
Submitted:
02 February 2023
Posted:
03 February 2023
You are already at the latest version
Abstract
The reaction of mercaptoacetic acid esters with pentachloro-2-nitro-1,3-butadiene provides appropriate precursors for the synthesis of 2,3,4-trisubstituted benzo[h]quinolines. These heterocycles are easily accessible via a single-step reaction with 1-naphthyl- or 1-anthracenylamine, respectively. Due to steric bulk and high electron density ring closure to benzo[h]quinolines takes place, exclusively. Such highly substituted annelated pyridine systems can be modified in subsequent, selective reactions to build up new N-heterocycles with promising microbiological properties. Antibacterial and antiproliferative assays against four cell mammalian cell lines demonstrate that some of the sulfur-substituted benzo[h]quinolines analogs display potent phenotypic bioactivities in the single-digit micromolar range.
Keywords:
synthetic methods
; 2-nitroperchlorobutadiene
; benzoquinolines
; cyclization
; amines
; sulfides
; nucleophilic substitution
; oxidation
; medicinal chemistry
1. Introduction
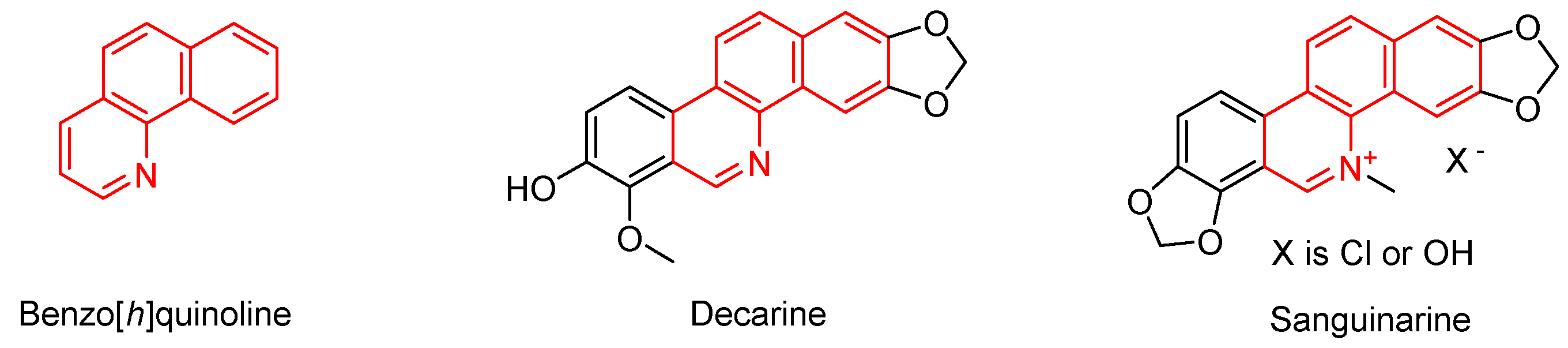
Previous articles in the field of polyhalogenated nitrobutadienes have already demonstrated the enormous potential of pentachloro-2-nitro-1,3-butadiene (1) as a precursor for the “click synthesis” of highly functionalized (hetero)cyclic as well as acyclic compounds [1,2]. The corresponding syntheses that we have developed up to now always start with the attack of an appropriate nucleophile at the activated terminal carbon atom of the nitrodichlorovinyl group within 1 to undergo a vinylic substitution reaction. Thus, in case of sulfur nucleophiles, the corresponding thioperchlorobutadiene derivatives are easily accessible [3,4]. In this paper, we describe the unexpected reaction of thioperchloronitrobutadienes to 2,3,4-trisubstituted benzo[h]quinolines, tricyclic azaheterocycles with an unusual substitution pattern. Benzo[h]quinolines are important natural products, isolated from the stem wood of Zanthoxylum nitidum [5] or from the roots of Zanthoxylum capense [6]. Alkaloid decarine (Figure 1) shows high antibacterial activity against mycobacterial, Gram-positive and Gram-negative bacteria, and low cytotoxicity towards human macrophages [7]. Sanguinarine (Figure 1) is one of the most examined members in the class of natural 3,4-disubstituted benzo[h]quinoline compounds and has multiple application possibilities due to its broad scale of bioactivities, such as antibacterial activities [8], inhibition of enzyme activities [9], prevention and treatment of cancer [10] and mononucleosome-binding affinities [11]. In the field of plant protection sanguinarine was shown to improve the environmental stress resistance of a plant [12]. A panel of fluoro substituted benzo[h]quinolines as inhibitors of tolubutamide hydroxylation was used for phenotype analysis of human cytochrome P450 2C9 polymorphism [13]. Benzo[h]quinoline itself (Figure 1) has been suggested as part of an insecticide composition for controlling stored product insects [14]. Additionally, the parent azatricyclic system has been proposed as starting reagent for the synthesis of osmium and ruthenium complexes containing an N-heterocyclic carbene ligand [15].
These selected applications in combination with the unique substitution pattern of the novel benzo[h]chinolines presented herein indicate, how promising it is to continue with our efforts in this field of medicinal chemistry.
Until now it proved possible to synthesize 2,3,4-trisubsituted benzo[h]quinolines to our best knowledge by three ways (Scheme 1):
- -
- -intramolecular cyclization of the N-(2-alkyl-1-aryl-3-oxo-4,4,4-trifluoro-1-butenyl)-1-naphthylamines using polyphosphoric acid (PPA) at 120 °C leading to 3-alkyl-2-phenyl-4-(trifluoromethyl)benzo[h]quinoline derivatives [16].
- -
- -electrocyclization of 3-(naphthylamino)-2-alkene imines triggered by ultraviolet light leads to the regioselective synthesis of substituted benzo[h]quinolines giving good to high yields [17].
- -
- -heterocyclization of 1-aminonaphthalene with lithium enolates of 2-(fluoroacetyl)cy-cloalkanones in refluxing trifluoroacetic acid to fluoromethylbenzo[h]cyclopenta[c]quinolines (yield 51–61%) [18].
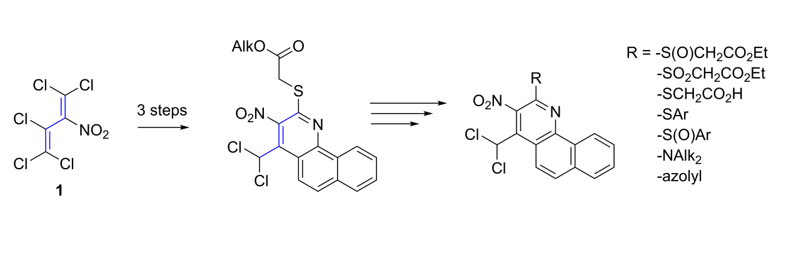
A literature review has revealed that many substituted benzo[h]quinolines are showing interesting microbiological properties [7,8,9,10,11,12,13,14]. With our new synthesis in hand starting from perchloro-2-nitro-1,3-butadiene derivatives 3a,b we were able to create uniquely trisubstituted benzo[h]quinolines 5a,b easily in gram scale. This synthetic route makes it feasible to introduce widely modifiable substituents in just one reaction step.
2. Results
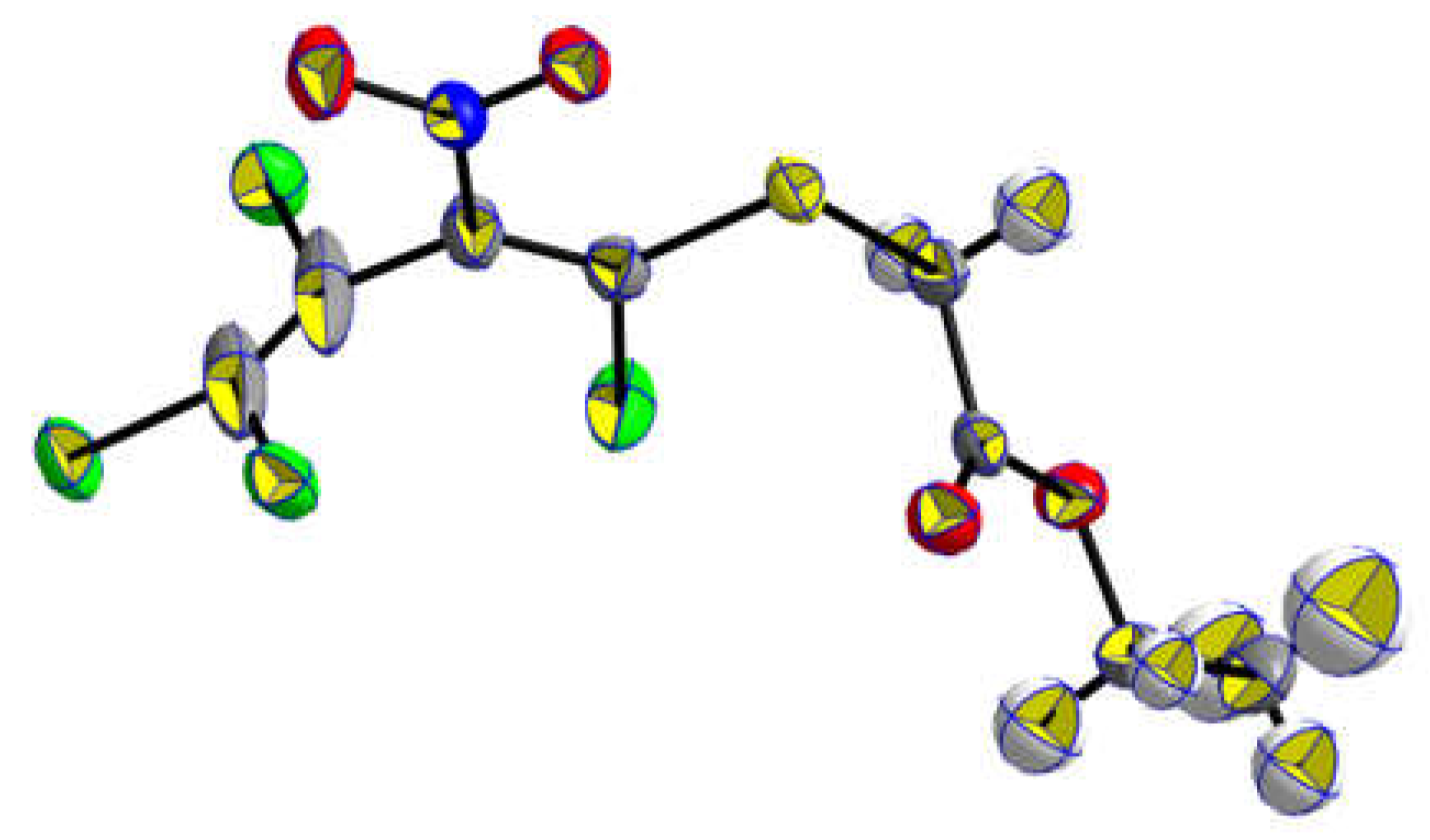
The solventless reaction of pentachloro-2-nitro-1,3-butadiene (1) with 1.5 equivalents of ethyl 2-mercaptoacetate (2a) at room temperature for 14 days furnished ethyl 2-(1,3,4,4-tetrachloro-2-nitrobuta-1,3-dienylthio) acetate (3a) as a single isomer in 93% yield and additionally, following the same procedure the methyl ester 3b in 83% yield (Scheme 2). The E-configuration of the sulfanyl-substituted C=C bond could be proved by X-ray analysis of a single crystal of thiodiene 3a (Figure 2).
Products 3a/b are versatile synthetic building blocks. Base promoted reaction of 3a/b with 1-naphthyl amine and subsequent cyclization led to the benzo[h]quinolines 5,6 (Scheme 2).
A conceivable mechanism for the cascade reaction leading to the benzo[h]quinolines 5 is presented in Scheme 3. Starting material 4 exists exclusively as E-isomer probably due to a strong hydrogen bond between the NH and NO2 groups. Initially, the (1E,2E)-imine A is formed from diene 4 upon enamine-imine tautomerism. Due to an almost free rotation around the central single bond of the side chain an intramolecular electrocyclization of A to give intermediate B as part of an equilibrium appears feasible. Subsequently, the dihydropyridine B is aromatized by elimination of HCl in the presence of triethylamine giving benzo[h]quinolines 5. A Friedel-Crafts substitution or a Michael addition appears less likely under the applied reaction conditions.
The structure of the benzo[h]quinoline 5a was also confirmed by an X-ray analysis (Figure 3).
The key step of the reaction path to form the 4-dichloromethyl-2-ethoxycarbonylmethylsulfanyl-3-nitro-benzo[h]quinolines 5 is a cyclization reaction. There are two possible ways to receive product 5. The first one was a one-pot reaction of the mercaptoacetates 3, 1-naphthylamine and triethylamine as base in THF with up to 33% yield (path A, scheme 2). Following synthetic route A it also proved possible to synthesize naphtho[h]quinoline 6 in 54% yield. To improve the yield we varied the base (without a base, triethylamine, N,N-dimethylaniline and pyridine), solvent (THF, methanol and chloroform), reaction temperature and time. The benzo[h]chinolines were formed in moderate yields when triethylamine or N,N-dimethylaniline were used as a base. Without a base the intermediate product 4 could be isolated in yields up to 93%. Reacting isolated 4 in a subsequent reaction with triethylamine (path B) led to the formation of 5 in the same very good yield. Therefore, it appeared advantageous to carry out the reaction in a two-step mode (Scheme 2).
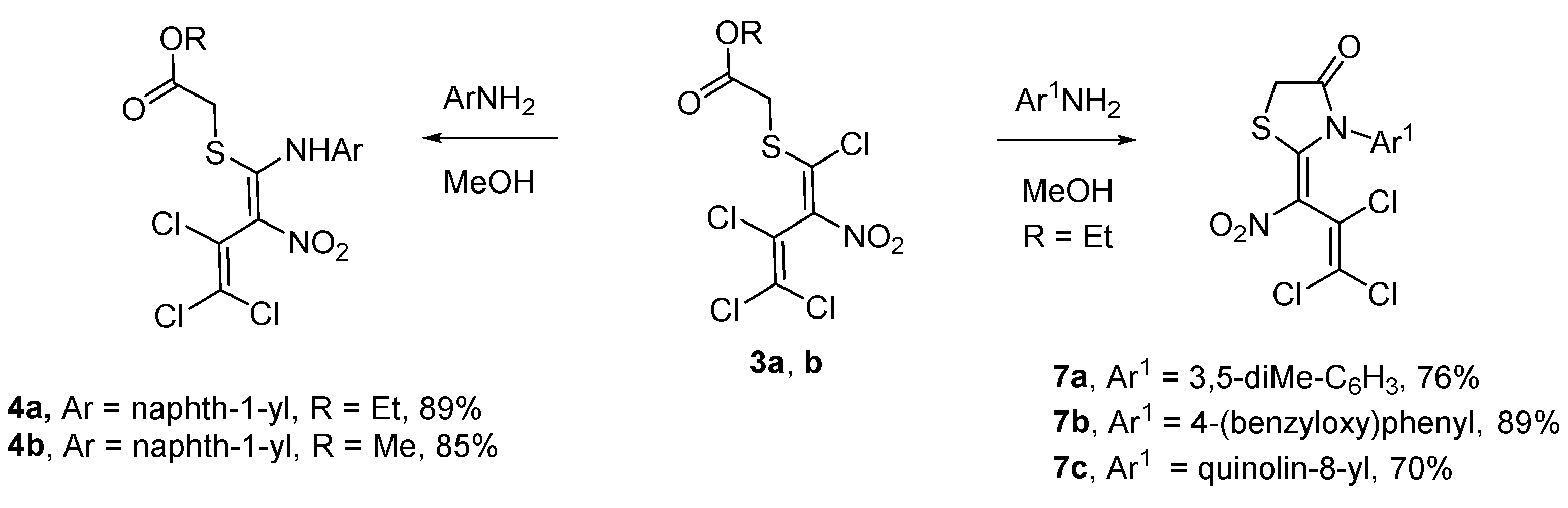
Since path B had been found to be the more efficient one, an attempt was made to synthesize more intermediate products under these reaction conditions. A variety of aromatic amines were selected (Scheme 4).
Interestingly, conversion of 3 with an one ring system like 3,5-dimethylaniline or 4-(benzyloxy)aniline in methanol afforded the Z-thiazolidinones 7a and 7b in 76% and 89% yield similarly to the literature [3], and not the open chain product 4. It is interesting that the 8-quinoline amine produces a Z-thiazolidinone 7c in 70% yield. In the case of thiazolidinone 7c two atropisomers (1: 0.9, 1H-NMR) were generated. This phenomenon is due to a less hindered rotation of the C–N bond of the former 8-quinoline amine unit. Unfortunately, there were no conversions to the desired product in the following cases: 5-aminonaphthalene-2-sulfonic acid, naphthalene-1,8-diamine, naphthalene-1,5-diamine and anthracen-1-amine.
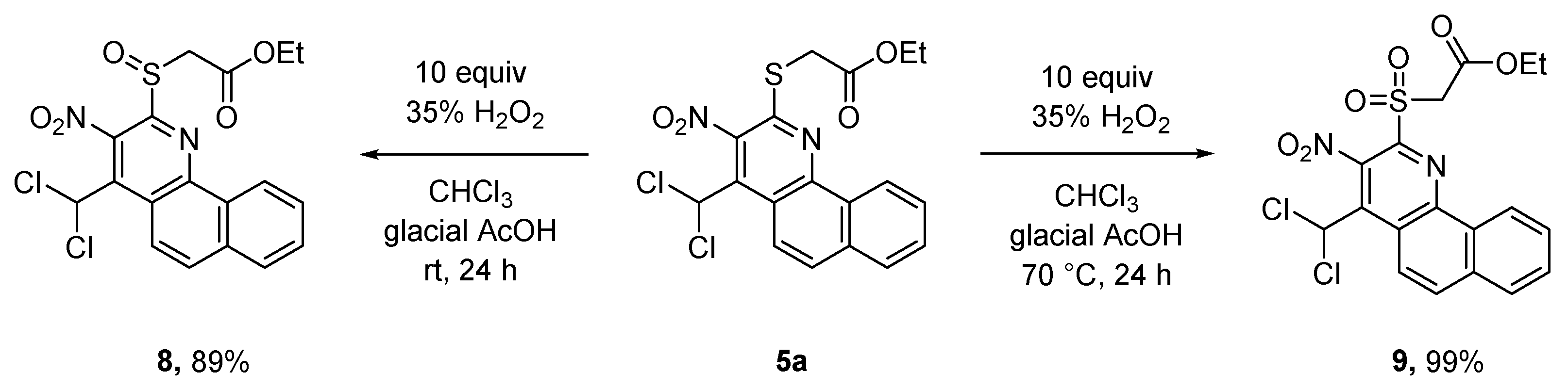
There is a variety of ways to modify benzo[h]quinolines 5. The sulfoxide group of 8 is a good leaving group and can therefore be used for substitution reactions.The reaction probably proceeds via an addition-elimination mechanism. This way, both S- (sulfide 10a–d) as well as N-substituents (amines 11a,b) can be introduced. Compound 5a is reacted with 35% hydrogen peroxide solution selectively to sulfoxide 8 and subsequently sulfone 9 in very good yields. The stepwise reaction is temperature controlled (Scheme 5).
Selective substitution reactions of the sulfoxide group of 8 succeeded well with different thiophenolates and morpholine (Scheme 6). The nucleophilicity of alkoholates proved to be too low for a reaction.
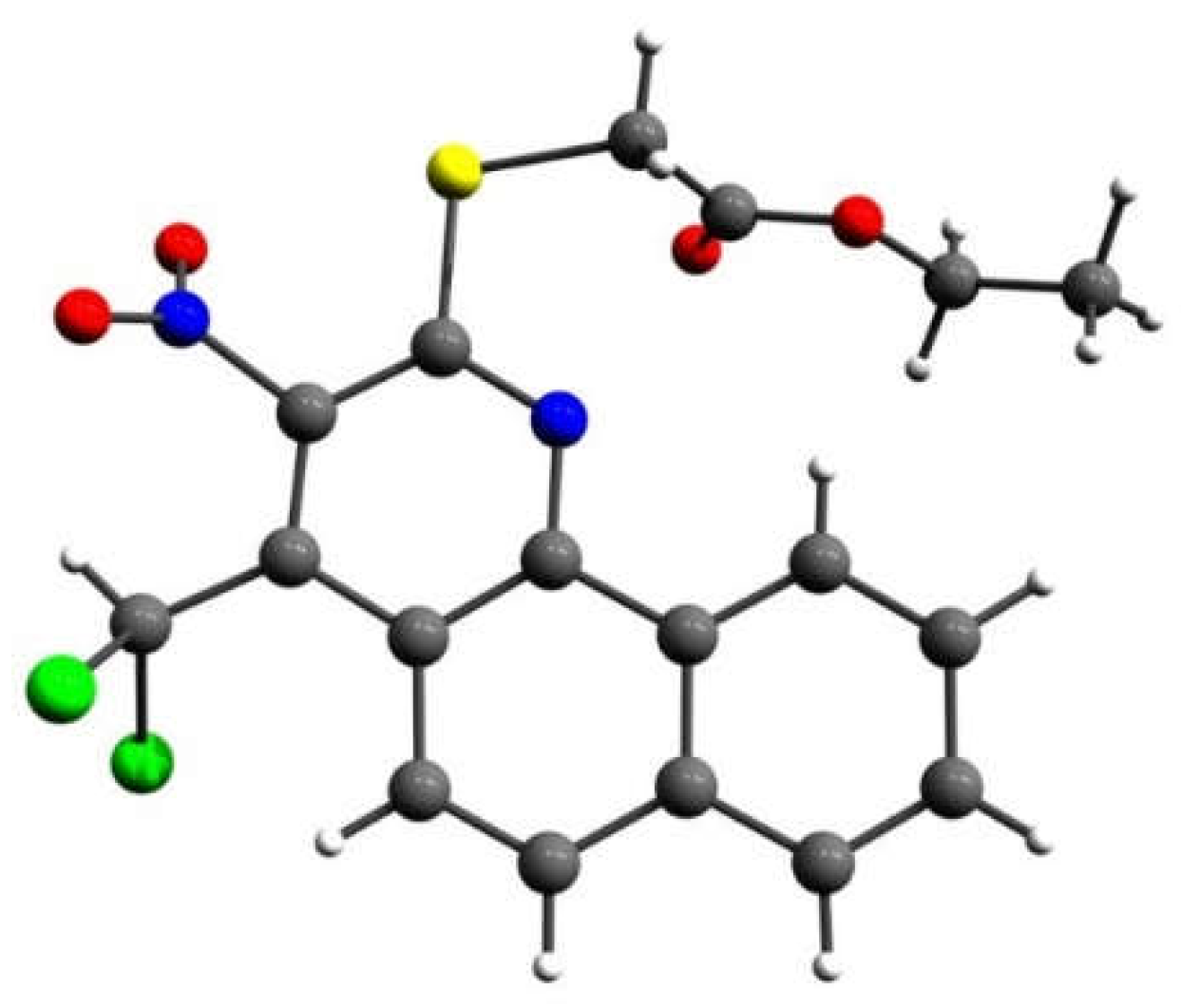
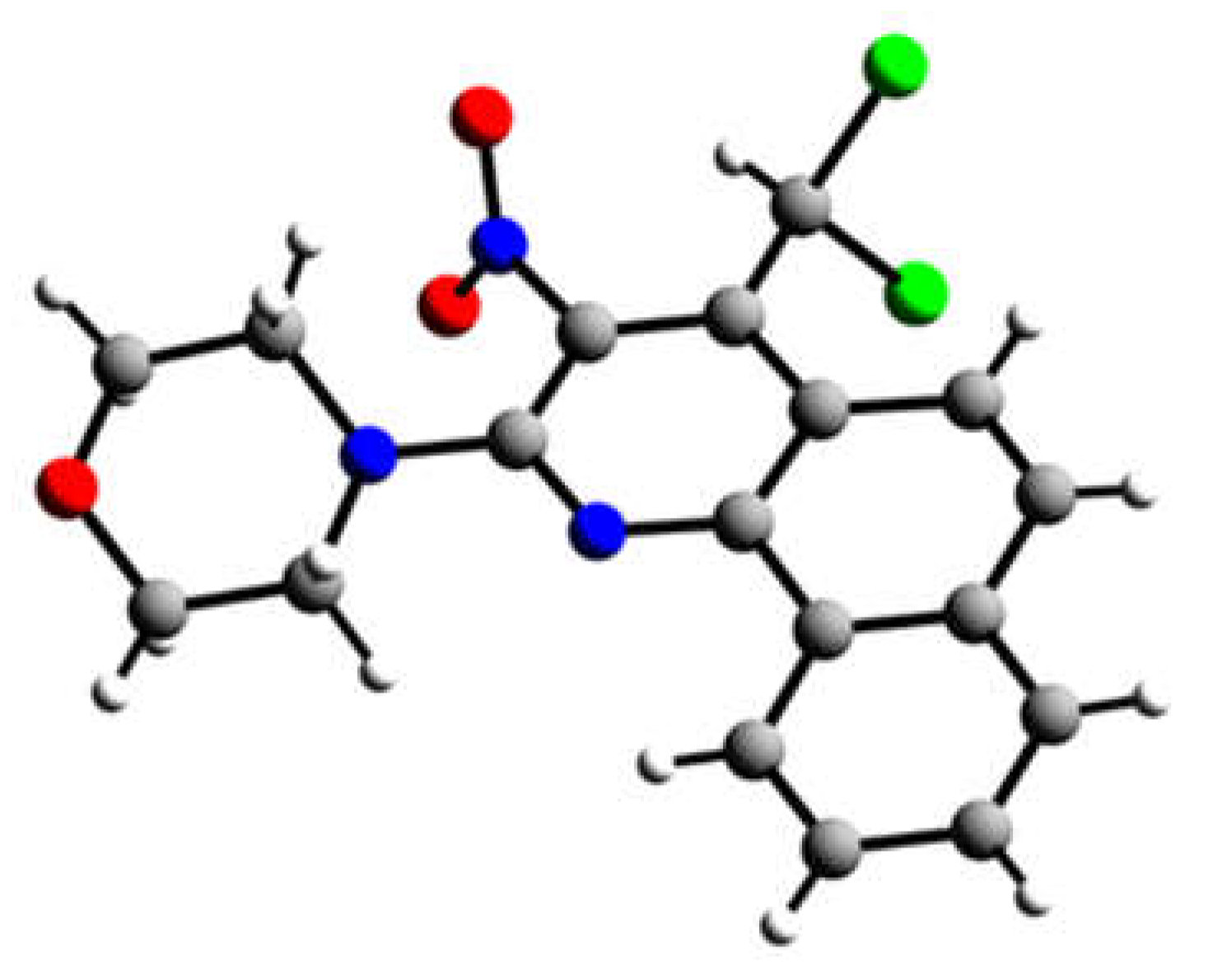
In case of quinoline 11b we were fortunate to carry out an X-Ray crystallographic analysis to prove the structural conclusions that we had drawn from the nmr spectra (see Figure 4).
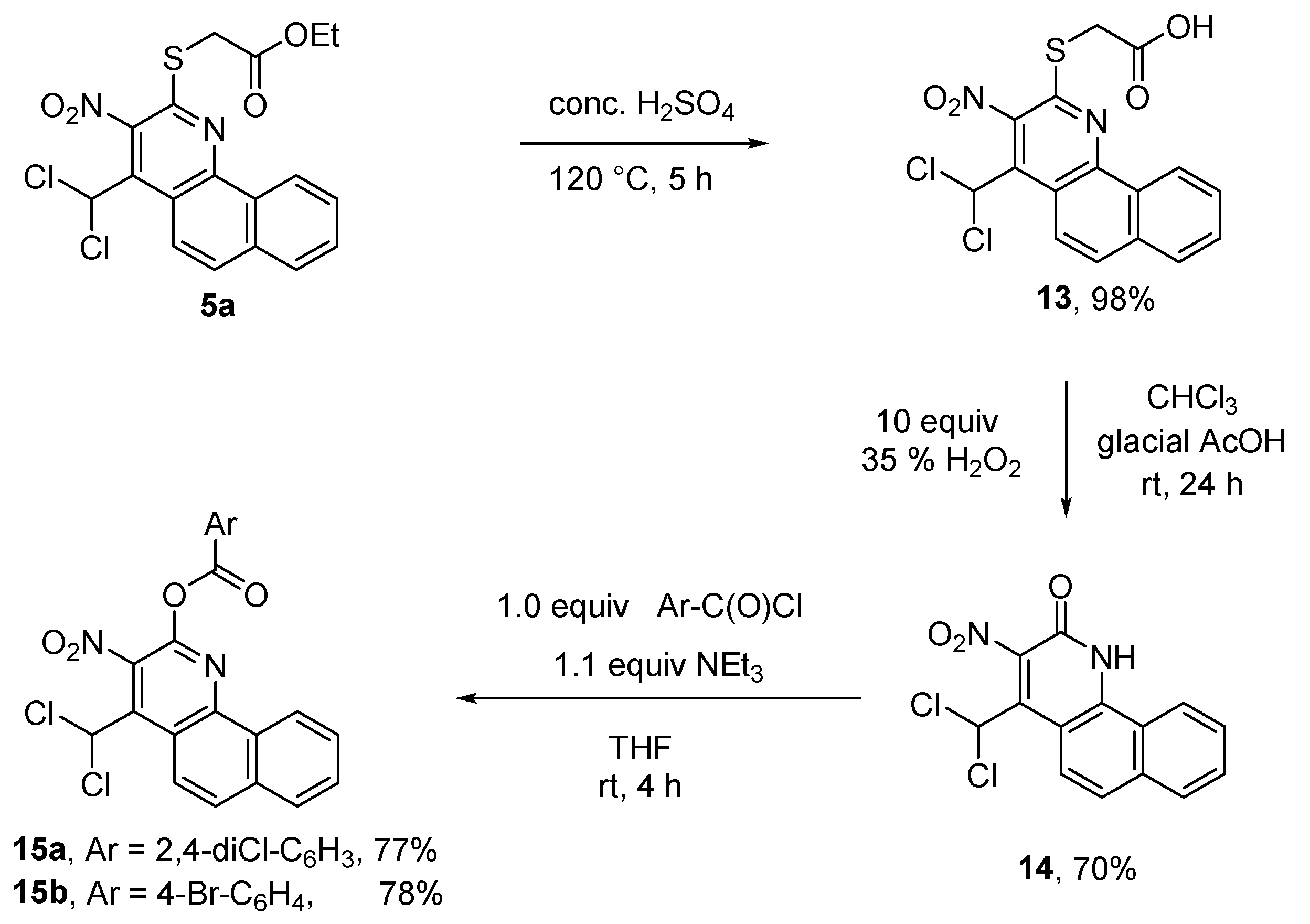
Another method to modify compound 5a is the almost quantitative acidic cleavage of its ester group to 2-((4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl)thio)acetic acid (13). This molecule deserves interest because of its high activity against methicillin-resistant Staphylococcus aureus (MRSA) in biological tests. Acid 13 can be hydrolyzed into lactam 14 in 70% yield, followed by esterification with benzoyl chlorides giving 15a and 15b in 77% and 78% yield, respectively (Scheme 7).
Additionally, a convenient way to the synthesis of 2-substituted azolylquinolines in three steps, starting from nitrodiene 1 was developed. At first, diene 1 was reacted with azoles such as benzotriazole, 1,2,4-triazole and pyrazole under formation of 1,1-diazolyl-2-nitrotrichlorobutadienes 16a–c in good yields (76-92%). The transamination of compounds 16 with an equimolar amount of 1-naphthylamine was running smoothly in ether at -30 °C to rt, already, furnishing 1-azolyl-1-(naphthylamino)dienes 17 (86–90%). Treatment of butadienes 17 with triethylamine as base in CHCl3 again led to the formation of 2,3,4-trisubstituted benzo[h]quinolines 18, in 36-87% yield. Change of solvent (CH2Cl2, DMSO, MeOH or Et2O) and base (NaOH or NaHCO3) decreased the yields of quinoline 18a to 9-55% (Scheme 8). The heterocycles in 2 position are also known to be excellent leaving groups.
Because of the diverse bioactivities reported for benzo[h]quinolines before, a selection of the newly prepared analogs shown above was characterized in phenotypic cellular assays. Their antibiotic activities were measured by growth inhibition assays against one Gram-positive pathogen, a Methicillin-resistant Staphylococcus aureus (MRSA), and two Gram-negative pathogens, i.e. Escherichia coli and Pseudomonas aeruginosa. None of the compounds could inhibit the Gram-negative strains at concentrations of 50 µM, but some analogs were active against MRSA (Table 1): Notably, compounds 8, 12 and 13 prevented the the growth of MRSA with minimal inhibitory concentrations (MICs) of 17.5, 1.2 and 8.4 µM (corresponding to 7.7, 0.5 and 3.3 µg/ml, respectively). Thus, 12 was the most potent antibiotic among all tested analogs. Interestingly, the formal reduction of the sulfoxide in 12 to a sulfide, as in 10d, led to a complete loss of activity against MRSA. The importance of the naphth-1-yl residue in 8 was illustrated by the fact that its replacement by an anthracen-1-yl moiety, as present in 6, also led to an inactive compound. Also the sulfur substituent was crucial for activity, because nitrogen-substituted benzo[h]quinolines such as the amines 11a and 11b, or the triazols and pyrazol 18a–18c were inactive. Also the tested non-cyclized precursors such as 16c or 17a–17c had no activity.
In addition to probing effects on bacterial pathogens, the compound’s ability to interfere with the proliferation and/or viability of four mammalian cell lines was assessed by quantifying the mitochondrial dehydrogenase activity in a colorimetric assay with the tetrazolium dye WST-1. For this purpose, the cells were exposed to the compounds at varying concentrations for a time period of 5 days (for L929, KB-3-1, and MCF-7 cells) or 24 h (for FS4-LTM cells). By and large, similar activity trends were observed against mammalian cell lines: The sulfoxide 8 was most potent and inhibited the proliferation of all four cell lines with EC50’s of 2.5 - 3.8 µM. Compound 13, that differed from 8 by a hydrolysis of the ester to a carboxylic acid and an oxidation of the sulfide to a sulfoxide, had weaker activities with EC50’s of 6.9 - 45 µM. In contrast, the aliphatic substituent at the sulfoxide in 8 could be replaced by an aromatic p-fluorophenyl group, because 12 inhibited the proliferation of L929, KB-3-1, and FS4-LTM cells with EC50’s of 3.3, 3.1, amd 4.4 µM, respectively. Compound 10a, differing from 12 by the oxidation state at sulfur, displayed significantly reduced activities in L929 and FS4-LTM cells, but was more potent in KB-3-1 cells.
In summary, the phenotypic cellular assays demonstrate that a distinct subset of the benzo[h]quinolines synthesized in this study possess high bioactivities against mammalian cell lines and the bacterial pathogen MRSA.
3. Materials and Methods
3.1. General Remarks
Solvents and reagents were used as received from commercial sources without further purification. TLC was performed with Merck aluminum-backed TLC plates with silica gel 60, F254. Flash column chromatography was performed with Macherey–Nagel silica gel 60 M (0.040–0.063 mm) with appropriate mixtures of petroleum ether (PE, boiling range 60–70 °C) and ethyl acetate as eluents. Melting points were determined in capillary tubes with a Büchi B-520. FTIR spectra were recorded with a Bruker “Alpha-T” spectrometer with solid compounds measured as KBr pellets. ATR-IR spectra were measured on the same instrument with a Bruker “Alpha Platinum ATR” single reflection diamond ATR module. 1H NMR and 13C NMR spectra at 600 and 150 MHz, respectively, were recorded with an “Avance III” 600 MHz FTNMR spectrometer (Bruker, Rheinstetten, Germany). 1H NMR and 13C NMR spectra at 400 and 100 MHz, respectively, were recorded with an “Avance” 400 MHz FT-NMR spectrometer (also Bruker). 1H and 13C NMR spectra were referenced to the residual solvent peak: CDCl3, δ = 7.26 (1H) and 77.0 ppm (13C); [D6]DMSO, δ = 2.50 (1H), and 39.7 ppm (13C). The NMR-spectra of the newly synthesized compounds could be found in the Supplementary Materials. Mass spectra were obtained with a Hewlett–Packard MS 5989B spectrometer, usually in direct mode with electron impact (70 eV). For chlorinated and brominated compounds, all peak values of molecular ions and fragments refer to the isotope 35Cl. High resolution mass spectra were recorded with a Waters mass spectrometer “VG Autospec” (EI), with a WATERS mass spectrometer “Q-Tof Premier” coupled with a Waters “Acquity UPLC” (ESI), or with a Micromass mass spectrometer “LCT” coupled with a Waters “Alliance 2965 HPLC” (ESI) at the Institute of Organic Chemistry, Leibniz University of Hannover.
Pentachloro-2-nitro-1,3-butadiene (1). The product was prepared from 2H–penta chloro-1,3-butadiene in 53% yield (b.p. 69–71 °C / 1 mbar) according to the literature [19].
Ethyl {[(1E)-1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl]sulfanyl}acetate (3a). A mixture of pentachloro-2-nitrobuta-1,3-diene (1) (75.30 g, 280 mmol) and ethyl 2-mercaptoacetate (50.60 g, 421 mmol) was stirred for 14 d at room temperature (rt). Then pentane was added (2 × 150 mL), each time the two-phase mixture was stirred for an additional 15 min and most of the solvent decanted after the product had solidified. It was then filtered with suction and dried in vacuo. Yellowish solid, yield 92.98 g (93%), m.p. 114–115 °C. IR (KBr): νmax = 2991, 1731 (C=O), 1528 (NO2), 1291 (NO2), 1205, 1006, 758 cm-1. 1H NMR (400 MHz, CDCl3): δ = 4.47 (q, 2 H, J = 7.1 Hz, OCH2), 3.91 (s, 2 H, SCH2), 1.28 (t, 3 H, J = 7.1 Hz, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 166.5 (C=O), 156.4 (SCCl), 138.9 (CNO2), 128.8 (CCl), 120.7 (CCl2), 62.4 (OCH2), 37.3 (SCH2), 13.8 (CH3) ppm. EIMS: m/z (%) = 355 (3) [M]+, 317 (10) [M - Cl]+, 272 (15) [M - Cl - NO2]+, 270 (55) [M - CCl2]+, 224 (30) [M - CCl2 - NO2]+, 120 (45) [SCH2COOEt]+. HRMS (EI): m/z calcd. for C8H8Cl4NO4S [M + H]+ 353.8923; found 353.8923.
Methyl {[(1E)-1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl]sulfanyl}acetate (3b). The product was prepared according to acetate 3a from buta-1,3-diene (1) (4.00 g, 14.7 mmol) and methyl 2-mercaptoacete (2.34 g, 22.1 mmol). Yellowish solid, yield 4.18 g (83%), m.p. 183 °C. IR (ATR): νmax = 3003, 1742 (C=O), 1538 (NO2), 1295 (NO2), 1160, 990, 760 cm-1. 1H NMR (400 MHz, CDCl3): δ = 3.94 (s, 2 H, SCH2), 3.81 (s, 3 H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 167.3 (C=O), 155.8 (SCCl), 139.6 (CNO2), 129.3 (CCl), 120.8 (CCl2), 53.4 (SCH2), 37.3 (CH3) ppm. EIMS: m/z (%) = 337 (65) [M]+, 233 (15) [M - SCH2COOCH3]+, 187 (45) [M - SCH2COOCH3 - NO2]+, 151 (50) [M - SCH2COOCH3 - CCl2]+, 105 (95) [SCH2COOCH3]+. HRMS (ESI): calcd. for C7H6Cl4NO4S [M + H]+: 339.87662; found 339.87687.
Ethyl {[(1E)-3,4,4-trichloro-1-(naphth-1-ylamino)-2-nitrobuta-1,3-dien-1-yl]sulfanyl}-acetate (4a). A solution of ethyl {[(1E)-1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl]sulfanyl}acetate (3a) (5.00 g, 14.0 mmol) and 1-naphthylamine (4.00 g, 28.0 mmol) in methanol (20 mL) was stirred for 3 h at rt, the precipitated product filtered off and washed with aqueous HCl (18%, 20 mL), water (20 mL) and cold methanol (20 mL). Yellowish solid, 5.73 g (89%), m.p.: 118–119 °C. IR (KBr): νmax = 2986, 1741, 1556, 1470, 1393, 1182, 829 cm-1. 1H NMR (400 MHz, CDCl3): δ = 12.16 (s, 1 H, NH), 8.03–7.93 (m, 3 H), 7.62–7.50 (m, 4 H), 4.98 (q, J = 7.1 Hz, 2 H), 2.95 (s, 2 H), 1.12 (t, J = 7.1 Hz, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 166.6 (C=O), 158.3 (NCS), 134.2 (Cq), 133.0 (Cq), 131.3 (Cq), 128.8 (CH), 128.7 (CH), 128.2 (CCl), 127.7 (CH), 127.3 (CH), 125.4 (CH), 123.6 (CCl2), 122.9 (CH), 122.2 (CNO2), 121.5 (CH), 62.1 (OCH2), 34.5 (SCH2), 13.9 (CH3) ppm. EIMS: m/z (%) = 462 (2) [M + H]+, 424 (2) [M - Cl]+, 414 (5) [M - NO2]+, 379 (5) [M - NO2 - Cl]+, 343 (10) [M - NO2 - Cl - Cl]+, 252 (10) [M - NO2 - Cl - naphthyl]+, 143 (100) [naphthylamine]+. HRMS (ESI): m/z calcd. for C18H15Cl3N2O4SNa [M + Na]+ 482.9710; found 482.9714.
Methyl {[(1E)-3,4,4-trichloro-1-(naphth-1-ylamino)-2-nitrobuta-1,3-dien-1-yl]sulfanyl}-acetate (4b). The product was prepared according to acetate 4a from acetate 3b (0.600 g, 1.759 mmol) and 1-naphthylamine (0.500 g, 3.519 mmol). Yellowish solid, yield: 0.668 g (85%), m.p. 110 °C. IR (ATR): νmax = 2905, 1745, 1553, 1527, 1390, 1170, 801 cm-1. 1H NMR (400 MHz, CDCl3): δ = 12.14 (s, 1 H, NH), 8.01 (d, J = 7.4 Hz, 1 H), 7.94 (d, J = 7.4 Hz, 1 H), 7.89 (d, J = 8.0 Hz, 1 H), 7.66–7.60 (m, 3 H), 7.52 (t, J = 8.0 Hz, 1 H), 3.53 (s, 1 H, CH3), 2.97 (s, 1 H, CH2) ppm. 13C-NMR (100 MHz, CDCl3): δ = 161.8 (C=O), 152.9 (NCS), 123.6 (2C, Cq, CH), 123.5 (CH), 123.2 (CNO2), 123.1 (CCl), 122.6 (CH), 122.1 (CH), 120.2 (CH), 118.2 (CCl2), 117.8 (CH), 116.3 (CH), 47.6 (CH3), 29.1 (CH2) ppm. EIMS: m/z (%) = 447 (25) [M + H]+, 351 (20) [M - Cl - COOCH3]+, 296 (20) [M - NO2 - SCH2COOCH3]+, 225 (40) [M - 2 Cl - NO2 - SCH2COOCH3]+ 143 (55) [naphthyl]+. HRMS (ESI): m/z calcd. for C17H13Cl3N2O4SNa [M + Na]+ 468.9554; found 468.9560.
Ethyl {[4-dichloromethyl-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetate (5a). To a solution of 4a (2.00 g, 4.3 mmol) in chloroform (20 mL) triethylamine (0.870 g, 0.60 mmol) was added at 0 °C and the reaction mixture stirred for 1 d at rt. The solvent was removed in vacuo and the residue dissolved in methanol (5 mL). The precipitated product was filtered off with suction and washed with aqueous HCl (18%, 20 mL), water (20 mL) and methanol (30 mL) and then dried in vacuo to give 5a, yellow solid. Yield 1.70 g (93%), m.p. 171–172 °C. IR (KBr): νmax = 3042, 2989, 1744, 1534, 1334, 831, 735 cm-1. 1H NMR (400 MHz, CDCl3): δ = 9.16 (d, J = 7.2 Hz, 1 H), 8.65 (d, J = 9.6 Hz, 1 H), 7.95 (d, J = 9.6 Hz, 1 H), 7.94 (d, J = 7.2 Hz, 1 H), 7.82–7.74 (m, 2 H), 7.20 (s, 1 H, CHCl2), 4.22 (q, J = 7.2 Hz, 2 H, OCH2), 4.20 (s, 2 H, SCH2), 1.28 (t, J = 7.2 Hz, 3 H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 168.6 (C=O), 149.0 (Cq), 147.9 (Cq), 139.1 (Cq), 135.7 (Cq), 134.3 (Cq), 130.4 (Cq), 130.3 (CH), 128.8 (CH), 128.0 (CH), 127.9 (CH), 125.7 (CH), 122.3 (CH), 119.6 (Cq), 63.4 (CHCl2), 62.1 (OCH2), 33.8 (SCH2), 14.2 (CH3) ppm. EIMS: m/z (%) = 424 (12) [M]+, 379 (5) [M - OEt]+, 355 (8) [M - Cl - Cl]+, 303 (25) [M- SCH2CO2Et]+, 192 (15) [M - HCl - HCl - SCH2CO2Et - NO2]+. HRMS (ESI): m/z calcd. for C18H14Cl2N2O4SNa [M + Na]+ 446.9944; found 446.9947.
Methyl {[4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetate (5b). The product was prepared according to benzo[h]quinoline 5a from acetate 4b (0.300 g, 0.67 mmol) and triethylamine (0.068 g, 0.126 mmol). Yellowish solid, yield 0.207 g (75%), m.p. 263 °C. IR (ATR): νmax = 2951, 1747, 1533, 1331, 827, 731 cm-1. 1H NMR (400 MHz, CDCl3): δ = 9.13 (d, J = 7.2 Hz, 1 H), 8.65 (d, J = 9.4 Hz, 1 H), 7.95 (d, J = 9.4 Hz, 1 H), 7.94 (d, J = 7.2 Hz, 1 H), 7.81–7.75 (m, 2 H), 7.02 (s, 1 H, CHCl2), 4.19 (s, 2 H, SCH2), 3.77 (s, 3 H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 169.1 (o, 1C, C=O), 148.9 (Cq), 148.0 (Cq), 139.1 (Cq 135.8 (Cq), 134.3 (Cq), 130.4 (Cq), 130.3 (CH), 128.9 (CH), 128.0 (2C, CH), 125.5 (CH), 122.3 (CH), 199.7 (Cq), 63.3 (CHCl2), 52.9 (CH3), 33.6 (SCH2) ppm. EIMS: m/z (%) = 410 (55) [M]+, 304 (40) [M - SCH2COOCH3]+, 260 (15) [M - SCH2COOCH3 - NO2]+, 252 (100) [M - SCH2COOCH3 - C4H4]+. HRMS (ESI): m/z calcd. for C17H12Cl2N2O4SNa [M + Na]+ 432.9787; found 432.9793.
Ethyl {[4-(dichloromethyl)-3-nitronaphtho[2,3-h]chinolin-2-yl]sulfanyl}acetate (6). A solution of ethyl {[(1E)-1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl]sulfanyl}acetate (3a) (0.500 g, 1.4 mmol) and 1-aminoanthracene (0.270 g, 1.40 mmol) in anhydrous THF (20 mL) was stirred at rt for 16 h. Subsequently, triethylamine (0.141 g, 1.4 mmol) was added, the mixture stirred at rt for 1 d and the solvent removed in vacuo. During addition of methanol (2 mL) a solid was precipitating and filtered off with suction, washed with aqueous HCl (18%, 20 mL), water (20 mL), and cold (methanol (25 mL). The yellowish solid was dried in vacuo. Yield 0.377 g (54%), m.p. 224 °C. IR (ATR): νmax = 1734, 1548, 1533, 1337, 1149, 987 cm-1. 1H NMR (400 MHz, CDCl3): δ = 9.56 (s, 1 H), 8.60 (s, 1 H), 8.38 (d, J = 9.5 Hz, 1 H), 8.20–8.16 (m, 3 H), 8.06 (s, 1 H, CHCl2), 7.72–7.69 (m, 2 H), 4.45 (s, 2 H, SCH2), 4.09 (q, J = 14.2, 7.1 Hz, 2 H, OCH2), 1.06 (t, J = 7.1 Hz, 3 H, CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 168.8 (C=O), 149.1 (Cq), 148.4 (Cq), 139.6 (Cq), 135.2 (Cq), 133.5 (Cq), 131.9 (Cq), 130.8 (Cq), 130.0 (CH), 129.0 (CH), 128.2 (CH), 127.9 (CH), 127.4 (Cq), 127.2 (2C, CH), 125.8 (CH), 120.6 (CH), 119.5 (Cq), 64.3 (CHCl2), 61.5 (OCH2), 33.6 (SCH2), 14.2 (CH3) ppm. EIMS: m/z (%) = 474 (77) [M]+, 429 (7) [M - OEt]+, 386 (10) [M - CH3COOEt]+, 355 (60) [M - SCH2COOEt]+, 302 (100) [M - CH3COOEt - CHCl2]+. HRMS (ESI): m/z calcd. for C22H16Cl2N2O4SNa [M + Na]+ 497.0100; found 497.0106.
(2Z)-3-(3,5-dimethylphenyl)-2-(2,3,3-trichloro-1-nitroprop-2-en-1-yliden)-1,3-thiazolidin-4-one (7a). A mixture of ethyl {[(1E) -1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl]sulfanyl}acetate (3a) (0.500 g, 1.4 mmol) and 4-(benzyloxy)aniline (0.350 g, 2.9 mmol) in methanol (2.5 mL) was stirred for 7 d at rt while the product was precipitating. It was filtered off with suction, washed with cold methanol (25 mL) and dried in vacuo. Beige solid, yield 0.415 g (76%), m.p. 240 °C. IR (ATR): νmax = 2983, 1756, 1595, 1369, 1287, 679 cm-1. 1H NMR (400 MHz, [D6]DMSO): δ = 7.14 (s, 1 H), 6.99 (s, 2 H), 4.16 (q, 2 H, J = 18.4 Hz, CH2), 2.31 (s, 6 H, CH3) ppm. 13C NMR (100 MHz, [D6]DMSO): δ = 173.7 (C=O), 165.7 (SCN), 138.7 (Cq), 138.4 (Cq), 134.0 (Cq), 131.7 (CH), 128.2 (CCl), 126.2 (CH), 124.6 (CH), 121.1 (CNO2), 120.9 (CCl2), 32.4 (CH2), 20.5 (2C, CH3) ppm. EIMS: m/z (%) = 391 (3) [M+], 356 (7) [M - Cl]+, 264 (30) [M - NO2 - CCl2]+, 247 (100) [M - NO2 - 2 CH3 - 2 Cl]+, 228 (25) [M - NO2 - CCl2 - Cl]+, 202 (70) [M - CCl2 - Ph(CH3)2]+, 104 (60) [Ph(CH3)2]+. HRMS (ESI): m/z calcd. for C14H11Cl3N2O3SNa [M + Na]+ 414.9448; found 414.9453.
(2Z)-3-[4-(benzyloxy)phenyl]-2-(2,3,3-trichloro-1-nitroprop-2-en-1-yliden)-1,3-thiazolidin-4-one (7b). The product was prepared according to thiazolidin-4-one 7a from acetate 3a (1.00 g, 2.80 mmol), 4-(benzyloxy)aniline hydrochloride (1.39 g, 7.02 mmol) and sodium hydroxide (0.28 g, 7.02 mmol) in methanol (10 mL) and stirred for 7 d at rt. Beige solid, yield 1.174 g (89%), m.p. 201 °C. IR (ATR): νmax = 2923, 1754, 1523, 1363, 1229, 743 cm-1. 1H NMR (400 MHz, [D6]DMSO): δ = 7.46 (d, J = 6.8 Hz, 2 H), 7.40 (d, J = 6.8 Hz, 1 H), 7.30 (d, J = 6.8 Hz, 1 H), 7.33 (t, J = 6.8 Hz, 1 H), 7.31 (d, J = 7.6 Hz, 2 H), 7.12–7.09 (m, 2 H), 5.17 (d, J = 8.2 Hz, 2 H, OCH2), 4.13 (q, J = 18.3 Hz, 2 H, SCH2) ppm. 13C NMR (100 MHz, [D6]DMSO): δ = 174.5 (C=O), 166.3 (SCN), 159.8 (Cq), 137.2 (Cq), 130.5 (CH), 129.1 (CH), 128.9 (2C, CH), 128.5 (Cq), 128.4 (CH), 128.1 (2C, CH), 127.4 (CCl), 121.7 (2C, CNO2, CCl2), 115.7 (CH), 115.6 (CH), 69.9 (OCH2), 32.8 (SCH2) ppm. EIMS: m/z (%) = 469 (10) [M+], 435 (20) [M - Cl]+, 378 (45) [M - Bz]+, 326 (50) [M - OBz - Cl]+, 313 (100) [M - OBz - NO2]+. HRMS (ESI): m/z calcd. for C19H13Cl3N2O4S [M+] 469.9656; found 469.9583.
(2Z)-3-(quinolin-8-yl)-2-(2,3,3-trichloro-1-nitroprop-2-en-1-yliden)-1,3-thiazolidin-4-one (7c). The product was synthesized according to thiazolidin-4-one 7a from acetate 3a (0.500 g, 1.40 mmol) and quinoline-8-amine (0.60 g, 4.20 mmol) in methanol (10 mL) by stirring for 6 h at rt. Orange solid, mixture of two rotamers, ratio (1: 0.9). Yield 0.404 g (70%), m.p. 197 °C. IR (ATR): νmax = 2978, 1747, 1519, 1374, 1285, 754 cm-1. Major Isomer: 1H NMR (600 MHz, [D6]DMSO): δ = 8.92 (dd, J = 4.2, 1.6 Hz, 1 H), 8.54 (ddd, J = 9.5, 8.2, 1.4 Hz, 1 H), 8.22 (dd, J = 8.2, 1.3 Hz, 1 H), 7.95 (dd, J = 7.3, 1.3 Hz, 1 H), 7.80 (dd, J = 7.4, 4.0 Hz, 1 H), 7.66 (dd, J = 8.3, 4.2 Hz, 1 H), 4.46 (q, J = 18.7 Hz, 2 H, CH2) ppm. 13C NMR (150 MHz, [D6]DMSO): δ = 174.0 (C=O), 166.2 (NCS), 151.6 (CH), 142.6 (Cq), 136.8 (CH), 131.2 (Cq), 131.1 (CH), 129.2 (Cq), 129.1 (CH), 127.8 (CCl), 126.2 (CH), 122.8 (CH), 121.3 (CNO2), 120.9 (CCl2), 32.1 (CH2) ppm. Minor Isomer: 1H NMR (600 MHz, [D6]DMSO): δ = 8.90 (dd, J = 8.2, 1.6 Hz, 1 H), 8.54 (ddd, J = 9.5, 8.2, 1.4 Hz, 1 H), 8.18 (dd, J = 8.2, 1.3 Hz, 1 H), 8.00 (dd, J = 7.3, 1.3 Hz, 1 H), 7.76 (dd, J = 7.4, 4.2 Hz, 1 H), 7.66 (dd, J = 8.3, 4.2 Hz, 1 H), 4.35 (q, J = 18.7 Hz, 2 H, CH2) ppm. 13C NMR (150 MHz, [D6]DMSO): δ = 174.0 (C=O), 166.8 (NCS), 151.6 (CH), 143.0 (Cq), 136.7 (CH), 131.3 (Cq), 131.2 (CH), 129.3 (Cq), 129.1 (CH), 128.7 (CCl), 126.2 (CH), 122.7 (CH), 121.6 (CNO2), 120.2 (CCl2), 32.3 (CH2) ppm. EIMS: m/z (%) = 414 (1) [M+], 369 (100) [M - NO2]+, 299 (15) [M - NO2 - Cl - Cl]+, 228 (15) [M - C3NO2Cl3]+, 128 (40) [quinoline]+. HRMS (ESI): m/z calcd. for C15H8Cl3N3O3S [M]+ 414.9346; found 414.9356.
Ethyl {[4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl]sulfinyl}acetate (8). Ethyl {[4-dichloromethyl-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetate (5a) (1.00 g, 2.350 mmol) was suspended in a mixture of chloroform (10 mL) and glacial acetic acid (10 mL). Aqueous hydrogen peroxide solution (35%, 1.5 mL) was added and the mixture stirred at rt for 2 d. Then ice (50 g) was added, the reaction solution stirred for an additional 5 min, followed by extraction with chloroform (3 × 60 mL). The organic phase was washed with water (2 × 50 mL) and dried (sodium sulfate). After evaporation of the solvent the obtained solid was purified by column chromatography (petroleum ether/ethyl acetate, 2:1) and finally dried in vacuo. Yellowish solid, yield 0.921 g (89%), m.p. 90 °C. IR (ATR): νmax = 2987, 1723, 1538, 1334, 1215, 775 cm-1. 1H NMR (400 MHz, [D6]DMSO): δ = 9.24 (d, J = 7.4 Hz, 1 H), 8.71 (d, J = 9.4 Hz, 1 H), 8.43 (d, J = 9.4 Hz, 1 H), 8.22 (d, J = 7.4 Hz, 1 H), 8.14 (s, 1 H, CHCl2), 8.00–7.92 (m, 2 H), 4.64 (d, J = 14.2 Hz, 1 H, SCH2), 4.41 (d, J = 14.2 Hz, 1 H, SCH2), 4.08 (q, J = 7.1 Hz, 2 H, OCH2), 1.06 (t, J = 7.1 Hz, 3 H, CH3) ppm. 13C NMR (100 MHz, [D6]DMSO): δ = 165.2 (C=O), 153.3 (Cq), 146.7 (Cq), 139.2 (Cq), 136.1 (Cq), 133.6 (Cq), 132.0 (CH), 131.1 (CH), 129.7 (Cq), 128.9 (CH), 128.5 (CH), 125.2 (CH), 122.9 (Cq), 121.5 (CH), 63.7 (CHCl2), 61.5 (OCH2), 57.9 (SCH2), 13.7 (CH3) ppm. EIMS: m/z (%) = 440 (12) [M+], 323 (75) [M - NO2 - Cl - Cl]+, 304 (20) [M - SOCH2COOEt]+, 226 (75) [M - NO2 - Cl - SOCH2COOEt]+, 191 (100) [M - NO2 - Cl - Cl - SOCH2COOEt]+. HRMS (ESI): m/z calcd. for C18H15Cl2N2O5S [M + H]+ 441.0073; found 441.0076.
Ethyl {[4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl]sulfonyl}acetate (9). Ethyl {[4-dichloromethyl-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetate (5a) (0.200 g, 0.47 mmol) was suspended in a mixture of chloroform (4 mL) and glacial acetic acid (4 mL). Aqueous hydrogen peroxide solution (35%, 0.3 mL) was added and the mixture stirred at 70 °C for 6 h. Then ice (50 g) was added, the reaction mixture stirred for an additional 5 min, followed by extraction with chloroform (6 × 60 mL). The organic phase was washed with water (2 × 50 mL), dried with sodium sulfate, and the solvent removed in vacuo. Yellowish solid, yield 0.212 g (99%), m.p. 154 °C. IR (ATR): νmax = 3043, 1745, 1553, 1344, 1263, 749 cm-1. 1H NMR (400 MHz, [D6]DMSO): δ = 9.18 (d, J = 8.0 Hz, 1 H), 8.64 (d, J = 9.4 Hz, 1 H), 8.46 (d, J = 9.4 Hz, 1 H), 8.18 (d, J = 8.0 Hz, 1 H), 8.06 (s, 1 H, CHCl2), 7.97–7.88 (m, 2 H), 5.35 (s, 2 H, SCH2), 3.89 (q, J = 7.1 Hz, 2 H, OCH2), 0.80 (t, J = 7.1 Hz, 3 H, CH3) ppm. 13C NMR (100 MHz, [D6]DMSO): δ = 162.5 (C=O), 145.7 (Cq), 144.9 (Cq), 137.4 (Cq), 135.9 (Cq), 133.7 (CH), 133.6 (Cq), 131.4 (CH), 129.4 (Cq), 129.3 (CH), 128.6 (CH), 125.5 (CH), 124.1 (Cq), 121.5 (CH), 63.5 (CHCl2), 61.7 (OCH2), 56.8 (SCH2), 13.4 (CH3) ppm. EIMS: m/z (%) = 456 (20) [M+], 411 (10) [M - OEt]+, 363 (100) [M - OEt - NO2]+, 318 (45) [M - CH2COOEt - NO2]+, 177 (70) [benzo[h]quinoline]+. HRMS (ESI): m/z calcd. for C18H14Cl2N2O6SNa [M + Na]+ 478.9842; found 478.9847.
4-(Dichloromethyl)-3-nitro-2-(phenylsulfanyl)benzo[h]quinoline (10a). The product was synthesized according to benzo[h]quinoline 5a from quinoline 8 (0.200 g, 0.45 mmol), diisopropyl-N-ethylamine (0.058 g, 0.076 mmol) and benzenethiol (0.050 g, 0.45 mmol). Yellowish solid, yield 0.185 g (99%), m.p. 235 °C. IR (ATR): νmax = 3062, 1729, 1538, 1324, 1247, 714 cm-1. 1H NMR (400 MHz, CDCl3): δ = 8.59 (d, J = 9.6 Hz, 1 H), 8.28 (d, J = 8.0 Hz, 1 H), 7.89 (d, J = 9.6 Hz, 1 H), 7.85 (d, J = 8.0 Hz, 1 H), 7.71–7.68 (m, 3 H), 7.58–7.48 (m, 4 H), 7.17 (s, 1 H, CHCl2) ppm. 13C-NMR (100 MHz, CDCl3): δ = 150.5 (Cq), 147.8 (Cq), 136.3 (2C, CH), 136.2 (Cq), 135.2 (Cq), 134.0 (Cq), 130.5 (Cq), 130.0 (CH), 129.9 (CH 129.5 (2C, CH), 128.9 (CH), 128.6 (Cq), 127.8 (CH), 127.7 (CH), 125.5 (CH), 122.1 (CH), 199.6 (Cq), 63.5 (CHCl2) ppm. EIMS: m/z (%) = 414 (20) [M+], 304 (10) [M - SC6H5]+, 298 (15) [M - 2Cl - NO2]+, 186 (25) [M - SC6H5 - 2Cl - NO2]+. HRMS (MS): m/z calcd. for C20H12Cl2N2O2S [M+] 413.9991; found 413.9996.
4-(Dichloromethyl)-2-(naphth-1-ylsulfanyl)-3-nitrobenzo[h]quinoline (10b). The product was prepared according to benzo[h]quinoline 5a from quinoline 8 (0.200 g, 0.45 mmol), diisopropyl-N-ethylamine (0.058 g, 0. 076 mmol) and 2-naphthylthiol (0.072 g, 0.45 mmol). Yellowish solid, yield 0.059 g (28%), m.p. 254 °C. IR (ATR): νmax = 3053, 1538, 1323, 1220, 787, 712 cm-1. 1H-NMR (400 MHz, CDCl3): δ = 8.59 (d, J = 9.2 Hz, 1 H), 8.24 (s, 1 H), 8.15 (d, J = 8.4 Hz, 1 H), 7.97 (d, J = 8.4 Hz, 1 H), 7.96 (d, J = 8.0 Hz, 1 H), 7.89 (d, J = 9.2 Hz, 1 H), 7.87 (d, J = 9.0 Hz, 1 H), 7.81 (d, J = 8.0 Hz, 1 H), 7.69 (d, J = 9.0 Hz, 1 H), 7.64–7.56 (m, 3 H), 7.28 (t, J = 8.0 Hz, 1 H), 7.20 (s, 1 H, CHCl2) ppm. 13C-NMR (100 MHz, CDCl3): δ = 150.3 (Cq), 147.8 (Cq), 139.1 (Cq), 135.9 (CH), 135.2 (Cq), 134.0 (Cq), 133.8 (Cq), 133.6 (Cq), 132.3 (CH), 130.4 (Cq), 130.0 (CH), 128.9 (CH), 128.8 (CH), 128.1 (CH), 127.9 (CH), 127.8 (CH), 127.6 (CH), 127.4 (CH) 126.7 (CH), 126.0 (Cq), 125.5 (CH), 122.1 (CH), 199.7 (Cq), 63.5 (CHCl2) ppm. EIMS: m/z (%) = 464 (40) [M+], 365 (35) [M - NO2 - C4H4]+, 349 (15) [M - NO2 - 2Cl]+, 336 (10) [M - naphthyl]+, 191 (40) [M - NO2 - 2Cl - naphthyl]+, 127 (100) [naphthyl]+. HRMS (ESI): m/z calcd. for C24H14Cl2N2O2S [M]+ 464.0148; found 464.0153.
2-[(4-Chlorophenyl)sulfanyl]-4-(dichloromethyl)-3-nitrobenzo[h]quinoline (10c). The product was prepared according to benzo[h]quinoline 5a from quinoline 8 (0.200 g, 0.45 mmol), diisopropyl-N-ethylamine (0.058 g, 0. 076 mmol) and 4-chlorobenzenethiol (0.065 g, 0.45 mmol). Yellowish solid, yield 0.089 g (44%), m.p. 265 °C. IR (ATR): νmax = 1520, 1475, 1326, 1088, 822, 734 cm-1. 1H NMR (400 MHz, [D6]DMSO): δ = 8.52 (d, J = 9.2 Hz, 1 H), 8.18 (d, J = 7.8 Hz, 1 H), 8.16 (d, J = 9.2 Hz, 1 H), 8.10 (s, 1 H, CHCl2), 8.05 (d, J = 7.8 Hz, 1 H), 7.79 (t, J = 7.8 Hz, 1 H), 7.76 (d, J = 8.6 Hz, 2 H), 7.67 (d, J = 8.6 Hz, 2 H), 7.60 (t, J = 7.8 Hz, 1 H) ppm. 13C-NMR (100 MHz, [D6]DMSO): δ = 149.2 (Cq), 146.6 (Cq), 139.2 (Cq), 137.5 (2C, CH), 135.3 (Cq), 135.2 (Cq), 133.6 (Cq), 130.5 (CH), 129.6 (2C, CH), 129.5 (CH), 128.1 (2C, CH), 127.1 (Cq), 124.3 (CH), 121.3 (CH), 119.4 (Cq), 64.1 (CHCl2) ppm. EIMS: m/z (%) = 447 (45) [M+], 348 (45) [M - C5H4Cl]+, 304 (10) [M - SC6H4Cl]+, 235 (60) [M - SC6H4Cl - 2Cl]+, 191 (25) [M - SC6H4Cl - 2Cl - NO2]+, 177 (100) [benzo[h]quinoline]+. HRMS (ESI): m/z calcd. for C20H11Cl3N2O2S [M+] 447.9601; found 447.9607.
4-(Dichloromethyl)-2-[(4-fluorophenyl)sulfanyl]-3-nitrobenzo[h]quinoline (10d). To a solution of ethyl {[4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl]sulfinyl}acetate (8) (0.400 g, 0.90 mmol) in dichloromethane (10 mL) diisopropyl-N-ethylamine (0.116 g, 0.90 mmol) was added under stirring. The solution was cooled to 0 °C and treated with 4-fluorobenzenethiol (0.115 g, 0.90 mmol). The reaction mixture was stirred for 3.5 h at rt. Subsequently, the reaction mixture was acidified with aqueous HCl (18%, 1 mL), extracted with chloroform (3 × 30 mL), washed with water (1 × 50 mL) and dried with calcium chloride. After evaporation of the solvent the obtained solid was purified by column chromatography (petroleum ether/ethyl acetate, 10:1). The solvents were removed in vacuo to receive a yellowish solid, yield 0.296 g (74%), m.p. 267 °C. IR (ATR): νmax = 1522, 1488, 1328, 1220, 827, 744 cm-1. 1H NMR (600 MHz, CDCl3): δ = 8.61 (d, J = 9.3 Hz, 1 H), 8.30 (d, J = 7.9 Hz, 1 H), 7.89 (d, J = 7.9 Hz, 1 H), 7.85 (d, J = 7.9 Hz, 1 H), 7.69 (t, J = 7.9 Hz, 1 H), 7.69–7.65 (m, 2 H), 7.55 (t, J = 7.9 Hz, 1 H), 7.26–7.22 (m, 2 H), 7.17 (s, 1 H, CHCl2) ppm. 13C NMR (150 MHz, CDCl3): δ = 164.0 (CF, 1JC,F = 250.9 Hz), 150.3 (Cq), 147.8 (Cq), 139.0 (Cq), 138.5 (CH, 3JC,F = 8.3 Hz), 135.3 (Cq), 134.1 (Cq), 130.4 (Cq), 130.1 (CH), 129.0 (CH), 127.9 (CH), 127.8 (CH), 125.3 (CH), 123.9 (Cq, 4JC,F = 3.4 Hz), 122.1 (CH), 119.8 (Cq), 116.7 (CH, 2JC,F = 22.0 Hz), 63.4 (CHCl2) ppm. EIMS: m/z (%) = 432 (30) [M+], 397 (8) [M - Cl]+, 333 (30) [M - Cl - F - NO2]+, 285 (20) [M - CCl2 - F - NO2]+. HRMS (EI): m/z calcd. for C20H11Cl2FN2O2S [M+] 431.9897; found 431.9902.
4-(Dichloromethyl)-3-nitro-2-(piperidin-1-yl)benzo[h]quinoline (11a). To a solution of ethyl {[4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl]sulfinyl}acetate (8) (0.127 g, 0.29 mmol) in dry toluene (10 mL) piperidine (0.025 g, 0.29 mmol) was added under stirring, followed by refluxing for an additional 6 h. Subsequently, the reaction mixture was cooled to rt, acidified with aqueous HCl (18%, 1 mL), extracted with chloroform (3 × 30 mL), washed with water (1 × 50 mL) and dried with calcium chloride. After evaporation of the solvent the obtained solid was purified by column chromatography (petroleum ether/ethyl acetate, 10:1). Red solid, yield 0.091 g (80%), m.p. 236 °C. IR (ATR): νmax = 2930, 2832, 1582, 1527, 1332, 1234, 727 cm-1. 1H NMR (400 MHz, CDCl3): δ = 9.07 (d, J = 7.6 Hz, 1 H), 8.54 (d, J = 9.2 Hz, 1 H), 7.87 (d, J = 7.6 Hz, 1 H), 7.76 (d, J = 9.2 Hz, 1 H), 7.73–7.65 (m, 2 H), 7.08 (s, 1 H, CHCl2), 3.53–3.50 (m, 4 H) 1.77–1.67 (m, 6 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 149.5 (Cq), 146.9 (Cq), 135.9 (Cq), 134.3 (Cq), 134.0 (Cq), 130.5 (Cq), 129.4 (CH), 127.8 (CH), 127.0 (CH), 125.7 (CH), 125.4 (CH), 122.6 (CH), 116.0 (Cq), 63.8 (CHCl2), 49.9 (2C, CH2), 25.7 (2C, CH2), 24.4 (CH2) ppm. EIMS: m/z (%) = 389 (100) [M+], 318 (60) [M - 2Cl]+, 304 (40) [M - piperidine]+. HRMS (MS): m/z calcd. for C19H17Cl2N3O2 [M]+ 389.0692; found 389.0699.
4-(Dichloromethyl)-2-(morpholin-4-yl)-3-nitrobenzo[h]quinoline (11b). The product was prepared according to benzo[h]quinoline 11a from quinoline 8 (0.200 g, 0.45 mmol) and morpholine (0.039 g, 0.45 mmol). The reaction mixture was stirred for 8 h at 70 °C. Purification by column chromatography (petroleum ether/ethyl acetate, 10:1) led to a red solid, yield 0.095 g (54%), m.p. 233 °C. IR (ATR): νmax = 2960, 2857, 1587, 1525, 1352, 1227, 734 cm-1. 1H NMR (400 MHz, [D6]DMSO): δ = 9.02 (d, J = 7.8 Hz, 1 H), 8.47 (d, J = 9.3 Hz, 1 H), 8.05 (d, J = 7.8 Hz, 1 H), 8.02 (d, J = 9.3 Hz, 1 H), 7.84–7.59 (m, 2 H), 7.83 (s, 1 H, CHCl2), 3.77 (t, J = 4.5 Hz, 4 H, CH2), 3.47 (t, J = 4.5 Hz, 4 H, CH2) ppm. 13C-NMR (100 MHz, [D6]DMSO): δ = 148.6 (Cq), 145.6 (Cq), 135.8 (Cq), 133.9 (Cq), 133.7 (Cq), 129.9 (CH), 129.4 (Cq), 128.0 (CH), 127.6 (CH), 126.5 (CH), 124.9 (CH), 121.7 (CH), 116.0 (Cq), 65.8 (2C, CH2), 64.4 (CHCl2), 48.7 (2C, CH2) ppm. EIMS: m/z (%) = 391 (100) [M+], 357 (10) [M - Cl]+, 321 (20) [M - 2Cl]+, 305 (40) [M - morpholine]+, 177 (65) [benzo[h]quinoline]+. HRMS (ESI): m/z calcd. for C18H15Cl2N3O3 [M]+ 391.0485; found 391.0490.
4-(Dichloromethyl)-2-[(4-fluorophenyl)sulfinyl]-3-nitrobenzo[h]quinoline (12). 4-(Dichloromethyl)-2-[(4-fluorophenyl)sulfanyl]-3-nitrobenzo[h]quinoline (10d) (0.200 g, 0.45 mmol) was dissolved in a mixture of chloroform (5 mL) and glacial acetic acid (5 mL). Under ice cooling aqueous hydrogen peroxide solution (35%, 0.3 mL) was added, followed by stirring for an additional 2 d at rt. Subsequently, ice (50 g) was added, the reaction mixture stirred for 5 min, extracted with chloroform (5 × 60 mL), the organic phase washed with water (2 × 50 mL) and dried (sodium sulfate). After evaporation of the solvent the obtained solid was purified by column chromatography (petroleum ether/ethyl acetate, 2:1). Yellowish solid, yield 0.196 g (95%), m.p. 187 °C. IR (ATR): νmax = 1585, 1542, 1353, 1223, 1090, 754 cm-1. 1H NMR (400 MHz, CDCl3): δ = 9.29 (d, J = 9.2 Hz, 1 H), 8.71 (d, J = 9.2 Hz, 1 H), 8.11 (d, J = 9.2 Hz, 1 H), 8.05 (d, J = 8.8 Hz, 1 H), 8.04 (d, J = 8.8 Hz, 1 H), 7.99–7.96 (m, 1 H), 7.87–7.85 (m, 2 H), 7.24–7.20 (m, 3 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 164.9 (CF, 1JC,F = 253.3 Hz), 153.7 (Cq), 148.2 (Cq), 138.2 (Cq, 3JC,F = 8.2 Hz), 138.1 (Cq), 136.0 (Cq), 134.0 (Cq), 131.9 (CHTh), 131.0 (CHTh), 130.6 (Cq), 128.8 (CH), 128.3 (CH), 128.2 (2 C, CH), 125.8 (CH), 123.3 (Cq, 4JC,F = 3.0 Hz), 122.0 (CH), 116.8 (CH, 2JC,F = 22.6 Hz), 62.5 (CHCl2) ppm. EIMS: m/z (%) = 446 (3) [M+], 403 (10) [M - NO2]+, 368 (20) [M - NO2 - Cl]+, 333 (8) [M - NO2 - 2Cl]+, 143 (100) [SOPhF]+. HRMS (ESI): m/z calcd. for C20H11Cl2FN2O3SNa [M + Na]+ 470.9744; found 470.9749.
{[4-(Dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetic acid (13). Ethyl {[4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetate (5a) (1.500 g, 3.527 mmol) was suspended in conc. sulfuric acid (25 mL) and stirred at 90 ° C for 5 h. Subsequently, ice-water (100 mL) was added under stirring. The reaction mixture was extracted with chloroform (4 × 50 mL) and washed with water (3 × 70 mL). The organic phase was dried with sodium sulfate and the solvent removed in vacuo. Yellowish solid, yield 1.379 g (99%), m.p. 204 °C. IR (ATR): νmax = 2119, 1705, 1524, 1359, 1221, 968, 751 cm-1. 1H NMR (600 MHz, [D6]DMSO): δ = 13.05 (bs, 1 H, OH), 9.13 (d, J = 8.0 Hz, 1 H), 8.52 (d, J = 9.6 Hz, 1 H), 8.17 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 8.07 (s, 1 H, CHCl2), 7.87 (d, J = 8.0 Hz, 1 H), 7.80 (d, J = 8.0 Hz, 1 H), 4.29 (s, 2 H, SCH2) ppm. 13C NMR (150 MHz, [D6]DMSO): δ = 170.1 (C=O), 149.6 (Cq), 147.2 (Cq), 139.8 (Cq), 135.3 (Cq) 134.2 (Cq), 130.9 (CH), 129.9 (Cq), 129.6 (CH), 128.7 (CH), 128.6 (CH), 125.8 (CH), 121.9 (CH), 119.2 (Cq), 64.4 (CHCl2), 34.0 (SCH2) ppm. EIMS: m/z (%) = 396 (20) [M+], 303 (25) [M - SCH2COOH]+, 252 (100) [M - SCH2COOH - C4H4]+, 177 (30) [benzo[h]quinoline]+. HRMS (ESI): m/z calcd. for C16H11Cl2N2O4S [M + H]+ 396.9811; found 396.9813.
4-(Dichloromethyl)-3-nitrobenzo[h]quinolin-2(1H)-one (14). To a suspension of {[4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetic acid (13) (0.840 g, 2.120 mmol) in a mixture of chloroform (15 mL) and glacial acetic acid (15 mL) aqueous hydrogen peroxide solution (35%, 1.2 mL) was added and stirred at rt for 2 d. Subsequently, water (50 mL) was added and the reaction mixture stirred for an additional 30 min. After extraction with chloroform (4 × 50 mL) the organic phase was dried with sodium sulfate and the solvent removed in vacuo. Yellowish solid, yield 0.475 g (70%), m.p. 286 °C. IR (ATR): νmax = 2923, 1654, 1537, 1350, 1216, 736 cm-1. 1H NMR (400 MHz, [D6]DMSO): δ = 8.90 (d, J = 8.0 Hz, 1 H), 8.33 (d, J = 9.2 Hz, 1 H), 8.01 (d, J = 8.0 Hz, 1 H), 7.88 (s, 1 H, CHCl2), 7.86 (d, J = 8.0 Hz, 1 H), 7.76 (t, J = 8.0 Hz, 1 H), 7.69 (t, J = 8.0 Hz, 1 H) ppm. 13C-NMR (100 MHz, [D6]DMSO): δ = 153.9 (Cq), 136.5 (Cq), 134.0 (Cq), 131.3 (Cq), 130.0 (Cq), 129.7 (CH), 128.4 (CH), 127.4 (CH), 123.9 (Cq), 123.8 (CH), 123.2 (CH), 121.9 (CH), 119.0 (Cq), 64.0 (CHCl2) ppm. EIMS: m/z (%) = 322 (100) [M+], 252 (25) [M - 2Cl]+, 229 (75) [M - Cl - NO2 - O]+, 207 (20) [M - 2Cl - NO2]+, 177 (60) [benzo[h]quinoline]+. HRMS (ESI): m/z calcd. for C14H8Cl2N2O3Na [M + Na]+ 344.9804; found 344.9816.
4-(Dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl 2,4-dichlorobenzoate (15a). To a solution of 4-(dichloromethyl)-3-nitrobenzo[h]quinolin-2(1H)-one (14) (0.086 g, 0.269 mmol) and 2,4-dichlorobenzoyl chloride (0.056 g, 0.269 mmol) in anhydrous THF (5 mL) under nitrogen atmosphere triethylamine (0.029 g, 0.290 mmol) was added at rt and stirred for 1 d. Subsequently, the reaction mixture was diluted with ice-water (30 mL) at stirring, extracted with chloroform (3 × 30 mL), washed with water (1 × 50 mL), saturated aqueous NaHCO3 solution (10 mL) and water (30 mL). The organic phase was dried with sodium sulfate, the solvent removed in vacuo, and the residue purified by column chromatography (petroleum ether/ethyl acetate, 3:1). Yellowish solid, yield 0.100 g (77%), m.p. 177 °C. IR (ATR): νmax = 2956, 1757, 1535, 1335, 1214, 1150, 732 cm-1. 1H NMR (400 MHz, CDCl3): δ = 9.09 (d, J = 7.8 Hz, 1 H), 8.64 (d, J = 9.4 Hz, 1 H), 8.05 (d, J = 8.5 Hz, 1 H), 8.02 (d, J = 9.4 Hz, 1 H), 7.92 (d, J = 7.8 Hz, 1 H), 7.76 (t, J = 7.8 Hz, 1 H), 7.70 (t, J = 7.8 Hz, 1 H), 7.54 (s, 1 H), 7.36 (d, J = 8.5 Hz, 1 H), 7.13 (s, 1 H, CHCl2) ppm. 13C NMR (100 MHz, CDCl3): δ = 161.0 (C=O), 147.1 (Cq), 145.4 (Cq), 140.5 (Cq), 137.7 (Cq), 136.8 (Cq), 134.1 (Cq), 133.7 (CH), 131.8 (CH), 130.6 (Cq), 130.6 (CH), 130.5 (Cq), 130.2 (CH), 128.2 (CH), 128.0 (CH), 127.5 (CH), 126.0 (CH), 125.3 (Cq), 121.9 (CH), 121.8 (Cq), 62.8 (CHCl2) ppm. EIMS: m/z (%) =493 (3) [M+], 172 (100) [COC6H3Cl2]+. HRMS (ESI): C21H10Cl4N2O4: m/z calcd. for [M+] 493.9389; found 493.9397.
4-(Dichloromethyl)-3-nitrobenzo[h]quinolin-2-yl 4-bromobenzoate (15b). The product was prepared according to benzo[h]quinoline 15a from quinoline 14 (0.100 g, 0.31 mmol), 4-bromobenzoyl chloride (0.068 g, 0.31 mmol) and triethylamine (0.034 g, 0.34 mmol). The reaction mixture was stirred for 4 h. Yellow solid, yield 0.122 g (78%), m.p. 239 °C. IR (ATR): νmax = 2924, 1754, 1558, 1508, 1398, 1227, 1056, 752 cm-1. 1H NMR (400 MHz, CDCl3): δ = 9.14 (d, J = 8.0 Hz, 1 H), 8.71 (d, J = 9.3 Hz, 1 H), 8.08 (d, J = 9.3 Hz, 1 H), 8.07 (d, J = 8.5 Hz, 2 H), 7.98 (d, J = 8.0 Hz, 1 H), 7.82 (t, J = 8.0 Hz, 1 H), 7.76 (t, J = 8.0 Hz, 1 H), 7.71 (d, J = 8.5 Hz, 2 H), 7.20 (s, 1 H, CHCl2) ppm. 13C NMR (100 MHz, CDCl3): δ = 163.0 (C=O), 147.1 (Cq), 145.7 (Cq), 137.6 (Cq), 134.1 (Cq), 132.4 (Cq), 132.3 (2C, CH), 132.2 (2C, CH), 130.6 (CH), 130.5 (Cq), 130.2 (Cq), 130.1 (CH), 128.2 (CH), 128.0 (CH), 126.7 (Cq , 126.0 (CH), 121.9 (CH), 121.7 (Cq), 62.8 (CHCl2) ppm. EIMS: m/z (%) = 505 (75) [M+], 392 (100) [M - Cl - Br]+. HRMS (MS): m/z calcd. for C21H11Cl2N2O4Br [M+] 503.9274; found 503.9280.
1,1-Bis(benzotriazol-1-yl)-2-nitro-3,4,4-trichlorobuta-1,3-diene (16a). The product was prepared according to the published literature [20] from the nitrodiene 1 and 1H-benzotriazole. Yield: 76%. All spectral data were in accordance with the literature.
1,1-Bis(1H-1,2,4-triazol-1-yl)-2-nitro-3,4,4-trichlorobuta-1,3-diene (16b). The product was synthesized according to a previously published procedure [21] from nitrodiene 1 and 1H-1,2,4-triazole. Yield: 92%. All spectral data were in accordance with the literature.
1,1-Bis(1H-pyrazol-1-yl)-2-nitro-3,4,4-trichlorobuta-1,3-diene (16c). To a solution of 2.72 g (40.00 mmol) of 1H-pyrazole in diethyl ether (50 mL) at 0 °C was added a solution of nitrodiene 1 (2.71 g, 10.00 mmol) in ether (5 mL). The resulting mixture was stirred at 0 °C for 1 h and at rt for an additional 20 h. The solvent was removed and then cold water (50 mL) was added with stirring. The resulting precipitate was filtered off with suction, washed with water (2 × 20 mL) and cold methanol (5 mL) and dried in vacuo to give 16c. Yellow solid, yield 3.01 g (90%), m.p. 133–134 °C. IR (KBr): νmax = 3102, 1653, 1532 (NO2), 1394, 1314 (NO2), 957, 772 cm-1. 1H NMR (400 MHz, CDCl3): δ = 7.93 (d, J = 1.3 Hz, 1 H), 7.87 (d, J = 1.3 Hz, 1 H), 7.56 (d, J = 3.0 Hz, 1 H), 7.51 (d, J = 2.8 Hz, 1 H), 6.63–6.58 (m, 2 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 146.0 (CH), 145.4 (CH), 138.3 (C1), 132.1 (CH), 131.8 (CH), 129.7 (C-NO2), 129.0 and 121.1 (C2Cl3), 111.0 (2 CH) ppm. EIMS: m/z (%) = 333 (2) [M+], 298 (100) [M - Cl]+, 263 (10) [M - 2Cl], 252 (37) [M - Cl - NO2]+, 217 (55) [M - 2Cl - NO2]+. HRMS (ESI): m/z calcd. for C10H6Cl3N5O2Na [M + Na]+ 355.9479; found 355.9485.
(E)-1-(1H-Benzotriazol-1-yl)-1-(naphth-1-ylamino)-3,4,4-trichloro-2-nitrobuta-1,3-diene (17a). The product was obtained according to a previously published procedure [22] from bisazole 16a and 1-naphthylamine. Yield: 90%. All spectral data were in accordance with the literature.
(E)-1-(1H-1,2,4-Triazol-1-yl)-1-(naphth-1-ylamino)-3,4,4-trichloro-2-nitrobuta-1,3-diene (17b). 1-Naphthylamine (1.50 g, 10.5 mmol) was added to a suspension of the bisazole 16b (3.37 g, 10.0 mmol) in MeOH (40 mL) at 0 °C within 2 min. The resulting mixture was stirred at 0 °C for 1 h, and was then kept at rt overnight. Subsequently, the supernatant liquid was concentrated to a volume of about 15 mL, cooled to 10 °C and then treated with aqueous HCl (5%, 80 mL). The mixture was stirred for an additional 20 min. The formed precipitate was collected on a suction filter, washed with water (2 × 20 mL) and cold MeOH (1 × 10 mL), and then finally dried under reduced pressure. Yellow solid, yield: 3.53 g (86%), m.p. 114–115 °C. IR (KBr): νmax = 3129, 1620, 1575 (NO2), 1334 (NO2), 1262, 1179, 773 cm-1. 1H NMR (400 MHz, CDCl3): δ = 11.73 (br s, 1 H, NH), 8.04 (d, J = 8.3 Hz, 1 H), 8.00 (s, 1 H, CHN), 7.89 (d, J = 7.3 Hz, 1 H), 7.87 (s, 1 H, CHN), 7.78 (d, J = 8.3 Hz, 1 H), 7.72–7.55 (m, 2 H), 7.26 (t, J = 7.7 Hz, 1 H), 6.91 (d, J = 7.3 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 153.0 (CHN), 147.8 (C1), 144.6 (CHN), 134.0 (NH-Cq), 130.6 (Cq), 129.2 (CH), 128.9 (Cq), 128.8 (CH), 128.5 (Cq), 128.1 (CH), 127.3 (CH), 125.1 (CH), 122.5 (CH), 121.0 (CH), 120.6 (Cq), 118.8 (C-NO2) ppm. EIMS: m/z (%) = 409 (35) [M+], 374 (1) [M - Cl]+, 327 (6) [M - Cl - HNO2]+, 214 (24), 143 (74), 133 (100). HRMS (ESI): m/z calcd. for C16H11Cl3N5O2 [M + H]+ 409.9973; found 409.9978.
(E)-1-(1H-Pyrazol-1-yl)-1-(naphthalen-1-ylamino)-3,4,4-trichloro-2-nitrobuta-1,3-diene (17c). To a suspension of the bisazole 16c (3.35 g, 10.0 mmol) in diethyl ether (40 mL) was added 1-aminonaphthalene (1.50 g, 10.5 mmol) at - 30 °C within 5 min. The resulting mixture was stirred at - 30 °C for 4 h, then kept at rt overnight. The mixture was concentrated to a volume of about 10 mL by means of a rotary evaporator. Subsequently, after addition of cold water (200 mL), aqueous HCl (37%, 5 mL) was added dropwise. After 20 min stirring, the mixture was extracted with chloroform (3 × 70 mL). The combined organic layers were washed with brine (150 mL) and dried with calcium chloride. After evaporation of the solvent the crude product was purified by means of column chromatography (petroleum ether/ethyl acetate, 3:1). Evaporation of all solvents gave nitrodiene 17c as a light-brown solid. Yield 3.69 g (90%), m.p. 177–179 °C. IR (KBr): νmax = 3222, 1617, 1571 (NO2), 1485, 1360 (NO2), 1084, 757 cm-1. 1H NMR (400 MHz, CDCl3): δ = 11.91 (br s, 1 H, NH), 8.11 (d, J = 8.2 Hz, 1 H), 7.89 (d, J = 8.1 Hz, 1 H), 7.75 (d, J = 8.3 Hz, 1 H), 7.67 (ddd, J = 8.2, 7.2, 1.1 Hz, 1 H), 7.59 (ddd, J = 8.2, 7.0, 1.1 Hz, 1 H), 7.58 (d, J = 2.0 Hz 1 H, CH-N), 7.37 (d, J = 2.4 Hz, 1 H, CHN), 7.24 (t, J = 7.8 Hz, 1 H), 6.78 (d, J = 7.3 Hz, 1 H), 6.23 (dd, J = 2.4, 2.0 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 150.6 (C1), 143.3 (CHN), 134.0 (NH-Cq), 131.6 (Cq), 131.4 (CH), 128.7 (CH), 128.3 (CH), 128.2 (Cq), 127.9 (CH), 127.7 (Cq), 127.1 (CH), 125.2 (CH), 122.1 (Cq), 121.44 (CH), 121.2 (CH), 118.1 (C-NO2), 109.1 (CH) ppm. EIMS: m/z (%) = 408 (2) [M+], 372 (4) [M - HCl]+, 362 (5) [M - NO2]+, 327 (3) [M - NO2 - HCl]+, (7), 292 (4) [M - 2 Cl - NO2]+, 169 (100). HRMS (ESI): m/z calcd. for C17H11Cl3N4O2Na [M + Na]+ 430.9840; found 430.9845.
2-(1H-Benzotriazol-1-yl)-4-(dichloromethyl)-3-nitrobenzo[h]quinoline (18a). Triethylamine (202 mg, 2.00 mmol) was added to a solution of nitrodiene 17a (461 mg, 1.00 mmol) in anhydrous chloroform (20 mL) at 0 °C within 2 min. The resulting mixture was stirred at 0 °C for 1 h, then kept at rt overnight. Subsequently, the supernatant liquid was evaporated in vacuo, and MeOH (10 mL) was added to the residue. The mixture was stirred for 10 min, the formed precipitate filtered off with suction, washed successively with aqueous HCl (5%, 1 × 10 mL), water (2 × 10 mL) and MeOH (1 × 5 mL), and then dried under reduced pressure. Yellowish solid, yield: 369 mg (87%), m.p. 260–262 °C. IR (KBr): νmax = 3007, 1578, 1542 (NO2), 1459, 1359 (NO2), 1205, 741 cm-1. 1H NMR (600 MHz, [D6)DMSO): δ = 9.08 (d, J = 7.9 Hz, 1 H), 8.72 (d, J = 9.3 Hz, 1 H), 8.53 (d, J = 8.3 Hz, 1 H), 8.39 (d, J = 9.4 Hz, 1 H), 8.33 (ddd, J = 8.3, 0.8, 0.8 Hz, 1 H), 8.22 (dd, J = 7.3, 1.2 Hz, 1 H), 8.17 (br s, 1 H, CHCl2), 7.96 (ddd, J = 7.3, 7.3, 1.4 Hz, 1 H), 7.95–7.91 (m, 2H), 7.69 (ddd, J = 8.3, 7.1, 1.0 Hz, 1 H) ppm. 13C NMR (150 MHz, [D6]DMSO): δ = 146.0 (Cq), 145.5 (Cq), 137.7 (Cq), 137.5 (Cq), 134.6 (C-NO2), 134.0 (Cq), 131.8 (Cq), 131.2 (CH), 131.1 (CH), 130.7 (CH), 129.9 (Cq), 129.1 (CH), 128.7 (CH), 126.4 (CH), 125.1 (CH), 121.6 (CH), 121.5 (Cq), 120.3 (CH), 113.4 (CH), 64.1 (CHCl2) ppm. EIMS: m/z (%) = 423 (12) [M+], 395 (4) [M - N2], 349 (7) [M - N2 - NO2], 302 (20), 279 (10), 266 (10), 240 (12), 92 (100). HRMS (ESI): m/z calcd. for C20H11Cl2N5O2Na [M + Na]+ 446.0182; found 446.0187.
2-(1H-1,2,4-Triazol-1-yl)-4-(dichloromethyl)-3-nitro-benzo[h]quinoline (18b). The product was prepared according to quinoline 18a from nitrodiene 17b (411 mg, 1.00 mmol) and triethylamine (202 mg, 2.00 mmol). Beige solid, yield: 135 mg (36%), m.p. 247–249 °C. IR (KBr): νmax = 3008, 1547 (NO2), 1501, 1364 (NO2), 1198, 1009, 732 cm-1. 1H NMR (600 MHz, [D6]DMSO): δ = 9.89 (s, 1 H, 1JC,H = 222 Hz, CHN), 9.27 (d, J = 8.1 Hz, 1 H), 8.63 (d, J = 9.3 Hz, 1 H), 8.44 (s, 1 H, 1JC,H = 209 Hz, CHN), 8.32 (d, J = 9.3 Hz, 1 H), 8.15 (d, J = 8.3 Hz, 1 H), 8.05 (br s, 1 H, CHCl2), 7.92 (ddd, J = 7.8, 7.1, 1.0 Hz, 1 H), 7.87 (ddd, J = 8.1, 7.1, 1.1 Hz, 1 H) ppm. 13C NMR (150 MHz, [D6]DMSO): δ = 153.9 (CHN), 145.6 (Cq), 145.5 (CHN), 137.2 (Cq), 136.5 (Cq), 133.9 (Cq), 132.9 (C-NO2), 131.04 (CH), 130.98 (CH), 129.6 (Cq), 128.7 (CH), 128.5 (CH), 125.8 (CH), 121.6 (Cq), 121.5 (CH), 64.0 (CHCl2) ppm. EIMS: m/z (%) = 373 (100) [M+], 338 (2) [M - Cl]+, 327 (7) [M - NO2]+, 303 (20), 292 (10) [M - NO2 - Cl]+, 280 (45), 269 (15), 253 (25). HRMS (ESI): m/z calcd. for C16H10Cl2N5O2 [M + H]+ 374.0206; found 374.0211.
2-(1H-Pyrazol-1-yl)-4-(dichloromethyl)-3-nitro-benzo[h]quinoline (18c). The product was prepared according to quinoline 18a from nitrodiene 17c (410 mg, 1.00 mmol) and triethylamine (202 mg, 2.00 mmol). Light brown solid, yield 291mg (78%), m.p. 197–199 °C. IR (KBr): νmax = 3006, 1545 (NO2), 1502, 1395, 1359 (NO2), 1260, 739 cm-1. 1H NMR (400 MHz, CDCl3): δ = 9.14–9.09 (m, 1 H), 8.77 (dd, J = 2.7, 0.6 Hz, 1 H, 1JC,H = 194 Hz, CH-N), 8.69 (d, J = 9.3 Hz, 1 H), 7.99 (d, J = 9.3 Hz, 1 H), 7.96–7.92 (m, 1 H), 7.81 (dd, J = 1.6, 0.6 Hz, 1 H, CH=N), 7.79 (ddd, J = 7.2, 7.1, 1.7 Hz, 1 H), 7.79 (ddd, J = 7.3, 7.3, 1.6 Hz, 1 H), 7.11 (s, 1JC,H = 179 Hz, 1 H, CHCl2), 6.60 (dd, J = 2.7, 1.6 Hz, 1JC,H = 179 Hz, 1 H) ppm. 13C NMR (100 MHz, CDCl3): δ = 146.1 (Cq), 143.7 (CH=N), 138.3 (Cq), 136.8 (Cq), 134.0 (Cq), 132.8 (C-NO2), 130.3 (Cq), 130.2 (CH), 129.3 (CH), 129.0 (CH), 128.1 (CH), 128.0 (CH), 125.2 (CH), 122.2 (CH), 120.7 (Cq), 108.8 (CH), 63.2 (CHCl2) ppm. EIMS: m/z (%) = 372 (100) [M+], 355 (2) [M - OH]+, 326 (10) [M - NO2]+, 291 (24) [M - NO2 - Cl]+, 279 (23), 256 (25), 243 (12) [M - NO2 - CHCl2]+. HRMS (ESI): m/z calcd. for C16H10Cl2N4O2Na [M + Na]+ 395.0073; found 395.0079.
Crystal Data
X-Ray structure analysis for ethyl {[(1E)-1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl]sulfanyl}acetate C8H7Cl4NO4S (3a), M = 355.01 g mol–1: A suitable single crystal of the title compound was selected under a polarization microscope and mounted in a glass capillary (d = 0.3 mm). The crystal structure was determined by X-ray diffraction analysis using graphite monochromated Mo-Kα radiation (0.71073 Å) [T = 223(2) K], whereas the scattering intensities were collected with a single crystal diffractometer (STOE IPDS II). The crystal structure was solved by Direct Methods using SHELXS and refined using alternating cycles of least squares refinements against F2 (SHELXL). All non-H atoms were located in Difference Fourier maps and were refined with anisotropic displacement parameters. The H positions were determined by a final Difference Fourier Synthesis [23].
C8H7Cl4NO4S (3a) crystallized in the triclinic space group P1 (no. 2), lattice parameters a = 7.685(1) Å, b = 8.189(2) Å, c = 12.236(2) Å, α = 77.66(1) °, β = 76.20(1) °, γ = 68.33(1) °, V = 688.2(2) Å3, Z = 2, dcalc. = 1.713 g cm–3, F(000) = 356 using 2558 independent reflections and 191 parameters. R1 = 0.0568 [I > 2σ(I)], wR2 = 0.1527 [I > 2σ(I)], goodness of fit on F2 = 1.043, residual electron density 1.145 and –1.098 e Å–3. Further details of the crystal structure investigations have been deposited with the Cambridge Crystallographic Data Center, CCDC 1583680. Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (Fax: +44(1223)-336033; e-mail: fileserv@ccdc.ac.uk or http://www.ccdc.cam.ac.uk).
X-Ray structure analysis for ethyl {[4-dichloromethyl-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetate C18H14Cl2N2O4S (5a), M = 425.27 g mol–1: A suitable single crystal of the title compound was selected under a polarization microscope and mounted in a glass capillary (d = 0.3 mm). The crystal structure was determined by X-ray diffraction analysis using graphite monochromated Mo-Kα radiation (0.71073 Å) [T = 223(2) K], whereas the scattering intensities were collected with a single crystal diffractometer (STOE IPDS II). The crystal structure was solved by Direct Methods using SHELXS and refined using alternating cycles of least squares refinements against F2 (SHELXL). All non-H atoms were located in Difference Fourier maps and were refined with anisotropic displacement parameters. The H positions were determined by a final Difference Fourier Synthesis [23].
C18H14Cl2N2O4S (5a) crystallized in the monoclinc space group P21/n (no. 14), lattice parameters a = 11.504(2) Å, b = 11.520(2) Å, c = 14.139(2) Å, β = 101.17(1) °, V = 1838.3(5) Å3, Z = 4, dcalc. = 1.537 g cm–3, F(000) = 872 using 3257 independent reflections and 295 parameters. R1 = 0.0642 [I > 2σ(I)], wR2 = 0.1298 [I > 2σ(I)], goodness of fit on F2 = 1.032, residual electron density 1.123 and –1.042 e Å–3. Further details of the crystal structure investigations have been deposited with the Cambridge Crystallographic Data Center, CCDC 1583675. Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (Fax: +44(1223)-336033; e-mail: fileserv@ccdc.ac.uk or http://www.ccdc.cam.ac.uk).
X-Ray structure analysis for 4-(dichloromethyl)-2-(morpholin-4-yl)-3-nitrobenzo[h]quinoline C18H15Cl2N3O3 (11b), M = 392.23 g mol–1: A suitable single crystal of the title compound was selected under a polarization microscope and mounted in a glass capillary (d = 0.3 mm). The crystal structure was determined by X-ray diffraction analysis using graphite monochromated Mo-Kα radiation (0.71073 Å) [T = 223(2) K], whereas the scattering intensities were collected with a single crystal diffractometer (STOE IPDS II). The crystal structure was solved by Direct Methods using SHELXS and refined using alternating cycles of least squares refinements against F2 (SHELXL). All non-H atoms were located in Difference Fourier maps and were refined with anisotropic displacement parameters. The H positions were determined by a final Difference Fourier Synthesis [23].
C18H15Cl2N3O3 (11b) crystallized in the monoclinc space group P21/c (no. 14), lattice parameters a = 7.1031(9) Å, b = 22.053(2) Å, c = 11.362(1) Å, β = 99.70(1) °, V = 1754.3(4) Å3, Z = 4, dcalc. = 1.485 g cm–3, F(000) = 808 using 3328 independent reflections and 295 parameters. R1 = 0.0528 [I > 2σ(I)], wR2 = 0.1378 [I > 2σ(I)], goodness of fit on F2 = 1.075, residual electron density 0.335 and –0.577 e Å–3. Further details of the crystal structure investigations have been deposited with the Cambridge Crystallographic Data Center, CCDC 1583679. Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (Fax: +44(1223)-336033; e-mail: fileserv@ccdc.ac.uk or http://www.ccdc.cam.ac.uk).
Antibacterial assays
Overnight cultures of the bacteria were grown aerobically at 37 °C in Müller Hinton broth with added 1% glucose and pH 7.2 for Gram-negative strains, or with Trypticase soy yeast extract medium (TSY – 30 g/l Trypticase soy broth, 3 g/L yeast extract, pH 7.2) for Gram-positive strains. The cultures were adjusted to an OD600 nm of 0.001, which resulted in a final start OD600 nm of 0.0005 in the test. 25 μL of test culture was added to 25 μL of a serial dilution of the test compounds in the appropriate medium for the different strains in accordance with standardized procedures in 384 well plates. For screening purposes, the residual absorbance in % was tested at compound concentrations of 5 and 50 µM. For selected compounds, concentration-dependent growth inhibition curves were recorded from stock solutions in DMSO at final concentrations of 100, 50, 25, 12.5, 6,25, 3.125, 1.56, 0.78, 0.39, 0.2 µM. As positive control compounds, Linezolid (both MRSA strains) Ciprofloxacin (E. faecium, E. coli), and Amikacin (P. aeruginosa) were applied. The highest DMSO concentration in the assay was 1%, which had no apparent effect on the growth of the bacteria. After an incubation time of 18 h at 37 °C under moist conditions, the optical density at 600 nm was measured with a Fusion Universal Microplate Analyzer (Perkin–Elmer, Waltham, USA). The lowest concentration that completely suppressed growth defined the MIC values. The following bacterial strains were used: Gram-negative: Escherichia coli (DSM 1116) and Pseudomonas aeruginosa PA7 (DSM 24068). Gram-positive: Staphylococcus aureus MRSA (clinical isolate, RKI 11-02670) and Staphylococcus aureus MRSA (DSM 11822). The MIC values were determined by curve fitting with Sigma Plot.
Antiproliferative assays
The effect of compounds on cell viability was probed with a WST-1 test using the procedure of Ishiyama et al. [24] as modified by Sasse et al [25]. The following cell lines were used: mouse fibroblast cell line L929 (DSM ACC 2), human cervix carcinoma cell line KB-3-1 (DSM ACC 158) and human breast cancer cell line MCF-7 (DSM ACC 115). In addition, the conditional immortalized human fibroblast cell line FS4-LTM (InScreenex, Braunschweig, Germany) was used without doxycyclin to induce primary cell-like behavior (Pub. No.: US2011/0189142 A2). Briefly, the subconfluent cells were washed with Earle’s Balanced Salt Solution (Gibco) without Ca and Mg, trypsinized and re-suspended in Dulbecco’s modified eagle’s medium that contained 5% fetal bovine serum (FBS; L929, KB-3-1, FS4-LTM) or Roswell Park Memorial Institute medium that contained 5% FBS, 0.5% Minimum Essential Medium Non-Essential Amino Acids, Gibco (MEM NEAA), 0.5% GlutaMAX (Gibco) and insulin at 5 μg/mL (MCF-7). 25 µl of serial dilutions of the test compounds (100-0.2 µM), that were made with a pipetting robot (epMotion, Eppendorf, Hamburg, Germany), were added to 25 µl aliquots of a cell suspension (1500 cells for KB3-1 and L929, 3000 cells for MCF-7 and 7500 cells for FS4-LTM) in 384 well microtiter plates. Blank and solvent controls were incubated under identical conditions. After an incubation period of 5 days (for L929, KB-3-1, and MCF-7) or 24 h (for FS4-LTM), 3 μL WST-1 (ready to use solution by Roche) was added. The incubation time of the plates at 37 °C varied between the cell lines from 20 min for KB-3-1, 30 min for L929, 1 h for FS4-LTM to 2 h for MCF-7, before measuring absorbance at 450 nm (reference 600 nm) with an Infinite 200 PRO plate reader (Tecan, Männedorf, Switzerland). As positive control compounds, Auranofin and Staurosporin were applied. The absorbance of the solvent control was set to 100%. The EC50 values were determined with Sigma Plot.
4. Conclusions
Starting from the versatile building block pentachloro-2-nitro-1,3-butadiene 1 a synthetic protocol for the efficient preparation of new 2,3,4-trisubstituted benzo[h]quinolines 5–6 was developed via the intermediates 4 in a two-step process. Various modifications on quinolines 5 such as oxidations and nucleophilic substitutions were investigated. In the process, new benzo[h]quinolines 8, 9, 11–15 and 18 with a unique substitution pattern consisting of a nucleophilic S, N or O-unit on the position 2, nitro group on the position 3 and the dichloromethyl group on the position 4 at the pyridine ring were synthetized.
The phenotypic cellular assays demonstrated that some of the benzo[h]quinolines synthesized herein possess high bioactivities against mammalian cell lines and the bacterial pathogen MRSA. Although the underlying molecular mechanism or target is unknown so far, the fact that the activity was not ubiquituous across the whole series, but depended on distinct substitution patterns found in some analogs, suggests that it is not due to unspecific effects. In addition, the overlapping, but non-parallel effects against bacteria and mammalian cells imply that it might be possible to find compounds that selectively target bacteria vs. eucaryotic cells. For their future synthesis, an efficient access has been established by this study.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figures S1–S30: 1H–NMR, 13C–NMR.
Author Contributions
Conceptualization, V.A.Z. and D.E.K.; synthesis and spectroscopic identification of the synthesized compounds, S.K. and V.A.Z.; evaluation of the biological activity of the synthesized compounds, M.B. and B.K.; writing—original draft, S.K., V.A.Z., M.G. and M.B.; writing—review & editing, D.E.K., V.A.Z. and M.B; project administration, D.E.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank G. Dräger (Leibniz University Hannover, Germany) for extensive HRMS measurements, and M. Weigert (Clausthal University of Technology) for technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of all compounds are available from the authors (V.A.Z.).
References
- Zapol’skii, V.A.; Bilitewski, U.; Kupiec, R.K.; Ramming, I.; Kaufmann, D.E. Chemistry of Polyhalogenated Nitrobutadienes, 16. Polyhalonitrobutadienes as Versatile Building Blocks for the Biotargeted Synthesis of Substituted N-Heterocyclic Compounds. Molecules 2020, 25, 2863. [Google Scholar] [CrossRef]
- Zapol’skii, V.A.; Berneburg, I.; Bilitewski, U.; Dillenberger, M.; Becker, K.; Jungwirth, S.; Shekhar, A.; Krueger, B.; Kaufmann, D.E. Chemistry of polyhalogenated nitrobutadienes, 17: Efficient synthesis of persubstituted chloroquinolinyl-1H-pyrazoles and evaluation of their antimalarial, anti-SARS-CoV-2, antibacterial, and cytotoxic activities. Beilstein J. Org. Chem. 2022, 18, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Zapol’skii, V.A.; Namyslo, J.C.; Gjikaj, M.; Kaufmann, D.E. Chemistry of polyhalogenated nitrobutadienes, 14: Efficient synthesis of functionalized (Z)-2-allylidenethiazolidin-4-ones. Beilstein J. Org. Chem., 2014, 10, 1638–1644. [Google Scholar] [CrossRef]
- Zapol’skii, V.A.; Namyslo, J.C.; Adam, A.E.W.; Kaufmann, D.E. Chemistry of Polyhalogenated Nitrobutadienes, 1: A New Synthesis of Perfunctionalized 3-Amino-4-nitrothiophenes. Heterocycles 2004, 63, 6, 1281–1298. [Google Scholar] [CrossRef]
- Chen, J.-J.; Lin, Y.-H.; Day, S.-H.; Hwang, T.-L.; Chen, I.-S. New benzenoids and anti-inflammatory constituents from Zanthoxylum nitidum. Food Chemistry 2011, 125, 282–287. [Google Scholar] [CrossRef]
- Luo, X.; Pedro, L.; Milic, V.; Mulhovo, S.; Duarte, A.; Duarte, N.; Ferreira, M.-J. U. Antibacterial Benzofuran Neolignans and Benzophenanthridine Alkaloids from the Roots of Zanthoxylum capense. Planta Med 2012, 78, 148–153. [Google Scholar] [CrossRef]
- Zhang, H.-l.; Gan, X.-q.; Fan, Q.-f.; Yang, J.-j.; Zhang, P.; Hu, H.-b.; Song, Q.-s. Chemical constituents and anti-inflammatory activities of Maqian (Zanthoxylum myriacanthum var. pubescens) bark extracts. Scientific Reports, 2017, 7, 45805. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Zhang, Z.; Xu, Y.; Chen, Y.; Li, C. Sanguinarine, a major alkaloid from Zanthoxylum nitidum (Roxb.) DC., inhibits urease of Helicobacter pylori and jack bean: Susceptibility and mechanism. J. Ethnopharmacol. 2022, 295, 115388. [Google Scholar] [CrossRef]
- Karlgren, M.; Vildhede, A.; Norinder, U.; Wisniewski, J.R.; Kimoto, E.; Lai, Y.; Haglund, U.; Artursson, P. Classification of Inhibitors of Hepatic Organic Anion Transporting Polypeptides (OATPs): Influence of Protein Expression on Drug–Drug Interactions. J Med Chem 2012, 55, 4740–4763. [Google Scholar] [CrossRef]
- Ullah, A.; Ullah, N.; Nawaz, T.; Aziz, T. Molecular mechanisms of Sanguinarine in cancer prevention and treatment. Anticancer Agents Med Chem. 2022 Aug 31. Online ahead of print. [CrossRef]
- Selvi, B.R.; Pradhan, S.K.; Shandilya, J.; Das, C.; Sailaja, B.S.; Shankar, G.N.; Gadad, S.S.; Reddy, A.; Dasgupta, D.; Kundu, T.K. Sanguinarine interacts with chromatin, modulates epigenetic modifications, and transcription in the context of chromatin. Chem Biol 2009, 16, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Nakanishi, M. Composition for improving resistance to environmental stress of plant and method for improving resistance to environmental stress of plant. EP2759201 A1, 30 July 2014. [Google Scholar]
- Matsuyama, N.; Kato, T.-a.; Kimura, K.; Mizutani, T.; Saeki, K.-i. Phenotype analysis of human cytochrome P450 2C9 polymorphism using a panel of fluorine-substituted benzo[h]quinolines as inhibitors of tolubutamide hydroxylation. Journal of Health Science 2006, 52, 821–824. [Google Scholar] [CrossRef]
- Lee, H.S.; Yang, J.Y.; Jeon, J.H. Insecticide composition for controlling stored product insects. Korea, Republic of, KR2016094534 A 2016-08-10.
- Esteruelas, M.A.; Fernández-Alvarez, F.J.; Oñate. E. Osmium and Ruthenium Complexes Containing an N-Heterocyclic Carbene Ligand Derived from Benzo[h]quinoline. Organometallics 2007, 26, 5239–5245. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Duarte, S.H.G.; Zanatta, N.; Martins, M.A.P. Regiospecific synthesis of 3-alkyl-2-aryl-4-trifluoromethylbenzo[h]quinolines by intramolecular cyclization of N-(2-alkyl-1-aryl-3-oxo-4,4,4-trifluorobut-1-en-1-yl)-1-naphthylamines. Synthesis 2002, 8, 1037–1042. [Google Scholar] [CrossRef]
- Campos, P.J.; Añón, E.; Malo, M.C.; Tan, C.-Q.; Rodríguez, M.A. Synthesis of substituted benzoquinolines by the irradiation of 3-amino-2-alkene imines. Tetrahedron 1998, 54, 6929–6938. [Google Scholar] [CrossRef]
- Karpenko, N.S.; Flyakova, V.I.; Matochkina, E.G.; Kodess, M.I.; Pashkevich, K.I. Synthesis of 6-fluoroalkylbenzo[h]cyclopenta[c]quinoline and -benzo[c]phenanthridine derivatives. Russ. Chem. Bull. 2003, 52, 1215–1216. [Google Scholar] [CrossRef]
- Potkin, V.I. , Zapol’skii, V.A., Kaberdin, R.V. Nitration of 2-H-pentachloro-1,3-butadiene. Doklady of the National Academy of Sciences of Belarus, 1996, 40, 1, 68–71. [Google Scholar]
- Zapol’skii, V.A.; Namyslo, J.C.; de Meijere, A.; Kaufmann, D.E. Chemistry of polyhalogenated nitrobutadienes, 10: Synthesis of highly functionalized heterocycles with a rigid 6-amino-3-azabicyclo[3.1.0]hexane moiety. Beilstein J. Org. Chem. 2012, 8, 621–628. [Google Scholar] [CrossRef]
- Zapol’skii, V. A., Potkin, V. I., Nechai, N. I., Kaberdin, R. V., Pevzner, M. S. Azolyl Derivatives of Nitrohalobutadienes. II. Synthesis and Some Reactions of 1,1-Bis(3,5-dimethylpyrazol-1-yl) and 1,1-Bis(1,2,4-triazol-1-yl)-2-nitrotrihalo-1,3-butadienes. Russ. J. Org. Chem. 1997, 33, 1632–1637.
- Zapol’skii, V. A., Potkin, V. I., Nechai, N. I., Kaberdin, R. V., Pevzner, M. S. Azolyl Derivatives of Nitrohalobutadienes. I. Reaction of 1,1-Bis(benzotriazol-1-yl)-2-nitrotrihalo-1,3-budadienes with N–, N,N–, and N,O–Nucleophiles. Russ. J. Org. Chem., 1997, 33, 1461–1467.
- Sheldrick, G.M. Crystal structure refinement with SHELXL, Acta Crystallogr. 2015, C71, 3–8. [CrossRef]
- Ishiyama, M.; Tominaga, H.; Shiga, M.; Sasamoto, K.; Ohkura, Y.; Ueno, K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol. Pharm. Bull. 1996, 19, 1518–1520. [Google Scholar] [CrossRef]
- Sasse, F.; Steinmetz, H.; Schupp, T.; Petersen, F.; Memmert, K.; Hofmann, H.; Heusser, C.; Brinkmann, V.; Matt, P. v.; Höfle, G.; Reichenbach, H. Argyrins, immunosuppressive cyclic peptides from myxobacteria. I. Production, isolation, physico-chemical and biological properties. J. Antibiot. 2002, 55, 543–551. [Google Scholar] [CrossRef]
Figure 1.
Structures of benzo[h]quinoline and two of its natural derivatives.
Scheme 1.
Three synthetic ways to 2,3,4-trisubsituted benzo[h]quinolines.
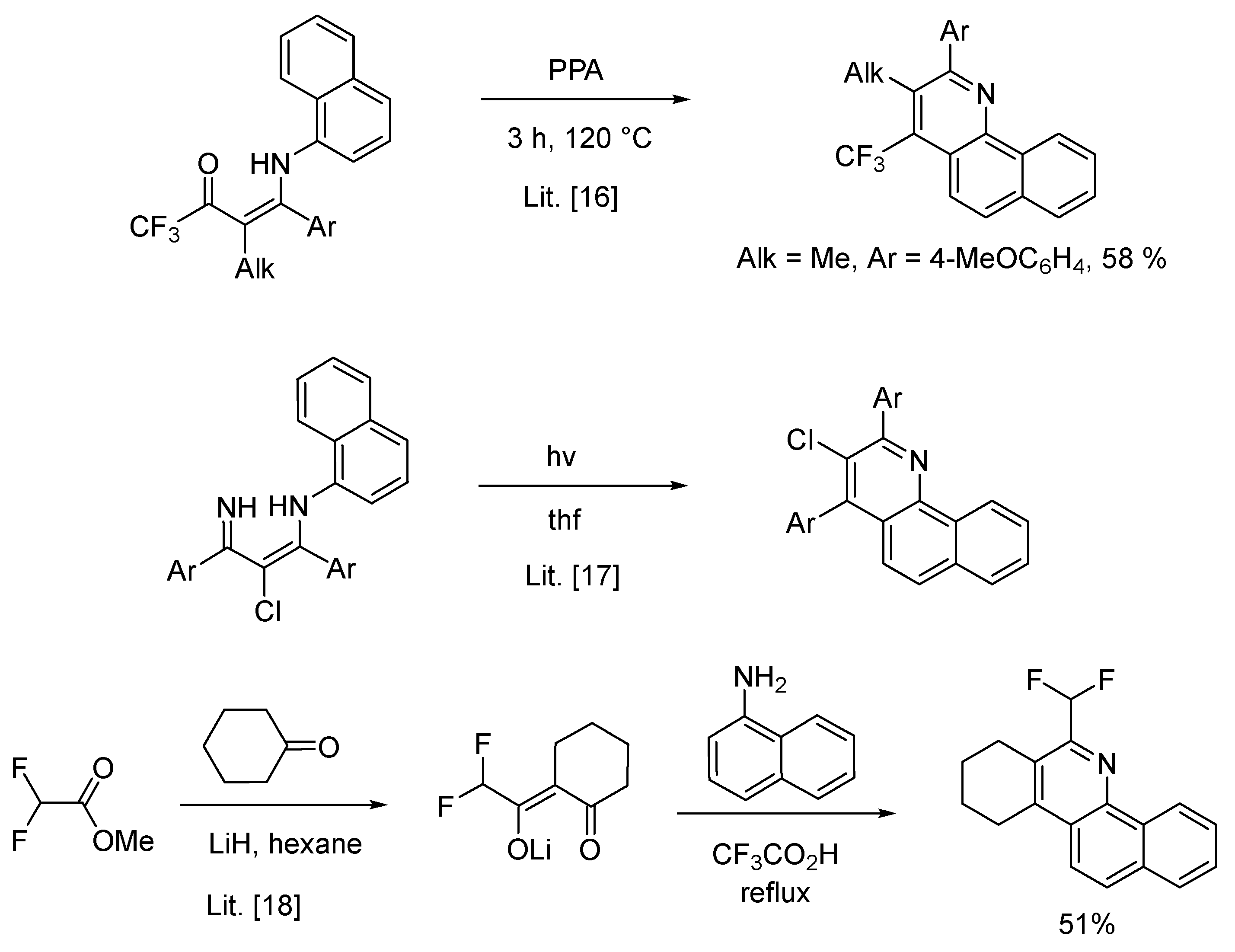
Figure 2.
X-ray single crystal structure of ethyl {[(1E)-1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl]sulfanyl}acetate (3a).
Figure 2.
X-ray single crystal structure of ethyl {[(1E)-1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl]sulfanyl}acetate (3a).
Scheme 2.
A new way to the 2,3,4-trisubstituted benzo[h]quinolines.
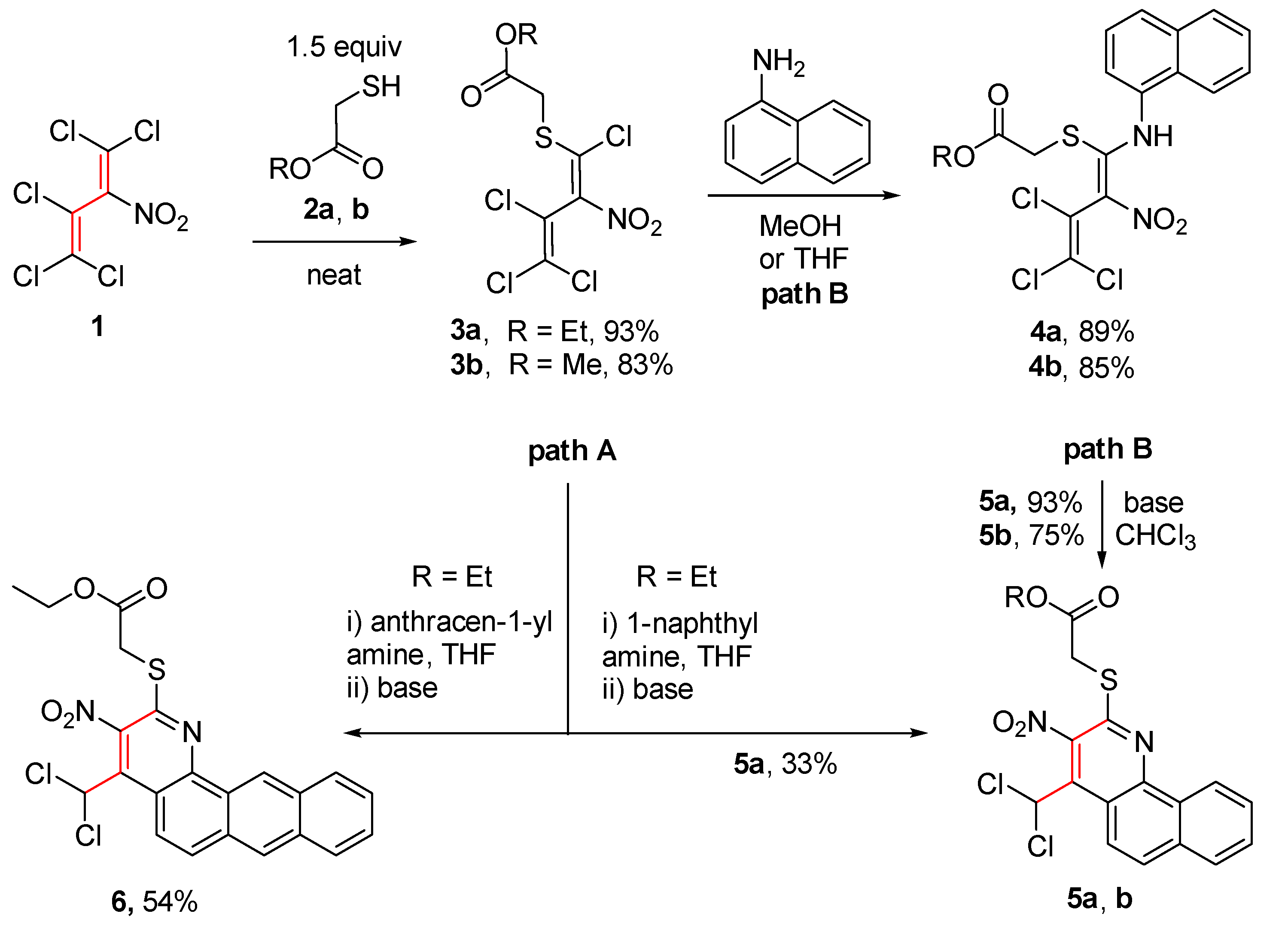
Scheme 3.
Assumed mechanism for the formation of benzo[h]quinolines 5.
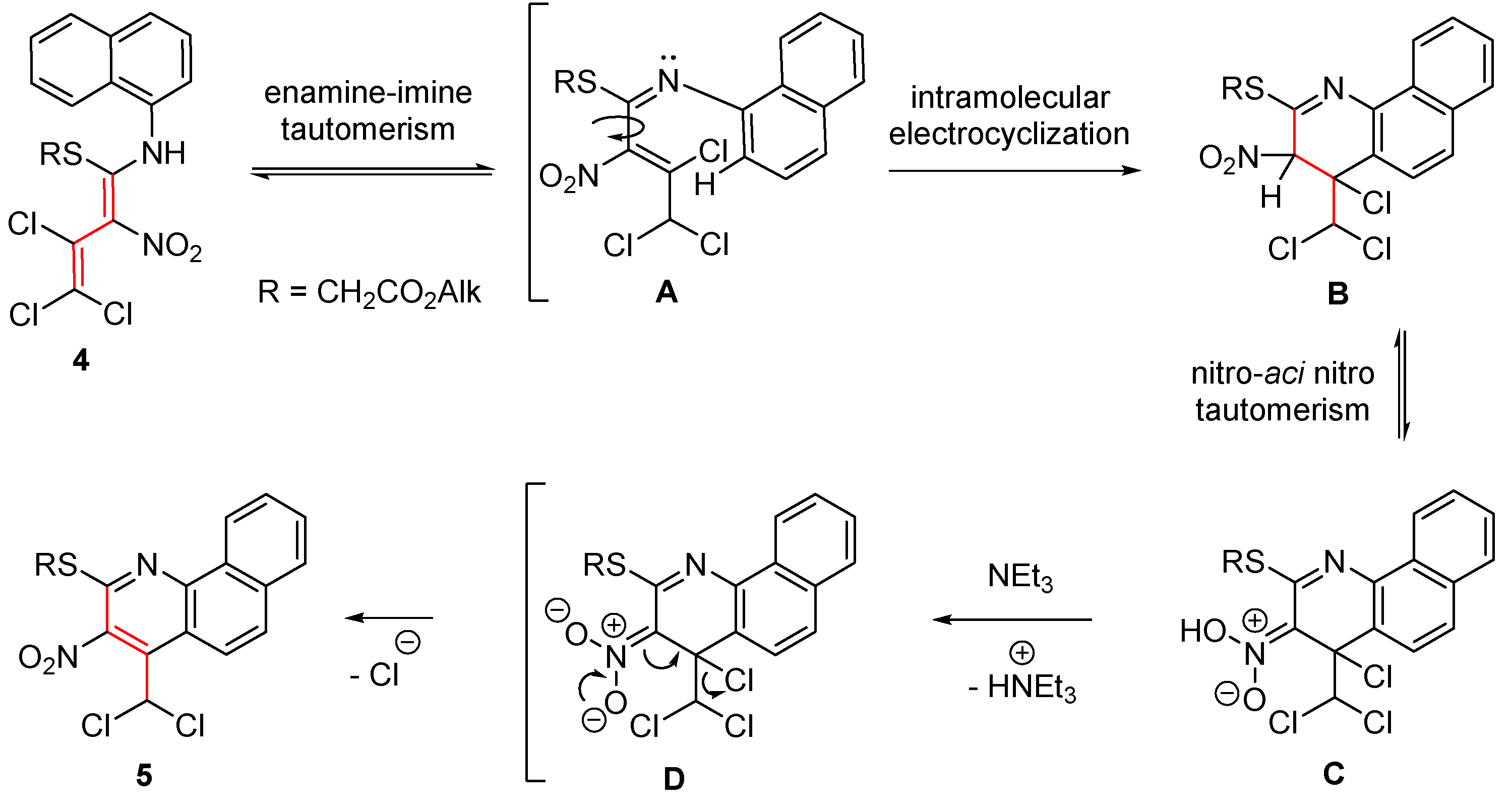
Figure 3.
X-ray single crystal structure of ethyl {[4-dichloromethyl-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetate (5a).
Figure 3.
X-ray single crystal structure of ethyl {[4-dichloromethyl-3-nitrobenzo[h]quinolin-2-yl]sulfanyl}acetate (5a).
Scheme 4.
Synthetic pathways to aminothiodienes 4a–b and thiazolidinones 7a–c.
Scheme 5.
Oxidation of sulfide 5a to sulfoxide 8 and sulfone 9.
Scheme 6.
Substitution reactions of sulfoxide 8 with S– and N–nucleophiles.
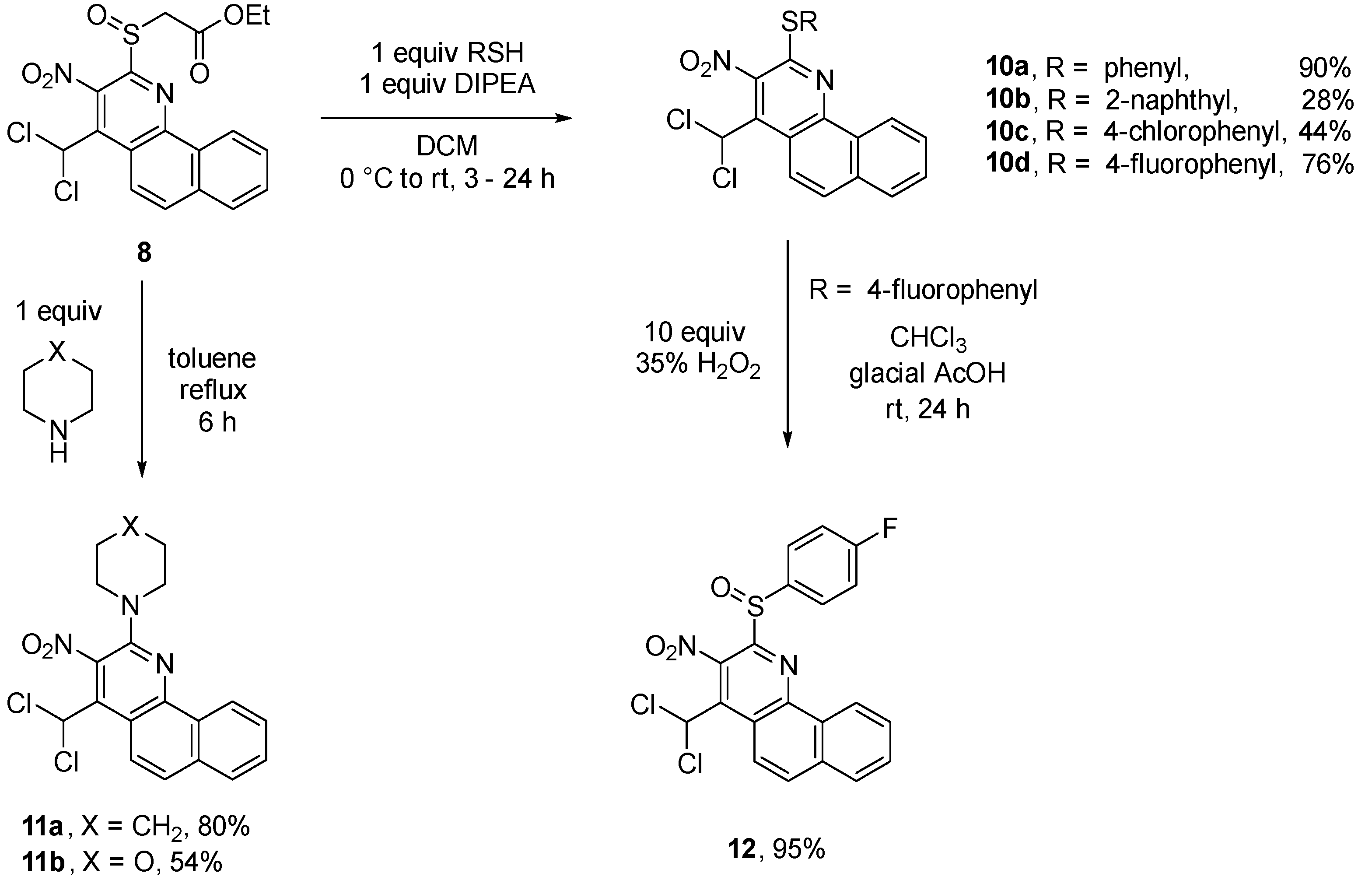
Figure 4.
X-ray single crystal structure of 4-(dichloromethyl)-2-(morpholin-4-yl)-3-nitrobenzo[h]quinoline (11b).
Figure 4.
X-ray single crystal structure of 4-(dichloromethyl)-2-(morpholin-4-yl)-3-nitrobenzo[h]quinoline (11b).
Scheme 7.
Chemical transformations of sulfide 5a to acid 13, quinolinone 14 and ester 15a, b.
Scheme 8.
Formation of benzo[h]quinolines 18a–c starting from nitrobutadiene 1.
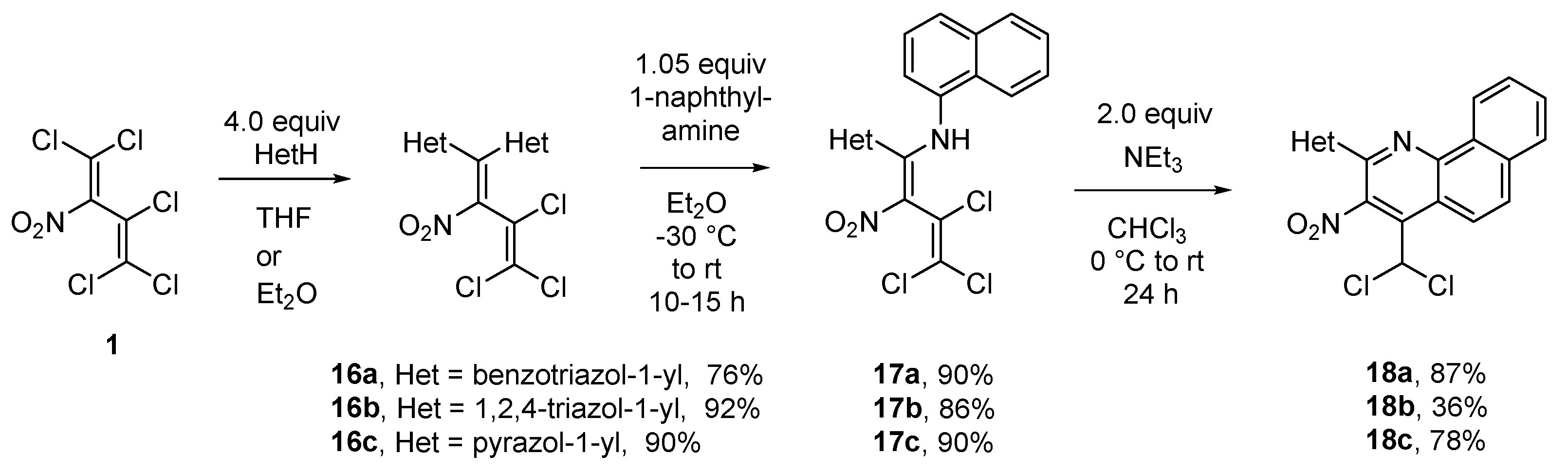

Table 1.
Antibacterial and antiproliferative activities of selected benzo[h]quinolines.
| Antibacterial assays | Antiproliferative assaysc | |||||
|---|---|---|---|---|---|---|
| Compound | MRSAa %growth | MRSAb MIC | L929 | KB-3-1 | MCF-7 | FS4-LTM |
| 6 | 112.4 | n.t. | n.t. | n.t. | n.t. | n.t. |
| 8 | 1.0 | 17.5 | 2.5 | 3.8 | 3.5 | 3.4 |
| 10d | 98.4 | n.t. | >50 | <0,1 | >50 | >50 |
| 11a | 95.6 | n.t. | >50 | >50 | 19.0 | >50 |
| 11b | 123.3 | n.t. | 27.0 | 10.0 | 50.0 | >50 |
| 12 | 2.0 | 1.2 (0.5) | 3.3 | 3.1 | >50 | 4.4 |
| 13 | 1.0 | 8.4 (3.3) | 45.0 | 6.9 | 17.0 | 15.7 |
| 16c | 125.6 | n.t. | n.t. | n.t. | n.t. | n.t. |
| 17a | 119.6 | n.t. | n.t. | n.t. | n.t. | n.t. |
| 17b | 112.0 | n.t. | n.t. | n.t. | n.t. | n.t. |
| 17c | 101.1 | n.t. | n.t. | n.t. | n.t. | n.t. |
| 18a | 95.1 | n.t. | n.t. | n.t. | n.t. | n.t. |
| 18b | 107.0 | n.t. | n.t. | n.t. | n.t. | n.t. |
| 18c | 110.5 | n.t. | n.t. | n.t. | n.t. | n.t. |
a) Strain DSM 11822; % growth at compound concentration of 5 µM; b) strain RKI 11-02670, minimal inhibitory concentrations in µM (µg/ml values in brackets) c) EC50 values in µM. .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



