
Submitted:
02 February 2023
Posted:
03 February 2023
You are already at the latest version
Abstract
The human immunodeficiency virus 1 (HIV-1) viral protease (PR) is one of the most studied viral enzymes, and approval of drugs targeting its catalytic activity opened the door to the develop-ment of highly active antiretroviral therapy (HAART). Despite the fact that its crucial role in viri-on maturation is well characterized, an increasing body of research is starting to focus on its abil-ity to cleave host cell proteins, based on recent advances in proteomics and genomics technologies. Such findings are apparently in contrast with the dogma of HIV-1 PR activity being restricted to the interior of nascent virions, and suggest catalytic activity within the host cell environment. Given the limited amount of PR present in the virion at the time of infection, it is tempting to specu-late that such events mainly occur during viral late gene expression, mediated by newly synthe-sized Gag-Pol polyprotein precursors, rather than at a very early stage of infection, before pro-viral integration. Among cellular targets of HIV-1 PR, three major clusters can be identified: pro-teins involved in viral and cellular translation, those controlling cell survival, and restriction fac-tors responsible for innate/intrinsic antiviral responses. Indeed, by cleaving host cell translation initiation factors HIV-1 PR can impair cap-dependent translation, thus promoting IRES-mediated translation of late viral transcripts and viral production, while by targeting several apoptotic fac-tors it modulates cell survival, thus promoting immune evasion and viral dissemination. Addi-tionally, HIV-1 PR counteracts restriction factors incorporated in the virion that would otherwise interfere with nascent virus vitality. Thus, HIV-1 PR appears to modulate host cell function at dif-ferent times and locations during its life cycle, to ensure efficient viral persistency and propaga-tion. This kind of PR-mediated host cell modulation is found in a plethora of different viruses and HIV-1 is no exception, and although we are far from having a complete picture, it is clear that the PR has a multifaceted role in interfering with host machineries to better suit viral replication, and is a field that needs to be explored further.
Keywords:
HIV-1 PR
; host factors
; host cell shut-off
; protease
; antiviral therapy
; cell death
; apoptosis
1. Introduction
The human immunodeficiency virus 1 (HIV-1) - the causative agent of the acquired immunodeficiency syndrome (AIDS) - is one of the most infamous viruses known to man. Since its adaptation to humans at the beginning of the 20th century [1], more than 84 million people have been infected, resulting in one of the largest epidemics in human history [2]. Virus isolation and characterization, together with the exponential growth in emergence of AIDS cases, ignited the quest to discover effective antiviral treatments. The first drugs to be discovered were inhibitors of the reverse transcriptase, however these alone were not able to control the infection completely, due to the rapid selection of resistant strains and high toxicity [3,4,5]. Of paramount importance in the fight against AIDS was the discovery of new antivirals capable of inhibiting the viral protease (PR), an essential enzyme encoded in the HIV-1 genome, that catalyzes the maturation of the nascent virion: a great target for therapeutic compounds development. Approval of PR inhibitors allowed to implement a combination therapy entailing the administration of multiple drugs against different viral targets, that ultimately led to a better control the infection and granted HIV-1 infected people with a life comparable to that of uninfected individuals [6,7,8].
1.1. HIV-1 PR structure and function
HIV-1 PR is a homodimeric aspartic protease composed of two 99-aminoacids monomers (Figure 1). Each monomer contains one α-helix and nine β-strands, four of which are antiparallel sheets and make up the highly-stable dimer interface, which in turn forms the active site [9]. The latter is characterized by a hydrophobic core and contains the Asp-Thr-Gly catalytic triad, common among different aspartic proteases at positions 25-27/25’-27’. Near the active site two important domains can be found: the so called “fireman’s grip” - a network of hydrogen bonds that supports the core’s rigidity - and the flaps that cover the active site (Figure 1) [10,11]. The flaps are composed by flexible β-hairpins, and control the substrate or inhibitor access to the active site depending on their conformation, making them an essential element for the modulation of the enzymatic activity. These can be found in a number of different conformations that can be summarized in: closed, semi-open and open; such forms exist in an equilibrium but it appears that the semi-open state is predominant [12]. It is thought that the flaps open to accommodate the substrate or inhibitor and then close when the PR forms a complex with such molecules and several models have been proposed for their opening and closing. For example it is believed that the curling of the flaps’ tips in the hydrophobic walls of the active site creates enough space for the substrate to fit in, and in this conformation the active site acquires a negative charge that would facilitate the interaction with positively charged substrates; finally once the substrate is in position the flaps can extend over it, thus closing the active site and allowing the enzymatic cleavage to occur [13]. Another possibility is that this curled intermediate acts as a transitioning conformation between the closed and semi-open state [12]. In another model instead, the interaction with the ligand makes the flaps opening more frequent and stable, and when the substrate is in the correct position in the active site, the flaps switch to a closed state until cleavage occurs [14].
1.2. Sequence specificity
A consensus cleavage sequence for the HIV-1 PR has yet to be defined. Indeed, its target sequences present in the viral Gag and Gag-Pol polyproteins differ significantly from one another and are structurally asymmetric. Studies on such cleavage sites showed that the PR obtains its substrate specificity by recognizing shape and volume rather than a specific amino acid stretch. Therefore, cleavage is not dictated by the sequence itself but is influenced by both substrate dynamics and interactions of the PR and the ligand outside of the active site cleft. Moreover, it has been theorized that H2O molecules and hydrogen bonds facilitate the substrate tridimensional structure recognition by the PR and, interestingly, all the substrates derived from the protease cleavage show an extended and asymmetric β-strand conformation while bound to the active site, giving rise to a consensus envelope called “substrate envelope”. Furthermore, the interaction with the ligand happens both via the active site and the substrate groove (S-groove) - an active pocket present in the PR - that allows the enzyme to bind up to 24 residues [15]. While there may not be a clear consensus for the cleavage site, evidence shows that HIV-1 PR may have some preferences in the cleavage sequence: the two amino-acid residues before and after the cleavage site (P2 and P2’) are thought to be more important in recognition than the residues beyond this region, and negatively charged, hydrophobic and β-branched residues are preferred in these positions; whereas in P1 and P1’ there is a preference for large non-β branched hydrophobic and aromatic amino acids. Pro, Gly and basic residues are mainly ill-favored in all the four positions while Arg has never been observed in P2, the same is true for Pro in P1 that can instead be present in P1’ [16,17,18].
1.3. PR activation and virion maturation
PR dimerization is instrumental in modulating the activation of its catalytic activity, and dictates the place and timing of nascent virion maturation. Precursor dimers form when the protease is still embedded in the Gag-Pol polyprotein, but are less stable and therefore poorly active. Although PR precursor monomers can self-interact, it has been postulated that they reach a catalytically active conformation in only 3-5% of the cases, subsequently triggering an ordered cascade of events [19]. Due to the poor activity of the precursor dimer, the first proteolytic cuts are intramolecular and result in the release of free enzyme from the polyprotein context. Cleavage occurs at the SP1/NC interface (an internal site within the transframe region), and at the transframe/PR interface (Figure 2a). Next, the enzyme is able to remove the RT domain from its C-term, thus releasing itself from the polyprotein and acquiring a mature conformation with increased stability and activity. Finally, fully active PR dimers can start to act intermolecularly on their targets on the Gag polyprotein (Figure 2b) [20]. This activation process is therefore highly concentration dependent and is fundamental to avoid the untimely formation of mature PR dimers before the recruitment of a sufficient amount of Gag-Pol to the cell membrane, and consequent budding of the immature virion - that up to this stage is composed of approximately 2400 Gag molecules and 120 Gag-Pol molecules [18]. Before the action of the PR, the proto-HIV-1 particle is constituted by a spherical or semi-spherical shell of Gag precursor where the matrix protein (MA) domain lines the viral envelope, the capsid protein (CA) domain forms protein-protein lattice contacts, and the nucleocapsid protein (NC) domain binds the viral genome; both the immature and mature capsid are formed by hexameric units, however these lattices change drastically upon maturation and proteolytic cleavage by the viral PR (Figure 2c). This maturation process is a complex concert of different events: first, the cleavage between the spacer peptide 1 (SP1) and NC separates the latter - bound to the viral genome - from the MA-CA lattice, afterwards, MA is separated from the CA lattice, followed by cleavage at the spacer peptide 2 (SP2)/p6 interface that frees the NC from the p6 region. The final cuts are at the N-term of the spacer peptides, SP1 and SP2, releasing them from CA and NC (Figure 2) [18].
2. Cellular targets of HIV-1 Protease
Despite HIV-1 PR role in viral maturation has always been recognized as a crucial event in the virus life cycle, its involvement in cleavage of cellular factors has been greatly debated. Detection of HIV-1 polyprotein processing on the cytoplasm of HIV-1 infected cells 5 days post infection, suggested a possible activity on host cell targets at late times [21]. However, since an infectious virion brings only 120 copies of PR inside the host cell, the dependency on dimerization for PR activation makes it unlikely to have high protease activity in infected cells before the strong de novo Gag-Pol precursor synthesis [18]. Therefore, while it is difficult to hypothesize a massive activity on host factors during the early phase of infection (pre-integration), there is growing evidence that the HIV-1 PR is involved in the modulation of host cell functions at later stages of infection (post-integration). While the lack of a clear consensus cleavage sequence makes it extremely difficult to predict its possible targets inside the cell, a number of studies have applied high throughput approaches such as affinity tagging and purification mass spectrometry to identify possible viral protease targets [16,22], some of which have been extensively validated in follow-up studies, suggesting that HIV-1 PR targets a number of cellular factors to modulate host cell function, thus promoting viral replication and persistence.

Table 1.
HIV-1 PR cleavage sites validated in cell culture systems.
| Protein | Cleavage sequence |
|---|---|
| HIV-1 cleavage sites | |
| Gag (MA-CA) | SQNY^PIVQ [17] |
| Gag (CA-p2) | ARVL^AEAM [17] |
| Gag (p2-NC) | ATIM^MQRG [17] |
| Gag (NC-p1) | RQAN^FLGK [17] |
| Gag (p1-p6) | PGNF^LQSR [17] |
| Gag-Pol (TF-PR) | SFNF^PQIT [17] |
| Gag-Pol (PR-RT) | TLNF^PISP [17] |
| Gag-Pol (RT-RH) | AETF^YVDG [17] |
| Gag-Pol (RH-IN) | RKIL^FLDG [17] |
| Host protein cleavage sites | |
| RIPK1 | PQVL^YQNN [23] |
| NDR1 | KDWV^FINY [24] |
| NDR2 | KDWV^FlNY [24] |
| proCaspase8 | PKVF^FIQA [25] |
| eIF4GI | KIIA^TVLM [26] |
| ATVL^MTED [26] | |
| RFSA^LQQA [26] | |
| eIF3d | RRNM^LQFN [22] |
| Bcl2 | RRDF^AEMS [27] |
| PABP | GFVS^FERH [28] |
| PRVM^STQR [28] | |
| GCN2 | GQDY^VETV [29] |
| NF-κB1 | HYGF^PTYG [30] |
| TBK1 | SNTL^VEMT [31] |
In one of the largest HIV-1 interactome studies, Jäger et. al identified 497 viral-host protein interactions from transfected HEK293T and Jurkat cells. Among them, 67 proteins have been shown to interact with the PR (Figure 3) [22]. Similarly, in a following proteomics-based study, researchers were able to identify more than 140 putative cleavage sites for HIV-1 PR from Jurkat cell lysates incubated in vitro with bacterially expressed PR [16].
2.1. Role of the protease in protein synthesis modulation
A number of studies contributed to shine a light on the possible multifaceted role of HIV-1 PR in different steps of its viral life cycle and paved the way to in depth characterization of its cellular targets (Figure 4). The best characterized host-cell process targeted by HIV-1 PR is arguably protein synthesis: during infection several factors involved in cap-dependent translation are cleaved, resulting in a decrease of cellular cap-dependent translation, in favor of late Internal ribosome entry site (IRES)-dependent viral protein production [26,28,29,33,34]. This is also supported by a strong reduction in cellular translation observed following exogenous expression of PR in COS-7 cells [26]. Indeed, HIV-1 RNA genome harbors one IRESs within the 5’ LTR [35,36], and one in the gag gene (Figure 5) [37], which are especially important for translation of late gene products. Nucleotides 1-270 from the gag transcript have been shown to retain cap dependent translation initiation properties [35], therefore, since all spliced and unspliced HIV-1 transcripts possess the same 289 nt long 5'UTR, they could all internally initiate translation. However, the latter has been observed only from the gag, tat, vpr, vpu, vif , and nef leaders, and their activity has been shown to be highly variable [38]. While during the first 48 hours of the viral life cycle translation of viral transcripts is heavily cap-dependent, after that time point IRES-mediated translation importantly contributes to viral proteins synthesis [33]. Furthermore, addition of recombinant PR to rabbit reticulocytes for in vitro translation assays causes a 10-fold reduction of in vitro cap dependent protein synthesis while barely affecting IRES-driven translation, and a 4-fold increase in translation of a synthetic mRNA encoding for the HIV-1 5’ leader sequence, gag and pr [26].
In addition, HIV-1 infection of human leukemia T cells C8166, resulted in a decrease of total protein synthesis starting from 2 days post infection; such phenomenon is associated with both a decrease of eukaryotic translation initiation factor 4G (eIF4G) levels and the appearance of eIF4G cleavage products, and is completely inhibited by treatment with Saquinavir - thus proving the ability of the PR to cleave eIF4G [26]. Based on experiments performed incubating recombinant PR with HeLa cells extracts, eIF4G cleavage site was identified between residues 678-9, 681-2, and 1086-7, physically separating the eIF4E and eIF3 binding moieties of eIF4G, and functionally impairing its ability to participate in cap dependent translation (Figure 6) [26]. A further contribution to cap-dependent cellular translation inhibition is the HIV-1 PR-mediated cleavage of the poly(A) binding protein (PABP) at positions 237/238 and 477/478 (Figure 6). Such cleavage has been observed both in HIV-1 infected MT-2 cells and in BHK-21 cells transiently expressing the PR, and takes place in a saquinavir sensitive fashion [28]. Therefore, HIV-1 PR catalytic activity further contributes to host cell cap-dependent translation shut off, inhibiting poly(A)-dependent initiation of translation by disrupting the synergy between the poly(A) tail and the cap in cellular mRNAs [34].
Additionally, HIV-1 infection also stimulates cleavage of translation factor eIF3d which, upon transient expression of PR in HEK 293T cells, is specifically targeted between residues 114 and 115, with an efficiency close to the one of the Gag-Pol processing [22]. The physiological significance of eIF3d cleavage might extend from the cap-dependent host cell translation shut-off described above, since its knockdown boosts infectivity upon single round infection with VSV-G pseudotyped HIV-1 particles, and promotes accumulation of reverse transcription products [22], thus suggesting an antiviral role for eIF3d during the early stages of viral infection, before proviral integration (Figure 4). It has been hypothesized that eIF3d is cleaved upon viral infection by PR present in incoming virions, possibly explaining an earlier study which showed that a 1 hour pre-treatment of H9 cells with a HIV-1 PR inhibitor (UK-88,947) strongly inhibited HIV-1 DNA synthesis 18 hours post infection, thus suggesting the requirement of HIV-1 PR for optimal viral genome RT process and integration [39]. However, the importance of HIV-1 PR catalytic activity during the early phase of the viral life cycle has been heavily debated, and the role of incoming PR during HIV-1 infection is still questionable [40,41,42,43,44]. Apart from factors directly involved in translation initiation, HIV-1 PR-1 is able to cleave another protein responsible for the regulation of cellular translation: the antiviral kinase general control non-derepressible-2 (GCN2). GCN2 phosphorylates eIF2, hence halting AUG dependent translation in response to several stimuli - such as amino acid or serum deprivation, UV light irradiation and viral infection [29]. During the late phase of infection, the abundant production of viral mRNAs could trigger GCN2 activation, thus resulting in the inhibition of both cap- and IRES-dependent protein synthesis, with a detrimental effect on viral replication. Accordingly, infection of PBMC with HIV-1 results in a 45% reduction in GCN2 at 4- and 5-days post infection, which can be prevented by the addition of saquinavir. In addition, GCN2 cleavage is observed as early as 4 hours post transfection in BHK-21 cells transfected with an exogenous plasmid encoding for the HIV-1 PR [45]. By cleaving GCN2, HIV-1 PR prevents a generalized inhibition of AUG-dependent translation that would negatively impact on the viral life cycle (Figure 4). Therefore, HIV-PR is able to target multiple levels of the translation process: on the one hand it causes a decrease in cap-dependent cellular protein production, thus favoring IRES-mediated translation of unspliced viral transcripts, thanks to cleavage of eIF4G; on the other one it ensures abundant viral products translation by preserving AUG dependent translation – thanks to cleavage of GCN2.
2.2. PR and apoptosis
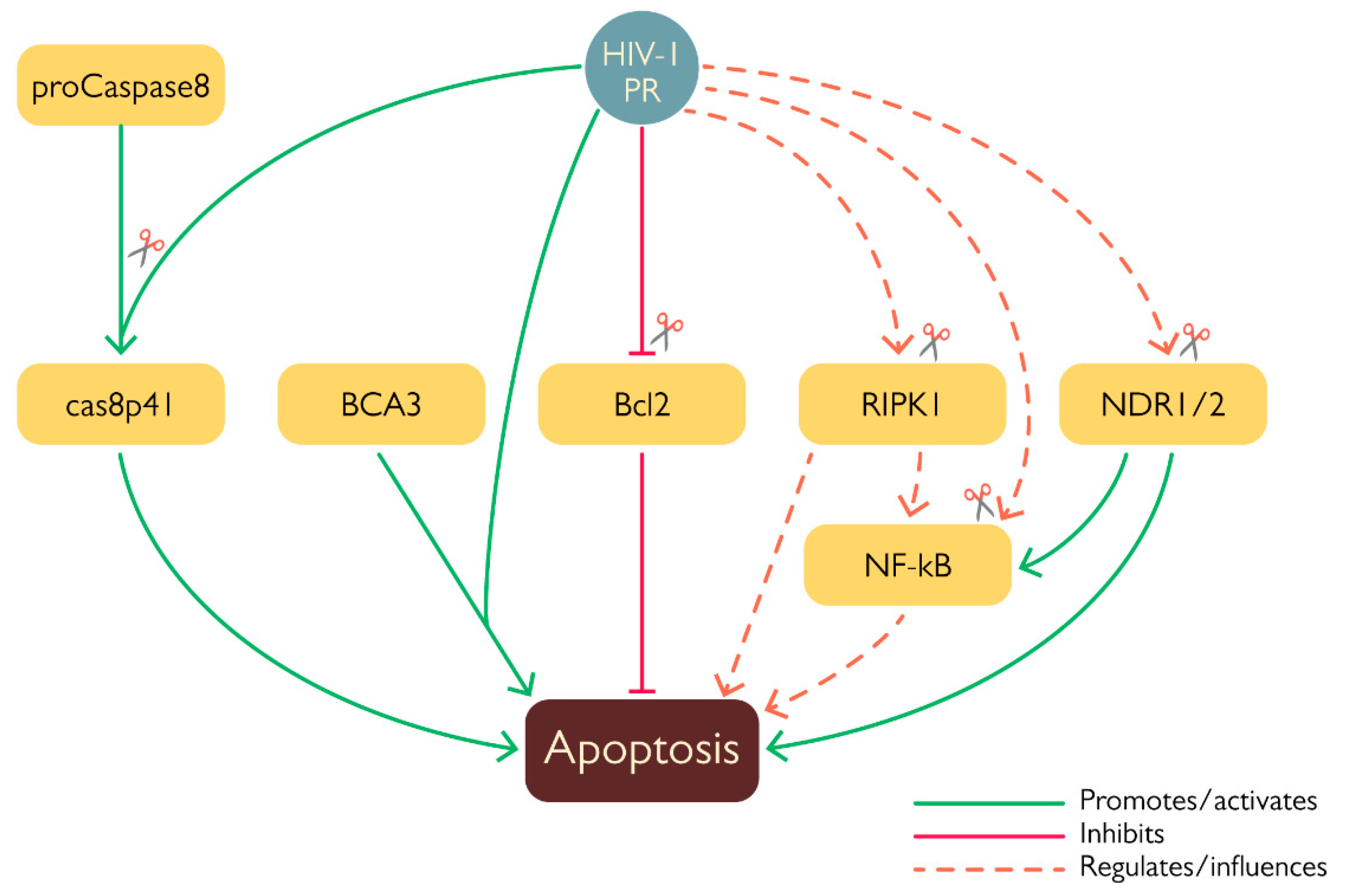
HIV-1 modulates host cell survival at several levels thanks to the action of viral proteins such as Tat, Env, Nef, Vpr and Vpu [46,47], all of which are endowed with both stimulatory and inhibitory properties, likewise, there are growing evidences that HIV-1 PR is similarly involved in modulation of programmed cell death (Figure 7). Given its remarkable effect on translation (see above), it is not surprising that exogenous expression of HIV-1 PR is highly toxic in a number cellular systems [48,49,50]. In addition, HIV-1 PR has been shown stimulate apoptosis by several mechanisms, such as via cleavage of anti-apoptotic factor Bcl-2 [27], procaspase 8 [25], and of a number of mitochondrial proteins [51], highlighting its ability to interfere with more than one cell death pathway. The first proof of the involvement of HIV-1 PR in modulation of apoptosis came from identification of Bcl-2 amongst its targets. Cleavage was first characterized in vitro, and confirmed by Western Blot analysis of COS-7 cells 24 h after having been transfected to co-express Bcl-2 and HIV-1 PR [27]. Cleavage of Bcl-2 likely promotes apoptosis activation, seen that its overexpression in HIV-1 infected lymphocytes protected cells from apoptosis [27]. In this context, another important target of the HIV-1 PR is procaspase 8, whose cleavage generates the peculiar cas8p41 fragment, which exogenous expression in cells is able to induce apoptosis in a caspase 9 and Bak/Bax dependent fashion, similarly to what is observed upon transient expression of HIV-1 in cells. However the cas8p41 fragment is not detectable in all cells exhibiting infection-induced apoptosis, highlighting how HIV-1 possesses more than one mechanism to induce this kind of programmed cell death [25]. HIV-1 PR also destabilizes mitochondrial membranes integrity, when expressed in HeLa cells the viral protease localizes in the mitochondria and in vitro experiments showed its ability to cleave a number of mitochondrial proteins such as Tom22, VDAC and ANT. Furthermore the PR was shown to interact with the Breast carcinoma-associated protein 3 (BCA3) in the mitochondria [51]; this cellular protein is known to interact with several cellular factors such as PKAc, Nf-κB, p73 and the apoptosis inducing factor (AIF), however, its role in the cell’s physiology remains largely unknown. Despite the fact that interaction between HIV-1 and BCA3 does not result in cleavage, co-expression of the two proteins in HEK 293T cells is associated to increased apoptosis [51]. Interestingly BCA3 is incorporated in HIV-1 virus like particles (VLPs) [52], this incorporation has no effect on viral infectivity and may be mediated by the interaction between BCA3 C-term domain and PKAc which is incorporated into the virion [53]. It has been observed that BCA3 mutants lacking the C-terminal domain do not get incorporated into nascent virions, advancing the hypothesis that PKAc acts as a target for this cellular protein [52].
Less clear is the role in apoptosis modulation of the HIV-1 PR-mediated cleavage of NF-κB1, a member of the Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) family. Indeed, NF-κB can act both as an antiapoptotic agent as well as a promoter of cell death [54]. HIV-1 PR specifically cleaves NF-κB1 (also known as p105), the cytoplasmatic precursor of the NF-κB subunit p50; cleavage occurs between residues F412 and P413, and has been observed both in human T-cells extracts treated with HIV-1 PR and in COS cells transiently co-expressing p105 and Pol. In doing so the PR increases the levels of readily available NF-κB and facilitates its translocation into the nucleus [30]. In the absence of additional experimental evidences, at the moment it is extremely difficult to speculate the exact outcome of such particular cleavage, due to the complexity of NF-kB regulation although it very likely represents an additional way through which HIV-1 exerts its host-modulating activity via the PR.
As alluded to above, HIV-1 PR is also capable of suppressing programmed cell death pathways, one way in which it can do it is by targeting the nuclear Dbf2-related kinases NDR1 and NDR2 [24,31]. Their cleavage was observed in HEK293T cells transfected with HIV-1NL4-3 proviral DNA, where expression of the PR resulted in processing of almost 50% of endogenous NDR1. These kinases have been associated with different cellular mechanisms, such as morphological changes, cell cycle, apoptosis and innate immunity [55]; for example NDR1/2 are activated by death receptor and their knockdown reduces cell death and apoptosis [46]. Interestingly, NDR1 negatively affects the TLR9-mediated response and the lack of this kinase has been associated with an enhanced pro-inflammatory cytokine production induced by CpG in vivo, likewise, NDR2 also seems to influence the TLR9 response and a reduced activity of NDR2 correlates with a stronger secretion of IL-6 mediated by CpG [55]. Furthermore NDR1/2 act by promoting the ubiquitination of MEKK2 via Smurf1 thus stimulating the production of TNF-α, IL17, IL6 and the activation of NF-κB. Moreover, mice lacking NDR1 and infected with E. coli produce higher levels of such cytokines and have a higher mortality rate [56,57]. NDR1 is also able to promote the production of STAT1 inside cells, hence enhancing the interferon mediated immunity. Additionally, knockdown/overexpression of NDR1 showed a direct involvement in antiviral defense, since NDR1 deficient macrophages from mice showed a decreased production of pro-inflammatory cytokines and ISG expression, while the opposite has been observed when NDR1 is overexpressed in murine RAW264.7 cells [58]. Cleavage of NDR1 and NDR2 in their hydrophobic domain ablated their transphosphorylating activity but did not affect their autophosphorylation, furthermore, it altered the cellular distribution of the truncated portion of NDR2 that was shown to abnormally migrate into the nucleus. What is more is that these enzymes were shown to be incorporated inside the virion along with other cellular kinases, like ERK2/MAPK and C-PKA, which are reported to be involved in infectivity modulation [24]. The role of NDR1 and NDR2 inside the cell is complex and still not completely characterized, but seen their involvement in apoptosis induction and innate immunity responses, it is fair to think that their cleavage by HIV-1 PR and consequent alteration of their activity serves as another layer of immune evasion employed by the virus.
Similarly, HIV-1 PR is also able to cleave the receptor interacting kinases RIPK1 and RIPK2 [23,31], which are potentially implicated in cell survival and innate response. Indeed, RIPK1 is involved in apoptosis and necroptosis activation, while RIPK2 is involved in the activation of MAVS. However, although both kinases are efficiently cleaved in vitro, only endogenous RIPK1 is cleaved in a Jurkat cell line upon doxycycline mediated induction of HIV-1 PR and in HEK293T infected with a VSVg pseudotyped HIV-1 NL4.3 and Sup-T1 cells infected with replication-competent HIV-1 NL4.3, at 24 and 48 hours post infection respectively. In all three cases treatment with a PR inhibitor prevented the cleavage of the cellular protein, moreover inhibition of either reverse transcription or integration completely abrogated RIPK1 cleavage after infection of Sup-T1 cells with HIV-1 NL4.3, proving that RIPK1 is mainly cleaved by post-integration expressed PR rather than by the one present in incoming virions. RIPK1 cleavage was placed between residues 462-463 thanks to mass spectrometry experiments, with the result of separating its kinase domain and death domain. Therefore, despite the overall levels of endogenous RIPK1 was not reduced, the emergence of such defective RIPK1 forms likely promotes cell survival of HIV-1 infected cell [23].
2.3. Effect of PR on innate defenses
Likewise, the TANK-binding kinase 1 (TBK1) was shown to be cleaved by HIV-1 PR, first via a cell free Alpha Screen assay aimed at identifying potential new kinase targets of the viral PR, and subsequently in HEK 293T cells transiently expressing Gag-Pol and TBK1 [31]. This particular kinase has a fundamental role in IFN type I and III signaling and the cleavage between residues L683 and V684 prevents its phosphorylation, thus suppressing its ability to activate IRF3. Interestingly treatment of T7 cells harboring latently infected HIV-1 with Bryostatin-1 – an agent capable of strongly reactivating HIV-1 from latency - and the PR inhibitor Indinavir, resulted in an increased IFNβ1 transcription and upregulation of ISG-15 mRNA compared to untreated cells, suggesting a possible role of HIV-1 PR as a negative regulator of IFN-1 and the innate immune response [31].
Finally, as seen with the interaction with BCA3, cleavage is not the only mechanism by which the HIV-1 PR is able to exert its host-modulation action. Indeed the PR is able to downregulate the protein level of RIG-I in a proteolysis-independent way [59]. RIG-I is a pattern recognition receptor (PRR) involved in the recognition of viral ssRNAs, that causes the subsequent activation of antiviral defenses. The interaction between RIG-I and HIV-1 PR causes the relocalization of the former in the insoluble membrane fraction and consequent lysosomal elimination [59], thus contributing to the complex interplay between HIV-1 and its host.
2.3. Cleavage of virion-incorporated restriction factors
Remarkably, HIV-1 PR targets two restriction factors that are incorporated into the virion to interfere with virus fitness (Figure 4): YTHDF3 and APOBEC3H haplotype II splice variant 200 [60,61,62]. The N6-methyladenosine (m6A) reader protein YTHDF3 is incorporated into the nascent virion via NC interaction and is subsequently cleaved by the PR [60]. YTHDF3 recognizes RNA molecules bearing the m6A modification and can promote either their translation or degradation. HIV-1 infection upregulates the appearance of the m6A modification, whose presence in viral RNAs is linked to enhanced nuclear export and increased viral protein production. NC-mediated incorporation of YTHDF3 was observed by production and analysis of virus like particles generated by co-transfection of Gag and FLAG-YTHDF3 constructs in HEK293T cells. Cleavage was investigated in virions produced in A3R5-Rev-GFP T cells infected with the NL4-3 HIV-1 strain that was harvested 12 days post infection. Analysis on both viral and cell lysates showed that the former presented a majority of shorter processed YTHDF3 compared to the full-length version that was instead detected in cell lysates. YTHDF3 is believed to act as a restriction factor for HIV-1 since its knockout in A3R5-Rev-GFP cells and CD4+ T cells resulted in a higher susceptibility to infection, whereas its overexpression negatively impacted viral infectivity [60] and therefore its cleavage by HIV-1 PR plays a role in incrementing the virus fitness.
APOBEC3H haplotype II splice variant 200 (A3H-II SV200) is similarly cleaved by HIV-1 PR in the nascent virion. Other members of the APOBEC3 family such as APOBEC3D, APOBEC3F and APOBEC3G have a strong anti-HIV-1 effect and are incorporated into the virion where they hypermutate and inactivate the viral genome catalyzing the deamination of C to U [63]. Their antiviral activity is usually counteracted by HIV-1 accessory protein Vif (Viral infectivity factor) which recruits the host E3 ubiquitin ligase complex on APOBEC3 proteins thus promoting their degradation [63]. In addition, A3H-II SV200 - one of the APOBEC3 family members most susceptible to genetic variability in the population – is cleaved by the PR. In particular haplotype II is one of the few functional variants of A3H and can be found in 82% of HIV-1 pandemic areas population in sub-Saharan Africa; additional variability of this protein comes from the possible splice variants of this allele, among which the SV200 is the most active in restricting HIV-1 infectivity. Cleavage of A3H-II SV200 was detected in Vif-deficient virions harvested from infected HEK293T cells transfected with a A3H-II SV200 construct. This factor was confined inside the viral particle, the PR-mediated cleavage likely involves its C-term region and decreases its activity [61]. Therefore, despite the fact that HIV-1 is able to potently counteract the action of the APOBEC3 proteins by the activity of Vif, it appears that A3H-II SV200 is further specifically targeted by HIV-1 PR.
Lastly, the HIV-1 PR was shown to target Lyric (also named metadherin or AEG-1), a protein involved in several cellular pathways like NF-κB, Ras, Wnt, PI3K, tumor growth and metastatization. Lyric directly interacts with Gag and is incorporated in the nascent virion where it is cleaved by the PR. Experiments with infected MT-4 cells showed a 6-fold enrichment of Lyric in virions compared to cell lysates. Furthermore, virion-incorporated lyric lacked the C-term portion, while the full length protein was recovered in PR-deficient virions and cells [62]. Although the meaning of this interaction is still unknown, it has been reported that expression of Lyric correlated with an increased Gag expression and viral infectivity, and it possibly plays a role in HIV-associated neurocognitive disorder (HAND) [64].
2.4. PR-mediated cytoskeleton modulation
Nearly every virus evolved mechanisms to interact with the cytoskeleton to some extent, for example exploiting it for viral replication or heavily altering its properties upon viral-induced cell transformation [65], likewise, HIV-1 interacts with it in several different ways, one of which is via PR-mediated cleavage of cytoskeletal proteins. Indeed the viral protease was demonstrated to cleave several cytoskeletal proteins: vimentin for example is cleaved after PR microinjection in human skin fibroblast with subsequent alteration of nuclear morphology and chromatic condensation [66]. Actin was also cleaved in vivo in A3.01 T lymphocytes infected with HIV-1 (LAV-1BRU), but the protease was only able to cleave circulating globular Actin and not the cytoskeletal fraction [67,68]. Intriguingly, HIV-1 greatly depends on actin in early steps of the replication cycle, from attachment and fusion of the virion to the cellular membrane, to nuclear localization of the pre-integration complex [69]. Notably, disruption of the Actin cytoskeleton by cytochalasin D inhibits viral entry and reverse transcription. Furthermore, Actin has also a role in later stages of the infection, where actin-depolymerizing agents were shown to inhibit HIV-1 assembly [65]. However, even though many aspects of the close-knit relationship between HIV-1 infection and the cytoskeleton are well characterized and involve many of the virus proteins [70], the functional relevance of the cleavage of cytoskeletal proteins by the PR is still unclear; it may serve as a way for the virus to better navigate the host cell, or yet another way the virus has evolved to interfere with more complex regulatory pathways.
3. Conclusions
HIV-1 PR activity was long thought to only be restricted to immature virions during or after budding to catalyze maturation of nascent viral particles, but as we reported in this review that is just a small part of the whole picture. This viral enzyme has been proven to interfere with several host physiological processes with the aim of facilitating the progression of viral infection, and although there is a growing body of evidence on this matter, the depth of what is unknown is still baffling. Open questions still remain in relation to the functional role of several reported PR targets, as well as their exact cleavage location and timing in infected cells. Moreover, these studies are far from perfect and present some limitations, for example the use cell lines that are not naturally targeted by the virus or the employment of laboratory-adapted viral strains of HIV-1, as well as transient transfection do not depict an accurate model of HIV-1 infection, studies on primary cell lines would be required to confirm some of these results and understand their effect on CD4+ lymphocytes and monocytes. In light of these facts, it is clear that there still is a huge gap in knowledge in understanding how the HIV-1 infection affects a host cell, and PR activity in this regard may have been grossly underestimated and needs to be researched thoroughly.
References
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS Pandemic. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- IN DANGER: UNAIDS Global AIDS Update 2022; Geneva, 2022.
- Groopman, J.E. Zidovudine Intolerance. Rev. Infect. Dis. 1990, 12 Suppl 5, S500-6. [Google Scholar] [CrossRef]
- Larder, B.A.; Darby, G.; Richman, D.D. HIV with Reduced Sensitivity to Zidovudine (AZT) Isolated During Prolonged Therapy. Science (80-. ). 1989, 243, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Hochhauser, D.; Harris, A.L. Drug Resistance. Br. Med. Bull. 1991, 47, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Tomasselli, A.G.; Thaisrivongs, S.; Heinrikson, R.L. Discovery and Design of HIV Protease Inhibitors as Drugs for Treatment of Aids; 1996; Volume 2, ISBN 1559386932. [Google Scholar]
- Collier, A.C.; Coombs, R.W.; Schoenfeld, D.A.; Bassett, R.L.; Timpone, J.; Baruch, A.; Jones, M.; Facey, K.; Whitacre, C.; McAuliffe, V.J.; et al. Treatment of Human Immunodeficiency Virus Infection with Saquinavir, Zidovudine, and Zalcitabine. AIDS Clinical Trials Group. N. Engl. J. Med. 1996, 334, 1011–1017. [Google Scholar] [CrossRef]
- Simon, V.; Ho, D.D.; Abdool Karim, Q. HIV/AIDS Epidemiology, Pathogenesis, Prevention, and Treatment. Lancet 2006, 368, 489–504. [Google Scholar] [CrossRef]
- Yang, H.; Nkeze, J.; Zhao, R.Y. Effects of HIV-1 Protease on Cellular Functions and Their Potential Applications in Antiretroviral Therapy. Cell Biosci. 2012, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Mager, P.P. The Active Site of HIV-1 Protease. Med. Res. Rev. 2001, 21, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Konvalinka, J.; Kräusslich, H.G.; Müller, B. Retroviral Proteases and Their Roles in Virion Maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.; Chen, Z.; Wang, G.; Shao, Q.; Shi, J.; Zhu, W. Structural Insights into HIV-1 Protease Flap Opening Processes and Key Intermediates. RSC Adv. 2017, 7, 45121–45128. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Schiffer, C.A. Curling of Flap Tips in HIV-1 Protease as a Mechanism for Substrate Entry and Tolerance of Drug Resistance. Structure 2000, 8, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Trylska, J.; Tozzini, V.; Chang, C.E.A.; McCammon, J.A. HIV-1 Protease Substrate Binding and Product Release Pathways Explored with Coarse-Grained Molecular Dynamics. Biophys. J. 2007, 92, 4179–4187. [Google Scholar] [CrossRef] [PubMed]
- Laco, G.S. HIV-1 Protease Substrate-Groove: Role in Substrate Recognition and Inhibitor Resistance. Biochimie 2015, 118, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Impens, F.; Timmerman, E.; Staes, A.; Moens, K.; Ariën, K.K.; Verhasselt, B.; Vandekerckhove, J.; Gevaert, K. A Catalogue of Putative HIV-1 Protease Host Cell Substrates. Biol. Chem. 2012, 393, 915–931. [Google Scholar] [CrossRef] [PubMed]
- Lawal, M.M.; Sanusi, Z.K.; Govender, T.; Maguire, G.E.M.; Honarparvar, B.; Kruger, H.G. From Recognition to Reaction Mechanism: An Overview on the Interactions between HIV-1 Protease and Its Natural Targets. Curr. Med. Chem. 2018, 25, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Potempa, M.; Swanstrom, R. The Choreography of HIV-1 Proteolytic Processing and Virion Assembly. J. Biol. Chem. 2012, 287, 40867–40874. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Louis, J.M.; Aniana, A.; Suh, J.Y.; Clore, G.M. Visualizing Transient Events in Amino-Terminal Autoprocessing of HIV-1 Protease. Nature 2008, 455, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Agniswamy, J.; Sayer, J.M.; Weber, I.T.; Louis, J.M. Terminal Interface Conformations Modulate Dimer Stability Prior to Amino Terminal Autoprocessing of HIV-1 Protease. Biochemistry 2012, 51, 1041–1050. [Google Scholar] [CrossRef]
- Kaplan, A.H.; Swanstrom, R. Human Immunodeficiency Virus Type 1 Gag Proteins Are Processed in Two Cellular Compartments. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 4528–4532. [Google Scholar] [CrossRef]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global Landscape of HIV-Human Protein Complexes. Nature 2012, 481, 365–370. [Google Scholar] [CrossRef]
- Wagner, R.N.; Reed, J.C.; Chanda, S.K. HIV-1 Protease Cleaves the Serine-Threonine Kinases RIPK1 and RIPK2. Retrovirology 2015, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Devroe, E.; Silver, P.A.; Engelman, A. HIV-1 Incorporates and Proteolytically Processes Human NDR1 and NDR2 Serine-Threonine Kinases. Virology 2005, 331, 181–189. [Google Scholar] [CrossRef]
- Nie, Z.; Bren, G.D.; Vlahakis, S.R.; Schimnich, A.A.; Brenchley, J.M.; Trushin, S.A.; Warren, S.; Schnepple, D.J.; Kovacs, C.M.; Loutfy, M.R.; et al. Human Immunodeficiency Virus Type 1 Protease Cleaves Procaspase 8 In Vivo. J. Virol. 2007, 81, 6947–6956. [Google Scholar] [CrossRef]
- Ventoso, I.; Blanco, R.; Perales, C.; Carrasco, L. HIV-1 Protease Cleaves Eukaryotic Initiation Factor 4G and Inhibits Cap-Dependent Translation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 12966–12971. [Google Scholar] [CrossRef] [PubMed]
- Strack, P.R.; Frey, M.W.; Rizzo, C.J.; Cordova, B.; George, H.J.; Meade, R.; Ho, S.P.; Corman, J.; Tritch, R.; Korant, B.D. Apoptosis Mediated by HIV Protease Is Preceded by Cleavage of Bcl-2. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 9571–9576. [Google Scholar] [CrossRef]
- Álvarez, E.; Castelló, A.; Menéndez-Arias, L.; Carrasco, L. HIV Protease Cleaves Poly(A)-Binding Protein. Biochem. J. 2006, 396, 219–226. [Google Scholar] [CrossRef] [PubMed]
- del Pino, J.; Jiménez, J.L.; Ventoso, I.; Castelló, A.; Muñoz-Fernández, M.Á.; de Haro, C.; Berlanga, J.J. GCN2 Has Inhibitory Effect on Human Immunodeficiency Virus-1 Protein Synthesis and Is Cleaved upon Viral Infection. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Rivière, Y.; Blank, V.; Kourilsky, P.; Israël, A. Processing of the Precursor of NF-ΚB by the HIV-1 Protease during Acute Infection. Nature 1991, 350, 625–626. [Google Scholar] [CrossRef] [PubMed]
- Jeremiah, S.S.; Miyakawa, K.; Matsunaga, S.; Nishi, M.; Kudoh, A.; Takaoka, A.; Sawasaki, T.; Ryo, A. Cleavage of TANK-Binding Kinase 1 by HIV-1 Protease Triggers Viral Innate Immune Evasion. Front. Microbiol. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Amorim, R.; Costa, S.M.; Cavaleiro, N.P.; da Silva, E.E.; da Costa, L.J. HIV-1 Transcripts Use IRES-Initiation under Conditions Where Cap-Dependent Translation Is Restricted by Poliovirus 2A Protease. PLoS One 2014, 9, e88619. [Google Scholar] [CrossRef] [PubMed]
- Castelló, A.; Franco, D.; Moral-López, P.; Berlanga, J.J.; Álvarez, E.; Wimmer, E.; Carrasco, L. HIV-1 Protease Inhibits Cap-and Poly(A)-Dependent Translation upon EIF4GI and PABP Cleavage. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Brasey, A.; Lopez-Lastra, M.; Ohlmann, T.; Beerens, N.; Berkhout, B.; Darlix, J.-L.; Sonenberg, N. The Leader of Human Immunodeficiency Virus Type 1 Genomic RNA Harbors an Internal Ribosome Entry Segment That Is Active during the G2/M Phase of the Cell Cycle. J. Virol. 2003, 77, 3939–3949. [Google Scholar] [CrossRef] [PubMed]
- Vallejos, M.; Carvajal, F.; Pino, K.; Navarrete, C.; Ferres, M.; Huidobro-Toro, J.P.; Sargueil, B.; López-Lastra, M. Functional and Structural Analysis of the Internal Ribosome Entry Site Present in the MRNA of Natural Variants of the HIV-1. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Shen, X.; Egan, M.A.; Pierson, T.C.; Walker, C.M.; Siliciano, R.F. The Human Immunodeficiency Virus Type 1 Gag Gene Encodes an Internal Ribosome Entry Site. J. Virol. 2001, 75, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Plank, T.D.M.; Whitehurst, J.T.; Kieft, J.S. Cell Type Specificity and Structural Determinants of IRES Activity from the 5’ Leaders of Different HIV-1 Transcripts. Nucleic Acids Res. 2013, 41, 6698–6714. [Google Scholar] [CrossRef]
- Baboonian, C.; Dalgleish, A.; Bountiff, L.; Gross, J.; Oroszlan, S.; Rickett, G.; Smith-Burchnell, C.; Troke, P.; Merson, J. HIV-1 Proteinase Is Required for Synthesis of pro-Viral DNA. Biochem. Biophys. Res. Commun. 1991, 179, 17–24. [Google Scholar] [CrossRef]
- Jacobsen, H.; Ahlborn-laake, L.; Gugel, R.; Mous, J.A.N. Progression of Early Steps of Human Immunodeficiency Virus Type 1 Replication in the Presence of an Inhibitor of Viral Protease A-I. 1992, 66, 5087–5091.
- Nagy, K.; Young, M.; Baboonian, C.; Merson, J.; Whittle, P.; Oroszlan, S. Antiviral Activity of Human Immunodeficiency Virus Type 1 Protease Inhibitors in a Single Cycle of Infection: Evidence for a Role of Protease in the Early Phase. J. Virol. 1994, 68, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.H.; Manchester, M.; Smith, T.; Yang, Y.L.; Swanstrom, R. Conditional Human Immunodeficiency Virus Type 1 Protease Mutants Show No Role for the Viral Protease Early in Virus Replication. J. Virol. 1996, 70, 5840–5844. [Google Scholar] [CrossRef]
- Uchida, H.; Maeda, Y.; Mitsuya, H. HIV-1 Protease Does Not Play a Critical Role in the Early Stages of HIV-1 Infection. Antiviral Res. 1997, 36, 107–113. [Google Scholar] [CrossRef]
- Stefanidou, M.; Herrera, C.; Armanasco, N.; Shattock, R.J. Saquinavir Inhibits Early Events Associated with Establishment of HIV-1 Infection: Potential Role for Protease Inhibitors in Prevention. Antimicrob. Agents Chemother. 2012, 56, 4381–4390. [Google Scholar] [CrossRef] [PubMed]
- Monette, A.; Valiente-Echeverría, F.; Rivero, M.; Cohen, E. ´ A.A.; Lopez-Lastra, M. Dual Mechanisms of Translation Initiation of the Full-Length HIV-1 MRNA Contribute to Gag Synthesis. PLoS One 2013, 8, 68108. [Google Scholar] [CrossRef] [PubMed]
- Vichalkovski, A.; Gresko, E.; Cornils, H.; Hergovich, A.; Schmitz, D.; Hemmings, B.A. NDR Kinase Is Activated by RASSF1A/MST1 in Response to Fas Receptor Stimulation and Promotes Apoptosis. Curr. Biol. 2008, 18, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, M.L. Apoptosis as an HIV Strategy to Escape Immune Attack. Nat. Rev. Immunol. 2003, 3, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Baum, E.Z.; Bebernitz, G.A.; Gluzman, Y. Isolation of Mutants of Human Immunodeficiency Virus Protease Based on the Toxicity of the Enzyme in Escherichia Coli. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 5573–5577. [Google Scholar] [CrossRef] [PubMed]
- M’Barek, N. Ben; Audoly, G.; Raoult, D.; Gluschankof, P. HIV-2 Protease Resistance Defined in Yeast Cells. Retrovirology 2006, 3. [Google Scholar] [CrossRef] [PubMed]
- Konvalinka, J.; Litterst, M.A.; Welker, R.; Kottler, H.; Rippmann, F.; Heuser, A.M.; Kräusslich, H.G. An Active-Site Mutation in the Human Immunodeficiency Virus Type 1 Proteinase (PR) Causes Reduced PR Activity and Loss of PR-Mediated Cytotoxicity without Apparent Effect on Virus Maturation and Infectivity. J. Virol. 1995, 69, 7180–7186. [Google Scholar] [CrossRef] [PubMed]
- Rumlová, M.; Křížová, I.; Keprová, A.; Hadravová, R.; Doležal, M.; Strohalmová, K.; Pichová, I.; Hájek, M.; Ruml, T. HIV-1 Protease-Induced Apoptosis. Retrovirology 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Rumlová, M.; Křížová, I.; Zelenka, J.; Weber, J.; Ruml, T. Does BCA3 Play a Role in the HIV-1 Replication Cycle? Viruses 2018, 10. [Google Scholar] [CrossRef]
- Cartier, C.; Hemonnot, B.; Gay, B.; Bardy, M.; Sanchiz, C.; Devaux, C.; Briant, L. Active CAMP-Dependent Protein Kinase Incorporated within Highly Purified HIV-1 Particles Is Required for Viral Infectivity and Interacts with Viral Capsid Protein. J. Biol. Chem. 2003, 278, 35211–35219. [Google Scholar] [CrossRef]
- Baichwal, V.R.; Baeuerle, P.A. Apoptosis: Activate NF-ΚB or Die? Curr. Biol. 1997, 7, R94–R96. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Ong, N.; An, H.; Zheng, Y. The Emerging Roles of NDR1/2 in Infection and Inflammation. Front. Immunol. 2020, 11, 534. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Ma, X.; Cheng, H.; Jiang, W.; Xu, X.; Zhang, Y.; Zhang, Y.; Guo, Z.; Yu, Y.; Xu, H.; et al. Stk38 Protein Kinase Preferentially Inhibits TLR9-Activated Inflammatory Responses by Promoting MEKK2 Ubiquitination in Macrophages. Nat. Commun. 2015 61 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Ma, X.; Wang, D.; Li, N.; Gao, P.; Zhang, M.; Zhang, Y. Hippo Kinase NDR2 Inhibits IL-17 Signaling by Promoting Smurf1-Mediated MEKK2 Ubiquitination and Degradation. Mol. Immunol. 2019, 105, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qin, Q.; Wu, C.; Li, H.; Shou, J.; Yang, Y.; Gu, M.; Ma, C.; Lin, W.; Zou, Y.; et al. Downregulated NDR1 Protein Kinase Inhibits Innate Immune Response by Initiating an MiR146a-STAT1 Feedback Loop. Nat. Commun. 2018 91 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-Mediated Antiviral Signaling Is Inhibited in HIV-1 Infection by a Protease-Mediated Sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef]
- Jurczyszak, D.; Zhang, W.; Terry, S.N.; Kehrer, T.; Bermúdez González, M.C.; McGregor, E.; Mulder, L.C.F.; Eckwahl, M.J.; Pan, T.; Simon, V. HIV Protease Cleaves the Antiviral M6A Reader Protein YTHDF3 in the Viral Particle. PLOS Pathog. 2020, 16, e1008305. [Google Scholar] [CrossRef]
- Ebrahimi, D.; Richards, C.M.; Carpenter, M.A.; Wang, J.; Ikeda, T.; Becker, J.T.; Cheng, A.Z.; McCann, J.L.; Shaban, N.M.; Salamango, D.J.; et al. Genetic and Mechanistic Basis for APOBEC3H Alternative Splicing, Retrovirus Restriction, and Counteraction by HIV-1 Protease. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Engeland, C.E.; Oberwinkler, H.; Schumann, M.; Krause, E.; Muller, G.A.; Krausslich, H.-G. The Cellular Protein Lyric Interacts with HIV-1 Gag. J. Virol. 2011, 85, 13322–13332. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E.; Yoshikawa, R.; Nakano, Y.; Misawa, N.; Koyanagi, Y.; Sato, K. Impacts of Humanized Mouse Models on the Investigation of HIV-1 Infection: Illuminating the Roles of Viral Accessory Proteins in Vivo. Viruses 2015, 7, 1373–1390. [Google Scholar] [CrossRef]
- Ye, X.; Zhang, Y.; Xu, Q.; Zheng, H.; Wu, X.; Qiu, J.; Zhang, Z.; Wang, W.; Shao, Y.; Xing, H.Q. HIV-1 Tat Inhibits EAAT-2 through AEG-1 Upregulation in Models of HIV-Associated Neurocognitive Disorder. Oncotarget 2017, 8, 39922–39934. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Koyuncu, O.O.; Enquist, L.W. Subversion of the Actin Cytoskeleton during Viral Infection. Nat. Rev. Microbiol. 2011, 9, 427–439. [Google Scholar] [CrossRef]
- Honer, B.; Shoeman, R.L.; Traub, P. Human Immunodeficiency Virus Type 1 Protease Microinjected into Cultured Human Skin Fibroblasts Cleaves Vimentin and Affects Cytoskeletal and Nuclear Architecture. J. Cell Sci. 1991, 100, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Shoeman, R.L.; Sachse, C.; Höner, B.; Mothes, E.; Kaufmann, M.; Traub, P. Cleavage of Human and Mouse Cytoskeletal and Sarcomeric Proteins by Human Immunodeficiency Virus Type 1 Protease: Actin, Desmin, Myosin, and Tropomyosin. Am. J. Pathol. 1993, 142, 221–230. [Google Scholar] [PubMed]
- Adams, L.D.; Tomasselli, A.G.; Robbins, P.; Moss, B.; Heinrikson, R.L. HIV-1 Protease Cleaves Actin During Acute Infection of Human T-Lymphocytes. AIDS Res. Hum. Retroviruses 1992, 8, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.; Naghavi, M.H. The Host Cytoskeleton: A Key Regulator of Early HIV-1 Infection. FEBS J. 2022. [Google Scholar] [CrossRef]
- Matarrese, P.; Malorni, W. Human Immunodeficiency Virus (HIV)-1 Proteins and Cytoskeleton: Partners in Viral Life and Host Cell Death. Cell Death Differ. 2005 121 2005, 12, 932–941. [Google Scholar] [CrossRef]
Figure 1.
3D structure of the HIV-1 PR (PDB ID 6O48) visualized as a ribbon diagram with functional domains highlighted in different colors [12]: protease flaps in green, flap elbows in turquoise, fulcrum in orange, cantilever in yellow, dimer interface in pale magenta and active site in red.
Figure 1.
3D structure of the HIV-1 PR (PDB ID 6O48) visualized as a ribbon diagram with functional domains highlighted in different colors [12]: protease flaps in green, flap elbows in turquoise, fulcrum in orange, cantilever in yellow, dimer interface in pale magenta and active site in red.
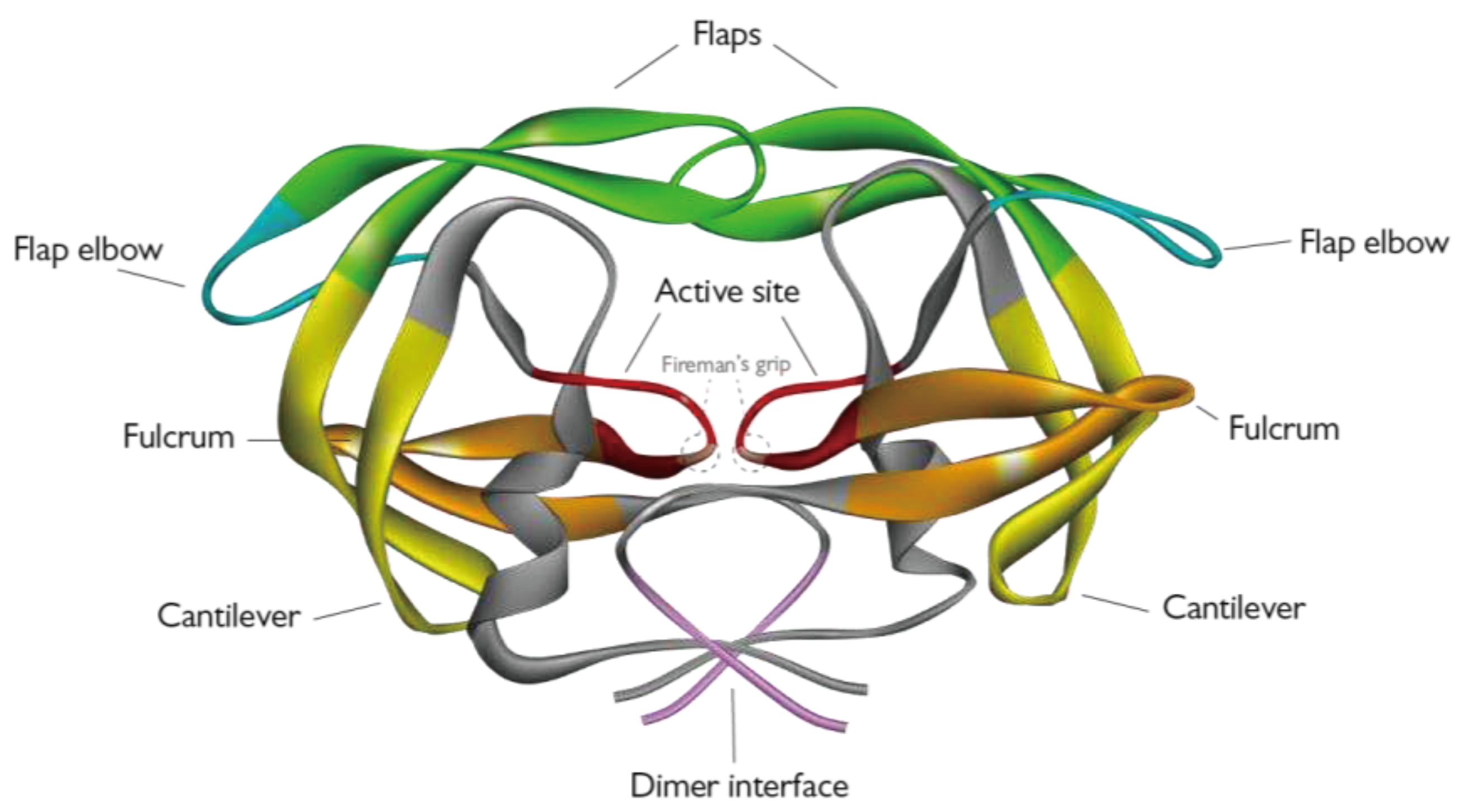
Figure 2.
Processing of HIV-1 polyproteins by the viral protease. (a) Ordered processing of GagPol by the poorly-active PR precursor still embedded in the polyprotein context. The first cleavage events aim at separating the PR from the polyprotein at its N-term, later events finally free the protease from the rest of the precursor. (b) Processing of Gag by the mature PR. Maturation happens in a precise and ordered fashion, with the first cleavage happening between NC and Sp1, then MA/CA and sp2/p6 and lastly CA and NC are separated from the spacer peptides. (c) Schematic representation of virion maturation.
Figure 2.
Processing of HIV-1 polyproteins by the viral protease. (a) Ordered processing of GagPol by the poorly-active PR precursor still embedded in the polyprotein context. The first cleavage events aim at separating the PR from the polyprotein at its N-term, later events finally free the protease from the rest of the precursor. (b) Processing of Gag by the mature PR. Maturation happens in a precise and ordered fashion, with the first cleavage happening between NC and Sp1, then MA/CA and sp2/p6 and lastly CA and NC are separated from the spacer peptides. (c) Schematic representation of virion maturation.
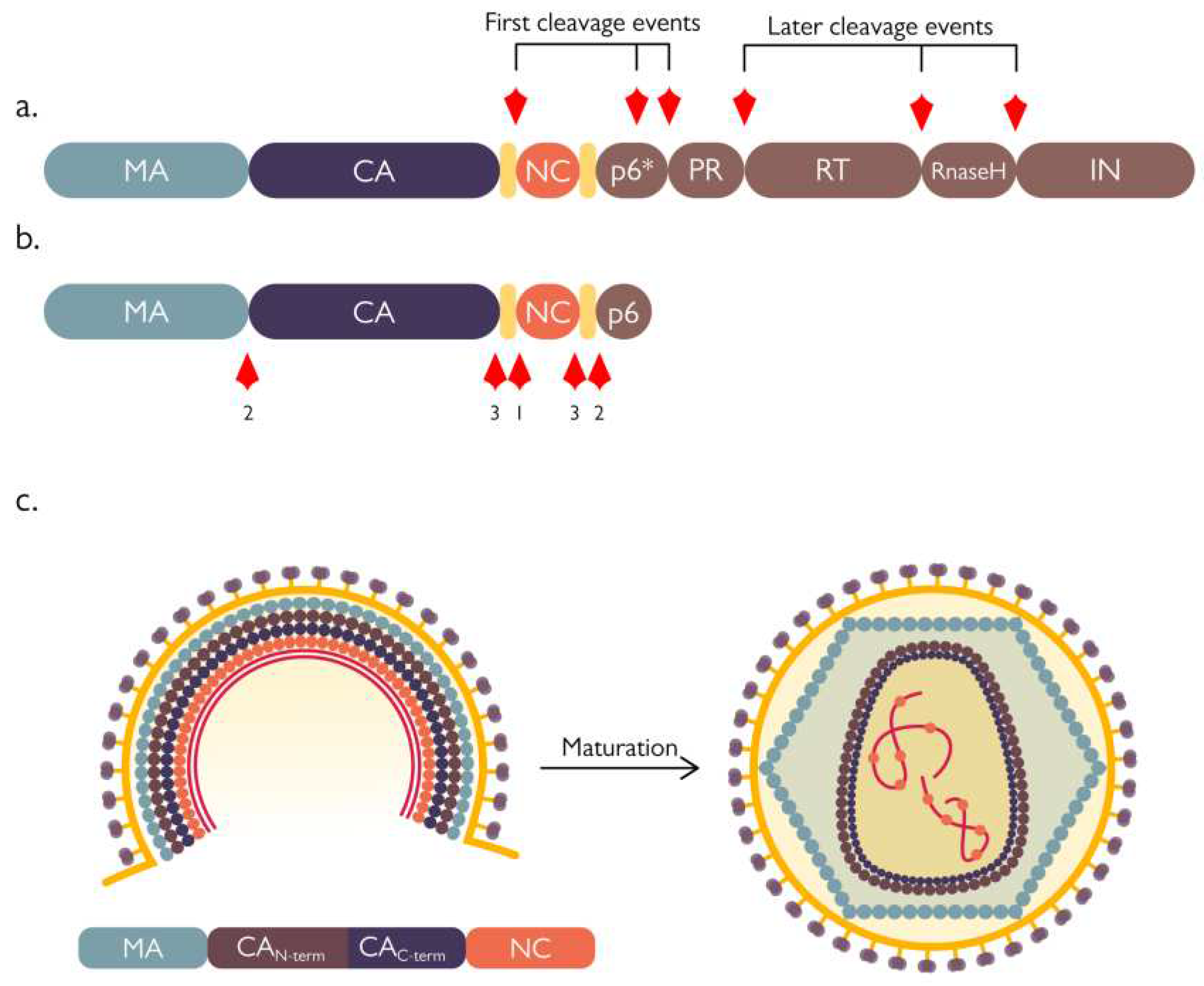
Figure 3.
Graph of identified HIV-1 PR interactors from Jäger et. al, represented by STRING [32].
Figure 3.
Graph of identified HIV-1 PR interactors from Jäger et. al, represented by STRING [32].
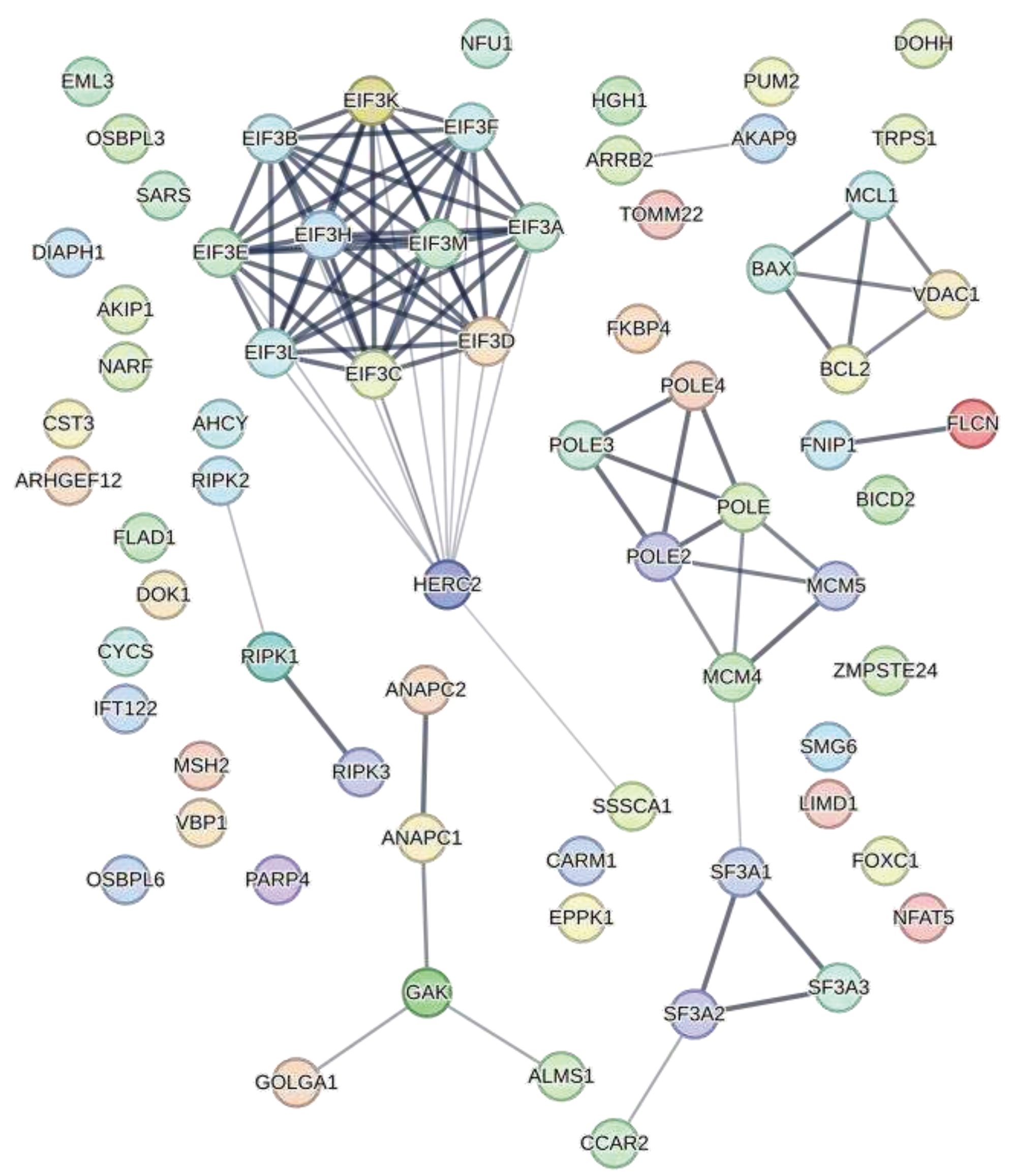
Figure 4.
HIV-1 replication cycle with targets cleaved by the HIV-1 protease. The exact timing and location of the activity of HIV-1 PR are still not completely characterized. It is speculated that due to its possible anti RT activity eIF3D may be an early target of the PR, inactivating this factor may serve as a way to escape antiviral defenses of the cell and hindering cap-dependent translation. The other intracellular targets are most likely cleaved by post-integration synthetized PR, among these targets there are several proteins involved in protein synthesis that are responsible for either cap-dependent translation initiation (eIF4G, PABP) or translation regulation (GCN2). The other major protein cluster that is targeted by the viral enzyme represents proteins involved in cell death and the innate defenses of the cell (Bcl2, Procaspase8, NF-κB1, NDR1/2, RIPK1, TBK1). Lastly, HIV-1 PR was shown to cleave host proteins that are incorporated into the nascent virion, two of which exert an antiviral activity (A3H SV200, YTHDF3), while the specific function of the other two (NDR1/2, Lyric) is still not known.
Figure 4.
HIV-1 replication cycle with targets cleaved by the HIV-1 protease. The exact timing and location of the activity of HIV-1 PR are still not completely characterized. It is speculated that due to its possible anti RT activity eIF3D may be an early target of the PR, inactivating this factor may serve as a way to escape antiviral defenses of the cell and hindering cap-dependent translation. The other intracellular targets are most likely cleaved by post-integration synthetized PR, among these targets there are several proteins involved in protein synthesis that are responsible for either cap-dependent translation initiation (eIF4G, PABP) or translation regulation (GCN2). The other major protein cluster that is targeted by the viral enzyme represents proteins involved in cell death and the innate defenses of the cell (Bcl2, Procaspase8, NF-κB1, NDR1/2, RIPK1, TBK1). Lastly, HIV-1 PR was shown to cleave host proteins that are incorporated into the nascent virion, two of which exert an antiviral activity (A3H SV200, YTHDF3), while the specific function of the other two (NDR1/2, Lyric) is still not known.

Figure 5.
Schematic representation of IRESs location in the HIV-1 genome and transcripts.
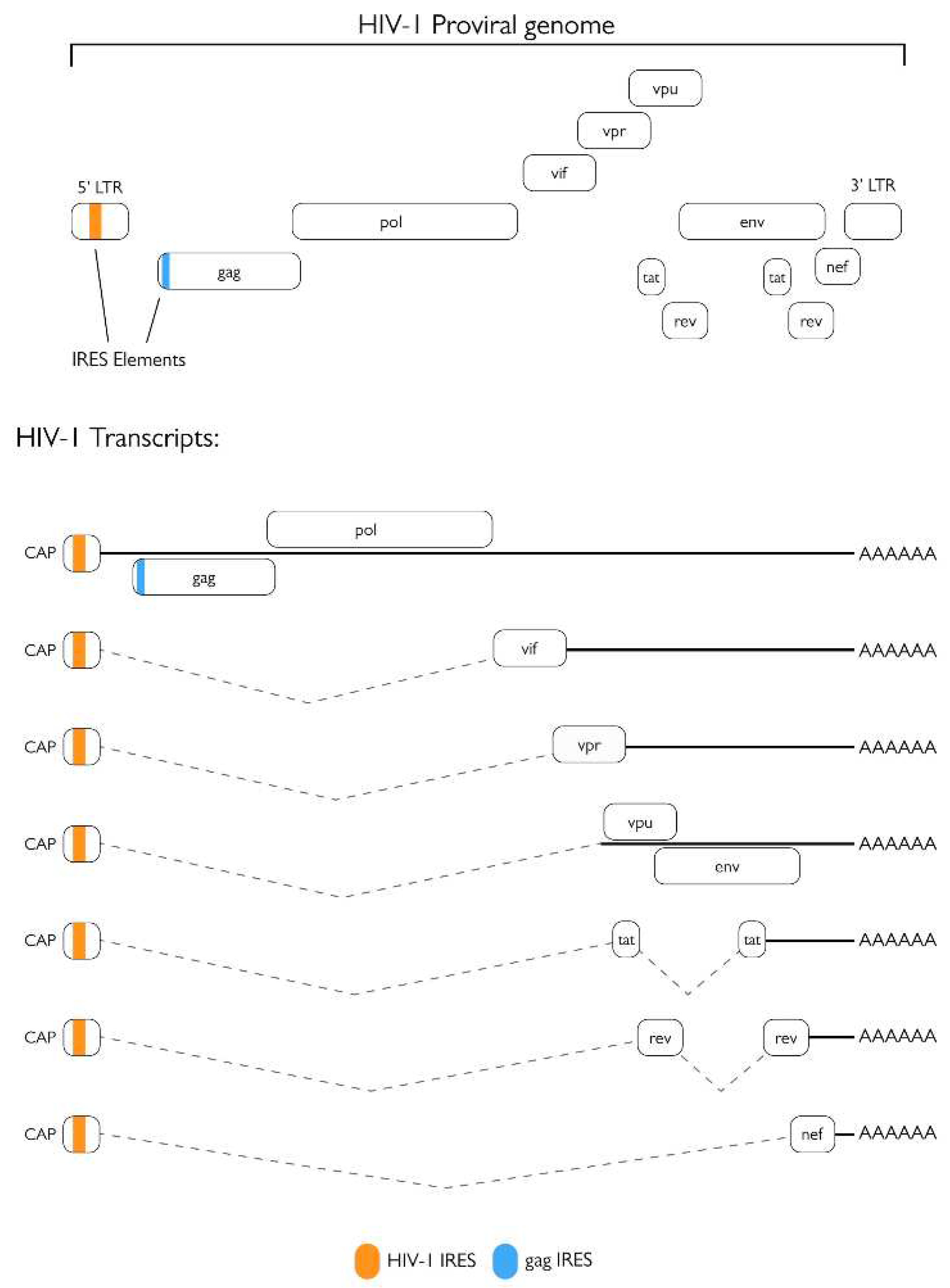
Figure 6.
Schematic representation of eukaryotic 43S preinitiation complex with HIV-1 PR cleavage targets.
Figure 6.
Schematic representation of eukaryotic 43S preinitiation complex with HIV-1 PR cleavage targets.
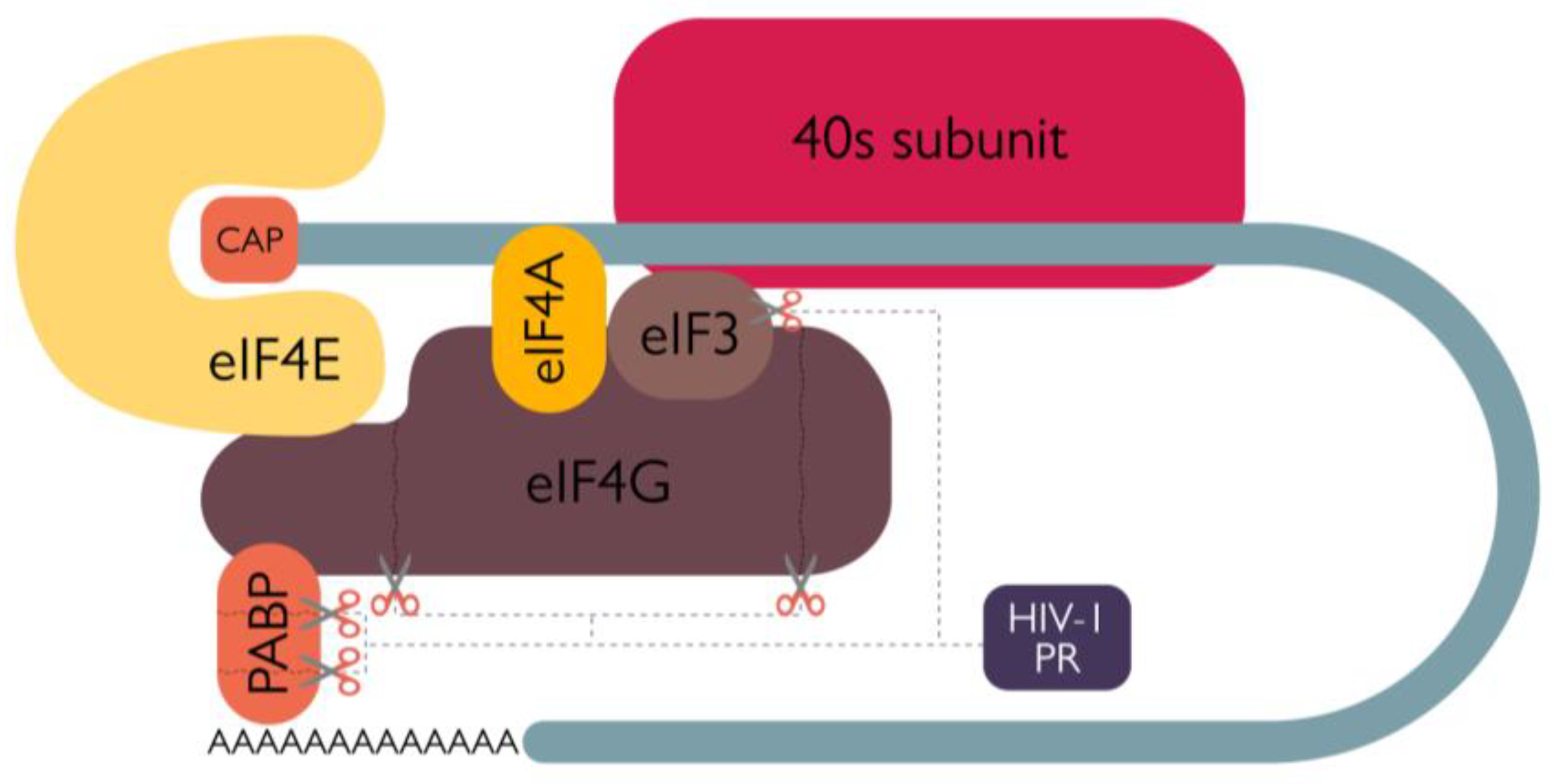
Figure 7.
HIV-1 PR modulation of apoptosis via cleavage or interaction with cellular factors. The viral protease is able to cleave several proteins involved in apoptosis regulation, by cleaving the Procaspase8 into its cas8p41 fragment the viral enzyme is able to promote programmed cell death, likewise cleavage and consequent inhibition of Bcl-2 also contributes apoptosis activation. Interestingly the non-proteolytic interaction between the PR and BCA3 was also shown to increase apoptosis. Furthermore HIV-1 PR cleaves several other proteins involved in activation and regulation of apoptosis, in this case the consequences of their cleavage is not as clear and fits in the complex scheme of HIV-1 dependent regulation of programmed cell death.
Figure 7.
HIV-1 PR modulation of apoptosis via cleavage or interaction with cellular factors. The viral protease is able to cleave several proteins involved in apoptosis regulation, by cleaving the Procaspase8 into its cas8p41 fragment the viral enzyme is able to promote programmed cell death, likewise cleavage and consequent inhibition of Bcl-2 also contributes apoptosis activation. Interestingly the non-proteolytic interaction between the PR and BCA3 was also shown to increase apoptosis. Furthermore HIV-1 PR cleaves several other proteins involved in activation and regulation of apoptosis, in this case the consequences of their cleavage is not as clear and fits in the complex scheme of HIV-1 dependent regulation of programmed cell death.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



