temporal plasticity in the placental mitochondrial landscape, there is also considerable heterogeneity in mitochondrial content, structure and function across the distinct cell populations and structures within the placenta.
In humans, the syncytiotrophoblast (STB) cellular layer (also referred to as the syncytium) of the chorionic villi, serves as the barrier between the circulations of the gestational parent and the fetus. The syncytium has long been considered the metabolic workhorse of the placenta, due to its direct roles in gestational parent-fetal nutrient/gas exchange exchange and hormone production. However, recent findings suggest that primary human placental villous cytotrophoblasts (CTBs) – cells that terminally differentiate and fuse into the overlying syncytium - have higher mitochondrial content and respiration rates than syncytiotrophoblast (STB) in culture. Mitochondria in CTBs are large and oval shaped with defined cristae, while mitochondria in differentiated STB are smaller and round with undefined cristae, which may reflect cell-specific roles and metabolic properties for these respective mitochondrial pools [31]. Functionally, mitochondria are important for steroid biosynthesis via their role in isoprenoid precursor synthesis and, in particular, mitochondria found in the STBs constitutively produce progesterone to sustain pregnancy. It is thought that mitochondrial size and cristae structural changes occurs during CTB to STB differentiation to support improved cholesterol import into the STB mitochondria, supporting efficient production of progesterone [32,33,34,35].
A third trophoblast cell population - the extravillious cytotrophoblasts (EVTs) - demonstrate high energy demands associated with their functionality, namely invading the uterine decidua and remodelling of the uterine vasculature. Using the immortalized EVT cell line (HTR-8/SVneo) in vitro, paracrine signalling from placenta-derived mesenchymal stem cells has been shown to increase expression of mitochondrial proteins important for ATP production, such as ATP-binding cassette sub-family B member 10(ABCB10) and mitochondrial Ca+ uniporter (MCU), the latter of which also acts as a cellular glucose sensor [36,37]. Furthermore, compared to the villous counterparts, the EVTs exhibit increased mitochondrial respiration and produce more ATP [38]. Moreover, activation of mitochondrial quality control process, mitophagy and balancing mitochondrial dynamics is known to be essential in successful EVT invasion, required for proper placentation [36,37].
In summary, mitochondria play a central role in ensuring proper development and functioning of the human placenta, supporting the development and growth of the fetus and successful pregnancy outcomes. Conversely, dysregulation of mitochondrial content, structure and/or function can have negative consequences for placentation and pregnancy health, and has been identified as a significant contributor to the pathophysiology of all placenta-mediated obstetric complications, including preeclampsia (PE) - one of the most prevalent and devastating of the placenta-mediated diseases [37].
3. Preeclampsia
PE is one of the most common and serious placenta-mediated diseases in humans, contributing to approximately 76,000 gestational parent and 500,000 fetal/neonatal deaths per year worldwide [3,39]. PE is characterized by de novo hypertension after 20 weeks of gestation, accompanied by proteinuria and/or evidence of end-organ damage in the gestational parent [40]. Fetal growth restriction is a common comorbidity, particularly in cases of early onset PE, estimated to occur in ~ 39% of PE cases [41]. Adverse neonatal health outcomes associated with PE include periventricular or intraventricular hemorrhage or subdural and cerebral hemorrhage, hypoxic-ischemic encephalopathy or periventricular leukomalacia, stillbirth, and perinatal death [40], in many cases the iatrogenic results accompanying medically-indicated preterm delivery. Aside from the immediate health risks associated with a PE diagnosis (i.e. HELLP, eclampsia), individuals who have had PE are seven times more likely to have a recurrence of PE in subsequent pregnancies, particularly with early onset disease [42]. Further, these individuals are at increased risk for cardiovascular disease in later life, with some estimates indicating the average age of first cardiac events as early as 38 years of age [42].
The underlying cause of PE is not entirely clear, however placental dysfunction is recognized as a critical component of the pathophysiology. In a healthy pregnancy, uterine spiral arteries are remodelled by the EVT population, modifying them into large-diameter vessels that are no longer responsive to vasopressors in the circulation of the gestational parent; modifications aimed at increasing blood flow into the intervillous space of the placenta to promote fetal growth and development [43]. In the dogmatic two-stage model of PE pathophysiology [44], it is proposed that this vital aspect of placentation becomes compromised, with shallow invasion of the EVTs and insufficient remodelling of the uterine spiral arteries [45,46,47,48]. This poor vascular remodelling results in placental ischemic-reperfusion injury; the high speed, pulsatile blood flow into the placenta causing damage to the delicate villous structures, promoting thrombus formation and compromising adequate gestational parent-fetal exchange of oxygen and nutrients. This injury is supported by findings of STB necrosis, damage and destruction of syncytial microvilli structures, hyperplastic and apoptotic CTBs, and bulged endoplasmic reticulum cisternae and mitochondria [9,49,50]. Damaged trophoblasts are proposed to respond to the structural damage and oxidative stress through the up-regulation and release of pro-inflammatory cytokines and anti-angiogenic factors, along with extracellular vesicles, apoptotic debris and cell-free fetal DNA into the systemic circulation of the gestational parent [51,52,53]. In the second stage of the PE pathophysiology model, these placenta-derived factors illicit a heightened systemic immune response and endothelial dysfunction in the gestational parent, precipitating in the clinical manifestation of the disease [54,55]. While there is certainly robust evidence to support this model of PE pathophysiology, it is important to note that not all cases of PE demonstrate evidence of insufficient invasion and uterine spiral artery remodelling, nor do all PE placentas demonstrate structural or molecular evidence of ischemia-reperfusion type injury [4].
4. Subclasses of Disease in Preeclampsia
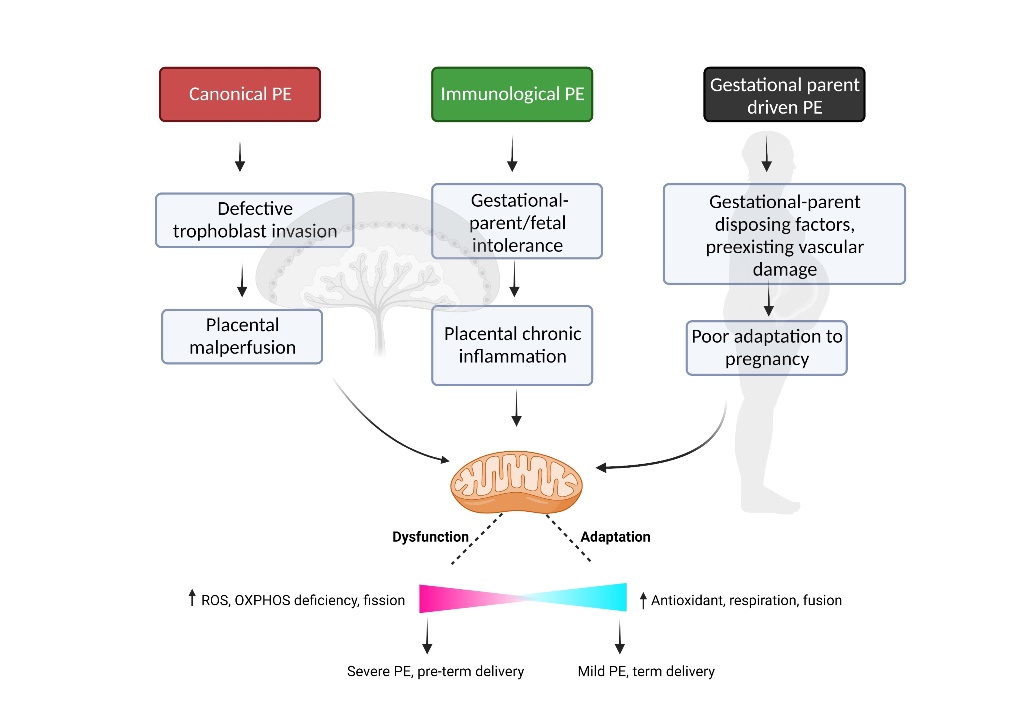
PE is a heterogeneous disorder that demonstrates considerable variability in the timing of disease onset, presentation and severity of symptoms, gestational parent and fetal health outcomes, and pathological evidence of placental disease - indicative of multiple disease subtypes with divergent underlying etiology and pathophysiology. In this vein, the scientific community has embarked on several lines of inquiry attempting to identify and characterize distinct subtypes of PE [4,56,57,58,59,60,61]. Discovery-driven approaches using robust -omics datasets are particularly useful when trying to understand complex diseases with heterogeneous presentation and are proving useful in gaining insight into the presence of distinct PE subclasses. Using clustering approaches, groups of PE patients who share commonality in (epi-)genome [62], transcriptome [4] or proteome [59] profiles in relevant tissues (i.e. blood, placenta) have been identified. Our group has contributed to this body of work, completing multi-scale profiling of a robust sample of PE patients that spanned the clinical spectrum of disease [4,63,64]. Through integration of clinical, placental transcriptome and histopathology profiles, our group uncovered at least three clinically relevant PE subclasses (Table 1) with divergent forms of placental disease, or lack thereof, which are highly supported by the growing body of research in this domain (see [65] for a detailed review on this topic by the Global Pregnancy Collaboration).
4.1. Canonical PE Subclass
The largest PE subclass characterized by our previous work demonstrates a phenotype which tightly aligns with the traditional framework of PE pathophysiology. These patients have small placentas with lesions of maternal vascular malperfusion (MVM) and gene expression profiles consistent with hypoxia-reperfusion injury - including upregulation of hypoxia-inducible factors and anti-angiogenic molecules commonly associated with PE. Clinically, these patients have an earlier onset and more severe form of gestational parent disease and are more likely to experience preterm delivery (<34 weeks) [4,63].
4.2. Immunological PE Subclass
Heightened systemic inflammation has long been identified as a key component of PE pathophysiology [66,67,68], and more recently it is becoming clear that an immune-driven subclass of PE exists [69,70,71]. This subclass of PE patients demonstrates evidence of chronic inflammatory insults at the utero-placenta interface, with heightened immune cell infiltration, a propensity for gestational parent-fetal HLA-discordance, heightened placental expression of pro-inflammatory cytokines and chemokines, and placental lesions consistent with chronic inflammation and gestational parent-fetal interface disturbances [4].
4.3. Gestational Parent-Driven PE Subclass
Despite being widely identified as a placenta-mediated disease, historical reports have long described a handful of PE cases with little evidence of placental disease [4,63,72]—a PE subclass driven in large part by underlying and predisposing factors in the gestational parent, which compromise appropriate physiological adaptation to pregnancy. This subclass of PE patients demonstrates normally grown placentas with gene expression profiles that mimic healthy tissues and little histological evidence of pathology. Fetuses in this subclass demonstrate normal growth trajectories and gestational parent symptoms are relatively mild with a later onset (>37 weeks gestation) [4,63]. Collectively, this phenotype suggests minimal placental involvement, but rather predisposing gestational parental factors as the primary drivers of PE pathophysiology.
While there is convincing evidence supporting distinct mechanistic pathways driving placental and/or gestational parent disease across PE subclasses, there is likewise convincing evidence to suggest that mitochondrial dysfunction may be a central point of convergence within the pathophysiology of PE across all subclasses of patients.

Table 2.
Overview of clinical features and mechanistic pathways involved in the pathophysiology of distinct PE subclasses.
Table 2.
Overview of clinical features and mechanistic pathways involved in the pathophysiology of distinct PE subclasses.
| Canonical PE | Immunological PE | Gestational-parent driven PE |
|
|---|---|---|---|
|
Onset and severity of disease |
|
|
|
| Pathogenesis |
|
|
|
| Genes expressed in placenta |
|
|
|
| Placenta pathology |
|
|
|
| Potential risk factors |
|
|
|
5. Placental Mitochondrial Dysfunction in Preeclampsia
Compromised placental mitochondrial biogenesis, structure, and functionality contribute to placental dysfunction in PE. Below, the existing evidence for placental mitochondrial dysfunction as a key component of PE pathophysiology - as a singular clinical diagnosis - is reviewed (Table 1).
5.1. Mitochondrial Content
The evidence related to mitochondrial content in cases of PE is not clear cut. Decreased placental mitochondrial DNA content and citrate synthase protein activity have been described in cases of PE, compared to healthy term controls, coupled to a downregulation of genes that regulate mitochondrial biogenesis, such as PGC1-a, NRF1 and Tfam [73]. It should be noted, however, that when PE placentas are compared against gestationally-age matched controls, the reported difference in citrate synthase levels are no longer apparent, and an increase in mitochondrial complex II and III protein levels are observed [74]. In a subsequent prospective case control study, placental mtDNA copy number was in fact found to be elevated in cases of preterm/severe PE, compared to both term PE and healthy term controls, possibly indicative of a compensatory up-regulation of mitochondrial biogenesis in the face of severe placental disease [75]. Systemically, lower cell-free (membrane bound and non-membrane bound) plasma mtDNA content has been measured in the circulation of individuals with PE in the third trimester, compared to gestational-age matched healthy controls [76]. However, others have reported elevated whole blood, plasma and serum mtDNA content in cases of PE compared to health controls, with the mtDNA content elevations significantly associated with female fetal sex according to one of these studies [77,78,79]. On its own, the evidence surrounding placental mitochondrial content in the context of PE does not appear to be highly informative - however the evidence suggests the timing of disease onset, severity of disease, gestational sampling time and fetal sex may all influence the necessary interpretations of mitochondrial content measurements.
5.2. Mitochondrial Structure and Dynamics
To maintain mitochondrial homeostasis in the face of environmental stress, mitochondria go through dynamic morphological changes via two processes, fusion and fission. The fusion of separate mitochondria is driven by outer mitochondrial membrane proteins mitofusins 1 and 2 (Mfn1 and 2), and the inner membrane protein optic atrophy 1 (OPA1) [80]. The benefits of fusion have been linked to the diffusion of metabolites, protein and mtDNA across the network of mitochondria, enabling mitochondria to produce energy more efficiently. Fission, on the other hand, permits the separation of damaged portions of mitochondria for degradation via mitochondrial autophagy (i.e. mitophagy), and is mediated by in large part by mitochondrial fission 1 (FIS1) and dynamin-1-like (DNM1L) proteins [81].
Evidence for dysregulated mitochondrial dynamics (fission and fusion) has been collected from placenta tissues of pregnancies impacted by PE. Term PE placenta tissues demonstrate decreased mRNA expression of the fusion protein Mfn2, coupled to decreased ATP content, when compared to gestational age-matched controls [82]. While not explicitly examined in that study, similar decreases in Mfn2 expression have been associated with mitochondrial fragmentation in both human and rodent models of pulmonary arterial hypertension [83]. Structurally, placental mitochondria from cases of severe/early onset PE demonstrate swelling, impaired cristae and fragmentation when visualized with electron microscopy [84,85,86,87]. Fission processes are also likely impacted in early onset PE cases, with reported measurements of increased DNM1 expression relative to term controls [73]. Interestingly, cultured 1st trimester immortalized human placenta cells (TEV-1) likewise exhibit decreased expression of Mfn2, decreased mitochondrial membrane potential, decreased ATP production, mitochondrial fragmentation and mitophagy when exposed to hypoxic insults in vitro [82] - similar to what is proposed to occur within the context of PE pathophysiology. Damaged mitochondrial clearance via mitophagy is described in cases of PE, particularly those with severe/early onset disease [88]. This finding is paralleled by an increase in placental expression of mitophagy-related proteins, such as PTEN-induced putative kinase 1 (PINK1), Parkin, B-cell lymphoma-2 (BCL-2) interacting protein 3 (BNIP3) and BNIP3 Like (BNIP3L), when compared to preterm controls [73,88].
5.3. Mitochondrial Function—Respiration and ATP Generation
The mitochondria primarily act as an ATP generation hub through their ability to perform oxidative respiration. Altered placental mitochondrial respiration is widely reported in cases of PE, however the direction of this functional change varies across studies. It should be noted that while performing mitochondrial functionality assays using fresh samples (immediately after collection) is ideal; however fresh tissue collection can pose a difficulty in multi-site clinical studies or when performing retrospective studies, especially in pregnancy research. Unfortunately, freezing of samples can lead to damage of mitochondrial membranes, uncoupling ETC activity and loss of cytochrome c. Thus, historical results must be interpreted with caution when frozen tissue was used. To mitigate such issues, newly reported techniques that restore electron transfer components lost during freeze/thaw and correct for membrane permeabilization, have demonstrated considerable success, preserving 90-95% of the maximal respiratory capacity in frozen samples, and may serve as tremendously useful for future research in this area (see the article for method [89]).
In cryopreserved placental samples collected from cases of early onset PE (onset at < 34 wk), a considerable decrease in mitochondrial respiration is described, when compared to health term controls [87]. Supporting these findings, enzymes critical to the glycolytic pathway have been found to be upregulated in placentas from PE pregnancies, indicative of suppressed mitochondrial respiration [73]. Muralimanoharan et al. more specifically described decreased mitochondrial complex-I and complex-III activity in isolated mitochondria from frozen PE term placentas, when compared to gestational age matched controls, functional findings that were coupled to decreased expression of respiratory complex proteins [85].
In contrast, others have described an increase in placental mitochondrial respiration activity in cases of PE. For example, Vishnyakova et al. [86] demonstrated a modest increase in complex-I mediated respiration, with no change in complex-II mediated respiration, when studying freshly isolated mitochondria from placentas of early onset PE patients compared to healthy term controls. It should be noted, however, this same study found no measurable differences in mitochondrial activity in late onset PE samples, when compared to gestational age-matched controls - highlighting the importance of considering the temporal changes of mitochondrial content and activity across pregnancy when interpreting this data. However, others have reported an increase in complex-I and complex-I+II mediated respiration in term PE permeabilized placenta tissue that occurred in conjunction with increases in the expression of respiratory protein complexes, when gestational age was appropriately matched in the healthy control group [74]. Importantly though, the reserve respiratory capacity - the amount of extra ATP that can be produced by OXPHOS with an energetic demand - was reduced in these same PE samples [74], suggesting these mitochondria were likely functioning close to their maximal capacity. These findings may also imply that PE cases that do reach term may do so because of a unique capacity to induce compensatory increases in mitochondrial function.
There are several possible reasons for the high degree of study finding discrepancies on this topic. One possible interpretation may be that those placentas capable of initiating a compensatory increase in mitochondrial respiratory function at times of cellular stress, can limit the severity of placental disease and allow the pregnancy to progress to term. Conversely, placentas unable to initiate this response succumb to a more severe form of placental disease and dysfunction, initiating an earlier onset and more severe form of PE. Alternatively, these discrepancies may in fact be further evidence of distinct subclasses of PE, with divergent underlying pathophysiology and placental disease. It should be noted that in all studies described, no efforts to identify distinct PE subclasses were attempted, aside from the clinical distinction of early vs late onset disease.
5.4. Mitochondrial Function—Redox Homeostasis and Apoptosis
Mitochondrial complexes I and III are among the major sites of cellular ROS generation [90], particularly under hypoxic conditions [91,92]. Oxidative stress, due to excessive ROS generation, is a well described hallmarks of PE pathophysiology [9,93]. For instance, elevated levels of oxidative stress markers such as 8-oxoDG, an indicator of DNA damage, and lipid peroxides in the placenta and maternal plasma in PE cases have been reported [94,95]. Elevated levels of ROS, such as H2O2, have been measured in both the placentas and circulating serum of PE patients compared to healthy controls, however it is not entirely clear where the systemic ROS is originally generated [96]. Evidence of increased mitochondrial lipid peroxidation in PE placentas corresponds to high levels of lipid peroxide in the circulation of the gestational parent [95,97,98,99]. Moreover, higher levels of dichlorofluorescin (DCFH) oxidants, reactive nitric oxide metabolites such as NO2- and NO3-, and protein carbonyl has been observed in the placental tissue mitochondria from preterm/severe PE patients compared to term healthy controls; implying a major contribution of mitochondrial ROS in the oxidative stress observed in PE [100].
Coupled to increased ROS generation, there is also evidence of adaptive changes in antioxidant capacity in the context of PE. For example, mitochondrial superoxide dismutase (mnSOD2) enzymatic activity is decreased in preterm/severe PE placentas compared to gestational age matched controls, while its mRNA levels have been shown to be upregulated [74,101]. Some studies suggest that there is an overall increase in antioxidant capacity of PE placenta through activation of other antioxidant enzymes present in cytoplasm and mitochondria such as glutathione peroxidase (GPx) [73,74]. This eludes that an adaptive response is required in order to maintain placental function in the oxidative stress condition.
Intracellular stress such as oxidative damage can activate mitochondria mediated caspase-9 apoptotic pathway [102]. Intracellular stress induces activation of proapoptotic proteins Bax and Bak that inactivate anti-apoptotic Bcl-2 and Bcl-xL proteins, leading to opening of mitochondrial membrane permeability pores [103]. PE Placentas show increased presence of apoptotic syncytial knots with evidence of oxidative DNA damage [104,105,106]. Holland et al., showed that the ratio of BAX/BCL2 protein expression was increased in preterm/severe PE placentas compared to gestational age-matched controls [74]. On the other hand, in term PE placentas, the ratio of BAX/BCL2 expression was relative to term controls while the levels of antioxidant enzymes increased [74]. These findings suggest that increased antioxidant activity in PE placentas may keep oxidative stress at bay and in turn reduce cellular apoptosis and facilitate reaching term pregnancy.
6. Mitochondrial Dysfunction—A Point of Convergence across Preeclampsia Subclasses
To date, most research focused on understanding placental mitochondrial function and dysfunction in PE combines all cases together or uses the clinical classification scheme of “early-onset” vs. “late-onset”. While this foundational work has been tremendously helpful in gaining an appreciation for the central role of mitochondrial dysfunction in PE, it is important to note that even within these clinically distinct groups of patients, there remains a high degree of heterogeneity in: 1) the types of placental disease and damage noted at the time of pathological review; 2) circulating concentrations of placenta-derived biomarkers of disease (i.e. sFLT, sENG); 3) placental gene expression profiles; and 4) impact on fetal growth profiles. Collectively, this data would indicate that even within these clinically distinct populations, their very likely exists different underlying etiology and pathophysiology, and detailed profiling work conducted by our group [4,61,63,64] and others [65], certainly supports this conclusion. Hence, to gain a true appreciate for the role of mitochondrial dysfunction in the establishment and progression of PE, it is important to consider the underlying disease processes at play.
6.1. Canonical PE Subclass - Ischemia-Reperfusion Induced Mitochondrial Dysfunction
The historical dogma of PE pathophysiology includes defective EVT invasion of the decidua and impaired spiral artery remodelling, leading to hypoxia-reoxygenation events and oxidative damage in the placenta [107,108]. The canonical PE subclass previously described by our group demonstrates gene-expression, histopathology and clinical findings aligned to this disease model [4]. Data collected in rodent models of PE established by surgical reductions in uterine blood flow - the RUPP model - demonstrate a 30% increase in placental H2O2 production, compared to sham treated controls [109], highly indicative of placental mitochondrial redox dysfunction. Likewise, in primary human trophoblast cells, hypoxic culture conditions (2-5% O2) increase the production of mitochondrial ROS, triggering HIF1-α-mediated expression of the anti-angiogenic PE marker sFLT-1 - a circulating vascular endothelial growth factor signaling inhibitor [110,111]. Increase in sFLT-1 not only causes systemic endothelial dysfunction in the gestational parent but also induces further mitochondrial dysfunction and oxidative stress in placenta [112]. A recent study of placentas from early onset ‘canonical PE’ with evidence of ischemia-reperfusion injury, demonstrated that activation of mitochondrial unfolded protein response UPRmt leads to a decrease in the expression of mitochondrial quality control protease Caseinolytic peptidase P (CLPP) [87]. In vitro, knockdown of CLPP in BeWo trophoblast cell line led to increased mitochondrial fission and reduced respiration, mimicking findings of canonical human PE placentas. Recently another study demonstrated that chronic hypoxia in ovine pregnancy induces ER and cytoplasmic UPR, however, activation of mitochondrial UPR was not investigated [113].
6.2. Immunological PE Subclass - Inflammation Induced Mitochondrial Dysfunction
Preeclampsia has long been associated with a pro-inflammatory state, however a clearer identification of PE cases that demonstrate profound immune dysregulation at the utero-placenta interface, in the absence of traditional hallmarks of ischemia-reperfusion injury, has helped to clarify the existence of this unique subclass of PE patients [4,74,114,115]. Due to the understudied nature of this PE subclass in general, our current state of knowledge on the mechanisms through which chronic inflammation at this interface establishes placental dysfunction, and more specifically what role placental mitochondria may play in the placental pathophysiology within this subclass, are not currently well understood. However, a wealth of literature has identified mitochondrial dysfunction as a common feature of many chronic inflammatory diseases in non-pregnant populations, as well as within the aging process [116,117,118]. Inflammatory mediators has been shown to alter the activities of respiratory complexes, ATP production, mitochondrial membrane potential and its morphology [118]. Proinflammatory cytokine such as TNF-α has been shown to be an inducer of both cytoplasmic and mitochondrial ROS generation [119,120,121,122]. A recent study suggests that inflammation induced by TNF-α causes reverse electron flow through mitochondrial complex I and thus, stimulates mitochondrial ROS production [122].
Increased levels of TNF-α have been measured in placentas of PE cases [114,123,124], and profiling work carried out by our group demonstrated a very high placental gene expression of both TNF-α and TNF-α receptors uniquely within the ‘immune-driven PE subclass’ samples [4]. Interestingly, daily infusions of TNF-α (50 ng/day) in a pregnant rat model is capable of eliciting PE clinical features, including hypertension and decreases in both glomerular filtration rate and renal plasma flow [125]. In vitro treatment of first trimester placental explants with TNF-α (10ng/ml) compromises key aspects of placental function, including migratory and invasive properties [126]. In JEG3 trophoblast cell line, TNF-α treatment (0.2-5.0ng/ml) prevented its integration into endothelial networks and tube formation [127]. While there is certainly strong evidence to suggest that TNF-α, in addition to other pro-inflammatory signalling molecules, can initiate placental dysfunction and contribute to adverse pregnancy outcomes, the specific role of placental mitochondria, or mitochondrial dysfunction, in the context of chronic inflammation in pregnancy is not yet as clear. In vitro, TNF-α treatment in primary human trophoblasts, isolated from female offspring origin from overweight or obese pregnant individuals, caused decreased expression of mitochondrial complex-I subunit NDUFA4 and Iron-Sulfur Cluster Assembly Enzyme (ISCU) and inhibited mitochondrial respiration [128]. In an LPS-induced mouse model of intrauterine inflammation, placental transcriptomic analysis demonstrated dysregulated expression of upstream regulators of mitochondrial function. Further, metabolomics analysis showed alterations in TCA cycle and increase in acylcarnitine levels, known to induce mitochondrial dysfunction and oxidative stress [129,130]. Collectively, this body of work that is still in its infancy, certainly suggest that inflammation alone can initiate mitochondrial dysfunction in placental tissues, and as such would lend support to the hypothesis that mitochondrial dysfunction may in fact be a common point of convergence in the establishment of placental disease across multiple PE subclasses.
6.3. Gestational Parent Driven PE - Predisposing Factors and Mitochondrial Dysfunction
Contrary to the traditional dogma of PE pathophysiology, in which placental dysfunction is central to disease establishment, there have long been reports of PE cases with minimal evidence of placental disease [4,131,132]. Our group has confirmed the existence of this patient population through detailed clinical and placental profiling, concluding this group of patients likely arrive at a PE diagnosis (hypertension and end organ damage) as a result of poor gestational-parent adaptation to pregnancy, rather than placental dysfunction [4]. In healthy pregnancies, the process of placentation and normal placental function results in the release of some level of placenta-mediated factors into the gestational-parents circulation, including: antiangiogenic factors (i.e. sFLT-1), cytokines and chemokines, microparticles/microvesicles, exesomes, apoptotic bodies and cell free fetal DNA (to name a few) [133,134,135,136]. In a healthy pregnancy the gestational parent responds to these signalling factors in a way that promotes appropriate adaptation to pregnancy. However, predisposing risk factors (i.e. diabetes, obesity, advanced age, kidney disease, systemic lupus, antiphospholipid syndrome, chronic hypertension) may prevent the appropriate adaptive response in the gestational parent and instead may trigger systemic pathophysiology, including a heightened immune response, endothelial damage and dysfunction and the development of hypertension. As discussed in the previous section, a pro-inflammatory environment may be a potent trigger for mitochondrial dysfunction. Further, the dysregulated role of mitochondria within the context of endothelial dysfunction and hypertension has already been widely described, even in the context of pregnancy (see review [17,137,138] for more details). As such, while there may be a subclass of PE patients in which placental mitochondrial dysfunction may not be a major contributing factor to disease, systemic mitochondrial dysfunction in the gestational parent is still likely of major concern - and this is likely true of all subclasses of PE described. In fact, elevated mutational loads of circulating cell free (ccf)-mtDNA have been identified in blood (buffy coat) collected from 3rd trimester patients with PE and gestational hypertension alike. As ccf-mtDNA is often regarded as a marker of systemic inflammation, this result suggests that dysregulated mitochondria from gestational parent origin is a likely contributor to PE pathophysiology [139]. In vitro studies have shown that treatment of human umbilical vascular endothelial cells (HUVECs) with serum of PE patients or exogenous placenta-mediated factors (sFLT-1 or angiotensin II type 1 autoantibodies), leads to decreased mitochondrial respiratory capacity, increased superoxide formation and induced a glycolytic phenotype [79,140,141]. Thus, with or without the direct contribution of placental disease, systemic mitochondrial dysfunction within the gestational parent is likely another common point of convergence of pathophysiology across all PE subclasses.
Collectively, the current literature supports a role for mitochondrial dysfunction, both within the placenta and within gestational parent tissues, in the pathophysiology of PE. The underlying etiology of disease may in fact be different across PE subclasses, but we propose that dysregulation of mitochondrial biogenesis and function may be a common feature to all cases of PE [Figure 1], and as such serve as an important therapeutic target in the identification of effective treatments for this common and debilitating disorder of pregnancy.
7. Potential for Mitochondrial Targeting Therapies in Preeclampsia
Currently, there are no highly effective treatments for PE. Antihypertensive drugs are used to manage clinical symptoms in the gestational parent, but these treatments are not aimed at targeting the source of disease and as such these treatments neither serve to prolong the pregnancy nor ameliorates adverse impacts of PE on fetal growth profiles [142]. As there is sufficient evidence to implicate mitochondrial dysfunction in the establishment and/or progression of PE, consideration should be given to how therapeutic strategies aimed at improving mitochondrial function may be applied in cases of PE [6,8]. In this section some of the most recent advancements on this topic will be discussed (Figure 2).
7.1. Resveratrol
Several studies have shown that mitochondrial complex-I inhibitors may provide benefits in disease conditions by reducing superoxide generation through complex-I. Resveratrol (3,4,5-trihydroxy-trans-stilbene) is one of them. It is a plant-based polyphenol found in some fruits such as grapes, berries, and peanuts. It has antioxidant, anti-inflammatory and anti-microbial properties. Studies also suggest that it enhances mitochondrial homeostasis by activating the master regulator of mitochondrial biogenesis, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1ɑ) (see the following review for more information [143]). Notably, it reduces blood pressure in hypertensive rodent models [144]. Resveratrol administration also improved uterine artery blood flow velocity and increased fetal weight in a pregnant mouse model of uterine blood flow defect [145]. Similarly, resveratrol has shown some beneficial effects in various in vitro PE models. In one study, first-trimester HTR-8/SVneo trophoblast cell lines were treated with angiotensin II, TNF-α, protein kinase C (PKC) activator, and/or phorbol-12-myristate-13-acetate (PMA) for 24hrs. These treatments resulted in increased expression of sFLT1 - a major contributor to gestational parent endothelial dysfunction in PE. However, co-treatment with resveratrol significantly reduced the expression of this potent antiangiogenic factor and did not demonstrate any cytotoxic effect on the cells [146]. Interestingly, in a randomized control trial carried out with 400 patients diagnosed with PE, the use of resveratrol as an adjuvant treatment alongside oral nifedipine (an anti-hypertensive drug commonly prescribed in severe PE) was shown to further improve both systolic and diastolic blood pressure compared to Nifedipine treatment alone. With the resveratrol-combination therapy, time required for controlling blood pressure was reduced and time before developing new hypertension was delayed; thus, indicating a beneficial effect of resveratrol [147]. Interpretation of these results must be taken with caution, however, as there was no resveratrol alone in the study arm, rather the patients all received nifedipine as well. Thus, it is not clear based on these study results alone that resveratrol may serve as an effective solo therapy.
7.2. Metformin
Metformin is an effective treatment for type 2 diabetes that lowers blood glucose levels and reduces insulin resistance. It has anti-inflammatory and antioxidant effects [148,149]. Metformin has been shown to increase the expression of mitochondrial Sirtuin 3, an NAD+ dependent deacetylase, that regulates oxidative phosphorylation and activates mitochondrial antioxidant mnSOD2 [149,150]. It also activates adenosine 3′,5′-monophosphate (AMP) protein kinase (AMPK), a major regulator of mitochondrial fission, mitophagy and biogenesis [151,152]. Metformin has been used during pregnancy to improve pregnancy outcomes in individuals with obesity, gestational diabetes, type 2 diabetes and polycystic ovary syndrome. Interestingly, metformin has been shown to reduce the production of the anti-angiogenic factors sFLT1 and sENG (endoglin) in both villous cytotrophoblasts and endothelial cells in culture [153]. Moreover, 3rd trimester placental villous explants collected from PE patients treated with metformin in culture demonstrated a decrease in the secretion of sFLT1 and sENG, and co-treatment with succinate blocked this effect, indicating metformin-mediated regulation of sFLT1 and sENG expression is likely mediated via mitochondrial function [153]. In a PE mouse model induced via high fat diet, metformin administration was also shown to reduced blood pressure and increase circulating levels of vascular) endothelial growth factor (VEGF) [154]. The data in humans collected to date, however, is not as clear. A number of clinical trials, observational cohort studies and subsequent meta-analyses have been carried out to assess the ability of metformin treatment to reduce the establishment of PE in individuals at high risk, with mixed results [155,156,157,158,159]. Collectively, the data suggest that metformin may prove to be beneficial for a specific subset of patients, specifically those that have polycystic ovary syndrome (PCOS) [156,157,158]. Moreover, metformin treatment in the context of gestational diabetes has been associated with low birth weights, indicating a potential fetal risk with this form of treatment [6,160].
7.3. MitoQ and MitoTEMPO
MitoQ is a synthetic ubiquinone that is intracellularly converted into ubiquinol by the enzymatic activity of mitochondrial complex II. Ubiquinol serves to neutralize free radicals and can be continuously recycled to maintain a potent antioxidant presence in the mitochondria. Similarly, MitoTEMPO, which can specifically be targeted to the mitochondria via a bound lipophilic cation, contains piperidine nitroxide (Tempo) that mimics superoxide dismutase and catalyzes the dismutation of the superoxide radical into molecular oxygen and hydrogen peroxide. These mitochondria-targeted antioxidants have shown therapeutic promise in the RUPP rodent model of PE, where they have been shown to significantly reduce the maternal blood pressure and increase both placental and fetal weights. [109]. In another study, MitoTEMPO improved placental mtDNA stability and mitochondrial dynamics in a PE like rat model [161]. In cell culture, it reduced mitochondrial ROS production in HUVECs treated with plasma from PE patients. Both MitoQ and mitTEMPO has been tested in humans and found to be tolerable with no adverse effect [162,163,164,165]. MitoQ has been reported to reduce liver inflammation in patients with chronic Hepatitis C Virus (HCV) infection [163]. On the other hand, MitoTEMPO has been shown to improve cutaneous vascular conductance (CVC) in patients with chronic kidney disease [164]. However, caution should be taken when considering therapeutic application of such antioxidants in pregnancy as one of the rodent studies indicate that MitoQ administration early in pregnancy could hinder normal placenta development by reducing physiological ROS levels [166].
7.4. Nicotinamide (NAM)
Nicotinamide or NAM is a derivative of vitamin B3 and has antioxidant and anti-inflammatory properties [167,168]. NAM is a known cellular NAD+ donor and NAD+ level is critical for maintenance of OXPHOS function, mitochondrial biogenesis, and homeostasis [169]. NAD+ is also important for DNA damage response, inflammation, and oxidative stress pathways as it functions as a cofactor for certain enzymes such PARPs and Sirtuins [170]. The therapeutic potential of NAM treatment has been investigated using three different rodent models of PE: RUPP, sFLT1 overexpression and ASB4 (Ankiryn-repeat-and-SOCS-box-containing-protein) knockout mice. In all three models, NAM administration (500 mg/kg per day) was shown to decrease blood pressure, proteinuria and prevent glomerular endotheliosis in the mothers. Further, NAM treatment increased fetal and placental weight, prolonged pregnancy duration and increased the number of live births. NAM also decreased HIF1-alpha expression; and increased the levels of NAM, nicotinamide adenine dinucleotide (NAD+) and ATP in fetal brains [171]. As NAM is an NAD+ donor, it is likely that the observed beneficial effects were through improving mitochondrial function at various maternal or fetal tissue levels including placenta. Thus, more research needs to be done to understand the mechanism. Moreover, there is a safety concern regarding the use of NAM at high dose during pregnancy. Studies performed in rats showed that NAM administration at 500 mg - 4 g/kg/day dose is associated with liver toxicity [172,173]. Maternal NAM intake led to DNA hypomethylation in rat placenta and fetal liver [174]. Indeed, in humans, NAM intake at 300 mg/day was shown to elevate plasma levels of homocysteine, an indicator for methyl donor consumption due to NAM methylation [175]. However, there are additional NAD+ donors that can be examined for their efficacy in improving mitochondrial and placental function, and ultimately pregnancy outcomes, such as nicotinamide riboside (NR), nicotinamide mononucleotide (NMN), dihydronicotinamide riboside (NRH) - all of which may likely demonstrate a more appropriate safety profile for use in pregnancy.
Future Perspective
Findings from animal and human studies indicate that mitochondrial dysfunction is a central component to the establishment of both placental disease and gestational parent disease in PE. The degree of mitochondrial dysfunction and inherent ability to adapt to cellular stress signals may dictate the severity of placental or gestational parent tissue damage and ultimately pregnancy outcome. Novel therapeutic interventions aimed at targeting such common disease features will likely prove to be highly effective at the population level regardless of their subclass in a clinical setting. But there are still many hurdles to overcome in this domain. Using animal models and human studies, future research should aim to 1) identify specific aspects of mitochondrial dysfunction that are common across PE and should be the focus of therapies moving forward; 2) determine the critical window when mitochondrial dysfunction occurs during PE pathogenesis in order to maximize therapeutic benefits 3) vigorously test the safety profiles of these treatments, as any novel treatments must not interfere with placental or fetal development.
Author Contributions
Conceptualization, F.J., S.A.B. and K.J.M.; writing—original draft preparation, F.J., G.V.; writing—review and editing, F.J., A.G., S.A.B. and K.J.M.; visualization, F.J.; supervision, S.A.B. and K.J.M.; funding acquisition, S.A.B. and K.J.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Canadian Institutes of Health Research (CIHR) Project Grant #PJT-153055 to S.A.B. and K.J.M. F.J. is supported by Frederick Banting and Charles Best Canada Graduate Scholarship from CIHR (FRN-167027).
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bauer, M.K., et al., Fetal growth and placental function. Mol Cell Endocrinol, 1998. 140(1-2): p. 115-20. [CrossRef]
- Gude, N.M., et al., Growth and function of the normal human placenta. Thromb Res, 2004. 114(5-6): p. 397-407. [CrossRef]
- Rana, S., et al., Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ Res, 2019. 124(7): p. 1094-1112.
- Leavey, K., et al., Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension, 2016. 68(1): p. 137-47. [CrossRef]
- Griffiths, S.K. and J.P. Campbell, Placental structure, function and drug transfer. Continuing Education in Anaesthesia Critical Care & Pain, 2014. 15(2): p. 84-89.
- Aye, I., et al., Placental energy metabolism in health and disease-significance of development and implications for preeclampsia. Am J Obstet Gynecol, 2022. 226(2S): p. S928-S944. [CrossRef]
- Spinelli, J.B. and M.C. Haigis, The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol, 2018. 20(7): p. 745-754. [CrossRef]
- Hebert, J.F. and L. Myatt, Placental mitochondrial dysfunction with metabolic diseases: Therapeutic approaches. Biochim Biophys Acta Mol Basis Dis, 2021. 1867(1): p. 165967. [CrossRef]
- Holland, O., et al., Review: Placental mitochondrial function and structure in gestational disorders. Placenta, 2017. 54: p. 2-9. [CrossRef]
- Burton, G.J., H.W. Yung, and A.J. Murray, Mitochondrial - Endoplasmic reticulum interactions in the trophoblast: Stress and senescence. Placenta, 2017. 52: p. 146-155. [CrossRef]
- Constancia, M., et al., Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature, 2002. 417(6892): p. 945-8. [CrossRef]
- Constancia, M., et al., Adaptation of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter systems. Proc Natl Acad Sci U S A, 2005. 102(52): p. 19219-24. [CrossRef]
- Sferruzzi-Perri, A.N., et al., An obesogenic diet during mouse pregnancy modifies maternal nutrient partitioning and the fetal growth trajectory. FASEB J, 2013. 27(10): p. 3928-37. [CrossRef]
- Hay, W.W., Jr., Energy and substrate requirements of the placenta and fetus. Proc Nutr Soc, 1991. 50(2): p. 321-36. [CrossRef]
- Fisher, J.J., et al., Placental mitochondria and reactive oxygen species in the physiology and pathophysiology of pregnancy. Clin Exp Pharmacol Physiol, 2020. 47(1): p. 176-184. [CrossRef]
- Bartho, L.A., et al., Mitochondrial transformations in the aging human placenta. Am J Physiol Endocrinol Metab, 2020. 319(6): p. E981-E994. [CrossRef]
- Correia, Y., et al., Placental mitochondrial function as a driver of angiogenesis and placental dysfunction. Biol Chem, 2021. 402(8): p. 887-909. [CrossRef]
- Burton, G.J. and A.L. Fowden, The placenta: a multifaceted, transient organ. Philos Trans R Soc Lond B Biol Sci, 2015. 370(1663): p. 20140066.
- Jauniaux, E., et al., Polyol concentrations in the fluid compartments of the human conceptus during the first trimester of pregnancy: maintenance of redox potential in a low oxygen environment. J Clin Endocrinol Metab, 2005. 90(2): p. 1171-5. [CrossRef]
- Bloxam, D.L. and P.M. Bobinski, Energy metabolism and glycolysis in the human placenta during ischaemia and in normal labour. Placenta, 1984. 5(5): p. 381-94. [CrossRef]
- Holland, O.J., et al., Changes in mitochondrial respiration in the human placenta over gestation. Placenta, 2017. 57: p. 102-112. [CrossRef]
- Jauniaux, E., et al., Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol, 2000. 157(6): p. 2111-22. [CrossRef]
- Drose, S. and U. Brandt, Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol, 2012. 748: p. 145-69. [CrossRef]
- Mannaerts, D., et al., Oxidative stress in healthy pregnancy and preeclampsia is linked to chronic inflammation, iron status and vascular function. PLoS One, 2018. 13(9): p. e0202919. [CrossRef]
- Bardin, N., P. Murthi, and N. Alfaidy, Normal and pathological placental angiogenesis. Biomed Res Int, 2015. 2015: p. 354359. [CrossRef]
- Basu, J., et al., Placental Oxidative Status throughout Normal Gestation in Women with Uncomplicated Pregnancies. Obstet Gynecol Int, 2015. 2015: p. 276095. [CrossRef]
- Sferruzzi-Perri, A.N., et al., Placental mitochondria adapt developmentally and in response to hypoxia to support fetal growth. Proc Natl Acad Sci U S A, 2019. 116(5): p. 1621-1626. [CrossRef]
- Sesso, A., et al., Mitochondrial swelling and incipient outer membrane rupture in preapoptotic and apoptotic cells. Anat Rec (Hoboken), 2012. 295(10): p. 1647-59. [CrossRef]
- Huppertz, B., et al., Apoptosis cascade progresses during turnover of human trophoblast: analysis of villous cytotrophoblast and syncytial fragments in vitro. Lab Invest, 1999. 79(12): p. 1687-702.
- Levy, R. and D.M. Nelson, To be, or not to be, that is the question. Apoptosis in human trophoblast. Placenta, 2000. 21(1): p. 1-13. [CrossRef]
- Kolahi, K.S., A.M. Valent, and K.L. Thornburg, Cytotrophoblast, Not Syncytiotrophoblast, Dominates Glycolysis and Oxidative Phosphorylation in Human Term Placenta. Sci Rep, 2017. 7: p. 42941. [CrossRef]
- De los Rios Castillo, D., et al., Atypical cristae morphology of human syncytiotrophoblast mitochondria: role for complex V. J Biol Chem, 2011. 286(27): p. 23911-9.
- Martinez, F., M. Kiriakidou, and J.F. Strauss, 3rd, Structural and functional changes in mitochondria associated with trophoblast differentiation: methods to isolate enriched preparations of syncytiotrophoblast mitochondria. Endocrinology, 1997. 138(5): p. 2172-83. [CrossRef]
- Martinez, F., et al., Multiple functions of syncytiotrophoblast mitochondria. Steroids, 2015. 103: p. 11-22. [CrossRef]
- Watson, A.L., et al., Susceptibility of human placental syncytiotrophoblastic mitochondria to oxygen-mediated damage in relation to gestational age. J Clin Endocrinol Metab, 1998. 83(5): p. 1697-705.
- Seok, J., et al., Mitochondrial Dynamics in Placenta-Derived Mesenchymal Stem Cells Regulate the Invasion Activity of Trophoblast. Int J Mol Sci, 2020. 21(22). [CrossRef]
- Seok, J., et al., Human placenta-derived mesenchymal stem cells induce trophoblast invasion via dynamic effects on mitochondrial function. J Cell Physiol, 2021. 236(9): p. 6678-6690. [CrossRef]
- LeBleu, V.S., et al., PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol, 2014. 16(10): p. 992-1003, 1-15. [CrossRef]
- Hogan, M.C., et al., Maternal mortality for 181 countries, 1980-2008: a systematic analysis of progress towards Millennium Development Goal 5. Lancet, 2010. 375(9726): p. 1609-23.
- ACOG Practice Bulletin No. 202: Gestational Hypertension and Preeclampsia. Obstet Gynecol, 2019. 133(1): p. 1.
- Weiler, J., S. Tong, and K.R. Palmer, Is fetal growth restriction associated with a more severe maternal phenotype in the setting of early onset pre-eclampsia? A retrospective study. PLoS One, 2011. 6(10): p. e26937. [CrossRef]
- Mastrolia, S.A., et al., Single vs. Recurrent Episodes of Preeclampsia-population–based Epidemiological and Clinical Characteristics. Maternal-Fetal Medicine, 2021. 3(3): p. 190-196. [CrossRef]
- Hutchinson, E.S., et al., Utero-placental haemodynamics in the pathogenesis of pre-eclampsia. Placenta, 2009. 30(7): p. 634-41. [CrossRef]
- Roberts, J.M. and C.A. Hubel, The two stage model of preeclampsia: variations on the theme. Placenta, 2009. 30 Suppl A(Suppl A): p. S32-7. [CrossRef]
- Lyall, F., S.C. Robson, and J.N. Bulmer, Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction: relationship to clinical outcome. Hypertension, 2013. 62(6): p. 1046-54.
- Aardema, M.W., et al., Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta, 2001. 22(5): p. 405-11. [CrossRef]
- Khong, T.Y., et al., Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br J Obstet Gynaecol, 1986. 93(10): p. 1049-59. [CrossRef]
- Espinoza, J., et al., Normal and abnormal transformation of the spiral arteries during pregnancy. J Perinat Med, 2006. 34(6): p. 447-58. [CrossRef]
- Jones, C.J. and H. Fox, An ultrastructural and ultrahistochemical study of the human placenta in maternal pre-eclampsia. Placenta, 1980. 1(1): p. 61-76. [CrossRef]
- Longtine, M.S., et al., Villous trophoblast apoptosis is elevated and restricted to cytotrophoblasts in pregnancies complicated by preeclampsia, IUGR, or preeclampsia with IUGR. Placenta, 2012. 33(5): p. 352-9. [CrossRef]
- Redman, C.W., I.L. Sargent, and A.C. Staff, IFPA Senior Award Lecture: making sense of pre-eclampsia - two placental causes of preeclampsia? Placenta, 2014. 35 Suppl: p. S20-5.
- Tannetta, D., et al., Update of syncytiotrophoblast derived extracellular vesicles in normal pregnancy and preeclampsia. J Reprod Immunol, 2017. 119: p. 98-106. [CrossRef]
- Nikuei, P., et al., Accuracy of Soluble Endoglin for Diagnosis of Preeclampsia and its Severity. Iran Biomed J, 2017. 21(5): p. 312-30. [CrossRef]
- von Dadelszen, P., L.A. Magee, and J.M. Roberts, Subclassification of preeclampsia. Hypertens Pregnancy, 2003. 22(2): p. 143-8. [CrossRef]
- McCartney, C.P., Pathological Anatomy of Acute Hypertension of Pregnancy. Circulation, 1964. 30: p. SUPPL 2:37-42. [CrossRef]
- Xiong, X. and W.D. Fraser, Impact of pregnancy-induced hypertension on birthweight by gestational age. Paediatr Perinat Epidemiol, 2004. 18(3): p. 186-91. [CrossRef]
- Redman, C.W. and A.C. Staff, Preeclampsia, biomarkers, syncytiotrophoblast stress, and placental capacity. Am J Obstet Gynecol, 2015. 213(4 Suppl): p. S9 e1, S9-11. [CrossRef]
- Redman, E.K., et al., Clinical Course, Associated Factors, and Blood Pressure Profile of Delayed-Onset Postpartum Preeclampsia. Obstet Gynecol, 2019. 134(5): p. 995-1001. [CrossRef]
- McElrath, T.F., et al., Circulating microparticle proteins obtained in the late first trimester predict spontaneous preterm birth at less than 35 weeks' gestation: a panel validation with specific characterization by parity. Am J Obstet Gynecol, 2019. 220(5): p. 488 e1-488 e11. [CrossRef]
- Powers, R.W., et al., Low placental growth factor across pregnancy identifies a subset of women with preterm preeclampsia: type 1 versus type 2 preeclampsia? Hypertension, 2012. 60(1): p. 239-46.
- Benton, S.J., et al., The clinical heterogeneity of preeclampsia is related to both placental gene expression and placental histopathology. Am J Obstet Gynecol, 2018. 219(6): p. 604 e1-604 e25. [CrossRef]
- Wilson, S.L., et al., Mining DNA methylation alterations towards a classification of placental pathologies. Hum Mol Genet, 2018. 27(1): p. 135-146. [CrossRef]
- Leavey, K., S.A. Bainbridge, and B.J. Cox, Large scale aggregate microarray analysis reveals three distinct molecular subclasses of human preeclampsia. PLoS One, 2015. 10(2): p. e0116508. [CrossRef]
- Leavey, K., D. Grynspan, and B.J. Cox, Both "canonical" and "immunological" preeclampsia subtypes demonstrate changes in placental immune cell composition. Placenta, 2019. 83: p. 53-56.
- Roberts, J.M., et al., Subtypes of Preeclampsia: Recognition and Determining Clinical Usefulness. Hypertension, 2021. 77(5): p. 1430-1441. [CrossRef]
- Conrad, K.P. and D.F. Benyo, Placental cytokines and the pathogenesis of preeclampsia. Am J Reprod Immunol, 1997. 37(3): p. 240-9. [CrossRef]
- Harmon, A.C., et al., The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond), 2016. 130(6): p. 409-19. [CrossRef]
- Moffett, A. and F. Colucci, Uterine NK cells: active regulators at the maternal-fetal interface. J Clin Invest, 2014. 124(5): p. 1872-9. [CrossRef]
- Hiby, S.E., et al., Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med, 2004. 200(8): p. 957-65. [CrossRef]
- Triche, E.W., et al., Maternal-fetal HLA sharing and preeclampsia: variation in effects by seminal fluid exposure in a case-control study of nulliparous women in Iowa. J Reprod Immunol, 2014. 101-102: p. 111-119. [CrossRef]
- Kenny, L.C. and D.B. Kell, Immunological Tolerance, Pregnancy, and Preeclampsia: The Roles of Semen Microbes and the Father. Front Med (Lausanne), 2017. 4: p. 239. [CrossRef]
- Myatt, L. and J.M. Roberts, Preeclampsia: Syndrome or Disease? Curr Hypertens Rep, 2015. 17(11): p. 83.
- Vangrieken, P., et al., Placental Mitochondrial Abnormalities in Preeclampsia. Reprod Sci, 2021. 28(8): p. 2186-2199. [CrossRef]
- Holland, O.J., et al., Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis, 2018. 9(12): p. 1150. [CrossRef]
- Pandey, D., et al., Mitochondrial DNA copy number variation - A potential biomarker for early onset preeclampsia. Pregnancy Hypertens, 2021. 23: p. 1-4. [CrossRef]
- Cushen, S.C., et al., Reduced Maternal Circulating Cell-Free Mitochondrial DNA Is Associated With the Development of Preeclampsia. J Am Heart Assoc, 2022. 11(2): p. e021726. [CrossRef]
- Marschalek, J., et al., Maternal serum mitochondrial DNA (mtDNA) levels are elevated in preeclampsia - A matched case-control study. Pregnancy Hypertens, 2018. 14: p. 195-199. [CrossRef]
- Qiu, C., et al., A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int J Mol Epidemiol Genet, 2012. 3(3): p. 237-44.
- McCarthy, C. and L.C. Kenny, Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci Rep, 2016. 6: p. 32683. [CrossRef]
- Westrate, L.M., et al., Mitochondrial morphological features are associated with fission and fusion events. PLoS One, 2014. 9(4): p. e95265. [CrossRef]
- Youle, R.J. and A.M. van der Bliek, Mitochondrial fission, fusion, and stress. Science, 2012. 337(6098): p. 1062-5.
- Yu, J., et al., Downregulation of Mitofusin 2 in Placenta Is Related to Preeclampsia. Biomed Res Int, 2016. 2016: p. 6323086. [CrossRef]
- Ryan, J.J., et al., PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med, 2013. 187(8): p. 865-78. [CrossRef]
- Salgado, S.S. and M.K.R. Salgado, Structural changes in pre-eclamptic and eclamptic placentas--an ultrastructural study. J Coll Physicians Surg Pak, 2011. 21(8): p. 482-6.
- Muralimanoharan, S., et al., MIR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta, 2012. 33(10): p. 816-23. [CrossRef]
- Vishnyakova, P.A., et al., Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci Rep, 2016. 6: p. 32410. [CrossRef]
- Yung, H.W., et al., Noncanonical mitochondrial unfolded protein response impairs placental oxidative phosphorylation in early-onset preeclampsia. Proc Natl Acad Sci U S A, 2019. 116(36): p. 18109-18118. [CrossRef]
- Ausman, J., et al., Ceramide-induced BOK promotes mitochondrial fission in preeclampsia. Cell Death Dis, 2018. 9(3): p. 298. [CrossRef]
- Acin-Perez, R., et al., A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J, 2020. 39(13): p. e104073. [CrossRef]
- Cadenas, E. and K.J. Davies, Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med, 2000. 29(3-4): p. 222-30. [CrossRef]
- Guzy, R.D., et al., Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab, 2005. 1(6): p. 401-8. [CrossRef]
- Hernansanz-Agustin, P., et al., Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol, 2017. 12: p. 1040-1051. [CrossRef]
- Gupta, S., A. Agarwal, and R.K. Sharma, The role of placental oxidative stress and lipid peroxidation in preeclampsia. Obstet Gynecol Surv, 2005. 60(12): p. 807-16. [CrossRef]
- Wiktor, H., et al., Oxidative DNA damage in placentas from normal and pre-eclamptic pregnancies. Virchows Arch, 2004. 445(1): p. 74-8. [CrossRef]
- Llurba, E., et al., A comprehensive study of oxidative stress and antioxidant status in preeclampsia and normal pregnancy. Free Radic Biol Med, 2004. 37(4): p. 557-70. [CrossRef]
- Aris, A., et al., Potential biomarkers of preeclampsia: inverse correlation between hydrogen peroxide and nitric oxide early in maternal circulation and at term in placenta of women with preeclampsia. Placenta, 2009. 30(4): p. 342-7. [CrossRef]
- Wang, Y. and S.W. Walsh, Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta, 1998. 19(8): p. 581-6. [CrossRef]
- Rafeeinia, A., et al., Serum copper, zinc and lipid peroxidation in pregnant women with preeclampsia in gorgan. Open Biochem J, 2014. 8: p. 83-8. [CrossRef]
- Shaker, O.G. and N.A. Sadik, Pathogenesis of preeclampsia: Implications of apoptotic markers and oxidative stress. Hum Exp Toxicol, 2013. 32(11): p. 1170-8.
- Padmini, E., S. Lavanya, and V. Uthra, Preeclamptic placental stress and over expression of mitochondrial HSP70. Clin Chem Lab Med, 2009. 47(9): p. 1073-80. [CrossRef]
- Wiktor, H. and M. Kankofer, Superoxide dismutase activity in normal and preeclamptic placentas. Ginekol Pol, 1998. 69(12): p. 915-8.
- Wu, F., F.J. Tian, and Y. Lin, Oxidative Stress in Placenta: Health and Diseases. Biomed Res Int, 2015. 2015: p. 293271. [CrossRef]
- Brunelle, J.K. and A. Letai, Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci, 2009. 122(Pt 4): p. 437-41. [CrossRef]
- Allaire, A.D., et al., Placental apoptosis in preeclampsia. Obstet Gynecol, 2000. 96(2): p. 271-6.
- Leung, D.N., et al., Increased placental apoptosis in pregnancies complicated by preeclampsia. Am J Obstet Gynecol, 2001. 184(6): p. 1249-50. [CrossRef]
- Fogarty, N.M., A.C. Ferguson-Smith, and G.J. Burton, Syncytial knots (Tenney-Parker changes) in the human placenta: evidence of loss of transcriptional activity and oxidative damage. Am J Pathol, 2013. 183(1): p. 144-52.
- Hung, T.H., et al., Hypoxia-reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res, 2002. 90(12): p. 1274-81.
- Sanchez-Aranguren, L.C., et al., Endothelial dysfunction and preeclampsia: role of oxidative stress. Front Physiol, 2014. 5: p. 372. [CrossRef]
- Vaka, V.R., et al., Role of Mitochondrial Dysfunction and Reactive Oxygen Species in Mediating Hypertension in the Reduced Uterine Perfusion Pressure Rat Model of Preeclampsia. Hypertension, 2018. 72(3): p. 703-711. [CrossRef]
- Covarrubias, A.E., et al., AP39, a Modulator of Mitochondrial Bioenergetics, Reduces Antiangiogenic Response and Oxidative Stress in Hypoxia-Exposed Trophoblasts: Relevance for Preeclampsia Pathogenesis. Am J Pathol, 2019. 189(1): p. 104-114.
- Nagamatsu, T., et al., Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: an implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology, 2004. 145(11): p. 4838-45. [CrossRef]
- Jiang, Z., et al., A Role of sFlt-1 in Oxidative Stress and Apoptosis in Human and Mouse Pre-Eclamptic Trophoblasts. Biol Reprod, 2015. 93(3): p. 73. [CrossRef]
- Tong, W., et al., Chronic Hypoxia in Ovine Pregnancy Recapitulates Physiological and Molecular Markers of Preeclampsia in the Mother, Placenta, and Offspring. Hypertension, 2022. 79(7): p. 1525-1535. [CrossRef]
- Peracoli, J.C., M.V. Rudge, and M.T. Peracoli, Tumor necrosis factor-alpha in gestation and puerperium of women with gestational hypertension and pre-eclampsia. Am J Reprod Immunol, 2007. 57(3): p. 177-85. [CrossRef]
- Raghupathy, R., Cytokines as key players in the pathophysiology of preeclampsia. Med Princ Pract, 2013. 22 Suppl 1: p. 8-19. [CrossRef]
- Clayton, S.A., et al., Mitochondria as Key Players in the Pathogenesis and Treatment of Rheumatoid Arthritis. Front Immunol, 2021. 12: p. 673916. [CrossRef]
- Green, D.R., L. Galluzzi, and G. Kroemer, Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science, 2011. 333(6046): p. 1109-12. [CrossRef]
- Lopez-Armada, M.J., et al., Mitochondrial dysfunction and the inflammatory response. Mitochondrion, 2013. 13(2): p. 106-18. [CrossRef]
- Blaser, H., et al., TNF and ROS Crosstalk in Inflammation. Trends Cell Biol, 2016. 26(4): p. 249-261. [CrossRef]
- Fiers, W., et al., More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene, 1999. 18(54): p. 7719-30. [CrossRef]
- Kim, J.J., et al., TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ, 2010. 17(9): p. 1420-34.
- Roca, F.J., et al., Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science, 2022. 376(6600): p. eabh2841. [CrossRef]
- Wang, Y. and S.W. Walsh, TNF alpha concentrations and mRNA expression are increased in preeclamptic placentas. J Reprod Immunol, 1996. 32(2): p. 157-69.
- I, C.W., et al., Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J Reprod Immunol, 2017. 123: p. 40-47.
- Alexander, B.T., et al., Tumor necrosis factor-alpha-induced hypertension in pregnant rats results in decreased renal neuronal nitric oxide synthase expression. Am J Hypertens, 2002. 15(2 Pt 1): p. 170-5. [CrossRef]
- Guo, X., et al., Upregulation of VEGF by small activating RNA and its implications in preeclampsia. Placenta, 2016. 46: p. 38-44. [CrossRef]
- Xu, B., et al., TNF-alpha inhibits trophoblast integration into endothelial cellular networks. Placenta, 2011. 32(3): p. 241-6. [CrossRef]
- Muralimanoharan, S., et al., Sexual dimorphism in miR-210 expression and mitochondrial dysfunction in the placenta with maternal obesity. Int J Obes (Lond), 2015. 39(8): p. 1274-81. [CrossRef]
- Lien, Y.C., et al., Intrauterine Inflammation Alters the Transcriptome and Metabolome in Placenta. Front Physiol, 2020. 11: p. 592689. [CrossRef]
- Batchuluun, B., et al., Elevated Medium-Chain Acylcarnitines Are Associated With Gestational Diabetes Mellitus and Early Progression to Type 2 Diabetes and Induce Pancreatic beta-Cell Dysfunction. Diabetes, 2018. 67(5): p. 885-897.
- Odegard, R.A., et al., Risk factors and clinical manifestations of pre-eclampsia. BJOG, 2000. 107(11): p. 1410-6. [CrossRef]
- Redman, C., Pre-eclampsia: A complex and variable disease. Pregnancy Hypertens, 2014. 4(3): p. 241-2. [CrossRef]
- Clark, D.E., et al., A vascular endothelial growth factor antagonist is produced by the human placenta and released into the maternal circulation. Biol Reprod, 1998. 59(6): p. 1540-8. [CrossRef]
- Levine, R.J., et al., Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med, 2004. 350(7): p. 672-83. [CrossRef]
- Mor, G., et al., Inflammation and pregnancy: the role of the immune system at the implantation site. Ann N Y Acad Sci, 2011. 1221(1): p. 80-7. [CrossRef]
- Abu-Raya, B., et al., Maternal Immunological Adaptation During Normal Pregnancy. Front Immunol, 2020. 11: p. 575197. [CrossRef]
- Tang, X., et al., Mitochondria, endothelial cell function, and vascular diseases. Front Physiol, 2014. 5: p. 175.
- Kirkman, D.L., et al., Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am J Physiol Heart Circ Physiol, 2021. 320(5): p. H2080-H2100. [CrossRef]
- Ricci, C.A., et al., Mitochondria-mediated Maternal-fetal Interactions and Consequences of Mitochondrial Dysregulation Indicate New Roles for Mitochondria in Hypertensive Pregnancies. medRxiv, 2021: p. 2021.12.18.21268029.
- Sanchez-Aranguren, L.C., et al., Soluble Fms-Like Tyrosine Kinase-1 Alters Cellular Metabolism and Mitochondrial Bioenergetics in Preeclampsia. Front Physiol, 2018. 9: p. 83. [CrossRef]
- Deer, E., et al., Vascular endothelial mitochondrial oxidative stress in response to preeclampsia: a role for angiotension II type 1 autoantibodies. Am J Obstet Gynecol MFM, 2021. 3(1): p. 100275. [CrossRef]
- Duley, L., Pre-eclampsia and the hypertensive disorders of pregnancy. Br Med Bull, 2003. 67: p. 161-76. [CrossRef]
- Uriho, A., et al., Effects of resveratrol on mitochondrial biogenesis and physiological diseases. Advances in Traditional Medicine, 2021. 21(1): p. 1-14. [CrossRef]
- Bonnefont-Rousselot, D., Resveratrol and Cardiovascular Diseases. Nutrients, 2016. 8(5). [CrossRef]
- Poudel, R., et al., Effects of resveratrol in pregnancy using murine models with reduced blood supply to the uterus. PLoS One, 2013. 8(5): p. e64401. [CrossRef]
- Cudmore, M.J., et al., Resveratrol inhibits the release of soluble fms-like tyrosine kinase (sFlt-1) from human placenta. Am J Obstet Gynecol, 2012. 206(3): p. 253 e10-5. [CrossRef]
- Ding, J., et al., Efficacy of resveratrol to supplement oral nifedipine treatment in pregnancy-induced preeclampsia. Endocr Connect, 2017. 6(8): p. 595-600. [CrossRef]
- Giusti, L., et al., The Protective Action of Metformin against Pro-Inflammatory Cytokine-Induced Human Islet Cell Damage and the Mechanisms Involved. Cells, 2022. 11(15). [CrossRef]
- Sung, J.Y., et al., Metformin mitigates stress-induced premature senescence by upregulating AMPKalpha at Ser485 phosphorylation induced SIRT3 expression and inactivating mitochondrial oxidants. Mech Ageing Dev, 2022. 206: p. 111708.
- Ansari, A., et al., Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell, 2017. 16(1): p. 4-16.
- Feng, J., et al., Mitochondria as an important target of metformin: The mechanism of action, toxic and side effects, and new therapeutic applications. Pharmacol Res, 2022. 177: p. 106114. [CrossRef]
- Herzig, S. and R.J. Shaw, AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol, 2018. 19(2): p. 121-135. [CrossRef]
- Brownfoot, F.C., et al., Metformin as a prevention and treatment for preeclampsia: effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am J Obstet Gynecol, 2016. 214(3): p. 356 e1-356 e15. [CrossRef]
- Wang, F., et al., Effect of Metformin on a Preeclampsia-Like Mouse Model Induced by High-Fat Diet. Biomed Res Int, 2019. 2019: p. 6547019. [CrossRef]
- Alqudah, A., et al., Risk of pre-eclampsia in women taking metformin: a systematic review and meta-analysis. Diabet Med, 2018. 35(2): p. 160-172. [CrossRef]
- Glueck, C.J., et al., Height, weight, and motor-social development during the first 18 months of life in 126 infants born to 109 mothers with polycystic ovary syndrome who conceived on and continued metformin through pregnancy. Hum Reprod, 2004. 19(6): p. 1323-30. [CrossRef]
- Glueck, C.J., et al., Effects of metformin-diet intervention before and throughout pregnancy on obstetric and neonatal outcomes in patients with polycystic ovary syndrome. Curr Med Res Opin, 2013. 29(1): p. 55-62. [CrossRef]
- De Leo, V., et al., The administration of metformin during pregnancy reduces polycystic ovary syndrome related gestational complications. Eur J Obstet Gynecol Reprod Biol, 2011. 157(1): p. 63-6. [CrossRef]
- Cluver, C.A., et al., Use of metformin to prolong gestation in preterm pre-eclampsia: randomised, double blind, placebo controlled trial. BMJ, 2021. 374: p. n2103. [CrossRef]
- Thompson, J.D. and D. Diers, Management of nursing intensity. Nurs Clin North Am, 1988. 23(3): p. 473-92. [CrossRef]
- Long, J., et al., Mitochondria Targeted Antioxidant Significantly Alleviates Preeclampsia Caused by 11beta-HSD2 Dysfunction via OPA1 and MtDNA Maintenance. Antioxidants (Basel), 2022. 11(8).
- Gane, E.J., et al., The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int, 2010. 30(7): p. 1019-26. [CrossRef]
- Snow, B.J., et al., A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord, 2010. 25(11): p. 1670-4.
- Kirkman, D.L., et al., Role of mitochondria-derived reactive oxygen species in microvascular dysfunction in chronic kidney disease. Am J Physiol Renal Physiol, 2018. 314(3): p. F423-F429. [CrossRef]
- Mason, S.A., et al., Effect of mitochondrial-targeted antioxidants on glycaemic control, cardiovascular health, and oxidative stress in humans: A systematic review and meta-analysis of randomized controlled trials. Diabetes Obes Metab, 2022. 24(6): p. 1047-1060.
- Yang, Y., et al., The Potent Antioxidant MitoQ Protects Against Preeclampsia During Late Gestation but Increases the Risk of Preeclampsia When Administered in Early Pregnancy. Antioxid Redox Signal, 2021. 34(2): p. 118-136. [CrossRef]
- Ayla, S., et al., Doxorubicin induced nephrotoxicity: protective effect of nicotinamide. Int J Cell Biol, 2011. 2011: p. 390238. [CrossRef]
- Wahlberg, G., et al., Protective effect of nicotinamide against nephropathy in diabetic rats. Diabetes Res, 1985. 2(6): p. 307-12.
- Rongvaux, A., et al., Reconstructing eukaryotic NAD metabolism. Bioessays, 2003. 25(7): p. 683-90. [CrossRef]
- Canto, C., K.J. Menzies, and J. Auwerx, NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab, 2015. 22(1): p. 31-53. [CrossRef]
- Li, F., et al., Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc Natl Acad Sci U S A, 2016. 113(47): p. 13450-13455. [CrossRef]
- Li, D., et al., Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats. Br J Nutr, 2013. 110(12): p. 2156-64. [CrossRef]
- Mahmoud, Y.I. and A.A. Mahmoud, Role of nicotinamide (vitamin B3) in acetaminophen-induced changes in rat liver: Nicotinamide effect in acetaminophen-damged liver. Exp Toxicol Pathol, 2016. 68(6): p. 345-54.
- Tian, Y.J., et al., Maternal nicotinamide supplementation causes global DNA hypomethylation, uracil hypo-incorporation and gene expression changes in fetal rats. Br J Nutr, 2014. 111(9): p. 1594-601. [CrossRef]
- Sun, W.P., et al., Comparison of the effects of nicotinic acid and nicotinamide degradation on plasma betaine and choline levels. Clin Nutr, 2017. 36(4): p. 1136-1142. [CrossRef]
Figure 1.
Mitochondrial Dysfunction as a Common Feature across all PE subclasses. PE disease driven by either placental malperfusion, chronic inflammation at the utero-placental interface and/or poor gestational-parent adaptation likely all demonstrate altered mitochondrial respiration, increase in ROS production and mitochondrial fragmentation within the placenta and across numerous gestational-parent tissues. The inherent ability for the mitochondria to adaptation during pregnancy to various pathological insults may in part dictate the clinical outcome of the pregnancy, with high degrees of mitochondrial adaptation associated with a milder clinical presentation of the disease, prolonged gestation, and improved fetal growth profiles. Figure was created by Biorender.com.
Figure 1.
Mitochondrial Dysfunction as a Common Feature across all PE subclasses. PE disease driven by either placental malperfusion, chronic inflammation at the utero-placental interface and/or poor gestational-parent adaptation likely all demonstrate altered mitochondrial respiration, increase in ROS production and mitochondrial fragmentation within the placenta and across numerous gestational-parent tissues. The inherent ability for the mitochondria to adaptation during pregnancy to various pathological insults may in part dictate the clinical outcome of the pregnancy, with high degrees of mitochondrial adaptation associated with a milder clinical presentation of the disease, prolonged gestation, and improved fetal growth profiles. Figure was created by Biorender.com.
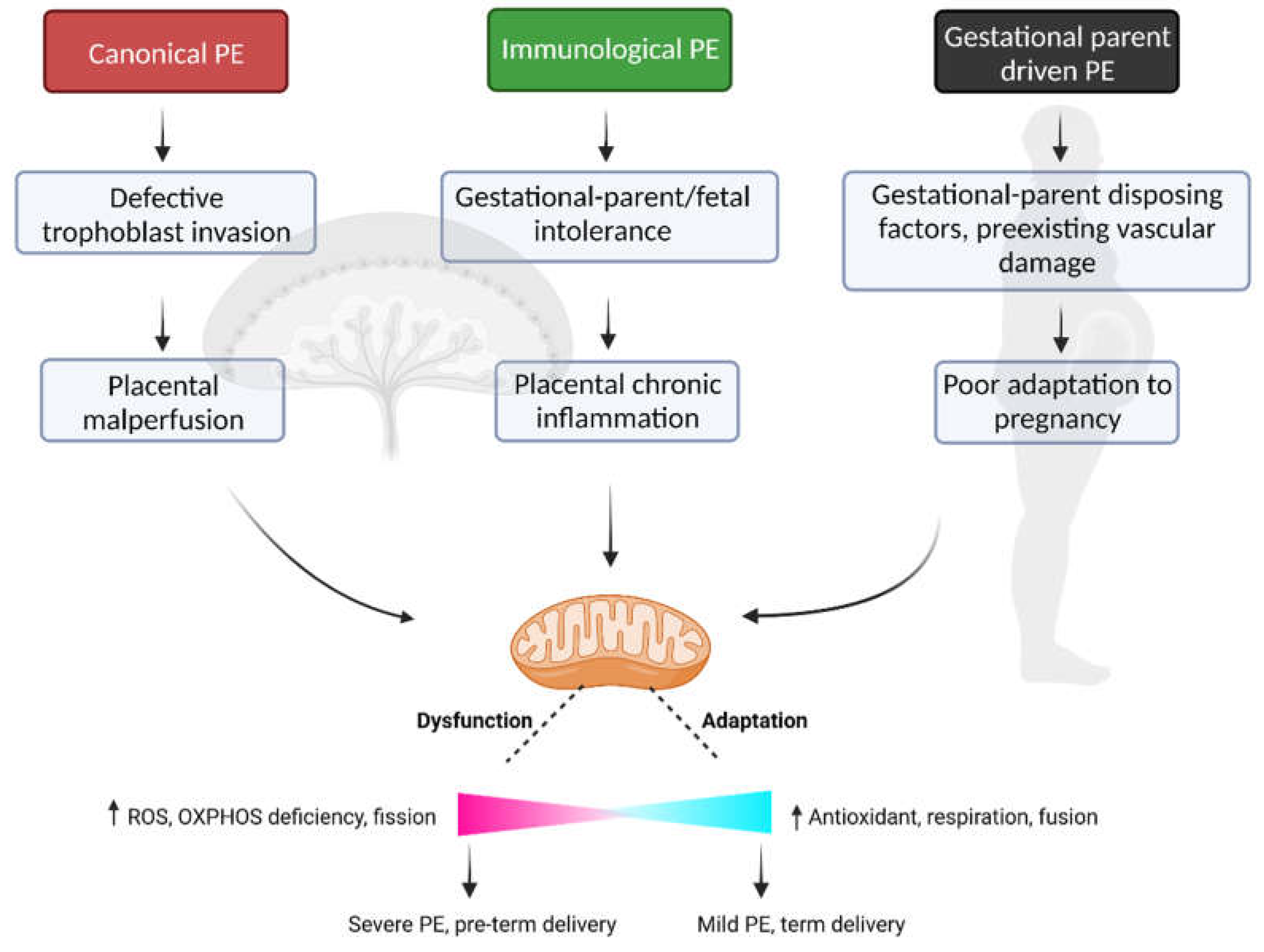
Figure 2.
Mitochondria targeting treatments in preeclampsia. Among the drugs and nutraceuticals tested in PE studies, the most promising ones that target mitochondria are metformin, resveratrol, nicotinamide (NAM) and mitoQ/mitoTEMPO. Metformin, resveratrol, and NAM function on multiple pathways that improve mitochondrial biogenesis, function, quality control (by mitophagy), and reduces ROS; thereby establishing mitochondrial homeostasis under pathological conditions. MitoQ/mitoTEMPO act by reducing mitochondrial ROS which eventually improve mitochondrial homeostasis. Figure was created by Biorender.com.
Figure 2.
Mitochondria targeting treatments in preeclampsia. Among the drugs and nutraceuticals tested in PE studies, the most promising ones that target mitochondria are metformin, resveratrol, nicotinamide (NAM) and mitoQ/mitoTEMPO. Metformin, resveratrol, and NAM function on multiple pathways that improve mitochondrial biogenesis, function, quality control (by mitophagy), and reduces ROS; thereby establishing mitochondrial homeostasis under pathological conditions. MitoQ/mitoTEMPO act by reducing mitochondrial ROS which eventually improve mitochondrial homeostasis. Figure was created by Biorender.com.
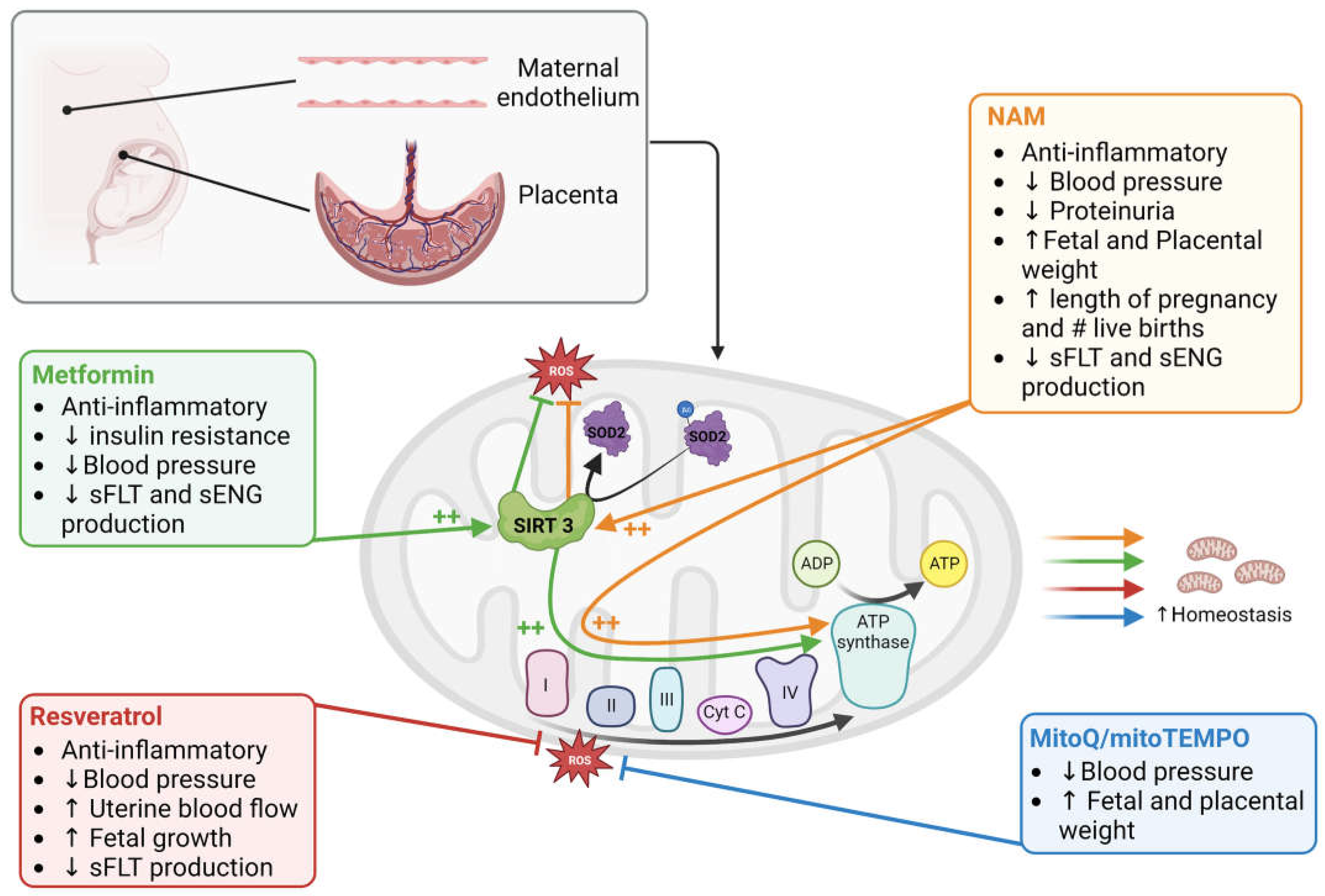
Table 1.
overarching trends of placental mitochondria content, structure/dynamics and function in healthy pregnancies (early and late) and PE pregnancies (early onset vs late onset).
Table 1.
overarching trends of placental mitochondria content, structure/dynamics and function in healthy pregnancies (early and late) and PE pregnancies (early onset vs late onset).
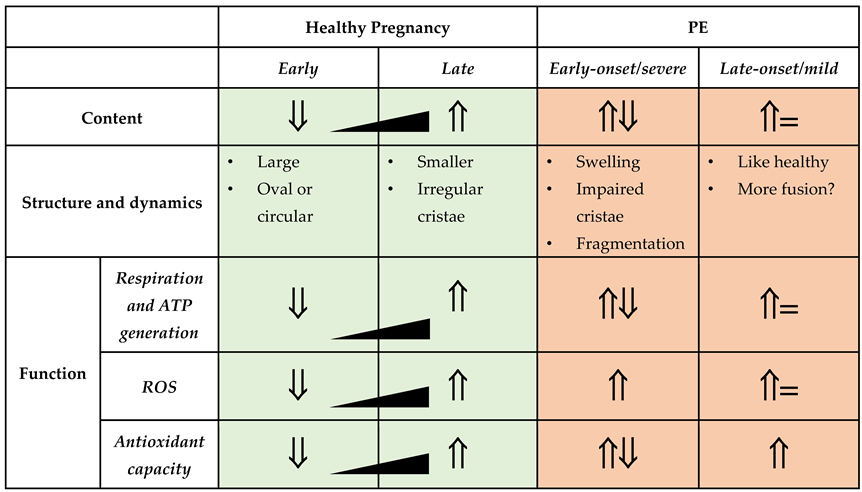 |
* In healthy pregnancy, comparison was between early and late gestation. In PE complicated pregnancy, comparison was with healthy term or preterm pregnancy. The symbol “=” was used when the finding was similar to healthy control.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



