
Submitted:
03 February 2023
Posted:
07 February 2023
You are already at the latest version
Abstract
The integrity of the genome is crucial for the survival of all living organisms. However, genomes need to adapt to survive certain pressures and for this propose use several mechanisms to diversify. Chromosomal instability (CIN) is one of the main mechanism leading to the creation of genomic heterogeneity by altering the number of chromosomes as well as changing their structures. In this review we will discuss the different chromosomal patterns and changes observed in speciation, in evolutional biology as well as during tumor progression. By nature, human genome shows induction of diversity during gametogenesis but as well during tumorigenesis that can conclude in drastic changes such as whole genome doubling to more discrete changes as indels as well as the recent discovered complex chromosomal rearrangement chromothripsis. More importantly, changes observed during speciation are strikingly similar to the genomic evolution observed during tumor progression and resistance to therapy. The different origins of CIN will be treated as the importance of double strand breaks (DSB) or the consequences of micronuclei. We will also explain the mechanisms behind the controlled DSBs and recombination of homologous chromosomes observed during meiosis, to explain how errors lead to similar pattern observed during tumorigenesis. Then, we will also list several diseases associated to CIN resulting in fertility issue, miscarriage, genetic rare diseases and cancer. Understanding better chromosomal instability as a whole is primordial for the understanding of mechanisms leading to tumor progression.
Keywords:
Chromosomal instability
; Cancer
; Genome evolution
; Speciation
; Structural variant
; Meiosis
; micronuclei
1. Introduction
Genome is organized into chromosomes that provide the structure for faithful transmission of the hereditary information. Maintenance of genome integrity is essential for the survival of all living organisms. To accomplish this, cells have developed intricate DNA repair mechanisms to maintain genome integrity, however, aberrant dynamic of the genome or repair of DNA damage, such as double-strand breaks (DSBs), can lead to genome instability, including chromosomal anomalies and mutations, which can promote tumorigenesis over an individual’s lifetime [1]. Tumorigenesis is the complex evolutionary process of loss of cell identity and gain of characteristics, with different stages as metastasis and with a base of accumulation of different forms of genome instability. New technologies have uncovered new chromosomal abnormalities linked with cancer progression [2], providing new insight into the plasticity and the instability of the genome as well as on the mechanisms underlying the maintenance of modifications of chromosome structure, that provides us new perspective to understand the instability behind of tumorigenesis.
Germ cells reflect the evolutionary history and future potential of a specie as they give rise to the formation of gametes. Genomic instability in germ cells can lead to chromosomal anomalies, resulting in sterility or genetic diseases in the offspring [3,4,5], but can also result in stable and heritable rearranged genomic constitutions such as mutations or chromosomal changes [6,7]. Thus, chromosomal instability plays a crucial role in evolution, providing a new advantage for organisms adapting to changing environments. These changes can include from changes in chromatin conformation and reorganization to a wide range of factors, single nucleotide polymorphisms, or large-scale structural variants such as inversions, translocations, fusions, and fissions, which can result in variations in the number and morphology of chromosomes [5,6,8,9]. Over millions of years, the accumulation of these changes can lead to speciation.
This review summarizes the relationship between genome and chromosome instability associated with cancer, infertility, and evolution, highlighting the main difference in time scale. A proper understanding of the underlying biology will lead to more effective cancer treatment and improved understanding of evolution.
2. Different Pattern of Structure Variants and Chromosomal Instability
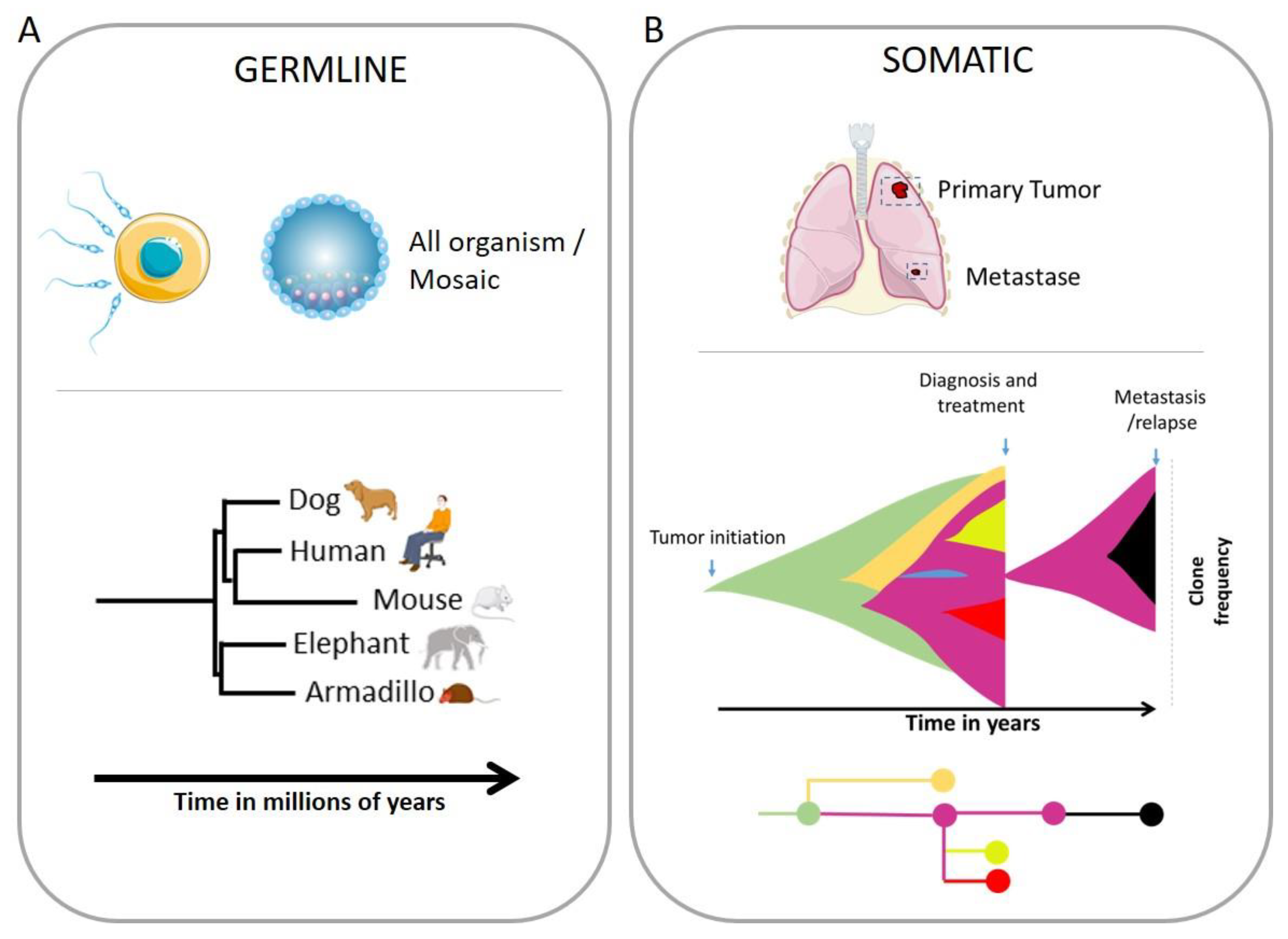
Chromosomal instability (CIN) is seen in different forms, more or less complex and result in structural and numerical chromosomal abnormalities. Some of them are compensated and don’t lead to major changes in global expression but can affect some small part of the genome and disrupting some important genes with massive consequences, as in the predisposition to produce cancer during certain translocations or during complex chromosomal rearrangement (Figure 1).
Chromosomal numerical changes are known as copy number alterations (CNA) or copy number variations (CNV), indicating a difference when compared to the patient’s normal cells. Loss of heterozygosity (LOH) is defined as an allelic imbalance because one of the two parental alleles gets lost.
2.1. Aneuploidy
The simplest forms of CIN are the gain or loss of chromosome/s or chromosome arms, named aneuploidy, leading to an unbalanced number of chromosomes (Figure 1). The most visible case is the Down syndrome, also referred as Trisomy 21, where all cells from the body have an extra full or portion [10] of the chromosome 21. Other extreme case of aneuploidy is the gain in ploidy where cells can duplicate the entire genome and conclude in a whole genome doubling (WGD). It has been described that cancer cells can proceed to several round of WGD during their progression [11].
2.2. Common Chromosomal Changes
The four main types of structural chromosomal aberrations are deletion, duplication, inversion, and translocation (Figure 1). Deletion is the loss of some full or part of the chromosome. Translocation is when a piece of one chromosome breaks off and attaches to another chromosome. Duplication happens when a part of the chromosome double and results in having extra genetic material. An inversion in a chromosome occurs when a segment breaks off, turns 180 degrees and reattaches within the same chromosome.
2.3. Indels
Indels are a term to refer to a small insertion or deletion in the genome (Figure 1). The genetic variations measure from 1 base pair (commonly known as SNPs or single nucleotide polymorphism) to 10 000 base pairs in length. Indels larger than 1 kb are referred to as copy number variations that typically have arisen through amplification or duplication events or through deletion events resulting from two distant DSBs followed by fusion of the DNA ends [12].
2.4. Complex Genomic Rearrangements
Structural chromosome abnormalities can also be observed as complex chromosomal rearrangement (CCR) which involves more than two breakpoints and including one or more chromosomes. CCRs are not only important causes of fertility issues but also largely involved in cancer. Chromothripsis, a CCR example, comprises hundreds of chromosomal rearrangements in a localized region of one or few chromosomes, leading to a new configuration of this one [13,14] (Figure 1). Chromoplexy, in another hand, tends to involved less pieces of chromosome but implicate more chromosomes [15]. Other forms of complex genomic rearrangements include breakage-fusion-bridge cycles, which lead to complex duplications and inversions of genomic regions [13].
2.5. ecDNA
Extrachromosomal DNA (Figure 1) refers to DNA that is not located on a chromosome, but instead exists as a separate, circular entity, and invisible by light microscopy but detectable by biophysical methods or sequencing [16]. ecDNA can range in size, often being 1-3 Mb or larger, can contain one or multiple full genes and regulatory regions and forms lacking a centromere or a telomere [16]. ecDNA are frequently observed across many cancer types and often in high copy numbers and can be associated to the amplifications of several oncogenes such as EGFRvIII [17] or MYC [18].
3. Chromosomal Instability during Evolution
Adaptive evolution is the ability of genetic changes to adapt to relative fitness through selection. species modify their genome to survive and diverge into new lineages. To be hereditable and then transmitted to the next generation, such changes need to be done through the fecundation and the genetic diversification of the gametes. Genome modification can be through mutations by increasing the genetic diversity or can be more drastic as observed with changes in structural variants (SV) and number of chromosomes.
Figure 2.
Phylogenetic trees from evolution specie (A) and from tumors (B). (A) During evolution, changes in the genome need to be germline, meaning that all cells own the same genetic background and this genetic is hereditary. For this purpose, those changes need to appear during the fecundation or during gametogenesis. All organisms in earth share a common ancestor. During time, accumulation of genomic changes led to diversification in specific species. (B) Tumor initiation involves changes in the genomes. One cell gains genomic insults, resulting in the formation of tumors. Because of genomic instability, tumors cells derive in diverse clones with their own genetic changes. During tumor progression and metastasis, specific clones will survive to the new environment and to the therapeutic pressure.
Figure 2.
Phylogenetic trees from evolution specie (A) and from tumors (B). (A) During evolution, changes in the genome need to be germline, meaning that all cells own the same genetic background and this genetic is hereditary. For this purpose, those changes need to appear during the fecundation or during gametogenesis. All organisms in earth share a common ancestor. During time, accumulation of genomic changes led to diversification in specific species. (B) Tumor initiation involves changes in the genomes. One cell gains genomic insults, resulting in the formation of tumors. Because of genomic instability, tumors cells derive in diverse clones with their own genetic changes. During tumor progression and metastasis, specific clones will survive to the new environment and to the therapeutic pressure.
3.1. SNPs
Human genome contains 4 to 5 million Single nucleotide polymorphisms (SNPs) making the diversity of our specie [19]. This genetic variation has specific distribution across the globe [19]. Similar to human, SNP are present in all species [20].
Because of genomic diversity, species can have different abilities to survive disasters due to genomic diversity. Point mutations play a major role in creating genomic diversity. Specific genetic variants such as Single nucleotide polymorphisms (SNPs) can be crucial in short-term resistance to specific pressures. For example, in the case of Chernobyl accident and the major release of radioactive material to the environment, it only took 30 years for the frog population to select and shift to dark skin coloration population to adapt to local radiation [21]. Other extreme case is the human response to Black Death, a pandemic cause by the Y. Pestis infection that devastated Afro-Eurasia, killing up to 30–50% of the human population [22]. Ancient DNA reveals rapid natural selection during this pandemic [22,23]. This study [22] shows that individuals carrying specific genetic immune defenses variants had much better odd of surviving infection. In just a few generations, the survivors selected specific variants that responded efficiently to Y. pestis at a speed and an intensity never observed before in human genomes [22,23]. However, the protection afforded by those variants could have come at a cost, as some of those variants have deleterious effect in modern area by being associated with auto-immune diseases such as rheumatoid arthritis or Crohn’s disease [22,24].
3.2. Genome Size Variability and Chromosomal Instability
Genome size varies greatly in time and support the diversification and adaptation of the specie. In early time of evolution and diversification, events of translocations and whole genome doubling were primordial to create such diversity of life. Recent studies demonstrated the occurrence of two whole genome duplications in the lineage that precedes the ancestor of vertebrates [25,26] (Figure 3). Some fishes duplicated a third time their genome in view to increase the genome size [26,27,28,29,30], having an important evolutionary role in term of genomic variability and plasticity. However, changes in ploidy do not appear to represent a major source of genome size variation in birds or mammals [27]. Plant kingdoms also present great variability in genome size and events of WGD [31].
Gain and loss of some chromosomes are also indispensable during evolution. For example, in case of the well-studied Equus phylogeny through the last 4.5 million of years, it was observed a diversification of at least several dozens of recognized genera. Equine phylogeny has shown important SNP but also demonstrate extremely divergent chromosomal structure, both in number (gain and loss) or in chromosomal rearrangement. As such numbers of chromosomes are really diverse (Horses (E. baballus) n= 32, Zebras n=16-22, African Ass n= 31, and Asian Ass n= 26) [32].
Genomic diversity is not only observed as change in number of full chromosomes but also involve modification in their structures. Chromosomal structure variants as events of translocation, deletion, duplication, inversion, and also complex chromosomal rearrangements such as chromothripsis (see below) have shaped the history of many evolutionary lineages [26,29,32,33,34].
3.3. Chromosomal Variance in Human
Single nucleotide polymorphisms are common is human and explain the diversity of phenotype in our specie. However, structural variants (SV) also exist in human and represent 0.1% of variants [35]. In fact, SVs are common in healthy individuals. A study [35] mapping and characterizing the structural variation in 17,795 human genomes has shown that 2% of individuals carry ultra-rare megabase-scale structural variants with nearly half of which are balanced (translocations and inversions) or complex rearrangements. On average, individuals carry 2.9 rare structural variants that alter coding regions and 19.1 rare noncoding deletions; these variants affect the dosage or structure of 4.2 genes [35]. Structural variants in normal individual are predominantly deletion, mobile-element insertion and duplication but also present complex rearrangements.
Another study [36] analyzing the complex landscape of 433,371 SVs, has shown that SVs are responsible for 25–29% of all rare protein-truncating events per genome [36]. In their study they found 3.9% of individual carrying very large (over one megabase) rare SVs [36].
Recurrent inversion is characterized by large inversion of part of chromosomes (Figure 1). It is estimated than an average of 11.6 Mbps is flipped in a normal genome [37]. Recurrent inversions are flanked by retrotransposons or by segmental duplications that often exceed the length of sequencing reads or library inserts. This technical challenge made that such genetic variation class is underexplored and might represent more than 0.6% of the genome [37]. Recurrent inversions exhibit a sex-chromosomal bias and co-localize with genomic disorder critical regions.
4. Chromosomal Instability in Cancer
In the past decade, thousands of patient tumors have been sequenced and have revealed that cancer genomes are highly complex and heterogeneous. Sequencing tumors and metastatic sites from the same patients has allowed to draw the phylogenetic tree of the tumors and to determine the early and late events [11,38,39,40,41,42]. Something very intriguing is the similarity and parallelisms between genomic complexity history of tumors and genome evolutionary changes. Drastic events such as chromosome rearrangements and gain/loss of chromosomes both draw the evolution of them to adapt and progress to new environment and pressure. Whole genome doubling in tumorigenesis [11,38,39,40,41] is also an early event supporting the creation of genome diversity as observed early before the diversification leading to vertebrates (Figure 3) or as observed in plants [31]. Interestingly tumor cells may occur multiple times to the duplication of their genome during tumor development, leading to highly complex cancer genomes [11,43].
Tumors arose from one cell that will generate a plethora of diverse clones with independent genomic alterations (Figure 1B). Tumorigenesis involves modification in the genome and mutations are key in the process. However, chromosomal instability is indispensable for the generations of diversity and is one of the hallmark of cancer [44]. 80-97% of all tumor present detectable CIN [43,45]. Besides the classical and simple genomic aberrations as mutations, deletions, translocations and insertions, cancer cells can present complex chromosomal rearrangements such as chromothripsis and chromoplexy [15]. Chromothripsis is a chromosomal instability phenomenon, a genomic catastrophe, where hundreds of chromosomal rearrangements occur during one single event in a localized region of one or few chromosomes. This type of chromosomal rearrangement is highly frequent in cancer with a prevalence of 49% to up to 80% [46]. Chromothripsis is associated with the formation of circular extrachromosomal DNA (ecDNA) [47,48] as well as with segmental deletion. Chromothripsis increased with whole-genome duplications in most cancer types [49].
New study claims that those drastic change arose form mitotic catastrophes that rapidly shape new genetic distinct clones. The analysis of these phylogenetic trees revealed that complex structural events, including chromothripsis, are major drivers [46,50]. Chromosomal instability is even enriched in metastases of several cancer types [51,52].
As performed for mutations [53], different chromosomal instability patterns are observed in cancer upon the origin of insults and can lead to a specific signature [43,45]. Some signatures predict drug response and identify new drug targets [45].
Chromosomal instability is primordial for tumor progression and support tumor progression and metastasis. In fact, tumors harboring high level of CNA have worst prognosis than high mutation rate tumors because of their ability to escape the immune system [54]. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies [55]. Tumor aneuploidy predicts survival following immunotherapy across multiple cancers and elevated aneuploidy score is an independent and complementary predictor of overall survival for patients with low tumor mutational burden tumors [56]. Furthermore, CIN also appear to be indispensable for the success of metastasis. Tumor cells with high CIN level activate the pro-inflammatory pathway cGAS-STING to support their survival and growth [51].
5. Possible Origins of Chromosomal Instability
There are several potential causes of CIN, including genetic mutations, errors in cell division, and exposure to certain environmental factors. Thereafter, we delve into some of them.
5.1. Mitotic-Cell Cycle Errors
Mitosis is a delicate event that must be executed with high fidelity to ensure genomic integrity. However, errors in cell division can contribute to the development of malignant karyotypes in cancer [59]. This can be due to a variety of factors, including defects in the spindle assembly checkpoint (SAC), cohesion defects, problems with kinetochore attachment, or an increase in the number of centrosomes [59].
Issues during the mitosis can conclude to gross gain or loss of chromosome such as during mis-segregation where a full chromosome goes to the wrong daughter cell. Importantly, lagging chromosome during the mitosis can produce the formation of micronuclei. In fact, if a chromosome is late during the nuclear envelope (NE) formation and is not clustered with the other chromosomes, a proper NE will form around this one, forming a separate small nucleus close to the main nucleus, called micronuclei (MN). Micronuclei can contain a full chromosome or a chromosome fragment [60]. MN are in fact a major source of DNA damage and chromosomal instability due to NE rupture [61].
5.2. Transient Nuclear Envelope Rupture (NER)
Recent work has demonstrated the existence of transient Nuclear Envelope Rupture (NER) during interphase as origin of several form of chromosomal instability. The NE under certain stress can collapse, exposing the genome to the harmful cytoplasm for several minutes to the main nucleus or even permanently in the case of micronuclei [60]. Such unexpected events have drastic consequences on genome evolution as NER can lead to complex and simple chromosomic events such as chromothripsis, extended above [48,62,63,64,65,66,67].
NER happens on micronuclei, where the envelope is fragile and tends to disrupt without possibility of proper repair [61,68]. In another case, NER happens in cells with chromatin bridge, when cells experiencing telomere fusion connect two daughter cells. This implies the generation of additional tension forces affecting the NE during movement, leading to NER that can last up to two minutes [67]. Cells during migration can also experience NER of their nuclei [69,70,71,72]. Interestingly, immune attack can fail and can contribute to mutagenesis. Cytotoxic T lymphocytes often fail to kill target cells during a one/one conjugation and can lead to transient NER, leading to DNA damage [73]. Such ‘failed’ attack then participate to the generation of genomic diversity ad progression toward resistance.
The mechanisms responsible for the genomic aberrations linked with NER are still under debate. Some authors have demonstrated that in the cytoplasm the unprotected DNA can be attacked by DNAses such as TREX1 [74], by the immune DNA mutator APOBEC [65] or can break due to mechanical forces during migration [64]. Furthermore, the loss of compartmentalization can affect the replication inside the micronuclei, provoking a desynchronization with the main nucleus. Thus, the main nucleus may start the mitosis too early for the DNA trapped inside the micronuclei, which is not folded nor protected and can lead to its pulverization within the cytoplasm, resulting in chromothripsis [62,64]. Another explanation is that micronuclei accumulate large amounts of RNA:DNA hybrids that can lead to DNA fragmentation due to pathological DNA base excision repair [75].
5.3. Double-Stranded Breaks
Double-stranded breaks (DSBs) are a type of DNA damage that occur when both strands of the DNA helix are broken, a chromosome break. They can be caused by several factors such as exposure to ionizing radiation, reactive oxygen species, and certain chemical agents, or by the activity of endogenous enzymes like topoisomerases, helicases and primases. DSBs are a common type of DNA damage, and are considered as the most dangerous as they can lead to chromosomal aberrations, genomic instability, and cell death if not repaired properly. The repair of DSBs is a complex process and it can be accomplished mainly by non-homologous end joining (NHEJ) or homologous recombination (HR) pathways. The choice of repair pathway depends on the cell cycle stage, the type of DNA damage, and the cell type. Some of the genetic causes of CIN associated with DSBs include mutations in genes that are involved in maintaining the integrity of chromosomes, such as the BRCA1 and BRCA2 genes, which are involved in the HR pathway and are associated with an increased risk of breast and ovarian cancer. Other genetic causes of CIN include mutations in genes that act as sensors of DNA damage, such as the ATM, ATR, and DNA-PK genes. But in case of repair failure, cells risk having a broken chromosome that can lead to the formation of micronuclei. In fact, a recent study using CRISPR-Cas9 gene editing has shown that a single DSB can lead to a cascade of events resulting in the formation of micronuclei and chromosome bridges [76]. Such simple DSBs can be amplified into far more extensive genomic alterations during subsequent mitosis, leading to a myriad of genomic diversity.
5.4. Expression of Meiotic-Specific Proteins
Meiotic-specific proteins are a group of proteins that are specifically expressed and play important roles during the meiotic stage of the cell cycle. However, in some cases, meiotic-specific proteins can be expressed during mitosis as a result of genetic mutations, chromosomal abnormalities, or other disturbances in the cell cycle. This can lead to chromosomal instability, micronuclei formation, cell death, or even cancer. These events can occur in normal somatic cells or in cancer cells, the expression of meiotic proteins during mitosis has been associated with chromosomal instability, DNA damage, and aneuploidy, which are hallmarks of cancer [77,78,79,80,81]. Importantly, a study has revealed that overexpression of the meiotic cohesin Rec8 in mitotic fission yeast cells leads to the uniparental disomy of chromosomes and chromosomal instability (CIN) in this organism [82]. Another meiotic gene is PRDM9, described below in more detail. A study made with 1879 cancer samples across 39 distinct cancer types discovered that PRDM9 was unexpectedly present in 20% of these tumors, even after applying strict gene homology adjustments [83].
5.5. Tumorigenesis Drive CIN
Cancer cells have a tendency to accumulate a variety of mechanisms that result in genomic instability. This instability allows cancer cells to evolve and accumulate errors, which enables them to survive and progress to increase their fitness. One of the most commonly mutated genes in cancer is TP53, also known as the “guardian of the genome” [49]. When TP53 is mutated or deleted, cells lose their ability to correct errors, leading to genomic diversity and allowing the cells to survive. For example, in mouse models, TP53 LOH leads to accumulation of deletions, genome doubling, and the emergence of gains and amplification [84].
6. Germ Cells and Meiotic Program as Drivers for Genome Evolution
Mitosis is the most common cell division program and takes place in somatic tissues, but not the only one. Meiosis is a specific cell division that only takes place in the germ line of organisms with sexual reproduction. Germ line is composed by pluripotent and mitotic self-renewing stem cells which finally trigger the meiotic program to sustain gamete production, both oogenesis (egg production) and spermatogenesis (sperm production) (Figure 4A). Meiosis comprises two consecutive rounds of cell divisions, meiosis I and meiosis II, with the peculiarity that only a single round of S-phase genome replication takes place in meiosis I and not during meiosis II. As result, the formation of four haploid daughter cells, each containing only one set of chromosomes (half the number present in the original cell) (Figure 4A). After fertilization, the whole set of chromosomes that define the specie is recovered [85,86].
Therefore, unlike the rest of somatic tissues, germ cells are the only ones that contribute genetically to descent and then information contained is passed from generation to generation. In this regard, protection of genome information of this tissue is essential for perpetuation of species. Then, it is possible that germ cells possess unique strategies to protect the genetic information. In addition, meiotic program is highly regulated as defects are linked with infertility, miscarries and congenital diseases. But at the same time, meiosis is linked with a generation of genome diversity with the final goal of breeding diversity in the offspring for an adaptative response to the environment and increasing the fitness of the species. The first level of genetic shuffling and generation of new alleles combination is coming from the reduction in chromosome number and random assortment of them during meiosis and the fertilization of gametes after sexual reproduction from different parents. A second level implies a molecular mechanism that involves programmed breaks point along DNA, repairing with and linking homologous chromosomes forming the crossing overs, essential for proper chromosome segregation at meiosis I. As a consequence, an exchange of material takes place, generating new combination of genetic information.
Therefore, germ cells contain the genetic information that reflects the evolutionary history and holds the potential for future developments of species. Understanding how the genome is organised in germ cells, the maintaining of genome stability along its self-renewing and during the meiotic program, and during chromosome segregation, is fundamental to understanding fertility and its impact on genetic diversity and evolution of species, and might highlight similar mechanisms in cancer.
6.1. Chromosomal Axis and Synapsis in Genome Stability and Evolution
Two rounds of cell divisions, meiosis I and meiosis II, are required to produce gametes. Before entering meiosis, germ cells are typically diploid and possess two copies of each chromosome, called homologous chromosomes. A single round of DNA replication results in four copies, each consisting of two homologous chromosomes made up of two sister chromatids. The sister chromatids of each homolog are attached along replication by ring-shaped protein complexes called cohesins. During meiosis I, the homologous chromosomes segregate, while during meiosis II, in a similar way to mitosis, sister chromatids segregate, resulting in four products with half the genetic information (Figure 4A,B) [88].
The special segregation of homologous chromosomes during Meiosis I is achieved through a mechanism involving both synapsis, the pairing of two homologous chromosomes, and DNA recombination between them (Figure 4B). These two relevant events take place during prophase I, maybe the most important phase from meiosis which lasts the longest. Prophase I is broken down into four stages called leptonema, zygonema, pachynema, and diplonema according to these events and the associated dynamics of meiotic chromosomes, as there are dramatic chromosomal movements and chromatin remodelling during this phase [88]. In short, the joining of homologous chromosomes begins during leptonema with DNA organization into loops anchored to a chromosomal axis formed by a protein scaffold structure along the chromosomes, made of cohesin (REC8 or RAD21L in mammals) and specific proteins (the HORMA domain-containing proteins) constituting the axial element of the synaptonemal complex (SC) along each homologous chromosome [89,90,91,92] (Figure 4B). After specific homologous recognition takes place, the synapsis or polymerization of central elements (SYCP proteins) between both axes occurs [93,94]. Simultaneously during leptonema, meiotic recombination starts with the creation of DSBs (double-stranded breaks) by the SPO11 endonuclease protein [85]. During zygonema, DSBs are repaired resulting in synapsis and exchange of genetic material between homologous chromosomes, leading to crossovers (COs) (Figure 4B) [95]. Along the next phase, pachynema, chromosomes finish completely synapsis, forming bivalent structures, and recombination is completed and COs are resolved. During diplonema, the chromosomes begin to condense and become more compact in preparation for cell division. In this phase the SC and cohesin begins to release leading to the chiasmata, the visual and physical manifestation of attached homologous chromosomes, where the recombination occurred. This allows accurate segregation of homologous chromosomes during anaphase I. Later on, gametes are obtained with the completion of cohesin release from the arms of the sister chromatids during anaphase II.
Defects in these events can result in nondisjunction, producing aneuploid gametes with abnormal numbers of chromosomes which leads to aneuploidy and sterility [96]. Overall, errors in gametogenesis leading to aneuploidy is a relatively common occurrence in humans, with estimates suggesting that it occurs in 10-30% of cases, particularly increasing sharply with maternal age [96].
However, alteration of normal ploidy can have a beneficial effects, such as accelerating genetic diversification and rapid adaptation. Studies done in unicellular organism have shown that rapid adaptation to stress environments is often characterized by aneuploid states, which are also associated with changings in gene expression and an increase in mutational rates and accumulation of them to increase fitness [97,98,99]. Accordingly, a study in Arabidopsis has shown that the REC8-cohesin complex is linked to multigenic adaptive evolution in autopolyploid and whole genome duplication [100]. Robertsonian (Rb) chromosome fusions, expanded upon at a later point, are another example. Further of their impact in fertility, their consequent secondary defects in synapsis leads to chromosomal incompatibilities between divergent lineages, thus contributing to chromosomal speciation, as it has been described in the Alai mole vole (Ellobius alaicus) in the family Cricetidae [101].
6.2. Impact of Three-Dimensional Chromatin Structure on the Control of Gene Expression
During gametogenesis, further to the Synaptonemal Complex (SC) configuration between homologous chromosomes, genome undergoes a 3D reorganization, and insulator proteins such as CTFC and cohesin complexes play a role in organizing chromatin into compartments (open/closed chromatin), topologically associated domains (TADs), and DNA loops. This organization ultimately affects both the physical arrangement of chromatin and the regulation of gene activity [102,103,104] (Figure 4C,D). This reveals that spatial conformation of genomes is given by a close interplay between gene function and chromatin organisation. Research in mice has shown that the chromatin architecture is reprogrammed during the development of germ cells [105]. Therefore, it is not surprising that recent studies have shown that functional plasticity under higher-order genomic organization is passed on to offspring, ultimately contributing to the formation of new allelic variants on which natural selection and evolution can act [104].
Importantly, it is known that the repair of DSBs occurs in the context of this axial chromosomal structure [106,107]. A relatively low level of compartmentalization during prophase I has been linked to the specific events that occur during this phase, such as chromosomal movements, chromatin condensation, and the formation and repair of DSBs. Later on during gametogenesis, there is a close interplay between increasing compartmentalization and differential gene expression. But recent discoveries have improved our understanding of the link between the dynamics of chromatin organization and its role in mouse spermatogenesis beyond gene expression. Briefly, chromatin conformation plays a crucial role in the generation and resolution of DSBs, which have significant implications for evolution, as discussed above [104].
In this sense, large-scale chromosomal changes can also result in inherited diseases, genome instability, and cancer by altering the expression of genes in those reorganized genomic regions. Along with this view, it has been reported that disorders of domain architecture and chromatin organization caused by inversions, fusions, or insertions can lead to the activation of oncogenes or the emergence of new gene functions [108,109,110]. Hence, similar underlying mechanisms are responsible for both evolutionary changes in the genome and genetic instability diseases.
6.3. DNA Recombination and Repair as a Mayor Source for Evolution
DNA recombination is a central process during meiosis that leads to a physical attachment between homologous chromosomes. This step is called Crossing-over (CO) and is a universal feature that is necessary for faithful chromosome segregation during meiosis and sexual reproduction, maintaining the genome integrity of species. Additionally, meiotic recombination shuffles alleles along homologous chromosomes, leading to genetic diversity in sexual gametes and offspring [111]. In this sense, understanding the mechanisms and factors involved in meiotic CO formation and resolution can provide insight into biodiversity, but if not well-regulated, it can lead to repair errors resulting in sterility or miscarriages. On the other hand, proper regulation of new meiotic CO formation and resolution could be adaptive, supporting the evolution of species or the emergence of new ones.
6.3.1. Recombinational Hotspots as a Factor for Genome Stability and Evolution
DNA recombination begins with programmed DSBs mediated by an endonuclease called SPO11. This protein is highly conserved across eukaryotic evolution [85,107]. Along with SPO11, other proteins are involved in the control and formation of DSBs, such as PRDM9, MEI4, REC114, or HORMAD1 in mice [106,107,112]. Due to its importance, this process is highly regulated throughout the meiotic cell cycle, particularly at leptotene and prophase I. Breaks are concentrated at the called recombination hotspots, where SPO11 and other factors are involved in the formation and repair of DNA breaks in the early stages of meiosis, and it’s highly regulated including total number and genome distribution of DNA breaks [6]. Recombination hotspots have been found in localized short regions of hundreds to thousands of base pairs in many species including yeast, plants, and all vertebrates studied to date [113]. However, in some species such as nematodes, the recombination landscape appears more uniform and lacks these hotspots [114]. It has been described between 150 meiotic DSBs per cell in humans and around 200-300 in mice [115,116]. It is known that human hotspots are normally 1–2 kb in size, spaced 50–100 kb apart, accounting for no more than 20% of the genome, and only ~5% to 30% (depending on the organism) go on to produce COs [117]. Centromeres are normally recombination deserts, whereas in (sub)telomeres recombination rates increase. Mirroring what has been described in mice [118], hotspots are localized in genic and intergenic regions likewise. This suppression of recombination around centromeres has been described as not related with the presence of the heterochromatic domains, which are typically associated with satellite DNA in this area, at least in horse spermatocytes [119]. Some studies also unveil the importance of specific DNA motifs and epigenetic modifications in determining recombination hotspots [120,121].
6.3.2. Factors and Complexes Defines Recombinational Hotspots
- Cohesin complexes and Synaptonemal Complex axial elements
As previously mentioned, the way genomes are packaged is likely another factor influencing species-specific differences in recombination rates. The mean number of DSBs per cell is influenced by DNA compaction through SC length and DNA loop size (Figure 4E). There is a close interplay between SC assembly, the organization of DNA loops, and the formation of DSBs [86,122]. It has been described that chromosomes with longer DNA loops and shorter SC axes showed a reduced number of DSBs in early prophase I. An explanation could be that shorter SC axes could anchor less DNA loops, but longer, providing less scaffold for the formation of DSBs and recombination rates. Likewise, loop sizes were inversely correlated with CO density [6,86,123] (Figure 4E).
Recent studies have highlighted the importance of certain factors in shaping genomic architecture and subsequently, evolution. Cohesin-mediated DNA loops are organized along the SC axial scaffold, anchoring as long DNA loops and establishing a physical first level that defines hotspots [93,124] (Figure 4D). Cohesin complexes, HORMADs, and SC proteins correlate with these recombination hotspots, as reflected by the interactions observed at shorter distances (2.5–4.5 Mbp). In this sense, a recent paper found that a majority of the cohesin complex during spermatogenesis is localized within promoter regions of genes located in DNA loops away from the axes [104] (Figure 4C). In this regard, meiotic-specific cohesin provides an integrated structural and functional framework for the 3D organization in germ cells and spermatocytes in mice, manifesting a fine-tuned balance among recombination, chromatin remodeling, architectural proteins, and gene expression during spermatogenesis [104].
- PRDM9
Among species with recombination hotspots, locations are specified by PRDM9 (PR domain 9) binding. PRDM9 is a specific early meiosis protein expressed only in testes and ovaries [83]. This protein presents a zinc finger (ZnF) motif which recognizes specific DNA sequences in the genome and adds H3K4me3 and H3K36me3 marks at nearby nucleosomes close to DSBs in early meiosis, recruiting the DSB repair machinery and SPO-11 in early stages of meiosis and determining recombination hotspots [125,126]. In terms of evolution, PRDM9-ZnF motif recognizes a high variability of DNA motifs, which modification can be translated into a redistribution of recombination hotspots [125]. In this sense, different distributions of DSBs have been described in humans with different PRDM9 alleles [127] and in mice [128], providing new clues on a factor that generates new DSB sites, which could be a source of genetic instability and indirectly have important consequences in evolution. PRDM9 is not found in all vertebrates, it is absent in certain birds, canids, and fishes, however, orthologs of PRDM9 have been identified in some of these groups [8]. Interestingly, mammals with PRDM9 have the fastest evolving zinc fingers (ZF) array [117,129]. Then, these mammals generate novel sets of hotspots leading to rapid turnover in the recombination landscape between populations and species, and thus having a role in the evolution rate. The selection pressure to explain the fast evolution of the PRDM9 ZF remains unclear. It could be explained as an assurance of recombination hotspots and to avoid inefficient DSB repair leading to reduced fertility or sterility [128,130].
On the other hand, in species without PRDM9, the rapid evolution of recombination hotspots is not seen. For example, species of birds lacking an ortholog of PRDM9, the locations of recombination hotspots are conserved over long evolutionary time scales, reducing evolution over time [8]. Species that do not use PRDM9 and have more stable recombination locations tend to direct recombination in promoter-like features, as opposed to species that use PRDM9 and experience rapid turnover of hotspot landscapes, which tend to recombine away from promoters and coding-protein sequences, even in some subfamilies of transposable elements in mammals by ZF domain recognition [131]. These different dynamics have important consequences for genome structure and, therefore, evolution [132,133]. As expected, recombination hotspots have higher rates of point mutations, insertions, deletions, and translocations. Therefore, directing recombination to non-coding regions might be an advantage in avoiding genome instability. In this regard, recombination at transcription-related loci could uncouple coding regions and its regulatory elements, leading to transcriptional regulation of genes or negative epistasis [134].
- Chromosome organization as a modulator of recombination landscape
The normal progression of the meiotic cell cycle involves reshuffling of the genome, which is mediated by programmed DSBs and provides diversity among organisms with new combinations of alleles on which natural selection can act. However, sometimes large-scale genomic changes, such as inversions, translocations, fusions, and fissions, occur, leading to infertility or providing new sources of variation on which natural selection can act and pushing speciation. Several genetic and mechanistic factors, such as PRDM9 in mammals [126], affect the recombination landscape and the combinatorial effect of chromosomal rearrangements, leading to speciation.
On the other hand, chromosomal rearrangements can play an important role in further modifying both the structure and regulation of genes located near the affected regions, as proposed by the “suppressed recombination” model [135]. This is because recombination is suppressed within rearranged segments, inversions, or chromosomal fusions, resulting in a reduction of COs within these regions [136]. As a result, there is a reduction in gene flow around reorganized genomic regions, and chromosomal reorganization could lead to a combination of alleles, polymorphisms, or haplotypes that provide a certain degree of selective advantage or accumulate genetic incompatibilities, ultimately contributing to evolution and species divergence in the long term. As a result of chromosome reorganizations, there is an alteration of the recombination maps, leading to genetic differentiation across genomes, referred to as “genomic islands of divergence” [137,138]. Evidence for this has been described in mammals, such as between human and macaque lineages, where recombination patterns have undergone rearrangements that could be favouring adaptive alleles in the immune response [139]. Other studies have described similar reductions in recombination landscapes in mice due to Robertsonian translocations, also related to alterations in epigenetic signatures for heterochromatinization [135,140].
Future comparative research on recombination hotspot landscapes, speciation genes, and different chromosomal rearrangements, and how these changes in chromosome structures affect recombination, should be conducted to enhance our understanding of the main pathways for genome reshuffling and evolution.
6.3.3. Pathways of Repair of Meiotic DSB to Chiasma Formation
Not all breaks mediated by SPO11 are resolved by CO formation and subsequent chiasma formation, point of contact and the physical link, between homologs. DSBs lead to an orchestrated DNA damage response mediated by the proteins ATM (ataxia telangiectasia mutated) and ATR (ATM-Rad3-related) due to the phosphorylation of histone H2AX on serine 139 (γH2AX) [141]. After several coordinated steps involving DNA resection, which leads to 3′ strand overhangs where monomers of RPA are located and displaced by the recombinase RAD51 and/or DMC1, forming nucleoprotein filaments that catalyze strand invasion. Different repair pathways can mediate the repair of the processed breaks with two possible final outcomes: COs leading to the chiasmata connecting both homologous chromosomes or non-COs (NCOs) generating gene conversion without a connection between chromosomes. To assure homeostasis and correct chromosome segregation during the first meiotic division, CO assurance in mammals is carefully controlled and the total number is relatively constant among cells [116]. Along with this view, several studies in different organisms have revealed that the number of meiotic DSBs exceeds the number of chiasmata, indicating that the majority of DSBs are resolved as NCOs [116,142]. A highly regulated genetic control determines the fate and distribution of COs. This is known as CO interference, which reflects the restriction on a new CO to take place near another established CO. This restriction decreases with chromosomal length, leading to a spaced distribution across chromosome axes [86,122]. All of these aspects, from meiotic pre-replication to DSB formation to controlled resolution until COs, are relevant for understanding the diversity of genomes at the individual level but also the genetic variation leading to evolutionary change.
6.3.4. Defects in Meiotic Recombination Leads to Chromosome Rearrangements
While it has been reported that disruptions of chromosome architecture such as inversions, fusions, or translocations are associated with genetic instability and cancer due to oncogene activation and novel gene functions, there is a parallelism in organism evolution due to intra or interchromosomal alterations in the germ line or during the meiosis program, which can alter normal segregation patterns and contribute to evolution [8,107]. Otherwise, failures in the meiotic program can also lead to aneuploidy, infertility, genetic abnormalities, and congenital disease [143,144].
- Large chromosomal rearrangements as drivers for speciation
One chromosomal rearrangement that affects the recombination landscape and is linked to miscarriages, infertility, or aneuploid descendance in humans and pushes evolution is the Robertsonian (Rb) fusions or translocations, the most common balanced chromosomal rearrangement in nature. Rb translocations typically involve two acrocentric chromosomes, homologous or non-homologous, and result in the fusion of both long arms to form a single metacentric chromosome, plus the loss of both short arms. Aneuploidy and loss of genetic information are responsible for miscarriages and aneuploid offspring in humans. However, recent findings indicate that the situation is not as straightforward, and Rb fusions may also have an impact on the structure of the genome in germ cells, which could lead to functional and evolutionary consequences [145]. Indeed, Rb fusions can significantly change the 3D arrangement of chromatin in spermatocytes, potentially leading to new interactions between domains and exposing them to different regulatory environments that may impact gene expression and regulation, having implications for both fertility and evolution [109]. Additionally, the redistribution of COs across chromosomal arms in Rb mice was consistent with the “suppressed recombination” model, showing low recombination rates at Rb fusions, also affecting both chromosomal axis length and recombination [145].
This results in the existence of a gametic barrier due to the fixation of chromosomal rearrangements in a population, which allows for a genetic distinction between species. Genome sequencing analysis has indicated that these evolutionary rearrangements are much more numerous than initially estimated [146,147]. An example can be seen in the human and chimpanzee genomes, which were initially estimated to differ only in 9 chromosomal inversions and one fusion in 1980 [148], but later, in 2005, 93 supplementary evolutionary rearrangements were described, ranging from 12 kb to 1 Mb [149]. In this sense, more than 245 large rearrangements including translocations, inversions, fusions, and deletions have been identified in the divergence between mice and humans [150], and several duplications involved in human-specific adaptive qualities have been recently characterized between the human and non-human primate genome [151].
- Microchromosomes
Microchromosomes, or chromosomes that are smaller in size than a typical chromosome, refer to chromosomes that have undergone structural changes, such as deletions, duplications, or inversions, resulting in a smaller size than normal, and sometimes a proper NE will form around [61]. These changes can lead to a loss of genetic material. Micro chromosomes are not found in humans but are highly express in normal cells from birds and reptiles.
Bird and reptile karyotypes are striking in their differences, as in addition of normal size mammalian chromosomes, they are generally characterized by the presence of well-conserved tiny microchromosomes, but little is known about the progression of meiosis in them. These are gene-rich and highly conserved between species of birds and reptiles. It seems that levels of DNA double-strand break (DSB) formation in reptiles are lower compared to mammals, suggesting that low recombination rates are a distinctive feature of reptiles [152,153]. Oppositely, micronuclei tend to clump into a central compartment at interphase and during mitosis and meiosis, leading to stronger inter-chromosomal interactions between microchromosomes rather than macrochromosomes, probably facilitating homology search and DSB formation that is maintained even in germ cells [153]. This suggests functional coherence. Many translocations of micronuclei and fusions with each other or with larger chromosomes have taken place independently in different clades such as turtles, snakes, and lizards, or others have been lost, leading to diversity and speciation. In mammals, they have completely disappeared. The work by Waters and colleagues [153] has shed light on the contribution of micronuclei in evolution by comparing the genomes of different birds and reptiles, as well as mammals and an amphioxus, providing evidence that microchromosomes are highly conserved ancient animal chromosomes, whereas macrochromosomes have undergone multiple lineage-specific rearrangements, especially in mammals, probably due to its higher level of DSB formation.
- Whole genome duplications and gain in chromosomes
Beyond rearrangements of genomic segments throughout evolution, large-scale genomic duplication may occur, but more rarely, involving a macroscopic portion of the genome or even, the complete genome, also called Whole Genome Duplications (WGDs).
WGD has a significant impact on an organism’s physical characteristics, and it is highly likely that it causes infertility and reduces the organism’s fitness, which can negatively impact the organism’s short-term survive [154]. Therefore, most WGD events do not result in viable outcomes, and they do not become fixed in the population. However, under specific conditions, WGD can provide an immediate evolutionary advantage in extreme environmental conditions [155,156]. It is now understood that WGD has played a significant role in evolution. WGD can reduce the risk of extinction for the affected lineage by alleviating its intrinsic disadvantages, and it can also help boost the biological fitness of the organism or create new species. This is because ancient WGDs often occurred during periods of environmental turmoil or extinction events [156,157]. Such advantages could explain why so many cancers present one to several rounds of WGD [11,154,156].
While WGD events are relatively common in plants, such as Arabidopsis, and have also been observed in the yeast Saccharomyces [158,159], identifying WGD pairs or quartets in vertebrates is more difficult. It is by now widely accepted that two rounds of WGD happened at the origin of the vertebrate lineage about 500–550 millions of years ago [160,161] (Figure 2). However, how these global-scale WGD events affected the gene regulatory network is not yet fully understood. It has been proposed that a main vertebrate WGD played a crucial role in the organization of multicellularity by influencing the interaction of proteins, the selection and retention of genes, and the balance of gene dosage [162].
Many tumors present rounds of WGD, but often stabilized to a near 3N karyotype [11]. Some genes are selected to offer advantages. Interestingly, genes that have undergone duplication are three times more likely to be involved in cancer and autosomal dominant diseases, as they are more susceptible to dominant deleterious mutations [163]. Duplicated genes have stricter requirements for maintaining proper dosage balance [164], are normally involved in signaling, development, and transcriptional regulation and they are represented in gene ontology categories associated with organism’s level of complexity [163,165,166]. Therefore, duplicated genes resulting from stabilized WGD tend to be essential, evolutionarily conserved genes, and are related to increased redundancy and complexity, allowing for an advantage under stressful or disturbed environments compared to non-WGD organisms [160,165]. This parallelism between WGD in evolution and the high incidence of WGD in cancer, both strive for increasing fitness to overcome non WGD. But it is also possible that the high incidence of cancer in human population due to WGD is caused by adverse and polluted environmental conditions. However, further analysis is needed to fully understand the molecular mechanisms responsible for the advantage of WGD genomes over non-duplicated ones in cancer or evolution.
- Chromothripsis
Traditionally, studies on evolution have treated genome alterations as a result of many incremental single steps. However, recent research has discovered that genomes can be altered in a single, large catastrophic event, leading to complex genomic rearrangements. This phenomenon is called chromothripsis [14]. Chromothripsis occurs in a single cell and results in the breaking and rearranging of one or multiple segments of chromosomes in a random manner (Figure 1). It was initially discovered in tumors, but it has also been identified in individuals with congenital malformations, developmental disorders, or seemingly balanced chromosomal rearrangements [167,168]. However, it has also been described in healthy individuals [169]. Furthermore, chromothripsis may also have a role in evolution as a source of new genetic combinations. Different hypotheses have been proposed, such as the idea that derived chromosomal rearrangements can restrict gene flow through suppression of recombination, or facilitate rapid changes in patterns of genes unrelated to recombination suppression. Chromothripsis could also lead to the formation of new gene linkage blocks and new chimeric genes, as well as disruption of the cis-regulatory machinery for gene expression [170]. One proposed explanation for the connection between the various causes of chromothripsis and the limited scope of genomic changes it causes is that the affected chromosome(s) may be placed in a micronucleus [61,171]. The formation of micronuclei can happen due to a failure in chromosome segregation, but also can be caused by various types of stress during any stage of the cell cycle, and it can persist for multiple cell cycles before being eliminated or incorporated back into the main nucleus [171].
6.3.5. Repetitive DNA Elements as Contributors for Chromosome Evolution and Speciation
To fully understand how mammalian genomes are shaped and how speciation occurs, it is important to consider the structural organization of genomes. Research has shown that certain breakpoint regions tend to cluster in specific genomic features, such as duplications or repetitive elements [172,173], tandem repeats [174,175], and transposable elements [174,176]. Changes in the presence or length of these DNA elements can alter the chromatin state, which in turn can affect gene expression [3,139]. In an interesting paper published by Capilla et al. [6] on rodents, a family characterized by high chromosomal variability between clades, they found an association between evolutionary breakpoint regions and active chromatin state landscapes.
A repetitive DNA element that plays a role in driving genome evolution is the large tandem repeat known as satellite DNA (satDNA) [177]. satDNA has also been linked to various genetic disorders, such as cancer and developmental abnormalities [178]. The presence of satDNA remains a prevalent concern for various species, including both vertebrates and invertebrates. To establish comparative studies of genome divergences, satDNA is still an ongoing issue in sequencing techniques due to genome assembly and annotation. Recent studies have shown that satDNA can vary in abundance, size, and organization within a single genome, and it can also differ considerably among closely related species [177]. This dynamic behaviour has led to speculations about the role satDNA plays in genome plasticity and chromosome rearrangements. Some studies suggest that satDNA could act as a source of genetic variation and evolution, through mechanisms such as non-identical CO, replication errors, and non-allelic homologous recombination. These mechanisms could result in changes in number and reorganization of chromosomes and genetic diversity [177,179].
Transposable elements, also known as transposons, play a significant role in chromosome evolution and speciation. They can act as drivers of DNA breaks and changes in chromatin conformation, both of which can compromise genomic stability [180]. However, they can also promote adaptability in a population through changes in gene expression or rapid chromosome restructuring [181,182]. An example of this is the gibbon genome, where the insertion of the retro-transposon LAVA in genes involved in cell cycle progression and chromosome segregation has led to a high rate of chromothripsis-related rearrangements and the emergence of different gibbon lineages with highly rearranged chromosomes [183,184]. Furthermore, ZF domain from PRDM9 tends to recognise transposable elements in mammals. However, further research is needed to completely understand the role of repetitive DNA elements in the evolution of genomes [185].
6.3.6. Telomeres
Telomeres have also been described as a driver for evolution. An interesting study reveals a unique feature of marsupial meiosis, a non-traditional model mammal, the detection of alternative lengthening of telomeres (ALT) during prophase I spermatogenesis [152]. They propose a telomeric elongation model in dunnarts, a family of marsupials native to Australia and New Guinea, that implies telomerase activity, telomere transcription and alternative lengthening of telomeres (ALT) [186]. Opposite, telomere length remains stable through spermatogenesis or oogenesis in human, mice, or any other mammal [187,188,189,190]. The open chromatin state of dunnart telomeres is coupled with heterologous telomeric associations in early pachytene, which supports the homologous recombination-based mechanism in ALT mediated by RPA and RAD51, searching and inducing inter-telomere recombination [152,191,192]. Interestingly, ALT was initially described in cancer cells but further than in marsupial meiosis, it has also been detected in non-cancer pluripotent stem cells, to maintain long telomeres that are heterogeneous in size [193,194]. This suggests that the mechanisms used to maintain long, heterogeneous telomeres in evolution may also be present in cancer. Supporting this idea, dasyurids have genetic predisposition to lymphomas [195]. Then, further exploration of ALT for telomere homeostasis in dasyurids in the germline could provide new insight in the high incidence of tumors reported in this clade, and stablish a correlation.
7. CIN during Fecundation and First Mitosis
Around 50%–70% of human cleavage embryos are aneuploid. Most aneuploid embryos do not develop to term, making aneuploidy in embryos a leading cause of miscarriages and infertility. Errors during meiosis explain a great portion of aneuploidy and CIN in early embryos. However, aneuploidy is also thought to arise during the mitotic divisions of the embryo during early development. A recent study [196] shows that improper clustering of parental genomes with nucleoli in each pronucleus during fecundation can lead to lagging chromosomes and the formation of micronuclei.
8. Disease Associated to Chromosomal Instability
It has been describing a variety of genetic diseases associated with CIN. Next, we detail some of them.
8.1. Fertility
The association between fertility and chromosomal instability is complex and multi-factorial. CIN can lead to infertility in several ways. For example, abnormal chromosomes can cause problems during meiosis, leading to the production of aneuploidy sperm or eggs. If these sperm or eggs are used in fertilization, the resulting embryos will have chromosomal abnormalities, leading to miscarriage or developmental problems. CIN can lead also to infertility affecting the mitotic self-renewing cells from the germ line, that ultimately give rise the gametes. One example is the inversion of chromosome 9 in human, present in 2.6% of the population and it is associated to recurrent pregnancy loss [197]. With the use of new genome sequencing techniques, we are now able to explore the complexity of chromosomal abnormalities in recurrent miscarriage, providing a deeper understanding of the underlying causes and potential targeted treatment options.
8.2. Rare Disease
Rare diseases are defined as diseases that affect a small percentage of the population. Many rare diseases are caused by genetic mutations, and some may be related to CIN. Some specific syndromes associated with CIN:
8.2.1. Impairment of DNA Damage Repair Pathways
Fanconi anemia (FA) is a recessive disease that is associated with growth retardation, congenital malformations, bone marrow failure, and a high risk of cancer. FA is a model syndrome of genome instability and is caused by a deficiency in DNA interstrand crosslink repair resulting in chromosome breakage. Cancer derived from FA present of high numbers of structural variants [198]. FA is caused by biallelic mutations in at least 12 different genes, some of which form a nuclear core complex that involves the ubiquitination of the central FANCD2 protein. This process promotes the accumulation of FANCD2 in nuclear repair foci, along with BRCA1, BRCA2, and RAD51. Biallelic mutations in BRCA2 and FANCD2 can lead to a severe FA-like phenotype. The FA genes and FANC2 are involved in DNA repair functions that are associated with the resolution of DNA crosslinks and stalled replication forks [199]. Although genetic instability caused by mutations in the FANC genes can be detrimental for most FA patients, around 20% appear to benefit from it, as it increases the chance of somatic reversion of their constitutional mutations. This can lead to improved bone marrow function through intragenic crossover, gene conversion, back mutation, and compensating mutations [200,201].
Another rare disease associated to DNA repair is the Bloom syndrome, a genetic disorder that leads to developmental abnormalities, and an increased risk of developing cancer. It is caused by mutations in the BLM gene, which is involved in the repair of DNA damage.
Another rare disease is the Nijmegen breakage syndrome, a genetic disease that leads to increased susceptibility to infections, developmental delays, and an increased risk of cancer. It is caused by mutations in the NBS1 gene, which is also involved in the repair of DNA damage.
8.2.2. Germile SV and CCR
Structural variants (SV) represent 0.1% of variants [35] but account for 4 to 11.2% of deleterious coding alleles, a disproportionate contribution highlighting the major role of chromosomal instability in human disease [35]. Furthermore, SVs are responsible for 25–29% of all rare protein-truncating events per genome [36] making germline CIN an underestimated cause of rare disease. Importantly, autism and schizophrenia were recently linked with specific SV [35,202,203,204].
In children, much more than in adults, fusion proteins play an important role in driving tumorigenesis, and many different fusions have been identified as potential driver events in pediatric CNS neoplasms [57]. In pediatric cancer, 11% of known clinically relevant fusions detected at a high confidence level are germline [205].
Because of the tremendous advances obtained in sequencing tools, complex genomic rearrangements are also discovered to be an underestimated cause of rare diseases [206].
9. Concluding Remarks
Investigating and understanding the high degree of plasticity of genomes has altered our perception of how genomes originate, remain stable, and evolve in cancer cells and during the speciation process. With the development of new multidisciplinary approaches that combine sequencing technologies, bioinformatics tools, genomic approaches, and experimental methods has made it possible to study the functional and structural basis of the genomes of various organisms, strengthening the links between genome expression, genome stability, and 3D genome architecture. Nowadays, we are closer to being able to predict the consequences in developmental biology, such as cancer, fertility, and evolution, due to altered genomes, as reproducible patterns are revealed. It is common to find new studies focusing on the dynamics and plasticity of genome architecture within the framework of integrative analysis, its causes and consequences, and finding similarities between processes such as cancer evolution and macroevolution leading to speciation, with the main difference being the time scale. It is clear that the importance of genome-level events emphasizes the need for a new conceptual framework that integrates information based on genome organization. Alternatively, the spatial organization and folding of chromosomes within the nucleus plays a crucial role in regulating gene expression, and this has important ramifications for evolution or the development of cancer.
Here, we have summarized how different large-scale chromosome reorganizations take place and their consequences. From understanding the factors involved to conducting mechanistic and functional studies, we will gain new insights into the mechanisms responsible for these processes, providing an overview of similarities and differences in their sequential evolution steps.
Author Contributions
M.C.-P. and V.C. have contributed to the structure, literature search, figure design and writing of this review. All authors have read and agreed to the published version of the manuscript.
Funding
V.C. is funded by the Spanish association against the Cancer AECC investigator grant (INVES20033COMA) and from the MSCA IF grant Onco-inflammation 101026137. V.C. is funded from the Fundación Vencer el Cancer. M.C.-P. is funded from the MSCA IF grant coDNAres 101023902.
Acknowledgments
Figures 2 and 3 use some contents from Bioincons.com and are issues from DBCLS and Servier upon the license CC-BY 3.0 and 4.0. We would like to thanks Dr. Mark W. Bierner form University of Texas at Austin as well as Kepa Izaguirre to give the opportunity to give a class on evolution and medicine from the BIO 370 Evolution upper-level undergraduate course as part of the study abroad program. This class initiate this paper in the attempt to compare the evolution of genome from macroevolution to cancer.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Heng, H.H. The genome-centric concept: resynthesis of evolutionary theory. Bioessays 2009, 31, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Liu, G.; Bremer, S.; Ye, K.J.; Stevens, J.; Ye, C.J. Clonal and non-clonal chromosome aberrations and genome variation and aberration. Genome 2006, 49, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Larkin, D.M.; der Wind, A.E.-V.; Bourque, G.; Tesler, G.; Auvil, L.; Beever, J.E.; Chowdhary, B.P.; Galibert, F.; Gatzke, L. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 2005, 309, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Barton, N.H. Chromosomal speciation and molecular divergence--accelerated evolution in rearranged chromosomes. Science 2003, 300, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Gatinois, V.; Puechberty, J.; Geneviève, D.; Lefort, G. Chromothripsis: potential origin in gametogenesis and preimplantation cell divisions. A review. Fertility and sterility 2014, 102, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Capilla, L.; Garcia Caldés, M.; Ruiz-Herrera, A. Mammalian Meiotic Recombination: A Toolbox for Genome Evolution. Cytogenet Genome Res 2016, 150, 1–16. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Human molecular genetics 2011, 20, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, K.; Oliveros, W.; Gerhardinger, C.; Andergassen, D.; Maass, P.G.; Rinn, J.L.; Melé, M. Cis and trans effects differentially contribute to the evolution of promoters and enhancers. Genome biology 2020, 21, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, Y.; Koshiba-Takeuchi, K. Significance of whole-genome duplications on the emergence of evolutionary novelties. Brief Funct Genomics 2018, 17, 329–338. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Lyle, R.; Dermitzakis, E.T.; Reymond, A.; Deutsch, S. Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet 2004, 5, 725–738. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nature genetics 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.P.; Petersen, B.L.; Johansen, I.E.; Niu, Y.; Yang, Z.; Chamberlain, C.A.; Met, O.; Wandall, H.H.; Frodin, M. INDEL detection, the ‘Achilles heel’ of precise genome editing: a survey of methods for accurate profiling of gene editing induced indels. Nucleic Acids Res 2020, 48, 11958–11981. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.L.; Zhang, C.Z.; Pellman, D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu Rev Genet 2015, 49, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Bafna, V.; Mischel, P.S. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer 2019, 19, 283–288. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef]
- Storlazzi, C.T.; Fioretos, T.; Surace, C.; Lonoce, A.; Mastrorilli, A.; Strombeck, B.; D’Addabbo, P.; Iacovelli, F.; Minervini, C.; Aventin, A.; et al. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet 2006, 15, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, G.; Yang, W.; Jin, W.; Gong, J.; Xu, X.; Niu, X. Animal-SNPAtlas: a comprehensive SNP database for multiple animals. Nucleic Acids Res 2022. [Google Scholar] [CrossRef]
- Burraco, P.; Orizaola, G. Ionizing radiation and melanism in Chornobyl tree frogs. Evol Appl 2022, 15, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Klunk, J.; Vilgalys, T.P.; Demeure, C.E.; Cheng, X.; Shiratori, M.; Madej, J.; Beau, R.; Elli, D.; Patino, M.I.; Redfern, R.; et al. Evolution of immune genes is associated with the Black Death. Nature 2022, 611, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Enard, D. Ancient DNA reveals rapid natural selection during the Black Death. Nature 2022, 611, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Bubonic plague left lingering scars on the human genome. Nature 2022. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.Z.; Ocampo Daza, D. A new look at an old question: when did the second whole genome duplication occur in vertebrate evolution? Genome Biol 2018, 19, 209. [Google Scholar] [CrossRef] [PubMed]
- Sacerdot, C.; Louis, A.; Bon, C.; Berthelot, C.; Roest Crollius, H. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol 2018, 19, 166. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc Natl Acad Sci USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef]
- Glasauer, S.M.; Neuhauss, S.C. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol Genet Genomics 2014, 289, 1045–1060. [Google Scholar] [CrossRef]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef]
- Berthelot, C.; Brunet, F.; Chalopin, D.; Juanchich, A.; Bernard, M.; Noel, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun 2014, 5, 3657. [Google Scholar] [CrossRef]
- Clark, J.W.; Donoghue, P.C.J. Whole-Genome Duplication and Plant Macroevolution. Trends Plant Sci 2018, 23, 933–945. [Google Scholar] [CrossRef]
- Jonsson, H.; Schubert, M.; Seguin-Orlando, A.; Ginolhac, A.; Petersen, L.; Fumagalli, M.; Albrechtsen, A.; Petersen, B.; Korneliussen, T.S.; Vilstrup, J.T.; et al. Speciation with gene flow in equids despite extensive chromosomal plasticity. Proc Natl Acad Sci USA 2014, 111, 18655–18660. [Google Scholar] [CrossRef]
- Du, K.; Stock, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat Ecol Evol 2020, 4, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Bejerano, P.; Royo, C.; Torres-Perez, R.; Grimplet, J.; Fernandez, L.; Franco-Zorrilla, J.M.; Lijavetzky, D.; Baroja, E.; Martinez, J.; Garcia-Escudero, E.; et al. Catastrophic Unbalanced Genome Rearrangements Cause Somatic Loss of Berry Color in Grapevine. Plant Physiol 2017, 175, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Abel, H.J.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; Reeves, C.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alfoldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Porubsky, D.; Hops, W.; Ashraf, H.; Hsieh, P.; Rodriguez-Martin, B.; Yilmaz, F.; Ebler, J.; Hallast, P.; Maria Maggiolini, F.A.; Harvey, W.T.; et al. Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell 2022, 185, 1986–2005. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nature genetics 2015, 47, 1047–1055. [Google Scholar] [CrossRef]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat Commun 2020, 11, 496. [Google Scholar] [CrossRef]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of copy number alterations in human cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarro, L.; Couturier, D.L.; Liu, L.; Schneider, M.; et al. A pan-cancer compendium of chromosomal instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef]
- Voronina, N.; Wong, J.K.L.; Hubschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The landscape of chromothripsis across adult cancer types. Nat Commun 2020, 11, 2320. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Page, D.C.; Campbell, P.J.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nature genetics 2019, 51, 705–715. [Google Scholar] [CrossRef]
- Consortium, I.T.P.-C.A.o.W.G. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun 2019, 10, 3835. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Gronroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Lukow, D.A.; Sausville, E.L.; Suri, P.; Chunduri, N.K.; Wieland, A.; Leu, J.; Smith, J.C.; Girish, V.; Kumar, A.A.; Kendall, J. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev Cell 2021, 56, 2427–2439 e2424. [Google Scholar] [CrossRef]
- Spurr, L.F.; Weichselbaum, R.R.; Pitroda, S.P. Tumor aneuploidy predicts survival following immunotherapy across multiple cancers. Nature genetics 2022, 54, 1782–1785. [Google Scholar] [CrossRef] [PubMed]
- Roosen, M.; Ode, Z.; Bunt, J.; Kool, M. The oncogenic fusion landscape in pediatric CNS neoplasms. Acta Neuropathol 2022, 143, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Sweet-Cordero, E.A.; Biegel, J.A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev 2018, 32, 620–638. [Google Scholar] [CrossRef]
- Gauthier, B.R.; Comaills, V. Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation. Int J Mol Sci 2021, 22, 7281. [Google Scholar] [CrossRef]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, N.T.; Zhang, C.Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Chatzipli, A.; Dananberg, A.; Chu, K.; Toufektchan, E.; Klimczak, L.J.; Gordenin, D.A.; Campbell, P.J.; de Lange, T. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nature genetics 2020, 52, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Hatch, E.M. Nuclear Membrane Rupture and Its Consequences. Annu Rev Cell Dev Biol 2020, 36, 85–114. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Schultz, S.W.; Bellanger, A.; Jones, C.M.; Petersen, L.I.; Raiborg, C.; Skarpen, E.; Pedurupillay, C.R.J.; Kjos, I.; Kip, E.; et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat Cell Biol 2020, 22, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef]
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep 2016, 17, 2632–2647. [Google Scholar] [CrossRef]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, B.R.; Lorenzo, P.I.; Comaills, V. Physical Forces and Transient Nuclear Envelope Rupture during Metastasis: The Key for Success? Cancers (Basel) 2021, 14, 83. [Google Scholar] [CrossRef] [PubMed]
- Weigelin, B.; den Boer, A.T.; Wagena, E.; Broen, K.; Dolstra, H.; de Boer, R.J.; Figdor, C.G.; Textor, J.; Friedl, P. Cytotoxic T cells are able to efficiently eliminate cancer cells by additive cytotoxicity. Nat Commun 2021, 12, 5217. [Google Scholar] [CrossRef] [PubMed]
- Nader, G.P.F.; Aguera-Gonzalez, S.; Routet, F.; Gratia, M.; Maurin, M.; Cancila, V.; Cadart, C.; Palamidessi, A.; Ramos, R.N.; San Roman, M. Compromised nuclear envelope integrity drives TREX1-dependent DNA damage and tumor cell invasion. Cell 2021, 184, 5230–5246 e5222. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Stokasimov, E.; Cui, Y.; Pellman, D. Breakage of cytoplasmic chromosomes by pathological DNA base excision repair. Nature 2022, 606, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nature genetics 2021. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, J.W.; Koster, J.; Lodder, P.; Repping, S.; Hamer, G. Massive expression of germ cell-specific genes is a hallmark of cancer and a potential target for novel treatment development. Oncogene 2018, 37, 5694–5700. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S.; Salmina, K.; Hausmann, M.; Scherthan, H. The role of meiotic cohesin REC8 in chromosome segregation in gamma irradiation-induced endopolyploid tumour cells. Exp Cell Res 2009, 315, 2593–2603. [Google Scholar] [CrossRef]
- Feichtinger, J.; Larcombe, L.; McFarlane, R.J. Meta-analysis of expression of l(3)mbt tumor-associated germline genes supports the model that a soma-to-germline transition is a hallmark of human cancers. Int J Cancer 2014, 134, 2359–2365. [Google Scholar] [CrossRef]
- Kalejs, M.; Ivanov, A.; Plakhins, G.; Cragg, M.S.; Emzinsh, D.; Illidge, T.M.; Erenpreisa, J. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer 2006, 6, 6. [Google Scholar] [CrossRef]
- Lindsey, S.F.; Byrnes, D.M.; Eller, M.S.; Rosa, A.M.; Dabas, N.; Escandon, J.; Grichnik, J.M. Potential role of meiosis proteins in melanoma chromosomal instability. Journal of skin cancer 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Folco, H.; Chalamcharla, V.R.; Sugiyama, T.; Thillainadesan, G.; Zofall, M.; Balachandran, V.; Dhakshnamoorthy, J.; Mizuguchi, T.; Grewal, S.I. Untimely expression of gametogenic genes in vegetative cells causes uniparental disomy. Nature 2017, 543, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Houle, A.A.; Gibling, H.; Lamaze, F.C.; Edgington, H.A.; Soave, D.; Fave, M.J.; Agbessi, M.; Bruat, V.; Stein, L.D.; Awadalla, P. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res 2018, 28, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Baslan, T.; Morris, J.P.t.; Zhao, Z.; Reyes, J.; Ho, Y.J.; Tsanov, K.M.; Bermeo, J.; Tian, S.; Zhang, S.; Askan, G.; et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature 2022, 608, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Kleckner, N.; Storlazzi, A.; Zickler, D. Coordinate variation in meiotic pachytene SC length and total crossover/chiasma frequency under conditions of constant DNA length. Trends in Genetics 2003, 19, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Herrera, A.; Vozdova, M.; Fernández, J.; Sebestova, H.; Capilla, L.; Frohlich, J.; Vara, C.; Hernández-Marsal, A.; Sipek, J.; Robinson, T.J.; et al. Recombination correlates with synaptonemal complex length and chromatin loop size in bovids-insights into mammalian meiotic chromosomal organization. Chromosoma 2017, 126, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, H. Meiosis: an overview of key differences from mitosis. Cold Spring Harb Perspect Biol 2015, 7. [Google Scholar] [CrossRef]
- Bannister, L.A.; Reinholdt, L.G.; Munroe, R.J.; Schimenti, J.C. Positional cloning and characterization of mouse mei8, a disrupted allele of the meiotic cohesin Rec8. Genesis 2004, 40, 184–194. [Google Scholar] [CrossRef]
- Revenkova, E.; Eijpe, M.; Heyting, C.; Hodges, C.A.; Hunt, P.A.; Liebe, B.; Scherthan, H.; Jessberger, R. Cohesin SMC1 beta is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat Cell Biol 2004, 6, 555–562. [Google Scholar] [CrossRef]
- Winkel, K.; Alsheimer, M.; Öllinger, R.; Benavente, R. Protein SYCP2 provides a link between transverse filaments and lateral elements of mammalian synaptonemal complexes. Chromosoma 2009, 118, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; De La Fuente, R.; Leu, N.A.; Baumann, C.; McLaughlin, K.J.; Wang, P.J. Mouse SYCP2 is required for synaptonemal complex assembly and chromosomal synapsis during male meiosis. J Cell Biol 2006, 173, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Henderson, K.A.; Keeney, S. Synaptonemal complex formation: where does it start? BioEssays 2005, 27, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Page, S.L.; Hawley, R.S. The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol 2004, 20, 525–558. [Google Scholar] [CrossRef]
- Romanienko, P.J.; Camerini-Otero, R.D. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell 2000, 6, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Hassold, T.; Hunt, P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2001, 2, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, A.C.; Berman, J. Shift and adapt: the costs and benefits of karyotype variations. Curr Opin Microbiol 2015, 26, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.P.; Martinez, D.A.; Sakthikumar, S.; Anderson, M.Z.; Berlin, A.; Gujja, S.; Zeng, Q.; Zisson, E.; Wang, J.M.; Greenberg, J.M.; et al. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res 2015, 25, 413–425. [Google Scholar] [CrossRef]
- Kaya, A.; Mariotti, M.; Tyshkovskiy, A.; Zhou, X.; Hulke, M.L.; Ma, S.; Gerashchenko, M.V.; Koren, A.; Gladyshev, V.N. Molecular signatures of aneuploidy-driven adaptive evolution. Nature Communications 2020, 11, 588. [Google Scholar] [CrossRef]
- Morgan, C.; Knight, E.; Bomblies, K. The meiotic cohesin subunit REC8 contributes to multigenic adaptive evolution of autopolyploid meiosis in Arabidopsis arenosa. PLoS Genet 2022, 18, e1010304. [Google Scholar] [CrossRef]
- Matveevsky, S.; Bakloushinskaya, I.; Tambovtseva, V.; Atsaeva, M.; Grishaeva, T.; Bogdanov, A.; Kolomiets, O. Nonhomologous Chromosome Interactions in Prophase I: Dynamics of Bizarre Meiotic Contacts in the Alay Mole Vole Ellobius alaicus (Mammalia, Rodentia). Genes 2022, 13, 2196. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Vara, C.; Paytuví-Gallart, A.; Cuartero, Y.; Le Dily, F.; Garcia, F.; Salvà-Castro, J.; Gómez, H.L.; Julià, E.; Moutinho, C.; Aiese Cigliano, R.; et al. Three-Dimensional Genomic Structure and Cohesin Occupancy Correlate with Transcriptional Activity during Spermatogenesis. Cell Rep 2019, 28, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Alavattam, K.G.; Maezawa, S.; Sakashita, A.; Khoury, H.; Barski, A.; Kaplan, N.; Namekawa, S.H. Attenuated chromatin compartmentalization in meiosis and its maturation in sperm development. Nat Struct Mol Biol 2019, 26, 175–184. [Google Scholar] [CrossRef] [PubMed]
- de Massy, B. Initiation of meiotic recombination: how and where? Conservation and specificities among eukaryotes. Annu Rev Genet 2013, 47, 563–599. [Google Scholar] [CrossRef] [PubMed]
- Lam, I.; Keeney, S. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb Perspect Biol 2014, 7, a016634. [Google Scholar] [CrossRef] [PubMed]
- Bompadre, O.; Andrey, G. Chromatin topology in development and disease. Curr Opin Genet Dev 2019, 55, 32–38. [Google Scholar] [CrossRef]
- Farré, M.; Robinson, T.J.; Ruiz-Herrera, A. An Integrative Breakage Model of genome architecture, reshuffling and evolution: The Integrative Breakage Model of genome evolution, a novel multidisciplinary hypothesis for the study of genome plasticity. Bioessays 2015, 37, 479–488. [Google Scholar] [CrossRef]
- Franke, M.; Gómez-Skarmeta, J.L. An evolutionary perspective of regulatory landscape dynamics in development and disease. Curr Opin Cell Biol 2018, 55, 24–29. [Google Scholar] [CrossRef]
- Veller, C.; Kleckner, N.; Nowak, M.A. A rigorous measure of genome-wide genetic shuffling that takes into account crossover positions and Mendel’s second law. Proc Natl Acad Sci USA 2019, 116, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bourbon, H.-M.; de Massy, B. Functional conservation of Mei4 for meiotic DNA double-strand break formation from yeasts to mice. Genes & development 2010, 24, 1266–1280. [Google Scholar]
- Lange, J.; Yamada, S.; Tischfield, S.E.; Pan, J.; Kim, S.; Zhu, X.; Socci, N.D.; Jasin, M.; Keeney, S. The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 2016, 167, 695–708 e616. [Google Scholar] [CrossRef] [PubMed]
- Rockman, M.V.; Kruglyak, L. Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet 2009, 5, e1000419. [Google Scholar] [CrossRef]
- Barlow, A.L.; Benson, F.E.; West, S.C.; Hulten, M.A. Distribution of the Rad51 recombinase in human and mouse spermatocytes. The EMBO journal 1997, 16, 5207–5215. [Google Scholar] [CrossRef] [PubMed]
- Cole, F.; Kauppi, L.; Lange, J.; Roig, I.; Wang, R.; Keeney, S.; Jasin, M. Homeostatic control of recombination is implemented progressively in mouse meiosis. Nat Cell Biol 2012, 14, 424–430. [Google Scholar] [CrossRef]
- Myers, S.; Bowden, R.; Tumian, A.; Bontrop, R.E.; Freeman, C.; MacFie, T.S.; McVean, G.; Donnelly, P. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 2010, 327, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Smagulova, F.; Gregoretti, I.V.; Brick, K.; Khil, P.; Camerini-Otero, R.D.; Petukhova, G.V. Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature 2011, 472, 375–378. [Google Scholar] [CrossRef]
- Cappelletti, E.; Piras, F.M.; Badiale, C.; Bambi, M.; Santagostino, M.; Vara, C.; Masterson, T.A.; Sullivan, K.F.; Nergadze, S.G.; Ruiz-Herrera, A.; et al. CENP-A binding domains and recombination patterns in horse spermatocytes. Sci Rep 2019, 9, 15800. [Google Scholar] [CrossRef]
- Brick, K.; Smagulova, F.; Khil, P.; Camerini-Otero, R.D.; Petukhova, G.V. Genetic recombination is directed away from functional genomic elements in mice. Nature 2012, 485, 642–645. [Google Scholar] [CrossRef]
- Parvanov, E.D.; Petkov, P.M.; Paigen, K. Prdm9 controls activation of mammalian recombination hotspots. Science 2010, 327, 835. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zickler, D.; Kleckner, N.; Zhang, L. Meiotic crossover patterns: obligatory crossover, interference and homeostasis in a single process. Cell cycle 2015, 14, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Zickler, D.; Kleckner, N. Meiotic chromosomes: integrating structure and function. Annual review of genetics 1999, 33, 603. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Caballero, C.; Herrán, Y.; Sánchez-Martín, M.; Suja, J.Á.; Barbero, J.L.; Llano, E.; Pendás, A.M. Identification and molecular characterization of the mammalian α-kleisin RAD21L. Cell cycle 2011, 10, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Baudat, F.; Buard, J.; Grey, C.; Fledel-Alon, A.; Ober, C.; Przeworski, M.; Coop, G.; De Massy, B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 2010, 327, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Mihola, O.; Trachtulec, Z.; Vlcek, C.; Schimenti, J.C.; Forejt, J. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 2009, 323, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Pratto, F.; Brick, K.; Khil, P.; Smagulova, F.; Petukhova, G.V.; Camerini-Otero, R.D. Recombination initiation maps of individual human genomes. Science 2014, 346, 1256442. [Google Scholar] [CrossRef] [PubMed]
- Smagulova, F.; Brick, K.; Pu, Y.; Camerini-Otero, R.D.; Petukhova, G.V. The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes & development 2016, 30, 266–280. [Google Scholar]
- Oliver, P.L.; Goodstadt, L.; Bayes, J.J.; Birtle, Z.; Roach, K.C.; Phadnis, N.; Beatson, S.A.; Lunter, G.; Malik, H.S.; Ponting, C.P. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS genetics 2009, 5, e1000753. [Google Scholar] [CrossRef]
- Davies, B.; Hatton, E.; Altemose, N.; Hussin, J.G.; Pratto, F.; Zhang, G.; Hinch, A.G.; Moralli, D.; Biggs, D.; Diaz, R. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 2016, 530, 171–176. [Google Scholar] [CrossRef]
- Myers, S.; Freeman, C.; Auton, A.; Donnelly, P.; McVean, G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nature genetics 2008, 40, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Duret, L.; Galtier, N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annual review of genomics and human genetics 2009, 10, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Janoušek, V.; Munclinger, P.; Wang, L.; Teeter, K.C.; Tucker, P.K. Functional organization of the genome may shape the species boundary in the house mouse. Molecular biology and evolution 2015, 32, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Baker, Z.; Schumer, M.; Haba, Y.; Bashkirova, L.; Holland, C.; Rosenthal, G.G.; Przeworski, M. Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. Elife 2017, 6, e24133. [Google Scholar] [CrossRef] [PubMed]
- Capilla, L.; Medarde, N.; Alemany-Schmidt, A.; Oliver-Bonet, M.; Ventura, J.; Ruiz-Herrera, A. Genetic recombination variation in wild Robertsonian mice: on the role of chromosomal fusions and Prdm9 allelic background. Proceedings of the Royal Society B: Biological Sciences 2014, 281, 20140297. [Google Scholar]
- Faria, R.; Navarro, A. Chromosomal speciation revisited: rearranging theory with pieces of evidence. Trends in ecology & evolution 2010, 25, 660–669. [Google Scholar]
- Feulner, P.G.; Chain, F.J.; Panchal, M.; Huang, Y.; Eizaguirre, C.; Kalbe, M.; Lenz, T.L.; Samonte, I.E.; Stoll, M.; Bornberg-Bauer, E. Genomics of divergence along a continuum of parapatric population differentiation. PLoS genetics 2015, 11, e1004966. [Google Scholar] [CrossRef]
- Sodeland, M.; Jorde, P.E.; Lien, S.; Jentoft, S.; Berg, P.R.; Grove, H.; Kent, M.P.; Arnyasi, M.; Olsen, E.M.; Knutsen, H. “Islands of Divergence” in the Atlantic cod genome represent polymorphic chromosomal rearrangements. Genome biology and evolution 2016, 8, 1012–1022. [Google Scholar] [CrossRef]
- Ullastres, A.; Farré, M.; Capilla, L.; Ruiz-Herrera, A. Unraveling the effect of genomic structural changes in the rhesus macaque-implications for the adaptive role of inversions. BMC genomics 2014, 15, 1–13. [Google Scholar] [CrossRef]
- Dumas, D.; Britton-Davidian, J. Chromosomal rearrangements and evolution of recombination: comparison of chiasma distribution patterns in standard and Robertsonian populations of the house mouse. Genetics 2002, 162, 1355–1366. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.-X. γ-H2AX-a novel biomarker for DNA double-strand breaks. In vivo 2008, 22, 305–309. [Google Scholar]
- Murakami, H.; Keeney, S. Regulating the formation of DNA double-strand breaks in meiosis. Genes & development 2008, 22, 286–292. [Google Scholar]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: mechanisms and new insights into an age-old problem. Nature Reviews Genetics 2012, 13, 493–504. [Google Scholar] [CrossRef]
- Sasaki, M.; Lange, J.; Keeney, S. Genome destabilization by homologous recombination in the germ line. Nature reviews Molecular cell biology 2010, 11, 182–195. [Google Scholar] [CrossRef]
- Vara, C.; Paytuví-Gallart, A.; Cuartero, Y.; Álvarez-González, L.; Marín-Gual, L.; Garcia, F.; Florit-Sabater, B.; Capilla, L.; Sanchéz-Guillén, R.A.; Sarrate, Z. The impact of chromosomal fusions on 3D genome folding and recombination in the germ line. Nature communications 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Britton-Davidian, J.; Catalan, J.; da Graça Ramalhinho, M.; Ganem, G.; Auffray, J.-C.; Capela, R.; Biscoito, M.; Searle, J.B.; da Luz Mathias, M. Rapid chromosomal evolution in island mice. Nature 2000, 403, 158–158. [Google Scholar] [CrossRef] [PubMed]
- Dutrillaux, B. Chromosomal evolution in primates: tentative phylogeny from Microcebus murinus (Prosimian) to man. Human genetics 1979, 48, 251–314. [Google Scholar] [CrossRef]
- Yunis, J.J.; Sawyer, J.R.; Dunham, K. The striking resemblance of high-resolution G-banded chromosomes of man and chimpanzee. Science 1980, 208, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Newman, T.L.; Tuzun, E.; Morrison, V.A.; Hayden, K.E.; Ventura, M.; McGrath, S.D.; Rocchi, M.; Eichler, E.E. A genome-wide survey of structural variation between human and chimpanzee. Genome research 2005, 15, 1344–1356. [Google Scholar] [CrossRef] [PubMed]
- Pevzner, P.; Tesler, G. Human and mouse genomic sequences reveal extensive breakpoint reuse in mammalian evolution. Proceedings of the National Academy of Sciences 2003, 100, 7672–7677. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.Y.; Harshman, L.; Nelson, B.J.; Penn, O.; Cantsilieris, S.; Huddleston, J.; Antonacci, F.; Penewit, K.; Denman, L.; Raja, A. The evolution and population diversity of human-specific segmental duplications. Nature ecology & evolution 2017, 1, 1–10. [Google Scholar]
- Marín-Gual, L.; González-Rodelas, L.; Pujol, G.; Vara, C.; Martín-Ruiz, M.; Berríos, S.; Fernández-Donoso, R.; Pask, A.; Renfree, M.B.; Page, J.; et al. Strategies for meiotic sex chromosome dynamics and telomeric elongation in Marsupials. PLoS Genet 2022, 18, e1010040. [Google Scholar] [CrossRef]
- Waters, P.D.; Patel, H.R.; Ruiz-Herrera, A.; Álvarez-González, L.; Lister, N.C.; Simakov, O.; Ezaz, T.; Kaur, P.; Frere, C.; Grützner, F.; et al. Microchromosomes are building blocks of bird, reptile, and mammal chromosomes. Proc Natl Acad Sci USA 2021, 118. [Google Scholar] [CrossRef]
- Comai, L. The advantages and disadvantages of being polyploid. Nat Rev Genet 2005, 6, 836–846. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat Rev Genet 2009, 10, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat Rev Genet 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Crow, K.D.; Wagner, G.P. What is the role of genome duplication in the evolution of complexity and diversity? Molecular biology and evolution 2005, 23, 887–892. [Google Scholar] [CrossRef]
- Kellis, M.; Birren, B.W.; Lander, E.S. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 2004, 428, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H.; Shields, D.C. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387, 708–713. [Google Scholar] [CrossRef]
- Mottes, F.; Villa, C.; Osella, M.; Caselle, M. The impact of whole genome duplications on the human gene regulatory networks. PLoS Comput Biol 2021, 17, e1009638. [Google Scholar] [CrossRef]
- Ohno, S. The enormous diversity in genome sizes of fish as a reflection of natureˈs extensive experiments with gene duplication. Transactions of the American Fisheries Society 1970, 99, 120–130. [Google Scholar] [CrossRef]
- D’Antonio, M.; Ciccarelli, F.D. Integrated analysis of recurrent properties of cancer genes to identify novel drivers. Genome biology 2013, 14, 1–17. [Google Scholar] [CrossRef]
- Singh, P.P.; Arora, J.; Isambert, H. Identification of ohnolog genes originating from whole genome duplication in early vertebrates, based on synteny comparison across multiple genomes. PLoS computational biology 2015, 11, e1004394. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; McLysaght, A. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proceedings of the National Academy of Sciences 2010, 107, 9270–9274. [Google Scholar] [CrossRef] [PubMed]
- Acharya, D.; Ghosh, T.C. Global analysis of human duplicated genes reveals the relative importance of whole-genome duplicates originated in the early vertebrate evolution. BMC genomics 2016, 17, 1–14. [Google Scholar] [CrossRef]
- Huminiecki, L.; Heldin, C.H. 2R and remodeling of vertebrate signal transduction engine. BMC biology 2010, 8, 1–21. [Google Scholar] [CrossRef]
- Cai, H.; Kumar, N.; Bagheri, H.C.; von Mering, C.; Robinson, M.D.; Baudis, M. Chromothripsis-like patterns are recurring but heterogeneously distributed features in a survey of 22,347 cancer genome screens. BMC genomics 2014, 15, 1–13. [Google Scholar] [CrossRef]
- Weckselblatt, B.; Rudd, M.K. Human structural variation: mechanisms of chromosome rearrangements. Trends in Genetics 2015, 31, 587–599. [Google Scholar] [CrossRef] [PubMed]
- De Pagter, M.S.; Van Roosmalen, M.J.; Baas, A.F.; Renkens, I.; Duran, K.J.; Van Binsbergen, E.; Tavakoli-Yaraki, M.; Hochstenbach, R.; Van Der Veken, L.T.; Cuppen, E. Chromothripsis in healthy individuals affects multiple protein-coding genes and can result in severe congenital abnormalities in offspring. The American Journal of Human Genetics 2015, 96, 651–656. [Google Scholar] [CrossRef]
- Pellestor, F.; Gatinois, V. Chromoanagenesis: a piece of the macroevolution scenario. Molecular Cytogenetics 2020, 13, 1–9. [Google Scholar] [CrossRef]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Cooper, D.N. Structural divergence between the human and chimpanzee genomes. Human genetics 2007, 120, 759–778. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Bourque, G. Recovering genome rearrangements in the mammalian phylogeny. Genome research 2009, 19, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Farré, M.; Bosch, M.; López-Giráldez, F.; Ponsà, M.; Ruiz-Herrera, A. Assessing the role of tandem repeats in shaping the genomic architecture of great apes. PLoS One 2011, 6, e27239. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Sandig, C.; Goidts, V.; Hameister, H. Breakpoint analysis of the pericentric inversion between chimpanzee chromosome 10 and the homologous chromosome 12 in humans. Cytogenetic and Genome Research 2005, 108, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.; Harris, R.A.; Mootnick, A.R.; Milosavljevic, A.; Martin, D.I.; Rocchi, M.; Capozzi, O.; Archidiacono, N.; Konkel, M.K.; Walker, J.A. Centromere remodeling in Hoolock leuconedys (Hylobatidae) by a new transposable element unique to the gibbons. Genome biology and evolution 2012, 4, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA evolution: old ideas, new approaches. Current opinion in genetics & development 2018, 49, 70–78. [Google Scholar]
- Ferreira, D.; Meles, S.; Escudeiro, A.; Mendes-da-Silva, A.; Adega, F.; Chaves, R. Satellite non-coding RNAs: the emerging players in cells, cellular pathways and cancer. Chromosome Research 2015, 23, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Larracuente, A.M. The organization and evolution of the Responder satellite in species of the Drosophila melanogaster group: dynamic evolution of a target of meiotic drive. BMC evolutionary biology 2014, 14, 1–12. [Google Scholar] [CrossRef]
- Nazaryan-Petersen, L.; Bertelsen, B.; Bak, M.; Jønson, L.; Tommerup, N.; Hancks, D.C.; Tümer, Z. Germline chromothripsis driven by L1-mediated retrotransposition and Alu/Alu homologous recombination. Human mutation 2016, 37, 385–395. [Google Scholar] [CrossRef]
- Klein, S.J.; O’Neill, R.J. Transposable elements: genome innovation, chromosome diversity, and centromere conflict. Chromosome Research 2018, 26, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.N.; Vandewege, M.W.; Ray, D.A. Mammalian transposable elements and their impacts on genome evolution. Chromosome Research 2018, 26, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.; Alan Harris, R.; Gnerre, S.; Veeramah, K.R.; Lorente-Galdos, B.; Huddleston, J.; Meyer, T.J.; Herrero, J.; Roos, C.; Aken, B. Gibbon genome and the fast karyotype evolution of small apes. Nature 2014, 513, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.J.; Held, U.; Nevonen, K.A.; Klawitter, S.; Pirzer, T.; Carbone, L.; Schumann, G.G. The flow of the gibbon LAVA element is facilitated by the LINE-1 retrotransposition machinery. Genome biology and evolution 2016, 8, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Louzada, S.; Lopes, M.; Ferreira, D.; Adega, F.; Escudeiro, A.; Gama-Carvalho, M.; Chaves, R. Decoding the role of satellite DNA in genome architecture and plasticity—An evolutionary and clinical affair. Genes 2020, 11, 72. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: models, mechanisms and implications. Nature reviews genetics 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Bailey, S.M.; Okuka, M.; Muñoz, P.; Li, C.; Zhou, L.; Wu, C.; Czerwiec, E.; Sandler, L.; Seyfang, A.; et al. Telomere lengthening early in development. Nat Cell Biol 2007, 9, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Reig-Viader, R.; Brieño-Enríquez, M.A.; Khoriauli, L.; Toran, N.; Cabero, L.; Giulotto, E.; Garcia-Caldés, M.; Ruiz-Herrera, A. Telomeric repeat-containing RNA and telomerase in human fetal oocytes. Hum Reprod 2013, 28, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Reig-Viader, R.; Garcia-Caldés, M.; Ruiz-Herrera, A. Telomere homeostasis in mammalian germ cells: a review. Chromosoma 2016, 125, 337–351. [Google Scholar] [CrossRef]
- Reig-Viader, R.; Vila-Cejudo, M.; Vitelli, V.; Buscà, R.; Sabaté, M.; Giulotto, E.; Caldés, M.G.; Ruiz-Herrera, A. Telomeric repeat-containing RNA (TERRA) and telomerase are components of telomeres during mammalian gametogenesis. Biol Reprod 2014, 90, 103. [Google Scholar] [CrossRef]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Ingles, E.D.; Deakin, J.E. Telomeres, species differences, and unusual telomeres in vertebrates: presenting challenges and opportunities to understanding telomere dynamics. Aims Genetics 2016, 3, 001–024. [Google Scholar] [CrossRef]
- Hoang, S.M.; O’Sullivan, R.J. Alternative Lengthening of Telomeres: Building Bridges To Connect Chromosome Ends. Trends Cancer 2020, 6, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. Linking Telomere Regulation to Stem Cell Pluripotency. Trends Genet 2017, 33, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Canfield, P.J.; Hartley, W.J.; Reddacliff, G.L. Spontaneous proliferations in Australian marsupials--a survey and review. 2. Dasyurids and bandicoots. J Comp Pathol 1990, 103, 147–158. [Google Scholar] [CrossRef]
- Cavazza, T.; Takeda, Y.; Politi, A.Z.; Aushev, M.; Aldag, P.; Baker, C.; Choudhary, M.; Bucevicius, J.; Lukinavicius, G.; Elder, K.; et al. Parental genome unification is highly error-prone in mammalian embryos. Cell 2021, 184, 2860–2877 e2822. [Google Scholar] [CrossRef]
- Nonaka, T.; Takahashi, M.; Nonaka, C.; Enomoto, T.; Takakuwa, K. The analysis of chromosomal abnormalities in patients with recurrent pregnancy loss, focusing on the prognosis of patients with inversion of chromosome (9). Reprod Med Biol 2019, 18, 296–301. [Google Scholar] [CrossRef]
- Webster, A.L.H.; Sanders, M.A.; Patel, K.; Dietrich, R.; Noonan, R.J.; Lach, F.P.; White, R.R.; Goldfarb, A.; Hadi, K.; Edwards, M.M.; et al. Genomic signature of Fanconi anaemia DNA repair pathway deficiency in cancer. Nature 2022, 612, 495–502. [Google Scholar] [CrossRef]
- Webster, A.L.H.; Sanders, M.A.; Patel, K.; Dietrich, R.; Noonan, R.J.; Lach, F.P.; White, R.R.; Goldfarb, A.; Hadi, K.; Edwards, M.M.; et al. Genomic signature of Fanconi anaemia DNA repair pathway deficiency in cancer. Nature 2022. [Google Scholar] [CrossRef]
- Gregory, J.J., Jr.; Wagner, J.E.; Verlander, P.C.; Levran, O.; Batish, S.D.; Eide, C.R.; Steffenhagen, A.; Hirsch, B.; Auerbach, A.D. Somatic mosaicism in Fanconi anemia: evidence of genotypic reversion in lymphohematopoietic stem cells. Proc Natl Acad Sci USA 2001, 98, 2532–2537. [Google Scholar] [CrossRef]
- Gross, M.; Hanenberg, H.; Lobitz, S.; Friedl, R.; Herterich, S.; Dietrich, R.; Gruhn, B.; Schindler, D.; Hoehn, H. Reverse mosaicism in Fanconi anemia: natural gene therapy via molecular self-correction. Cytogenet Genome Res 2002, 98, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Werling, D.M.; Brand, H.; An, J.Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nature genetics 2018, 50, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Kleiber, M.L.; Whitney, J.; Maile, M.S.; Hong, O.; Chapman, T.R.; Tan, S.; Tandon, P.; et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 2018, 360, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Howrigan, D.P.; Merico, D.; Thiruvahindrapuram, B.; Wu, W.; Greer, D.S.; Antaki, D.; Shetty, A.; Holmans, P.A.; Pinto, D.; et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nature genetics 2017, 49, 27–35. [Google Scholar] [CrossRef]
- van Belzen, I.A.E.M.; Cai, C.; van Tuil, M.; Badloe, S.; Strengman, E.; Janse, A.; Verwiel, E.T.; van der Leest, D.F.M.; Kester, L.; Molenaar, J.J.; et al. Systematic discovery of gene fusions in pediatric cancer by integrating RNA-seq and WGS. bioRxiv, 2008; 2021.2008.2031.458342. [Google Scholar] [CrossRef]
- Schuy, J.; Grochowski, C.M.; Carvalho, C.M.B.; Lindstrand, A. Complex genomic rearrangements: an underestimated cause of rare diseases. Trends Genet 2022, 38, 1134–1146. [Google Scholar] [CrossRef]
Figure 1.
Examples of some structural variants and chromosomal anomalies.
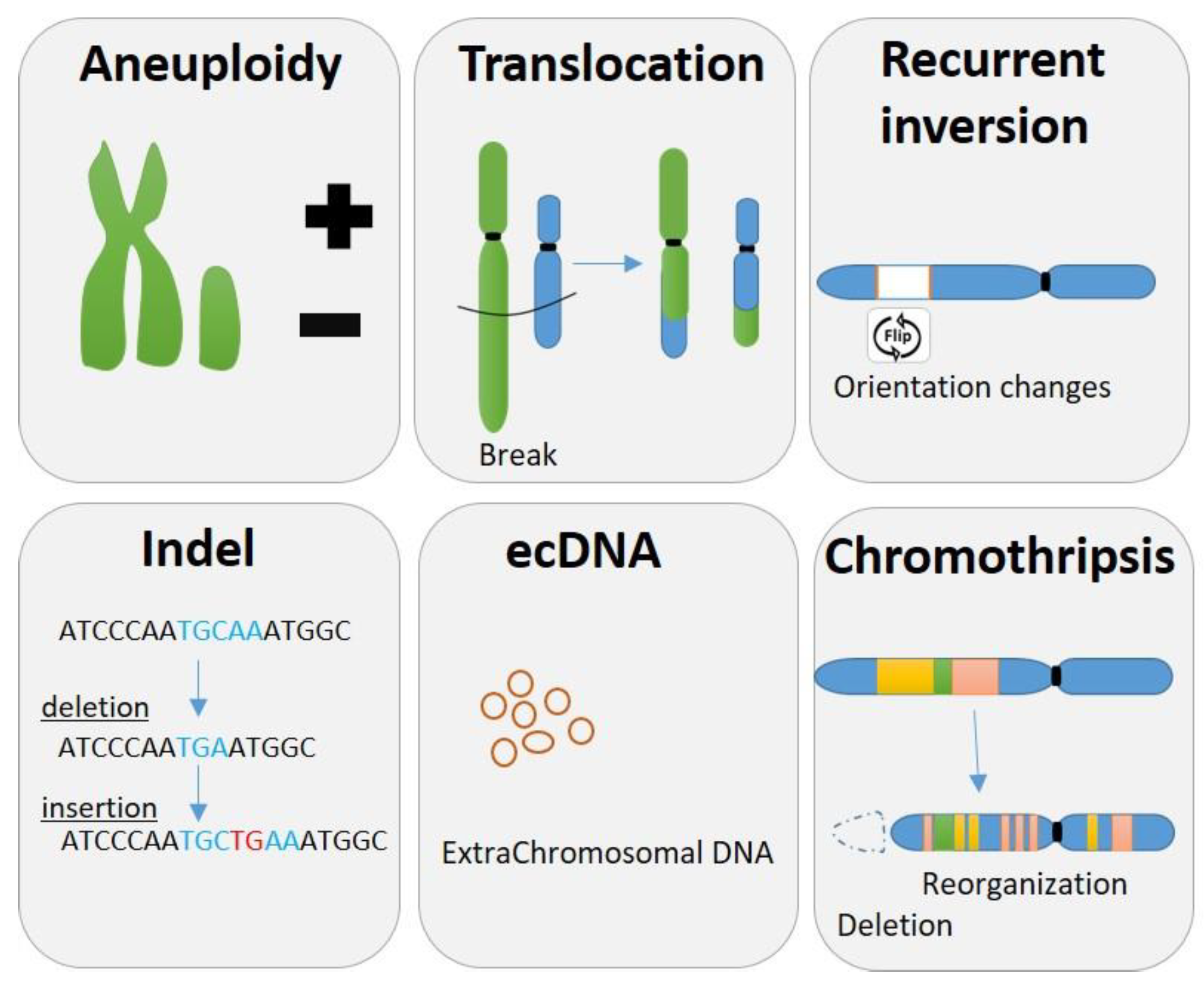
Figure 3.
Reconstructed evolutionary history of karyotypes from Chordata to Amniota. A simplified species tree of the Chordata is shown, with WGD events depicted by red stars. The eight lineages represented from left to right are mammals, birds, teleost fish, holostocean fish (gar), cartilaginous fish, cyclostomes (lamprey, hagfish), tunicates (ciona), and cephalochordates (amphioxus). Important events of chromosomal rearrangement such as translocation and fusion are shown (green chromosome with blue arm translocation). Adapted from [26].
Figure 3.
Reconstructed evolutionary history of karyotypes from Chordata to Amniota. A simplified species tree of the Chordata is shown, with WGD events depicted by red stars. The eight lineages represented from left to right are mammals, birds, teleost fish, holostocean fish (gar), cartilaginous fish, cyclostomes (lamprey, hagfish), tunicates (ciona), and cephalochordates (amphioxus). Important events of chromosomal rearrangement such as translocation and fusion are shown (green chromosome with blue arm translocation). Adapted from [26].
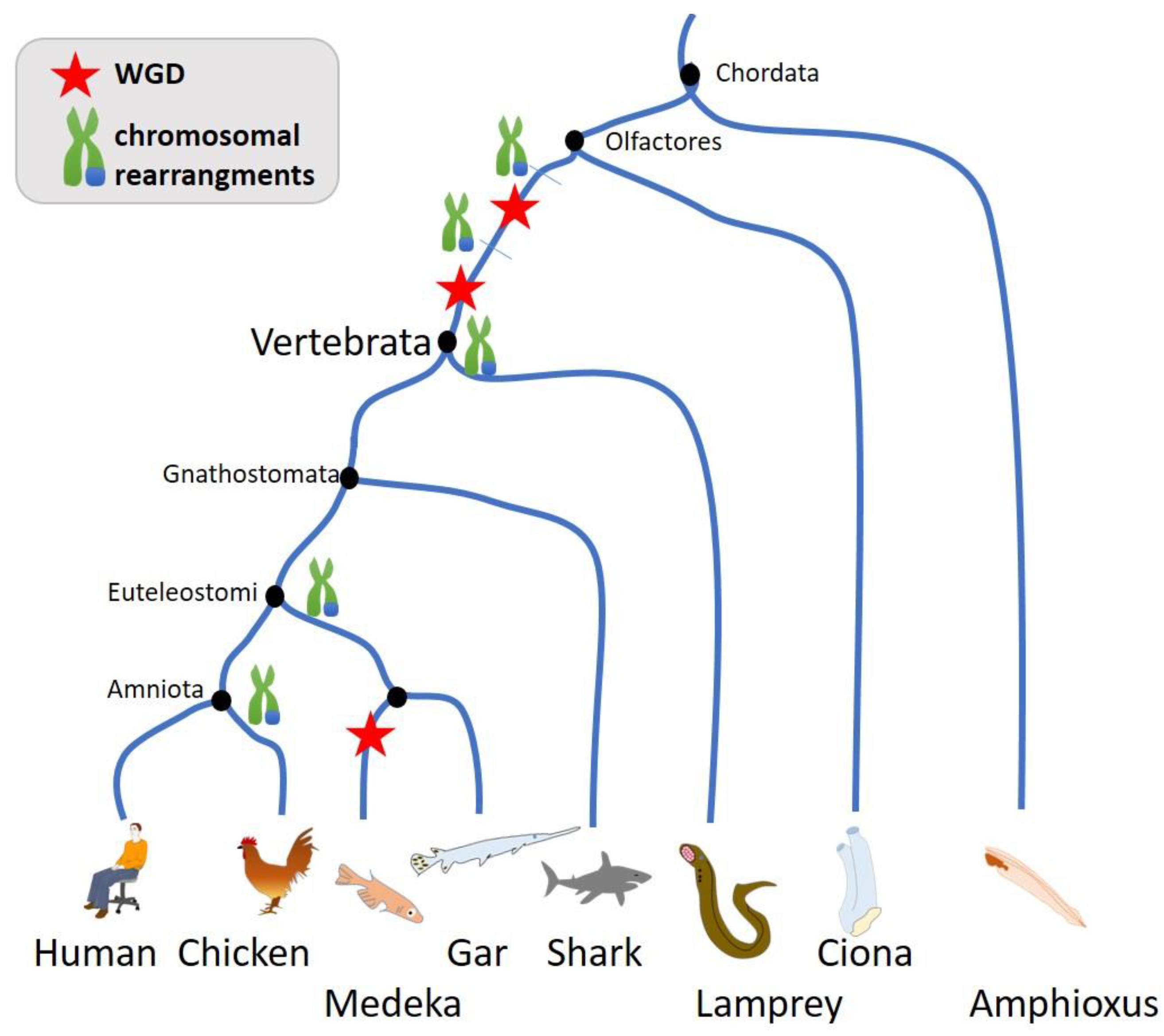
Figure 4.
Meiosis, key processes and factors involved. (A) Different phases and key processes for Meiosis I and Meiosis II, from meiotic replication to the formation of haploid gametes. The use of only one pair of homologous chromosomes (blue and green) and their replication to sister chromatids (purple and light green) have been used for simplification. In Anaphase I, homologous chromosomes segregates, and in Anaphase II, sisters are pulled apart. Key processes during Prophase I (dashed line) are detailed in B, where different phases are represented. (B) Along meiotic DNA replication, cohesin complexes are loaded leading to SCC (sister chromatid cohesion). From leptonema to pachytene, sister chromatids begin to condense along a proteinaceous structure formed by cohesin complexes, as REC8, and axial elements, as HORMAD1. Meiotic chromosomes are organized as DNA loops anchored to the axial elements, where proteins involved in the formation of DSBs, as PRDM9 and SPO11, are recruited. These breaks are repaired where its resection leading to 3′ ends is key, covered by RPA and then by the recombinases RAD51 and DMC1, at zygonema. These two proteins catalyse strand invasion and will help for the homologous chromosomes’ recognition. In the meantime, that homologous recombination takes place, synapsis of homologous chromosomes occurs, with the polymerization of the central elements SYPs between both axials, leading to the Synaptonemal Complex (SC) formation. By pachynema, SC is completed and recombination intermediates can be resolved either as COs or non-COs. The resolution of COs is mediated by MLH1 proteins that will lead ultimately to chiasma formation. (C) cohesin occupancy mediates a role in transcription regulation by bringing two DNA regions together. (D) Schematic representation for distinct chromosomal localisation pattern for cohesin mediating: SCC between sisters, loops and TADs formation, or transcription regulation. (E) The length of chromosomal axes and the size of DNA loops are inversely related, resulting in different numbers of DSBs formed by the protein SPO11. Figure adapted from [87].
Figure 4.
Meiosis, key processes and factors involved. (A) Different phases and key processes for Meiosis I and Meiosis II, from meiotic replication to the formation of haploid gametes. The use of only one pair of homologous chromosomes (blue and green) and their replication to sister chromatids (purple and light green) have been used for simplification. In Anaphase I, homologous chromosomes segregates, and in Anaphase II, sisters are pulled apart. Key processes during Prophase I (dashed line) are detailed in B, where different phases are represented. (B) Along meiotic DNA replication, cohesin complexes are loaded leading to SCC (sister chromatid cohesion). From leptonema to pachytene, sister chromatids begin to condense along a proteinaceous structure formed by cohesin complexes, as REC8, and axial elements, as HORMAD1. Meiotic chromosomes are organized as DNA loops anchored to the axial elements, where proteins involved in the formation of DSBs, as PRDM9 and SPO11, are recruited. These breaks are repaired where its resection leading to 3′ ends is key, covered by RPA and then by the recombinases RAD51 and DMC1, at zygonema. These two proteins catalyse strand invasion and will help for the homologous chromosomes’ recognition. In the meantime, that homologous recombination takes place, synapsis of homologous chromosomes occurs, with the polymerization of the central elements SYPs between both axials, leading to the Synaptonemal Complex (SC) formation. By pachynema, SC is completed and recombination intermediates can be resolved either as COs or non-COs. The resolution of COs is mediated by MLH1 proteins that will lead ultimately to chiasma formation. (C) cohesin occupancy mediates a role in transcription regulation by bringing two DNA regions together. (D) Schematic representation for distinct chromosomal localisation pattern for cohesin mediating: SCC between sisters, loops and TADs formation, or transcription regulation. (E) The length of chromosomal axes and the size of DNA loops are inversely related, resulting in different numbers of DSBs formed by the protein SPO11. Figure adapted from [87].
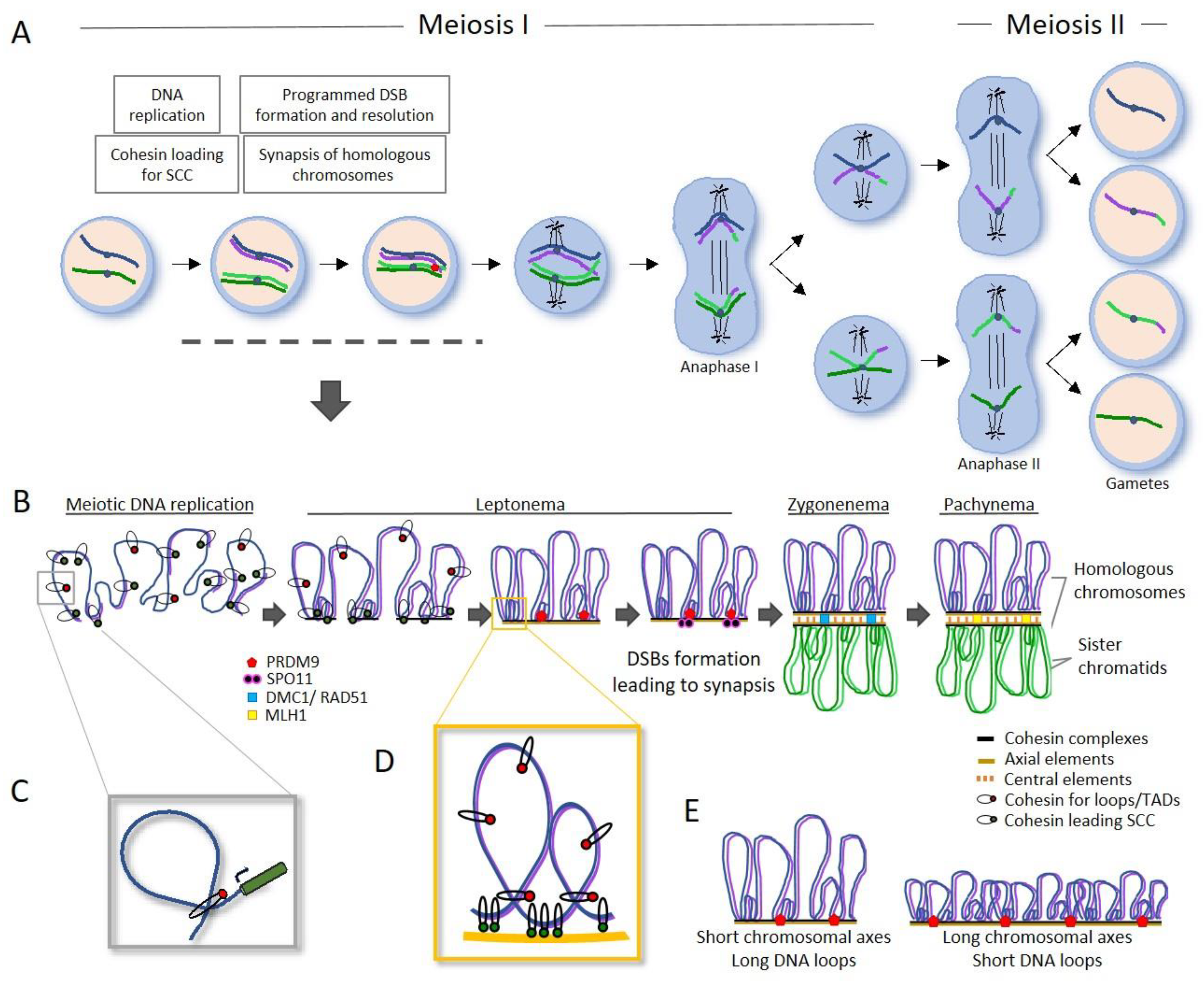
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



