
Submitted:
04 February 2023
Posted:
06 February 2023
You are already at the latest version
Abstract
A pandemic happens when a novel influenza A virus is able to infect and transmit efficiently to a new, distinct host species. Although the exact timing of pandemics is uncertain, it is known that both viral and host factors play a role in their emergence. Species-specific interactions between the virus and the host cell determine the virus tropism. These include binding and entering cells, replicating the viral RNA genome within the host cell nucleus, assembling, maturing, and releasing the virus to neighbouring cells, tissues, or organs before transmitting it between individuals. Influenza has a vast and antigenically varied reservoir. In wild aquatic birds, the infection is typically asymptomatic. Avian influenza virus (AIV) can cross into new species, and occasionally, it can acquire the ability to transmit from human to human. A pandemic might occur if a new influenza virus acquires enough adaptive mutations to maintain transmission between people. This review highlights the key determinants AIV must achieve to initiate a human pandemic and describes how AIV mutates to establish tropism and stable human adaptation. Understanding the tropism of AIV may be crucial in preventing virus transmission in humans and may help design vaccines, antivirals and therapeutic agents against the virus.
Keywords:
influenza A virus
; avian influenza virus
; virus tropism in human
; antiviral
; vaccine
1. Introduction
Influenza is a contagious acute respiratory illness caused by the influenza virus via direct infection, mainly in the respiratory tract, that usually exhibits symptoms like fever, headache, chills, myalgia, dry cough and sore throat [1,2]. Influenza viruses are a group of segmented, negative-sense RNA viruses that are classified into four types: A, B, C, and D [3]. These viruses belong to the Orthomyxoviridae family, which includes significant pathogens for humans and animals. Influenza A viruses can infect various host species, whereas influenza B and C mainly infect humans, and the new type of influenza D is known to circulate in cattle [4]. Influenza A and B can cause a predictable seasonal flu epidemic in humans every year. However, the influenza A virus (IAV) from zoonotic reservoirs can sometimes cause a deadly pandemic [5].
The natural reservoir of IAV is birds, where the virus can adapt and transmit to mammalian hosts like humans, pigs and poultry [6]. Transmission of IAV from birds to humans has been associated with high pathogenicity of the new novel virus that causes high severity and mortality. The deadliest pandemic in recent history, the influenza pandemic 1918 (H1N1) or Spanish Flu, which was most likely caused by an avian influenza virus (AIV), killed between 50 and 100 million people worldwide [7]. Additionally, a subtype of AIV, H5N1, was also reported to infect humans and caused a high mortality rate, estimated at more than 50% of infected patients [8]. Avian influenza is an infectious disease caused by AIV in poultry, commonly called the fowl plague. Human avian influenza is an acute respiratory tract infection caused by certain strains of subtypes of AIV. In 1878, a severe fowl plague in chickens was first reported in Italy. It was then confirmed to be caused by a virus identified as the influenza A virus in 1955. In 1981, such a disease was officially named avian influenza at the first international avian influenza conference [9]. AIV subtypes that have caused human avian influenza in the past and present include H1, H2, H3, H5, H6, H7, H9, and H10 [10].
AIV enters the respiratory tract through the nose, mouth, or even eyes in humans and continuously travels along the respiratory tract to search for suitable host cells [4]. Types of human cells infected by AIV are respiratory epithelial cells [11,12], intestinal epithelial cells [13,14], immune cells [15,16] and nerve cells [17]. AIV does not typically transmit directly to humans and is assumed to require consecutive adaptation before transmitting efficiently among humans [18]. AIV tropism in humans depends on various host and viral factors for efficient replication, which means that without proper interaction with the human host cell, AIV cannot replicate. The AIV tropism determinants can be found at each replication step, from viral entry into cells to virus progeny release from infected cells [19]. This review discusses the key determinants of AIV tropism in humans by both virus and host factors. Understanding AIV's tropism may be essential for preventing and controlling the virus' transmission across humans as well as enhancing the development of antivirals and therapeutics against the virus.
2. Influenza A Virus Structure
IAV virions are enveloped, pleiomorphic particles in the shape of spheres with a diameter of 100 nm, or filaments with a diameter of 100 nm to 30 µm [20]. They are composed of three main subviral components: an envelope, a layer of matrix 1 (M1) proteins, and a viral ribonucleoprotein (vRNP) core. Their outer layer (envelope) consists of a lipid bilayer membrane derived from the host cell membrane and acquired during the budding process; it contains two integral transmembrane glycoproteins forming spikes on the surface of the virus: hemagglutinin (HA) and neuraminidase (NA); and a transmembrane ion channel matrix 2 (M2) protein. IAVs are divided into different subtypes based on their surface glycoproteins HA and NA. To date, there are 18 different HA (H1-H18) and 11 NA (N1—N11) identified proteins, which results in 198 IAV potential subtype combinations [21]. Underlying the viral lipid membrane is a protein layer composed of the inner surface envelope M1 protein, which forms the shell, and serves as a structural support for viral particles, and the nuclear export protein (NEP/NS2), which contributes to nuclear export regulation [22,23].
The IAV genome is made of eight segments of negative-sense, single-stranded viral RNA (vRNA), identified from 1 to 8, which coded for the proteins polymerase basic 2 (PB2), polymerase basic 1 (PB1), polymerase acidic (PA), HA, nucleoprotein (NP), NA, M (M1 and M2) and non-structural protein (NS) (NS1 and NEP/NS2), respectively (Figure 1). They are packaged into a single virion with nucleoprotein to form a separate vRNP complex [24,25]. The genomic segments vary in size and encode 17 viral proteins [26] (Table 1). These proteins are grouped into 3 categories: the proteins of the polymerase complex, the surface glycoproteins and the proteins encoded by the internal gene segments. Each genomic RNA molecule is covered with NP and associated with an RNA-dependent RNA polymerase (RdRp) complex consisting of the PA, PB1 and PB2 proteins. The viral gene segments organize themselves in a similar fashion for all orthomyxoviruses. Each vRNA segment has a coding region, flanked at both sides by untranslated regions (UTRs) with a range of 19 to 58 nucleotides [27]. These contain regions specific to each gene segment, segment-specific noncoding regions (ssNCRs) as well as a conserved sequence common to all vRNAs: the 12 nucleotides of the 3 'end and the 13 of the 5' end are conserved for all the gene segments of all the influenza A strains. Partially complementary, they combine to form a secondary structure, hairpin or corkscrew, found at vRNP level [28,29].
3. Zoonotic Transmission of Influenza A Viruses
IAV evolution, the vast biodiversity of host species, and the host adaptation mechanisms (tissue tropism, host immune response, host receptor adaptation) enabled the virus to infect a wide variety of avian and mammalian animal species, which increases the probability of cross-species transmission events, from which they can spread to humans [30,43]. As illustrated in Figure 2, IAV has the ability to infect swine, avian, feline, equine, and canine, as well as humans and other mammalian species [44].
3.1. Swine
Pigs are excellent mixing vessels for IAV of different host origins as they are highly susceptible to co-infections with both avian and human influenza viruses due to the mixed sialic acid receptor (α-2,3 and α-2,6) distribution on the glycocalyx of epithelial cells lining the porcine trachea [43,45]. It is unknown if swine IAV infections existed prior to 1918 when swine IAV was clinically identified in the context of the 1918 pandemic [46]. Since then, swine IAV has been consistently recognized as a serious disease with significant economic and public health implications [45]. Swine IAV H1N1 was initially isolated by Shope and Lewis in 1930 [10]. Since then, IAV of various subtypes (H1N1, H1N2 and H3N2) has been isolated from pigs around the world, with some subtypes causing enzootic infections and others causing only localized outbreaks with no sustained spread [47].
3.2. Equine
Equine influenza is a respiratory viral disease which can affect all Equidae (horses, ponies, donkeys, etc.), as well as animals resulting from their crossbreeding (mules) [48,49]. It is highly contagious and spreads rapidly; an unvaccinated population has an almost 100% infection rate [50]. Mortality is low, except in young or weakened equids [49]. It is hypothesized that avian influenza strains were the progenitors of equine influenza viruses (EIVs) [51]. Two IAV subtypes are known to infect horses: H7N7 and H3N8. It appears that the H7N7 subtype is no longer in circulation since; the last epidemic occurred in the late 1970s. Since then, the H3N8 subtype has been the most isolated from sick horses [52].
3.3. Canine and Feline
Canine, feline influenzas are contagious acute respiratory illnesses in dogs and cats, caused by equine influenza virus H3N8, swine influenza virus H3N2 and H1N1, as well as the low pathogenic avian influenza virus (LPAIV) H7N2 and high pathogenic avian influenza virus (HPAIV) H5N1 [53,54]. As a result of co-infections with various avian, swine, and human IAV, other subtypes, primarily of avian origin or resulting from genetic reassortments, have also been isolated from cats and dogs with respiratory illness (i.e., H5N6, H5N2, H3N1) [53]. It has been reported in 2011 transmission of the 2009 H1N1 IAV from humans to cats, causing severe pneumonia in cats [55]. In 2016, the New York City Department of Health and Mental Hygiene (NYC DOHMH) documented the first cat-to-human transmission of IAV H7N2 [56].
4. Avian Influenza Virus
Avian influenza viruses (AIVs) can adapt to many host habitats thanks to the vast diversification of birds (more than 10,000 species) [57]. All AIVs are thought to have originated from migratory birds that near wetlands or other aquatic habitats, particularly those belonging to the orders Anseriformes (waterfowl) and Charadriiformes (shorebirds) [58]. Along their migration route, they spread and interchange various viral strains, which causes antigenic drift and genome reassortment, and can result in the emergence of novel influenza strains [8]. AIVs are classified into 16 HA and 9 NA subtypes based on the antigenic variation of their surface glycoproteins, which can yield up to 144 possible HA/NA combinations identified in birds, and two additional subtypes isolated from bats (H17N10, H18N11) [59,60]. Nearly all AIVs subtypes are present in wild aquatic birds and can spread sporadically to domestic poultry and/or mammals through their saliva, faeces, and nasal secretions [61]. In poultry, the pathogenicity of AIV is classified into two phatotypes: low-pathogenic avian influenza (LPAI) and high-pathogenic avian influenza (HPAI) [62]. Only six subtypes of avian influenza viruses have been reported to cause infections in humans: H3 (H3N8), H5 (HPAI H5N1, H5N6 and H5N8), H6, H7, H9 (LPAI H9N2), and H10 viruses [63,64] (Table 2). It is hypothesized that AIV spreads from poultry to humans primarily through direct contact with sick poultry or surfaces and objects contaminated by their faeces or secretions. Another speculation holds that AIV infects pigs first, and then the infection spreads to humans via contact with the infected pigs' secretions, blood, skin, and fur [65].
5. Key determinants of AIV tropism in humans: virus and host factors
Interaction between host and virus highlights some of the host factors that limit AIV replication at each stage in humans. However, mutations in AIV counteract these restrictions to enable efficient replication in human cells. This article discusses the key determinants of AIV's ability to replicate efficiently in humans, from both virus and host factors, in each stage of virus replication (Figure 3).
5.1. Attachment
The interaction of viral transmembrane envelope proteins; hemagglutinin (HA) and neuraminidase (NA), with host cell surface receptors during attachment and internalization is the critical determinant of AIV tropism in humans.
5.1.1. HA and host SA
HA is initially expressed as a precursor protein of HA0 before being cleaved into a mature HA1-HA2 complex by a host protease [88]. The mature HA1-HA2 complex consists of two domains, which are the globular head domain composed of HA1 (containing receptor binding site, RBS) and the stalk domain, primarily composed of HA2 (containing fusion peptide) [5]. The HA1 binds to cell surface receptors and triggers viral internalization via endocytosis, whereas the HA2 facilitates virus-host cell membrane fusion via alteration of pH in the endosome [89]. The RBS in HA1 is relatively shallow and comprises at least four amino acids: Y98, W153, H183, and Y195, which are conserved throughout all AIV subtypes (H1-H16 subtypes). These amino acids are encircled by 130-loop, 150-loop, 190-helix and 220-loop structures [90,91]. Although all these loop and helix structures are present in all strains, their length and amino acid composition vary, and these differences are typically critical determinants in recognizing the type of receptor for binding [92].
The most established and well-known host cell surface receptor that binds to the RBS in HA1 is sialic acid (SA), specifically N-acetylneuraminic acid (Neu5Ac or also known as α2,6 SA) and N-glycolylneuraminic acid (Neu5Gc or also known as α2,3 SA), located on the terminal glycans of host transmembrane proteins [4]. Generally, AIV prefers to bind α2,3 SA, while human influenza viruses prefer to bind α2,6 SA [92]. Studies of SA distribution in humans revealed that α2,6 SA is more abundant in the upper respiratory tract (including nasopharynx and mucus-producing cells), whereas α2,3 and α2,6 SA are equally prevalent in the lower respiratory tract, which includes the trachea, lungs, and bronchus [93]. Despite the presence of α2,3 SAs in the respiratory tract, AIV transmission into humans remains inefficient. This is possibly due to a lack of α2,3 SAs and effective host innate immune responses in the upper respiratory tract, which prevents AIV from traveling to the lower respiratory tract that contains more α2,3 SAs [93,94]. However, the previous cases of AIV transmission in humans occurred as a result of direct contact or exposure to infected animals, causing a high viral load that can travel deeper into the lower respiratory tract while escaping the host antiviral response, allowing AIV to bind to an acceptable amount of α2,3 SAs and then lead to a viral pathological effect [44,95].
To achieve efficient and stable transmission between humans, AIV needs to switch binding preference from α2,3 SA to α2,6 SA via amino acid substitution at the RBS of HA1 [96]. Initially, the RBS must have distinct amino acids to accommodate the different conformations of α2,3 SA and α2,6 SA (Shi et al., 2014). In the past influenza pandemics, at least two adaptive substitutions in RBS were required to shift the preference from α2,3 SA to α2,6 SA: E190D and G225D for H1N1 (1918 and 2009), Q226L and G228S for H2N2 (1957) and H3N2 (1968) [92]. In H1N1, E190D and G225D substitutions influence receptor-binding specificity via two slightly distinct but correlated mechanisms: mutations within the 190-helix (located at the top of the RBS) improve stability via hydrogen bonding and remove side chains that potentially inhibit α2,6 SA binding, while mutations within the 220-loop (located toward the RBS base) influence preferential adaptation from α2,3 SA to α2,6 SA [97]. In H2N2 and H3N2, the Q226L substitution established a hydrophobic environment incompatible with the α2,3 SA's hydrophilic glycosidic oxygen but complementary to the α2,6 SA's hydrophobic C6 atom, resulting in preferential binding to human receptors [98]. Furthermore, the G228S formed a hydrogen bond with SA, thus enhancing HA's affinity for the α2,6 SA [99]. Despite being different variants, the broad effect of Q226L/G228S in H2 and H3 appears to be similar to G225D in H1, where adaptive variants act in close proximity to the SA glycosidic linkage to promote binding to α2,6 SA while reducing preference for α2,3 SA through steric hindrance [97].
The possibility of a pandemic, along with evidence of partial human adaptation, has triggered the curiosity of researchers about the further adaptive mutations that may be required for AIVs to fully 'jump' the species barrier into humans, especially in the case of H5N1, H7N9, H6N1, H9N2 and H10N8. According to studies, H5N1 could establish persistent human-to-human and full airborne transmission via Q226L, N224, or G228S mutations [97]. However, current natural H5N1 and variants have been reported to preserve dominant binding to α2,3 SA, showing that only partial adaptation to α2,6 SA has occurred. Furthermore, from 2016 to 2020, there was a significant decline in H5N1 human infections, lowering the probability of such evolution and the risk of a new pandemic [69]. Early H7N9 isolates were reported to possess a Q226L substitution linked to pandemic-related receptor specificity switch mutation but retained significant binding to α2,3 SA [96]. According to de Vries et al., the variant requires simultaneous substitutions of three amino acids for complete α2,6 SA switch specificity, either V186G/K-K193T-G228S or V186N-N224K-G228S [100]. Meanwhile, in the 5th wave of human infection by H7N9, Pu et al. discovered six more substitutions in the HA1 RBS that may contribute to the α2,6 SA switch specificity: A118T, S123N, A131V, R136K, L173I, and M232I [101]. This wave was worse than previous and following waves, spreading the fastest from September 2016 to April 2017 with 623 confirmed cases [102]. Thus, current circulating H7N9 viruses have attracted a lot of attention due to the increased potential for a pandemic. In the cases of H6N1, H9N2, and H10N8, these viruses have a low risk of infecting humans and cause only mild symptoms. Nonetheless, their pandemic risks must not be overlooked. H6N1 and H10N8 could achieve complete human-type receptor specificity by only substituting G225D, Q226L, or G228S [90,100]. Meanwhile, H9N2 has been reported to be able to reassort with other circulating AIVs, including H5N1, H7N3, and H7N9 [97].
5.1.2. NA and host SA
Another AIV glycoprotein that interacts with SA, NA, has been chiefly focused on its role in the exit of progeny virus from infected cells, where recent growing data support an essential role of NA during the virus attachment and internalization process [5]. The catalytic activity of NA removes sialylated 'decoy' receptors on mucin, cilia, and glycocalyx, allowing the virus to travel smoothly across the cell surface and efficiently access functional receptors on the surface of the host cell [103]. In contrast with the catalytic activity of NA, it was proposed that NA also interacts with SA through direct receptor binding. The NA binding site is either at the same catalytic site or near it and is referred to as the second binding site. Although the biological role of receptor binding via NA is still unclear, it is thought to be critical for viral entry.
Substitution of D151G, D151A, D151N or T148I near the NA active site in H3N2 [104,105] or G147R in N1 NA [106] correlates with the receptor binding acquisition. The fact that the entry and infection of AIV having NA D151G in MDCK cells can be blocked by NA inhibitors [107] supports the crucial role of NA active site-associated receptor binding in virus entry. Concerning the complementary and opposing effects of HA and NA on SA binding, the relative activity of the two proteins must be balanced, preventing one's role from overshadowing the other's and preserving the capacity to successfully infect and release from cells [5]. The HA avidity for α2,6 SA must be high enough to allow binding before the NA may cleave the receptor in order to attach and enter the host cell. In contrast, HA binding to α2,6 SA cannot be too strong because the NA must be able to cleave the receptor to release new progeny virions and avoid aggregation at the cell surface [103]. Moreover, a functional balance between HA affinity and NA enzymatic activity with SA is necessary to facilitate airborne transmission between humans [4].
5.2. Membrane Fusion
Following viral internalization via endocytosis, the early endosomes containing virus particles interact with microtubules for retrograde traffic towards the microtubule's organizing centres adjacent to the cellular nucleus via dynein motor proteins [108]. Upon reaching the nucleus, acidification of the late endosome due to the host intracellular degradation system [109] then activates the proton channel function of AIV matrix protein 2 (M2) to bring protons into the virion and triggers a conformational change in HA, allowing virus-host cell membrane fusion [110]. Based on previous studies focused on virus-cell membrane fusion, several key determinants of AIV tropism in humans are highlighted in this replication step; HA, host proteases and pH of the host.
5.2.1. HA and host proteases
At an early stage, viral HA is translated as a fusion-inactive precursor to avoid premature fusion and HA activation along the secretory pathway [111]. The proteolytic cleavage of the HA precursor (HA0) by the HA-processing proteases into HA1 and HA2 subunits generate the fusion peptide at the N terminus of HA2, therefore gaining the ability to facilitate membrane fusion [3]. Hence, uncleaved HA is unable to result in membrane fusion. Since HA-processing proteases are not encoded in the virus genome, AIV utilizes proteases from the host cell for HA cleavage. The availability of appropriate host proteases is crucial for efficient virus replication and the severity of the infection [112]. Due to the specificity of the cleavage site, only proteases originating from specific tissues, organs or species can cleave and activate HA0, therefore showing that the cleavage process is a key determinant of the tropism and pathogenicity of AIV in different types of tissues, organs and species. Based on previous studies, there are at least two types of human host proteases that can cleave HA0 of AIV; transmembrane serine proteases and secreted serine proteases [111].
The discovery of specific type II transmembrane serine proteases (TTSPs) capable of cleaving and activating HA from human-adapted subtypes has sparked a lot of interest in influenza studies. The transmembrane protease serine-2 (TMPRSS2) from the TTSP family and human airway trypsin-like protease (HAT) have been reported to accelerate trypsin-independent influenza virus propagation in vitro [111]. Both proteases are expressed in the human lung, suggesting that they might play an important role in influenza infection in vivo [113,114,115]. Other members of the TTSP family have also been discovered to cleave and activate the viral HA since the discovery of these two proteases. Chaipan et al. reported that TMPRSS4 could cleave and activate the HA from the 1918 pandemic influenza virus [116] while Okumura et al. discovered that mosaic serine protease large-form and a splice variant, TMPRSS13, can cleave and activate the HA from HPAI strains, giving an alternative to furin cleavage [117].
The HA is also being cleaved by a few secreted trypsin-like proteases. Secreted proteases that cleave HA from the H3 subtype (H3N2) include cellular trypsin, porcine mast cell tryptase, tryptase Clara, and tryptase TC30 [3]. On the other hand, human mast cell tryptase was found incapable of activating HA [118]. Blood proteases such as plasmin, urokinase, plasma kallikrein, and thrombin also have been found to cleave and activate HA [3]. The cleavage of HA from various subtypes and strains by these blood proteases indicated diversity in cleavage within a particular subtype. This is fascinating because all of the subtypes and strains studied have a monobasic cleavage site with little variation in the cleavage site region.
The involvement of co-infecting bacteria in HA activation has also been explored, as they may provide an additional supply of HA-cleaving proteases. Certain strains of Staphylococcus aureus were found to secrete proteases that may activate HA either directly or indirectly, and the inoculation of this protease with the virus improved pathogenicity in vivo [6,111,119]. It is exciting for future research to extend this investigation to determine if any other commonly found co-infecting bacteria give an additional supply of HA-cleaving proteases. Overall, it is unclear if these proteases are present in their active form in the respiratory system during infection, but they clearly have the ability to activate HA from human-adapted subtypes and thus, may play a role in influenza infection in vivo.
The cleavage site sequence linking HA1 and HA2 is a crucial determinant for the pathogenicity and tropism of AIV in humans. Highly pathogenic avian influenza (HPAI) strains have a polybasic sequence at the HA cleavage site that allows for intracellular cleavage by ubiquitous subtilisin-like proteases such as furin [120]. Because the host protease responsible for cleavage activation is widespread, the HPAI strains are not restricted to a particular tissue in this condition, which is thought to be one of the main reasons for the enhanced virulence. Conversely, low pathogenic avian influenza (LPAI) strains feature a monobasic cleavage site that is cleaved by trypsin-like serine proteases that are either secreted into the extracellular space or present at the plasma membrane. As a result, the localisation of these proteases appears to limit the tissue tropism of LPAI. Although not well understood in avian species, equivalent host proteases for AIV in humans are thought to be primarily found in the respiratory system [121].
Moreover, N-glycosylation of the HA of AIV, which involves carbohydrate side chains linked to asparagine residues, is believed to significantly influence the proteolytic activation of HA [3]. The glycosylation at asparagine of HA can sterically block or limit the protease access to the cleavage site and modulate HA activation and AIV virulence [122]. The deglycosylation or the loss of the linked carbohydrate side chains due to an asparagine mutation at position 11, enables the protease-independent HA activation and a shift to higher pathogenicity. In contrast, in an HPAIV A/Mallard/Huadong/S/2005(H5N1) virus, the removal of the same glycosylation site resulted in significantly delayed HA cleavability and lowered virus fitness in vivo and in vitro, while the experimentally modified glycosylation site was restored after a few viral replication cycles [123].
5.2.2. HA and pH of the host
Aside from host proteases and carbohydrate modification on HA, the membrane fusion activity of HA is influenced by the pH of the host cell. The association between HA and pH of the host cell is always attributed to HA activation pH and HA stability. HA activation pH is the pH value at which membrane fusion occurs due to HA's irreversible conformational change. In comparison, HA stability is defined as the ability of HA to resist inactivation after exposure to an extreme pH value [124]. All subtypes and species have an HA activation pH of 5.0 to 6.0, with avian viruses (H5N1, H7N7, H7N9, H9N2) having an average HA activation pH of 5.7 and human viruses (H1N1, H3N2) trending lower with an average of 5.4. Before infecting human airway epithelial cells, AIV virions must cross the respiratory mucosa, which has a pH as low as 5.5 [4,125]. At the low pH condition outside the targeted cell (as in the respiratory mucosa), HA of AIV will be activated prematurely. This will result in the irreversible conformational change of HA, which will then cause the inactivation of the virions [124].
In order to avoid premature activation, the HA activation pH of AIV needs to be in the range between 5.0 and 5.5, slightly lower than the pH of respiratory mucosa, as this will increase HA stability for efficient transmission in humans. Avian H5N1 mutants with activated stabilized HA at pH 5.2 demonstrated efficient replication in the upper respiratory tract and airborne transmissibility in ferrets, similar to human influenza viruses [126]. The H17, Y17, H1062, and H1112 amino acid substitutions are likely the causes of the stabilized HA in H5N1 and other AIV subtypes [127]. It is generally recognised that electrostatic interactions between amino acids at interfaces, including hydrogen bonds, salt bridges, and van der Waals interactions, have a significant role in HA's stability, flexibility, and functionality. For example, it was proposed that at low pH, the protonated H1062 establishes a repulsive force with the K512 at the stalk, which may be crucial for the folding of the helix A following the development of the extended intermediate [127]. This repulsive force is lost when one of these amino acids is changed to a neutral amino acid, which lowers the fusion’s pH and stabilizes the HA.
5.3. Nuclear Import of vRNP
Upon membrane fusion, the newly released vRNP from the cytoplasm need to be imported into the nucleus since the replication and transcription of the viral genome by the viral RNA polymerase take place inside there. In comparison to the earlier viral entry steps, the import of vRNP into the nucleus is highly dependent on the host cell machinery [128]. The nuclear import can be facilitated by factors such as heat shock protein 90 (HSP90) [129], heat shock protein 40 (Hsp40/DnaJB1) [130], phospholipid scramblase 1 (PLSCR1) [131], translation elongation factor 1 delta (eEF1D) [132], nucleoporin 85 [133], and importin α [134], which tend to result in efficient and enhanced IAV replication. Among host machineries involved in nuclear import of vRNP, interaction between importin α and viral NP is considered as key determinant of AIV tropism in humans [134,135].
5.3.1. NP and host importin α
Protein trafficking from the cytoplasm into the nucleus is a facilitated process driven by importin family members, which are divided into importin α and importin β based on their function [136]. Importin α recognizes nuclear localization signal (NLS) from molecules that leads to recruitment of importin β that facilitates molecules into the nucleus through nuclear pore complex (NPC) [128]. IAV hacks this host machinery by equipping NP (which is a complex as vRNP) with NLS that is recognized by importin α as an NLS-carrying cargo protein, and thus results in vRNP being transported into the nucleus by importin β [128].
Importin α is further categorized into a variety of isoforms, with the isoforms of α1, α3, α4, α5, α6 and α7 being found in both avian and human hosts [134]. According to Gabriel et al., AIV prefer on utilizing importin α3 (in avian host), whereas adapted human AIV prefer on utilizing both importin α3 and importin α7 (in human host) to start transport of viral proteins into nucleus [134]. The fact that importin α7 is more widely distributed in the upper and lower respiratory tracts of humans than importin α3 suggests that the adapted human AIV may be under selective pressure to prefer importin 7 [137]. Hence, a retain preference to importin α3 and an additional preference towards importin α7 is needed by AIV to allow efficient nuclear import in human cells [134,137,138].
The adaptation of H7N7 NP towards importin α7 via a substitution from asparagine to lysine at location 319 (N319K) promotes viral growth and enhances pathogenicity in human cells [134]. This N319K substitution was also found in human isolates H5N1 [139], and therefore highlighting the possibility of this substitution in enhancing the nuclear import of vRNP and efficient replication of AIV in humans.
5.4. Replication and Transcription of Viral RNA
Because of the acidification of the late endosome and the irreversible conformational change of HA, the fusion peptide hidden inside HA is exposed and inserted into the target membrane, causing the viral and host membranes to merge [4]. Subsequently, the virus releases the viral vRNPs containing viral RNA, NP and RNA polymerases (PB1, PB2 and PA) from the M1 layer into the cytosol, and then the vRNPs travel into the nucleus and proceed with viral RNA replication and transcription [128].
5.4.1. PB2 and host ANP32A
The replication of viral RNA takes place in the nucleus, where it comprises two-step processes. In the first step, the complementary RNA intermediate (cRNA) is synthesized from the viral RNA template, while in the second step, the progeny vRNA is synthesized from the cRNA template [140]. The initiation starts when the RNA polymerase generates a pppApg dinucleotide at position 1 (3’ terminal UC of the vRNA template) that serves as a primer for replication, with the priming loop acting as a stabilizer for the initiation complex. Following the initiation, the priming loop is truncated, stimulating the nascent cRNA's elongation. In order to avoid cRNA degradation, the nascent cRNA released from the RNA polymerase binds to the new NP. It assembles with the terminal end of the cRNA and the newly synthesized RNA polymerase to form a complementary RNP (cRNP) complex. In contrast to cRNA synthesis, where pppApg is produced at position 1 of the vRNA template, pppApg during vRNA synthesis is produced at locations 4 and 5 of the cRNA template. The dimerisation between the cRNP RNA polymerase and a regulatory polymerase causes the priming loop to change position and facilitates the backtracking of the cRNA template. This contributes to the realignment of pppApg from positions 4 and 5 to positions 1 and 2 before acting as a primer to initiate vRNA synthesis [141]. As soon as the vRNA product leaves the cRNP-resident polymerase's active site after elongation, the 5' end is most likely grabbed by a stabilizing polymerase, starting the assembly of the vRNA with NP into vRNP [142].
Since AIV polymerases conduct genome replication poorly in humans [4], there are many mutations for the host adaptation, the best known of which is E627K in the PB2 that interacts with a host protein, acidic nuclear phosphoprotein 32 family member A (ANP32A) [143]. ANP32A belongs to a group of nuclear proteins involved in several cellular processes, including messenger RNA export and the regulation of transcription [144]. It was proposed that the residue at location 627 of PB2 interacts with the host ANP32A to recruit a second polymerase for nascent vRNP formation [145]. The dependency of viral polymerase on ANP32A is essential to cross the host barrier from an avian to a human host [142]. Initially, avian ANP32A possesses a unique 33-amino acid insertion needed to support PB2 627E-containing polymerase. However, the lack of this insertion in human ANP32A restricts the AIV polymerase from replicating [146]. Interestingly, the E627K mutation in PB2 causes AIV polymerase activity to be supported by the shorter ANP32, resulting in AIV replication in humans [142]. All human circulating viruses of the 20th century carried the E627K mutation in the PB2, including the 1918 H1N1 pandemic virus, followed by the 1957 H2N2 and 1968 H3N2 pandemic viruses [7]. Alternatively, the 2009 pandemic virus retains E627 in PB2 but with mutations of G590S and Q591R in PB2, which are close to position 627, explains how it replicated efficiently in humans [4,147].
5.4.2. PA and host RNA polymerase II
Since influenza virus RNA polymerase cannot synthesize and methylate cap structures as nonsegmented negative-sense RNA viruses do, viral RNA polymerases snatch the cap-1 structure from the nascent host mRNA during viral transcription in the nucleus [142]. The transcription starts when the viral RNA polymerase interacts with the carboxy-terminal domain (CTD) of host RNA polymerase II (Pol II) that stabilizes the PB2 cap-binding and PA endonuclease domains before PB2 binds to the 5’ cap of nascent host mRNA and PA cleaves about 10 - 14 bases downstream from the cap [140]. Following that, the 3' end of the cap base pairs with the 3' end of the viral RNA template, which initiates elongation and duplex-winding until the viral RNA polymerase stops on a uridine sequence near the 5' end of the viral RNA template. This terminates transcription by the addition of a poly(A) tail, which generates viral pre-mRNA [142].
The interaction with host factors during viral genome transcription highlights PA potential as a key determinant of AIV tropism in humans. The PA protein is structurally divided into the major domains of N-terminal (PA-N) and C-terminal (PA-C) [148]. The PA-N domain comprises the cap-snatching endonuclease and protease active sites, while the PA-C domain forms the core structure of the polymerase complex with PB1. The PA-C also has a site for 5' vRNA binding and a site for host Pol II interaction, which together constitute the PA arch that functions to grab 5'-capped primers from nascent Pol II transcripts for transcription of viral mRNA. According to Arai et al., PA-C localized mutations, S388R and A448E, contribute to the higher polymerase activity of H5N1 and expand the host range into humans [148]. PA residue 388 is a vital element of the 5' vRNA binding site, located within the PA arch and the neighbouring PB1 β-hairpin domain. Initially, S388 was involved in a hydrogen bond network with the PB1 protein; however, substituting R388 excluded the residue from the network. This suggests that the S388R substitution in PA creates flexibility at this location and potential association with the vRNA promoter. Therefore, S388R may influence viral polymerase activity by modifying the interaction between the polymerase complex and the viral RNA promoter. Subsequently, PA residue 448 is found in helix α16 of the polymerase complex structure that interacts with the PB1 domain, and this residue also directly interacts with the host CTD Pol II [149]. The A448E substitution forms a new hydrogen bond to N444, possibly stabilizing the helix α16 structure. Thus, it is suggested that the substitution of A448E may influence polymerase activity by altering the main structure of the polymerase complex or interacting with host Pol II. In the case of H7N9, the substitution of amino acid at location 409 from asparagine (N) to serine (S) increases the polymerase activity of the virus to replicate efficiently in humans and mammals [150]. The interaction between 409S and the PB1-N terminal region may be the cause of the increase in polymerase activity, and as a result, this interaction may also enhance H7N9's ability for human adaptation [150].
5.5. Maturation of Viral mRNA
After transcription, the viral mRNAs leave the nucleus to be transported to the host ribosome for translation. However, the viral pre-mRNA of M and NS goes through splicing (using host machinery), where introns (noncoding sequences) must be cut out to precisely link exons, the actual coding sequences, to generate mature mRNA before translation [40]. Influenza viruses hijack host splicing machinery to block host mRNA translation while utilizing the host spliceosome to allow translation of specific spliced influenza virus mRNA.
5.5.1. M and host huTRA2A
Many host splicing regulators have been recognized as having a role in IAV mRNA splicing regulation, and one of them, host human transformer-2 protein homolog alpha (huTRA2A), has been reported as a determinant of AIV's ability to adapt to humans [151]. huTRA2A is one of the host splicing regulators, along with splicing factor 2 (SF2), heterogeneous nuclear ribonucleoprotein K (hnRNP K) and influenza virus NS1 binding protein (NS1-BP), that has been linked to the splicing of IAV mRNA [152]. SF2, hnRNP K and NS1-BP all promote virus replication in both humans and avians, but huTRA2A promotes virus replication in humans while inhibiting virus replication in avians [151].
As reported by Zhu et al., due to the RNA-recognition motifs of huTRA2A, it is able to bind the intronic splicing silencer (ISS) motif (GAAARGARR) of human IAV non-structural (NS) and AIV matrix (M) genes to stop mRNA splicing [151]. However, the binding generates two different outcomes. The inhibition of human NS mRNA splicing by huTRA2A produces a small but balanced ratio of NS1/NEP, which eventually increases viral polymerase activity and thus promotes human virus replication. On the other hand, the inhibition of AIV M mRNA splicing by huTRA2A leads to an imbalance in M2/M1 expression (less M2 and the same amount of M1), which in turn restricts AIV replication in humans, possibly because of the M2 role in virus budding. The results were confirmed by knocking down huTRA2A, which decreases human IAV replication while increasing AIV replication in human A549 cells. Human NS gene mutations 234G/236G and AIV M gene mutation 334M, both of which result in the loss of the ISS motif, have the same effect as the knockdown, decreasing human IAV replication while increasing AIV replication in human A549 cells. These findings indicate that huTRA2A is one of the determinants for AIV to cross the human barrier and that the AIV M gene substitution 334M is an important adaptive mutation that leads to a balance in M2/M1 expression for efficient replication in humans.
5.6. Assembly and Trafficking of Viral Proteins
Translation of IAV mRNA is carried out by cytosolic ribosomes (for PA, PB1, PB2, NP, NS1, NS2 and M1 transcripts) and endoplasmic reticulum-associated ribosomes (for HA, NA and M2 transcripts). All nascent proteins translated from the cytosolic ribosome will be imported back into the nucleus, while nascent HA, NA and M2 are trafficked through the Golgi to the plasma membrane. Once the copies of vRNA are generated in the nucleus, nascent NP binds to them, followed by the assembly of nascent PA, PB1 and PB2, forming vRNP [128]. Then, the vRNPs are exported from the nucleus with the assistance of M1 and NS2/NEP before being trafficked to the plasma membrane, where HA, NA and M2 are all located at the budding boundary. During the assembly and trafficking of new viral proteins, SERTA domain containing 3 (SERTAD3) is among the host factors that inhibit AIV replication in humans.
5.6.1. RdRp complex and host SERTAD3
SERTAD3 is a transcription factor belonging to the SERTA family. In normal cells, SERTAD3 knockdown significantly reduces cell’s growth rate, whereas SERTAD3 overexpression causes oncogenic transformation, implying a role in cellular growth [153]. Regarding this protein's role in infection and immune response, SERTAD3 interacts with the African swine fever virus protein MGF360-16R [154] and is involved in the late-phase response of toll-like receptor (TLR) signaling [155].
According to transcriptomic data from infected A549 cells, SERTAD3 was the most significantly expressed gene among the four SERTAD family members in response to H5N1 infection [156], indicating SERTAD3 may function as an interferon-stimulated gene (ISG). Similar to many other ISGs, Sun et al. postulated that SERTAD3 might be involved in antiviral signaling by participating in retinoic acid-inducible gene I (RIG-I)-like receptor (RLR) signaling [157]. Not long after, they discovered that SERTAD3, rather than playing a role in RLR signaling, suppresses AIV replication by interacting with the PB1, PB2, and PA and blocking the assembly of the functional RdRp complex. To demonstrate that SERTAD3 does not merely bind any overexpressed protein, NA and NP were utilized as negative controls, and no interaction was observed between SERTAD3 and either NA or NP. Additionally, immunofluorescence staining of SERTAD3 with PB1, PB2, or PA following virus infection revealed that all three of these proteins co-localized with SERTAD3 in the nucleoli [157].
In addition, a peptide containing eight amino acids (labelled as AD-C) found in the C-terminal activation domain of SERTAD3 is important in limiting AIV replication [157]. The deletion mutants missing the C-terminal activation domain lost the inhibitory activity of SERTAD3 (increased AIV replication), and overexpression of the AD-C reduced H5N1 minireplicon activity. We speculate that if AIV acquires an adaptive mutation that neutralizes this 8-amino acid peptide, SERTAD3's inhibitory function during the assembly of the RdRP complex will be blocked, allowing AIV to replicate in humans efficiently.
5.7. Release
When all of the new viral components are situated at the budding boundary within the membrane, IAV alters the structure of the membrane to initiate bud formation and, finally, the scission of the new virion envelope from the membrane, allowing it to infect other host cells [128]. Multiple studies have suggested that the involvement of M1 increases budding efficiency and shape uniformity of the newly produced virion envelope, and that the expression of HA and NA is sufficient to induce budding and facilitate the virus release [158,159,160]. Adaptation of these three viral proteins, HA, NA, and M1, which interact with the host factors, G protein subunit beta 1 (GNB1) and sialic acid receptor, promotes the efficient release of AIV from infected human cells.
5.7.1. M1 and host GNB1
M1 appears to be involved in the viral membrane-bending process during the release step, as its recruitment to the membrane budding site, which oligomerizes upon reaching the membrane, and the formation of the curvature structure by these oligomers have significantly promoted bud formation [128]. The recruitment of M1 to the membrane budding site is believed to be mediated by GNB1 [161]. GNB1 is a G protein subunit that mediates transmembrane signaling pathways that initiate membrane ruffling-like processes that result in virus budding [161].
As reported by Li et al., there is likely no interaction between host human GNB1 and AIV M1, indicating that M1 is not being recruited by GNB1 to the budding site [161]. However, they discovered that human GNB1, specifically in conjunction with the AIV reassorted mutant (H5N6 with H9N2 virus-derived M1), stimulated virus budding as GNB1 binds to M1 to form a complex before cotransporting to the viral assembly and budding site. Hence, the reassortment of genes involving AIV M1 in association with the host GNB1 successfully initiates efficient budding and release in human cells.
5.7.2. NA and host SA
As soon as the recently assembled virion buds, its release is heavily reliant on the enzymatic activity of NA [128]. NA is a homotetramer, and its homotetramer structure is important for forming a deep pocket containing the active site at the globulin head [162]. NA promotes viral release by cleaving the glycosidic linkage that connects sialic acid (SA) to the penultimate galactose via the action of residues at the active site [128]. By eliminating the SA, NA blocks HA from binding to the surface membrane, thereby enabling effective virus release during budding.
AIV release in humans appears to be inefficient due to aggregation of the new virion at the surface membrane, which is most likely caused by the very low enzymatic activity of NA [163]. The low enzymatic activity of NA is most probably because of the lack of specificity between the shape of the active site and the substrate [164]. Like HA, AIV NA's active site is specific to α2,3 SA, and this specificity excludes human SA with α2,6-linkage [103]. To properly release AIV from the human surface membrane, the virus must modify its NA active site to become specific to the SA via α2,6-linkage, hence enhancing NA enzymatic activity. A study comparing fatal and conjunctivitis cases of H7N7 infection in humans discovered that four residue substitutions in NA, N308S, A346V, T442A, and P458S, improve NA enzymatic activity, resulting in efficient virus release and an increase in virus titers [165]. Except for residue 458, which is located at the C-terminus of NA [166], all of the substituted residues are located near the active site of NA [167,168,169] and may affect the enzymatic activity.
Another aspect that contributes to the efficient release of AIV from the human surface membrane is the balance of HA-NA activity [5]. As HA of AIV has a poor binding with α2,6 SA, substitution in the second SA binding site in NA of the new virion enables the enzymatic activity of NA to be compatible with the poor HA-SA binding, thus leading to efficient release and replication in humans [162].
6. Conclusions
Many subtypes of AIV have been confirmed to infect humans, including H3N8, H5N1, H5N6, H5N8, H6N1, H7N1, H7N2, H7N3, H7N4, H7N7, H7N9, H9N2, H10N3, H10N7 and H10N8 [170,171]. Among these were the highly virulent subtypes H5N1 and H7N9, which caused high mortality in humans. Since its first appearance, H5N1 has been linked to more than 800 cases and 400 deaths globally, whereas H7N9 has been responsible for more than 1500 human cases and 600 deaths worldwide [172]. Most AIV subtype infections were caused mainly by exposure to infected poultry, suggesting that human-to-human transmission of AIV is still ineffective. However, given what we currently know about the rapid adaptation of AIV, efficient human-to-human transmission is possible, and it is just a matter of time before it happens.
Therefore, we have highlighted the key determinants for AIV tropism in humans according to the most frequently reported virus and host factors. Based on that, we suggest the minimal requirement for efficient AIV transmission between humans that could initiate new pandemics: (i) adaptation of receptor binding preference of HA to α2,6 SA during attachment; (ii) optimum HA stability during membrane fusion; (iii) the improvement of efficiency during the import of vRNP into the nucleus (iv) the increase of polymerase activity and flexibility during transcription and replication; (v) the balance in M2/M1 expression after M segment splicing; (vi) efficient assembly of the RdRP complex; (vii) cotransport of the M1-GNB1 complex to the budding site; (viii) a functional balance between HA affinity and NA enzymatic activity with SA.
It is necessary to be prepared for the worst since we cannot predict what types of mutations may occur in the future that could potentially cause pandemics. Understanding the mechanisms that underlie adaptive changes and the circumstances in which they can be acquired will enhance our ability to assess the public health risks posed by AIV and predict the source of future pandemics. It will also reveal intricate details of virus-host interactions that can lead to the development of novel vaccines, antiviral agents and effective therapeutic strategies.
Author Contributions
Conceptualization, U.A.B. and J.E.K.; Writing—original draft preparation, U.A.B., L.A. and F.K.; writing—review and editing, P.H., S.A.K. and J.E.K.; supervision, J.E.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: pathophysiology and epidemiology. Crit Care 2019, 23, 258. [Google Scholar] [CrossRef] [PubMed]
- Kien, F.; Ma, H.L.; Bruzzone, R.; Poon, L.L.; Nal, B. Definition of the cellular interactome of the highly pathogenic avian influenza H5N1 virus: identification of human cellular regulators of viral entry, assembly, and egress. Hong Kong Med J 2016, 22, 10–12. [Google Scholar] [PubMed]
- Kido, H.; Takahashi, E.; Kimoto, T. Role of host trypsin-type serine proteases and influenza virus-cytokine-trypsin cycle in influenza viral pathogenesis. Pathogenesis-based therapeutic options. Biochimie 2019, 166, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat Rev Microbiol 2019, 17, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Cui, Q.; Rong, L. Competitive Cooperation of Hemagglutinin and Neuraminidase during Influenza A Virus Entry. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Bottcher-Friebertshauser, E.; Klenk, H.D.; Garten, W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog Dis 2013, 69, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. The 1918 Influenza Pandemic and Its Legacy. Cold Spring Harb Perspect Med 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Blagodatski, A.; Trutneva, K.; Glazova, O.; Mityaeva, O.; Shevkova, L.; Kegeles, E.; Onyanov, N.; Fede, K.; Maznina, A.; Khavina, E.; et al. Avian Influenza in Wild Birds and Poultry: Dissemination Pathways, Monitoring Methods, and Virus Ecology. Pathogens 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.-X.; Jing, Y.; Xiao-Hua, L.; Zhou, B.-P.; Yu-Xin, S.; Zhi-Yong, Z.; Yu-Shen, Z.; Ying-Ying, D.; Xian-Gui, R.; Yang, G.; et al. Human Avian Influenza A H5N1, H7N9, H10N8 and H5N6 Virus Infection. In Diagnostic Imaging of Emerging Infectious Diseases; Lu, P.-X., Zhou, B.-P., Eds.; Springer Netherlands: Dordrecht, 2016; pp. 29–56. [Google Scholar] [CrossRef]
- Goneau, L.W.; Mehta, K.; Wong, J.; L'Huillier, A.G.; Gubbay, J.B. Zoonotic Influenza and Human Health-Part 1: Virology and Epidemiology of Zoonotic Influenzas. Curr Infect Dis Rep 2018, 20, 37. [Google Scholar] [CrossRef]
- Daidoji, T.; Kajikawa, J.; Arai, Y.; Watanabe, Y.; Hirose, R.; Nakaya, T. Infection of Human Tracheal Epithelial Cells by H5 Avian Influenza Virus Is Regulated by the Acid Stability of Hemagglutinin and the pH of Target Cell Endosomes. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Ching, R.H.H.; Chan, S.K.H.; Nicholls, J.M.; Sachs, N.; Clevers, H.; Peiris, J.S.M.; Chan, M.C.W. Tropism, replication competence, and innate immune responses of influenza virus: an analysis of human airway organoids and ex-vivo bronchus cultures. Lancet Respir Med 2018, 6, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Minodier, L.; Charrel, R.N.; Ceccaldi, P.E.; van der Werf, S.; Blanchon, T.; Hanslik, T.; Falchi, A. Prevalence of gastrointestinal symptoms in patients with influenza, clinical significance, and pathophysiology of human influenza viruses in faecal samples: what do we know? Virol J 2015, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Li, C.K.; Li, Z.; Gao, R.; Liang, Q.; Zhang, Y.; Dong, L.; Zhou, J.; Dong, J.; Wang, D.; et al. Avian influenza A(H5N1) viruses can directly infect and replicate in human gut tissues. J Infect Dis 2010, 201, 1173–1177. [Google Scholar] [CrossRef]
- Cline, T.D.; Beck, D.; Bianchini, E. Influenza virus replication in macrophages: balancing protection and pathogenesis. J Gen Virol 2017, 98, 2401–2412. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Liu, Y.; Sia, S.F.; Peiris, J.S.M.; Lau, Y.L.; Tu, W. Avian influenza virus directly infects human natural killer cells and inhibits cell activity. Virol Sin 2017, 32, 122–129. [Google Scholar] [CrossRef]
- Ng, Y.P.; Yip, T.F.; Peiris, J.S.M.; Ip, N.Y.; Lee, S.M.Y. Avian influenza A H7N9 virus infects human astrocytes and neuronal cells and induces inflammatory immune responses. J Neurovirol 2018, 24, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.; Lindskog, C.; Engholm, E.; Blixt, O.; Waldenstrom, J.; Munster, V.; Lundkvist, A.; Olsen, B.; Jourdain, E.; Ellstrom, P. Characterization of avian influenza virus attachment patterns to human and pig tissues. Sci Rep 2018, 8, 12215. [Google Scholar] [CrossRef]
- Nomaguchi, M.; Fujita, M.; Miyazaki, Y.; Adachi, A. Viral tropism. Front Microbiol 2012, 3, 281. [Google Scholar] [CrossRef]
- Moreira, E.A.; Yamauchi, Y.; Matthias, P. How Influenza Virus Uses Host Cell Pathways during Uncoating. Cells 2021, 10. [Google Scholar] [CrossRef]
- Pineo, R. Four Flu Pandemics: Lessons that Need to Be Learned. Journal of Developing Societies 2021, 37, 398–448. [Google Scholar] [CrossRef]
- Bremaud, E.; Favard, C.; Muriaux, D. Deciphering the Assembly of Enveloped Viruses Using Model Lipid Membranes. Membranes (Basel) 2022, 12. [Google Scholar] [CrossRef]
- Nayak, D.P.; Hui, E.K.; Barman, S. Assembly and budding of influenza virus. Virus Res 2004, 106, 147–165. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 Suppl 4, D49–53. [Google Scholar] [CrossRef]
- Noda, T. Native morphology of influenza virions. Front Microbiol 2011, 2, 269. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.P.; Gordon, M.L. An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates. Virus Genes 2022, 58, 255–269. [Google Scholar] [CrossRef]
- Li, X.; Gu, M.; Zheng, Q.; Gao, R.; Liu, X. Packaging signal of influenza A virus. Virology Journal 2021, 18, 36. [Google Scholar] [CrossRef]
- Keller, M.W.; Rambo-Martin, B.L.; Wilson, M.M.; Ridenour, C.A.; Shepard, S.S.; Stark, T.J.; Neuhaus, E.B.; Dugan, V.G.; Wentworth, D.E.; Barnes, J.R. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Sci Rep 2018, 8, 14408. [Google Scholar] [CrossRef]
- Skelton, R.M.; Huber, V.C. Comparing Influenza Virus Biology for Understanding Influenza D Virus. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Mostafa, A.; Abdelwhab, E.M.; Mettenleiter, T.C.; Pleschka, S. Zoonotic Potential of Influenza A Viruses: A Comprehensive Overview. Viruses 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Huddleston, J.A.; Brownlee, G.G. The sequence of RNA segment 1 of influenza virus A/NT/60/68 and its comparison with the corresponding segment of strains A/PR/8/34 and A/WSN/33. Nucleic Acids Research 1983, 11, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.N.; Chen, G.W.; Chen, C.J.; Kuo, R.L.; Shih, S.R. Computational analysis and mapping of novel open reading frames in influenza A viruses. PLoS One 2014, 9, e115016. [Google Scholar] [CrossRef]
- Pasricha, G.; Mukherjee, S.; Chakrabarti, A.K. Apoptotic and Early Innate Immune Responses to PB1-F2 Protein of Influenza A Viruses Belonging to Different Subtypes in Human Lung Epithelial A549 Cells. Advances in Virology 2018, 2018, 5057184. [Google Scholar] [CrossRef] [PubMed]
- Chiem, K.; Martinez-Sobrido, L.; Nogales, A.; DeDiego, M.L. Amino Acid Residues Involved in Inhibition of Host Gene Expression by Influenza A/Brevig Mission/1/1918 PA-X. Microorganisms 2021, 9, 1109. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. Journal of Virology 2015, 89, 6442–6452. [Google Scholar] [CrossRef]
- Hao, W.; Wang, L.; Li, S. Roles of the Non-Structural Proteins of Influenza A Virus. Pathogens 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Q.; Liu, T.; Chang, G.; Sun, Z.; Gao, Z.; Wang, F.; Zhou, H.; Liu, R.; Zheng, M.; et al. Host Interaction Analysis of PA-N155 and PA-N182 in Chicken Cells Reveals an Essential Role of UBA52 for Replication of H5N1 Avian Influenza Virus. Frontiers in Microbiology 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Dawson, W.K.; Lazniewski, M.; Plewczynski, D. RNA structure interactions and ribonucleoprotein processes of the influenza A virus. Briefings in Functional Genomics 2017, 17, 402–414. [Google Scholar] [CrossRef]
- Wen, F.; Wan, X.-F. Influenza Neuraminidase: Underrated Role in Receptor Binding. Trends in Microbiology 2019, 27, 477–479. [Google Scholar] [CrossRef]
- Dubois, J.; Terrier, O.; Rosa-Calatrava, M. Influenza viruses and mRNA splicing: doing more with less. mBio 2014, 5, e00070-00014. [Google Scholar] [CrossRef]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Current Opinion in Virology 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Paragas, J.; Talon, J.; O'Neill, R.E.; Anderson, D.K.; Garcı́a-Sastre, A.; Palese, P. Influenza B and C Virus NEP (NS2) Proteins Possess Nuclear Export Activities. Journal of Virology 2001, 75, 7375–7383. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasan, C.C.; Thomas, M.; Kaushik, R.S.; Wang, D.; Li, F. Influenza A in Bovine Species: A Narrative Literature Review. Viruses 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Borkenhagen, L.K.; Salman, M.D.; Ma, M.J.; Gray, G.C. Animal influenza virus infections in humans: A commentary. Int J Infect Dis 2019, 88, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Janczewski, T.A.; Fanning, T.G. Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos Trans R Soc Lond B Biol Sci 2001, 356, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Hennig, C.; Graaf, A.; Petric, P.P.; Graf, L.; Schwemmle, M.; Beer, M.; Harder, T. Are pigs overestimated as a source of zoonotic influenza viruses? Porcine Health Manag 2022, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Rosanowski, S.M.; Carpenter, T.E.; Adamson, D.; Rogers, C.W.; Pearce, P.; Burns, M.; Cogger, N. An economic analysis of a contingency model utilising vaccination for the control of equine influenza in a non-endemic country. PLOS ONE 2019, 14, e0210885. [Google Scholar] [CrossRef]
- Sack, A.; Cullinane, A.; Daramragchaa, U.; Chuluunbaatar, M.; Gonchigoo, B.; Gray, G.C. Equine Influenza Virus—A Neglected, Reemergent Disease Threat. Emerg Infect Dis. 25(6):1185-1191 2019, 25. [Google Scholar] [CrossRef]
- Singh, R.K.; Dhama, K.; Karthik, K.; Khandia, R.; Munjal, A.; Khurana, S.K.; Chakraborty, S.; Malik, Y.S.; Virmani, N.; Singh, R.; et al. A Comprehensive Review on Equine Influenza Virus: Etiology, Epidemiology, Pathobiology, Advances in Developing Diagnostics, Vaccines, and Control Strategies. Frontiers in Microbiology 2018, 9. [Google Scholar] [CrossRef]
- Oladunni, F.S.; Oseni, S.O.; Martinez-Sobrido, L.; Chambers, T.M. Equine Influenza Virus and Vaccines. Viruses 2021, 13, 1657. [Google Scholar] [CrossRef]
- Kessler, S.; Harder, T.C.; Schwemmle, M.; Ciminski, K. Influenza A Viruses and Zoonotic Events—Are We Creating Our Own Reservoirs? Viruses 2021, 13, 2250. [Google Scholar] [CrossRef]
- Borland, S.; Gracieux, P.; Jones, M.; Mallet, F.; Yugueros-Marcos, J. Influenza A Virus Infection in Cats and Dogs: A Literature Review in the Light of the "One Health" Concept. Front Public Health 2020, 8, 83. [Google Scholar] [CrossRef]
- Frymus, T.; Belák, S.; Egberink, H.; Hofmann-Lehmann, R.; Marsilio, F.; Addie, D.D.; Boucraut-Baralon, C.; Hartmann, K.; Lloret, A.; Lutz, H.; et al. Influenza Virus Infections in Cats. Viruses 2021, 13, 1435. [Google Scholar] [CrossRef]
- Driskell, E.A.; Jones, C.A.; Berghaus, R.D.; Stallknecht, D.E.; Howerth, E.W.; Tompkins, S.M. Domestic Cats Are Susceptible to Infection With Low Pathogenic Avian Influenza Viruses From Shorebirds. Veterinary Pathology 2013, 50, 39–45. [Google Scholar] [CrossRef]
- Lee, C.T.; Slavinski, S.; Schiff, C.; Merlino, M.; Daskalakis, D.; Liu, D.; Rakeman, J.L.; Misener, M.; Thompson, C.; Leung, Y.L.; et al. Outbreak of Influenza A(H7N2) Among Cats in an Animal Shelter With Cat-to-Human Transmission—New York City, 2016. Clinical Infectious Diseases 2017, 65, 1927–1929. [Google Scholar] [CrossRef]
- Causey, D.; Edwards, S.V. Ecology of Avian Influenza Virus in Birds. The Journal of Infectious Diseases 2008, 197, S29–S33. [Google Scholar] [CrossRef]
- Hurt, A.C.; Hansbro, P.M.; Selleck, P.; Olsen, B.; Minton, C.; Hampson, A.W.; Barr, I.G. Isolation of avian influenza viruses from two different transhemispheric migratory shorebird species in Australia. Arch Virol 2006, 151, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-T.; Linster, M.; Mendenhall, I.H.; Su, Y.C.F.; Smith, G.J.D. Avian influenza viruses in humans: lessons from past outbreaks. British Medical Bulletin 2019, 132, 81–95. [Google Scholar] [CrossRef]
- Yang, H.; Carney, P.J.; Chang, J.C.; Stevens, J. Molecular characterization and three-dimensional structures of avian H8, H11, H14, H15 and swine H4 influenza virus hemagglutinins. Heliyon 2020, 6. [Google Scholar] [CrossRef]
- Abdelwhab, E.-S.M.; Veits, J.; Mettenleiter, T.C. Genetic changes that accompanied shifts of low pathogenic avian influenza viruses toward higher pathogenicity in poultry. Virulence 2013, 4, 441–452. [Google Scholar] [CrossRef]
- Wang, Z.; Loh, L.; Kedzierski, L.; Kedzierska, K. Avian Influenza Viruses, Inflammation, and CD8+ T Cell Immunity. Frontiers in Immunology 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Terregino, C.; Aznar, I.; Guajardo, I.M.; et al. Avian influenza overview March - June 2022. Efsa j 2022, 20, e07415. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Sun, H.; Gao, F.; Luo, K.; Huang, Z.; Tong, Q.; Song, H.; Han, Q.; Liu, J.; Lan, Y.; et al. Human infection of avian influenza A H3N8 virus and the viral origins: a descriptive study. The Lancet Microbe 2022, 3, e824–e834. [Google Scholar] [CrossRef]
- Lu, P.; Zhao, Q. Highly Pathogenic Avian Influenza. In Radiology of Infectious Diseases: Volume 1, Li, H., Ed.; Springer Netherlands: Dordrecht, 2015; pp. 157–189. [Google Scholar] [CrossRef]
- Gubareva, L.V.; McCullers, J.A.; Bethell, R.C.; Webster, R.G. Characterization of Influenza A/HongKong/156/97 (H5N1) Virus in a Mouse Model and Protective Effect of Zanamivir on H5N1 Infection in Mice. The Journal of Infectious Diseases 1998, 178, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Yu, W.C.; Leung, C.W.; Cheung, C.Y.; Ng, W.F.; Nicholls, J.M.; Ng, T.K.; Chan, K.H.; Lai, S.T.; Lim, W.L.; et al. Re-emergence of fatal human influenza A subtype H5N1 disease. The Lancet 2004, 363, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.-Y.; Qin, E.-D.; Wang, W.; Yu, J.; Liu, B.-H.; Hu, Y.; Hu, J.-F.; Cao, W.-C. Fatal Infection with Influenza A (H5N1) Virus in China. New England Journal of Medicine 2006, 354, 2731–2732. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO) (2022). Avian Influenza Weekly Update Number 875. Available online: https://www.who.int/docs/default-source/wpro---documents/emergency/surveillance/avian-influenza/ai_20221209.pdf?Status=Master&sfvrsn=22ea0816_21 (accessed on 21 January 2023).
- Ding, L.; Li, J.; Li, X.; Qu, B. Evolutionary and Mutational Characterization of the First H5N8 Subtype Influenza A Virus in Humans. Pathogens 2022, 11, 666. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.-H.; Yang, J.-R.; Wu, H.-S.; Chang, M.-C.; Lin, J.-S.; Lin, C.-Y.; Liu, Y.-L.; Lo, Y.-C.; Yang, C.-H.; Chuang, J.-H.; et al. Human infection with avian influenza A H6N1 virus: an epidemiological analysis. The Lancet Respiratory Medicine 2013, 1, 771–778. [Google Scholar] [CrossRef]
- Update: influenza activity--United States and worldwide, 2003-04 season, and composition of the 2004-05 influenza vaccine. MMWR Morb Mortal Wkly Rep 2004, 53, 547–552. [PubMed]
- Editorial team, C. Avian influenza A/(H7N2) outbreak in the United Kingdom. Weekly releases (1997–2007) 2007, 12, 3206. [Google Scholar] [CrossRef]
- Belser, J.A.; Pulit-Penaloza, J.A.; Sun, X.; Brock, N.; Pappas, C.; Creager, H.M.; Zeng, H.; Tumpey, T.M.; Maines, T.R. A Novel A(H7N2) Influenza Virus Isolated from a Veterinarian Caring for Cats in a New York City Animal Shelter Causes Mild Disease and Transmits Poorly in the Ferret Model. Journal of Virology 2017, 91, e00672-00617. [Google Scholar] [CrossRef]
- Tweed, S.A.; Skowronski, D.M.; David, S.T.; Larder, A.; Petric, M.; Lees, W.; Li, Y.; Katz, J.; Krajden, M.; Tellier, R.; et al. Human Illness from Avian Influenza H7N3, British Columbia. Emerging Infectious Disease journal 2004, 10, 2196. [Google Scholar] [CrossRef]
- Nguyen-Van-Tam, J.S.; Nair, P.; Acheson, P.; Baker, A.; Barker, M.; Bracebridge, S.; Croft, J.; Ellis, J.; Gelletlie, R.; Gent, N.; et al. Outbreak of low pathogenicity H7N3 avian influenza in UK, including associated case of human conjunctivitis. Weekly releases (1997–2007) 2006, 11, 2952. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Lee, R.T.C.; Gunalan, V.; Eisenhaber, F. The highly pathogenic H7N3 avian influenza strain from July 2012 in Mexico acquired an extended cleavage site through recombination with host 28S rRNA. Virology Journal 2013, 10, 139. [Google Scholar] [CrossRef]
- Tong, X.-C.; Weng, S.-S.; Xue, F.; Wu, X.; Xu, T.-M.; Zhang, W.-H. First human infection by a novel avian influenza A(H7N4) virus. Journal of Infection 2018, 77, 249–257. [Google Scholar] [CrossRef]
- Kurtz, J.; Manvell, R.J.; Banks, J. Avian influenza virus isolated from a woman with conjunctivitis. The Lancet 1996, 348, 901–902. [Google Scholar] [CrossRef]
- Fouchier, R.A.M.; Schneeberger, P.M.; Rozendaal, F.W.; Broekman, J.M.; Kemink, S.A.G.; Munster, V.; Kuiken, T.; Rimmelzwaan, G.F.; Schutten, M.; van Doornum, G.J.J.; et al. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proceedings of the National Academy of Sciences 2004, 101, 1356–1361. [Google Scholar] [CrossRef]
- Puzelli, S.; Rossini, G.; Facchini, M.; Vaccari, G.; Di Trani, L.; Di Martino, A.; Gaibani, P.; Vocale, C.; Cattoli, G.; Bennett, M.; et al. Human infection with highly pathogenic A(H7N7) avian influenza virus, Italy, 2013. Emerging Infectious Diseases 2014, 20, 1745–1749. [Google Scholar] [CrossRef]
- Gu, M.; Xu, L.; Wang, X.; Liu, X. Current situation of H9N2 subtype avian influenza in China. Veterinary Research 2017, 48, 49. [Google Scholar] [CrossRef] [PubMed]
- Shanmuganatham, K.; Feeroz, M.M.; Jones-Engel, L.; Walker, D.; Alam, S.; Hasan, M.; McKenzie, P.; Krauss, S.; Webby, R.J.; Webster, R.G. Genesis of avian influenza H9N2 in Bangladesh. Emerging Microbes & Infections 2014, 3, 1–17. [Google Scholar] [CrossRef]
- Wang, Y.; Niu, S.; Zhang, B.; Yang, C.; Zhou, Z. The whole genome analysis for the first human infection with H10N3 influenza virus in China. J Infect 2021. [Google Scholar] [CrossRef]
- Shoham, D. Review: Molecular evolution and the feasibility of an avian influenza virus becoming a pandemic strain––a conceptual shift. Virus Genes 2006, 33, 127–132. [Google Scholar] [CrossRef]
- Arzey, G.; Kirkland, P.; Arzey, K.E.; Frost, M.; Maywood, P.; Conaty, S.; Hurt, A.; Deng, Y.-M.; Iannello, P.; Barr, I.; et al. Influenza Virus A (H10N7) in Chickens and Poultry Abattoir Workers, Australia. Emerging Infectious Disease journal 2012, 18, 814. [Google Scholar] [CrossRef]
- Wohlbold, T.J.; Hirsh, A.; Krammer, F. An H10N8 influenza virus vaccine strain and mouse challenge model based on the human isolate A/Jiangxi-Donghu/346/13. Vaccine 2015, 33, 1102–1106. [Google Scholar] [CrossRef]
- Galloway, S.E.; Liang, B.; Steinhauer, D.A. Activation of the Hemagglutinin of Influenza Viruses. In Activation of Viruses by Host Proteases; Böttcher-Friebertshäuser, E., Garten, W., Klenk, H.D., Eds.; Springer International Publishing: Cham, 2018; pp. 3–26. [Google Scholar] [CrossRef]
- Shirvani, E.; Paldurai, A.; Varghese, B.P.; Samal, S.K. Contributions of HA1 and HA2 Subunits of Highly Pathogenic Avian Influenza Virus in Induction of Neutralizing Antibodies and Protection in Chickens. Front Microbiol 2020, 11, 1085. [Google Scholar] [CrossRef]
- Tzarum, N.; de Vries, R.P.; Peng, W.; Thompson, A.J.; Bouwman, K.M.; McBride, R.; Yu, W.; Zhu, X.; Verheije, M.H.; Paulson, J.C.; et al. The 150-Loop Restricts the Host Specificity of Human H10N8 Influenza Virus. Cell Rep 2017, 19, 235–245. [Google Scholar] [CrossRef]
- Xiong, X.; McCauley, J.W.; Steinhauer, D.A. Receptor binding properties of the influenza virus hemagglutinin as a determinant of host range. Curr Top Microbiol Immunol 2014, 385, 63–91. [Google Scholar] [CrossRef]
- Lazniewski, M.; Dawson, W.K.; Szczepinska, T.; Plewczynski, D. The structural variability of the influenza A hemagglutinin receptor-binding site. Brief Funct Genomics 2018, 17, 415–427. [Google Scholar] [CrossRef]
- Ciminski, K.; Chase, G.P.; Beer, M.; Schwemmle, M. Influenza A Viruses: Understanding Human Host Determinants. Trends Mol Med 2021, 27, 104–112. [Google Scholar] [CrossRef]
- Mifsud, E.J.; Kuba, M.; Barr, I.G. Innate Immune Responses to Influenza Virus Infections in the Upper Respiratory Tract. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Parvin, R.; Kabiraj, C.K.; Mumu, T.T.; Chowdhury, E.H.; Islam, M.R.; Beer, M.; Harder, T. Active virological surveillance in backyard ducks in Bangladesh: detection of avian influenza and gammacoronaviruses. Avian Pathol 2020, 49, 361–368. [Google Scholar] [CrossRef]
- Xu, Y.; Peng, R.; Zhang, W.; Qi, J.; Song, H.; Liu, S.; Wang, H.; Wang, M.; Xiao, H.; Fu, L.; et al. Avian-to-Human Receptor-Binding Adaptation of Avian H7N9 Influenza Virus Hemagglutinin. Cell Rep 2019, 29, 2217–2228 e2215. [Google Scholar] [CrossRef]
- Thompson, A.J.; Paulson, J.C. Adaptation of influenza viruses to human airway receptors. J Biol Chem 2021, 296, 100017. [Google Scholar] [CrossRef]
- Song, H.; Qi, J.; Xiao, H.; Bi, Y.; Zhang, W.; Xu, Y.; Wang, F.; Shi, Y.; Gao, G.F. Avian-to-Human Receptor-Binding Adaptation by Influenza A Virus Hemagglutinin H4. Cell Rep 2017, 20, 1201–1214. [Google Scholar] [CrossRef]
- Sriwilaijaroen, N.; Suzuki, Y. Host Receptors of Influenza Viruses and Coronaviruses-Molecular Mechanisms of Recognition. Vaccines (Basel) 2020, 8. [Google Scholar] [CrossRef]
- de Vries, R.P.; Peng, W.; Grant, O.C.; Thompson, A.J.; Zhu, X.; Bouwman, K.M.; de la Pena, A.T.T.; van Breemen, M.J.; Ambepitiya Wickramasinghe, I.N.; de Haan, C.A.M.; et al. Three mutations switch H7N9 influenza to human-type receptor specificity. PLoS Pathog 2017, 13, e1006390. [Google Scholar] [CrossRef]
- Pu, Z.; Xiang, D.; Li, X.; Luo, T.; Shen, X.; Murphy, R.W.; Liao, M.; Shen, Y. Potential Pandemic of H7N9 Avian Influenza A Virus in Human. Frontiers in Cellular and Infection Microbiology 2018, 8. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, H.; Wu, P.; Uyeki, T.M.; Feng, L.; Lai, S.; Wang, L.; Huo, X.; Xu, K.; Chen, E.; et al. Epidemiology of avian influenza A H7N9 virus in human beings across five epidemics in mainland China, 2013–17: an epidemiological study of laboratory-confirmed case series. The Lancet Infectious Diseases 2017, 17, 822–832. [Google Scholar] [CrossRef]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front Microbiol 2019, 10, 39. [Google Scholar] [CrossRef]
- Lin, Y.P.; Gregory, V.; Collins, P.; Kloess, J.; Wharton, S.; Cattle, N.; Lackenby, A.; Daniels, R.; Hay, A. Neuraminidase receptor binding variants of human influenza A(H3N2) viruses resulting from substitution of aspartic acid 151 in the catalytic site: a role in virus attachment? J Virol 2010, 84, 6769–6781. [Google Scholar] [CrossRef]
- Mohr, P.G.; Deng, Y.-M.; McKimm-Breschkin, J.L. The neuraminidases of MDCK grown human influenza A(H3N2) viruses isolated since 1994 can demonstrate receptor binding. Virology Journal 2015, 12, 67. [Google Scholar] [CrossRef]
- Hooper, K.A.; Bloom, J.D. A Mutant Influenza Virus That Uses an N1 Neuraminidase as the Receptor-Binding Protein. Journal of Virology 2013, 87, 12531–12540. [Google Scholar] [CrossRef]
- Gulati, S.; Smith, D.F.; Cummings, R.D.; Couch, R.B.; Griesemer, S.B.; St George, K.; Webster, R.G.; Air, G.M. Human H3N2 Influenza Viruses Isolated from 1968 To 2012 Show Varying Preference for Receptor Substructures with No Apparent Consequences for Disease or Spread. PLoS One 2013, 8, e66325. [Google Scholar] [CrossRef]
- Simpson, C.; Yamauchi, Y. Microtubules in Influenza Virus Entry and Egress. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Translational Neurodegeneration 2015, 4, 18. [Google Scholar] [CrossRef]
- Luo, M. Influenza virus entry. Adv Exp Med Biol 2012, 726, 201–221. [Google Scholar] [CrossRef]
- Hamilton, B.S.; Whittaker, G.R.; Daniel, S. Influenza virus-mediated membrane fusion: determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses 2012, 4, 1144–1168. [Google Scholar] [CrossRef]
- Garten, W.; Braden, C.; Arendt, A.; Peitsch, C.; Baron, J.; Lu, Y.; Pawletko, K.; Hardes, K.; Steinmetzer, T.; Bottcher-Friebertshauser, E. Influenza virus activating host proteases: Identification, localization and inhibitors as potential therapeutics. Eur J Cell Biol 2015, 94, 375–383. [Google Scholar] [CrossRef]
- Bahgat, M.M.; Blazejewska, P.; Schughart, K. Inhibition of lung serine proteases in mice: a potentially new approach to control influenza infection. Virol J 2011, 8, 27. [Google Scholar] [CrossRef]
- Barbier, D.; Garcia-Verdugo, I.; Pothlichet, J.; Khazen, R.; Descamps, D.; Rousseau, K.; Thornton, D.; Si-Tahar, M.; Touqui, L.; Chignard, M.; et al. Influenza A induces the major secreted airway mucin MUC5AC in a protease-EGFR-extracellular regulated kinase-Sp1-dependent pathway. Am J Respir Cell Mol Biol 2012, 47, 149–157. [Google Scholar] [CrossRef]
- Sakai, K.; Ami, Y.; Tahara, M.; Kubota, T.; Anraku, M.; Abe, M.; Nakajima, N.; Sekizuka, T.; Shirato, K.; Suzaki, Y.; et al. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J Virol 2014, 88, 5608–5616. [Google Scholar] [CrossRef]
- Chaipan, C.; Kobasa, D.; Bertram, S.; Glowacka, I.; Steffen, I.; Tsegaye, T.S.; Takeda, M.; Bugge, T.H.; Kim, S.; Park, Y.; et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J Virol 2009, 83, 3200–3211. [Google Scholar] [CrossRef]
- Okumura, Y.; Takahashi, E.; Yano, M.; Ohuchi, M.; Daidoji, T.; Nakaya, T.; Bottcher, E.; Garten, W.; Klenk, H.D.; Kido, H. Novel type II transmembrane serine proteases, MSPL and TMPRSS13, Proteolytically activate membrane fusion activity of the hemagglutinin of highly pathogenic avian influenza viruses and induce their multicycle replication. J Virol 2010, 84, 5089–5096. [Google Scholar] [CrossRef]
- Kido, H.; Okumura, Y.; Takahashi, E.; Pan, H.Y.; Wang, S.; Yao, D.; Yao, M.; Chida, J.; Yano, M. Role of host cellular proteases in the pathogenesis of influenza and influenza-induced multiple organ failure. Biochim Biophys Acta 2012, 1824, 186–194. [Google Scholar] [CrossRef]
- Tashiro, M.; Ciborowski, P.; Klenk, H.D.; Pulverer, G.; Rott, R. Role of Staphylococcus protease in the development of influenza pneumonia. Nature 1987, 325, 536–537. [Google Scholar] [CrossRef]
- Sun, X.; Belser, J.A.; Yang, H.; Pulit-Penaloza, J.A.; Pappas, C.; Brock, N.; Zeng, H.; Creager, H.M.; Stevens, J.; Maines, T.R. Identification of key hemagglutinin residues responsible for cleavage, acid stability, and virulence of fifth-wave highly pathogenic avian influenza A(H7N9) viruses. Virology 2019, 535, 232–240. [Google Scholar] [CrossRef]
- Scheibner, D.; Ulrich, R.; Fatola, O.I.; Graaf, A.; Gischke, M.; Salaheldin, A.H.; Harder, T.C.; Veits, J.; Mettenleiter, T.C.; Abdelwhab, E.M. Variable impact of the hemagglutinin polybasic cleavage site on virulence and pathogenesis of avian influenza H7N7 virus in chickens, turkeys and ducks. Sci Rep 2019, 9, 11556. [Google Scholar] [CrossRef]
- Gischke, M.; Ulrich, R.; O, I.F.; Scheibner, D.; Salaheldin, A.H.; Crossley, B.; Bottcher-Friebertshauser, E.; Veits, J.; Mettenleiter, T.C.; Abdelwhab, E.M. Insertion of Basic Amino Acids in the Hemagglutinin Cleavage Site of H4N2 Avian Influenza Virus (AIV)-Reduced Virus Fitness in Chickens is Restored by Reassortment with Highly Pathogenic H5N1 AIV. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Yin, Y.; Zhang, X.; Qiao, Y.; Wang, X.; Su, Y.; Chen, S.; Qin, T.; Peng, D.; Liu, X. Glycosylation at 11Asn on hemagglutinin of H5N1 influenza virus contributes to its biological characteristics. Vet Res 2017, 48, 81. [Google Scholar] [CrossRef]
- Russell, C.J.; Hu, M.; Okda, F.A. Influenza Hemagglutinin Protein Stability, Activation, and Pandemic Risk. Trends Microbiol 2018, 26, 841–853. [Google Scholar] [CrossRef]
- England, R.J.A.; Homer, J.J.; Knight, L.C.; Ell, S.R. Nasal pH measurement: a reliable and repeatable parameter. Clinical Otolaryngology & Allied Sciences 1999, 24, 67–68. [Google Scholar] [CrossRef]
- Baumann, J.; Kouassi, N.M.; Foni, E.; Klenk, H.D.; Matrosovich, M. H1N1 Swine Influenza Viruses Differ from Avian Precursors by a Higher pH Optimum of Membrane Fusion. J Virol 2016, 90, 1569–1577. [Google Scholar] [CrossRef]
- Mair, C.M.; Ludwig, K.; Herrmann, A.; Sieben, C. Receptor binding and pH stability - how influenza A virus hemagglutinin affects host-specific virus infection. Biochim Biophys Acta 2014, 1838, 1153–1168. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Ostbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front Immunol 2018, 9, 1581. [Google Scholar] [CrossRef]
- Naito, T.; Momose, F.; Kawaguchi, A.; Nagata, K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol 2007, 81, 1339–1349. [Google Scholar] [CrossRef]
- Batra, J.; Tripathi, S.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sambhara, S.; Lal, S.K. Human Heat shock protein 40 (Hsp40/DnaJB1) promotes influenza A virus replication by assisting nuclear import of viral ribonucleoproteins. Sci Rep 2016, 6, 19063. [Google Scholar] [CrossRef]
- Luo, W.; Zhang, J.; Liang, L.; Wang, G.; Li, Q.; Zhu, P.; Zhou, Y.; Li, J.; Zhao, Y.; Sun, N.; et al. Phospholipid scramblase 1 interacts with influenza A virus NP, impairing its nuclear import and thereby suppressing virus replication. PLOS Pathogens 2018, 14, e1006851. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, C.; Ren, C.; Zhang, S.; Gao, X.; Jin, M.; Chen, H.; Ma, W.; Zhou, H. Eukaryotic Translation Elongation Factor 1 Delta Inhibits the Nuclear Import of the Nucleoprotein and PA-PB1 Heterodimer of Influenza A Virus. Journal of Virology 2020, 95, e01391-01320. [Google Scholar] [CrossRef]
- Ling, Y.H.; Wang, H.; Han, M.Q.; Wang, D.; Hu, Y.X.; Zhou, K.; Li, Y. Nucleoporin 85 interacts with influenza A virus PB1 and PB2 to promote its replication by facilitating nuclear import of ribonucleoprotein. Front Microbiol 2022, 13, 895779. [Google Scholar] [CrossRef]
- Gabriel, G.; Klingel, K.; Otte, A.; Thiele, S.; Hudjetz, B.; Arman-Kalcek, G.; Sauter, M.; Shmidt, T.; Rother, F.; Baumgarte, S.; et al. Differential use of importin-α isoforms governs cell tropism and host adaptation of influenza virus. Nat Commun 2011, 2, 156. [Google Scholar] [CrossRef]
- Staller, E.; Barclay, W.S. Host Cell Factors That Interact with Influenza Virus Ribonucleoproteins. Cold Spring Harb Perspect Med 2021, 11. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Yamada, K.; Yoneda, Y. Importin α: a key molecule in nuclear transport and non-transport functions. J Biochem 2016, 160, 69–75. [Google Scholar] [CrossRef]
- Ninpan, K.; Suptawiwat, O.; Boonarkart, C.; Phuangphung, P.; Sathirareuangchai, S.; Uiprasertkul, M.; Auewarakul, P. Expression of importin-α isoforms in human nasal mucosa: implication for adaptation of avian influenza A viruses to human host. Virol J 2016, 13, 90. [Google Scholar] [CrossRef]
- Rodriguez-Frandsen, A.; Martin-Sancho, L.; Gounder, A.P.; Chang, M.W.; Liu, W.-C.; Jesus, P.D.D.; Recum-Knepper, J.v.; Dutra, M.S.; Huffmaster, N.J.; Chavarria, M.; et al. Viral Determinants in H5N1 Influenza A Virus Enable Productive Infection of HeLa Cells. Journal of Virology 2020, 94, e01410-01419. [Google Scholar] [CrossRef]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.-D.; Stech, J. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proceedings of the National Academy of Sciences 2005, 102, 18590–18595. [Google Scholar] [CrossRef]
- Wandzik, J.M.; Kouba, T.; Cusack, S. Structure and Function of Influenza Polymerase. Cold Spring Harb Perspect Med 2021, 11. [Google Scholar] [CrossRef]
- Fan, H.; Walker, A.P.; Carrique, L.; Keown, J.R.; Serna Martin, I.; Karia, D.; Sharps, J.; Hengrung, N.; Pardon, E.; Steyaert, J.; et al. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 2019, 573, 287–290. [Google Scholar] [CrossRef]
- Fodor, E.; Te Velthuis, A.J.W. Structure and Function of the Influenza Virus Transcription and Replication Machinery. Cold Spring Harb Perspect Med 2020, 10. [Google Scholar] [CrossRef]
- Soh, Y.S.; Moncla, L.H.; Eguia, R.; Bedford, T.; Bloom, J.D. Comprehensive mapping of adaptation of the avian influenza polymerase protein PB2 to humans. Elife 2019, 8. [Google Scholar] [CrossRef]
- Long, J.S.; Giotis, E.S.; Moncorge, O.; Frise, R.; Mistry, B.; James, J.; Morisson, M.; Iqbal, M.; Vignal, A.; Skinner, M.A.; et al. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 2016, 529, 101–104. [Google Scholar] [CrossRef]
- Camacho-Zarco, A.R.; Kalayil, S.; Maurin, D.; Salvi, N.; Delaforge, E.; Milles, S.; Jensen, M.R.; Hart, D.J.; Cusack, S.; Blackledge, M. Molecular basis of host-adaptation interactions between influenza virus polymerase PB2 subunit and ANP32A. Nat Commun 2020, 11, 3656. [Google Scholar] [CrossRef]
- Baker, S.F.; Ledwith, M.P.; Mehle, A. Differential Splicing of ANP32A in Birds Alters Its Ability to Stimulate RNA Synthesis by Restricted Influenza Polymerase. Cell Rep 2018, 24, 2581–2588 e2584. [Google Scholar] [CrossRef]
- Kirui, J.; Bucci, M.D.; Poole, D.S.; Mehle, A. Conserved features of the PB2 627 domain impact influenza virus polymerase function and replication. J Virol 2014, 88, 5977–5986. [Google Scholar] [CrossRef]
- Arai, Y.; Kawashita, N.; Elgendy, E.M.; Ibrahim, M.S.; Daidoji, T.; Ono, T.; Takagi, T.; Nakaya, T.; Matsumoto, K.; Watanabe, Y. PA Mutations Inherited during Viral Evolution Act Cooperatively To Increase Replication of Contemporary H5N1 Influenza Virus with an Expanded Host Range. J Virol 2020, 95. [Google Scholar] [CrossRef]
- Lukarska, M.; Fournier, G.; Pflug, A.; Resa-Infante, P.; Reich, S.; Naffakh, N.; Cusack, S. Structural basis of an essential interaction between influenza polymerase and Pol II CTD. Nature 2017, 541, 117–121. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamada, S.; Fukuyama, S.; Murakami, S.; Zhao, D.; Uraki, R.; Watanabe, T.; Tomita, Y.; Macken, C.; Neumann, G.; et al. Virulence-affecting amino acid changes in the PA protein of H7N9 influenza A viruses. J Virol 2014, 88, 3127–3134. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, R.; Yu, L.; Sun, H.; Tian, S.; Li, P.; Jin, M.; Chen, H.; Ma, W.; Zhou, H. Human TRA2A determines influenza A virus host adaptation by regulating viral mRNA splicing. Sci Adv 2020, 6, eaaz5764. [Google Scholar] [CrossRef]
- Esparza, M.; Bhat, P.; Fontoura, B.M. Viral-host interactions during splicing and nuclear export of influenza virus mRNAs. Curr Opin Virol 2022, 55, 101254. [Google Scholar] [CrossRef]
- Darwish, H.; Cho, J.M.; Loignon, M.; Alaoui-Jamali, M.A. Overexpression of SERTAD3, a putative oncogene located within the 19q13 amplicon, induces E2F activity and promotes tumor growth. Oncogene 2007, 26, 4319–4328. [Google Scholar] [CrossRef]
- Ramirez-Medina, E.; Vuono, E.A.; Velazquez-Salinas, L.; Silva, E.; Rai, A.; Pruitt, S.; Berggren, K.A.; Zhu, J.; Borca, M.V.; Gladue, D.P. The MGF360-16R ORF of African Swine Fever Virus Strain Georgia Encodes for a Nonessential Gene That Interacts with Host Proteins SERTAD3 and SDCBP. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Lin, B.; Dutta, B.; Fraser, I.D.C. Systematic Investigation of Multi-TLR Sensing Identifies Regulators of Sustained Gene Activation in Macrophages. Cell Syst 2017, 5, 25–37.e23. [Google Scholar] [CrossRef]
- Menachery, V.D.; Mitchell, H.D.; Cockrell, A.S.; Gralinski, L.E.; Yount, B.L.; Graham, R.L.; McAnarney, E.T.; Douglas, M.G.; Scobey, T.; Beall, A.; et al. MERS-CoV Accessory ORFs Play Key Role for Infection and Pathogenesis. mBio 2017, 8, e00665-00617. [Google Scholar] [CrossRef]
- Sun, N.; Li, C.; Li, X.F.; Deng, Y.Q.; Jiang, T.; Zhang, N.N.; Zu, S.; Zhang, R.R.; Li, L.; Chen, X.; et al. Type-IInterferon-Inducible SERTAD3 Inhibits Influenza A Virus Replication by Blocking the Assembly of Viral RNA Polymerase Complex. Cell Rep 2020, 33, 108342. [Google Scholar] [CrossRef]
- Chlanda, P.; Schraidt, O.; Kummer, S.; Riches, J.; Oberwinkler, H.; Prinz, S.; Kräusslich, H.G.; Briggs, J.A. Structural Analysis of the Roles of Influenza A Virus Membrane-Associated Proteins in Assembly and Morphology. J Virol 2015, 89, 8957–8966. [Google Scholar] [CrossRef]
- Lai, J.C.; Chan, W.W.; Kien, F.; Nicholls, J.M.; Peiris, J.S.; Garcia, J.M. Formation of virus-like particles from human cell lines exclusively expressing influenza neuraminidase. J Gen Virol 2010, 91, 2322–2330. [Google Scholar] [CrossRef]
- Yondola, M.A.; Fernandes, F.; Belicha-Villanueva, A.; Uccelini, M.; Gao, Q.; Carter, C.; Palese, P. Budding capability of the influenza virus neuraminidase can be modulated by tetherin. J Virol 2011, 85, 2480–2491. [Google Scholar] [CrossRef]
- Li, F.; Liu, J.; Yang, J.; Sun, H.; Jiang, Z.; Wang, C.; Zhang, X.; Yu, Y.; Zhao, C.; Pu, J.; et al. H9N2 virus-derived M1 protein promotes H5N6 virus release in mammalian cells: Mechanism of avian influenza virus inter-species infection in humans. PLoS Pathog 2021, 17, e1010098. [Google Scholar] [CrossRef]
- Dai, M.; McBride, R.; Dortmans, J.; Peng, W.; Bakkers, M.J.G.; de Groot, R.J.; van Kuppeveld, F.J.M.; Paulson, J.C.; de Vries, E.; de Haan, C.A.M. Mutation of the Second Sialic Acid-Binding Site, Resulting in Reduced Neuraminidase Activity, Preceded the Emergence of H7N9 Influenza A Virus. J Virol 2017, 91. [Google Scholar] [CrossRef]
- Sorrell, E.M.; Schrauwen, E.J.; Linster, M.; De Graaf, M.; Herfst, S.; Fouchier, R.A. Predicting 'airborne' influenza viruses: (trans-) mission impossible? Curr Opin Virol 2011, 1, 635–642. [Google Scholar] [CrossRef]
- Zanin, M.; Duan, S.; Wong, S.S.; Kumar, G.; Baviskar, P.; Collin, E.; Russell, C.; Barman, S.; Hause, B.; Webby, R. An Amino Acid in the Stalk Domain of N1 Neuraminidase Is Critical for Enzymatic Activity. J Virol 2017, 91. [Google Scholar] [CrossRef]
- de Wit, E.; Munster, V.J.; van Riel, D.; Beyer, W.E.; Rimmelzwaan, G.F.; Kuiken, T.; Osterhaus, A.D.; Fouchier, R.A. Molecular determinants of adaptation of highly pathogenic avian influenza H7N7 viruses to efficient replication in the human host. J Virol 2010, 84, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yang, H.; Guo, Z.; Yu, W.; Carney, P.J.; Li, Y.; Chen, L.M.; Paulson, J.C.; Donis, R.O.; Tong, S.; et al. Crystal structures of two subtype N10 neuraminidase-like proteins from bat influenza A viruses reveal a diverged putative active site. Proc Natl Acad Sci U S A 2012, 109, 18903–18908. [Google Scholar] [CrossRef]
- Bentley, D.R.; Brownlee, G.G. Sequence of the N2 neuraminidase from influenza virus A/NT/60/68. Nucleic Acids Res 1982, 10, 5033–5042. [Google Scholar] [CrossRef] [PubMed]
- Shtyrya, Y.A.; Mochalova, L.V.; Bovin, N.V. Influenza virus neuraminidase: structure and function. Acta Naturae 2009, 1, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Tewawong, N.; Vichiwattana, P.; Korkong, S.; Klinfueng, S.; Suntronwong, N.; Thongmee, T.; Theamboonlers, A.; Vongpunsawad, S.; Poovorawan, Y. Evolution of the neuraminidase gene of seasonal influenza A and B viruses in Thailand between 2010 and 2015. PLoS One 2017, 12, e0175655. [Google Scholar] [CrossRef]
- De Marco, M.A.; Delogu, M.; Facchini, M.; Di Trani, L.; Boni, A.; Cotti, C.; Graziosi, G.; Venturini, D.; Regazzi, D.; Ravaioli, V.; et al. Serologic Evidence of Occupational Exposure to Avian Influenza Viruses at the Wildfowl/Poultry/Human Interface. Microorganisms 2021, 9. [Google Scholar] [CrossRef]
- Wen, F.; Zhang, X.; Guo, J.; Liang, Z.; Cheng, Q.; Wang, C.; Yu, H.; Du, Y.; Huang, S.; Li, J.; et al. Emergence of H3N8 avian influenza viruses possessing tri-basic hemagglutinin cleavage sites in China. J Infect 2022, 85, e112–e114. [Google Scholar] [CrossRef]
- Philippon, D.A.M.; Wu, P.; Cowling, B.J.; Lau, E.H.Y. Avian Influenza Human Infections at the Human-Animal Interface. J Infect Dis 2020, 222, 528–537. [Google Scholar] [CrossRef]
Figure 1.
Influenza A virus virion structure and genome organization. (A) Schematic organization of a gene segment (vRNA). The IAV genes consist of 3 parts: 1. Untranslated regions (UTRs) comprise of the universal primers uni-12 (12nt) and the uni-13 (13nt) at the end of 3’ and 5’ of each segment, which is highly conserved among the eight gene segments and among all IAV viral strains, and the segment-specific noncoding regions (ssNCRs); 2. Packaging signals; and 3. Coding region. (B) IAV Genome organization: The eight negative-sense, single-stranded RNA segments of IAV genome code for PB2, PB1, PA, HA, NP, NA, M, and NS, respectively (Indicated in white boxes); The darker blue colour boxes at the end of each vRNA segment indicate the 3′ and 5′ untranslated regions (UTRs); The lighter blue colour boxes indicate the packaging signals. The numbers inside the blue boxes represent the nucleotide length for the UTRs and the packaging signals. (C) IAV virion structure: IAV comprises of 3 components: an envelope made of a lipid bilayer containing three transmembrane proteins HA, NA and M2 ion channel; a layer of the inner surface envelope M1; and a core made of eight vRNA segments, packaged together to form vRNP. (Created with BioRender.com.)
Figure 1.
Influenza A virus virion structure and genome organization. (A) Schematic organization of a gene segment (vRNA). The IAV genes consist of 3 parts: 1. Untranslated regions (UTRs) comprise of the universal primers uni-12 (12nt) and the uni-13 (13nt) at the end of 3’ and 5’ of each segment, which is highly conserved among the eight gene segments and among all IAV viral strains, and the segment-specific noncoding regions (ssNCRs); 2. Packaging signals; and 3. Coding region. (B) IAV Genome organization: The eight negative-sense, single-stranded RNA segments of IAV genome code for PB2, PB1, PA, HA, NP, NA, M, and NS, respectively (Indicated in white boxes); The darker blue colour boxes at the end of each vRNA segment indicate the 3′ and 5′ untranslated regions (UTRs); The lighter blue colour boxes indicate the packaging signals. The numbers inside the blue boxes represent the nucleotide length for the UTRs and the packaging signals. (C) IAV virion structure: IAV comprises of 3 components: an envelope made of a lipid bilayer containing three transmembrane proteins HA, NA and M2 ion channel; a layer of the inner surface envelope M1; and a core made of eight vRNA segments, packaged together to form vRNP. (Created with BioRender.com.)
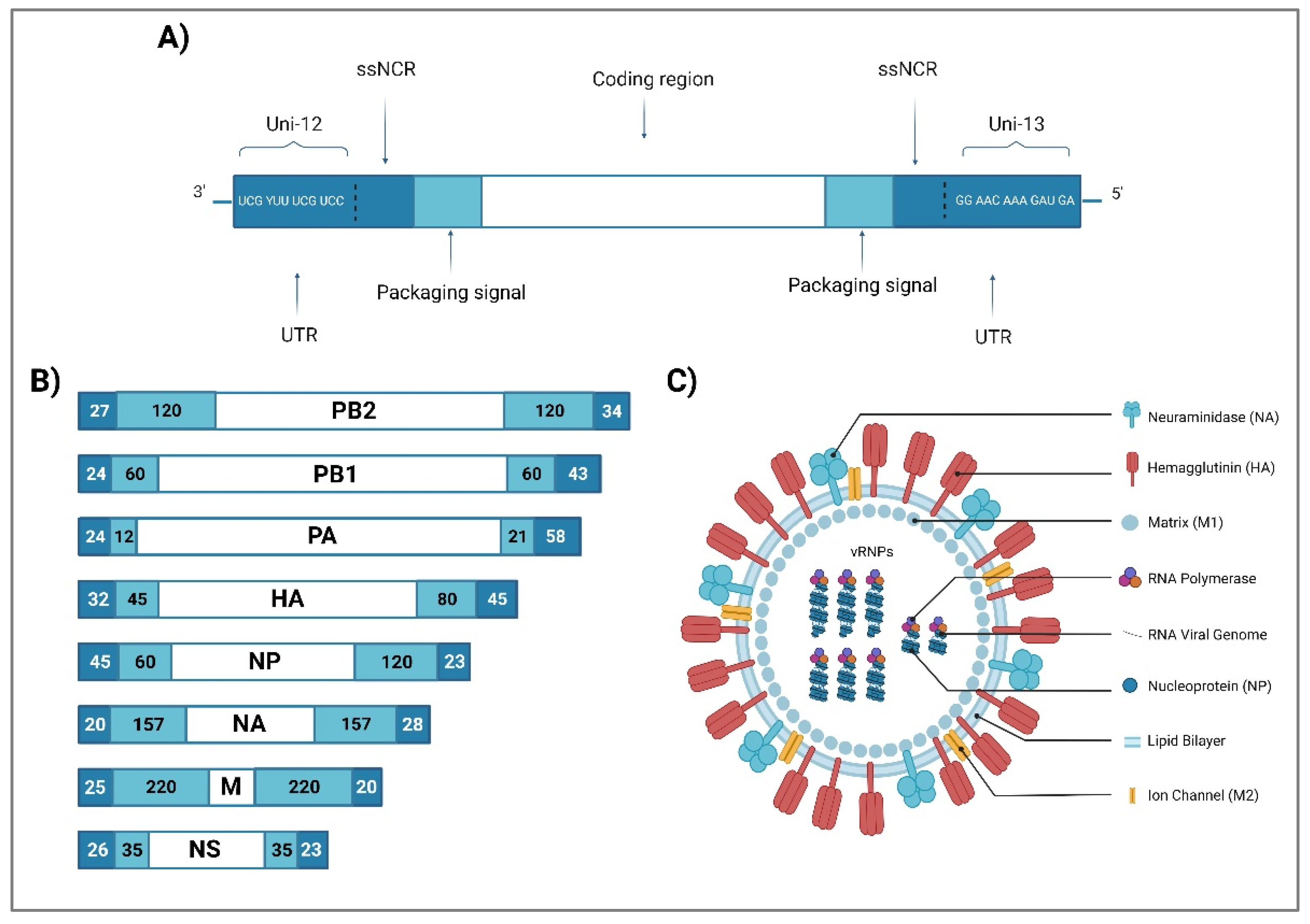
Figure 2.
Schematic diagram showing the host range of influenza A viruses. Reservoirs and interspecies transmission events of influenza A viruses and the subtypes involved in these events. Aquatic wild birds represent the natural reservoir of influenza A viruses, from which they can be transmitted to a variety of other hosts. Circled arrows represent continuous virus circulation among wild birds, domestic birds, domestic animals, bats, horses, pigs, and humans. (Created with BioRender.com).
Figure 2.
Schematic diagram showing the host range of influenza A viruses. Reservoirs and interspecies transmission events of influenza A viruses and the subtypes involved in these events. Aquatic wild birds represent the natural reservoir of influenza A viruses, from which they can be transmitted to a variety of other hosts. Circled arrows represent continuous virus circulation among wild birds, domestic birds, domestic animals, bats, horses, pigs, and humans. (Created with BioRender.com).
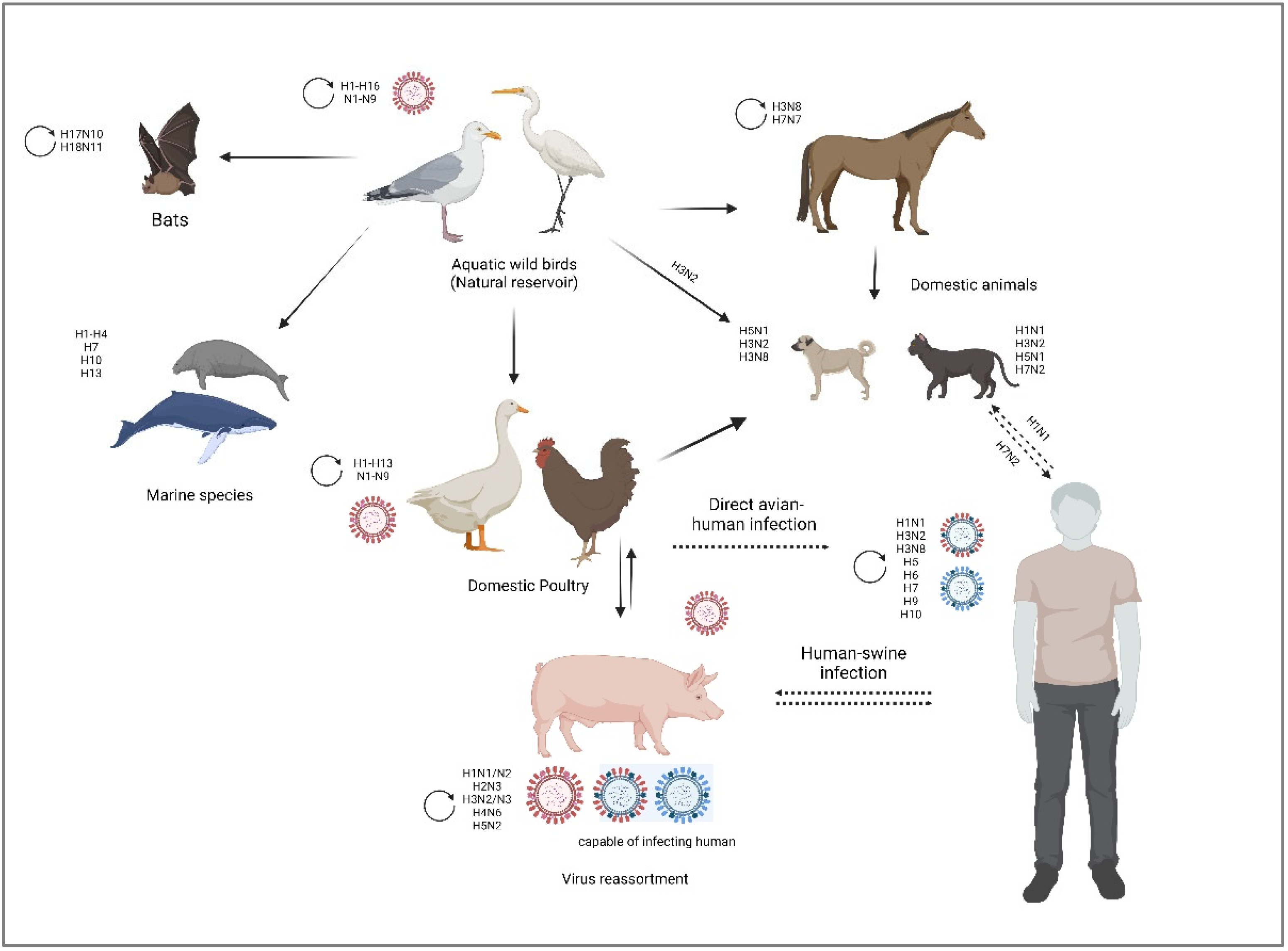
Figure 3.
IAV life cycle and the key determinants of AIV tropism in the human host. (1) Upon viral entry in the respiratory tract, AIV attaches to the host sialic acid (SA) receptor by interacting with HA-NA, thus inducing cell internalization via endocytosis. (2) The acidification of the late endosome (host pH) triggers a conformational change in cleaved HA (where HA is cleaved by host protease), which later causes membrane fusion and vRNP release into the cytoplasm. (3) Following that, vRNPs are transported into the nucleus via the host importin α/β-mediated nuclear import pathway. (4) In the nucleus, replication and transcription of viral RNA are proceeded by viral polymerases with the help of host RNA polymerase II and ANP32A. (5) The transcribed viral mRNA going a maturation process where the mRNA is spliced before the translation process. (6) Upon viral protein translation, NP binds to nascent viral RNA to prevent RNA degradation. PA, PB1 and PB2 are assembled as RdRp complex before binding with RNA-NP, thus generating nascent vRNP. Then, nascent vRNPs and all nascent viral proteins are trafficked to the budding membrane. (7) The bud formation is initiated by the interaction of M1 with host machinery before (8) the nascent virus is released by the interaction of HA-NA with the host SA receptor. (Created with BioRender.com.).
Figure 3.
IAV life cycle and the key determinants of AIV tropism in the human host. (1) Upon viral entry in the respiratory tract, AIV attaches to the host sialic acid (SA) receptor by interacting with HA-NA, thus inducing cell internalization via endocytosis. (2) The acidification of the late endosome (host pH) triggers a conformational change in cleaved HA (where HA is cleaved by host protease), which later causes membrane fusion and vRNP release into the cytoplasm. (3) Following that, vRNPs are transported into the nucleus via the host importin α/β-mediated nuclear import pathway. (4) In the nucleus, replication and transcription of viral RNA are proceeded by viral polymerases with the help of host RNA polymerase II and ANP32A. (5) The transcribed viral mRNA going a maturation process where the mRNA is spliced before the translation process. (6) Upon viral protein translation, NP binds to nascent viral RNA to prevent RNA degradation. PA, PB1 and PB2 are assembled as RdRp complex before binding with RNA-NP, thus generating nascent vRNP. Then, nascent vRNPs and all nascent viral proteins are trafficked to the budding membrane. (7) The bud formation is initiated by the interaction of M1 with host machinery before (8) the nascent virus is released by the interaction of HA-NA with the host SA receptor. (Created with BioRender.com.).
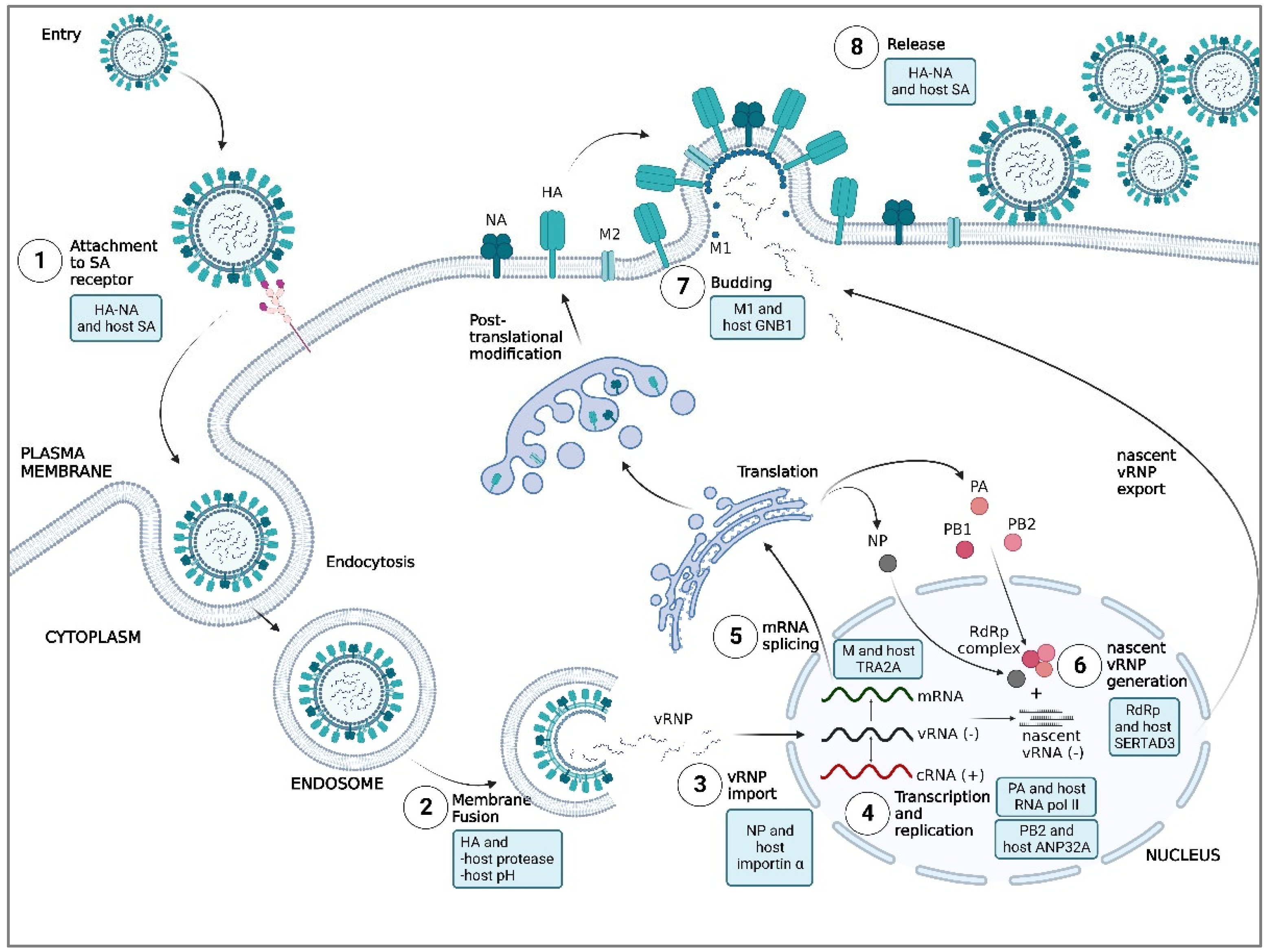

Table 1.
Influenza A virus gene segments and proteins size and functions.
| Segment | Segment size (nucleotides) | Proteins | Protein sequence (amino acids) | Viral function | Reference |
|---|---|---|---|---|---|
| 1 | 2341 | PB2 | 759 | RDRP 1 subunit: transcription initiation and cap-snatching mechanism | [30,31] |
| 2 | 2341 | PB1 | 757 | RDRP 1 subunit: replication and transcription of viral RNA segments; endonuclease activity | [24,32] |
| PB1-F2 2 | 87-90 | Proapoptotic activity contributes to the pathogenicity of IAVs | [30,33] | ||
| PB1-N40 2 | 718 | Regulation of PB1 expression and activity | [27,32] | ||
| 3 | 2233 | PA | 716 | RDRP 1 subunit: endonuclease, cleaves the capped RNA | [30] |
| PA-X 2 | 252 | Role in Influenza virus-induced host shutoff as it selectively degrades host RNAs and limits innate immune responses | [34,35] | ||
| PA-N155 2 | 562 | Role in viral replication | [36,37] | ||
| PA-N182 2 | 535 | Role in viral replication | [36,37] | ||
| 4 | 1778 | HA | 566 | Binding to cell receptor; Fusion of endosomal and viral membranes | [38] |
| 5 | 1565 | NP | 498 | Transcription/replication regulation, vRNP nuclear export | [24,38] |
| 6 | 1413 | NA | 454 | Sialidase activity (propagation of neovirions) | [38,39] |
| 7 | 1027 | M1 | 252 | Support of structure and internal viral architecture; Regulation of RNA nuclear export activity | [24,30,36] |
| M2 | 97 | Ion channel activity, as well as a role in virus uncoating and assembly | [24,30,36] | ||
| M42 2 | 99 | A functional alternative to M2 | [36,40] | ||
| 8 | 890 | NS1 | 215-237 | Inhibition of the antiviral response | [38,41] |
| NS3 2 | 194 | Unknown function | [30] | ||
| NS2/NEP | 121 | Nuclear export of new vRNP | [42] |
Table 2.
Summary of all human infections caused by the avian influenza viruses.
| AIV subtype | Year/Location | Cases/ fatalities | First isolated human strain | Reference |
|---|---|---|---|---|
| H3N8 | China/2022 | 2/0 | A/Henan/4-10/2022 A/Changsha/1000/2022 |
[64] |
| H5N1 | Hong Kong/1997 | 18/6 | A/Hong Kong/156/97 | [66] |
| Hong Kong/2003 | 2/1 | [67] | ||
| China/2003 | 1/1 | [68] | ||
| 21 countries/2003-Nov 2022 | 868/456 | [69] | ||
| H5N6 | Western Pacific Region /2014-Nov 2022 | 82/33 | A/Sichuan/26221/2014 | [69] |
| A(H5) * | Vietnam/Oct 2022 | 1/1 | [69] | |
| H5N8 | Russia/2020 | 7/0 | A/Astrakhan/3212/2020 | [70] |
| H6N1 | Taiwan/2013 | 1/0 | A/Taiwan/2/2013 | [71] |
| H7N2 | USA/2002 | 1/0 | A/New York/107/2003 | [72] |
| USA/2003 | 1/0 | [72] | ||
| UK/2007 | 4/0 | [73] | ||
| USA/2016 | 1/0 | [74] | ||
| H7N3 | Canada/2004 | 2/0 1/0 2/0 |
A/Canada/444/04 | [75] |
| UK/2006 | [76] | |||
| Mexico/2012 | [77] | |||
| H7N4 | China/2018 | 1/0 | A/Jiangsu/1/2018 | [78] |
| H7N7 | UK/1996 | 1/0 | A/England/268/96 | [79] |
| Netherlands/2003 | 89/1 | [80] | ||
| Italy/2013 | 3/0 | [81] | ||
| H7N9 | Western Pacific Region/2013-Nov 2022 | 1568/616 | A/Anhui/1/2013 | [69] |
| H9N2 | China/1998 | 5/0 | A/HK/1073/99 | [82] |
| Hong Kong/1999-2009 | 6/0 | [62] | ||
| Bangladesh/2011 | 1/0 | [83] | ||
| China/Dec 2015-Nov 2022 | 76/0 | [69] | ||
| Cambodia/Dec 2015-Nov 2022 | 2/0 | [69] | ||
| H10N3 | China/2021 | 2/0 | A/Jiangsu/428/2021 | [84] |
| H10N7 | Egypt/2004 | 2/0 2/0 |
A/Egypt/2004 | [85] [86] |
| Australia/2010 | ||||
| H10N8 | China/2013 | 3/2 | A/Jiangxi-Donghu/346/13 | [87] |
* The NA subtype could not be determined.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



