
Submitted:
31 January 2023
Posted:
07 February 2023
You are already at the latest version
Abstract
Human T-lymphotropic virus type 1 (HTLV-1) is associated with neurological, degenerative, and inflammatory diseases. Brazil has regions with high prevalence rates. The prevalence among relatives of positive people is high, and intrafamily transmission causes the conservation of the infection for different generations. The investigation of epidemiological and molecular factors may clarify the factors associated with the efficacy of intrafamilial transmission of HTLV-1. Objective: To analyze the epidemiological and molecular factors associated with intrafamilial transmission of HTLV-1 in an endemic area of Brazil. Material and Methods: A cross-sectional study investigated 72 relatives from 14 families with more than one infected. Epidemiological data were collected from all family members, and genetic sequencing was performed, phylogenetic analysis of 22 positive relatives. Results: The prevalence of HTLV-1 in families with infected people was 56.94% (41/72). Twelve vertical transmission routes and ten horizontal transmission routes were identified. Phylogenetic analysis demonstrated the absence of polymorphisms between infected mother and child sequences, while sequences of couples presented nucleotide divergence of up to 3%.among themselves. It was also identified in a family with an intrafamilial transmission with the occurrence of two viral strains. Conclusion: Molecular data confirmed intrafamilial transmission. They also indicated little genetic variability between the sequenced samples of family members and those deposited in GenBank used in this study. The absence of polymorphisms was observed in cases of vertical transmission, indicating a tendency to perpetuate identical sequences regardless of the time of acquisition of the infection.
Keywords:
HTLV
; Amazon
; Phylogeny
1. Introduction
Human T-lymphotropic virus type 1 (HTLV-1) is associated with lymphoproliferative, inflammatory, and degenerative diseases of the central nervous system [1]. However, it is estimated that approximately 10% of those infected develop any symptoms associated with infection. Thus, most remain asymptomatic, making them potential virus transmitters [2].
The occurrence of infection is endemic in specific regions of the world, and Brazil represents one of the highest rates of endemicity of infection in the world [3]. In the northern region of Brazil, the state of Pará is endemic. There have been investigations of riparians [4], blood donors [5], relatives of carriers [6], and passers-by from the metropolitan region of Belém [7] and different prevalences were found.
Transmission of the virus can occur through breastfeeding by an infected mother [8], transplacentally [9], sexual contact without condoms being more effective from man to woman [10], and contact with blood and blood products [11].
International and national studies have proven the high infection prevalence among families with virus carriers, especially in endemic areas [12,13]. Intrafamily transmission has a rate 18 times higher than transmission in the general population [5]. In areas of high endemicity in Japan, positive cases were identified in 38.5% of the relatives of the infected investigated [14] and 47.7% of the relatives of pregnant women with HTLV-1 [15].
Epidemiological characteristics such as age, gender, time of breastfeeding, and marital relationship, among others, are already established as risk factors associated with intrafamilial transmission of HTLV-1 [6,16,17]. However, epidemiological analyses associated with viral molecular characteristics may clarify the intrafamilial transmission process.
In an investigation based on the sequencing and analysis of the 5′LTR (Long terminal repeats) region of the viral genome of 28 index cases and 92 family members in Argentina, the prevalence rate found among family members was 31.52% (29/92). The analysis of LTR sequences and the presence of polymorphisms revealed a high prevalence of maternal and child transmission in these families [16]. In Brazil, a molecular analysis demonstrated that HTLV-1 isolates in a city in the northern region are highly preserved [18].
Information on the viral molecular characteristics involved in the intrafamily transmission process is scarce in Brazil and worldwide. Most studies on the evaluation of intrafamilial transmission are based only on epidemiological data, and viral molecular studies aim to know the rates of mutations and viral characterization. A study that simultaneously analyzes epidemiological data with viral molecular data is necessary to evaluate the efficacy of intrafamilial transmission of HTLV-1.
2. Results
We investigated 14 families with at least two family members positive for HTLV-1, totaling 72 family members studied. Of the total investigated, 56.94% (41/72) are infected with HTLV-1, and 43.06% (31/72) are not. Among the 31 uninfected family members, 58.06% (18/31) are women, and 41.94% (13/31) are men, aged between three and 86 years, mean of 29 years. Regarding breastfeeding, 38.71% (12/31) reported having received it during childhood, and condom use was affirmed by only 3.23% (1/31) of those investigated, 16.13% (5/31) reported being in a stable relationship, and none received a blood transfusion.
Among the 41 infected relatives, 60.98% (25/41) are women, and 39.02% (16/41) are men, ages ranging from three to 84 years, with a mean of 42 years. Regarding breastfeeding, 73.17% (30/41) people reported having been breastfed in childhood, condom use was reported by 21.95% (9/41) people, 70.73% (29/41) are in a stable relationship, and 14.63% (6/41) people have a history of receiving a blood transfusion. Epidemiological data d and all 72 family members investigated are summarized in Table 1.
The families were listed according to the order of entry of biological samples in the outpatient clinic, not following a chronological order. Based on the epidemiological data, family trees were built to understand the transmission routes better. A letter was assigned to each family member according to the number of relatives of the same family.
Only didactic criteria are represented in the family trees, some family members who, for some reason, could not be investigated. People who have sequenced samples are flagged with the corresponding lab number, as shown in Figure 1.
* Represents the relatives with sequenced samples followed by the corresponding number in the laboratory’s database. N.I.: A not investigated person.
Based on epidemiological data, vertical transmission was observed in 50% (7/14) of the families: FAM 5, FAM 11, FAM 13, FAM 119, FAM 151, FAM 156, and FAM 168. Sexual transmission occurred in 71.43% (10/14), except for families FAM5, FAM119, FAM151, and FAM168. In 21.49% (3/14) of the families, it is possible to observe the simultaneous occurrence of the two transmission routes: FAM 11, FAM 113 and FAM 156.
There was vertical transmission in 12 moments, with children aged 11 to 57 years, with a mean of 34 years. Mother-daughter transmission was transmitted in eight moments (5C->5E; 11E->11Q;13A->13F; 119A->119B; 151A->151B; 156A->156B; 168A->168B; 168A->168C) and mother to male child in four moments (5C->5F; 11A -> 11F; 11E->11N; 13A->13I). No significant difference was found (p=0.7165) between the efficacy of transmission with the sex of the breastfed child.
We observed 16 cases in which there was breastfeeding from an infected mother but without vertical transmission (5C->5G; 5C->5H; 11A->11G;11E->11M; 11E->11O; 13C->13D; 13C->13E; 13C->13E; 13A->13G; 13A->13H; 13A->13J; 13A->13K; 30B->30D; 30B->30E; 48C->48G; 48C->48H; 48C->48I), 50% (8/16) male and 50% (8/16) female. The age of the children ranged from three to 37 years, with a mean age of 19 years.
Regarding sexual transmission, it is suggested that there were ten 183occurrences (11F->11E; 13B->13A; 30C->30B; 43D->43C; 48D->48C; 110C->110B; 156B->156C; 158B->158A; 162B->162A; 167B->167A). It is inferable that sexual transmissions all occurred from the sense of a man to a woman due to the characteristic of infection with greater transmission efficacy in this sense [10]. Among the ten couples with both infected, the time of stable relationship ranged from three months to 40 years, with an average of 22 years; in addition to the report of non-use of condoms by 7/10 (70%) of these couples.
Five couples without sexual transmission were also observed, one of the partner’s positive for HTLV-1 and the other negative (5B->5C; 5D->5E; 11A->11B; 11C->11D; 119B->119C). The time of a stable relationship ranged from three to five years, with an average of four years. Of these couples, 80% (4/5) are positive women who did not transmit sexually to the spouse, and 20% (1/5) is a husband positive for HTLV-1 and a negative wife (11C->11D). In the latter case, the hypothesis of 11C was also formulated to be positive for HTLV-1. For unknown reasons, detecting the viral genome during the tests was impossible. Based on that marriage of 11C-11D, two of the couple’s three children, who were breastfed and are positive for the virus, are suggestive of vertical transmission of both from a common infected mother.
Analyzing cases with and without sexual transmission, no significant difference was found (p=0.7571) between stable relationship time and sexual transmission efficacy between spouses. Thus, there was no significant difference between couples without transmission (p=0.7389) between the sex of the spouse with the infection and the non-efficacy of transmission. No significant differences were found between the transmission route (vertical or sexual) with transmission effectiveness (p=0.6994).
Because of the unfeasibility of several samples for genetic sequencing, the analysis of viral genetic sequencing of the samples of 22 family members from 14 families was performed. Even the families that only sequenced a family sample, were included in the phylogenetic analysis aiming at a general evaluation of the sequences obtained.
All 22 sequenced samples from family members presented HTLV-1 subtype a A genotype (Transcontinental Cosmopolitan). A phylogenetic tree was constructed to analyze whether the samples from people from the same geographic region present greater similarity to each other compared to the 11 sequences deposited in the GenBank of other localities (Figure 2).
Another phylogenetic tree was created from the similarity of the 22 family samples, and only one sample was deposited in GenBank as an external group, demonstrating the similarity between the study families (Figure 3). The sequences that presented the greatest similarity were arranged in the same group (I, II, III, IV, V, VI, VII, or VIII).
Finally, a phylogenetic tree was constructed with samples of families with more than one person with a sequenced sample and without an external group to evaluate the similarity between family members (Figure 4).
An array of polymorphisms was created based on the degree of divergence in percentage between the analyzed sequences (S1). In general, nucleotide divergence rates were observed between the sequences of the 22 family members and the 11 deposited in GenBank used for 205 the construction of the phylogenetic tree in Figure 2.
Among the 11 sequences deposited in GenBank, even though they came from different regions of Brazil, the degree of nucleotide divergence ranged from 0 to 1% between them.
Comparing family sequences and those deposited at GenBank, rates ranged from 0 to 3%. In agreement with the data present in the literature on the low variability in HTLV-1 subgroups, even from different regions. Analyzing the family groups, a variation in the degree of polymorphisms between 1 and 3% could also be visualized, even though all families originated from the same region.
Among the sequences of the communicators of the same families, the rates ranged from 0 to 3%, being possible to observe the absence of divergence between mothers and children, resulting in identical sequences in cases of vertical transmission. In cases of horizontal transmission, rates of 1% nucleotide divergence were observed between couples.
3. Discussion
There are several epidemiological studies of the prevalence of HTLV in specific populations such as pregnant women, blood donors, injecting drug users, and sex workers, among others; where, in general, a prevalence between 0.1 and 22% is observed in these groups [19,20,21,22,23].
Among the areas already established as endemic to the occurrence of infection, the metropolitan region of Belém, located in the state of Pará, northern Brazil, has already been the target of several studies on the prevalence rates of infection in different populations. And in pregnant women who attended a Specialized Reference Unit for Women’s Health Care in Belém, a prevalence of 2.8% (13/479) [7] was found in passers-by in the city’s public places, a prevalence of 2% (21/1,059)[7], in different locations of cities near Belém was 0.35% (1/289) [24], among others.
The persistence of HTLV-1 among individuals from the same family makes intrafamilial transmission 18 times more effective within families than in the general population. Breastfeeding and prolonged sexual contact are the main factors associated with the maintenance of the virus within these family groups [6].
Studies suggest that breastfeeding from a carrier mother increases the chance of acquiring the infection in the infant by 14 times [25]. Some of the factors associated with the effectiveness of this transmission route are the period of breastfeeding greater than three months and maternal age greater than 35 years [25,26].
Although no significant differences were found regarding behaviors considered at risk for infection acquisition (stable relationship, non-use of condoms, and breastfeeding) among the infected and non-infected populations, it is possible to observe greater adherence to the factors considered at risk among the infected population. As it was possible to observe that most of the infected were in a stable relationship and were breastfed during childhood. As for the transmission routes within the families, there was no significant difference between the vertical and horizontal transmission routes and the effectiveness of the infection.
Regarding the viral genetic sequences obtained from the 22 relatives, today presented the HTLV-1 subtype A genotype (Cosmopolitan Transcontinental), according to other molecular studies conducted in the metropolitan region of the city of Belém [18], Pará [27] and other regions of Brazil [28,29].
Studies have already demonstrated the genetic stability of HTLV-1 according to viral evolution rate estimation calculations. In previous comparative investigations between family sequences, totally or virtually identical sequences have been demonstrated between several members of the same family over several generations [30]. In addition to the sequencing analysis of the LTR region, considered the most variable region of the genome, of relatives where only one base substitution [31] was found, suggesting the stable profile of the viral genome. In addition, studies suggest that, even in viruses from different geographical regions, the divergence between sequences is also considered low, below 10% [32].
Because it was not possible to obtain a higher number of infected people in the same family, in which some cases, it was only possible to obtain a sequence from each family, either due to the impossibility of contact, the non-attendance of other family members contacted, and factors associated with the COVID-19 pandemic; it was not possible to calculate the mutation rate between the sequenced samples. However, it is possible to observe, based on the matrix of polymorphisms, 100% similarity between some sequences of relatives or low divergence between them, showing the low genetic variability of the virus among people of the same family.
Regarding the analysis of polymorphisms, a significant difference was observed between the vertical and sexual transmission routes. Among the sequences obtained from cases of vertical transmission, there were no changes in the sequences between mother and child(s), with remaining identical sequences. In cases of sexual transmission, more polymorphisms were observed between the sequences obtained. These findings may suggest the reason for the greater efficacy of vertical transmission within families and the perpetuation of the virus within them. This can give viral genome stability in cases of breastfeeding acquisition, even over time.
It also showed the tendency of polymorphisms in cases of sexual transmission, which could be seen in cases of marital relationships with little time and already manifested polymorphisms among themselves, as was the case of Family 43.
In the phylogenetic tree constructed with 22 sequences of family members and 11 deposited in GenBank obtained from studies in other regions of Brazil (Figure 2), the 22 samples were grouped into two large groups (I and II). Showing that all family sequences were closely related, indicating that, even with a low degree of polymorphism, it is the same virus. The exception occurred with the three family members of Family 11 (FAM11), where two sequences (1385 -11L and 1615-11K) were listed in Group II and 1367 (11F) in Group I. The sequences of 11L and 11K showed 100% similarity to each other, confirming that there was a vertical transmission from the common infected mother, while the sequence of 1367 (11F) presented a certain degree of polymorphism compared to the other two, claiming to be a virus lineage with origin and evolution different from the other two. Confirming the simultaneous occurrence of two viral types in the same family.
In the phylogenetic tree constructed from the 22 sequences of communicators and an external group (Figure 3), it is noted that the samples were grouped into eight groups (I, II, III, IV, V, VI, VII, VIII, IX and X). The bootstrap values that supported the layout presented in the tree ranged from 80 to 100, depending on the sibling group. All family members belonging to the same family were disposed of in the same sibling group, except for Family 11, as previously mentioned, and in cases where only a sample of a family member was sequenced, the samples were separated from other groups with greater similarity to each other. The exception occurred in Group IV, in which the sequences of two relatives from different families grouped with the bootstrap value of 85 (939-FAM48 and 1367-FAM11). In the polymorphism matrix, it is possible to observe low nucleotide divergence between these sequences.
In the phylogenetic tree constructed only with samples of families with more than one sequenced communicant (Figure 4), sample 1367 (FAM11) behaved as an external group, showing that it presents a high degree of genetic and evolutionary divergence from the other samples, including those of people of the same family. Just as all the sibling groups formed are composed of people from the same family and all with the maximum bootstrap value, reinforcing viral genetic stability in people of the same family and supporting epidemiological data suggestive of intrafamilial transmission.
In this study, it was possible to analyze epidemiological and molecular data. In cases where genetic sequencing was possible, the inferences of occurrence of intrafamilial transmission and route of transmission were confirmed based on the epidemiological data described from each family. The data that supported this was the absence or low rate of divergence between sequences of communicators of the same family and the grouped disposition of these relatives in the phylogenetic trees built, stating that it is the same viral lineage within these families.
4. Materials and Methods
4.1. Study population
The study population consisted of HTLV-1 patients and their families treated from October 2015 to December 2019 as a reference unit for the diagnosis and multi-professional follow-up of people living with the virus in the city of Belém, Pará state, Northern Brazil.
In total, 72 people from 14 families were investigated. This study was approved by the Research Ethics Committee of the Center for Tropical Medicine of the Federal University of Pará (number 1,218,417). Family analyses with unfeasible samples for laboratory tests and genetic sequencing were excluded from family analyses.
4.2. DNA purification
From a blood sample, DNA was extracted from peripheral blood mononuclear cells (PBMC) using the commercial kit Wizard Genomic DNA Purification (Promega, USA) according to the manufacturer’s instructions.
Then the samples had the DNA integrity evaluated using electrophoresis. After this step, the samples of acceptable quality were stored at -20oC.
4.3. PCR
The Polymerase Chain Reaction (PCR) of 258 base pairs (bp) of the human β-globin gene, was performed according to previously published methodology [33].
Nested-PCR amplification of the pX gene was performed to detect the viral genome because it was considered a highly conserved region, amplifying in the two stages of PCR fragments of 259 bp and 159 bp, respectively, as previously described [34]. The samples positive in Nested-PCR were submitted to RFLP (Restrict Fragment Length Polymorphism) reaction to identify the viral type present in the sample (HTLV-1/2) [35].
In contrast to the high conservation of the pX region of the viral genome, the LTR region has a relatively high rate of mutations and is, therefore, commonly used in genetic sequencing for the evaluation of mutations with a subsequent formulation of phylogeny hypotheses. After confirmation of HTLV-1 infection, nested-PCR amplification of region 5′LTR was performed, generating fragments of 844bp and 744bp in the two PCR stages, respectively [36].
4.3. Sanger sequencing
The nested-PCR amplified fragments of the LTR region were purified using the QIAquick PCR purification (Qiagen, GER) kit according to the manufacturer’s guidelines. The fragments were sequenced directly on both tapes using the ABI PRISM Big Dye Terminator Sequencing kit (Applied Biosystems, EUA). Then the samples were sequenced using the ABI 5500 platform (thermofisher, USA)
4.3. Phylogeny
The nucleotide sequences were analyzed and edited through the GENEIOUS 4.8.5 program and aligned with each other and with sequences available in the Genbank database by the MAFFT v.7 programs [37].
The phylogenetic trees, as well as the distance matrix, were constructed through the IQTREE [38], and for bootstrap analysis, 1000 replicates were considered to generate greater reliability to the values of the groups, repeated the process 10 times. The editing of the trees was performed with FigTree v 1.1.4.
Calculating the genetic diversity rate between the sequences found and the sequences available in GenBank from BEAST v. 1.8 [39], using a total of 25 samples containing all HTLV-1 subtypes, generating the comparative trees. This analysis was based on the foundation of Bayes theorem, which establishes the probability relationship between different events. For statistical analyses, fisher’s chi-square and exact tests were used, adopting a confidence interval of p-value>0.05.
5. Conclusions
It was possible to conclude that the molecular data supported the suggestive occurrence of intrafamilial transmission based on epidemiological data. In addition, they indicate little genetic variability between the sequenced samples of family members and those deposited in GenBank used in this study, confirming the genetic stability of HTLV-1, especially in the transcontinental Cosmopolitan subtype and among communicators of the same family.
The absence of polymorphisms was observed in cases of vertical transmission, indicating a tendency to perpetuate identical sequences regardless of the time of infection acquisition. This may explain breastfeeding is the most effective way to maintain the virus within families. Genetic sequencing also confirmed the presence of two HTLV-1 strains in the same family with the occurrence of intrafamilial transmission.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Distance matrix based on the degree of nucleotide divergence in percentage between the 22 samples from family members and 11 deposited in GenBank.
Author Contributions
Conceptualization M.S.S., C.C.C.P., S.M.M.C. and A.F.S.N.; Data curation S.M.M.C. and A.F.S.N.; Methodology, C.C.C.P., S.M.M.C., M.N.L.R., L.S.C.C. and C.A.C.; Writing—review & editing C.C.C.P., S.M.M.C., M.S.S., C.C.C.P. and A.F.S.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Research Ethics Committee of the Center for Tropical Medicine of the Federal University of Pará (number 1,218,417).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
None.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tsukasaki, K.; Tobinai, K. Biology and treatment of HTLV-1 associated T-cell lymphomas. Best Pract. Res. Clin. Haematol. 2013, 26, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Futsch, N.; Mahieux, R.; Dutartre, H. HTLV-1, the Other Pathogenic Yet Neglected Human Retrovirus: From Transmission to Therapeutic Treatment. Viruses 2017, 10. [Google Scholar] [CrossRef]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed]
- de Morais, M.P.E.; Gato, C.M.; Maciel, L.A.; Lalwani, P.; Costa, C.A.; Lalwani, J.D.B. Prevalence of Human T-lymphotropic virus type 1 and 2 among blood donors in Manaus, Amazonas State, Brazil. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e80. [Google Scholar] [CrossRef]
- dos Santos, E.L.; Tamegão-Lopes, B.; Machado, L.F.A.; Ishak, M.d.O.G.; Ishak, R.; de Lemos, J.A.R.; Vallinoto, A.C.R. Molecular characterization of HTLV-1/2 among blood donors in Belém, State of Pará: first description of HTLV-2b subtype in the Amazon region. Rev. Soc. Bras. Med. Trop. 2009, 42, 271–276. [Google Scholar] [CrossRef]
- da Costa, C.A.; Furtado, K.C.Y.O.; Ferreira, L. de S.C.; Almeida, D. de S.; Linhares, A. da C.; Ishak, R.; Vallinoto, A.C.R.; de Lemos, J.A.R.; Martins, L.C.; Ishikawa, E.A.Y.; de Sousa, R.C.M.; de Sousa, M.S. Familial transmission of human T-cell lymphotrophic virus: silent dissemination of an emerging but neglected infection. PLoS Negl. Trop. Dis. 2013, 7, e2272. [CrossRef]
- Silva, I.C.; Pinheiro, B.T.; Nobre, A.F.S.; Coelho, J.L.; Pereira, C.C.C.; Ferreira, L. de S.C.; Almeida, C.P.S. de; Viana, M. de N. do S. de A.; Almeida, D.S. de; Falcão, J.R.; Santos, Y.C.V.D.; Araújo, M.W.L. de; Borges, M. da S.; Nascimento, L.D.; Valentim, L.S.; Casseb, J.S. do R.; Costa, C.A. da; Sousa, M.S. de Moderate endemicity of the human T-lymphotropic virus infection in the metropolitan region of Belém, Pará, Brazil. Rev. Bras. Epidemiol. 2018, 21, e180018. [CrossRef]
- Carneiro-Proietti, A.B.F.; Amaranto-Damasio, M.S.; Leal-Horiguchi, C.F.; Bastos, R.H.C.; Seabra-Freitas, G.; Borowiak, D.R.; Ribeiro, M.A.; Proietti, F.A.; Ferreira, A.S.D.; Martins, M.L. Mother-to-Child Transmission of Human T-Cell Lymphotropic Viruses-1/2: What We Know, and What Are the Gaps in Understanding and Preventing This Route of Infection. J. Pediatric Infect. Dis. Soc. 2014, 3 (Suppl. 1), S24–9. [Google Scholar] [CrossRef] [PubMed]
- Tezuka, K.; Fuchi, N.; Okuma, K.; Tsukiyama, T.; Miura, S.; Hasegawa, Y.; Nagata, A.; Komatsu, N.; Hasegawa, H.; Sasaki, D.; Sasaki, E.; Mizukami, T.; Kuramitsu, M.; Matsuoka, S.; Yanagihara, K.; Miura, K.; Hamaguchi, I. HTLV-1 targets human placental trophoblasts in seropositive pregnant women. J. Clin. Invest. 2020, 130, 6171–6186. [Google Scholar] [CrossRef]
- Kaplan, J.E.; Khabbaz, R.F.; Murphy, E.L.; Hermansen, S.; Roberts, C.; Lal, R.; Heneine, W.; Wright, D.; Matijas, L.; Thomson, R.; Rudolph, D.; Switzer, W.M.; Kleinman, S.; Busch, M.; Schreiber, G.B. Male-to-female transmission of human T-cell lymphotropic virus types I and II: association with viral load. The Retrovirus Epidemiology Donor Study Group. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 12, 193–201. [Google Scholar] [CrossRef]
- Satake, M.; Iwanaga, M.; Sagara, Y.; Watanabe, T.; Okuma, K.; Hamaguchi, I. Incidence of human T-lymphotropic virus 1 infection in adolescent and adult blood donors in Japan: a nationwide retrospective cohort analysis. Lancet Infect. Dis. 2016, 16, 1246–1254. [Google Scholar] [CrossRef]
- Mendes, M.S.T.; Costa, M.C.; Costa, I.M.C. Human T-cell lymphotropic virus-1 infection: three infected generations in the same family. Rev. Soc. Bras. Med. Trop. 2016, 49, 660–662. [Google Scholar] [CrossRef]
- Rosadas, C.; Taylor, G.P. Mother-to-Child HTLV-1 Transmission: Unmet Research Needs. Front. Microbiol. 2019, 10, 999. [Google Scholar] [CrossRef]
- Kajiyama, W.; Kashiwagi, S.; Ikematsu, H.; Hayashi, J.; Nomura, H.; Okochi, K. Intrafamilial transmission of adult T cell leukemia virus. J. Infect. Dis. 1986, 154, 851–857. [Google Scholar] [CrossRef]
- Take, H.; Umemoto, M.; Kusuhara, K.; Kuraya, K. Transmission routes of HTLV-I: an analysis of 66 families. Jpn J Cancer Res 1993, 84, 1265–1267. [Google Scholar] [CrossRef]
- Frutos, M.C.; Gastaldello, R.; Balangero, M.; Remondegui, C.; Blanco, S.; Otsuki, K.; Paulo Vicente, A.C.; Elías, D.; Mangeaud, A.; Nates, S.; Gallego, S. Silent dissemination of HTLV-1 in an endemic area of Argentina. Epidemiological and molecular evidence of intrafamilial transmission. PLoS ONE 2017, 12, e0174920. [Google Scholar] [CrossRef]
- Bandeira, L.M.; Uehara, S.N.O.; Puga, M.A.M.; Rezende, G.R.; Vicente, A.C.P.; Domingos, J.A.; do Lago, B.V.; Niel, C.; Motta-Castro, A.R.C. HTLV-1 intrafamilial transmission among Japanese immigrants in Brazil. J. Med. Virol. 2018, 90, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Nobre, A.F.S.; Almeida, D. de S.; Ferreira, L.C.; Ferreira, D.L.; Júnior, E.C.S.; Viana, M. de N. do S. de A.; Silva, I.C.; Pinheiro, B.T.; Ferrari, S.F.; Linhares, A. da C.; Ishikawa, E.A.; Sousa, R.C.M.; de Sousa, M.S. Low genetic diversity of the Human T-cell Lymphotropic Virus (HTLV-1) in an endemic area of the Brazilian Amazon basin. PLoS ONE 2018, 13, e0194184. [CrossRef] [PubMed]
- Opaleye, O.O.; Igboama, M.C.; Ojo, J.A.; Odewale, G. Seroprevalence of HIV, HBV, HCV, and HTLV among Pregnant Women in Southwestern Nigeria. J. Immunoassay Immunochem. 2016, 37, 29–42. [Google Scholar] [CrossRef]
- Pinto, M.T.; Slavov, S.N.; Valente, V.B.; Ubiali, E.M.A.; Covas, D.T.; Kashima, S. Evaluation of human T-lymphotropic virus prevalence/co-infection rates for a four-year period in a non-metropolitan blood center in Southeast Brazil. Rev. Soc. Bras. Med. Trop. 2016, 49, 232–236. [Google Scholar] [CrossRef]
- Forbi, J.C.; Odetunde, A.B. Human T-cell lymphotropic virus in a population of pregnant women and commercial sex workers in South Western Nigeria. Afr. Health Sci. 2007, 7, 129–132. [Google Scholar] [PubMed]
- Stufano, A.; Jahantigh, H.R.; Cagnazzo, F.; Centrone, F.; Loconsole, D.; Chironna, M.; Lovreglio, P. Work-Related Human T-lymphotropic Virus 1 and 2 (HTLV-1/2) Infection: A Systematic Review. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Berini, C.A.; Pando, M.A.; Bautista, C.T.; Eirin, M.E.; Martinez-Peralta, L.; Weissenbacher, M.; Avila, M.M.; Biglione, M.M. HTLV-1/2 among high-risk groups in Argentina: molecular diagnosis and prevalence of different sexual transmitted infections. J. Med. Virol. 2007, 79, 1914–1920. [Google Scholar] [CrossRef]
- de Lima, A.C.R.; Lopes, F.T.; de Oliveira Freitas, V.; Assad, M.N.; de Sousa, R.S.; Gonçalves, J.S.S.; Gomes, J.L.C.; Dos Santos, B.C.; Lima, C.N.C.; Abreu, I.N.; Dos Santos Brito, W.R.; Pereira, K.A.S.; da Silva Torres, M.K.; Lima, S.S.; Aben-Athar, C.Y.U.; Guerreiro, J.F.; Cayres Vallinoto, I.M.V.; Vallinoto, A.C.R.; Feitosa, R.N.M. Prevalence and Risk Factors for HTLV-1/2 Infection inRiverside and Rural Populations of the State of Pará. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Boostani, R.; Sadeghi, R.; Sabouri, A.; Ghabeli-Juibary, A. Human T-lymphotropic virus type I and breastfeeding; systematic review and meta-analysis of the literature. Iran. J. Neurol. 2018, 17, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.M.; Nduati, R.W.; Richardson, B.A.; Steele, M.S.; John-Stewart, G.C.; Mbori-Ngacha, D.A.; Kreiss, J.K.; Overbaugh, J. Longitudinal analysis of human immunodeficiency virus type 1 RNA in breast milk and of its relationship to infant infection and maternal disease. J. Infect. Dis. 2003, 187, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.A.; Lopes, I.G.L.; Maia, E.L.; Azevedo, V.N.; Machado, L.F.A.; Ishak, M.O.G.; Ishak, R.; Vallinoto, A.C.R. Molecular characterization of HTLV-1 among patients with tropical spastic paraparesis/HTLV-1 associated myelopathy in Belém, Pará. Rev. Soc. Bras. Med. Trop. 2006, 39, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Ishak, R.; de Oliveira Guimarães Ishak, M.; Vallinoto, A.C.R. The challenge of describing the epidemiology of HTLV in the Amazon region of Brazil. Retrovirology 2020, 17, 4. [Google Scholar] [CrossRef]
- Vieira, B.A.; Bidinotto, A.B.; Dartora, W.J.; Pedrotti, L.G.; de Oliveira, V.M.; Wendland, E.M. Prevalence of human T-lymphotropic virus type 1 and 2 (HTLV-1/-2) infection in pregnant women in Brazil: a systematic review and meta-analysis. Sci. Rep. 2021, 11, 15367. [Google Scholar] [CrossRef] [PubMed]
- Nerurkar, V.R.; Babu, P.G.; Song, K.J.; Melland, R.R.; Gnanamuthu, C.; Saraswathi, N.K.; Chandy, M.; Godec, M.S.; John, T.J.; Yanagihara, R. Sequence analysis of human T cell lymphotropic virus type I strains from southern India: gene amplification and direct sequencing from whole blood blotted onto filter paper. J. Gen. Virol. 1993, 74 Pt 12, 2799–2805. [Google Scholar] [CrossRef]
- Liu, H.F.; Vandamme, A.M.; Kazadi, K.; Carton, H.; Desmyter, J.; Goubau, P. Familial transmission and minimal sequence variability of human T-lymphotropic virus type I (HTLV-I) in Zaire. AIDS Res. Hum. Retroviruses 1994, 10, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Gallo, R.C.; Franchini, G. Low degree of human T-cell leukemia/lymphoma virus type I genetic drift in vivo as a means of monitoring viral transmission and movement of ancient human populations. J. Virol. 1992, 66, 2288–2295. [Google Scholar] [CrossRef] [PubMed]
- Greer, C.E.; Peterson, S.L.; Kiviat, N.B.; Manos, M.M. PCR amplification from paraffin-embedded tissues. Effects of fixative and fixation time. Am. J. Clin. Pathol. 1991, 95, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Tuke, P.W.; Luton, P.; Garson, J.A. Differential diagnosis of HTLV-I and HTLV-II infections by restriction enzyme analysis of “nested” PCR products. J. Virol. Methods 1992, 40, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Komurian-Pradel, F.; Pelloquin, F.; Sonoda, S.; Osame, M.; de The, G. Geographical subtypes demonstrated by RFLP following PCR in the LTR region of HTLV-I. AIDS Res. Hum. Retroviruses 1992, 8, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Seiki, M.; Hattori, S.; Hirayama, Y.; Yoshida, M. Human adult T-cell leukemia virus: complete nucleotide sequence of the provirus genome integrated in leukemia cell DNA. Proc Natl Acad Sci USA 1983, 80, 3618–3622. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
Figure 1.
Family tree of the transmission routes of the 14 families investigated.
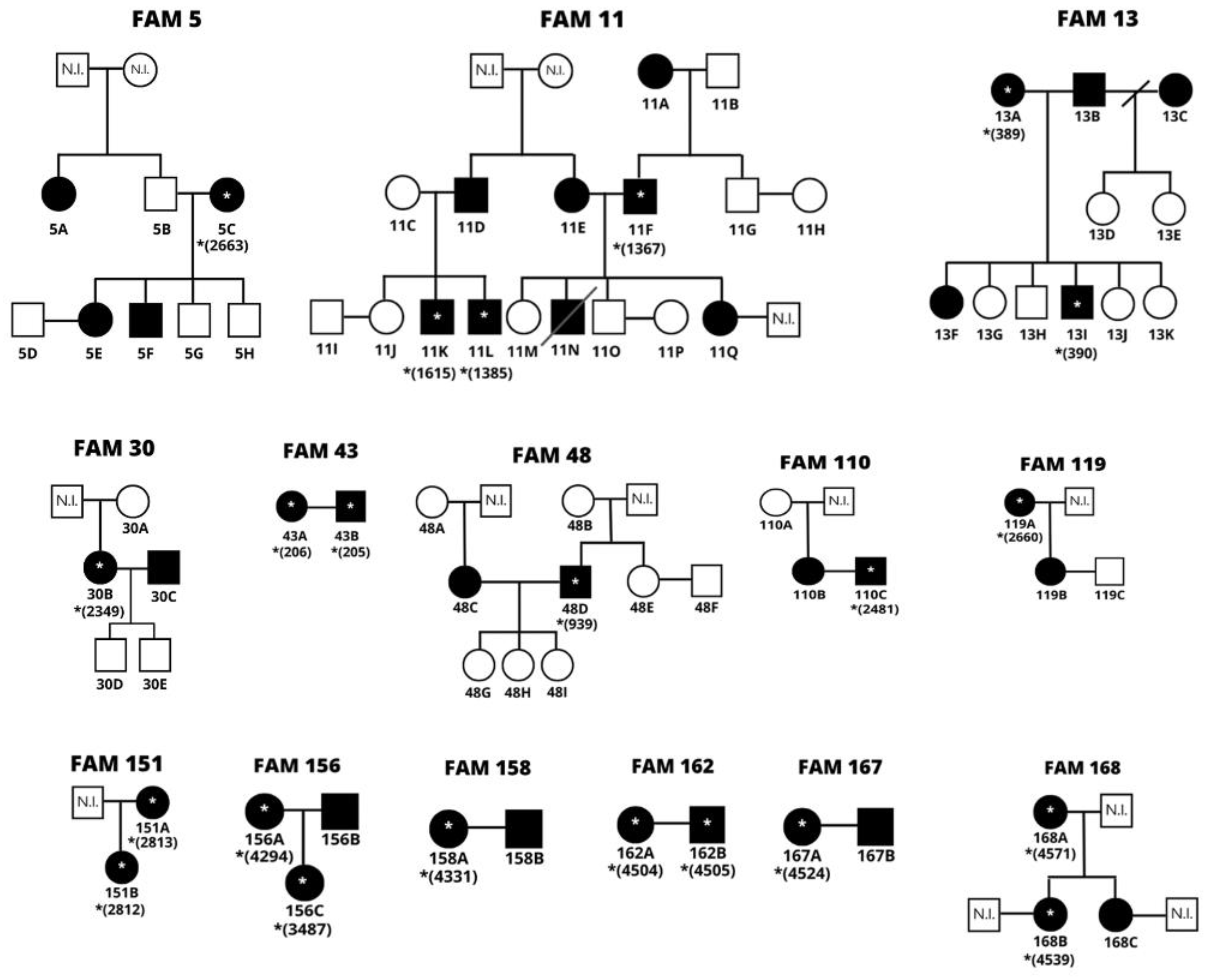
Figure 2.
Phylogenetic tree created from the sequencing of 22 relatives and 11 sequences deposited in GenBank.
Figure 2.
Phylogenetic tree created from the sequencing of 22 relatives and 11 sequences deposited in GenBank.
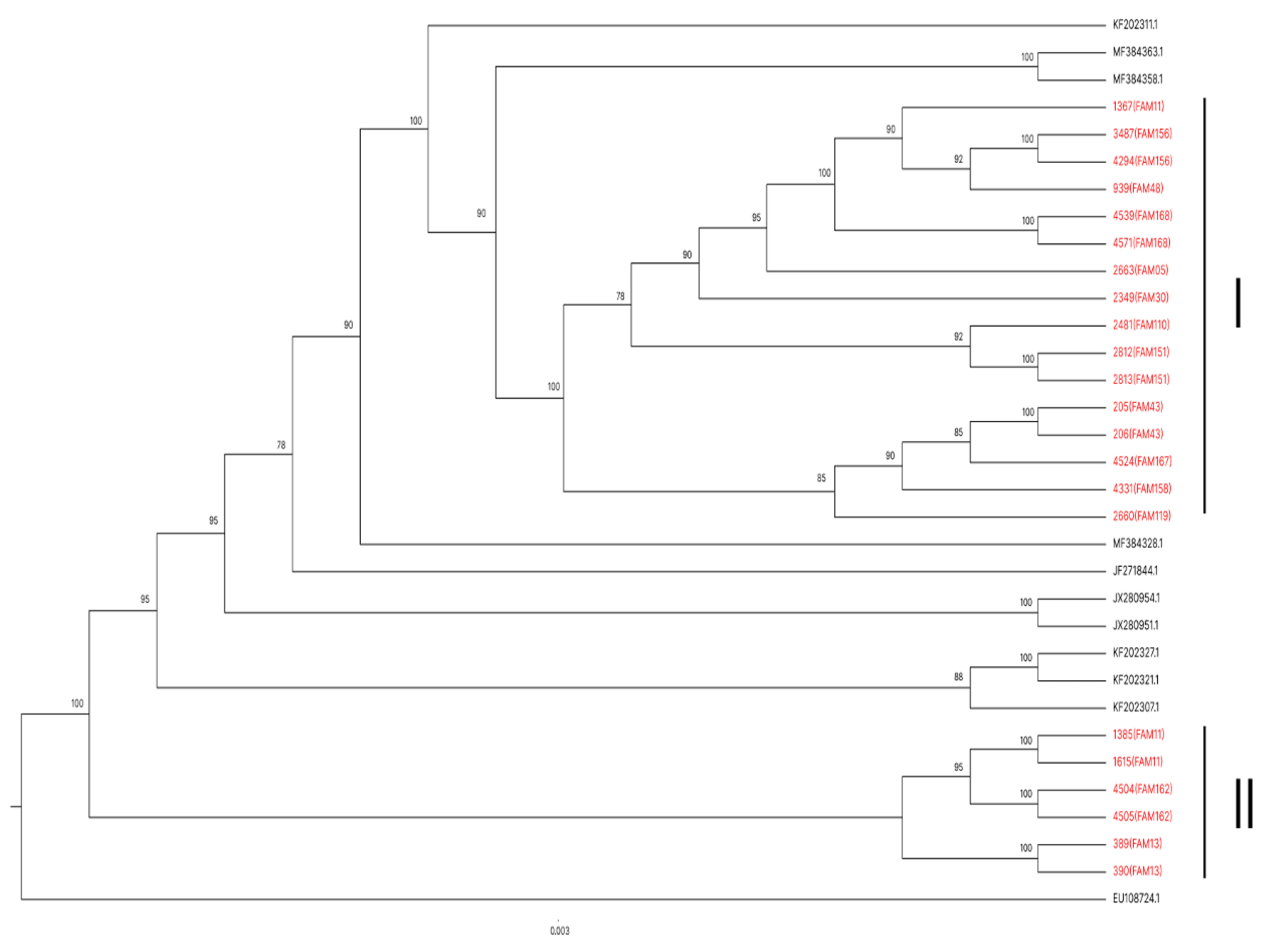
Figure 3.
Phylogenetic tree of the relationship between the 22 samples of Family 195 members and a sequence deposited in GenBank as an outgroup.
Figure 3.
Phylogenetic tree of the relationship between the 22 samples of Family 195 members and a sequence deposited in GenBank as an outgroup.
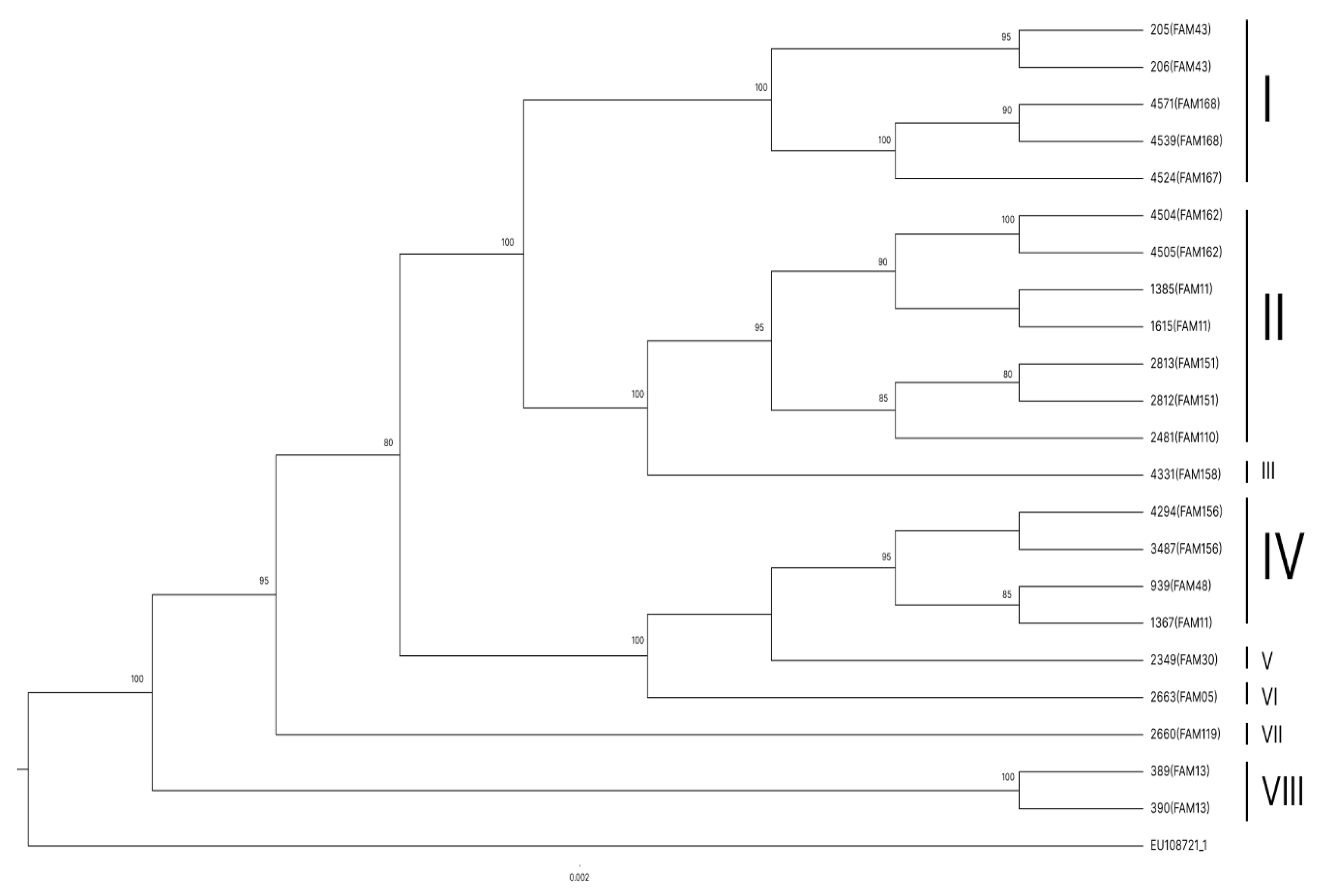
Figure 4.
The phylogenetic tree originated from the similar relationship of families with more than one sequenced sample on an outgroup.
Figure 4.
The phylogenetic tree originated from the similar relationship of families with more than one sequenced sample on an outgroup.


Table 1.
Epidemiological data of the 72 relatives of the 14 families.
| Uninfected | Infected | ||||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Gender | Female | 18 | 58,06 | 25 | 60,98 |
| Male | 13 | 41,94 | 16 | 39,02 | |
| Age (Years) | Minimum | 3 | - | 3 | - |
| Maximum | 86 | - | 84 | - | |
| Average | 29 | - | 42 | - | |
| Marital status | married | 5 | 16,13 | 29 | 70,73 |
| Single | 3 | 9,68 | 4 | 9,76 | |
| Widowed | 1 | 3,22 | 1 | 2,44 | |
| U.D. * | 22 | 70,97 | 7 | 17,07 | |
| Blood transfusion | Yes | 0 | 0 | 6 | 14,63 |
| history | No | 31 | 100 | 35 | 85,37 |
| Breastfeeding | Yes | 12 | 38,71 | 30 | 73,17 |
| history | No | 1 | 3,23 | - | - |
| Not sure | 18 | 58,06 | 11 | 26,83 | |
| Condom use | Yes | 1 | 3,23 | 9 | 21,95 |
| No | 4 | 12,9 | 24 | 58,54 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



