
Submitted:
03 February 2023
Posted:
03 February 2023
You are already at the latest version
Abstract
The human serotonin transporter (hSERT) terminates neurotransmission by removing serotonin from the synaptic cleft, which is an essential process plays an important role in depression. In addition to substrate serotonin, hSERT is also the target of drugs of abuse like cocaine and clinically used antidepressants such as escitalopram and paroxetine. To date, few studies attempt to investigate the unbinding mechanism underlying the orthosteric and allosteric modulation of hSERT. The high-resolution X-ray structure of hSERT resolved recently enables us to theoretically study the unbinding of the above four ligands against the S1 or S2 site of hSERT, by means of molecular docking, molecular dynamics (MD) and potential of mean force (PMF) simulations. We proved that for either the S1 or S2 site, the other three ligands (cocaine, escitalopram and paroxetine) are much more favorable than the original substrate serotonin, whether in kinetics along the unbinding pathways or in thermodynamics at the equilibrium states. Furthermore, the S1 site is much more favorable than the S2 site, for each ligand. Interestingly, inspection revealed that there are ~ 3Å lengths between the allosteric site of serotonin and cocaine, and an unseen un-binding pathway for escitalopram at the S1 site except for verification of the broadest trail.
Keywords:
hSERT
; comprehensive molecular dynamics (MD) simulation
; drug design
; MM/GBSA
1. Introduction
Depression is listed by the World Health Organization as one of the world’s leading disabilities, affecting over 280 million people[1]. Major depressive disorder is a serious mental health[2] condition with etiopathogenesis involving not only social and psychological factors[3,4], but also genetic and biological elements[5,6,7]. The human serotonin (5HT) transporter (SERT) terminates neurotransmission by removing serotonin from the synaptic cleft, an essential process associated with depression[8,9,10]. In addition to transport substrate serotonin, SERT is also the target of drugs of abuse such as cocaine, and clinically used antidepressants, such as escitalopram and paroxetine[11,12,13,14,15,16,17].
SERT belongs to the solute carrier 6 (SLC6) family, which includes neurotransmitter transporters such as dopamine transporter (DAT), γ-aminobutyric acid, norepinephrine and glycine[18]. In the mammalian brain, the expression of SLC6 neurotransmitter transporters plays a fundamental role in regulating neurotransmitter signaling via rapid reuptake of neurotransmitter molecules from the extracellular space into neurons and glial cells[19,20]. Cocaine is known to inhibit the activities of both SERT and DAT, which are homologous sequences[16,21]. Furthermore, cocaine has SLC6 neurotransmitter transporters as primary targets, which often leads to dependence and abuse. There residues Tyr95, Tyr175 and Ser438[16,21,22,23] located within the orthosteric 5HT-binding pocket in SERT are key interaction locations responsible for conservation among biogenic amine neurotransmitter transporters and potential selective recognition to selective serotonin reuptake inhibitors such as escitalopram and paroxetine in the treatment of depression. However, the properties associated with binding at the allosteric site of SERT remain unknown.
The highly conserved orthosteric substrate binding site (S1)[24], formed by transmembrane helices TM1, 3, 6, 8, and 10, is located in the center of SERT (Figure 1AB)[15,19,25,26,27]. Based on the recently resolved X-ray structure of SERT, in addition to the S1 site, an allosteric site (S2) was found[25,27,28]. The S2 site is formed by an aromatic pocket positioned in the scaffold domain in the extracellular vestibule, connected to the central S1 site via a short tunnel. The allosteric modulators such as escitalopram targeting the S2 site of SERT have come into the market as a medication for depression, and the S2 site together with the S1 site are extremely promising drug targets for design of future novel inhibitors. Furthermore, the site (S3) and site (S4) of docking serotonin may have been, in theory, found by investigating the substrate-bound X-ray structures of the dopamine transporter[29,30]. The S3 site was adjacent to the S2 site, and the S4 site was further toward the extracellular region. In recent decades, a number of drugs with potential selectivity for inhibiting the activity of SERT to treat depression have been continued discovered, via computational and structural analyses within high-resolution protein structures[8,12,14,27,30,31]. But while providing new opportunities for drug discovery of SERT, the patients were still suffering serious side-effects and delayed functions due to drug abuse[32,33,34,35]. Previous studies have focused on binding the affinity of drugs at the orthosteric site or allosteric site[15,16,31,36]. However, the lack of study of the unbinding mechanism of drugs from SERT limited our understanding of pharmacological implications of SERT inhibition.
In this study, we focused the mechanisms of each ligand interaction with SERT, and investigated the pathway of substrate and drugs dissociated from the S1/S2 site amid to provide spectrums of the molecular events involved in SERT. The high-resolution X-ray structure of SERT[27] resolved by Coleman et al. in 2016 was used as the initial structure for comprehensive molecular dynamics (MD) simulation. We investigated the molecular mechanism of the unbinding of substrate serotonin and drug of cocaine abuse and two antidepressants (escitalopram and paroxetine) (Figure 1C) against the S1 or S2 site of SERT. Serotonin, cocaine and escitalopram each cooperatively binds to S1 and S2 sites of SERT, while paroxetine only bound to the S1 site of SERT. Some interesting difference and similarity in the potential of mean force (PMF) simulations between SERT-serotonin, SERT-cocaine, SERT-escitalopram and SERT-paroxetine unbinding processes have been revealed. Based on our results, it can be concluded that the other three ligands, for either the S1 or S2 site, were much more favorable than the original substrate serotonin, and the distance between serotonin and cocaine at the allosteric site was ~ 3Å, and the pathway of escitalopram dissociated from the S1 site was an alternative maybe. Our work may aid in future further development of conformational selective inhibitors for the treatment of depression.
2. Results and Discussion
2.1. Hydrogen bonds analyses based on trajectories of classical MD
Hydrogen bonds are crucial for modeling protein folding, structure prediction, and complex formation[37,38]. inspection revealed that ligands (serotonin, cocaine, escitalopram and paroxetine) form different and similar interactions with SERT. The root-mean-square deviation (RMSD) of SERT Cα atoms (Figure 2) showed that the RMSD of SERT-escitalopram system was markedly higher than that of the binding original substrate serotonin system, compared with cocaine and paroxetine. This suggested that escitalopram enable to cause SERT structural instability, thereby inhibiting its activities. All the systems were stable after about 30ns during simulations, and therefore we chose the 30-100ns simulation trajectory for hydrogen bonds analyses[39,40].
For the S1 site, one stable hydrogen bond was formed for each complex. The N atom of the amine group of serotonin and hydroxyl oxygen of the Ser438 side chain formed a hydrogen bond with occupancy of 82.45%, and that of the Tyr (95 and 175) side chain form a hydrogen bond with occupancy of ~ 22.19% (Table S1). The carbonyl oxygen of cocaine benzoyl ester group and the oxygen atom of the phenol group of Tyr176 side chain formed a hydrogen bond with occupancy of 61.20%, and hydroxyl oxygen of the Ser438 side chain formed a hydrogen bond with occupancy of 11.86% (Table S3). The N atom connected to the benzofuran group of escitalopram and hydroxyl oxygen of the Ser438 side chain (the same donor as that for serotonin) formed a hydrogen bond with occupancy of 78.36% (Table S5). Finally, The N atom of the piperidine group of paroxetine and the oxygen atom of the Tyr95 main chain formed a hydrogen bond with occupancy of 99.52% (Table S7). This occupancy was the highest (approximating to 100%) among the above four hydrogen bonds, and the O atom connected to benzofuran group of paroxetine and hydroxyl oxygen of the Tyr175 side chain formed a hydrogen bond with occupancy of 8.43% (Table S7). Overall, the residues Tyr (95 and 175)[21,22] and Ser438[16,23] were the primary determinants of recognizing antidepressants upon binding the S1 site of SERT and Tyr176 may be a strong tool in designing the selectivity inhibitors towards SERT, which is consistent with previous studies.
In general, hydrogen bonds for the S2 site were much weaker (with much lower occupancy) than those for the S1 site, irrespective of the three complexes. Interestingly, serotonin, cocaine and escitalopram each formed a relatively stable hydrogen bond with the residue Arg104 of SERT, with occupancies of 25.63% (Table S2), 7.39% (Table S4) and 64.77% (Table S6), respectively.
In spite of the interaction of different residues with SERT at the S1 site and S2 site, these results suggest that for the three drugs, either the S1 site or S2 site has its own conserved residues; for example, Ser438 and Tyr were highly conserved at the S1 site, and Arg104 was conserved at S2 site. Those findings haves presented challenges for the design of selective inhibitors.
2.2. Binding free energies at the equilibrium states
The binding free energy is generally used in drug design to characterize the binding strength of a drug to its target. To further explore the interactions between the four studied ligands and SERT, the binding free energies (ΔG) of the newly designed four different complexes were estimated by the MM/GBSA method, based on the MD equilibrium trajectory (30-100ns). As shown in Table 1, the ΔG values of serotonin, cocaine, escitalopram and paroxetine at the S1 site were −28.38 kcal/mol, -46.42 kcal/mol, -51.47 kcal/mol and −51.34 kcal/mol, respectively. Further, the ΔG values of serotonin, cocaine, escitalopram at the S2 site were −18.88 kcal/mol, -39.60 kcal/mol and −38.76 kcal/mol, respectively.
After only considering the comparison of relative values (without absolute values taken into accout), the trend of the MM/GBSA results was consistent with the previous studies and the trend of experimental values. The results suggest in thermodynamics at the equilibrium states, the other three ligands (cocaine, escitalopram and paroxetine) were much more favorable for the S1 site than the original substrate serotonin. The same holds true for the S2 site (in the experiment, whether paroxetine can bind the S2 site wasn’t found). In general, the S1 site was much more favorable than the S2 site, for each ligand.
For a more detailed understanding of the interaction mechanisms between SERT and the four ligands, we further analyzed the effects of the energy component of ΔG in each system. As shown in Figure 3, the cocaine-binding affinity’s main contribution was van der Waals force (ΔEvdW), binding at the S1 site (-42.53 kcal/mol) and S2 (-39.47 kcal/mol) (Table S8), comparing the original substrate serotonin, and the solvation free energy (ΔGsol) was beneficial to cocaine competing for the target binding site of SERT. Between among bindings of the three drugs with SERT, the electrostatic interactions for escitalopram have very significant advantages. Overall, van der Waals interactions have fundamental roles in associations of cocaine, escitalopram and paroxetine with SERT, whether the S1 site or S2. Accordingly, in future drug designs towards SERT van der Waals interactions between inhibitors and SERT would be keys for the successful development of inhibitors, and Enhancing the electrostatic interaction of drugs may be a potential strategy for developing potent drugs.
Based on previous studies, for Drosophila dopamine transporter (dDAT), another important monoamine transporter in the neurotransmitter sodium symporter (NSS) family and closely related to SERT[42], cocaine (a drug of abuse) obviously hads higher competitiveness than its original substrate dopamine. In this respect, SERT and DAT showed the similar behavior for cocaine binding.
2.3. Potential of mean forces along the unbinding pathways
To explore inhibitors interactions with the corresponding target protein in kinetics along the unbinding pathways and to further understanding the mechanisms of each ligand studied in this work with SERT, we performed PMF simulation. The PMF results coincided perfectly well with the trend of our previous MM/GBSA results. As shown in Figure 2A, among the four ligands, the PMF of serotonin dissociated from the S1 site was the lowest (60.81 kcal/mol). As a drug of abuse, cocaine had a higher PMF of 74.56 kcal/mol. The PMFs for the two clinically used antidepressants, escitalopram and paroxetine, were 132.66 kcal/mol and 93.35 kcal/mol, respectively. Therefore, we can conclude that escitalopram was the most competitive for binding to SERT, with a PMF nearly twice of that for the substrate serotonin. In addition, our result for escitalopram was quite consistent with the result of 136.23 kcal/mol (when the S2 site is occupied by a second escitalopram) from the previous steered MD study by F Zhu et al.; however, it was much higher than their result of 111.97 kcal/mol (when the S2 site was empty), though we adopted a different method (including model, protocol, parameters, etc.) compared to theirs methods.
Figure 4.
Potentials of mean force (PMF) profiles of substrate/drugs dissociated from S1 and S2 sites of SERT. the cyan solid line represents that the substrate/drugs have escaped the protein via the channel. (A) The value of escitalopram dissociated from the S1 site (132.66 kcal/mol) was higher than that of paroxetine (93.35 kcal/mol), cocaine (74.56 kcal/mol) and serotonin (60.81 kcal/mol). The position of serotonin and cocaine dissociated from the S1 site to S2 site are represented a black and red circles with a and b representations, respectively. the value of serotonin and cocaine dissociated from the S2 site to away from protein was 10.15 kcal/mol and 17.18 kcal/mol respectively. (B) The PMF value of cocaine dissociated from the S2 site (31.24 kcal/mol) was higher than that of escitalopram (20.07 kcal/mol) and serotonin (10.36 kcal/mol). the length of the path to dissociate from the S2 site of cocaine was equal to that of escitalopram, and both of there was ~3 Å smaller than that of serotonin.
Figure 4.
Potentials of mean force (PMF) profiles of substrate/drugs dissociated from S1 and S2 sites of SERT. the cyan solid line represents that the substrate/drugs have escaped the protein via the channel. (A) The value of escitalopram dissociated from the S1 site (132.66 kcal/mol) was higher than that of paroxetine (93.35 kcal/mol), cocaine (74.56 kcal/mol) and serotonin (60.81 kcal/mol). The position of serotonin and cocaine dissociated from the S1 site to S2 site are represented a black and red circles with a and b representations, respectively. the value of serotonin and cocaine dissociated from the S2 site to away from protein was 10.15 kcal/mol and 17.18 kcal/mol respectively. (B) The PMF value of cocaine dissociated from the S2 site (31.24 kcal/mol) was higher than that of escitalopram (20.07 kcal/mol) and serotonin (10.36 kcal/mol). the length of the path to dissociate from the S2 site of cocaine was equal to that of escitalopram, and both of there was ~3 Å smaller than that of serotonin.
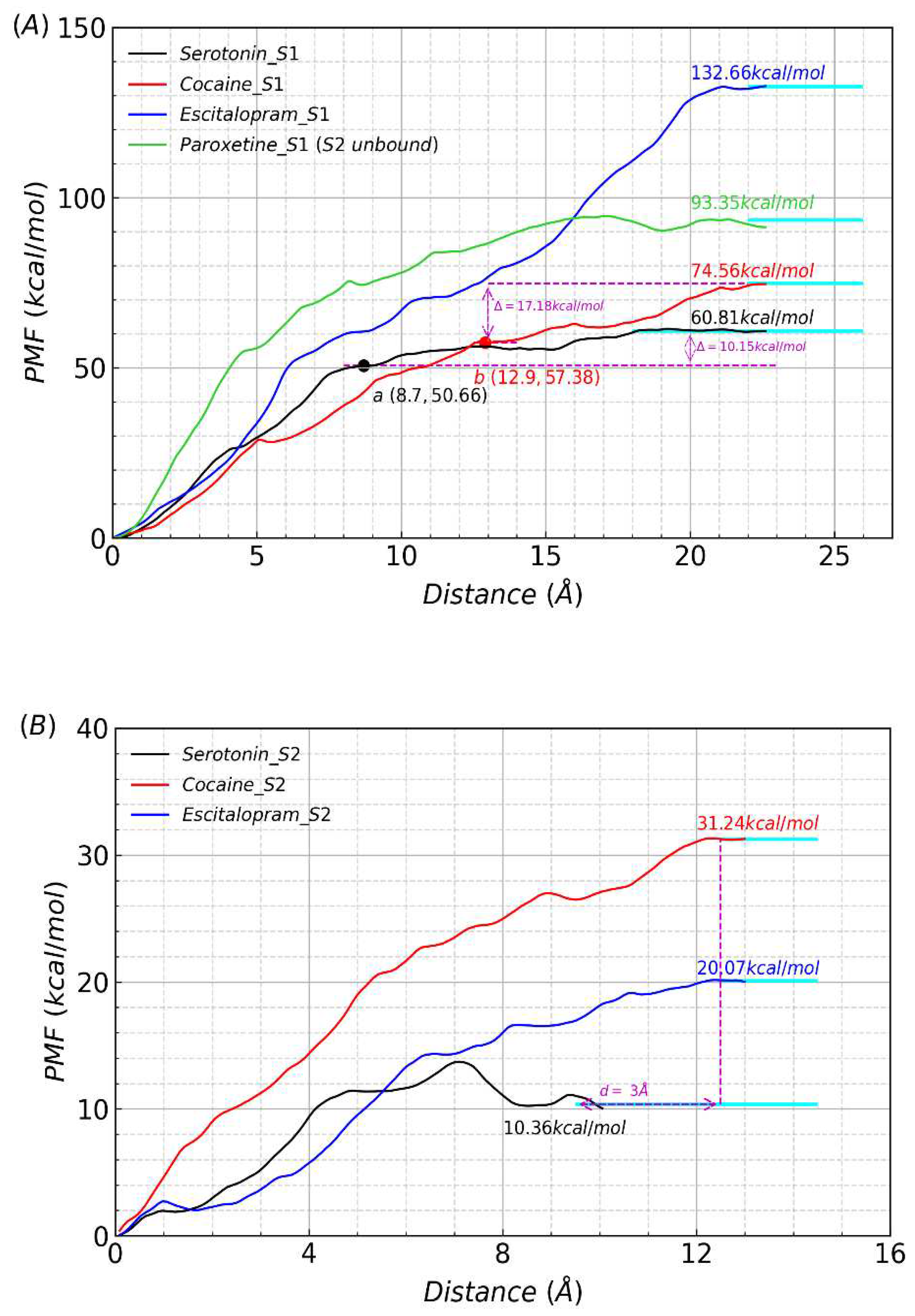
On the one hand, whether in kinetics along the unbinding pathways or in thermodynamics at the equilibrium states, for each ligand the PMF from the S2 site was much lower than that from the S1 site, which was reasonable in consideration of the relative positions of the two sites in SERT. On the other hand, limited to comparation only between the three ligands (serotonin, cocaine and escitalopram), the S2 site unbonded paroxetine, the situation was similar and different to that for the S1 site. Firstly, serotonin has the lowest PMF of 10.36 kcal/mol, among the three ligands. Secondly, our result of 20.07 kcal/mol for escitalopram is extremely in accordance with the result of 17.25 kcal/mol (when the S1 site is occupied by a second escitalopram) from the previous steered MD study by F Zhu et al., while much lower than their result of 33.72 kcal/mol (when the S1 site is empty). thirdly, the PMF value of cocaine dissociated from the S2 site (31.24 kcal/mol) was higher than that of escitalopram (20.07 kcal/mol) and serotonin (10.36 kcal/mol), as shown in Figure 2B.
In this article, based on the knowledge of structure of SERT and the mothed of umbrella-sampling all the steered MD of four systems were performed selecting the same three (K85, H143, E453, PDB code: 5I73) residues as the reference for the reaction coordinate. The pathways of ligands dissociated from the S1 site or S2 site were further investigated to gain some insights into the unbinding process. Based on the statistics of PMF windows counts (from the point at the binding site to the point of the absolute dissociation state which corresponds to the final platform of the PMF profile), the length for the dissociation pathway of serotonin from the S1 binding site (about 18 Å) was slightly shorter than that of other three ligands (cocaine, escitalopram and paroxetine), which was ~ 22 Å. This observation indicates that cocaine, escitalopram and paroxetine bound more deeply to the S1 site than serotonin. However, the length for the dissociation pathway of serotonin from the S2 binding site (about 12.5 Å) is slightly longer than that of cocaine or escitalopram, which is about 9.5 Å. Therefore, for the S2 binding site, cocaine or escitalopram is closer to the protein surface than serotonin. Investigating the structures of each window frame by frame with PMF values was to better understand unbinding processes of the four ligands. we found that the distances of serotonin and cocaine at the S1 site pulled to the S2 site were 8.7 Å (29th window) and 12.9 Å (43rd window), respectively (Figure 2A). But escitalopram at the S1 site directly moved without via the S1 site. Summarizing the above analysis, we predict that the lengths of pathways from the orthosteric site of SERT to the opening were ~ 18 Å for serotonin and ~ 22 Å for cocaine, escitalopram and paroxetine. Experimentally the size of SERT is ~ 55 Å[43] and the above speculations are valid. Similarly, it was presumed that the distances of pathways from the S2 site to an opening were ~ 9.5 Å for serotonin and ~ 12.5 Å for cocaine and escitalopram, respectively. The distances between serotonin and cocaine at the allosteric site were ~ 3 Å (Figure 2B). This maybe a further proof for the existence of another S2 site. But the PMF value of cocaine initially at the S1 site dissociated from the S2 site (17.18 kcal/mol) was still higher than that of serotonin initially at the S1 site (10.15 kcal/mol) (Figure 2A).Overall, the cocaine abuse, whether at the S1 site or S2, has a stronger binding ability beyond substrate serotonin and the depth-binding drug may be a novel respect for the treatment of depression in future.
2.4. Structural analysis and dissociative process of PMF simulations
During PMF simulations of the four complexes, SERT was always maintains stable and it underwent slight conformational changes; therefore, SERT could be a perfect background for further investigation of the ligands’ behaviors including movement relative to the protein. Thus, we investigated the intermediate snapshots of each system during our PMF simulations (a-f in Figure S2A-D). The unbinding pathways of the four ligands, except escitalopram at the orthosteric site, were similar; i.e., each ligand exported from the channel gradually with each step choosing the broadest space, which validates the reasonability of our definition for PMF reaction coordinates. Along the unbinding pathway, significant conformational changes (rotations) occurred for each ligand to fit different protein environment. Surprisedly, escitalopram at the S1 site was different from the other molecules, pulled out SERT directly along the MT8 shielding the S2 site departure from the channel (Figure 5A) (a-f in Figure S2C). Serotonin, cocaine, paroxetine, escitalopram (at the S2 site) were all via the broadest pathways from the orthosteric site of SERT to an opening between MT1b and MT6a (Figure 5B) (a-f in Figure S2A, B and D). The pathways is consistent with experiments[16,43]. Since serotonin and cocaine were dissociated from a similar opening, the previous presumption is rational that distances between serotonin and cocaine at the allosteric site were ~ 3Å.
In order to better understand the PMF values difference between serotonin or cocaine dissociated from the S1 site to the S2 site then leaving the channel and directly from the S2 site away, the corresponding structures were superimposed and analyzed the corresponding RMSDs of the protein Cα, when both ligands were at the S2 site during the PMF simulations. In the 1st window when the ligands began to be pulled away from the S1 site along the direction of intracellular to extracellular, the both RMSDs of 1st window increased slightly. Finally, it were constant as the ligands reached the S2 site, in the 29th window (1.75 Å) and the 43rd window (1.97Å), respectively (Figure 6, A and C). The variation of RMSDs were reasonable. Further, as substrate uptake into cells from the extracellular solution to the primary substrate site the transporter in an open-to-out conformation[44]. In theory, the observations may provide slight verification that the molecular activity phenomenon that conformation transitions between the outward-open to the occluded state[44,45]. Based on the RMSDs and compared the initial structures, protein both of all underwent slight conformational changes (Figure 6, B and D). Similarly, when ligands were pulled to the S2 site, the conformations of ligands occurred as an angle flip. Thus, considerations of variations between the conformational comparisons of structures of proteins and ligands could interpret the differences above values in our steered molecular dynamics simulations. In summary, we could conclude that the abuse drug cocaine was more competitive than the natural substrate serotonin, whether at the S1 site or S2, when SERT drive to reuptake.
3. Materials and Methods
3.1. Systems Preparation
Four simulation complexes were modeled. The complexes escitalopram-bound and paroxetine-bound were directly obtained from X-ray structures of SERT (PDB codes: 5I73 and 5I6X)[27], escitalopram simultaneously binding at the S1 site and S2 but paroxetine only at S1. Serotonin and cocaine (extracted from PDB code:6HIQ[46], 1Q72[47] respectively) were docked into the S1 site and S2 of SERT (PDB code: 5I73) in MOE[48] software: (i) Putting into SERT-escitalopram and dealing the structure with default databases. (ii)Selecting escitalopram atoms as the docking site and using wall constraint. (iii) 300 placement poses following 5 refinement poses. The RMSDs of the re-docked and co-crystalized ligand approximately were 1.60Å (serotonin bound S1), 1.87Å (serotonin bound S2), 1.07Å (cocaine bound S1) and 0.95Å (cocaine bound S2), respectively. The results is rational. The cholesterol (CLR) molecule contained in the experimental structure was reserved in consideration of its probable role in maintaining SERT’s conformation (Figure 1A-B). Three mutations: T110A, I291A, and T439S in the crystal structure of SERT ware mutated back to its native state using the mutagenesis tool in PyMOL. CHARMM-GUI was used to build the model of protein inserted into DOPC lipid bilayer (Figure S2). 128 × 128 phospholipid molecules (DOPC lipid) were arranged in the X and Y axis (XY dimension ratio was 1) and solvated with KCl (0.07 M), resulting in a box with approximate dimensions of 106 × 106 × 150 Å3 that contained around 150,000 atoms.
3.2. Conventional Molecular Dynamics Simulation
For each structure, energy-minimization was performed by using the Sander module of Amber 18 program[49,50,51,52,53] via a combined use of a 4000-step steepest descent calculation followed by a 36000-step conjugate gradient optimization, with a convergence criterion of 0.01 kcal·mol-1·Å-2, and the non-bonded cutoff distance was set to 10.0 Å. MD simulation was performed by using the Sander module of the Amber18 program package. SERT protein, DOPC liquids, K+ and Cl- ions, and TIP3P water were described using AMBER ff14SB[54], Lipid 14[55], and monovalent ion parameters for TIP3P water, respectively. The parameters of serotonin, cocaine, escitalopram, paroxetine and cholesterol were generated by Antechamber using GAFF2 Force Fields[56]. The partial charges of atoms of the ligands were calculated by using the restrained electrostatic potential-fitting (RESP) protocol implemented in the Multiwfn program[57], following the electrostatic potential (ESP) calculation at ab initio B3LYP/6-311+G(2d.p) level using Gaussian 09 program[58]. The electrostatic potential involved in the analyses was evaluated by Multiwfn based on the highly effective algorithm[59]. Sodium counter ions (Na+) were added to neutralize the solvated system. The solvated system was gradually heated to 298.15 K by the weak-coupling method[60] and equilibrated for 150 ps. During the MD simulations, a 10.0 Å non-bonded interaction cutoff was used and the non-bonded list was updated every 25 steps. The motion for the mass center of the system was removed every 1000 steps. The particle-mesh Ewald (PME) method[61] was used to treat long-range electrostatic interactions. The lengths of covalent bonds involving hydrogen atoms were fixed with the SHAKE algorithm[62], enabling the use of a 2-fs time step to numerically integrate the equations of motion. Finally, the production MD was kept running for 100ns with a periodic boundary condition in the NPT ensemble at T=298.15 K with Berendsen temperature coupling, and at P=1 atm with isotropic molecule-based scaling[63].
In the energy-minimization and heating procedures all of the ligands, protein and lipid atoms were restrained by a harmonic potential with a force constant of 2000kcal·mol-1·Å-2. The following NPT equilibration simulation divided into 6 steps: (1) 100ps, all of the ligand, protein and lipid atoms were restrained; (2) 100ps, the atoms of the protein were restrained; (3) 100ps, the mainchain atoms of the protein were restrained; (4) 100ps, the backbone atoms of the protein were restrained; (5) 100ps, the atoms of the protein were restrained; (6) 100ps, none of the systems was restrained. The harmonic potential used in equilibration simulation was the same as that used in energy-minimization and heating procedures.
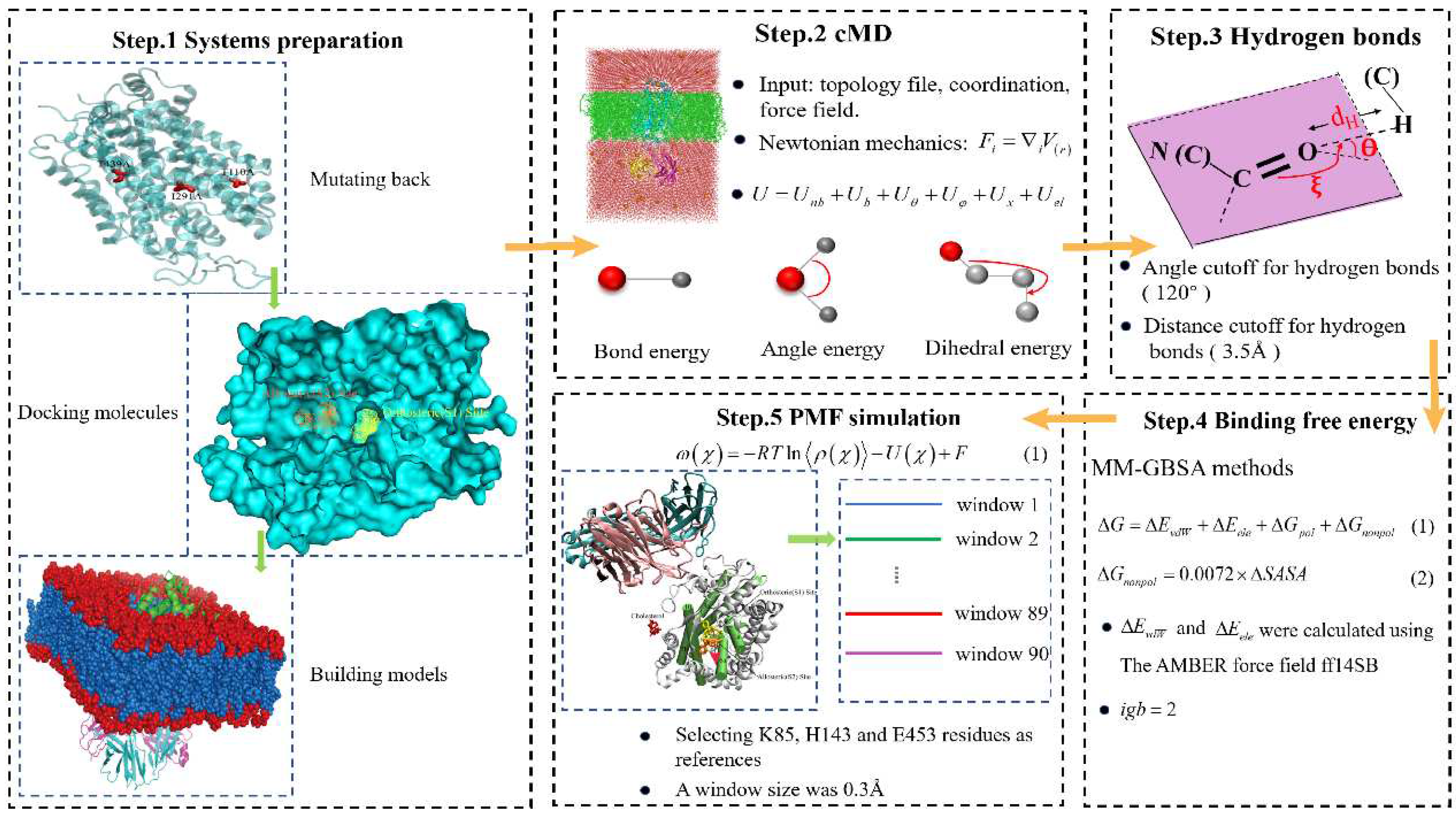
Hydrogen bonds analyses were based on the production simulation trajectory. The criterion for forming a hydrogen bond was that the distance between the heavy atoms was no larger than 3.5Å and the angle between the acceptor, hydrogen, and donor atoms was no smaller than 120 degree. The workflow of our work was showed as Figure 5.
Figure 7.
The workflow of our work. Firstly, three mutations of SERT are mutated back to its native state using the mutagenesis tool in PyMOL, and then serotonin molecules or cocaine are docked into two target pockets (S1 site and S2 site) of SERT in AutoDock Vina, and next CHARMM-GUI is employed to insert four complexes into DOPC lipid bilayer. Then, the conventional MD is performed for each structure. Next, Hydrogen bonds analyses is employed for the interactions of substrate and three drugs to SERT after systems has been stable. Next, the binding free energy is calculated with MM-GBSA method in thermodynamics at the equilibrium states. Finally, the PMF simulation is carried out by using umbrella-sampling MD simulation.
Figure 7.
The workflow of our work. Firstly, three mutations of SERT are mutated back to its native state using the mutagenesis tool in PyMOL, and then serotonin molecules or cocaine are docked into two target pockets (S1 site and S2 site) of SERT in AutoDock Vina, and next CHARMM-GUI is employed to insert four complexes into DOPC lipid bilayer. Then, the conventional MD is performed for each structure. Next, Hydrogen bonds analyses is employed for the interactions of substrate and three drugs to SERT after systems has been stable. Next, the binding free energy is calculated with MM-GBSA method in thermodynamics at the equilibrium states. Finally, the PMF simulation is carried out by using umbrella-sampling MD simulation.
3.3. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA)
MM/GBSA method[64] was employed to calculate the binding free energy(ΔG) between SERT and each ligand. The method has been successfully applied to rank the relative binding free energy of small molecules in membrane proteins[15,65,66,67,68,69]. In this work, using the 100 snapshots extracted from the last 80 ns equilibrium MD trajectory, ΔG was estimated as below.
The terms and represent the van der Waals and electrostatic interaction energies in the gas phase, respectively. and are the polar and nonpolar solvation energies, respectively. and were calculated using the AMBER force field ff14SB[36]. The free energy of polar solvation () was calculated by the modified GB model (), and solute and solvent dielectric constants were set to 2 and 80, respectively. The free energy of nonpolar solvation () was calculated by , and was estimated by the DOPC method with 1.4 Å Probe radii.
3.4. Steered Molecular Dynamics Simulation
In order to explore the free energy profiles for each complex, the potential of mean force (PMF) simulation was carried out by using umbrella-sampling[70] MD simulation. The classic PMF definition[71] can be represented by a function of reaction coordinate as below.
in which ρ(χ) is the probability density along the reaction coordinate χ, R is the gas constant, T is the simulation temperature, U(χ) is the biasing potential applied in the umbrella-sampling MD simulation, and F is the normalization constant. According to this approach, the reaction coordinate is usually divided into different regions, i.e., windows, and each window is sampled separately. A biasing (umbrella) potential, i.e., U(χ) is applied for each window in order to obtain nearly uniform sampling of the potential energy surface. In the present study, the reaction coordinate was defined as the distance from the mass center of the non-hydrogen atoms of each ligand to the mass center of the atoms of several residues of SERT. The total number of windows was 90, with a window size of 0.3 Å. The biasing force constant applied in different windows of umbrella-sampling was 10.0 kcal/(mol/Å2). For each umbrella-sampling window, the initial complex structure was selected from the last snapshot of the PMF simulations of the previous window. The selected structure for each window was first equilibrated for 200 ps and then kept running for 800 ps for production sampling. The frequency for data collection was set to 1 fs, which was the same as that of the time step of the umbrella-sampling MD. After the umbrella-sampling MD simulations for all windows were completed, the data collected from separate simulation windows were combined along the reaction coordinate. These data were then used to calculate the PMF for the whole binding process with the weighed histogram analysis method (WHAM)[72,73] using the code developed by Alan Grossfield (http://membrane.urmc.rochester.edu/?page_id=126).
4. Conclusions
In this work, combined MD and PMF simulations were employed to probe into the molecular mechanism of unbinding of substrate serotonin and drug of cocaine abuse and two antidepressants (escitalopram and paroxetine) against the S1or S2 sites in human SERT. For serotonin, cocaine and escitalopram, the hydrogen bonds for the S2 site were much weaker than those for the S1 site. Moreover, the identification of key residues Ser438, Tyr (95 and 175) and Arg104 was important for development of the selective and effective drugs of SERT, and further, Tyr176 may be the potency to design selectivity antidepressants upon binding the S1 site. In kinetics along the unbinding pathways and thermodynamics at the equilibrium states, cocaine, escitalopram and paroxetine (S2 unbound), whether at the S1 or S2, are much more favorable than the natural substrate serotonin and the inhibitory effect of drugs is more potent at the S1 site. Further, van der Waals interactions were beneficial to the affinities of the three drugs comparing substrate serotonin and electrostatic interactions were primary contributions for escitalopram. We predicted that distances of pathways from the S1 site to an opening, for serotonin and the three-mentioned drugs, were ~ 18 Å and ~ 22 Å, respectively. Furthermore, the distances between serotonin and cocaine at the allosteric site was ~ 3 Å. Continuing exploring the processes of unbinding four ligands against the two target pockets of SERT, this study better understands the interactions between SERT and drugs. The insights that the broadest pathway, from the central binding site of SERT to the opening between MT1b and MT6a, verified previous works, and maybe, in theory, provide slight proofs for the molecular activity phenomenon that conformation transitions between the outward-open to the occluded state. Our work would be beneficial for further studies of better inhibitors to treat depression.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Hydrogen bond analysis with the equilibrium trajectories of SERT- serotonin complex at S1 site; Table S2: Hydrogen bond analysis with the equilibrium trajectories of SERT- serotonin complex at S2 site; Table S3: Hydrogen bond analysis with the equilibrium trajectories of SERT- cocaine complex at S1 site; Table S4: Hydrogen bond analysis with the equilibrium trajectories of SERT- cocaine complex at S2 site; Table S5: Hydrogen bond analysis with the equilibrium trajectories of SERT- escitalopram complex at S1 site; Table S6: Hydrogen bond analysis with the equilibrium trajectories of SERT- escitalopram complex at S2 site; Table S7: Hydrogen bond analysis with the equilibrium trajectories of SERT- paroxetine complex at S1 site; Table S8. The energy component of binding free energy (MM/GBSA method); Figure S1: The model of the membrane protein of SERT; Figure S2: The intermediate snapshots of four system during the PMF simulation.
Author Contributions
Conceptualization, X.-G.T. and X.L.; formal analysis, X.-G.T., X.-F.L., X.L., M.-H.P., Y.-Q.W. and Y.-J.Z.; funding acquisition, X.-F.L., Y.-J.Z.; investigation, X.-G.T. and X.L.; methodology, X.-G.T., X.-F.L and Y.-J.Z. resources, X.-F.L. and Y.-J.Z.; software, X.-G.T.; data curation, X.-G.T., M.-H.P. and Y.-Q.W.; writing—original draft preparation, X.-G.T.; writing—review and editing, X.-G.T., X.-F.L., X.L., M.-H.P., Y.-Q.W. and Y.-J.Z.; supervision, X.-F.L. and Y.-J.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No.11904036 (XL)), (Grant No.12175081 (YZ)) and Fundamental Research Funds for the Central Universities CCNU22 QN004 (YZ).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet gastroenterology & hepatology 2020, 5, 245–266. [Google Scholar]
- Coccaro, E.F. New Hope for Patients with Major Depressive Disorder? 2019, 381, 980-981. [CrossRef]
- Recourt, K.; de Boer, P.; Zuiker, R.; Luthringer, R.; Kent, J.; van der Ark, P.; Van Hove, I.; van Gerven, J.; Jacobs, G.; van Nueten, L. The selective orexin-2 antagonist seltorexant (JNJ-42847922/MIN-202) shows antidepressant and sleep-promoting effects in patients with major depressive disorder. Translational psychiatry 2019, 9, 1–10. [Google Scholar]
- Saltiel, P.F.; Silvershein, D.I. Major depressive disorder: mechanism-based prescribing for personalized medicine. Neuropsychiatric disease and treatment 2015, 11, 875. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, D.J.; Frank, E.; Phillips, M.L. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. The Lancet 2012, 379, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Caspi, A.; Sugden, K.; Moffitt, T.E.; Taylor, A.; Craig, I.W.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef]
- Fuller, R.W.; Wong, D.T. Serotonin uptake and serotonin uptake inhibition. Annals of the New York Academy of Sciences 1990. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.L.; Lerner, A.; Rudnick, G.; Lesch, K.-P. Serotonin transporter: gene, genetic disorders, and pharmacogenetics. Molecular interventions 2004, 4, 109. [Google Scholar] [CrossRef]
- Chen, N. Reith ME, and Quick MW. Synaptic uptake and beyond: the sodium-and chloride-dependent neurotransmitter transporter family SLC6. Pflügers Arch 2004, 447, 519-531. [CrossRef]
- Sitte, H.H.; Freissmuth, M. Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends in pharmacological sciences 2015, 36, 41–50. [Google Scholar] [CrossRef]
- Niello, M.; Gradisch, R.; Loland, C.J.; Stockner, T.; Sitte, H.H. Allosteric modulation of neurotransmitter transporters as a therapeutic strategy. Trends in Pharmacological Sciences 2020, 41, 446–463. [Google Scholar] [CrossRef]
- Rothman, R.B.; Partilla, J.S.; Baumann, M.H.; Lightfoot-Siordia, C.; Blough, B.E. Studies of the biogenic amine transporters. 14. Identification of low-efficacy “partial” substrates for the biogenic amine transporters. Journal of Pharmacology and Experimental Therapeutics 2012, 341, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Hasenhuetl, P.S.; Bhat, S.; Freissmuth, M.; Sandtner, W. Functional selectivity and partial efficacy at the monoamine transporters: a unified model of allosteric modulation and amphetamine-induced substrate release. Molecular Pharmacology 2019, 95, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Fu, T.; Deng, S.; Yang, F.; Yang, J.; Zhu, F. Molecular mechanism for the allosteric inhibition of the human serotonin transporter by antidepressant escitalopram. ACS chemical neuroscience 2022, 13, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Gouaux, E. Illumination of serotonin transporter mechanism and role of the allosteric site. Science Advances 2021, 7, eabl3857. [Google Scholar] [CrossRef]
- Chan, M.C.; Selvam, B.; Young, H.J.; Procko, E.; Shukla, D. The substrate import mechanism of the human serotonin transporter. Biophysical journal 2022, 121, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Gether, U.; Andersen, P.H.; Larsson, O.M.; Schousboe, A. Neurotransmitter transporters: molecular function of important drug targets. Trends in pharmacological sciences 2006, 27, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, A.S.; Andersen, J.; Jørgensen, T.N.; Sørensen, L.; Eriksen, J.; Loland, C.J.; Strømgaard, K.; Gether, U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacological reviews 2011, 63, 585–640. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.; Blakely, R. Monoamine transporter gene structure and polymorphisms in relation to psychiatric and other complex disorders. The pharmacogenomics journal 2002, 2, 217–235. [Google Scholar] [CrossRef] [PubMed]
- Severinsen, K.; Koldsø, H.; Thorup, K.A.V.; Schjøth-Eskesen, C.; Møller, P.T.; Wiborg, O.; Jensen, H.H.; Sinning, S.; Schiøtt, B. Binding of mazindol and analogs to the human serotonin and dopamine transporters. Molecular pharmacology 2014, 85, 208–217. [Google Scholar] [CrossRef]
- Henry, L.K.; Field, J.R.; Adkins, E.M.; Parnas, M.L.; Vaughan, R.A.; Zou, M.-F.; Newman, A.H.; Blakely, R.D. Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. Journal of Biological Chemistry 2006, 281, 2012–2023. [Google Scholar] [CrossRef]
- Andersen, J.; Taboureau, O.; Hansen, K.B.; Olsen, L.; Egebjerg, J.; Strømgaard, K.; Kristensen, A.S. Location of the antidepressant binding site in the serotonin transporter. Journal of Biological Chemistry 2009, 284, 10276–10284. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Gouaux, E. Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nature structural & molecular biology 2018, 25, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Gradisch, R.; Szöllősi, D.; Niello, M.; Lazzarin, E.; Sitte, H.H.; Stockner, T. Occlusion of the human serotonin transporter is mediated by serotonin-induced conformational changes in the bundle domain. Journal of Biological Chemistry 2022, 298. [Google Scholar] [CrossRef]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Sinwel, D.; Hasenhuetl, P.S.; Saha, K.; Kumar, V.; Zhang, P.; Rankl, C.; Holy, M.; Sucic, S.; Kudlacek, O. Nanopharmacological force sensing to reveal allosteric coupling in transporter binding sites. Angewandte Chemie 2016, 128, 1751–1754. [Google Scholar] [CrossRef]
- Jørgensen, A.M.; Topiol, S. Driving forces for ligand migration in the leucine transporter. Chemical biology & drug design 2008, 72, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Topiol, S.; Bang-Andersen, B.; Sanchez, C.; Bøgesø, K.P. Exploration of insights, opportunities and caveats provided by the X-ray structures of hSERT. Bioorganic & Medicinal Chemistry Letters 2016, 26, 5058–5064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, G.; Fu, T.; Hong, J.; Li, F.; Yao, X.; Xue, W.; Zhu, F. The binding mode of vilazodone in the human serotonin transporter elucidated by ligand docking and molecular dynamics simulations. Physical Chemistry Chemical Physics 2020, 22, 5132–5144. [Google Scholar] [CrossRef]
- Millan, M.J. Dual-and triple-acting agents for treating core and co-morbid symptoms of major depression: novel concepts, new drugs. Neurotherapeutics 2009, 6, 53–77. [Google Scholar] [CrossRef]
- Stuivenga, M.; Giltay, E.J.; Cools, O.; Roosens, L.; Neels, H.; Sabbe, B. Evaluation of vilazodone for the treatment of depressive and anxiety disorders. Expert Opinion on Pharmacotherapy 2019, 20, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, P.; Siqueland, L.; Blaine, J.; Frank, A.; Luborsky, L.; Onken, L.S.; Muenz, L.R.; Thase, M.E.; Weiss, R.D.; Gastfriend, D.R. Psychosocial treatments for cocaine dependence: National Institute on Drug Abuse collaborative cocaine treatment study. Archives of general psychiatry 1999, 56, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Lambert, N.M.; McLeod, M.; Schenk, S. Subjective responses to initial experience with cocaine: an exploration of the incentive–sensitization theory of drug abuse. Addiction 2006, 101, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.A.B.; Plenge, P.; Andersen, J.; Eildal, J.N.; Kristensen, A.S.; Bøgesø, K.P.; Gether, U.; Strømgaard, K.; Bang-Andersen, B.; Loland, C.J. Structure–activity relationship studies of citalopram derivatives: examining substituents conferring selectivity for the allosteric site in the 5-HT transporter. British Journal of Pharmacology 2016, 173, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Wolynes, P.G. Biomolecules: where the physics of complexity and simplicity meet. Physics Today;(United States) 1994, 47. [Google Scholar] [CrossRef]
- García, A.E.; Sanbonmatsu, K.Y. α-Helical stabilization by side chain shielding of backbone hydrogen bonds. Proceedings of the National Academy of Sciences 2002, 99, 2782–2787. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, L.; Zhou, T.; Zeng, C.; Jia, Y.; Zhao, Y. A computational study of Tat–CDK9–Cyclin binding dynamics and its implication in transcription-dependent HIV latency. Physical Chemistry Chemical Physics 2020, 22, 25474–25482. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Zeng, C.; Zeng, C.; Zhao, Y. The TAR binding dynamics and its implication in Tat degradation mechanism. Biophysical Journal 2021, 120, 5158–5168. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, X.; Zheng, F.; Zhan, C.-G. Cocaine esterase–cocaine binding process and the free energy profiles by molecular dynamics and potential of mean force simulations. The Journal of Physical Chemistry B 2012, 116, 3361–3368. [Google Scholar] [CrossRef]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef]
- Coleman, J.A.; Yang, D.; Zhao, Z.; Wen, P.-C.; Yoshioka, C.; Tajkhorshid, E.; Gouaux, E. Serotonin transporter–ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 2019, 569, 141–145. [Google Scholar] [CrossRef]
- Singh, S.K.; Piscitelli, C.L.; Yamashita, A.; Gouaux, E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 2008, 322, 1655–1661. [Google Scholar] [CrossRef]
- Krishnamurthy, H.; Gouaux, E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 2012, 481, 469–474. [Google Scholar] [CrossRef]
- Polovinkin, L.; Hassaine, G.; Perot, J.; Neumann, E.; Jensen, A.A.; Lefebvre, S.N.; Corringer, P.-J.; Neyton, J.; Chipot, C.; Dehez, F. Conformational transitions of the serotonin 5-HT3 receptor. Nature 2018, 563, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Pozharski, E.; Moulin, A.; Hewagama, A.; Shanafelt, A.B.; Petsko, G.A.; Ringe, D. Diversity in hapten recognition: structural study of an anti-cocaine antibody M82G2. Journal of molecular biology 2005, 349, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Current topics in medicinal chemistry 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham III, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Computer Physics Communications 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz Jr, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. Journal of computational chemistry 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Frank, M.; Gutbrod, P.; Hassayoun, C.; von Der Lieth, C.-W. Dynamic molecules: molecular dynamics for everyone. An internet-based access to molecular dynamic simulations: basic concepts. Journal of Molecular Modeling 2003, 9, 308–315. [Google Scholar] [CrossRef]
- Karplus, M. Molecular dynamics of biological macromolecules: a brief history and perspective. Biopolymers: Original Research on Biomolecules 2003, 68, 350–358. [Google Scholar] [CrossRef]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: survey of methods for simulating the activity of proteins. Chemical reviews 2006, 106, 1589–1615. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of chemical theory and computation 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: the amber lipid force field. Journal of chemical theory and computation 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Man, V.H.; Yang, W.; Lee, T.-S.; Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. The Journal of Chemical Physics 2020, 153, 114502. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: a multifunctional wavefunction analyzer. Journal of computational chemistry 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, rev. Gaussian Inc, Wallingford 2009.
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Physical Chemistry Chemical Physics 2021, 23, 20323–20328. [Google Scholar] [CrossRef]
- Morishita, T. Fluctuation formulas in molecular-dynamics simulations with the weak coupling heat bath. The Journal of Chemical Physics 2000, 113, 2976–2982. [Google Scholar] [CrossRef]
- Toukmaji, A.; Sagui, C.; Board, J.; Darden, T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. The Journal of chemical physics 2000, 113, 10913–10927. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of computational physics 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.v.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. The Journal of chemical physics 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Accounts of chemical research 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Zheng, G.; Tu, G.; Yang, F.; Chen, Y.; Yao, X.; Li, X.; Xue, W.; Zhu, F. Exploring the binding mechanism of metabotropic glutamate receptor 5 negative allosteric modulators in clinical trials by molecular dynamics simulations. ACS chemical neuroscience 2018, 9, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Wang, P.; Li, B.; Li, Y.; Xu, X.; Yang, F.; Yao, X.; Chen, Y.Z.; Xu, F.; Zhu, F. Identification of the inhibitory mechanism of FDA approved selective serotonin reuptake inhibitors: an insight from molecular dynamics simulation study. Physical Chemistry Chemical Physics 2016, 18, 3260–3271. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Wang, P.; Tu, G.; Yang, F.; Zheng, G.; Li, X.; Li, X.; Chen, Y.; Yao, X.; Zhu, F. Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder. Physical Chemistry Chemical Physics 2018, 20, 6606–6616. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Xue, W.; Wang, P.; Yang, F.; Li, B.; Li, X.; Li, Y.; Yao, X.; Zhu, F. Exploring the inhibitory mechanism of approved selective norepinephrine reuptake inhibitors and reboxetine enantiomers by molecular dynamics study. Scientific reports 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Fu, T.; Zhang, X.; Yang, F.; Zheng, G.; Xue, W.; Chen, Y.; Yao, X.; Zhu, F. Differentiating physicochemical properties between NDRIs and sNRIs clinically important for the treatment of ADHD. Biochimica et Biophysica Acta (BBA)-General Subjects 2017, 1861, 2766–2777. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. Journal of Computational Physics 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kirkwood, J.G. Statistical mechanics of fluid mixtures. The Journal of chemical physics 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Berneche, S.; Roux, B. Energetics of ion conduction through the K+ channel. Nature 2001, 414, 73–77. [Google Scholar] [CrossRef]
- Roux, B. The calculation of the potential of mean force using computer simulations. Computer physics communications 1995, 91, 275–282. [Google Scholar] [CrossRef]
Figure 1.
Protein and ligand structures and related information used in this study. (A and B) Two target pockets of human SERT the orthosteric (S1) site (yellow stick) and allosteric (S2) site (orange stick) are confirmed in X-ray crystal structure. The human SERT and two 8B6 antibody molecules are colored in pink, cyan and white in cartoon representations, respectively. MT1, MT3, MT6 and MT10 are shown as green column, and cholesterol molecule is represented as red stick. Red arrow indicates the direction of the transit channel, from the intracellular to extracellular. (C) Chemical structures of the four ligands including their molecular names and non-standard names in PDB.
Figure 1.
Protein and ligand structures and related information used in this study. (A and B) Two target pockets of human SERT the orthosteric (S1) site (yellow stick) and allosteric (S2) site (orange stick) are confirmed in X-ray crystal structure. The human SERT and two 8B6 antibody molecules are colored in pink, cyan and white in cartoon representations, respectively. MT1, MT3, MT6 and MT10 are shown as green column, and cholesterol molecule is represented as red stick. Red arrow indicates the direction of the transit channel, from the intracellular to extracellular. (C) Chemical structures of the four ligands including their molecular names and non-standard names in PDB.
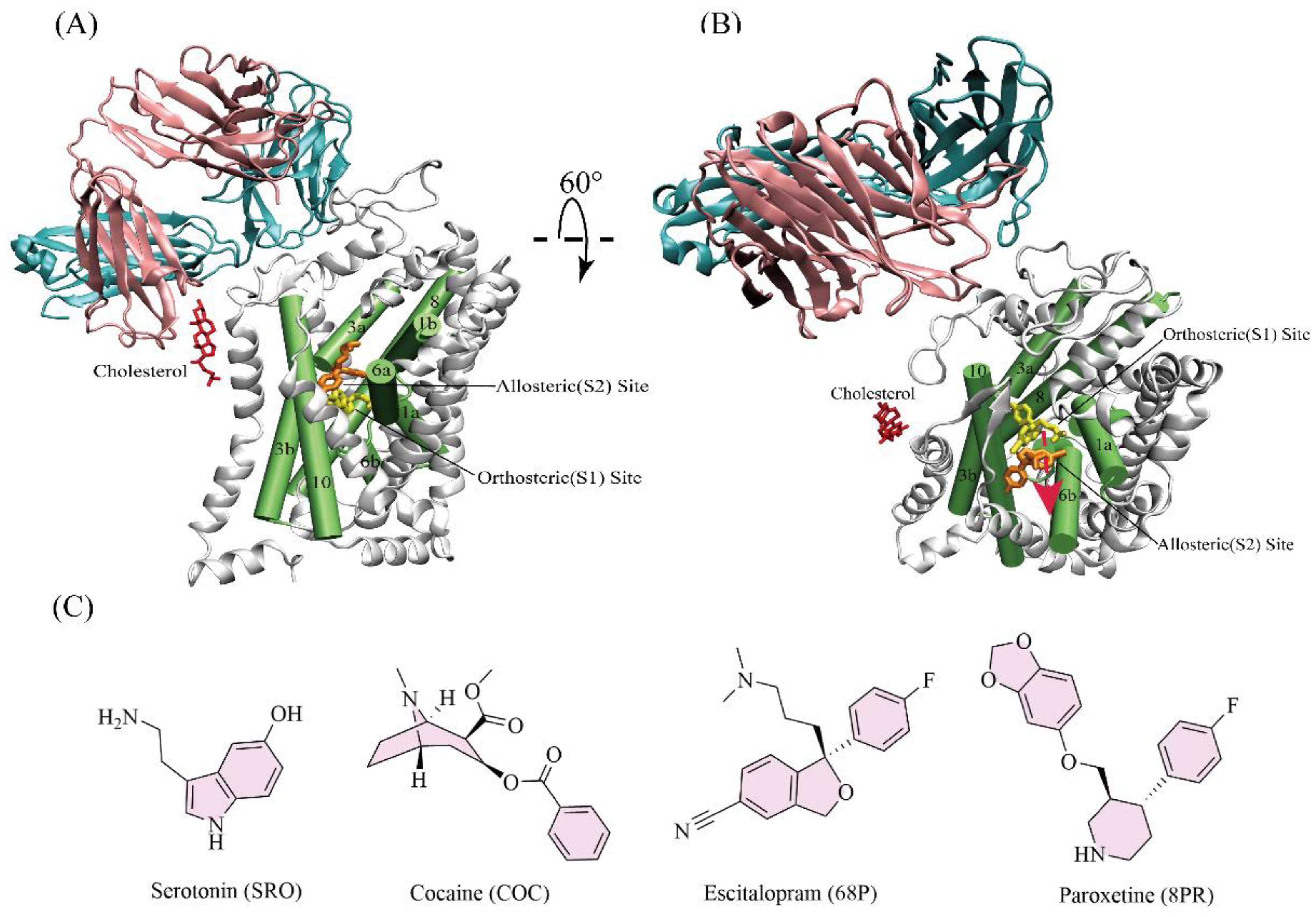
Figure 2.
The root-mean-square deviation (RMSD) of SERT Cα atoms. the RMSD of SERT-escitalopram system is markedly higher than that of the binding original substrate serotonin system, compared with cocaine and paroxetine, and the systems all are stable after about 30ns during the simulations.
Figure 2.
The root-mean-square deviation (RMSD) of SERT Cα atoms. the RMSD of SERT-escitalopram system is markedly higher than that of the binding original substrate serotonin system, compared with cocaine and paroxetine, and the systems all are stable after about 30ns during the simulations.
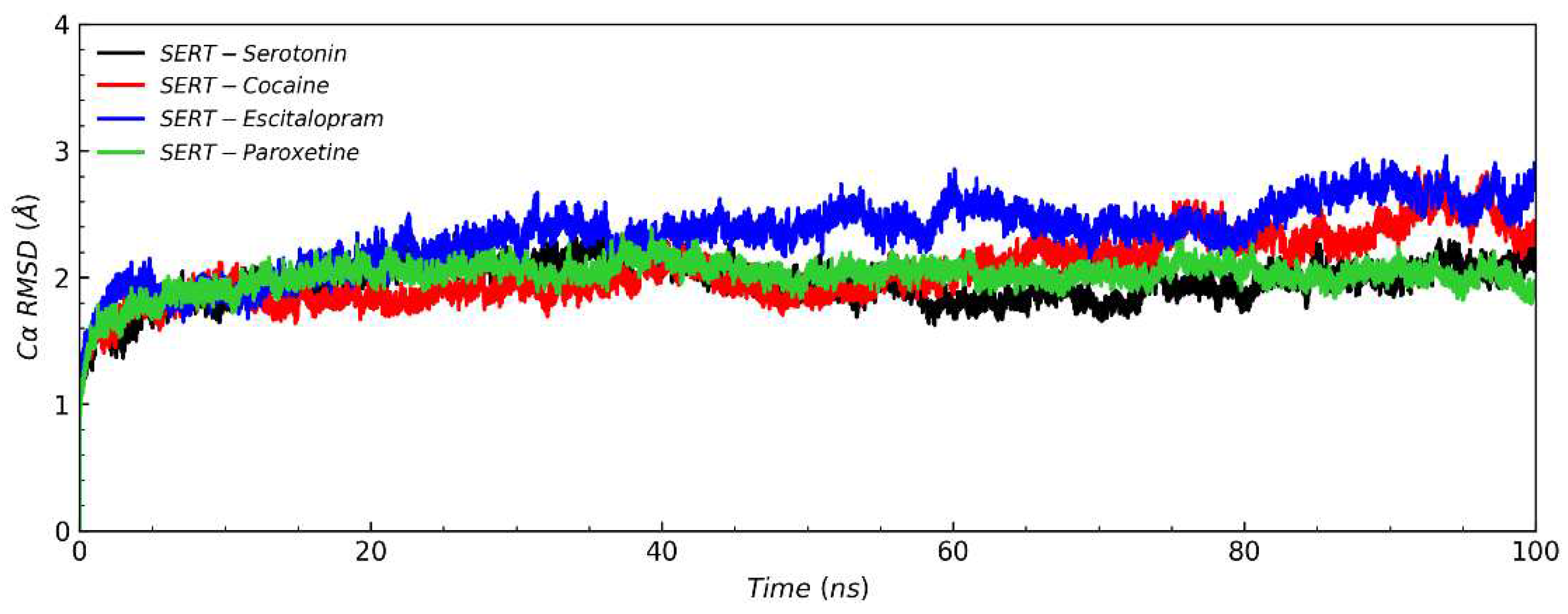
Figure 3.
The energy component of binding free energy (MM/GBSA method). ΔEvdW and ΔEele indicate the electrostatic and van der Waals interactions in the gas phase, respectively, and ΔGgas is the gas-phase energy (ΔEvdW and ΔEele). The solvation free energy (ΔGsol) can divide to polar (ΔGpol) and non-polar (ΔGnonpol). ΔGtotal denotes the total binding free energies. (A) ΔEvdW was favorable for associations of cocaine, escitalopram and paroxetine with SERT and ΔGsol was beneficial to cocaine competing for the target binding site of SERT. (B) Between among bindings of the three drugs with SERT, ΔEele for escitalopram have very significant advantages. ΔEele and ΔGsol of HERST-cocaine were -0.45 kcal/mol and 0.32 kcal/mol, respectively.
Figure 3.
The energy component of binding free energy (MM/GBSA method). ΔEvdW and ΔEele indicate the electrostatic and van der Waals interactions in the gas phase, respectively, and ΔGgas is the gas-phase energy (ΔEvdW and ΔEele). The solvation free energy (ΔGsol) can divide to polar (ΔGpol) and non-polar (ΔGnonpol). ΔGtotal denotes the total binding free energies. (A) ΔEvdW was favorable for associations of cocaine, escitalopram and paroxetine with SERT and ΔGsol was beneficial to cocaine competing for the target binding site of SERT. (B) Between among bindings of the three drugs with SERT, ΔEele for escitalopram have very significant advantages. ΔEele and ΔGsol of HERST-cocaine were -0.45 kcal/mol and 0.32 kcal/mol, respectively.
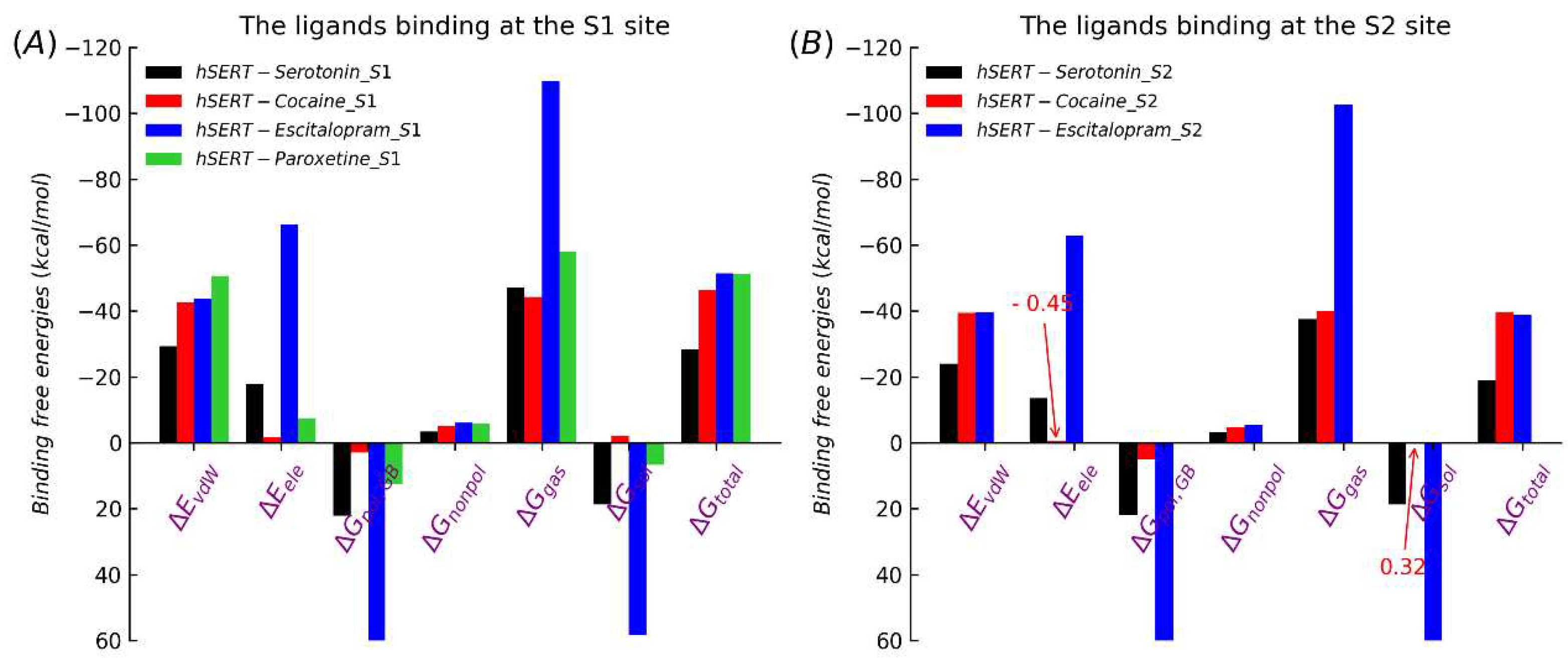
Figure 5.
Two unbinding pathways in this work. (A) escitalopram at S1 site dissociated from SERT directly along the MT8 shielding S2 site. (B) There is the broadest pathway where serotonin, cocaine, paroxetine and escitalopram (S2 bound) are all pulled towards a similar opening between MT1b and MT6a, whether at the S1 site or initially S2.
Figure 5.
Two unbinding pathways in this work. (A) escitalopram at S1 site dissociated from SERT directly along the MT8 shielding S2 site. (B) There is the broadest pathway where serotonin, cocaine, paroxetine and escitalopram (S2 bound) are all pulled towards a similar opening between MT1b and MT6a, whether at the S1 site or initially S2.
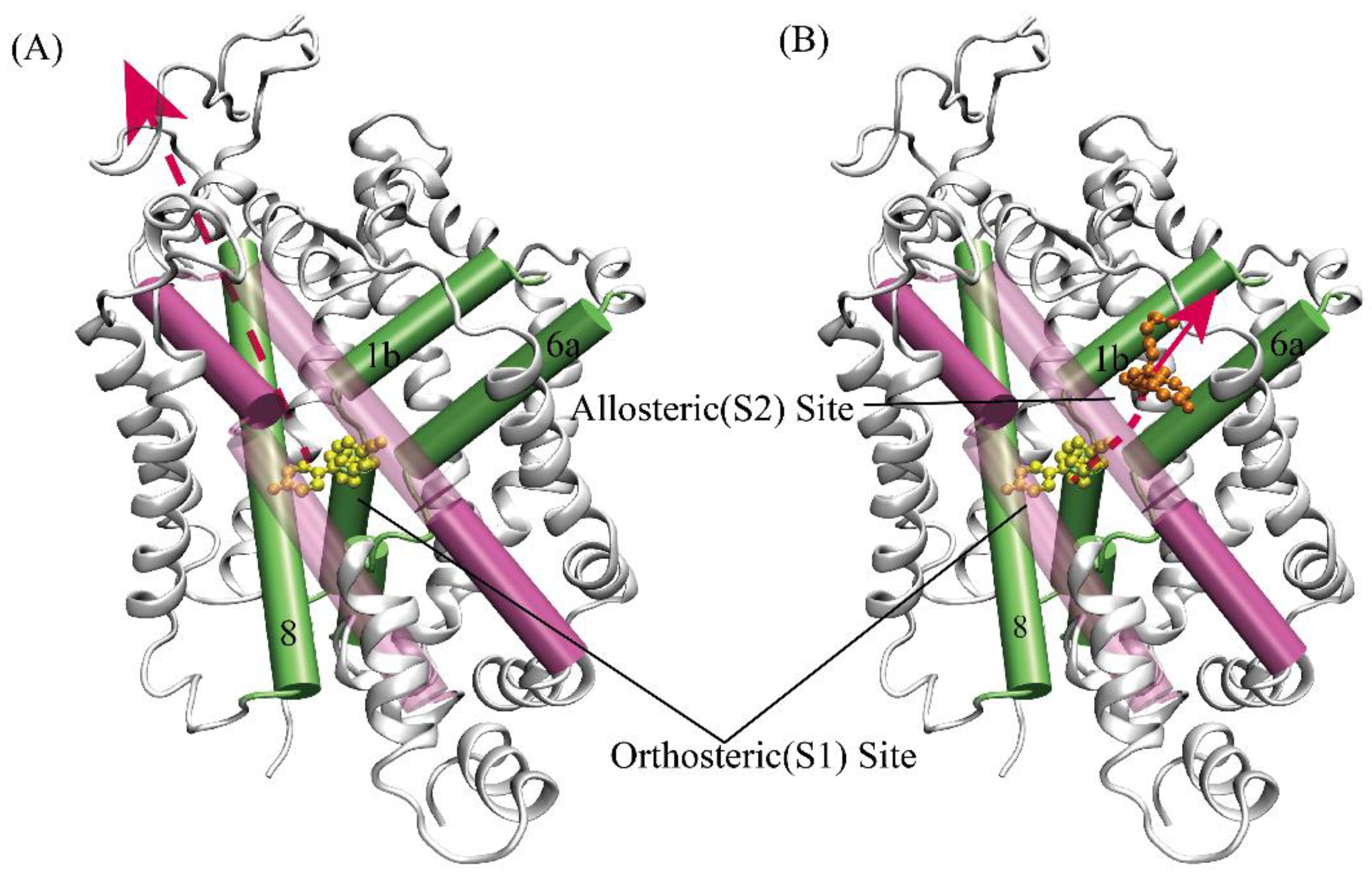
Figure 6.
Structure superposition at the S2 site and the RMSDs of the protein Cα in corresponding windows. (A, B) SERT-serotonin system. the structures of 0ns (the 1st window) and 29ns (the 29th window) were colored in magenta and green, respectively. (C, D) SERT-cocaine system. the structures of 0ns and 43ns (the 43rd window) were colored in yellow and blue, respectively. In the 1st window RMSDs of the protein Cα both increased slightly and kept stable in the 29th window (1.75 Å) and 43rd (1.97Å). Based on the RMSDs and compared the initial structures, protein both of all underwent slight conformational changes and the conformations of ligands occurred as an angle flip.
Figure 6.
Structure superposition at the S2 site and the RMSDs of the protein Cα in corresponding windows. (A, B) SERT-serotonin system. the structures of 0ns (the 1st window) and 29ns (the 29th window) were colored in magenta and green, respectively. (C, D) SERT-cocaine system. the structures of 0ns and 43ns (the 43rd window) were colored in yellow and blue, respectively. In the 1st window RMSDs of the protein Cα both increased slightly and kept stable in the 29th window (1.75 Å) and 43rd (1.97Å). Based on the RMSDs and compared the initial structures, protein both of all underwent slight conformational changes and the conformations of ligands occurred as an angle flip.
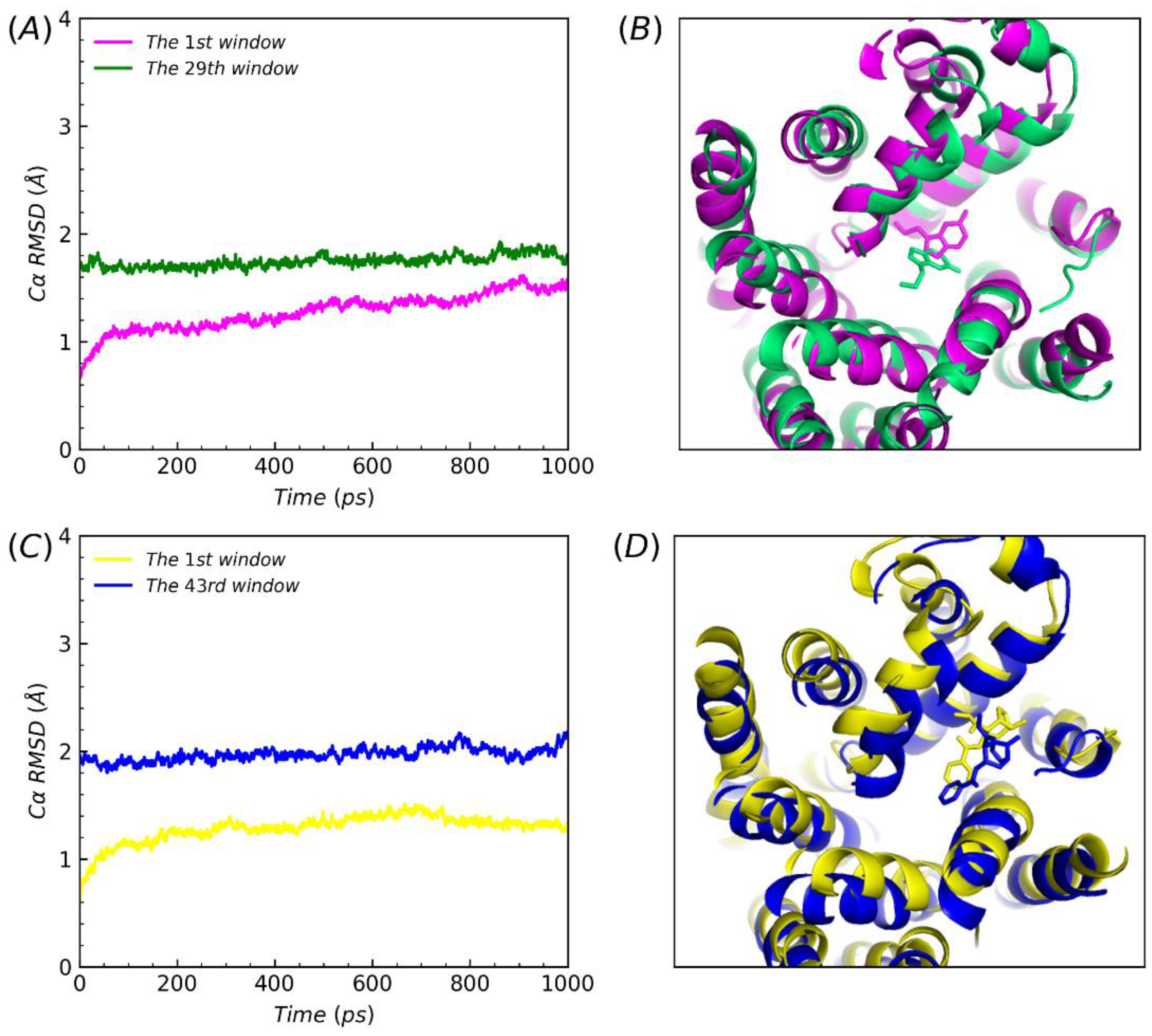

Table 1.
Calculated and Experimental Binding Free Energies of Studied substrate/Drugs to Targets.
| substrate/drug | target | ΔGcale (Kcal/mol)a | ΔGexp(Kcal/mol)b | Ki(nM)b |
|---|---|---|---|---|
| serotonin | SERT (S1)c | -28.38 | -6.62 | 14.2 μM[16] |
| SERT (S2)d | -18.88 | -5.90 | 48 μM | |
| cocaine | SERT (S1)c | -46.42 | f | |
| SERT (S2)d | -39.60 | |||
| escitalopram | SERT (S1)c | -51.47 | -10.23 | 32e[15] |
| SERT (S2)d | -38.76 | -7.16 | 5800 (IC50) | |
| paroxetine | SERT (S1)c | -51.34 | -10.40 | 24 |
aEstimated MM/GBSA binding free energy. bExperimental binding free energy (ΔGexp) based on Ki or IC50 values using ΔG = RTln(Ki) or ΔG ≈ RTln(IC50) (where R = 8.314 J/(mol·K), T = 310 K, IC50: half maximal inhibitory concentration)[31,41]. cSubstrate/Drug binding to the orthosteric (S1) site of SERT. dSubstrate/Drug binding to the allosteric (S2) site of SERT. ethe S2 site is empty, fNo experimental data reported.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



