
Submitted:
03 February 2023
Posted:
10 February 2023
You are already at the latest version
Abstract
Cardiopulmonary complications remain the major cause of mortality despite newer therapies and improvements in lifespan of patients with sickle cell disease (SCD). Inflammation has been identified as a major risk modifier in the pathogenesis of SCD associated cardiopulmonary complications in recent mechanistic and observational studies. In this review, we discuss recent cellular and molecular mechanisms of cardiopulmonary complications in SCD and summarize the most recent evidence from clinical and laboratory studies. We emphasize the role of inflammation in the onset and progression of these complications to better understand the underlying pathobiological processes. We also discuss future basic and translational research in addressing questions about the complex role of inflammation in the development of SCD cardiopulmonary complications, which may lead to promising therapies and reduce morbidity and mortality in this vulnerable population.
Keywords:
Sickle cell disease
; cardiopulmonary complications
; inflammation
; acute chest syndrome
; cardiac hypertrophy
; cardiac fibrosis
; diastolic dysfunction
; pulmonary hypertension
Introduction
Sickle cell disease (SCD) is the most common monogenic blood disorder, affecting approximately 100,000 Americans and millions more worldwide [1,2]. Cardiopulmonary complications are a major cause of morbidity and mortality in SCD, accounting for 32-70% of deaths [3,4,5]. Several pathophysiological processes, including anemia, hemolysis, endothelial dysfunction, and ventricular remodeling, may contribute to cardiopulmonary complications in SCD [3,4,5]. Although the etiology of cardiopulmonary complications in SCD is somewhat different from that in the general population, there are similarities in the cellular and molecular mechanisms that underlie the pathogenesis in both scenario, and that are beginning to gain prominence. Accumulating evidence has long identified chronic low-grade inflammation as a risk factor for the progression of myocardial infarction, ventricular hypertrophy, cardiac fibrosis, diastolic dysfunction, and pulmonary hypertension in the general population [6,7,8,9,10]. Recent mechanistic and observational studies on cardiopulmonary complications of SCD implicate inflammation as a major player in the onset and progression of cardiopulmonary complications in SCD [11,12,13,14,15,16]. These include several studies in animals and humans on the development of acute chest syndrome (ACS), cardiac hypertrophy, pulmonary hypertension (PH), cardiac fibrosis, and diastolic dysfunction [11,12,13,14,15]. These studies consistently suggested inflammatory pathways as a vital unifying mechanism that accompanies the structural and functional changes that occur at the onset and progression of these complications. Thus, integrating therapies that balance the pro-inflammatory and anti-inflammatory processes contributing to the chronic inflammatory state in SCD may provide opportunities for novel therapies that could be easily incorporated into the existing treatment options available to SCD patients. The recent advances in cellular and molecular mechanisms of cardiopulmonary complications of SCD, along with the complex interplay between inflammation and the unique cardiac pathology of SCD such as acute chest syndrome, pulmonary hypertension, diastolic dysfunction, and cardiac hypertrophy, are described in this review (Figure 1).
Inflammation and acute chest syndrome.
Acute chest syndrome (ACS) is a pulmonary complication of SCD and the second leading cause of mortality and morbidity in both adults and children with SCD [17]. It is defined as the presence of fever and/or new respiratory symptoms such as cough, chest pain and presence of a new pulmonary infiltrate on chest X-ray [17]. Risk factors for ACS include younger age, severe SCD genotypes (SS or Sβ0 thalassemia), lower fetal hemoglobin concentrations, inflammation, higher steady-state white blood cell counts, history of asthma, and tobacco smoke exposure [18,19]. The major causes known to trigger ACS include respiratory infection, pulmonary infarction or fat embolism, however no specific cause can be found in up to 30% of cases [18]. At a cellular level, an inciting trigger such as an infection permits increased adhesion of leukocytes (neutrophils) to the lung microvasculature, generation of cytokines, coupled with interactions with other cellular components such as platelets. This results in local hypoxemia and changes in rheology of the red blood cells (RBCs). This further facilitates interactions between RBCs, vascular endothelium and leukocytes resulting in increased oxidative stress, vaso-occlusion and tissue hypoxia. These events in turn result in additional recruitment of leukocytes and other cellular components to site thus amplifying the inflammatory cascade, resulting in a “vicious” cycle of lung injury and hypoxemia [20,21].
Evidence for heightened inflammation in the pulmonary microenvironment, during ACS comes from human studies which show that children with ACS have high levels of IL-6, IL-8, CCL2, and CCL3 in their sputum [12]. These cytokines, particularly CCL2 and CCL3, have been shown to recruit leukocytes, particularly neutrophils, via upregulation of platelet activating factor (PAF) and leukotriene B-4 (LTB4). The neutrophils firmly adhere to the endothelium and become activated as assessed by shedding of CD62L and upregulation of CD11b [22]. Upregulation of CD11b in arrested leukocytes enables their interaction with GPIbα expressed on platelets [23]. Arrested neutrophils can also interact with platelets via PSGL-1 on neutrophils binding to P-selectin on platelets. This is evidenced by autopsy studies which show presence of large neutrophil-platelet aggregates and platelet laden aggregates in pulmonary vasculature in patients with ACS [20,24]. Indeed, preclinical studies that inhibit P-selectin and GPIbα interactions show fewer leukocyte-platelet aggregates [25] highlighting the importance of neutrophil and platelet heterotypic interactions in pathogenesis of ACS. Furthermore, a study by Ghosh et al., in Townes sickle cell mouse model showed that P-selectin in both platelet and endothelium compartments played a dominant role in promoting heme-induced ACS in SCD [26].
Hemolysis is a pathological feature of SCD that releases free hemoglobin and heme into the circulation due to RBC sickling and lysis, leading to the activation of inflammatory signaling pathways and vascular inflammation [27,28,29,30]. The release of free heme and cell-free hemoglobin also results in activation of neutrophils and generation of neutrophil extracellular traps (NETs) [31], iron-based generation of reactive oxidative species (ROS) with subsequent oxidization of membrane lipids [32], depletion of nitric oxide [33], and endothelial cytoskeleton remodeling resulting in barrier dysfunction [34]. Furthermore, plasma free heme and other markers of hemolysis have been associated with increased odds of developing ACS in children with SCD [35]. Additionally, a mutation in the heme-oxygenase 1 (HMOX1) short (GT)n repeat promoter that confers stronger inducibility of HMOX-1, the rate-limiting enzyme that degrades heme, was associated with a reduction in the rate of hospitalization for ACS in children with SCD [36]. These studies were validated in both the Townes and Berkeley SCD mouse models using extracellular heme as a trigger for ACS. Heme exposure causes respiratory failure due to rapid hypoxemia and death, mimicking some of the events associated with ACS in SCD patients [37]. Treatment of SCD mice with D3T (3H-1,2-dithiole-3-thione), an activator of nuclear-factor erythroid 2 like 2 (NRF2), which controls HMOX1 expression, reduced lethality in a model of heme-induced ACS in SS mice [38]. Additionally, treatment with hemopexin, the plasma heme binding protein, abrogates lung injury and mortality in Chlorine (Cl2) inhalation model of inducing ACS [39]. These studies suggest that therapies that target the product (heme) or molecular consequence(s) of hemolytic pathways may offer protection from ACS in SCD.
Inflammation and pulmonary hypertension
Pulmonary hypertension (PH) is an independent risk factor for early death in SCD patients [40]. Its estimated prevalence, as assessed by right heart catheterization (RHC), is about 6-10% [41], although this estimate relied on an older definition used to diagnose PH. Per the most recent guidelines, PH is now defined as mean pulmonary artery pressure of >20 mm Hg in conjunction with pulmonary artery wedge pressure of ≤15 mm Hg and a pulmonary vascular resistance (PVR) of ≥3 Wood units (WU). A diagnosis of isolated postcapillary PH is made when PVR is <3 WU, whereas a PVR of ≥3 WU is supportive of pre-capillary PH [42]. In SCD, precapillary, post capillary and pulmonary thromboembolic PH or a combination can exist. Risk factors for PH include chronic intravascular hemolysis, pulmonary thrombosis or embolism, and heart failure [43,44].
The development of PH in SCD is complex and involves pulmonary vascular endothelial dysfunction, smooth muscle cell (SMC) proliferation and resistance to nitric oxide (NO) adventitial fibroblast accumulation, and inflammation. Interestingly, one of the unique features of PH in SCD is the presence of iron in pulmonary macrophages, a feature that is not seen in other forms of PH. An autopsy study of lung samples from SCD patients with PH and RV failure found peripheral monocytes and macrophages accumulating in the perivascular and alveolar regions of the lungs [45]. These macrophages had extensive iron accumulation concomitantly with the expression of HMOX1, ET-1, and IL-6 [45]. This suggests that, in pathological diseases with hemolysis such as SCD, circulating immune cells may be recruited into the lungs for heme degradation. However, this immune response may become maladaptive over time, as accumulated iron may contribute to oxidative stress, alter the redox balance, or induce transdifferentiation of resident lung macrophages and other alveolar cells. This underscores an important role for intravascular hemolysis in the pathogenesis of PH [46,47,48,49]. SCD is characterized by increased stress erythropoiesis as a compensatory mechanism for anemia, which increases the number of reticulocytes and younger RBCs in circulation. During hemolysis, these young RBCs released a large amount of arginase into the plasma [50]. This plasma arginase consumes plasma L-arginine, the substrate required for NO production by endothelial cells, and, in conjunction with the consumption of endothelial NO by cell-free plasma Hb, reduces NO bioavailability [51,52]. The depletion of NO affects intracellular calcium signaling that leads to dephosphorylation of myosin protein preventing smooth muscle relaxation [53]. The depletion of NO also results in leukocyte recruitment via increased surface expression of leukocyte adhesion proteins such as E-selectin, VCAM and ICAM-1 [54,55] and results in smooth muscle proliferation and vascular remodeling [56]. Heme related generation of ROS decreases availability and or activation of soluble guanylyl cyclase or its regulators such as cytochrome b5 reductase 3 (CYB5R3) which can result in poor vasodilation of pulmonary vasculature increasing risk of pulmonary hypertension [57,58].
Cell free plasma hemoglobin and heme can also independently activate platelets and neutrophils via TLR4 signaling mechanism [31] resulting in further inflammation. In addition, cell free hemoglobin also generates ROS which furthers endothelial dysfunction and activates the coagulation system [59]. Chronic hemolysis also promotes transition of pulmonary endothelial cells to a mesenchymal or smooth muscle cell type and contributes to vascular remodeling [60]. Thus, heme exposure results in pathological endothelial activation, increased recruitment of leukocytes and depletion of protective mechanisms that preserve vascular integrity.
Mechanistically, endothelial dysfunction results in increased production of vasoconstrictors such as endothelin-1 (ET-1). Heme related endothelial dysfunction can deplete peroxisome proliferator-activated receptor γ (PPARγ), which plays an active role in suppressing ET-1 production by regulating the level of microRNAs (miRs) such as miR-98 [55]. Lower levels of miR-98 associates with increased ET-1 production and endothelial dysfunction [52]. Exposure to heme also results in increased production of placenta growth factor (PIGF) by erythroid cells via the erythroid Krüppel-like factor (EKLF) [61] and NRF2-antioxidant response signaling [62]. PIGF is an angiogenic factor that activates endothelial cells to secrete ET-1 [63]. In an elegant study, overexpression of erythroid-specific PIGF in normal mice up to the levels seen in sickle cell mice resulted in an increase in the production of ET-1, which correlated with increased right ventricular pressure and pulmonary arteriolar thickening [64]. Elevated ET-1 and PlGF levels also correlate with severity of PH in patients with SCD [64]. PIGF was shown to activate expression of hypoxia-inducible factor 1α (HIF-1α), independently of hypoxia, which in turn can stimulate expression of ET-1, which is involved with the development and severity of PH in SCD [63].
Indeed, these cellular and molecular mechanisms have informed the current therapeutics usually used in patients with pulmonary hypertension such as endothelin receptor (ETR) antagonists (Bosentan, Ambrisetan), those which prevents the degradation of cyclic guanosine monophosphate (cGMP) (Riociguat and Sildenafil), vasodilators (Epoprostenol) and anticoagulant (warfarin) among others. Clinical trials using hemopexin, a scavenger molecule that removes heme from circulation are underway in humans and have shown promising results in murine models [65]. Unfortunately, trials with ETR antagonists [66], Riociguat [67], and Sildenafil [68,69] were either limited by small sample size or adverse side effects respectively underscoring the need to better understand the pathology and larger clinical trials researching PH in SCD.
Inflammation and pulmonary thrombosis/ embolism
Accumulating evidence from human studies discussed below suggests that inflammation is a risk factor for thrombosis. It is therefore not surprising that a retrospective study found that prevalence rates of venous thromboembolism (VTE) in adults with SCD was 25% and is associated with increased rates of recurrence and mortality [70,71]. Interestingly, the risk of pulmonary embolism (PE) is higher than risk of deep vein thrombosis (DVT) [71,72,73] suggesting that thrombosis may occur more ‘in situ’ in pulmonary vasculature of individuals with SCD. Risk factors for VTE include elevated leukocyte count [74], severe phenotype as defined by > 3 hospitalizations annually for vaso-occlusive crisis, presence of SCD variant genotypes, elevated tricuspid regurgitation jet velocity (TRJV) ≥2.5 m/s [70], elevated body mass index, and prior splenectomy [70,75,76]. Even in those with lower hospitalizations, the cumulative incidence rate of VTE was at 6.8% compared to 1.6% in individuals who had similar number of hospitalizations for asthma exacerbation [71] suggesting that intrinsic pathology in addition to external risk factors play a role in development of PE and/or DVT.
Several cellular and molecular pathways are perturbed in SCD that leads to a prothrombotic state. Chronic hemolysis results in release of intracellular molecules known as danger (or damage)-associated molecular patterns (DAMPs) [77]. For example, one of the most studied DAMPs or alarmins, high-mobility group box 1 (HMGB1), is significantly elevated in the plasma of SCD patients and mice at baseline compared to controls [78,79]. VOC episodes further increased HMGB1 levels in SCD patients, or acute sickling induced following hypoxia-reoxygenation in mice [78]. Furthermore, treating the TLR4 reporter cell line with plasma from SCD patients increased TLR4 receptor activity, suggesting that HMGB1 contributes to TLR4 signaling in SCD [78]. Elevated circulating HMGB1 is associated with platelet nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3 (NLRP3) activation, which is mediated through the TLR4 and Bruton tyrosine kinase signaling pathways [79,80]. DAMPs have also been implicated in endothelial dysfunction [81], activation and recruitment of leukocytes [31,77] and inflammation [77] which shift the balance to a more prothrombotic state in SCD. The characteristic changes in RBC rheology contributes to the formation of venous thrombi that have denser fibrin network and a friable thrombus [82]. In addition, red cell derived microparticles contribute to thrombin generation via activation of Factor XI. Indeed, red cell derived microparticles associate with increased markers of coagulation activation [83]. Activated platelets promote inflammasome activation and generation of EVs which can lead to formation of heterotypic aggregate and occlusion of the lung’s microvasculature [84]. DAMPs can also activate neutrophils and monocytes which can result in increased TF expression [85], NET formation, endothelial dysfunction and more inflammation [31] which have been linked to thrombus generation and propagation in non-SCD models [86]. Endothelial dysfunction from heme exposure results in the upregulation of adhesion molecules that attract neutrophils and platelets [31,34,81], and increased expression of TF and VWF which can contribute to pulmonary thrombosis [87].
The exact molecular mechanisms resulting in thrombus formation in lungs in SCD are not well studied and may involve mechanisms that either increases procoagulant proteins (such as TF, VWF, thrombin) [87,88], decreases anticoagulant proteins (like low protein C and S) [89,90], and/or decreases fibrinolysis [82]. There is some evidence that abrogation of TF using anti TF antibody reduces thrombus formation in sickle cell mouse model suggesting an important contribution of TF to thrombus generation in SCD. In addition, genetic or immunologic interventions that modulated expression of protein C and thrombin also diminished thrombus formation [91]. Data supporting the role of contact pathway leading to thrombosis in SCD is very sparse, however potential plausible sources include neutrophil nuclear content specifically nuclear DNA and histones which can initiate coagulation by activating Factor XII but also amplify thrombin-dependent factor XI activation [92], [86]. Partial support for this comes from observation that inducing neutropenia results in decreased thrombosis burden in an arterial thrombosis model [91]. Thus, several cellular and molecular mechanisms may be at play in pathogenesis of thrombosis.
Inflammation and reactive airway disease or airway hyper-activity (AHR)
Reactive airway disease or AHR is common among adults and children with SCD [93,94] and can occur independent of clinical asthma. Studies show that up to 77% of children demonstrate a positive methacholine challenge test (MCT) [93,95]. Interestingly, the severity of AHR correlates with high LDH, suggestive of a critical role played by hemolysis and disease severity [96]. Indeed, one study did show that AHR was more common in those with HbSS genotype and was predictive of increased risk of ACS and vaso-occlusive crisis [97]. From a pathophysiology perspective, AHR is characterized by bronchial hypersensitivity to stimuli, airway and lung inflammation, abnormal leukocyte recruitment, and airway and vessel wall thickening [98,99,100]. Chronic hemolysis and their byproducts may drive systemic inflammation and result in increased lung/airway inflammation. Indeed, in one study with SCD mice, even prior to sensitization with allergen, there was evidence of increased airway inflammation, increased lymphocytes in bronchoalveolar fluid (BAL), granulocyte-colony stimulating factor, interleukin 5 (IL-5), IL-7, and chemokine (C-X-C motif) ligand (CXCL)1 and lung T cell infiltration [101]. Mice exposed to specific allergen recapitulated specific features of asthma like phenotype including increased immunoglobulin E (IgE), IL-6, and IL-13 in serum and increased bronchial hyperresponsiveness to methacholine [101]. Another study corroborated the findings of increased IgE, and airway inflammation as evidenced by eosinophil infiltration, vessel wall thickening and increased concentrations of transforming growth factor beta (TGF-B) [102]. SCD mice with PIGF deficiency show decreased airway inflammation, leukotriene and IL-13 mediated immune responses suggesting an important role of PlGF signaling pathways in AHR [97]. Thus, multiple pathways are at work that makes individuals with SCD susceptible to allergens and environment pollutants.
Inflammatory mediators and cardiac hypertrophy
Both concentric and eccentric hypertrophy had been reported in children and adults with SCD [103]. These changes in cardiac structure and function start in early childhood and worsen with age [104,105].The enlargement of the heart begins as a compensatory myocardial remodeling in response to anemia [106]. The cardiomyocytes and capillary networks in the heart become enlarged to increase oxygen supply, leading to increase cardiac output at rest [106]. This elevated cardiac output also becomes exaggerated during exercise due to an increase in cardiac stroke volume in response to the increased oxygen consumption, indicating altered hemodynamics in SCD patients [106,107]. Restrictive cardiomyopathy can also co-exist with anemia-induced elevated cardiac output [104,105]. Morphological abnormalities of sickle RBCs such as polymerization and aberrant membrane transport properties, auto-oxidative ROS generation and ischemia-reperfusion injury, may also contribute to cardiac remodeling [108,109].
Studies have suggested that endothelial dysfunction and increased plasma markers of inflammation contribute to cardiac hypertrophy in SCD mouse models [110,111]. This may be due to the pre-activation of immune cells, including monocytes and endothelial cells in SCD [112,113,114,115]. There is also an increase in the mRNA and protein expression of pro-inflammatory markers such as TNF-α, IL-1, IL-6, MIP-1b and soluble endothelial adhesion molecules [114,116,117]. The heart, like the other organs is exposed to inflammatory insults from these circulating proinflammatory cytokines. Given that elevated systemic inflammation is associated with cardiac abnormalities in the general population [6], it is possible that the observed excessive systemic inflammation in individuals with SCD, could contribute to cardiac pathology in SCD.
In addition to pro-inflammatory cytokines, products of hemolysis such as heme and other DAMPs may further perturb the homeostatic state in the heart, thereby perpetuating the vicious cycle of inflammation and cardiac pathobiology described in the last paragraph. For instance, heme released into the circulation during hemolysis triggers several inflammatory pathways in SCD that contribute to organ damage [30,37,81]. In fact, a comparison of organ-specific expression patterns of HMOX1 in SCD mice treated with heme revealed that the heart has one of the highest expressions of HMOX1 [118]. This suggests that cardiac cells can uptake circulating heme and metabolize it. The potential problem with this process is that excess iron produced from heme breakdown and deposited in the heart can activate oxidative and apoptotic pathways. A recent study showed that heme-induced upregulation of HMOX1 promotes cardiac ferroptosis in SCD mice as well as the expression of cardiac hypertrophy genes [14], although T2* cardiac magnetic resonance imaging measurement of cardiac iron showed that iron overload is rare even in chronically transfused SCD patients [119,120]. Another study showed that increasing circulating heme significantly elevated plasma IL-6 and the expression of cardiac hypertrophy markers in Townes sickle cell mice [16]. These studies underscore the importance of hemolysis in the pathogenesis of cardiac hypertrophy. Another way that inflammation may contribute to cardiac pathology is through a complex interaction with the coagulation system. Increasing evidence in SCD has shown a link between vascular inflammation and hypercoagulation via activated intrinsic and extrinsic coagulation pathways, which may contribute to organ pathology [121,122]. A study by Sparkenbaugh et al. showed that short-term pharmacological inhibition of FXa in Berkeley sickle cell mice attenuated plasma IL-6 and cardiac hypertrophy [110]. Similarly, genetic inhibition of circulating FII in SCD mice improved right ventricle hypertrophy and dilatation, suggesting that increased thrombin generation or activity in SCD is a significant contributor to cardiac pathophysiology [111]. These studies suggest that perturbation in the coagulation system and the attendant vascular damage in SCD may contribute to inflammation, a key component of various mechanisms involved in cardiac dysfunction.
The inflammatory signaling pathway may also modify cardiac remodeling in SCD via the complex biological role played by PlGF. PlGF is an angiogenic cytokine that plays a role in the survival of endothelial cells and monocytes, and in cardiovascular health [115,123]. PlGF is crucial in the early inflammatory response needed for adaptive hypertrophic cardiac remodeling due to pressure overload [124,125]. Although studies have shown both beneficial and deleterious roles of PlGF in the heart [126,127,128], its expression is elevated in the plasma of SCD patients and linked with disease severity [64,114]. Furthermore, PlGF mRNA and protein expression were found to be elevated in the hearts of sickle cell mice at baseline and upon exposure to heme [129]. The role of PlGF in cardiopulmonary complications of SCD may be via an indirect effect on endothelial cells, fibroblasts, and monocytes in the heart, which are already primed for an exaggerated response due to a pro-inflammatory microenvironment mediated by cytokines such as IL-6. Nevertheless, no mechanistic studies have clearly delineated this intriguing role of PlGF in cardiac pathology in SCD by examining these cardiac cells individually.
Inflammation and diastolic dysfunction
Diastolic dysfunction occurs in both children and adults with SCD. It has been associated with anemia, older age, higher creatinine levels, exercise impairment, increased LV mass, low sleep or waking oxygen saturation, and diffuse myocardial fibrosis [11,103,130,131,132]. Though the causative sequence is not well defined, diastolic dysfunction is also an independent risk factor for death in SCD patients [104,133]. Increased doppler echocardiography ratio of mitral valve inflow (E) velocities (E) over peak early diastolic annular velocity (E′) > 8.2 has been shown to predict diastolic dysfunction in SCD patients [11,130,134]. Furthermore, diastolic dysfunction has been linked to the overexpression of interleukin-18 (IL-18), -L-fucosidase A2 (FUCA2), and thyroid hormone transporter (SLC16A2) in SCD patients' peripheral blood mononuclear cells (PBMCs) [11]. This finding was validated in mouse models, with results showing elevated expression of these genes in the myocardium of sickle cell mice compared to controls [11]. Although diastolic dysfunction in SCD involves multiple complex pathophysiological mechanisms, this report on IL-18 suggests the involvement of inflammation, which is chronic in SCD, either as a primary mechanism or as a secondary to cardiac remodeling due to the hyperdynamic state.
Inflammation and cardiac arrhythmia
Cardiac arrhythmia (CA) is defined as a disruption in the normal activation of the heart or an irregular heartbeat rhythm that is either too slow (60 beats per minute) or too fast (>100 beats per minute) [135]. Some forms of CA, including sinus arrhythmia, are considered to be benign; however, the presence of structural heart defects such as LV dysfunction or genetic arrhythmia syndromes, including long or short QT syndrome, increases the severity of CA and could result in heart failure or sudden cardiac death [135,136]. Cardiac arrythmias have been reported as the cause of death in 7.4% of in-hospital deaths in adult African Americans with sickle cell trait [137], and in 14% of SCD patients [138]. Cardiac arrhythmias have also been implicated in some sudden deaths recorded in SCD patients with a prolonged corrected QT interval (QTc), which is independently associated with an increased risk of death [107,139,140]. In a recent study, elevated expression of IL-18 in PBMCs of SCD patients was associated with longer QTc intervals and increased mortality risk [141]. Consequently, chronic inhibition of IL-18 binding protein in a sickle cell mouse model attenuated IL-18–mediated ventricular tachycardia and improved diastolic function, suggesting a link between cardiac inflammation and arrhythmias, in SCD [141].
Inflammation and cardiac fibrosis
Autopsy studies and studies in living SCD patients have shown the presence of both diffuse and transmural fibrosis [103,142,143]. The cellular and molecular mechanisms underlying cardiac fibrosis in SCD, are not completely understood. However, gene-expression profiles of heart tissue isolated from Berkeley sickle cell mice revealed elevated expression of genes involved in oxidative stress, angiogenesis, and TGF-β signaling, which correlated with imaging and histology data demonstrating diffuse cardiac fibrosis and diastolic dysfunction [144]. Another study in SCD mice showed that sustained neutralization of the IL-18 binding protein ameliorated cardiac fibrosis [141] (Figure 2). In a small observational study of SCD patients, early initiation of disease-modifying therapy such as hydroxyurea and chronic transfusion was shown to prevent diffuse myocardial fibrosis and diastolic dysfunction [145]. Although the underlying specific mechanism(s) of how these therapies ameliorate the development of cardiac fibrosis remains unknown, there is a need for detailed mechanistic studies that elaborate on the link between inflammation and other pathways that may be involved in the development of cardiac fibrosis in SCD.
Conclusions and future perspectives
Inflammation involves complex cellular and molecular pathways that have both beneficial and harmful effects, and it plays a major role in the pathogenesis of cardiopulmonary complications in SCD. Inflammation is also a common denominator in the development of ACS, cardiac fibrosis, PH, and diastolic dysfunction, as reviewed here (Table 1). Several studies have shown that chronic systemic inflammation as present in SCD contributes to a cascade of events that results in tissue injury, the generation of ROS, and endothelial dysfunction [146,147,148]. Additionally, inflammatory cells such as monocytes are in a state of pre-activation due to elevated levels of circulating inflammatory cytokines, which contribute to the chronic inflammation in SCD by amplifying the production and signaling of these cytokines [64,114,147]. Therefore, addressing cardiopulmonary manifestations of the disease by modulating inflammatory pathways using targeted therapies could offer a novel approach to reducing the risk of developing these complications. This would involve well-designed and detailed experiments that include investigating the role of genetic polymorphisms in the regulation of blood levels of circulating cytokines, inflammatory markers, and their signaling pathways in SCD. Additional investigation will also examine how genes encoding the production of inflammatory mediators are regulated and potentially silenced. Furthermore, a detailed examination of how the inflammatory SCD microenvironment modifies the functions of immune cells as well as the response of these immune cells. Identification of plasma inflammatory biomarkers with prognostic value for determining SCD patients at risk of cardiopulmonary complications may also be an additional direction of investigation that could be beneficial. The adoption and adaptation of advanced non-invasive imaging technology that provides both structural and functional information about the cardiopulmonary landscape in SCD patients may provide a novel approach to documenting cardiac remodeling in SCD patients. For example, hybrid positron emission tomography (PET) and magnetic resonance imaging (MRI) using the uptake of the radiotracer 18F-fluorodeoxyglucose (18F-FDG) have been used to identify and characterize vascular inflammation and different stages of atherosclerosis that are not yet detectable by other forms of structural imaging such as cardiac computed tomography and echocardiography [149,150]. This technique may be useful and could be adapted to SCD, where both inflammation and vascular dysfunction are present and linked with the development of cardiac hypertrophy, PH, and diastolic dysfunction. Because inflammation may precede the development of cardiopulmonary complications in SCD patients, extending the current guidelines for screening, diagnosis, and management of cardiopulmonary complications in SCD patients [42], by adding biomarkers of inflammation linked with cardiopulmonary complications. This may improve risk stratification and help with better identification of patients needing more urgent therapy, thus preventing the progression to irreversible organ damage.
Overall, early diagnosis of cardiopulmonary complication through early recognition and application of molecular risks factors before irreversible organ damage occurs would contribute to a better quality of life, as SCD patients are now living longer due to the availability of disease-modifying therapies.
Author Contributions
All authors drafted the review. HIH approved the final version of this review. All authors contributed to the article and approved the submitted version.
Funding
OTG is supported by the American Society of Hematology Scholar Award and the Parker B. Francis Fellowship in Pulmonary Research Award. H.I.H. is supported by grants from The National Institutes of Health (R01 HL138423, R01 HL156024 and 3R01 HL138423-05S).
Conflicts of Interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Piel, F.; Hay, S.; Gupta, S.; Weatherall, D.; Williams, T. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med 2013, 10, e1001484. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.; Patil, A.; Howes, R.; Nyangiri, O.; Gething, P.; Dewi, M.; Temperley, W.; Williams, T.; Weatherall, D.; Haya, S. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 2013, 381, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.; Lauder, N.; Jonassaint, J.; Telen, M.; Zhao, X.; Wright, E.; Gilliam, F.; De Castro, L. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am J Hematol 2010, 85, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M. Cardiovascular complications and risk of death in sickle-cell disease. Lancet 2016, 387, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, V.; Rosing, D.R.; Thein, S.L. Cardiovascular complications of sickle cell disease. Trends Cardiovasc Med 2020. [CrossRef]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am J Prev Cardiol 2020, 4, 100130. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Aday, A.W.; Rose, L.M.; Ridker, P.M. Residual Inflammatory Risk on Treatment With PCSK9 Inhibition and Statin Therapy. Circulation 2018, 138, 141–149. [Google Scholar] [CrossRef]
- Fang, L.; Ellims, A.H.; Beale, A.L.; Taylor, A.J.; Murphy, A.; Dart, A.M. Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy. Am J Transl Res 2017, 9, 5063–5073. [Google Scholar] [PubMed]
- Xiao, Z.; Kong, B.; Yang, H.; Dai, C.; Fang, J.; Qin, T.; Huang, H. Key Player in Cardiac Hypertrophy, Emphasizing the Role of Toll-Like Receptor 4. Front Cardiovasc Med 2020, 7, 579036. [Google Scholar] [CrossRef]
- Wang, R.R.; Yuan, T.Y.; Wang, J.M.; Chen, Y.C.; Zhao, J.L.; Li, M.T.; Fang, L.H.; Du, G.H. Immunity and inflammation in pulmonary arterial hypertension: From pathophysiology mechanisms to treatment perspective. Pharmacol Res 2022, 180, 106238. [Google Scholar] [CrossRef]
- Duarte, J.D.; Desai, A.A.; Sysol, J.R.; Abbasi, T.; Patel, A.R.; Lang, R.M.; Gupta, A.; Garcia, J.G.; Gordeuk, V.R.; Machado, R.F. Genome-Wide Analysis Identifies IL-18 and FUCA2 as Novel Genes Associated with Diastolic Function in African Americans with Sickle Cell Disease. PloS one 2016, 11, e0163013. [Google Scholar] [CrossRef] [PubMed]
- Allali, S.; de Montalembert, M.; Rignault-Bricard, R.; Taylor, M.; Brice, J.; Brousse, V.; Talbot, J.M.; Moulin, F.; Heilbronner, C.; Hermine, O.; Maciel, T.T. IL-6 levels are dramatically high in the sputum from children with sickle cell disease during acute chest syndrome. Blood advances 2020, 4, 6130–6134. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Kapetanaki, M.G.; Ghosh, S.; Villanueva, F.S.; Ofori-Acquah, S.F.; Kato, G.J. Heme Induces IL-6 and Cardiac Hypertrophy Genes Transcripts in Sickle Cell Mice. Frontiers in Immunology 2020. [CrossRef] [PubMed]
- Menon, A.V.; Liu, J.; Tsai, H.P.; Zeng, L.; Yang, S.; Asnani, A.; Kim, J. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood 2022, 139, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Karoor, V.; Swindle, D.; Pak, D.I.; Strassheim, D.; Fini, M.A.; Dempsey, E.; Stenmark, K.R.; Hassell, K.; Nuss, R.; Buehler, P.W.; Irwin, D.C. Evidence supporting a role for circulating macrophages in the regression of vascular remodeling following sub-chronic exposure to hemoglobin plus hypoxia. Pulmonary circulation 2021, 11, 20458940211056806. [Google Scholar] [CrossRef] [PubMed]
- Gbotosho, O.T.; Kapetanaki, M.G.; Ghosh, S.; Villanueva, F.S.; Ofori-Acquah, S.F.; Kato, G.J. Heme Induces IL-6 and Cardiac Hypertrophy Genes Transcripts in Sickle Cell Mice. Frontiers in immunology 2020, 11, 1910. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.P.; Styles, L.A.; Colangelo, L.H.; Wright, E.C.; Castro, O.; Nickerson, B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood 1997, 89, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.P.; Neumayr, L.D.; Earles, A.N.; Williams, R.; Lennette, E.T.; Dean, D.; Nickerson, B.; Orringer, E.; McKie, V.; Bellevue, R.; Daeschner, C.; Manci, E.A. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. The New England journal of medicine 2000, 342, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Bakshi, N.; Krishnamurti, L. Acute Chest Syndrome in Children with Sickle Cell Disease. Pediatr Allergy Immunol Pulmonol 2017, 30, 191–201. [Google Scholar] [CrossRef]
- Bennewitz, M.F.; Jimenez, M.A.; Vats, R.; Tutuncuoglu, E.; Jonassaint, J.; Kato, G.J.; Gladwin, M.T.; Sundd, P. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight 2017, 2, e89761. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Vichinsky, E. Pulmonary complications of sickle cell disease. N Engl J Med 2008, 359, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Reichel, C.A.; Rehberg, M.; Lerchenberger, M.; Berberich, N.; Bihari, P.; Khandoga, A.G.; Zahler, S.; Krombach, F. Ccl2 and Ccl3 mediate neutrophil recruitment via induction of protein synthesis and generation of lipid mediators. Arterioscler Thromb Vasc Biol 2009, 29, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Koltsova, E.K.; Sundd, P.; Zarpellon, A.; Ouyang, H.; Mikulski, Z.; Zampolli, A.; Ruggeri, Z.M.; Ley, K. Genetic deletion of platelet glycoprotein Ib alpha but not its extracellular domain protects from atherosclerosis. Thromb Haemost 2014, 112, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Anea, C.B.; Lyon, M.; Lee, I.A.; Gonzales, J.N.; Adeyemi, A.; Falls, G.; Kutlar, A.; Brittain, J.E. Pulmonary platelet thrombi and vascular pathology in acute chest syndrome in patients with sickle cell disease. Am J Hematol 2016, 91, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, M.A.; Novelli, E.; Shaw, G.D.; Sundd, P. Glycoprotein Ibalpha inhibitor (CCP-224) prevents neutrophil-platelet aggregation in Sickle Cell Disease. Blood Adv 2017, 1, 1712–1716. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Flage, B.; Weidert, F.; Ofori-Acquah, S.F. P-selectin plays a role in haem-induced acute lung injury in sickle mice. British journal of haematology 2019, 186, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The Worst Things in Life are Free: The Role of Free Heme in Sickle Cell Disease. Frontiers in immunology 2020, 11, 561917. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. The Journal of clinical investigation 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Kanias, T.; Kim-Shapiro, D.B. Hemolysis and cell-free hemoglobin drive an intrinsic mechanism for human disease. J Clin Invest 2012, 122, 1205–1208. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Ofori-Acquah, S.F. Erythroid DAMPs drive inflammation in SCD. Blood 2014, 123, 3689–3690. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef] [PubMed]
- Rank, B.H.; Carlsson, J.; Hebbel, R.P. Abnormal redox status of membrane-protein thiols in sickle erythrocytes. J Clin Invest 1985, 75, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.M.; Sysol, J.R.; Singla, S.; Smith, P.; Sandusky, G.E.; Wang, H.; Natarajan, V.; Dudek, S.M.; Machado, R.F. Cortactin loss protects against hemin-induced acute lung injury in sickle cell disease. Am J Physiol Lung Cell Mol Physiol 2022, 322, L890–L97. [Google Scholar] [CrossRef] [PubMed]
- Adisa, O.A.; Hu, Y.; Ghosh, S.; Aryee, D.; Osunkwo, I.; Ofori-Acquah, S.F. Association between plasma free haem and incidence of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. British journal of haematology 2013, 162, 702–705. [Google Scholar] [CrossRef]
- Bean, C.J.; Boulet, S.L.; Ellingsen, D.; Pyle, M.E.; Barron-Casella, E.A.; Casella, J.F.; Payne, A.B.; Driggers, J.; Trau, H.A.; Yang, G.; Jones, K.; Ofori-Acquah, S.F.; Hooper, W.C.; DeBaun, M.R. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood 2012, 120, 3822–3828. [Google Scholar] [CrossRef]
- Ghosh, S.; Adisa, O.A.; Chappa, P.; Tan, F.; Jackson, K.A.; Archer, D.R.; Ofori-Acquah, S.F. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. The Journal of clinical investigation 2013, 123, 4809–4820. [Google Scholar] [CrossRef]
- Ghosh, S.; Hazra, R.; Ihunnah, C.A.; Weidert, F.; Flage, B.; Ofori-Acquah, S.F. Augmented NRF2 activation protects adult sickle mice from lethal acute chest syndrome. British journal of haematology 2018, 182, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Alishlash, A.S.; Sapkota, M.; Ahmad, I.; Maclin, K.; Ahmed, N.A.; Molyvdas, A.; Doran, S.; Albert, C.J.; Aggarwal, S.; Ford, D.A.; Ambalavanan, N.; Jilling, T.; Matalon, S. Chlorine inhalation induces acute chest syndrome in humanized sickle cell mouse model and ameliorated by postexposure hemopexin. Redox Biol 2021, 44, 102009. [Google Scholar] [CrossRef]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994, 330, 1639–1644. [Google Scholar] [CrossRef]
- Parent, F.; Bachir, D.; Inamo, J.; Lionnet, F.; Driss, F.; Loko, G.; Habibi, A.; Bennani, S.; Savale, L.; Adnot, S.; Maitre, B.; Yaici, A.; Hajji, L.; O'Callaghan, D.S.; Clerson, P.; Girot, R.; Galacteros, F.; Simonneau, G. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med 2011, 365, 44–53. [Google Scholar] [CrossRef]
- Liem, R.I.; Lanzkron, S. T DC;DeCastro, L.; Desai, A.A.; Ataga, K.I.; Cohen, R.T.; Haynes, J.; Osunkwo, I.; Lebensburger, J.D.; Lash, J.P.; Wun, T.; Verhovsek, M.; Ontala, E.; Blaylark, R.; Alahdab, F.; Katabi, A.; Mustafa, R.A. American Society of Hematology 2019 guidelines for sickle cell disease: cardiopulmonary and kidney disease. Blood advances 2019, 3, 3867–3897. [Google Scholar] [PubMed]
- Adedeji, M.O.; Cespedes, J.; Allen, K.; Subramony, C.; Hughson, M.D. Pulmonary thrombotic arteriopathy in patients with sickle cell disease. Arch Pathol Lab Med 2001, 125, 1436–1441. [Google Scholar] [CrossRef]
- Wood, K.C.; Gladwin, M.T.; Straub, A.C. Sickle cell disease: at the crossroads of pulmonary hypertension and diastolic heart failure. Heart 2020, 106, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Redinus, K.; Baek, J.H.; Yalamanoglu, A.; Shin, H.K.H.; Moldova, R.; Harral, J.W.; Swindle, D.; Pak, D.; Ferguson, S.K.; Nuss, R.; Hassell, K.; Nozik-Grayck, E.; Palmer, A.F.; Fini, M.A.; Karoor, V.; Stenmark, K.R.; Buehler, P.W.; Irwin, D.C. An Hb-mediated circulating macrophage contributing to pulmonary vascular remodeling in sickle cell disease. JCI Insight.
- Buehler, P.W.; Swindle, D.; Pak, D.I.; Fini, M.A.; Hassell, K.; Nuss, R.; Wilkerson, R.B.; D'Alessandro, A.; Irwin, D.C. Murine models of sickle cell disease and beta-thalassemia demonstrate pulmonary hypertension with distinctive features. Pulm Circ 2021, 11, 20458940211055996. [Google Scholar] [CrossRef]
- Hsu, L.L.; Champion, H.C.; Campbell-Lee, S.A.; Bivalacqua, T.J.; Manci, E.A.; Diwan, B.A.; Schimel, D.M.; Cochard, A.E.; Wang, X.; Schechter, A.N.; Noguchi, C.T.; Gladwin, M.T. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 2007, 109, 3088–3098. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Barst, R.J.; Castro, O.L.; Gordeuk, V.R.; Hillery, C.A.; Kato, G.J.; Kim-Shapiro, D.B.; Machado, R.; Morris, C.R.; Steinberg, M.H.; Vichinsky, E.P. Pulmonary hypertension and NO in sickle cell. Blood 2010, 116, 852–854. [Google Scholar] [CrossRef]
- Minneci, P.C.; Deans, K.J.; Zhi, H.; Yuen, P.S.; Star, R.A.; Banks, S.M.; Schechter, A.N.; Natanson, C.; Gladwin, M.T.; Solomon, S.B. Hemolysis-associated endothelial dysfunction mediated by accelerated NO inactivation by decompartmentalized oxyhemoglobin. J Clin Invest 2005, 115, 3409–3417. [Google Scholar] [CrossRef]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M., Jr.; Gladwin, M.T. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.; Wang, X.; Tanus-Santos, J.; Hogg, N.; Cannon Rr Schechter, A.; MT, G. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Li, L.; Kitazawa, T. Cyclic GMP causes Ca2+ desensitization in vascular smooth muscle by activating the myosin light chain phosphatase. J Biol Chem 1997, 272, 5063–5068. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.B.; Souza, L.E.; Leonardo, F.C.; Costa, F.T.; Werneck, C.C.; Covas, D.T.; Costa, F.F.; Conran, N. Acute hemolytic vascular inflammatory processes are prevented by nitric oxide replacement or a single dose of hydroxyurea. Blood 2015, 126, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.J.; Chang, S.S.; Park, C.; Lee, C.M.; Benza, R.L.; Passineau, M.J.; Ma, J.; Archer, D.R.; Sutliff, R.L.; Hart, C.M.; Kang, B.Y. PPARgamma increases HUWE1 to attenuate NF-kappaB/p65 and sickle cell disease with pulmonary hypertension. Blood Adv 2021, 5, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Zuckerbraun, B.S.; Shiva, S.; Ifedigbo, E.; Mathier, M.A.; Mollen, K.P.; Rao, J.; Bauer, P.M.; Choi, J.J.; Curtis, E.; Choi, A.M.; Gladwin, M.T. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation 2010, 121, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.C.; Durgin, B.G.; Schmidt, H.M.; Hahn, S.A.; Baust, J.J.; Bachman, T.; Vitturi, D.A.; Ghosh, S.; Ofori-Acquah, S.F.; Mora, A.L.; Gladwin, M.T.; Straub, A.C. Smooth muscle cytochrome b5 reductase 3 deficiency accelerates pulmonary hypertension development in sickle cell mice. Blood advances 2019, 3, 4104–4116. [Google Scholar] [CrossRef]
- Potoka, K.P.; Wood, K.C.; Baust, J.J.; Bueno, M.; Hahn, S.A.; Vanderpool, R.R.; Bachman, T.; Mallampalli, G.M.; Osei-Hwedieh, D.O.; Schrott, V.; Sun, B.; Bullock, G.C.; Becker-Pelster, E.M.; Wittwer, M.; Stampfuss, J.; Mathar, I.; Stasch, J.P.; Truebel, H.; Sandner, P.; Mora, A.L.; Straub, A.C.; Gladwin, M.T. Nitric Oxide-Independent Soluble Guanylate Cyclase Activation Improves Vascular Function and Cardiac Remodeling in Sickle Cell Disease. Am J Respir Cell Mol Biol 2018, 58, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Moore, C.G.; Hillery, C.A.; Jones, S.; Whinna, H.C.; Strayhorn, D.; Sohier, C.; Hinderliter, A.; Parise, L.V.; Orringer, E.P. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica 2008, 93, 20–26. [Google Scholar] [CrossRef]
- Gonzales, J.; Holbert, K.; Czysz, K.; George, J.; Fernandes, C.; Fraidenburg, D.R. Hemin-Induced Endothelial Dysfunction and Endothelial to Mesenchymal Transition in the Pathogenesis of Pulmonary Hypertension Due to Chronic Hemolysis. Int J Mol Sci.
- Wang, X.; Mendelsohn, L.; Rogers, H.; Leitman, S.; Raghavachari, N.; Yang, Y.; Yau, Y.; Tallack, M.; Perkins, A.; Taylor, J.; Noguchi, C.; Kato, G. Heme-bound iron activates placenta growth factor in erythroid cells via erythroid Krüppel-like factor. Blood 124, 946–954. [CrossRef]
- Kapetanaki, M.G.; Gbotosho, O.T.; Sharma, D.; Weidert, F.; Ofori-Acquah, S.F.; Kato, G.J. Free heme regulates placenta growth factor through NRF2-antioxidant response signaling. Free radical biology & medicine 2019, 143, 300–308. [Google Scholar]
- Kalra, V.K.; Zhang, S.; Malik, P.; Tahara, S.M. Placenta growth factor mediated gene regulation in sickle cell disease. Blood reviews 2018, 32, 61–70. [Google Scholar] [CrossRef]
- Sundaram, N.; Tailor, A.; Mendelsohn, L.; Wansapura, J.; Wang, X.; Higashimoto, T.; Pauciulo, M.W.; Gottliebson, W.; Kalra, V.K.; Nichols, W.C.; Kato, G.J.; Malik, P. High levels of placenta growth factor in sickle cell disease promote pulmonary hypertension. Blood 2010, 116, 109–112. [Google Scholar] [CrossRef]
- Buehler, P.W.; Swindle, D.; Pak, D.I.; Ferguson, S.K.; Majka, S.M.; Karoor, V.; Moldovan, R.; Sintas, C.; Black, J.; Gentinetta, T.; Buzzi, R.M.; Vallelian, F.; Wassmer, A.; Edler, M.; Bain, J.; Schu, D.; Hassell, K.; Nuss, R.; Schaer, D.J.; Irwin, D.C. Hemopexin dosing improves cardiopulmonary dysfunction in murine sickle cell disease. Free radical biology & medicine 2021, 175, 95–107. [Google Scholar]
- Minniti, C.P.; Machado, R.F.; Coles, W.A.; Sachdev, V.; Gladwin, M.T.; Kato, G.J. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. British journal of haematology 2009, 147, 737–743. [Google Scholar] [CrossRef]
- Weir, N.A.; Conrey, A.; Lewis, D.; Mehari, A. Riociguat use in sickle cell related chronic thromboembolic pulmonary hypertension: A case series. Pulm Circ 2018, 8, 2045894018791802. [Google Scholar] [CrossRef]
- Machado, R.F.; Martyr, S.; Kato, G.J.; Barst, R.J.; Anthi, A.; Robinson, M.R.; Hunter, L.; Coles, W.; Nichols, J.; Hunter, C.; Sachdev, V.; Castro, O.; Gladwin, M.T. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol 2005, 130, 445–453. [Google Scholar] [CrossRef]
- Cramer-Bour, C.; Ruhl, A.P.; Nouraie, S.M.; Emeh, R.O.; Ruopp, N.F.; Thein, S.L.; Weir, N.A.; Klings, E.S. Long-term tolerability of phosphodiesterase-5 inhibitors in pulmonary hypertension of sickle cell disease. Eur J Haematol 2021, 107, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.P.; Streiff, M.B.; Haywood, C., Jr.; Nelson, J.A.; Lanzkron, S. Venous thromboembolism in adults with sickle cell disease: a serious and under-recognized complication. Am J Med 2013, 126, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Brunson, A.; Lei, A.; Rosenberg, A.S.; White, R.H.; Keegan, T.; Wun, T. Increased incidence of VTE in sickle cell disease patients: risk factors, recurrence and impact on mortality. Br J Haematol 2017, 178, 319–326. [Google Scholar] [CrossRef]
- Stein, P.D.; Beemath, A.; Meyers, F.A.; Skaf, E.; Olson, R.E. Deep venous thrombosis and pulmonary embolism in hospitalized patients with sickle cell disease. Am J Med 2006, 119, 897–e7. [Google Scholar] [CrossRef]
- Naik, R.P.; Streiff, M.B.; Haywood, C., Jr.; Segal, J.B.; Lanzkron, S. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J Thromb Haemost 2014, 12, 2010–2016. [Google Scholar] [CrossRef] [PubMed]
- Gollamudi, J.; Sarvepalli, S.; Vadaparti Binf, A.; Alin, T.; Little, J.A.; Nayak, L. Venous Thromboembolism in Sickle Cell Disease is Associated with Neutrophilia. Hemoglobin 2021, 45, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Srisuwananukorn, A.; Raslan, R.; Zhang, X.; Shah, B.N.; Han, J.; Gowhari, M.; Molokie, R.E.; Gordeuk, V.R.; Saraf, S.L. Clinical, laboratory, and genetic risk factors for thrombosis in sickle cell disease. Blood Adv 2020, 4, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Stanek, J.; Creary, S.; Dunn, A.; O'Brien, S.H. Prevalence and risk factors for venous thromboembolism in children with sickle cell disease: an administrative database study. Blood Adv 2018, 2, 285–291. [Google Scholar] [CrossRef]
- Mendonca, R.; Silveira, A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm Res 2016, 65, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wandersee, N.J.; Guo, Y.; Jones, D.W.; Holzhauer, S.L.; Hanson, M.S.; Machogu, E.; Brousseau, D.C.; Hogg, N.; Densmore, J.C.; Kaul, S.; Hillery, C.A.; Pritchard, K.A., Jr. Sickle cell disease increases high mobility group box 1: a novel mechanism of inflammation. Blood 2014, 124, 3978–3981. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Arora, T.; Wang, X.; Mendelsohn, L.; Nichols, J.; Allen, D.; Shet, A.S.; Combs, C.A.; Quezado, Z.M.N.; Thein, S.L. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood advances 2018, 2, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Murthy, P.; Durco, F.; Miller-Ocuin, J.L.; Takedai, T.; Shankar, S.; Liang, X.; Liu, X.; Cui, X.; Sachdev, U.; Rath, D.; Lotze, M.T.; Zeh, H.J., 3rd; Gawaz, M.; Weber, A.N.; Vogel, S. The NLRP3 inflammasome and bruton's tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochemical and biophysical research communications 2017, 483, 230–236. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef]
- Faes, C.; Ilich, A.; Sotiaux, A.; Sparkenbaugh, E.M.; Henderson, M.W.; Buczek, L.; Beckman, J.D.; Ellsworth, P.; Noubouossie, D.F.; Bhoopat, L.; Piegore, M.; Renoux, C.; Bergmeier, W.; Park, Y.; Ataga, K.I.; Cooley, B.; Wolberg, A.S.; Key, N.S.; Pawlinski, R. Red blood cells modulate structure and dynamics of venous clot formation in sickle cell disease. Blood 2019, 133, 2529–2541. [Google Scholar] [CrossRef]
- van Beers, E.J.; Schaap, M.C.; Berckmans, R.J.; Nieuwland, R.; Sturk, A.; van Doormaal, F.F.; Meijers, J.C.; Biemond, B.J.; group, C.s. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica 2009, 94, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Vats, R.; Brzoska, T.; Bennewitz, M.F.; Jimenez, M.A.; Pradhan-Sundd, T.; Tutuncuoglu, E.; Jonassaint, J.; Gutierrez, E.; Watkins, S.C.; Shiva, S.; Scott, M.J.; Morelli, A.E.; Neal, M.D.; Kato, G.J.; Gladwin, M.T.; Sundd, P. Platelet Extracellular Vesicles Drive Inflammasome-IL-1beta-Dependent Lung Injury in Sickle Cell Disease. Am J Respir Crit Care Med 2020, 201, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler Thromb Vasc Biol 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Solovey, A.; Kollander, R.; Shet, A.; Milbauer, L.C.; Choong, S.; Panoskaltsis-Mortari, A.; Blazar, B.R.; Kelm, R.J.; Jr Hebbel, R.P. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood 2004, 104, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Han, H.; Cruz, M.A.; Lopez, J.A.; Dong, J.F.; Guchhait, P. Haemoglobin blocks von Willebrand factor proteolysis by ADAMTS-13: a mechanism associated with sickle cell disease. Thromb Haemost 2009, 101, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Schnog, J.B.; Mac Gillavry, M.R.; van Zanten, A.P.; Meijers, J.C.; Rojer, R.A.; Duits, A.J.; ten Cate, H.; Brandjes, D.P. Protein C and S and inflammation in sickle cell disease. Am J Hematol 2004, 76, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Whelihan, M.F.; Lim, M.Y.; Mooberry, M.J.; Piegore, M.G.; Ilich, A.; Wogu, A.; Cai, J.; Monroe, D.M.; Ataga, K.I.; Mann, K.G.; Key, N.S. Thrombin generation and cell-dependent hypercoagulability in sickle cell disease. J Thromb Haemost 2016, 14, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Gavins, F.N.; Russell, J.; Senchenkova, E.L.; De Almeida Paula, L.; Damazo, A.S.; Esmon, C.T.; Kirchhofer, D.; Hebbel, R.P.; Granger, D.N. Mechanisms of enhanced thrombus formation in cerebral microvessels of mice expressing hemoglobin-S. Blood 2011, 117, 4125–4133. [Google Scholar] [CrossRef]
- Schulz, C.; Massberg, S. Demystifying the prothrombotic role of NETs. Blood 2017, 129, 925–926. [Google Scholar] [CrossRef]
- Leong, M.A.; Dampier, C.; Varlotta, L.; Allen, J.L. Airway hyperreactivity in children with sickle cell disease. J Pediatr 1997, 131, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Vendramini, E.C.; Vianna, E.O.; De Lucena Ethngulo, I.; De Castro, F.B.; Martinez, J.A.B.; Terra-Filho, J. Lung function and airway hyperresponsiveness in adult patients with sickle cell disease. Am J Med Sci 2006, 332, 68–72. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Barron-Casella, E.A.; Strunk, R.C.; Hamilton, R.G.; Casella, J.F.; DeBaun, M.R. Elevation of IgE in children with sickle cell disease is associated with doctor diagnosis of asthma and increased morbidity. J Allergy Clin Immunol 2011, 127, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Horst, J.; Strunk, R.C.; White, F.V.; DeBaun, M.R. Death due to asthma in two adolescents with sickle cell disease. Pediatr Blood Cancer 2011, 56, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Eiymo Mwa Mpollo, M.S.; Brandt, E.B.; Shanmukhappa, S.K.; Arumugam, P.I.; Tiwari, S.; Loberg, A.; Pillis, D.; Rizvi, T.; Lindsey, M.; Jonck, B.; Carmeliet, P.; Kalra, V.K.; Le Cras, T.D.; Ratner, N.; Wills-Karp, M.; Hershey, G.K.; Malik, P. Placenta growth factor augments airway hyperresponsiveness via leukotrienes and IL-13. The Journal of clinical investigation 2016, 126, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Postma, D.S.; Kerstjens, H.A. Characteristics of airway hyperresponsiveness in asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998, 158, S187–92. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D.G.; Irvin, C.G. Mechanisms of airway hyper-responsiveness in asthma: the past, present and yet to come. Clin Exp Allergy 2015, 45, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M.; Luyimbazi, J.; Xu, X.; Schofield, B.; Neben, T.Y.; Karp, C.L.; Donaldson, D.D. Interleukin-13: central mediator of allergic asthma. Science 1998, 282, 2258–2261. [Google Scholar] [CrossRef] [PubMed]
- Andemariam, B.; Adami, A.J.; Singh, A.; McNamara, J.T.; Secor, E.R.; Guernsey, L.A.; Thrall, R.S. The sickle cell mouse lung: proinflammatory and primed for allergic inflammation. Transl Res 2015, 166, 254–268. [Google Scholar] [CrossRef]
- Nandedkar, S.D.; Feroah, T.R.; Hutchins, W.; Weihrauch, D.; Konduri, K.S.; Wang, J.; Strunk, R.C.; DeBaun, M.R.; Hillery, C.A.; Pritchard, K.A. Histopathology of experimentally induced asthma in a murine model of sickle cell disease. Blood 2008, 112, 2529–2538. [Google Scholar] [CrossRef]
- Niss, O.; Fleck, R.; Makue, F.; Alsaied, T.; Desai, P.; Towbin, J.A.; Malik, P.; Taylor, M.D.; Quinn, C.T. Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia. Blood 2017, 130, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Niss, O.; Quinn, C.T.; Lane, A.; Daily, J.; Khoury, P.R.; Bakeer, N.; Kimball, T.R.; Towbin, J.A.; Malik, P.; Taylor, M.D. Cardiomyopathy With Restrictive Physiology in Sickle Cell Disease. JACC Cardiovasc Imaging 2016, 9, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res 2017, 121, 819–837. [Google Scholar] [CrossRef] [PubMed]
- Varat, M.A.; Adolph, R.J.; Fowler, N.O. Cardiovascular effects of anemia. American heart journal 1972, 83, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Hammoudi, N.; Lionnet, F.; Redheuil, A.; Montalescot, G. Cardiovascular manifestations of sickle cell disease. Eur Heart J 2020, 41, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- James, T.N. Homage to James B. Herrick: a contemporary look at myocardial infarction and at sickle-cell heart disease: the 32nd Annual Herrick Lecture of the Council on Clinical Cardiology of the American Heart Association. Circulation 2000, 101, 1874–1887. [Google Scholar] [CrossRef]
- Gbotosho, O.T.; Taylor, M.; Malik, P. Cardiac pathophysiology in sickle cell disease. J Thromb Thrombolysis 2021, 52, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Chandarajoti, K.; Mackman, N.; Key, N.S.; Pawlinski, R. Thrombin-independent contribution of tissue factor to inflammation and cardiac hypertrophy in a mouse model of sickle cell disease. Blood 2016, 127, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, P.I.; Mullins, E.S.; Shanmukhappa, S.K.; Monia, B.P.; Loberg, A.; Shaw, M.A.; Rizvi, T.; Wansapura, J.; Degen, J.L.; Malik, P. Genetic diminution of circulating prothrombin ameliorates multiorgan pathologies in sickle cell disease mice. Blood 2015, 126, 1844–1855. [Google Scholar] [CrossRef]
- Solovey, A.; Lin, Y.; Browne, P.; Choong, S.; Wayner, E.; Hebbel, R.P. Circulating activated endothelial cells in sickle cell anemia. The New England journal of medicine 1997, 337, 1584–1590. [Google Scholar] [CrossRef]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; Kiger, L.; Renard, J.M.; Larroque, C.; Le Clesiau, H.; Tedgui, A.; Bruneval, P.; Barja-Fidalgo, C.; Alexandrou, A.; Tharaux, P.L.; Boulanger, C.M.; Blanc-Brude, O.P. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Perelman, N.; Selvaraj, S.K.; Batra, S.; Luck, L.R.; Erdreich-Epstein, A.; Coates, T.D.; Kalra, V.K.; Malik, P. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood 2003, 102, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.K.; Giri, R.K.; Perelman, N.; Johnson, C.; Malik, P.; Kalra, V.K. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood 2003, 102, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Boasiako, C.; Donkor, E.S.; Sey, F.; Dzudzor, B.; Dankwah, G.B.; Otu, K.H.; Doku, A.; Dale, C.A.; Ekem, I. Levels of Soluble Endothelium Adhesion Molecules and Complications among Sickle Cell Disease Patients in Ghana. Diseases 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Sarray, S.; Saleh, L.R.; Lisa Saldanha, F.; Al-Habboubi, H.H.; Mahdi, N.; Almawi, W.Y. Serum IL-6, IL-10, and TNFalpha levels in pediatric sickle cell disease patients during vasoocclusive crisis and steady state condition. Cytokine 2015, 72, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Gbotosho, O.T.; Ghosh, S.; Kapetanaki, M.G.; Lin, Y.; Weidert, F.; Bullock, G.C.; Ofori-Acquah, S.F.; Kato, G.J. Cardiac expression of HMOX1 and PGF in sickle cell mice and haem-treated wild type mice dominates organ expression profiles via Nrf2 (Nfe2l2). British journal of haematology 2019, 187, 666–675. [Google Scholar] [CrossRef]
- Meloni, A.; Puliyel, M.; Pepe, A.; Berdoukas, V.; Coates, T.D.; Wood, J.C. Cardiac iron overload in sickle-cell disease. American journal of hematology 2014, 89, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.H.J.; Benites, B.D.; Fertrin, K.Y. Myocardial Iron Overload in Sickle Cell Disease: A Rare But Potentially Fatal Complication of Transfusion. Transfus Med Rev 2019, 33, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Sparkenbaugh, E.; Pawlinski, R. Interplay between coagulation and vascular inflammation in sickle cell disease. British journal of haematology 2013, 162, 3–14. [Google Scholar] [CrossRef]
- Chantrathammachart, P.; Mackman, N.; Sparkenbaugh, E.; Wang, J.G.; Parise, L.V.; Kirchhofer, D.; Key, N.S.; Pawlinski, R. Tissue factor promotes activation of coagulation and inflammation in a mouse model of sickle cell disease. Blood 2012, 120, 636–646. [Google Scholar] [CrossRef]
- Dewerchin, M.; Carmeliet, P. PlGF: a multitasking cytokine with disease-restricted activity. Cold Spring Harbor perspectives in medicine.
- Carnevale, D.; Cifelli, G.; Mascio, G.; Madonna, M.; Sbroggio, M.; Perrino, C.; Persico, M.G.; Frati, G.; Lembo, G. Placental growth factor regulates cardiac inflammation through the tissue inhibitor of metalloproteinases-3/tumor necrosis factor-alpha-converting enzyme axis: crucial role for adaptive cardiac remodeling during cardiac pressure overload. Circulation 2011, 124, 1337–1350. [Google Scholar] [CrossRef] [PubMed]
- Accornero, F.; van Berlo, J.H.; Benard, M.J.; Lorenz, J.N.; Carmeliet, P.; Molkentin, J.D. Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ Res 2011, 109, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Iwama, H.; Uemura, S.; Naya, N.; Imagawa, K.; Takemoto, Y.; Asai, O.; Onoue, K. Cardiac expression of placental growth factor predicts the improvement of chronic phase left ventricular function in patients with acute myocardial infarction. J Am Coll Cardiol 2006, 47, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Kolakowski, S., Jr.; Berry, M.F.; Atluri, P.; Grand, T.; Fisher, O.; Moise, M.A.; Cohen, J.; Hsu, V.; Woo, Y.J. Placental growth factor provides a novel local angiogenic therapy for ischemic cardiomyopathy. Journal of cardiac surgery 2006, 21, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Uemura, S.; Takeda, Y.; Samejima, K.; Matsumoto, T.; Hasegawa, A.; Tsushima, H.; Hoshino, E.; Ueda, T.; Morimoto, K.; Okamoto, K.; Okada, S.; Onoue, K.; Okayama, S.; Kawata, H.; Kawakami, R.; Maruyama, N.; Akai, Y.; Iwano, M.; Shiiki, H.; Saito, Y. Placental Growth Factor as a Predictor of Cardiovascular Events in Patients with CKD from the NARA-CKD Study. Journal of the American Society of Nephrology : JASN 2015, 26, 2871–2881. [Google Scholar] [CrossRef] [PubMed]
- Gbotosho, O.T.; Ghosh, S.; Kapetanaki, M.G.; Lin, Y.; Weidert, F.; Bullock, G.C.; Ofori-Acquah, S.F.; Kato, G.J. Cardiac expression of HMOX1 and PGF in sickle cell mice and haem-treated wild type mice dominates organ expression profiles via Nrf2 (Nfe2l2). British journal of haematology 2019, 187, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Sachdev, V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol 2012, 59, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.C.; Kirkham, F.J.; Redline, S.; Rosen, C.L.; Yan, Y.; Roberts, I.; Gruenwald, J.; Marek, J.; DeBaun, M.R. Left ventricular hypertrophy and diastolic dysfunction in children with sickle cell disease are related to asleep and waking oxygen desaturation. Blood 2010, 116, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Alsaied, T.; Niss, O.; Powell, A.W.; Fleck, R.J.; Cnota, J.F.; Chin, C.; Malik, P.; Quinn, C.T.; Taylor, M.D. Diastolic dysfunction is associated with exercise impairment in patients with sickle cell anemia. Pediatric blood & cancer 2018, 65, e27113. [Google Scholar]
- Sachdev, V.; Machado, R.F.; Shizukuda, Y.; Rao, Y.N.; Sidenko, S.; Ernst, I.; St Peter, M.; Coles, W.A.; Rosing, D.R.; Blackwelder, W.C.; Castro, O.; Kato, G.J.; Gladwin, M.T. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol 2007, 49, 472–479. [Google Scholar] [CrossRef]
- Sachdev, V.; Kato, G.J.; Gibbs, J.S.; Barst, R.J.; Machado, R.F.; Nouraie, M.; Hassell, K.L.; Little, J.A.; Schraufnagel, D.E.; Krishnamurti, L.; Novelli, E.M.; Girgis, R.E.; Morris, C.R.; Rosenzweig, E.B.; Badesch, D.B.; Lanzkron, S.; Castro, O.L.; Taylor JGt Hannoush, H.; Goldsmith, J.C.; Gladwin, M.T.; Gordeuk, V.R.; Walk, P.I. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation 2011, 124, 1452–1460. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.G. Cardiac Arrhythmias: Diagnosis, Symptoms, and Treatments. Cell Biochem Biophys 2015, 73, 291–296. [Google Scholar] [CrossRef] [PubMed]
- John, R.M.; Tedrow, U.B.; Koplan, B.A.; Albert, C.M.; Epstein, L.M.; Sweeney, M.O.; Miller, A.L.; Michaud, G.F.; Stevenson, W.G. Ventricular arrhythmias and sudden cardiac death. Lancet 2012, 380, 1520–1529. [Google Scholar] [CrossRef]
- Desai, R.; Bansod, S.; Patel, U. Nationwide prevalence and trends in acute cardiovascular events and in-hospital mortality among adult African Americans with sickle cell trait. Annals of hematology 2020, 99, 2207–2209. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.D.; Lauder, N.; Jonassaint, J.C.; Telen, M.J.; Zhao, X.; Wright, E.C.; Gilliam, F.R.; De Castro, L.M. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. American journal of hematology 2010, 85, 36–40. [Google Scholar] [CrossRef]
- Manci, E.A.; Culberson, D.E.; Yang, Y.M.; Gardner, T.M.; Powell, R.; Haynes, J., Jr.; Shah, A.K.; Mankad, V.N. Causes of death in sickle cell disease: an autopsy study. British journal of haematology 2003, 123, 359–365. [Google Scholar] [CrossRef]
- Darbari, D.; Kple-Faget, P.; Kwagyan, J.; Rana, S.; Gordeuk, V.; Castro, O. Circumstances of death in adult sickle cell disease patients. Am J Hematol 2006, 81, 858–863. [Google Scholar] [CrossRef]
- Gupta, A.; Fei, Y.D.; Kim, T.Y.; Xie, A.; Batai, K.; Greener, I.; Tang, H.; Ciftci-Yilmaz, S.; Juneman, E.; Indik, J.H.; Shi, G.; Christensen, J.; Gupta, G.; Hillery, C.; Kansal, M.M.; Parikh, D.S.; Zhou, T.; Yuan, J.X.; Kanthi, Y.; Bronk, P.; Koren, G.; Kittles, R.; Duarte, J.D.; Garcia, J.G.N.; Machado, R.F.; Dudley, S.C.; Choi, B.R.; Desai, A.A. IL-18 mediates sickle cell cardiomyopathy and ventricular arrhythmias. Blood 2021, 137, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.A.; Patel, A.R.; Ahmad, H.; Groth, J.V.; Thiruvoipati, T.; Turner, K.; Yodwut, C.; Czobor, P.; Artz, N.; Machado, R.F.; Garcia, J.G.N.; Lang, R.M. Mechanistic insights and characterization of sickle cell disease-associated cardiomyopathy. Circ Cardiovasc Imaging 2014, 7, 430–437. [Google Scholar] [CrossRef]
- James, T.N.; Riddick, L.; Massing, G.K. Sickle cells and sudden death: morphologic abnormalities of the cardiac conduction system. J Lab Clin Med 1994, 124, 507–520. [Google Scholar]
- Bakeer, N.; James, J.; Roy, S.; Wansapura, J.; Shanmukhappa, S.K.; Lorenz, J.N.; Osinska, H.; Backer, K.; Huby, A.C.; Shrestha, A.; Niss, O.; Fleck, R.; Quinn, C.T.; Taylor, M.D.; Purevjav, E.; Aronow, B.J.; Towbin, J.A.; Malik, P. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proceedings of the National Academy of Sciences of the United States of America 2016, 113, E5182–91. [Google Scholar] [CrossRef]
- Niss, O.; Detterich, J.; Wood, J.C.; Coates, T.D.; Malik, P.; Taylor, M.D.; Quinn, C.T. Early initiation of disease-modifying therapy can impede or prevent diffuse myocardial fibrosis in sickle cell anemia. Blood 2022, 140, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Platt, O.S. Sickle cell anemia as an inflammatory disease. The Journal of clinical investigation 2000, 106, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Marker, P.H.; Weber, J.P.; Hebbel, R.P.; Vercellotti, G.M. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood 2000, 96, 2451–2459. [Google Scholar] [CrossRef]
- Pathare, A.; Al Kindi, S.; Alnaqdy, A.A.; Daar, S.; Knox-Macaulay, H.; Dennison, D. Cytokine profile of sickle cell disease in Oman. American journal of hematology 2004, 77, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Nakamura, S.; Saito, Y.; Kosugi, M.; Magata, Y. What can be seen by 18F-FDG PET in atherosclerosis imaging? The effect of foam cell formation on 18F-FDG uptake to macrophages in vitro. J Nucl Med 2012, 53, 55–58. [Google Scholar] [CrossRef]
- Fernandez-Friera, L.; Fuster, V.; Lopez-Melgar, B.; Oliva, B.; Sanchez-Gonzalez, J.; Macias, A.; Perez-Asenjo, B.; Zamudio, D.; Alonso-Farto, J.C.; Espana, S.; Mendiguren, J.; Bueno, H.; Garcia-Ruiz, J.M.; Ibanez, B.; Fernandez-Ortiz, A.; Sanz, J. Vascular Inflammation in Subclinical Atherosclerosis Detected by Hybrid PET/MRI. J Am Coll Cardiol 2019, 73, 1371–1382. [Google Scholar] [CrossRef]
Figure 1.
A visual representation of the inflammatory mediators that may be involved in the cardiopulmonary complications of sickle cell disease. Interleukin 6 (IL-6), IL-5, IL-7, IL-18, IL-13, Tissue factor (TF), C-C motif chemokine ligand 2 (CCL2/3), Reactive oxygen species (ROS), nuclear-factor erythroid 2 like 2 (NRF2), heme-oxygenase 1 (HMOX1), Endothelin-1 (ET-1), Placental growth factor (PlGF), von Willebrand factor (VWF), Danger associated molecular patterns (DAMPs), Chemokine (C-X-C motif) ligand 1 (CXCL1), Factor II (FII), Factor Xa (Fxa) and Transforming growth factor (TGF-β).
Figure 1.
A visual representation of the inflammatory mediators that may be involved in the cardiopulmonary complications of sickle cell disease. Interleukin 6 (IL-6), IL-5, IL-7, IL-18, IL-13, Tissue factor (TF), C-C motif chemokine ligand 2 (CCL2/3), Reactive oxygen species (ROS), nuclear-factor erythroid 2 like 2 (NRF2), heme-oxygenase 1 (HMOX1), Endothelin-1 (ET-1), Placental growth factor (PlGF), von Willebrand factor (VWF), Danger associated molecular patterns (DAMPs), Chemokine (C-X-C motif) ligand 1 (CXCL1), Factor II (FII), Factor Xa (Fxa) and Transforming growth factor (TGF-β).
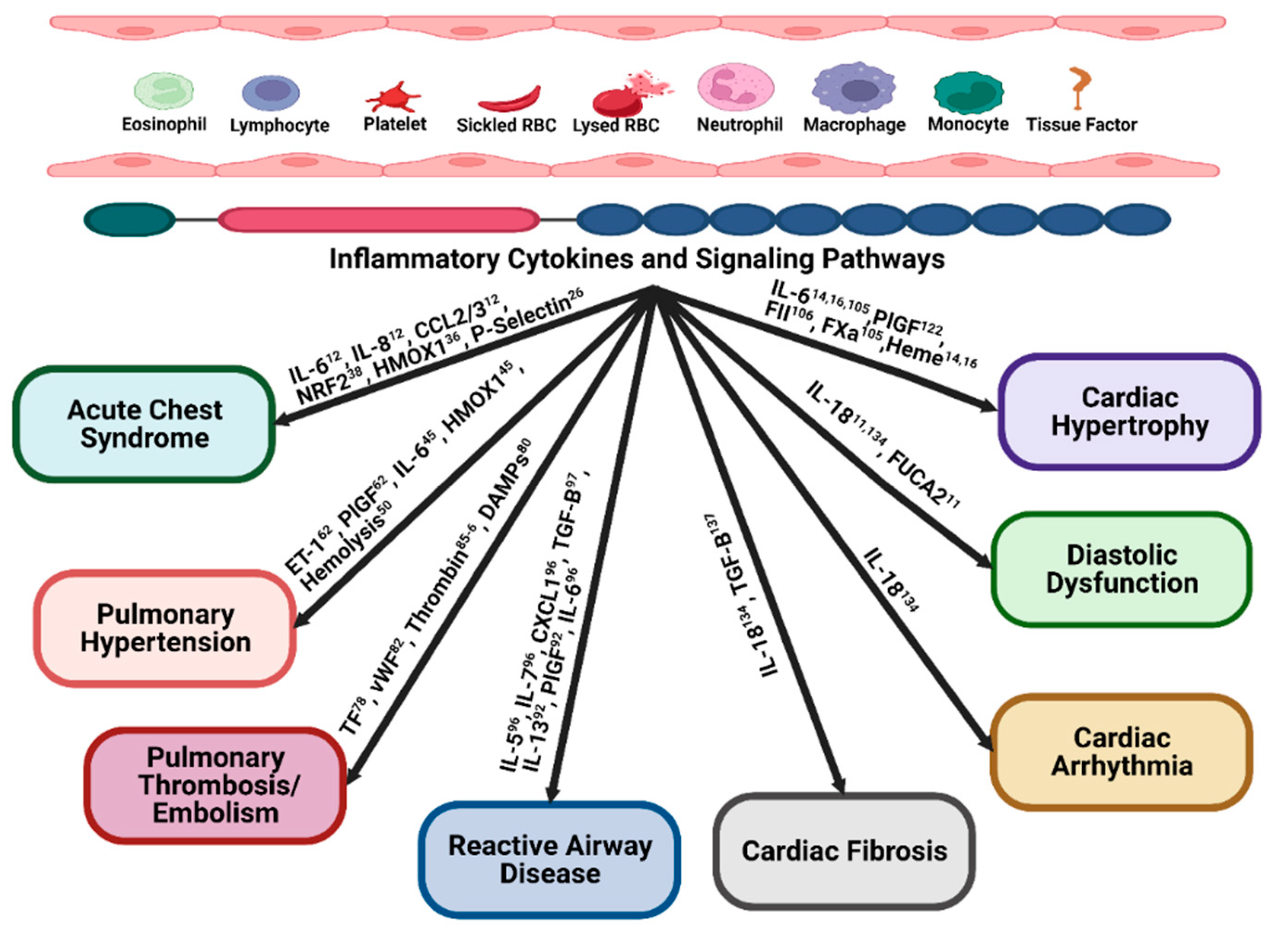
Figure 2.
Summary of the inflammatory mechanisms associated with cardiac fibrosis and diastolic dysfunction in animals with SCD (with the relevant reports that describe them) addressed in the current review manuscript. TGF-β: Transforming growth factor beta, IL-18: Interleukin-18, and FUCA2: -L-fucosidase A2. .
Figure 2.
Summary of the inflammatory mechanisms associated with cardiac fibrosis and diastolic dysfunction in animals with SCD (with the relevant reports that describe them) addressed in the current review manuscript. TGF-β: Transforming growth factor beta, IL-18: Interleukin-18, and FUCA2: -L-fucosidase A2. .


Table 1.
Summary of inflammatory mediators and potential novel treatments in cardiopulmonary complications in sickle cell Disease.
Table 1.
Summary of inflammatory mediators and potential novel treatments in cardiopulmonary complications in sickle cell Disease.
| Disease complication | Major contributors | Potential novel treatments that may target major inflammatory/anti-inflammatory pathways | Citations |
|---|---|---|---|
| 1. Acute Chest Syndrome (ACS) | Free heme, heme oxygenase (HMOX-1), neutrophils and platelet interactions, p-selectin | Glyco-protein Ibalpha inhibitor (CCP-224) [24] D3T (3H-1,2-dithiole-3-thione) [37] Hemopexin [38] |
Anea [24], Jimenez [25], Ghosh [26], Bean [36], Ghosh [37], Ghosh [38], Alishlash [39] |
| 2. Pulmonary hypertension | Endothelial dysfunction, hemolysis, decreased NO, increased placenta growth factor (PIGF), PPAR alpha and PPAR gamma | Hemopexin [38] BAY 54-654455 |
Jang [53], Wood [55], Gonzales [58], Hsu [47], Morris [50] Perelman [109], Selvaraj [110], Potoka [56], Buehler [63] |
| 3. Pulmonary thrombosis | NETs, DAMPs, tissue factor upregulation, lower protein S and C endothelial dysfunction | Anti-TF antibody | Sparkenbaugh [105] Whelihan [85] Faes [77] Solovey [82] |
| 4. Cardiac hypertrophy | ROS, endothelial dysfunction, hemolysis, hypercoagulation, PIGF, IL-6, heme, | Rivaroxaban [104] | Sparkenbaugh [104] Bakeer [137] Gbotosho [16] Menon [14] Arumugam [106] |
| 5. Diastolic dysfunction and cardiac arrhythmia | IL-18, FUCA-2 | Anti-IL-18 binding protein [133] | Duarte [11] Gupta [134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



