
Submitted:
01 February 2023
Posted:
03 February 2023
You are already at the latest version
Abstract
Untargeted metabolomics is a powerful tool for measuring and understanding complex biological chemistries. However, employment, bioinformatics and downstream analysis of mass spectrometry (MS) data can be daunting for inexperienced users. Numerous open-source and free-to-use data processing and analysis tools exist for various untargeted MS approaches, including liquid chro-matography (LC), but choosing the ‘correct’ pipeline isn’t straight-forward. This tutorial, in con-junction with a user-friendly online guide presents a workflow for connecting these tools to process, analyse and annotate various untargeted MS datasets. The workflow is intended to guide explor-atory analysis in order to inform decision-making regarding costly and time-consuming down-stream targeted MS approaches. We provide practical advice concerning experimental design, organisation of data and downstream analysis, and offer details on sharing and storing valuable MS data for posterity. The workflow is editable and modular, allowing flexibility for updated/ changing methodologies and increased clarity and detail as user participation becomes more common. Hence, the authors welcome contributions and improvements to the workflow via the online repository. We believe that this workflow will streamline and condense complex mass-spectrometry approaches into easier, more manageable, analyses thereby generating opportunities for researchers previously discouraged by inaccessible and overly complicated software.
Keywords:
metabolomics
; untargeted
; mass-spectrometry
; open-source
; bioinformatics
1. Introduction
With the advent of remote working, it became apparent that researchers conducting untargeted metabolomics analysis required resources to learn how to process mass spectrometry data remotely. After over a decade of experience with proprietary software, the challenge was to address a number of issues with current common practices and embrace the open-source approach to metabolomics data processing and analysis that can have a future legacy.
Notably, all software approaches discussed here are free, as the authors believe it is important that the discussed pipelines are accessible to all. This resource will not only address the growing need for tutorials on untargeted metabolomics workflows but will also improve over time so that it can be integrated into full multi-omics workflows, as highlighted by [1].
The newly developed workflow presented here converts mass spectrometry data, specifically from Waters (Wilmslow, UK) instruments to an open format for experiments in which there are no predefined molecules to be compared between two or more classes (groups) of samples. Untargeted metabolomics workflows seek to inform differences between two or more classes (groups) of samples.
Untargeted metabolomics is an increasingly popular tool for identifying perturbations within a metabolome and revealing phenotypic complexity in systems [1,2,3,4]. It is commonly the first part of a two-step research pipeline, where untargeted studies are used to gather information, identify the metabolome, and generate hypotheses. This is followed by targeted metabolomics which measures specific compounds and requires a priori knowledge of the whole metabolome [1,4,5].
There are many sources of variation throughout the process of metabolomics as well as several different methods and tools used by the community [6,7,8]. This variation can stem from sampling methods, machinery and model used, analytical methods employed and the deficit of standardised guidelines [9,10,11]. The aim is to provide a guide which will help to move towards standardised methodology and comparable research across the field of metabolomics. As a collaborative, developing and open-source resource, the hope is that it will be widely used.
Untargeted metabolomics can produce huge quantities of multi- dimensional data, which is difficult to visualise. This guide aims to navigate through all the data and jargon. It is worth noting, however, that an untargeted approach is intended for forming hypotheses, rather than being hypothesis-driven. The workflow aims to address the question:
Which compounds might be responsible for the difference in metabolomic fingerprint between the classes (groups) of samples?
There may not be a definitive difference or unquestionable compound identification from this workflow. Rather, it will direct further research and potential compounds to focus on for targeted analysis.
2. Results
2.1. How to use this guide
This tutorial guides the user through the untargeted metabolomics workflow that has been developed with some explanation of what each stage achieves. Further details are available in step-by-step guides on the associated website (https://untargeted-metabolomics-workflow.netlify.app/ accessed on 27 January 2023), which includes links to relevant open-source tools, and our own interoperable code where appropriate. This tutorial covers the steps required to process LC-ESI-MS data, however detailed instructions for processing MALDI-ToF-MS and DI-ESI-MS using similar open-source tools are also available on the associated website.
2.1.1. Stages of an untargeted metabolomics workflow
The workflow has been divided into stages. The following number codes are used in the online guide as well as in the R code and workflow diagram (for an abridged version of this diagram see Figure 1).
00. Overviews, workflow diagram & useful information
01. Metabolite extraction
02. Data acquisition (Mass Spectrometry)
03. Converting data to open format
04. Data pre-processing
05. Extracting & formatting peak table & metadata
06. Multivariate analysis (PCA) & further analysis (if applicable)
07. Putative metabolite identification
08. Archiving data & citing resources
Stages 01 and 02 are not covered in great detail in this documentation which focuses primarily on data processing and analysis.
2.1.3. Assumptions
For this workflow to function, the user must have:
- a basic understanding of R and RStudio, and of metabolomics technologies in general (experienced R users are also encouraged to consider tools such as the RforMassSpectrometry initiative https://rformassspectrometry.org accessed on 27 January 2023);
- access to the internet and remote access to raw data;
- used Waters mass spectrometers and MassLynx software to obtain data (or equivalent steps and outcomes from other instruments);
- access to a sample list from MassLynx and treatment information (i.e. metadata, though our code can help format this);
- files with unique identifiers, ideally in the format of the following example:
- experiment-identifier_001.raw
- experiment-identifier_002.raw
- experiment-identifier_003.raw
Dealing with technical replicates (each with their own .raw file) is addressed in the associated website (https://untargeted-metabolomics-workflow.netlify.app/ accessed on 27 January 2023).
2.1.4. Experimental structure
Difficulties in analysis and/ or workflows can arise from complexities in experimental structure. Many terms are used interchangeably in different contexts. Most tools for untargeted metabolomics are set up for one factor analysis with two or three levels e.g.
- case vs control
- wild-type vs transgenic line
- Strain 1 vs. strain 2 vs. strain 3
However, more complex experimental designs are quite often implemented e.g.
- Two factors with two or more levels in each such as +/- treatment for two strains
- Time course for one or two factors such as +/- treatment for two strains over three time points
To begin, consider the expectations of which groups of metabolite fingerprints may differ from one another, and to what extent. Think logically about the questions the analysis is required to answer and how the data may be classified.
- What are the biological replicates being analysed and are they independent of each other (or has the same organism/ population been sampled multiple times)?
- Are there technical replicates (i.e. repeated runs of the same sample)?
- Are Quality Control (QC) samples required? Are analytical standards needed? (See box 2.1.5)
- What groupings are required to answer the research questions outlined?
It is recommended to get the meta-data (e.g. treatment information) organised early (See 2.1.6).
2.1.5. Quality Control
Quality control (QC) can mean different things to researchers from different fields. For quality control of your mass spectrometry run, there are a few simple options for checking that there has not been subtle (or not so subtle) variation accumulating during the run. Decisions must be made on which one (or more) of these are necessary depending on the type of sample to be analysed and the MS techniques employed:
- Spike all prepared samples with a compound for which the m/z (and RT) is known and which is unlikely to be otherwise present in the experimental samples;
- Prepare a pooled QC sample from an aliquot of each of the samples and include this at regular intervals in the MS run;
- Include blanks and/ or extraction blanks at regular intervals in the MS run;
- Use lock mass calibration (for Waters instruments).
There are some basic data quality control steps you can take to limit errors during processing and analysis:
- Check file sizes of .raw files across the MS run;
- Check file sizes of converted .mzML files - reconvert any that are unexpected;
- Compare spectra between technical replicates
2.1.6. Nice, neat metadata for analysis
To process and analyse data using our workflow, two .csv files are required (these can be created in excel, R, google sheets etc. depending on preference) as long as the order and headings of the columns follow the pattern detailed below.
For samplelist.csv the following columns are required (which can be obtained from the MassLynx Sample List generated by the software that controls the MS run):
- “Filename”: this is a list of the filenames of the .mzml files (the part before the .mzml)
- “Filetext”: this is the name that has been manually added to the metadata of that sample in MassLynx (this can be found at $$ SampleID: in the _HEADER.txt file of the original .RAW folder if it is not already known)
- “MSFile” or an equivalent column that contains either “pos” or “neg” within it Any other columns will be ignored in this file
For treatments.csv at least two columns are required (but it can include as many as necessary to describe the metadata of your experiment):
- “Filetext”: this must contain all the distinct values of “Filetext” from
- samplelist.csv
- “Variable1”: the naming of this column is left to the user (but spaces are to be avoided: instead use “-” or “_"). For example, in an MS run comparing a wild-type to a control for example, this column could be named “treatment” and filled with “WT” and “C” as appropriate
- “Variable2” etc: further variables. This may include batch identifiers (for example if many samples were run over multiple days), treatments or environmental variables
These are kept in a folder with the .mzml data files. Examples can be found on the website at https://untargeted-metabolomics-workflow.netlify.app/03_conversion-to-open-format/05_samples-treatments/ accessed on 27 January 2023.
2.2. Metabolite extraction and data acquistion
Details of quenching, metabolite extraction or choice of mass spectrometry platform are not covered here, as they will likely be specific to the organism and/ or tissue involved and the questions being addressed. Figure 2 provides a conceptual overview of metabolite extraction and data acquisition from plant tissues. See [12,13,14,15] for introductory guidance and [16] for a specific metabolite extraction method appropriate to plant tissues for this workflow.
2.3. Converting data to open format using Proteowizard
Converting .RAW files (which contain a large amount of data and metadata about the run in separate files) to a more manageable format, such as .mzML (the standard open-data format for mass spectrometry [17]) is essential. .RAW files are specific to Waters software and are not compatible with many open-source tools. To convert .RAW to .mzML, Proteowizard software [18] is used. This comprises two applications: SeeMS and MSConvert.
SeeMS is useful for viewing chromatograms and spectra without access to proprietary software like MassLynx. MSConvert performs conversion of the MS data but depending on the type of MS used, different settings/ parameters in MSConvert may be required, detailed in the online step-by-step instructions to complete stage 03: (https://untargeted-metabolomics-workflow.netlify.app/03_conversion-to-open-format/03_msconvert-lcms/ accessed on 27 January 2023).
It is critically important to check the size of .mzML files once converted. They should all be similar. SeeMS can be used to check any that seem unusual and reconvert any with an incongruous file size (problems in conversion can arise, for instance from intermittent internet connection when converting files from a remote drive).
2.4. Preprocessing data
Untargeted metabolomics datasets can be huge! To get from compressed (but still huge) .mzML files to a tractable peak table that can be interrogated with multivariate statistics, it is necessary to do a little bit of data "tidying".
A peak table is a data-frame consisting of aligned spectra with concentration or intensity values against a set of features - mass to charge ratio (m/z) or m/z with retention time (RT). The file size will be dependent on sample number but will be smaller than the .mzML files.
Different downstream tools for multivariate statistics will require the peak table in slightly different formats, so the code included in this guide will help with formatting for some common uses (e.g. MetaboAnalyst one factor and two factor peak tables) as well as helping format treatment information as metadata so that peak tables can be interrogated.
Depending on the MS approach, different stages are involved but they broadly fall into:
- baseline correction and/ or noise reduction (estimating what part of the detected intensity is the sample and “cleaning away” or adjusting the spectra to show only the signal believed to be associated with the sample);
- normalisation and/ or standardisation (these can mean a range of different things to different people but broadly cover accounting for differences in sample volume or concentration or total intensity of the signal);
- grouping and peak picking (wave-form algorithms are used to determine which parts of the spectra constitute separate peaks utilising their m/z value);
- alignment or peak matching (assessing across samples to determine whether peaks with slightly different m/z values are the same peak so that samples can be compared more reliably).
By the end of this stage, data will be processed into a single table containing all the m/z and intensity values required for down-stream analysis. This stage relies on the use of open-source software (XCMS online [19] for LC-ESI-MS and MassUp [20] for MALDI-ToF-MS and DI-ESI-MS) to process the data. These provide user interfaces for well-documented R packages (XCMS [21] and MALDIquant [22] respectively) and provide the advantage of coping well with large datasets and, in the case of XCMS online, being run remotely.
For detailed instructions on pre-processing, consult stage 04 of our online guide (https://untargeted-metabolomics-workflow.netlify.app/04_data-preprocessing/ accessed on 27 January 2023).
To extract a peak table from pre-processed data, use code provided in stage 05 of our online guide (https://untargeted-metabolomics-workflow.netlify.app/05_extracting-formatting-peak-table/ accessed on 27 January 2023).
2.5. Multivariate analysis
There are often two key questions when analysing a new untargeted metabolomics dataset:
- Are the metabolomic fingerprints distinct classes (treatment groups) different from each other?
- Which features of the metabolomic fingerprint are causing them to be different from each other?
To answer the first question, data ordination, for instance using Principal Component Analysis (PCA), provides an unsupervised approach (the model is unaware of the classes to which the samples belong). A PERMANOVA can be used to provide statistical corroboration of patterns observed in the PCA. If clear differences between classes in the PCA are apparent, then pairwise differences between classes (treatment groups) can be investigated via exploring the loadings or using a pairwise analysis such as t-tests or volcano plots.
For a subsequent supervised analysis, OPLS-DA (orthogonal projections of latent structures) will accentuate the differences between any two classes, which will typically present “strong” differences between two randomly assigned classes. To limit false-positives it is important to consider the native separation in the data (i.e. through an unsupervised ordination, like PCA) for a robust biological justification for comparing two particular classes.
In the online guide, demonstration is given on how to perform these analyses using a free online platform and how to run some alternative code in R. MetaboAnalyst [23] is an online platform on which untargeted metabolomics data can be loaded, normalised, analysed and visualised. However, there is a strong emphasis on detailed statistics that may be more appropriate for targeted analyses, so the user must have a clear understanding of their objectives in choosing amongst the options.
MetaboAnalyst is interoperable with R and the underlying code can be accessed using the button at the top left of the “Results” page. Examples of figures produced with this approach can be found in Figure 3. The advantage of running the code is that the user can integrate it with other analyses (and formatting for figures). In contrast, the advantage of MetaboAnalyst is that it guides the user through the process and has some useful sense-checks and vignettes available.
Details can be found via the excellent tutorials and documentation provided by MetaboAnalyst [24].
It is also possible to analyse the same peak tables using SIMCA (Umetrics) or other proprietary softwares. However, it is much harder (and more costly) to use these remotely, and it is harder to document any analysis for sharing with other researchers. Other software worth considering includes MSDial, MetaboKit and MeV [25,26,27].
2.6. What are my metabolites?
It is very important to consider that this stage of the metabolomic process is not automated and can be incredibly time-consuming and challenging to do, so it is advisable that the preceding analysis has been adequately assessed for its effectiveness before committing time at this stage.
Annotating metabolomic features is challenging - there are some automated annotations included with e.g. XCMS that rely on the CAMERA package [28] amongst others. However, these often struggle with unusual experimental structures and/ or large datasets, or “unusual” (i.e. non-human) metabolites. Thus, reducing the number of metabolomic features to those that are causing a significant (in terms of reliability and magnitude) difference between two classes of samples is advisable.
To ascertain the identity of these features, comparing the m/z (or m/z at specific RT) values highlighted by multivariate analysis with databases of reference m/z and with experimental data from the literature (usually available in a publication or in repositories like MetaboLights [29] and Metlin [30]) is key.
Stage 07 of the online guide provides guidance on using a range of databases to help annotate “metabolites of interest” (https://untargeted-metabolomics-workflow.netlify.app/07_putative-metabolite-id/ accessed on 27 January 2023). These include:
- METLIN to search by m/z;
- KEGG PATHWAY and KEGG COMPOUND [31] to corroborate likelihood of detecting certain compounds in the study organism/ sample and to gain insight on biological function;
- Data repositories such as MetaboLights;
- Reporting Metabolomics Standards Initiative (MSI) identification levels (see also [36]).
2.7. Sharing metabolomics data
Metabolomics data from even a small study can be very large. It can also be very complex. But there are ways of sharing it with the wider scientific community (and indeed the public) without too much trouble. It is insufficient to only prepare a data availability statement or simply share graphs or peak tables.
Metabolomics data can be analysed in lots of different ways, so it is important to comply with the FAIR principles [37]:
- Findable
- Accessible
- Interoperable
- Reusable
Institution-based data repositories are an option, but they often require extra levels of support to submit large datasets and there is no guarantee that access to other researchers is feasible.
More useful is a field-specific repository where data will be made available together with other relevant data sets. Furthermore, these repositories provide guidance on appropriate data formatting, allowing it to be compatible with other published data to form part of potential future meta-analyses. Some journals will have specific guidelines on which repository to use [38].
Set aside time at the start of any project for submitting data to a repository. It is not optional!
2.7.1. MetaboLights repository
MetaboLights is a data repository specific to metabolomics studies [29]. Data from NMR, GC-MS, LC-MS, and MALDI amongst others, may be submitted.
The repository is maintained and curated by the European Bioinformatics Institute (EMBL-EBI) meaning that the data it holds is well-formatted and integrated with several other standardised databases and ontologies (ways of describing methods, data and metadata). This “future-proofs” the data stored, making it not only open-access but also more findable and reusable, as well as facilitating integration with other -omics data, if required.
MetaboLights has various stages of submission, validation and then curation by experts to make sure each submission has all the relevant metadata needed to recreate the analysis undertaken. Following curation, there is a review process and finally data can be added to the repository and made available.
Because of the curation process, there can be a significant lag between submission and data being available so early submission is advisable. However, once submitted, there is a reference that can be linked to any publication.
Account creation is required, after which, a video tutorial guide on using the submission portal is available. Additional hints and tips on this can be found on the associated website (https://untargeted-metabolomics-workflow.netlify.app/08_data-archiving-citation/02_metabolights/ accessed on 27 January 2023).
2.8. Citation of the tools used in the workflow
It is important to cite the version used and/ or the date accessed as these tools and repositories are regularly updated:
- Cite all R packages used in our functions (see function at start of each R code to produce list of references)
- Use citation(R) and RStudio.Version() to get the version information for R and RStudio that you have used for analysis;
- Proteowizard (SeeMS and MSConvert) citation;
- Up to date Metaboanalyst citation;
- Up to date XCMS online and METLIN citations;
- MassUp citation;
- MassBank citation (include access date);
- Up to date ECMDB citation (don’t forget any other organism specific metabolite databases used);
- Up to date KEGG citation (including BRITE, COMPOUND and PATHWAY);
- Up to date PubChem citation;
- Write a data availability statement in any publication that links to your archived data in MetaboLights.
Conclusions
At this point the choice in preparing and analysing metabolomics data is at the discretion of the research group. This guide is a useful starting point that leads the reader through an openly available, best-practice, pipeline. Complex data and analytical processes can be overwhelming, but by engaging in discussion forums, sharing ideas, troubleshooting, and having access to a community of like-minded researchers these processes can become more accessible and facilitate exploration of exciting biological questions.
Author Contributions
Conceptualization, E.J.P and D.D.C.; software, E.J.P, J.K.P., J.N.P., R.M.G.; resources, D.D.C.; writing—original draft preparation, E.J.P and K.C.B.; writing—review and editing, K.C.B., N.A., A.C., D.H., J.A.L., J.N.P., H.J.W., A.W.; supervision, H.J.W and D.D.C.; project administration, E.J.P.; funding acquisition, E.J.P and D.D.C. All authors have read and agreed to the published version of the manuscript
Funding
This work was funded by BBSRC, grant numbers BB/M011151/1, and BB/T010789/1. The project was supported by a small grant from the University of Sheffield Library's Unleash Your Data and Software Competition.
Data Availability Statement
No new data were created or analysed in this study. Research software described in this article is available at https://github.com/LizzyParkerPannell/Untargeted_metabolomics_workflow accessed on 27 January 2023. The associated online guide is available at https://untargeted-metabolomics-workflow.netlify.app/ accessed on 27 January 2023.
Acknowledgments
With thanks to Erika Hansson, Sophia van Mourik, Emily Magkourilou and Harry Wright for their feedback on the content of the website and to Tim Daniell and Giles Johnson for encouraging the sharing of the workflow. Many thanks to Neil Shephard and Robert Turner for their technical help and guidance in developing the website.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Allwood, J.W.; Williams, A.; Uthe, H.; van Dam, N.M.; Mur, L.A.J.; Grant, M.R.; Pétriacq, P. Unravelling Plant Responses to Stress—The Importance of Targeted and Untargeted Metabolomics. Metabolites 2021, 11(8), 558. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Cravatt, B.F.; Siuzdak, G. The expanding role of mass spectrometry in metabolite profiling and characterization. ChemBioChem 2005, 6(11), 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.M.; Ehmann, D.E.; Mills, S.D.; Perros, M.; Barrett, M.P. Untargeted metabolomics to ascertain antibiotic modes of action. Antimicrobial Agents and Chemotherapy 2016, 60(4), 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, A.; Gelzo, M.; Stornaiuolo, M.; Ruoppolo, M.; Castaldo, G. The evolving landscape of untargeted metabolomics. Nutrition, Metabolism and Cardiovascular Diseases 2021, 31(6), 1645–1652. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Jasbi, P.; Shi, X.; Turner, C.; Hrovat, J.; Liu, L.; Rabena, Y.; Porter, P.; Gu, H. Early Breast Cancer Detection Using Untargeted and Targeted Metabolomics. J. Proteome Res 2021, 20, 3133. [Google Scholar] [CrossRef]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies—Challenges and Emerging Directions. Journal of the American Society for Mass Spectrometry 2016, 27(12), 1897–1905. [Google Scholar] [CrossRef]
- Dudzik, D.; Barbas-Bernados, C.; García, A.; Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. a review. Journal of Pharmaceutical and Biomedical Analysis 2018, 147, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Rainer, J.; Vicini, A.; Salzer, L.; Stanstrup, J.; Badia, J.M.; Neumann, S.; Stravs, M.A.; Verri Hernandes, V.; Gatto, L.; Gibb, S.; Witting, M. A Modular and Expandable Ecosystem for Metabolomics Data Annotation in R. Metabolites 2022, 12, 173. [Google Scholar] [CrossRef] [PubMed]
- Blaženović, I.; Kind, T.; Ji, J.; Fiehn, O. Software tools and approaches for compound identification of LC-MS/MS data in metabolomics. Metabolites 2018, 8(2), 31. [Google Scholar] [CrossRef]
- Misra, B.B. New tools and resources in metabolomics: 2016–2017. Electrophoresis 2018, 39(7), 909–923. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Current Opinion in Biotechnology 2019, 55, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Jorge, T.F.; Mata, A.T.; António, C. Mass spectrometry as a quantitative tool in plant metabolomics. Philos Trans A Math Phys Eng Sci 2008, 374(2079), 20150370. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Su, X; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annual Review of Biochemistry 2017, 86(1), 277–304. [Google Scholar] [CrossRef] [PubMed]
- Pezzatti, J.; Boccard, J.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Picard, D.; González-Ruiz, V.; Rudaz, S. Implementation of liquid chromatography-high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: a tutorial. Anal. Chim. Acta 2020, 1105, 28–44. [Google Scholar] [CrossRef]
- Villate, A.; San Nicolas, M.; Gallastegi, M.; Aulas, P.-A.; Olivares, M.; Usobiaga, A.; Etxebarria, N.; Aizpurua-Olaizola, O. Metabolomics as a Prediction Tool for Plants Performance under Environmental Stress. Plant Sci. 2021, 303, 110789. [Google Scholar] [CrossRef] [PubMed]
- Austen, N.; Walker, H.J.; Lake, J.A.; Phoenix, G.K.; Cameron, D.D. The Regulation of Plant Secondary Metabolism in Response to Abiotic Stress: Interactions Between Heat Shock and Elevated CO2. Frontiers in Plant Science 2019, 10, 1463. [Google Scholar] [CrossRef]
- Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W.H.; Römpp, A.; Neumann, S.; Pizarro, A.D., Montecchi-Palazzi, L.; Tasman, N.; Coleman, M.; Reisinger, F.; Souda, P.; Hermjakob, H.; Binz, P-A.; Deutsch, E.W. mzML—a Community Standard for Mass Spectrometry Data. Mol. Cell. Proteomics 2011, 10(1), R110.000133. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24(21), 2534–2536. [Google Scholar] [CrossRef]
- Forsberg, E.; Huan, T.; Rinehart, D.; Benton, H.P.; Warth, B.; Hilmers, B.; Siuzdak, G. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat Protoc 2018, 13, 633–651. [Google Scholar] [CrossRef]
- López-Fernández, H.; Santos, H.M.; Capelo, J.L.; Fdez-Riverola, F.; Glez-Peña, D.; Reboiro-Jato, M. Mass-Up: an all-in-one open software application for MALDI-TOF mass spectrometry knowledge discovery. BMC Bioinformatics 2015, 16, 318. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O'Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching and identification. Analytical Chemistry 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Gibb, S.; Strimmer, K. MALDIquant: a versatile R package for the analysis of mass spectrometry data. Bioinformatics 2012, 28(17), 2270–2271. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucl. Acids Res. 2009, 37, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Metaboanalyst tutorials: available at https://dev.metaboanalyst.ca/docs/Tutorials.xhtml (accessed on 27/01/2023).
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; van der Gheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, P.; Teo, G.; Ow, J.R.; Lau, A.; Kaldis, P.; Tate, S.; Choi, H. MetaboKit: a comprehensive data extraction tool for untargeted metabolomics. Mol. Omics 2020, 16, 436. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.; Holton, K.; Nair, S.; Schlauch, D.; Sinha, R.; Quackenbush, J. MeV: MultiExperiment Viewer. In Biomedical Informatics for Cancer Research; Ochs, M., Casagrande, J., Davuluri, R., Eds.; Springer, Boston, MA. 2010; pp. 267-277. [CrossRef]
- Kuhl, C; Tautenhahn, R; Boettcher, C; Larson, T.R.; Neumann, S. CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Analytical Chemistry 2012, 84, 283–289. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Research 2020, 48(D1), D440–D444. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Analytical Chemistry 2018, 90(5), 3156–3164. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. KEGG Bioinformatics Resource for Plant Genomics and Metabolomics. In Plant Bioinformatics; Methods in Molecular Biology; Edwards, D., Eds. Humana Press, New York, NY, 2016; volume 1374. [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; Oda, Y.; Kakazu, Y.; Kusano, M.; Tohge, T.; et al. MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; Zaslavsky, L.; Zhang, J.; Bolton, E.E. PubChem 2023 update. Nucleic Acids Res. 2023, 51(D1), D1373–D1380. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Billington, R; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; Krummenacker, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Research 2014, 42(1), D459–D471. [Google Scholar] [CrossRef] [PubMed]
- The Metabolomics Workbench: available at https://www.metabolomicsworkbench.org/ (accessed on 27/01/2023).
- Sumner, L.W.; Lei, Z.; Nikolau, B.J.; Saito, K.; Roessner, U.; Trengove, R. Proposed quantitative and alphanumeric metabolite identification metrics. Metabolomics 2014, 10, 1047–1049. [Google Scholar] [CrossRef]
- Wilkinson, M.; Dumontier, M.; Aalbersberg, I.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data 3 2016, 160018. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; Heinemann, M.; Link, H.; Luo, J.; Neumann, S.; Nielsen, J.; Perez de Souza, L.; Saito, K.; Sauer, U.; Schroeder, F.C.; Schuster, S.; Siuzdak, G.; Skirycz, A.; Sumner, L.W.; Snyder, M.P.; Tang, H.; Tohge, T.; Wang, Y.; Wen, W.; Wu, S.; Xu, G.; Zamboni, N.; Fernie, A.R. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat Methods 2021, 18, 747–756. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Workflow diagram for processing and analysis of untargeted LC-MS metabolomics data. a) sample selection and preparation. b) Mass spectrometry analysis of samples. c) Conversion of data to open format. d) Data pre-processing and (e) production of a feature matrix with experimental information included. f) Statistical analysis for selection of features of interest and (g) identification of features of interest by comparison with literature and existing metabolite databases.
Figure 1.
Workflow diagram for processing and analysis of untargeted LC-MS metabolomics data. a) sample selection and preparation. b) Mass spectrometry analysis of samples. c) Conversion of data to open format. d) Data pre-processing and (e) production of a feature matrix with experimental information included. f) Statistical analysis for selection of features of interest and (g) identification of features of interest by comparison with literature and existing metabolite databases.
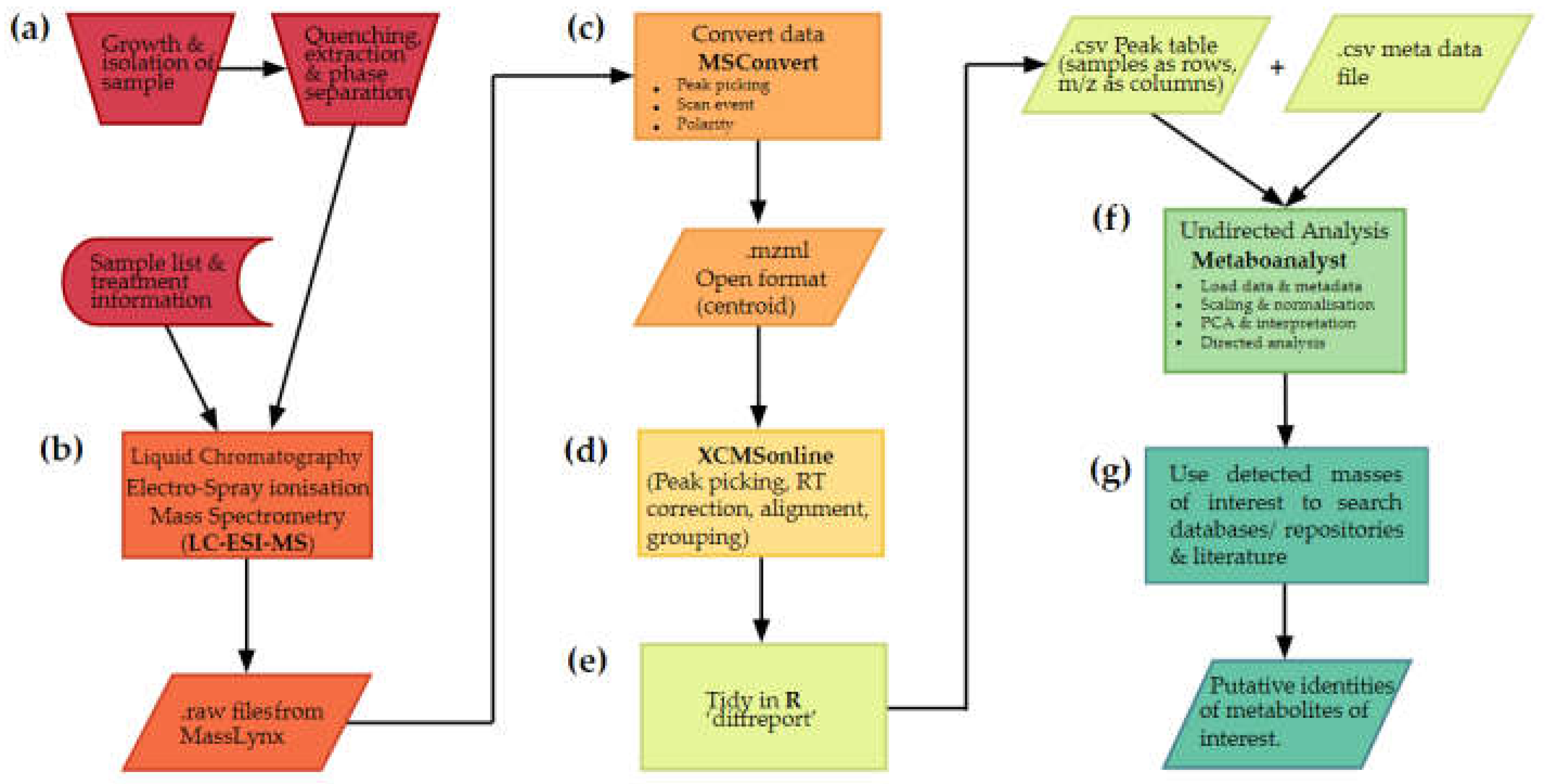
Figure 2.
Conceptual diagram of an untargeted metabolomics workflow, from leaf to mass spectrometry analysis. After sample harvest (a), metabolic reactions in a sample tissue must be first quenched (b; i.e. via liquid N immersion), the sample homogenised and the cell walls broken (c) to permit extraction of compounds within the cells using a range of solvents (d). Extracts may then be diluted and submitted to mass spectrometry analysis (e; e.g. UPLC-ESI-MS).
Figure 2.
Conceptual diagram of an untargeted metabolomics workflow, from leaf to mass spectrometry analysis. After sample harvest (a), metabolic reactions in a sample tissue must be first quenched (b; i.e. via liquid N immersion), the sample homogenised and the cell walls broken (c) to permit extraction of compounds within the cells using a range of solvents (d). Extracts may then be diluted and submitted to mass spectrometry analysis (e; e.g. UPLC-ESI-MS).
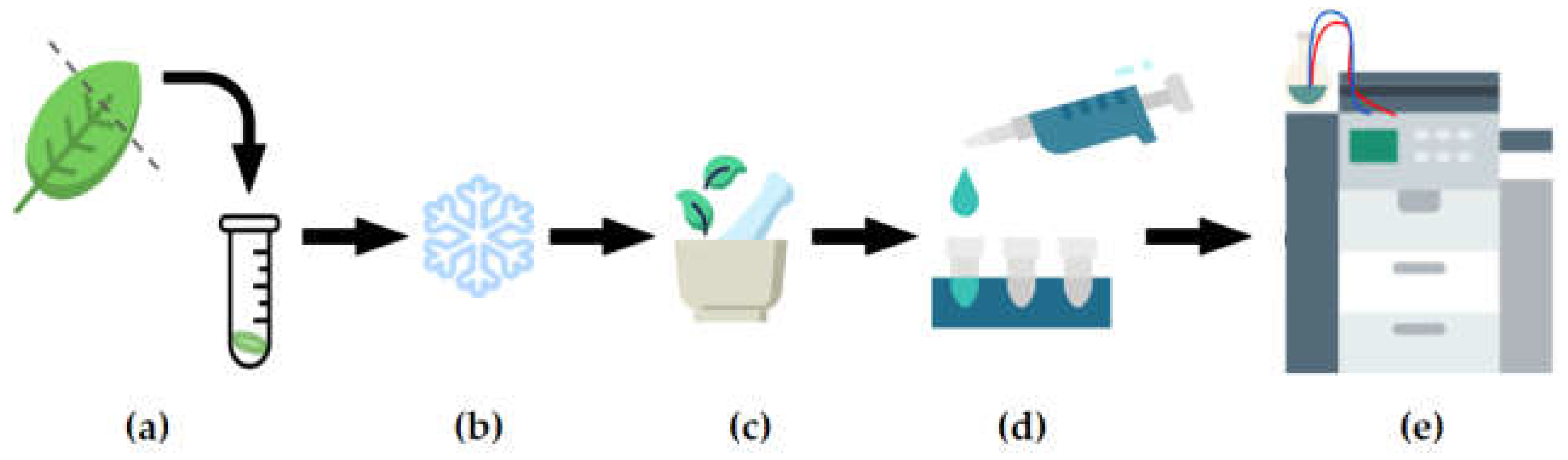
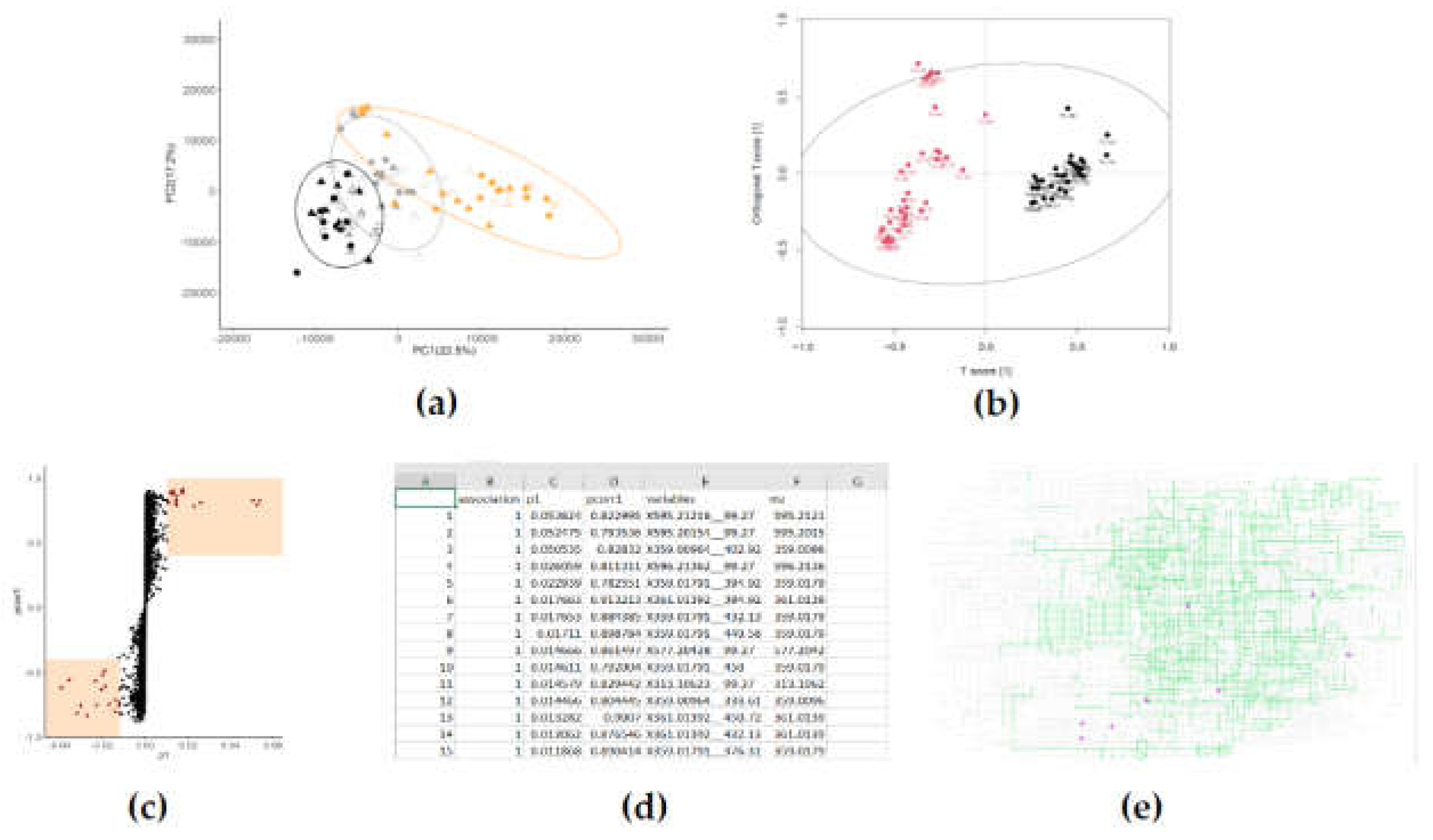
Figure 3.
Conceptual diagram of examples of multivariate analysis outputs of untargeted metabolomics analysis, all produced using open-source or freely available software. (a) Principal component analysis (PCA) 2-D scores plot produced with pcaMethods and ggplot2 packages in R; (b) OPLS-DA scores plot produced using the muma package in R; (c) scores plot created using ggplot2 package and data produced by the muma package in R; (d) example list of features of interest highlighted by an OPLS-DA using muma in R; (e) example of metabolites highlighted within a KEGG pathways global Esterichia coli metabolism map.
Figure 3.
Conceptual diagram of examples of multivariate analysis outputs of untargeted metabolomics analysis, all produced using open-source or freely available software. (a) Principal component analysis (PCA) 2-D scores plot produced with pcaMethods and ggplot2 packages in R; (b) OPLS-DA scores plot produced using the muma package in R; (c) scores plot created using ggplot2 package and data produced by the muma package in R; (d) example list of features of interest highlighted by an OPLS-DA using muma in R; (e) example of metabolites highlighted within a KEGG pathways global Esterichia coli metabolism map.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



