
Submitted:
08 February 2023
Posted:
10 February 2023
You are already at the latest version
Abstract
Chicory, a horticultural crop cultivated worldwide, presents many botanical varieties and local biotypes. Among these, Italian radicchio group cultivars of pure Cichorium intybus L. and interspecific hybrids with Cichorium endivia L., as the “Red of Chioggia” biotype which in turn includes several phenotypes. This study deals with a pipeline for the marker-assisted breeding of F1 hybrids: it presents the genotyping-by-sequencing results of four elite inbred lines using a RADseq approach and an original molecular assay based on CAPS markers for screening mutants with nuclear male-sterility in radicchio of Chioggia. Two thousand nine hundred fifty-three SNP-carrying RADtags were identified and used to compute the actual estimates of homozygosity, and overall genetic similarity and uniformity of the populations, as well as to determine their genetic distinctiveness and differentiation. Molecular data were further used to investigate the genomic distribution of the RADtags among the two Cichorium species, allowing their mapping in 1,131 and 1,071 coding sequences in chicory and endive, respectively. In parallel, an assay to screen the genotype at the male-sterility locus Cims-1 was developed to discriminate wild-type and mutant alleles of the causative gene myb80-like. Moreover, a RADtag mapped close to this genomic region proved the potential application of this method for future marker-assisted selection tools. Finally, combining the genotype information of the core collection, the best ten individuals from each inbred line were selected to compute the observed genetic similarity as a measure of uniformity as well as the expected homozygosity and heterozygosity estimates scorable by the putative progenies derived from selfing (pollen parent) and full-sibling (seed parent) or pair-wise crossing (F1 hybrids). This predictive approach was conducted as a pilot study for understanding the potential application of RADseq in improving molecular breeding strategies aimed at the development of inbred lines and F1 hybrids in leaf chicory.
Keywords:
chicory
; endive
; RADseq
; male-sterility
; molecular breeding
; SNPs
; inbred lines
; F1 hybrids
; predicted breeding value
1. Introduction
Cichorium intybus L., or chicory (2x=2n=18), is a perennial leafy vegetable belonging to the Asteraceae family. This species, cultivated worldwide for its adaptability to various environmental conditions, has been domesticated into the current vegetable products. It is an economically important European horticultural crop, and it is one of the most important among those cultivated in the Veneto region, north-eastern Italy, where it is differentiated into multiple different biotypes. The phenotypic variability observable in this species is well-represented by the local varieties of C. intybus originally from the Veneto region, where this crop took the traditional name “radicchio” [1,2,3,4,5]. Multiple chicory varieties are cultivated in this area, where their common ancestral biotype was first introduced during the 17th century, the “Late Red of Treviso”. During the following centuries, other biotypes differentiated by selection or due to interspecific mating with its related interfertile species, namely, Cichorium endivia L. (endive) [3,5]. Within the Cichorium genus, the compatible and crossable species C. endivia (2x=2n=18) is present, which is characterized by having a completely different mating habit from C. intybus. In the case of chicory, sporophytic self-incompatibility (SSI) causes an obligate allogamous mating system that makes the development of highly homozygous inbred lines difficult to achieve [4,6]; while in the case of endive, a prevalently autogamous species, cross-pollination is more difficult than in chicory, thus inhibiting the easy obtainment of F1 hybrids. Despite this, and as previously mentioned, it is possible to cross these two Cichorium species for the development of interspecific hybrids. In the past, by crossing these two species, biotypes of radicchio appeared, such as the “Red of Chioggia”, which now has economic importance and traditional value and can become a genetic variability resource for future breeding improvement plans in this crop [3,5,7,8]. Moreover, nuclear male-sterility (NMS) was found to be another efficient reproductive barrier observed in chicory, particularly in “Red of Chioggia” biotype varieties [9,10], capable of enhancing cross-pollination. NMS in Red of Chioggia is the result of a 4nt insertion (5’-AATT-3’) within the second exon of the myb80-like gene that, in the recessive homozygous state (msms), is responsible for the lack of pollen liberation from anthers[11]. The insertion causes a frameshift of the coding sequences, which, as a consequence, produces a shorter and nonfunctional MYB80-like protein due to the insurgence of a “stop” codon in between the second exon of the gene. Consequently, when in the recessive homozygous state, the male-sterile phenotype appears.
These two reproductive barriers (SSI and NMS), known for reducing self-pollination and enhancing heterozygosity in natural populations, can be useful or inhibitory in crop breeding depending on the aim. In fact, as they force out-breeding, thus reducing the frequency of homozygous progenies, these two reproductive barriers negatively influence the development of inbred parental lines, but they can be positively adopted to direct pair-wise crossings for the constitution of F1 hybrids. In particular, SSI in chicory, which is reported to be incompletely functional allowing low ratios of inbreeding [12,13,14,15], normally inhibits self-pollination and the constitution of inbred lines; while MS, also preventing self-pollination, can be exploited to direct cross-pollination between two genetically differentiated parental lines using one as pollen parent (male-fertile; Ms/-) and the other as seed parent (male-sterile; msms), paternal and maternal genotypes, respectively. Adopting this breeding strategy, highly heterozygous F1 hybrid progenies can be obtained by avoiding the harvesting of selfing-derived seeds from the male-sterile maternal line. Given the characteristics and possible implications of these two reproductive barriers, the adoption of molecular tools for screening breeding populations to select the desired genotypes would improve the breeding strategies of Cichorium crops by reducing the time and costs needed to obtain the target plant.
While the first biotypes of radicchio were selected by phenotypic mass selection, the current breeding strategies are supported by genotypic selection assisted by molecular markers and genomics as the main analytical tools. Currently, the newly released varieties of pure C. intybus as well as its interspecific biotypes with C. endivia are mainly F1 hybrids developed by Italian or European seed firms through large-scale single crosses between inbred lines selected according to their specific combining ability [5,16]. Thus, to maximize the heterosis phenomenon [17]), breeding programs of radicchio have significantly improved in recent years thanks to the use of more efficient molecular tools and analytical platforms [16,18,19,20,21]. To date in chicory, several linkage maps saturated with DNA markers and spanning the entire genome size have been produced and are available [18,19,21,22]. In particular, 29 selected simple sequence repeat (SSR) markers have demonstrated their potential in determining genetic similarities and differences in chicory populations for distinctiveness, uniformity and stability (DUS) testing and plant variety protection (PVP) [16]. In addition to SSR markers, one phenotype-related molecular marker associated with NMS in chicory has been recently developed that is based on allele-specific PCR (AS-PCR) for Myb80-like gene genotyping [11].
Breeding projects assisted by molecular markers require informative and reliable genotyping approaches, and the possibility of predicting a specific phenotype by characterizing the genotype of one specific locus can greatly help and speed the development of new varieties or the preservation or implementation of specific phenotypes. Currently, multiple genomic tools can be used [23], and the cost-effective aspect has become fundamental in the choice of which method to adopt for genotyping analyses. Among the various approaches that can be chosen, strategies based on genotyping-by-sequencing (GBS) demonstrated their suitability for the practical applications previously mentioned. There are many GBS-related techniques that differ in the sequencing platform, the chemistry they are based on, the type of output, data throughput and other aspects [24,25,26,27,28,29]. Among the available GBS approaches, restriction-site associated DNA sequencing (RADseq) [30] has demonstrated its potential use in different crop species for breeding, PVP and traceability purposes [16,31,32]. Among the several advantages provided by the adoption of this approach is the unnecessity of an already sequenced genome because a comparative analysis between the sequenced reads can be made even without mapping them onto chromosomes. This certainly makes RADseq a suitable method for partially or poorly unstudied species, even if the availability of a representative genome remains key helpful information for sequencing-based approaches.
Starting from these purposes, the final goal of this study is to provide a screening protocol for genotyping these crops and their interspecific hybrids for breeding purposes by providing a faster, cheaper, and more informative approach than the already available PCR-based markers for genotyping. Taking advantage of the recently published genomes of both chicory and endive [33], in this study, we evaluated the technical potential and robustness of a RADseq approach as a strategy for GBS-related analysis. In this framework, RAD sequencing has been adopted for the molecular characterization of four populations of the interspecific biotype “Red of Chioggia” of radicchio that derives from an ancestral crossing of C. intybus and C. endivia. In detail, the aims were to verify the suitability of the RADseq method for genotyping this crop, to establish its ability to determine the genetic distinctiveness and uniformity of four full-sibling (FS) lines and to hypothesize the average heterozygosity and genetic similarity obtainable in the progenies derived from specific crosses or self-pollination in future breeding plans. A representation of the canonical breeding schemes exploiting the NMS locus for breeding full-sibling and selfing-derived inbred lines is available in Figure 1. Highly homozygous lines can be subsequently used in pair-wise crossing systems between highly dissimilar inbred lines to produce F1 hybrid seeds.
In addition, parallel to the GBS approach and given the nucleotide sequence of the 4nt insertion (AATT) within the previously mentioned myb80-like male-sterile genotype, the adoption of a restriction enzyme that cuts depending on its exact sequence would provide a fast and reliable method for predicting male-fertile and male-sterile phenotypes before anthesis and even before flower development. With this aim, the Tru1I restriction enzyme was considered for its adoption in a CAPS marker essay able to distinguish the 3 possible genotypes and the related phenotypes (2 fertile: MsMs and Msms; 1 sterile: msms). Consequently, the investigation of associations between the GBS-derived RADtags and the CAPS results has been performed concerning male-sterility identification, aiming for future GBS-based marker-assisted selection (MAS) protocols without the need for further experiments.
2. Results
RAD sequencing output
After the gDNA was extracted and evaluated and the 96 plant samples initially considered were sequenced, two of them were excluded due to a high number of missing data and sequencing quality results. The molecular data for the remaining 94 samples were retained for bioinformatic analyses.
RAD sequencing produced approximately 180 million total raw reads with an average of 1.9 million reads per sample. After quality assessment and adapter trimming, the obtained reads were used to create a catalogue of consensus loci and then used for variant calling as a reference, as described in Stevanato et al. [31]. After variant calling, a starting pool of 9,351 SNPs was obtained, contained in 8,918 RADtags. In a subsequent filtering step, the RADtags presenting at least one missing value among the population were discarded to increase the stringency of the analysis, and the remaining 2,953 SNPs, contained in 2,917 RADtags, were used for the genetic statistics analyses to maintain proper representativeness.
Genetic statistics and AMOVA
Regarding the genetic statistics computed for each population, the number of observed alleles ranged from 1.36 to 1.56 (na), and that of the effective alleles (ne) was between 1.21 and 1.47. The observed (Ho) and expected (He) heterozygosity estimates were 12.09% and 11.93% minimum and 25.96% and 23.43% maximum, respectively, while the fixation index (F) ranged from 0.17 to -0.14. The percentages of polymorphic loci (PL%) and private alleles (PA%) were also computed and ranged from 56.48% to 36.23% for the first value and from 8.79% to 0.03% for the second value. All genetic statistics were calculated for each considered population and are reported in Table 1. The mean values were also computed for each statistic and are reported in Table 1.
Along with the genetic statistics for each population, the mean within-population expected (Hs) and total (Ht) heterozygosity, Wright’s F-statistics, and the gene flow (Nm) were also computed and reported in Table 2. Moreover, AMOVA was also computed to calculate the molecular variance within and among populations. The obtained results, shown in Table 3, indicate that 73.10% of the molecular variance is represented among populations and 26.90% within them. The computed probability is 0.001.
Genetic similarity (GS) estimates
From the GS analysis in all pairwise comparisons among the core collection, the resulting GS matrix (Supplementary Table 1) was used to compute the mean GS within and among each of the four populations represented in the core collection, together with the respective standard errors (±SE) (Table 4).
In detail, the genetic similarity ranged overall from 47.20% to 98.56% in single genotype estimations, with an average value of 65.53% among the core collection. The mean GS values within populations ranged from 82.33% (Pop1) to 91.05% (Pop4), while those among populations were between 54.08% (Pop3 vs. Pop4) and 76.60% (Pop1 vs. Pop2). Standard errors were also computed that were always |SE| < |0.05%| (Table 4).
From the GS matrix obtained from the analysis of the 94 samples of the core collection successfully sequenced, a UPGMA dendrogram was created that grouped samples into four distinct branches according to the samples’ population of origin. One exception was observed for sample “Pop1-06” (labelled in blue), which was grouped closer to Cluster B (labelled in red), even though it was part of Pop1 (Figure 2).
Considering the results shown in the UPGMA dendrogram, a comparison between Pop3, coloured in green, and the other three populations, respectively “Pop1”, “Pop2” and “Pop4”, was made that highlighted an observed GS of 54.66%, while that between the three populations located in one main branch of the dendrogram was 72.79%. For genetic similarity, observed homozygosity (Obs. Hom.) was also estimated, which presented the highest value in sample “Pop4-21” (Obs. Hom. = 94.48%), while the lowest was observed in sample “Pop1-03” (Obs. Hom. = 66.71%). Among the core collection, the mean observed homozygosity was 82.94%, while that within each population ranged from 74.04 ± 0.31% (Pop1) to 87.91 ± 0.13%.
The GS matrix was also used to perform a principal coordinate analysis (PCoA) that clustered samples in the chart depending on Dimensions 1 and 2, which represented 50.4% and 35.7% of the molecular variability, respectively (86.1% in total) (Figure 3). This analysis was based on the eigenvectors calculated starting from the genetic similarity matrix and highlighted the same four clusters previously identified in the UPGMA dendrogram.
In general, the GS results highlighted that all four populations clustered independently from the others, although genetic relatedness was observed between Pop1 and Pop2. In contrast, Pop3 and Pop4 formed unique clusters with their respective individuals.
Genetic structure reconstruction of the core collection
Regarding the investigation of the genetic structure of the radicchio core collection, the results obtained from STRUCTURE software were then analysed using STRUCTURE HARVESTER web software to determine the most likely value of K depending on the ΔK values. The best result observed was K = 4 (∆K = 40,925) (Supplementary Figure 1), which was plotted as a histogram to represent the membership of each individual to one or multiple identified ancestral groups.
What emerged from this analysis was that among the four considered populations, three of them presented high membership values to a specific ancestral genotype (Pop2 to Cluster-2; Pop3 to Cluster-3; Pop4 to Cluster-4), while Pop1 was admixed with a membership ratio of 50:50 between the fourth group Cluster-1 and Cluster-2 (Figure 4).
BLASTn-based RADtag mapping against Cichorium spp. exomes
The BLASTx-based approach used to predictively name the “hypothetical protein” annotations of C. intybus and C. endivia, showed partial results. Specifically, the obtained results associate 40.16% (17,559 among 43,721 CDSs) and 31.13% (16,150 among 51,881 CDSs) of the chicory and endive CDSs to be associated with the L. sativa proteome (11,383 CDS annotations are shared between chicory and endive proteomes; Figure 5). As a consequence, the remaining annotations of the two Cichorium exomes remain named as “hypothetical protein” (Supplementary Table 2 and Table 3). Given this, all 8,918 RADtags obtained from RADseq were used in a preliminary BLASTn investigation against the two Cichorium species genomes that showed 5,850 and 5,404 RADtags matching the C. intybus and C. endivia genomes, respectively. Among the RADtags mapping the entire genomes of the Cichorium species, 740 and 294 were specific for chicory and endive, respectively. These results highlighted the presence of genome-specific RADtags. A further investigation was then conducted with the 2,917 RADtags containing 2,953 SNPs sequenced for all 94 samples (no missing RADtags among any sample) to map them against the two newly annotated Cichorium exomes. In particular, the use of CDSs instead of entire genomic sequences aimed at providing predictive information about putative polymorphic positions within coding regions. One thousand three hundred eight and 1,255 RADtags matched 1,131 and 1,071 CDSs with an average identity of 98.00% and 97.23% in C. intybus and C. endivia, respectively (BLASTn results for chicory and endive are available in Supplementary Table 4 and Table 5). The results of the BLASTn and BLASTx analyses for chicory and endive are also reported in Figure 5, in which the shared results of the different computations are highlighted. In particular, 591 CDSs were successfully annotated using L. sativa proteome and were also matched by RADtags in both chicory and endive, while 55 and 52 CDS were annotated in both the Cichorium exomes, but were uniquely matched by RADtags in C. intybus and C. endivia. Moreover, RADtags matching exomes of chicory have been plotted in the corresponding linkage groups and reported in Figure 6. Similarly, in Supplementary Figure 2 the same plotting was made for the results against endive exome. This was made to give a graphical representation of the RADtags distribution across the genomes of the two species. The statistics of the BLASTn results are available in Table 5.
CAPS assay and myb80-like/RADtag association investigation
In breeding programs exploiting male-sterility, the characterization of NMS locus genotype is of main importance both to maintain the male-sterile lines in the breeding population by separating Msms heterozygous genotypes from the msms homozygous male-sterile ones and to properly select the maternal plants for the F1 hybrids constitution. For this reason, practical and easy-to-use screening methods would be helpful.
From the testing experiment of the CAPS essay on samples of known sterile/fertile phenotypes, the obtained results showed complete agreement between the expected and observed results. Specifically, samples known as “fertile”, thus having the Ms/- genotype, always presented one or 3 bands after AGE, while those samples having a “sterile” phenotype always presented 2 bands, as expected and described in the “Methods” paragraph. As shown in Figure 7, one band at ~300 bp height indicates no restriction events by Tru1I enzyme, thus homozygous male-fertile genotype (MsMs); while in the case of two bands present at ~230 bp and ~70 bp, complete digestion of both homologous myb80 alleles occurred (msms). Consequently, the presence of three bands in the AGE was the result of a partial restriction of the PCR products obtained, which reflects the heterozygous genotype Msms. Due to light exposure settings and the reduced size of the shorter restricted fragment, it is not always possible to identify the lowest band at ~70 bp.
After the evaluation of the CAPS essay efficiency and accuracy, a further investigation was performed to verify the presence of RADtags associated with the Myb80-like locus, which could be used in future studies as a predictive tool for determining the MS-locus genotype without using multiple experimental approaches. With this purpose, the results of the RADtag mapping analysis previously described have been used. From these results, it was possible to identify one RADtag matching close to the Myb80-like gene in both the C. intybus genome. From our findings, RADtag_2329 (SNV: A/G; nucleotide sequence: 5’-TGCAGGCGGTTCACACCATTTTAGGTGAGGTTGTTTCATTTACGATTTAC-3’) was found within a “probable amino acid permease 7” (AAP7-like) coding sequence at 13 Kbps from the initial codon of Myb80-like in chicory (CM042017, region: 1,450,846 to 1,449,760). RADtag matching position and genomic region annotations are reported in Figure 8. Given these mapping results, the CAPS assay for male-sterility identification was performed on the 94 samples successfully sequenced. As it was known that none of the sequenced samples was male-sterile, the obtained results agreed with our expectation, and only male-fertile genotypes (Ms/-) have been identified. In particular, the only population presenting heterozygous genotypes was Pop3, in which one disagreement between the MsG/msA association was observed, as one individual presented the Msms genotype at locus Myb80-like and the AA genotype for RADtag_2329. Considering that one mismatch was observed among the 94 genotyped samples (1.06%), this result has been attributed to a sequencing or raw read merging error. Data are reported in Figure 8.
Predicting progeny genotypes in planned selfing, full-sibling and pairwise crossing mating strategies
Consistent with the main aim of this study, concerning the identification of putative pairwise crossing, selfing or full-sibling mating strategies for breeding purposes, heterozygosity values of the putative progenies were computed, as described in the “Methods” below, selecting 10 putative parentals from each of the analysed populations. As reported in Supplementary Table 6, the quadrangular matrix 40×40 presents the genetic similarity values in all pairwise comparisons among the 40 selected parental individuals (below diagonal) and the expected progeny average heterozygosity values (above diagonal). Regarding the chosen parentals, the genotypes homozygosity and heterozygosity are reported (as in Supplementary Table 1). What emerges from this computation is that in case of high genetic similarity between parental individuals the computed progeny heterozygosity results, as expected, very low (selfing and full-sibling mating); while in the pairwise crossing-derived progenies, heterozygosity is increased compared to the parentals and, as hypothesized, the best pair-wise crossing results can be obtained from the most dissimilar and homozygous parentals. In Figure 9, the GS values computed within and among the 40 selected parentals are shown in combination with their respective homozygosity, which is represented as bubbles differing in size depending on the calculated standard deviation. In parallel, the plotting of the expected heterozygosity values of the hypothetical progenies is reported in Figure 10, where selfing-derived values of heterozygosity are shown to always be lower than those of the full-sibling progenies (especially in slightly uniform parental populations such as Pop1) and much lower than those of the pairwise crossing. One particular exception is represented by the pairwise crossing between Pop1 and Pop2, in which the plotted results are comparable with those obtained in the hypothetical full-sibling.
3. Discussion
The analysis performed in this study confirmed the suitability of the GBS method used. As supported by previous research [31,32,34,35], the restriction site-associated DNA sequencing method is a reliable tool for genotyping crop species. Genotyping-by-sequencing through RADseq and SNP markers has many possible applications for breeders in MAB, MAS, or PVP purposes. In comparison to other molecular PCR-based approaches, the advantages of RADseq consist of having not only the genotype information but also the availability of nucleotide sequences for each of the marker loci analysed, which can then be mapped in the respective or related crop’s genome. This can be made by requiring less time and with higher precision than using canonical PCR-based approaches, thus allowing the development of more reliable and informative screening assays. Moreover, given the high data throughput of the method adopted (1.8 million raw reads per sample; 9,351 SNVs and over 8 thousand RADtags), the identification of multiple discriminant alleles for different phenotypes would improve MAS approaches by combining multiple information in a single experiment [27]. These aspects, combined with the reduction in the analysis cost per sample and compared to PCR-based assays [36], make RADseq a suitable and effective analytical tool for both breeding and basic research applications.
In this study, we successfully analysed 94 individuals belonging to 4 distinct populations of chicory. The genetic statistics, based on almost three thousand SNP markers, demonstrated the good informativeness of the provided method. The ne, which is slightly lower than na, as expected, reflects a relevant variability (Total ne = 1.32; Total na = 1.46) within the observed populations and overall. In agreement with these findings, the F value was close to or below zero among the four populations, thus indicating random mating within them, which is consistent with the full-sibling nature of the analysed populations. Moreover, PL% indicates a relevant variability of both the molecular markers used in the analysis and the analysed populations. Notably, the observed number of private alleles (%PAmax = 8.79% in Pop3 and %PAtotal = 16.00%) reflects variable distinctiveness within and among the four populations analysed, which is important information that can be used for PVP and traceability purposes. Other findings that indicated the genetic variability within populations are the observed heterozygosity (Hotot = 17.89%) and the F-statistics reported in Table 2. Noteworthy, gene flow (Nm = 0.631) indicates a tendency of the population to inbreed, in agreement with the full-sibling nature of the populations analysed. Another result, obtained through the AMOVA analysis, reports that almost 75% of the total molecular variation occurs among populations (Table 3), which, combined with PA values and the other statistics already discussed, suggests a good distinctiveness of the four single FS lines used in this study, as well as a relatively high genetic uniformity of each population (82.33% < within GS < 91.05%). Moreover, as a confirmation of this hypothesis, both the results of the among-population GS and genetic structure separate individuals into four distinct clusters reflecting the core collection division into the four populations. Each of the individuals of the core collection is associated with its original population, but, as suggested by the UPGMA dendrogram (Figure 1), individuals from Pop1 and Pop2 are closely related. Additional information sustaining these findings was obtained from the STRUCTURE analysis, in which Pop1 was admixed with a 50% membership on average with Pop2’s Cluster-2 (Figure 4), thus suggesting that Pop1 is probably derived from Pop2. Moreover, the homozygosity results, combined with the within GS calculated for each population, highlighted different uniformity degrees, from highly uniform and homozygous populations (Pop4) to less uniform and heterozygous ones (Pop1). In this regard, the predictive analysis of the potentially obtainable progenies from selfing, full-sibling or pairwise crossing the best parental individuals selected from each population starting from the RADseq characterization highlights that parentals presenting high levels of homozygosity and within GS can be preferentially adopted for the constitution of F1 hybrid lines (e.g., Pop3 × Pop4), while those that do not match these requirements are more indicated for homozygosity increments, though self- or sibling mating strategies (e.g., Pop1). Moreover, highly related parentals, even those belonging to different populations, make cross-pollinations undesirable for constituting F1 hybrids due to the low obtainable heterozygosity, as demonstrated by the expected He (e.g., Pop1× Pop2) (Figure 7 and Figure 8).
As this study aims to provide a pipeline suitable for MAB, MAS and PVP, not only the possibility of overall genotypic characterization based on genome-wide platforms but also the locus-specific characterization of molecular markers related to specific traits is important. In this regard, the locus association between the RADtag_2329 and Myb80-like genomic regions is a possible example. Male-sterility predictive information can be adopted in future screening analyses aimed at the development of F1 hybrid cultivars by means of pair-wise crosses with highly differentiated pollen donors in order to maximize hybridity and heterozygosity, and so to exploit the potential manifestation of heterosis. In fact, from the identification of the msms genotypes, seed parental lines or clones can be selected to be crossed using fertile genotypes (MsMs) as pollinators. This strategy prevents maternal plants from developing inbred seeds, characterized by higher homozygosity, thus favouring the production of only F1 hybrid seeds derived from cross-pollination strategies, which have higher heterozygosity and potential heterotic vigour [17]. Parallel to this first promising result, the same goal can be achieved for other RADtags. The potential of RADseq methodology in identifying molecular markers associated with specific genes involved in phenotypic traits of agronomic interest has been highlighted by the RADtag mapping analysis results, consisting of 1,084 matches with both C. intybus and C. endivia exomes, which can be investigated in future studies for the validation of suitable molecular markers for MAS approaches. A further insight related to these results is that the BLASTn analysis against the newly available genomes of chicory and endive reported 5,110 RADtags (also considering those with missing values among one or more samples) shared among the two genomes, while 740 and 294 were specific for C. intybus and C. endivia, thus supporting the relatedness to these two species and the interspecific nature of the “Red of Chioggia” biotype to which all tested samples belong. This latter information provides useful knowledge for several purposes: interspecific hybrid identification, and phylogenesis and evolution studies of these two related crop species. Moreover, the possibility of investigating which of the identified SNPs were contained in expressed genomic regions (1,308 and 1,255 in chicory and endive, respectively) could provide a suitable starting point to develop future screening tools for MAS strategies in both Cichorium species, as well as genetic traceability tools for protecting cultivars from frauds or mislabelling (PVP). Another application in which this information can be used is the identification of suitable genetic resources to be introduced or genetic traits to be introgressed by specific breeding programs for each of these two crop plants, aimed at improving their environmental adaptability, resistance or tolerance to pests, and morphological characteristics of agronomic and commercial value.
In conclusion, combining a genome-wide characterization (i.e. MAB strategy) with genotype-specific information (e.g. MAS for male-sterility) allowed us to precisely assess the genetic value-added of parental inbreds and to design tailored pair-wise cross combinations for the development of true and highly heterozygous F1 hybrids in leaf chicory.
4. Materials and Methods
Plant Materials
Four full-sibling (FS) populations of Italian red chicory, biotype “Red of Chioggia”, for a total number of 96 individuals, were used in this study for RADseq-based genotyping. Populations belonging to separated inbred lines were selected to verify the homozygosity and genetic similarity within and among them, respectively. Genomic DNA was extracted for each sample using 100 mg of fresh leaf tissue using the DNeasy® 96 Plant Kit (Qiagen, Valencia, CA, USA) and following the protocols provided by the manufacturer. After DNA extraction and purification, DNA quality, quantity and integrity were evaluated using a NanoDrop 2000c UV‒Vis spectrophotometer (Thermo Fisher Scientific Inc., Pittsburgh, PA, USA), and agarose gel electrophoresis was prepared as a 1% agarose/1× TAE gel containing 1× Sybr Safe DNA stain (Life Technologies, Carlsbad, CA, USA). Moreover, the genomic DNA purification, quantification and quality evaluation protocols described above were also adopted to isolate the gDNA of 12 samples of known fertile/sterile phenotypes derived from three different segregating populations to be used for CAPS assay validation. Regarding the origin of these samples, four individuals belonging to a BC1 line with only heterozygous (Msms) and sterile homozygous (msms) individuals, four individuals from an F2 line with all three possible genotypes represented, and four individuals from a fertile BC1 population with heterozygous and fertile homozygous (MsMs) genotypes were adopted.
RADseq analysis and data analysis
Among the 96 gDNA samples obtained, 94 were successfully sequenced through the restriction site-associated DNA sequencing (RADseq) approach, while due to the high number of missing data, two samples were not considered in the analyses. RADseq analysis was performed using 1 μg of gDNA per sample and restricted using MseI enzyme (New England Biolabs, Ipswich, MA, USA); the procedure is described by Stevanato et al. [31]. For library preparation, digested DNA samples were diluted to a concentration of 3 ng/μL, and indexing, library preparation, sequencing, and bioinformatic analyses of the raw reads were performed according to the protocol described by Stevanato et al. [31]. Raw reads obtained through an Ion S5 sequencer (Thermo Fisher Scientific Inc., Waltham, MA, USA) were trimmed according to the restriction enzyme recognition motif. After quality assessment, all artefacts and Ns-containing reads were removed, and nucleotide variants were assessed using Stacks v2.41 software. [37]. SNPs with a sequence depth <4× and with more than two allelic variants were filtered out. Only biallelic SNPs were considered in this study.
Data analysis and genetic statistics
The obtained biallelic molecular marker data were analysed to calculate genetic statistics. The genetic similarity (GS) was computed using Rohlf’s simple matching coefficient in all pairwise comparisons, and the resulting GS matrix was used for the construction of an unweighted pair group method with arithmetic mean (UPGMA) dendrogram and a principal coordinate analysis (PCoA) using NTSys software v2.21 [38]. Moreover, the GenAlEx 6.5 [39] Excel macro was used to calculate the number of observed and effective alleles, the observed and expected heterozygosity estimates, the fixation index, the percentage of polymorphic loci, the number and percentage of private alleles of each population and identified overall, Wright’s F statistics, heterozygosity estimates and gene flow. AMOVA was also computed using GenAlEx software. Alongside the genetic statistics and similarity estimates, a Bayesian clustering algorithm implemented in STRUCTURE v.2.2 [40] was used to reconstruct the genetic structure of the core collection. The parameters adopted in this analysis consisted of numbers of founding groups ranging from 1 to 20, and 10 replicate simulations were conducted for each value of K based on a burn-in of 200,000 and a final run of 1,000,000 Markov Chain Monte Carlo (MCMC) steps. STRUCTURE HARVESTER [41] was used to estimate the most likely value of K based on the STRUCTURE software analysis results, and the estimates of membership were plotted as a histogram using an Excel spreadsheet.
BLASTn analysis and RADtag mapping
After the genetic statistics analysis, a further investigation was performed to verify which identified SNPs belonged to coding regions of the chicory genome.
Considering the newly published genomic assembly for both chicory (C. intybus L.) and endive (C. endivia L.), we considered these two datasets for the following BLASTn analysis (BLAST+ 2.11.0 package). The two assemblies were retrieved from NCBI using the accession numbers GCA_023525715.1 (chicory) and GCA_023376185.1 (endive). Although the two assemblies presented putative coding sequence (CDS) annotations (51,881 and 43,721 for endive and chicory, respectively), these were only reported as “hypothetical proteins” with no information about their genic function or gene ontology with other species. For this reason, and because our intention was the identification of the CDS-matching RADtags with possible phenotypic interaction/effect, further analysis was needed to putatively assess the nature of the CDS annotations. For this purpose, the translated exome of lettuce (Lactuca sativa L.; NCBI accession number: GCF_002870075.3) was adopted in a preliminary BLASTx analysis to annotate the two Cichorium CDSs using their translation as queries. For this analysis, UseGalaxy [42] implementing BLASTx (BLAST+ 2.11.0 package) software was used in two parallel computations for the two species considered.
After the Cichorium species genome annotation, two parallel BLASTn analyses were conducted to map the RADtags presenting no missing information among the core collection against the newly annotated CDSs. All obtained RADtags, both the completely shared and those missing in at least one genotype, were used in two BLASTn analyses against the entire genomic sequences (intra- and extragenic). In detail, the parameters adopted in these steps followed those described in Scariolo et al. [43], in which the following parameters were used: an E-value threshold ≤1.0 × 10−10 and a percentage of identity ≥80%, plus a minimum query coverage of 80%.
RADtag mapping and BLASTn analysis results were plotted in a Venn diagram using InteractiVenn web software [44] to highlight the shared CDSs annotated through L. sativa exome support, the CDSs shared between two exomes of the Cichorium species, plus the RADtag matches that were shared between the two reference exomes and those that were specific for one or the other. Moreover, two linkage maps have been created presenting C. intybus and C. endivia linkage groups that report the mapping region of the RADtags matching CDS in the exomes of the two species. Linkage maps were created using MapChart 2.32 [45]
Cross-strategy planning for breeding purposes
Considering the genotypes characterized through the RADseq approach, the GS estimates in all pairwise comparisons among the 94 samples successfully sequenced and analysed, as well as their observed homozygosity values, a further investigation of the most suitable crosses for different breeding purposes was performed. Specifically, the genotype information of the 10 selected individuals for each of the four populations was used to hypothesize the genetic estimates obtainable in putative progenies derived from selfing, full-sibling (within the population) and pairwise crossing (between different populations) breeding strategies. In particular, the possible application and aims of this investigation could be the obtainment of highly heterozygous and uniform F1 hybrids or the increment of homozygosity and uniformity within the populations considered in this study. To investigate the possible application of the GBS-derived information, the 40 selected genotypes were chosen for presenting high GS with individuals from the same population (within GS>90%, when possible) and the highest homozygosity. This was made to simulate the mating scenarios of inbreeding, aimed at obtaining highly uniform and homozygous putative parental lines, and out-breeding for developing F1 hybrids. Considering that the molecular markers analysed in this study are biallelic SNPs, the canonical Mendelian gene recombination expected frequencies were adopted for this computation, although the loci association was not considered as no information is available yet about the most common regions of crossing-over in chicory.
In Table 6, the expected heterozygosity frequency values associated with each genotype × genotype crossing combination are reported. By adopting the information reported in Table 6, the He of all pairwise crossing combinations was computed for each locus in the case of selfing, full-sibling or pairwise crossing breeding strategies.
CAPS screening essay for male-sterility identification
For the development of the CAPS essay for male-sterility characterization in chicory, research from Palumbo et al. has been followed. Two specific primers were designed by Primer3 software using as references the myb80 sequences retrieved from NCBI and published by Palumbo et al. [11] under the accession numbers MK285054.1 (sterile phenotype) and MK285053.1 (fertile phenotype). The designed primers were named CiMyb80_for (sequence: ACTGCGGTTGCTGGTCA) and CiMyb80_rev (sequence: CCCTGCTCATGCTCCTG) and were used for the amplification of a 302 bp long sequence containing a palindromic insertion (when present) of 4 nucleotides (AATT) that is responsible for the coding frameshift that causes the male-sterile phenotype. The initial primer testing phase was performed using the following protocol: gDNA amplifications were performed using a Veriti 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA, USA) in a total volume of 20 μL using 1x Platinum Multiplex PCR Master Mix (Thermo Scientific, Carlsbad, CA, United States), 5% GC Enhancer (Thermo Scientific), 0.25 μM of each primer, 30 ng of gDNA and sterile water to volume; the PCR thermal conditions were as follows: 5’ at 95 °C; 35 cycles at 95 °C for 30”, 56 °C for 30”, and 72 °C for 45”; and a final extension at 72 °C for 10’. After the PCR step, the obtained amplicons were digested using the Tru1I restriction enzyme (Thermo Scientific) that recognizes the 5’-AATT-3’ nucleotide motif that exactly matches the 4 nt insertion in the male-sterile allele (ms). Restriction reactions were performed adopting the following procedure: 10 µL of PCR product, 1x Buffer R, 3 U/µL of Tru1I restriction enzyme, and sterile water up to 30 µL final volume. Restriction reactions were performed using a Veriti® 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA, USA) for 16 h (overnight) at 65 °C. After amplicon restriction, digested products were evaluated by agarose gel electrophoresis (AGE) using 2% agarose/1 × TAE gels containing 1 × SYBR Safe DNA Gel Stain (Life Technologies) to separate the eventually restricted amplicon patterns. Specifically, undigested (1 band at 300 bp height – MsMs homozygous male-fertile genotype), partially digested (3 bands at 300 bp, 230 bp and 70 bp heights – Msms heterozygous male-fertile genotype) and completely restricted amplicons (2 bands at 230 bp and 70 bp heights – msms homozygous recessive male-sterile genotype) were expected. Samples used in the validation of the method described here were selected for having a known phenotype (Ms/-: fertile; msms: sterile) and genealogy.
5. Conclusions
To conclude, the genotyping method adopted in this study has demonstrated its informativeness and discriminative ability in distinguishing populations belonging to the same biotype and in finding genetic relationships among closely related breeding lines. The RADseq approach proved to be suitable for reconstructing the genetic structure of a breeding core collection, highlighting genomic information about the ancestral genotypes of different lines, and analysing interspecific hybrids. Moreover, through the use of related species’ assembled genomes, crop comparative analyses can be performed to exploit new genetic resources, which can be adopted for many breeding and traceability purposes, as already demonstrated in other studies [16,32,46,47]. Moreover, the CAPS marker for male-sterility characterization discussed here has proven its reliability and its possible application in breeding planning, as well as its associated RADtag_2329, which can be used in future wide-spectrum GBS approaches in this species.
More studies should be performed in the future to verify the informative and predictive potential of RAD sequencing in this crop, especially focusing on discriminative and phenotype-related markers to increase the possible application of this biotechnological tool. Nonetheless, the obtained results are encouraging for the possibility of developing high-throughput MAS and MAB screening assays to be adopted in next-generation breeding approaches. In addition to breeding aims, RADseq has another potential use related to phylogeny and interspecific hybrid characterization [48,49,50], which can provide informative knowledge on the interspecific biotypes of radicchio as the “Variegated of Castelfranco” or the “Red of Chioggia” studied in this work. From future studies related to this aspect, new interspecific crosses and their phenotypes could be developed that could improve breeding and genetic resource exploitation useful for this crop improvement.
Supplementary Materials
The following supporting information can be downloaded at Preprints.org: Figure S1: Graph representing the ∆K values resulting from Structure Harvester software analysis of STRUCTURE software results; Figure S2: Chromosome representation of C. endivia L. RADtags matching CDS among the 9 assembled linkage groups (LG1 to LG9) are reported; Table S1: Genetic similarity matrix (94x94) in all pairwise comparisons; homozygosity (Ho) and heterozygosity (Ho) are reported for each genotype; Table S2: BLASTx results of "hypothetical" annotated C. intybus proteome against L. sativa proteome; Table S3: BLASTx results of "hypothetical" annotated C. endivia proteome against L. sativa proteome; Table S4: BLASTn results of RADtags against annotated C. intybus exome; Table S5: BLASTn results of RADtags against annotated C. endivia exome; Table S6: Quadrangular matrix 40×40 presenting the GS values in all pairwise comparisons among the 40 selected parental individuals (below diagonal), the expected progeny average heterozygosity values (above diagonal) and the parental individual’s homozygosity and heterozygosity (two columns left side).
Author Contributions
Conceptualization, F.S. and G.B.; methodology, F.S.; software, F.S.; validation, F.S., S.F. and F.P.; formal analysis, F.S.; investigation, F.S.; resources, G.B.; data curation, F.S.; writing—original draft preparation, F.S.; writing—review and editing, F.P., S.F. and G.B.; visualization, F.S., F.P., S.F. and G.B.; supervision, G.B. and F.P.; project administration, G.B.; funding acquisition, G.B. All authors have read and agreed to the published version of the manuscript.
Funding
This study was performed within the Agritech National Research Center and received funding from the European Union Next-Generation EU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) – MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.4 – D.D. 1032 17/06/2022, CN00000022). In particular, our study represents an original paper related to Spoke 4 "Multifunctional and resilient agriculture and forestry systems for the mitigation of climate change risks" and a baseline for the fulfilment of the milestones within Task 4.1.1. titled “Next-generation genotyping and -omics technologies for the molecular prediction of multiple resilient traits in crop plants”. This manuscript reflects only the authors’ views and opinions, and neither the European Union nor the European Commission can be considered responsible for them.
Data Availability Statement
Not pertinent
Acknowledgments
This research was carried out by taking advantage of plant materials of leaf chicory kindly provided by Blumen Group SpA (Bologna, Italy).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bianchedi, A. I radicchi di Treviso: storia, coltivazione, forzatura e commercio. Ital. Agric. 1961, 98, 37–51. [Google Scholar]
- Pimpini, F.; Chillemi, G.; Lazzarin, R.; Bertolini, P.; Marchetti, C. Il Radicchio Rosso di Chioggia. Aspetti tecnici ed economici di produzione e conservazione; Veneto Agricoltura: Legnaro, Padova, 2001. [Google Scholar]
- Lucchin, M.; Varotto, S.; Barcaccia, G.; Parrini, P. Chicory and Endive. In Vegetables I: asteraceae, brassicaceae chenopodi-caceae; Prohens, J., Nuez, F., Eds.; Springer: New York, NY, 2008; Vol. 1, pp. 3–38. [Google Scholar]
- Barcaccia, G.; Ghedina, A.; Lucchin, M. Current Advances in Genomics and Breeding of Leaf Chicory (Cichorium intybus L.). Agriculture-Basel 2016, 6, 50. [Google Scholar] [CrossRef]
- Basso, A.; Scariolo, F.; Negrisolo, E.; Lucchin, M.; Barcaccia, G. Molecular Relationships and Genetic Diversity Analysis of Venetian Radicchio (Leaf Chicory, Cichorium intybus subsp. intybus var. sylvestre, 2n = 2x = 18) Biotypes. Diversity 2022, 14. [Google Scholar] [CrossRef]
- Varotto, S.; Pizzoli, L.; Lucchin, M.; Parrini, P. The incompatibility system in Italian red chicory (Cichorium intybus L). Plant Breed. 1995, 114, 535–538. [Google Scholar] [CrossRef]
- Barcaccia, G.; Varotto, S.; Soattin, M.; Lucchin, M.; Parrini, P. Genetic and molecular studies of sporophytic self-incompatibility in Cichorium intybus L. In Proceedings of Eucarpia meeting on Leafy Vegetables Genetics and Breeding; 2003.
- Barcaccia, G.; Pallottini, L.; Soattin, M.; Lazzarin, R.; Parrini, P.; Lucchin, M. Genomic DNA fingerprints as a tool for identifying cultivated types of radicchio (Cichorium intybus L.) from Veneto, Italy. Plant Breed. 2003, 122, 178–183. [Google Scholar] [CrossRef]
- Barcaccia, G.; Tiozzo Caenazzo, S. New male sterile Chicorium spp. mutant, parts or derivatives, where male sterility is due to a nuclear recessive mutation linked to a polymorphic genetic marker, useful for producing mutant F1 hybrids of Chicorium spp. WO2012163389-A1, 2012. [Google Scholar]
- Barcaccia, G.; Tiozzo Caenazzo, S. New male sterile mutant of leaf chicory, including Radicchio, used to produce chicory plants and seeds with traits such as male-sterility exhibiting cytological phenotype with shapeless, small and shrunken microspores in dehiscent anthers. 2014. US2014157448-A1, 2017. [Google Scholar]
- Palumbo, F.; Qi, P.; Pinto, V.B.; Devos, K.M.; Barcaccia, G. Construction of the First SNP-Based Linkage Map Using Genotyping-by-Sequencing and Mapping of the Male-Sterility Gene in Leaf Chicory. Front Plant Sci 2019, 10, 276. [Google Scholar] [CrossRef]
- Eenink, A.H. Gametic competition after mixed pollinations and double pollinations. In Compatibility and incompatibility in witloof-chicory (Cichorium intybus L.); Euphytica, 1982; Vol. 31, pp. 773–786. [Google Scholar]
- Eenink, A.H. Formation of self-seeds on a self-incompatible and a moderately self-compatible genotype after double and triple pollinations. In Compatibility and incompatibility in witloof chicory (Cichorium intybus L.); Euphytica, 1984; Vol. 33, pp. 161–167. [Google Scholar]
- Coppens d’Eeckenbrugge, G. The progamic phase in Cichorium intybus L. Pollen tube growth in the style, incompatibility reaction and gametophytic competition. Euphytica 1990, 48, 17–23. [Google Scholar] [CrossRef]
- Castaño, C.I.; Demeulemeester, M.A.C.; De Proft, M.P. Incompatibility reactions and genotypic identity status of five commercial chicory (Cichorium intybus L.) hybrids. Scientia Horticulturae 1997, 72, 1–9. [Google Scholar] [CrossRef]
- Patella, A.; Scariolo, F.; Palumbo, F.; Barcaccia, G. Genetic Structure of Cultivated Varieties of Radicchio (Cichorium intybus L.): A Comparison between F1 Hybrids and Synthetics. Plants (Basel) 2019, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.F. Dominance of Linked Factors as a Means of Accounting for Heterosis. Proc Natl Acad Sci U S A 1917, 3, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Cadalen, T.; Morchen, M.; Blassiau, C.; Clabaut, A.; Scheer, I.; Hilbert, J.L.; Hendriks, T.; Quillet, M.C. Development of SSR markers and construction of a consensus genetic map for chicory (Cichorium intybus L.). Molecular Breeding 2010, 25, 699–722. [Google Scholar] [CrossRef]
- Gonthier, L.; Blassiau, C.; Morchen, M.; Cadalen, T.; Poiret, M.; Hendriks, T.; Quillet, M.C. High-density genetic maps for loci involved in nuclear male sterility (NMS1) and sporophytic self-incompatibility (S-locus) in chicory (Cichorium intybus L., Asteraceae). Theor Appl Genet 2013, 126, 2103–2121. [Google Scholar] [CrossRef] [PubMed]
- Muys, C.; Thienpont, C.N.; Dauchot, N.; Maudoux, O.; Draye, X.; Van Cutsem, P. Integration of AFLPs, SSRs and SNPs markers into a new genetic map of industrial chicory (Cichorium intybus L. var. sativum). Plant Breed. 2014, 133, 130–137. [Google Scholar] [CrossRef]
- Galla, G.; Ghedina, A.; Tiozzo, S.C.; Barcaccia, G. Toward a First High-quality Genome Draft for Marker-assisted Breeding in Leaf Chicory, Radicchio (Cichorium intybus L.). In Plant Genomics; Abdurakhmonov, I.Y., Ed.; IntechOpen, 2016. [Google Scholar] [CrossRef]
- Ghedina, A.; Galla, G.; Cadalen, T.; Hilbert, J.L.; Caenazzo, S.T.; Barcaccia, G. A method for genotyping elite breeding stocks of leaf chicory (Cichorium intybus L.) by assaying mapped microsatellite marker loci. BMC Res Notes 2015, 8, 831. [Google Scholar] [CrossRef] [PubMed]
- Simko, I.; Jia, M.Y.; Venkatesh, J.; Kang, B.C.; Weng, Y.Q.; Barcaccia, G.; Lanteri, S.; Bhattarai, G.; Foolad, M.R. Genomics and Marker-Assisted Improvement of Vegetable Crops. Crit. Rev. Plant Sci. 2021, 40, 303–365. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front Plant Sci 2014, 5, 484. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.S.; Choi, S.C.; Jun, T.H.; Kim, C. Genotyping-by-Sequencing: a Promising Tool for Plant Genetics Research and Breeding. Horticulture Environment and Biotechnology 2017, 58, 425–431. [Google Scholar] [CrossRef]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity Arrays Technology: A Generic Genome Profiling Technology on Open Platforms. In Data Production and Analysis in Population Genomics: Methods and Protocols; Pompanon, F., Bonin, A., Eds.; Humana Press: Totowa, NJ, 2012; pp. 67–89. [Google Scholar] [CrossRef]
- Kim, C.; Guo, H.; Kong, W.; Chandnani, R.; Shuang, L.S.; Paterson, A.H. Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant Sci 2016, 242, 14–22. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Stevanato, P.; Broccanello, C.; Biscarini, F.; Del Corvo, M.; Sablok, G.; Panella, L.; Stella, A.; Concheri, G. High-Throughput RAD-SNP Genotyping for Characterization of Sugar Beet Genotypes. Plant Mol Biol Rep 2014, 32, 691–696. [Google Scholar] [CrossRef]
- Yu, Q.; Ling, Y.; Xiong, Y.; Zhao, W.; Xiong, Y.; Dong, Z.; Yang, J.; Zhao, J.; Zhang, X.; Ma, X. RAD-seq as an effective strategy for heterogenous variety identification in plants-a case study in Italian Ryegrass (Lolium multiflorum). BMC Plant Biol 2022, 22, 231. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Wang, S.; Wang, H.; Wang, A.; Jiang, F.; Liu, H.; Zhao, H.; Xu, D.; Zhang, Y. The genomes of chicory, endive, great burdock and yacon provide insights into Asteraceae palaeo-polyploidization history and plant inulin production. Mol Ecol Resour 2022, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 2007, 17, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Sica, P.; Galvao, A.; Scariolo, F.; Maucieri, C.; Nicoletto, C.; Pilon, C.; Sambo, P.; Barcaccia, G.; Borin, M.; Cabrera, M.; et al. Effects of Drought on Yield and Nutraceutical Properties of Beans (Phaseolus spp.) Traditionally Cultivated in Veneto, Italy. Horticulturae 2021, 7, 1–15. [Google Scholar] [CrossRef]
- Mir, R.R.; Hiremath, P.J.; Riera-Lizarazu, O.; Varshney, R.K. Evolving Molecular Marker Technologies in Plants: From RFLPs to GBS. In Diagnostics in Plant Breeding; Lübberstedt, T., Varshney, R.K., Eds.; Springer Netherlands: Dordrecht, 2013; pp. 229–247. [Google Scholar] [CrossRef]
- Rochette, N.C.; Catchen, J.M. Deriving genotypes from RAD-seq short-read data using Stacks. Nat Protoc 2017, 12, 2640–2659. [Google Scholar] [CrossRef] [PubMed]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System Version 2.1: Owner manual; Exeter Publishing: Setauket, New York, 2000. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Scariolo, F.; Palumbo, F.; Vannozzi, A.; Sacilotto, G.B.; Gazzola, M.; Barcaccia, G. Genotyping Analysis by RAD-Seq Reads Is Useful to Assess the Genetic Identity and Relationships of Breeding Lines in Lavender Species Aimed at Managing Plant Variety Protection. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Zhang, H.X.; Li, H.Y.; Li, Y.X. Identifying evolutionarily significant units for conservation of the endangered Malus sieversii using genome-wide RADseq data. Nordic Journal of Botany 2018, 36, e01733. [Google Scholar] [CrossRef]
- Bao, W.Q.; Ao, D.; Wuyun, T.N.; Wang, L.; Bai, Y.E. Development of 85 SNP markers for the endangered plant species Prunus mira (Rosaceae) based on restriction site-associated DNA sequencing (RAD-seq). Conserv. Genet. Resour. 2020, 12, 525–527. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 2016, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Z.; Du, X.; Yang, H.; Han, F.; Han, Y.; Yuan, F.; Zhang, L.; Peng, S.; Guo, E. A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L.) P. Beauv.] using RAD-seq. PLoS One 2017, 12, e0179717. [Google Scholar] [CrossRef] [PubMed]
- Basak, M.; Uzun, B.; Yol, E. Genetic diversity and population structure of the Mediterranean sesame core collection with use of genome-wide SNPs developed by double digest RAD-Seq. PLoS One 2019, 14, e0223757. [Google Scholar] [CrossRef]
Figure 1.
Breeding schemes exploiting male-sterile (msms) and male-fertile (Ms/-) genotypes in full-sibling and selfing strategies for inbred line development. FS: full sibling; S: selfing.
Figure 1.
Breeding schemes exploiting male-sterile (msms) and male-fertile (Ms/-) genotypes in full-sibling and selfing strategies for inbred line development. FS: full sibling; S: selfing.
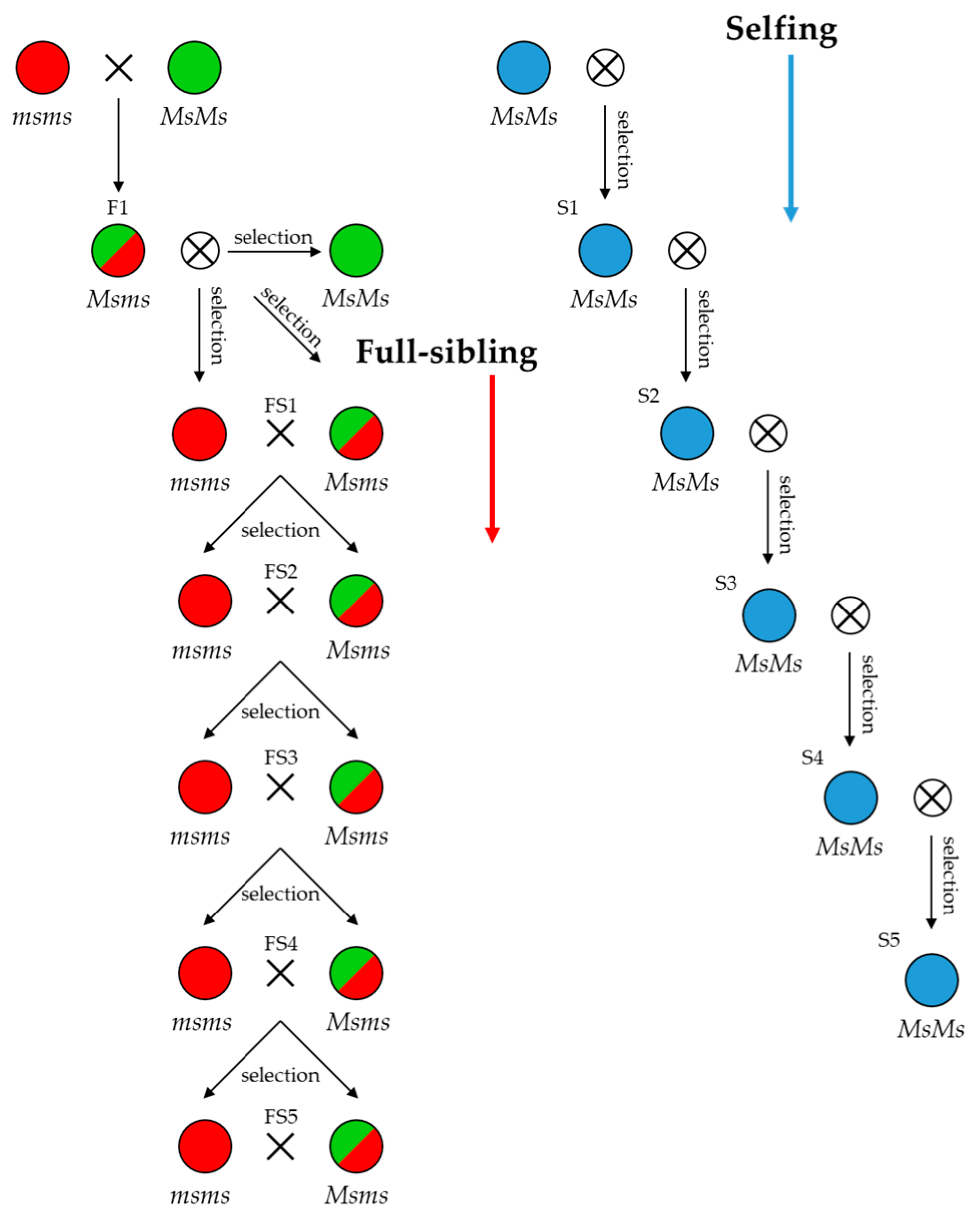
Figure 2.
UPGMA dendrogram based on Rohlf’s simple matching coefficient of genetic similarity. Colours indicate the four different populations analysed in the core collection.
Figure 2.
UPGMA dendrogram based on Rohlf’s simple matching coefficient of genetic similarity. Colours indicate the four different populations analysed in the core collection.
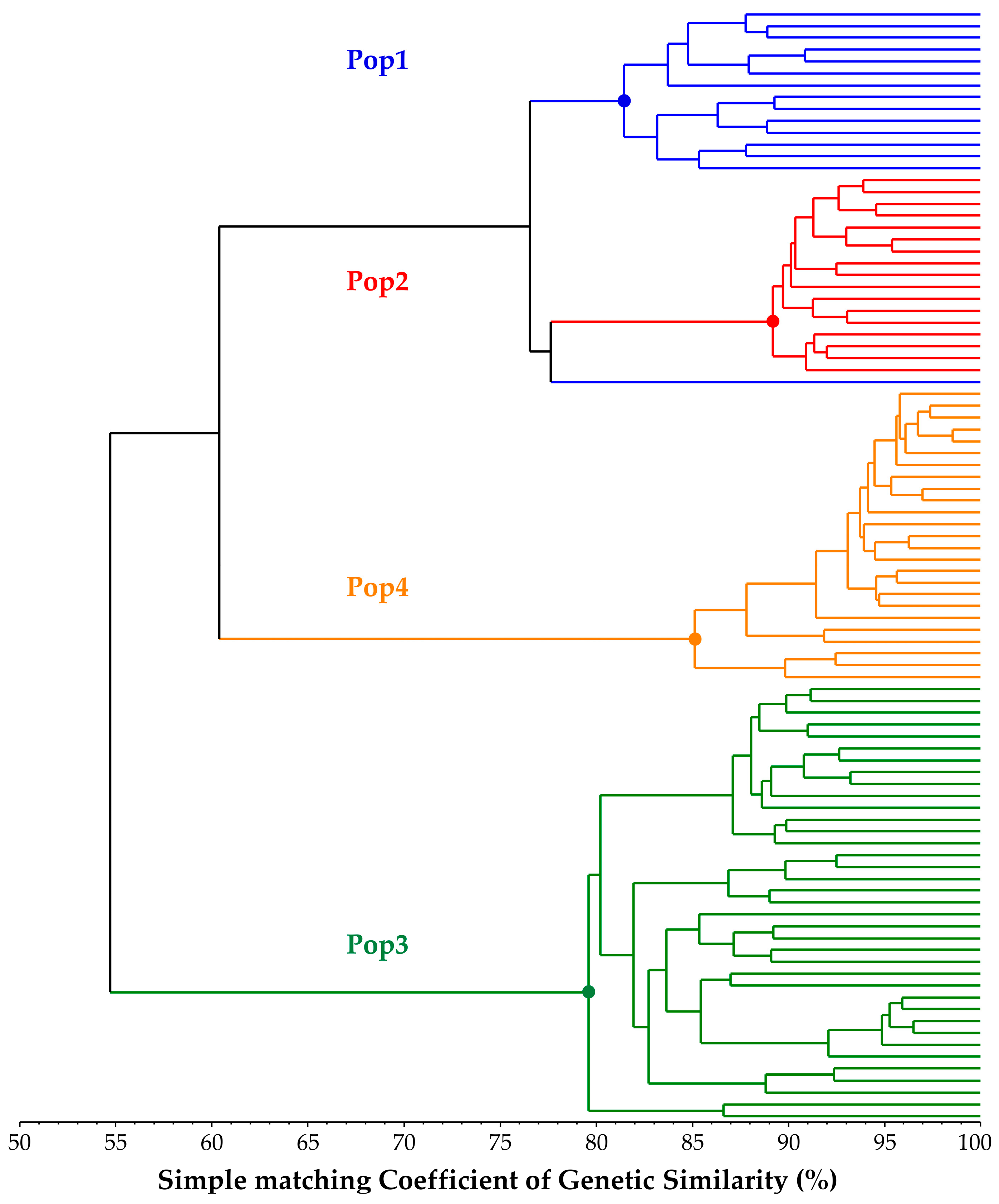
Figure 3.
Principal coordinate analysis grouping the four chicory populations of the core collection into 4 distinct clusters according to samples belonging to populations and the clusters identified in the UPGMA dendrogram. Colour labelling is maintained from the dendrogram illustrated in Figure 2.
Figure 3.
Principal coordinate analysis grouping the four chicory populations of the core collection into 4 distinct clusters according to samples belonging to populations and the clusters identified in the UPGMA dendrogram. Colour labelling is maintained from the dendrogram illustrated in Figure 2.
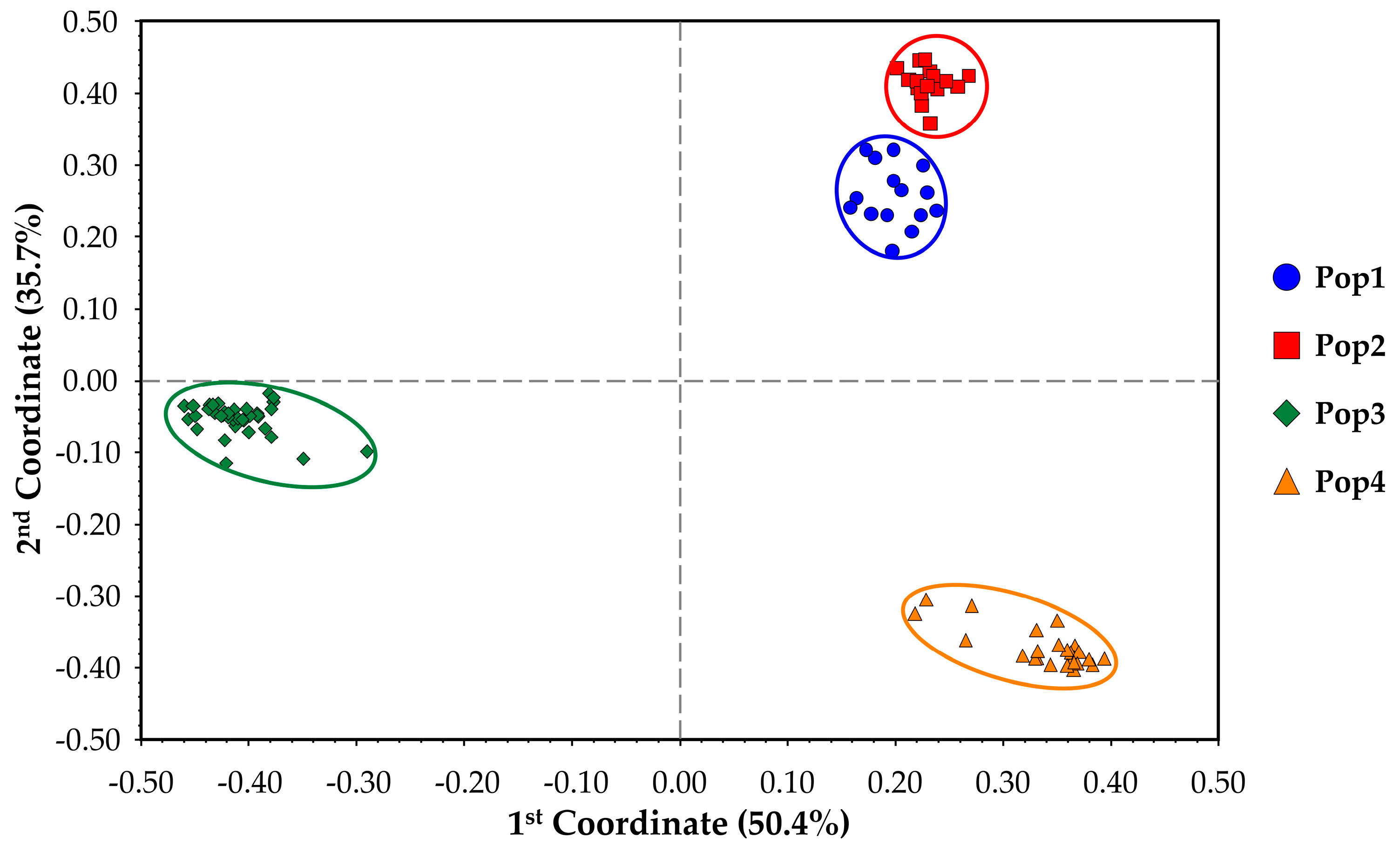
Figure 4.
Histogram representing the STRUCTURE software computed results for K=4. Different colour bars indicate the membership of each individual to a specifically labelled cluster, according to the same labelling adopted in the previous figures.
Figure 4.
Histogram representing the STRUCTURE software computed results for K=4. Different colour bars indicate the membership of each individual to a specifically labelled cluster, according to the same labelling adopted in the previous figures.
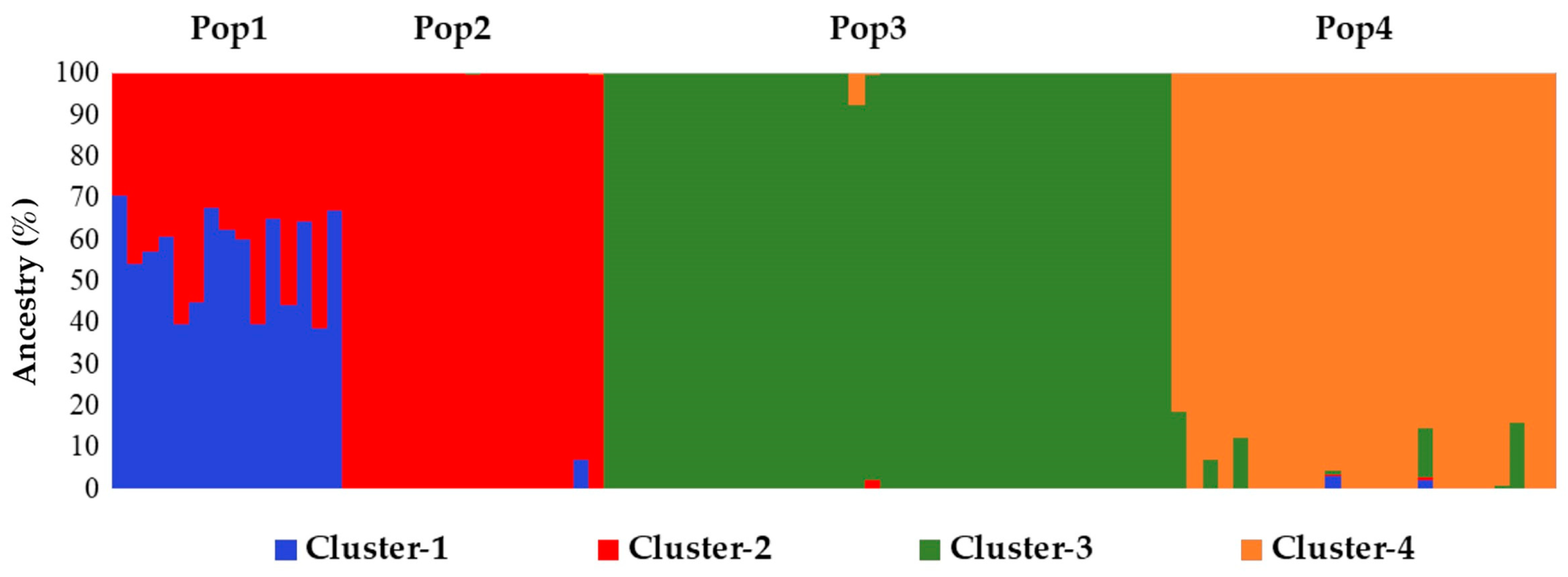
Figure 5.
Venn diagram representing the BLASTn and BLASTx results of the matched CDSs of chicory and endive exomes and the congruences between these results and the CDS annotations obtained using the L. sativa exome as a reference.
Figure 5.
Venn diagram representing the BLASTn and BLASTx results of the matched CDSs of chicory and endive exomes and the congruences between these results and the CDS annotations obtained using the L. sativa exome as a reference.
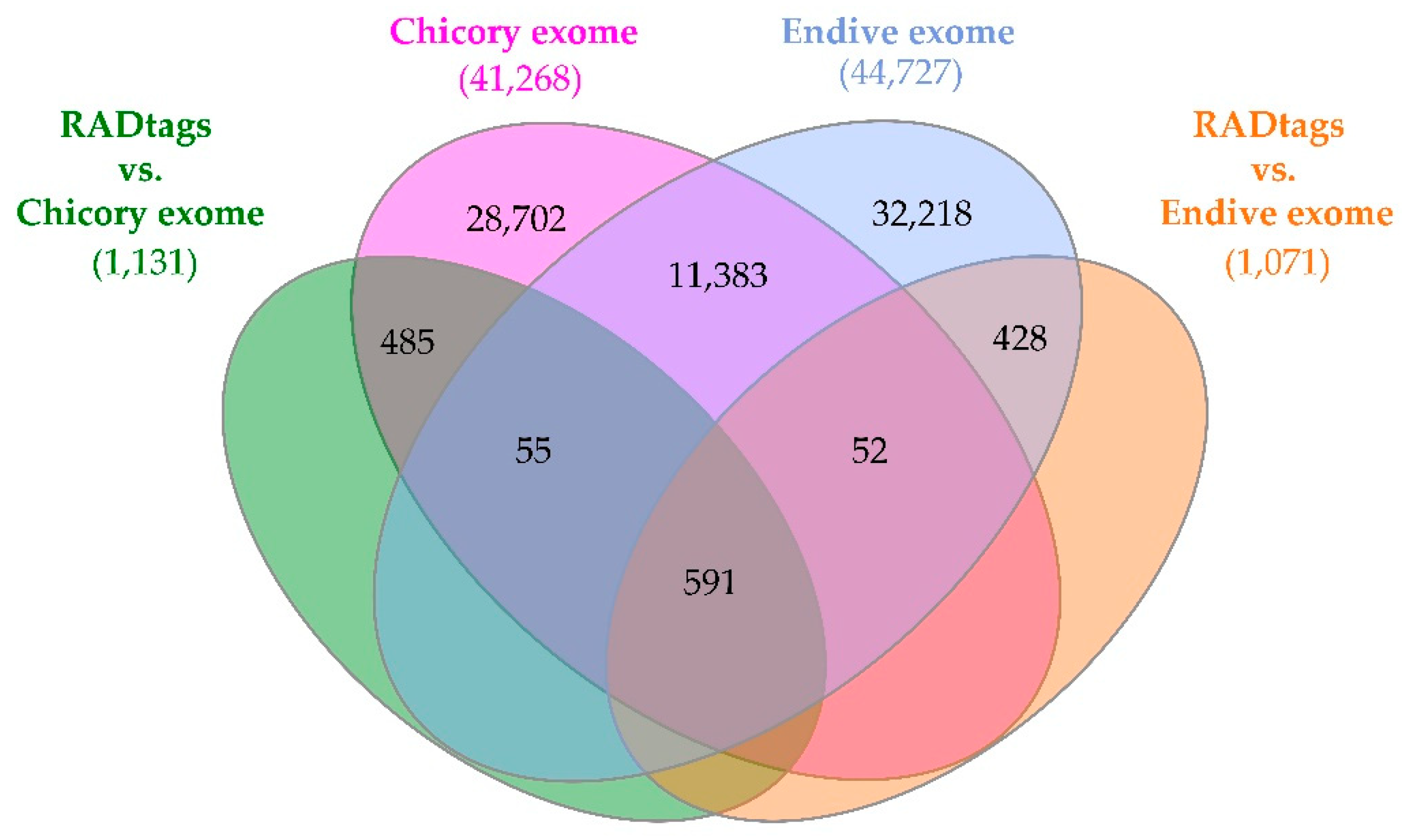
Figure 6.
Chromosome representation of C. intybus L. RADtags matching CDSs among the 9 assembled linkage groups (LG1 to LG9) are reported. Units on the left of each LG indicate 10,000,000 bps.
Figure 6.
Chromosome representation of C. intybus L. RADtags matching CDSs among the 9 assembled linkage groups (LG1 to LG9) are reported. Units on the left of each LG indicate 10,000,000 bps.

Figure 7.
Agarose gel of the male-sterility characterization CAPS marker representing the obtainable bands (white arrows) in the case of the three different observable genotypes at the Myb80-like gene (phenotypes): MsMs (fertile), Msms (fertile), and msms (sterile). (L: molecular weight ladder; white arrows indicate the expected band sizes of 300, 230 and 70 bp, top to bottom)
Figure 7.
Agarose gel of the male-sterility characterization CAPS marker representing the obtainable bands (white arrows) in the case of the three different observable genotypes at the Myb80-like gene (phenotypes): MsMs (fertile), Msms (fertile), and msms (sterile). (L: molecular weight ladder; white arrows indicate the expected band sizes of 300, 230 and 70 bp, top to bottom)
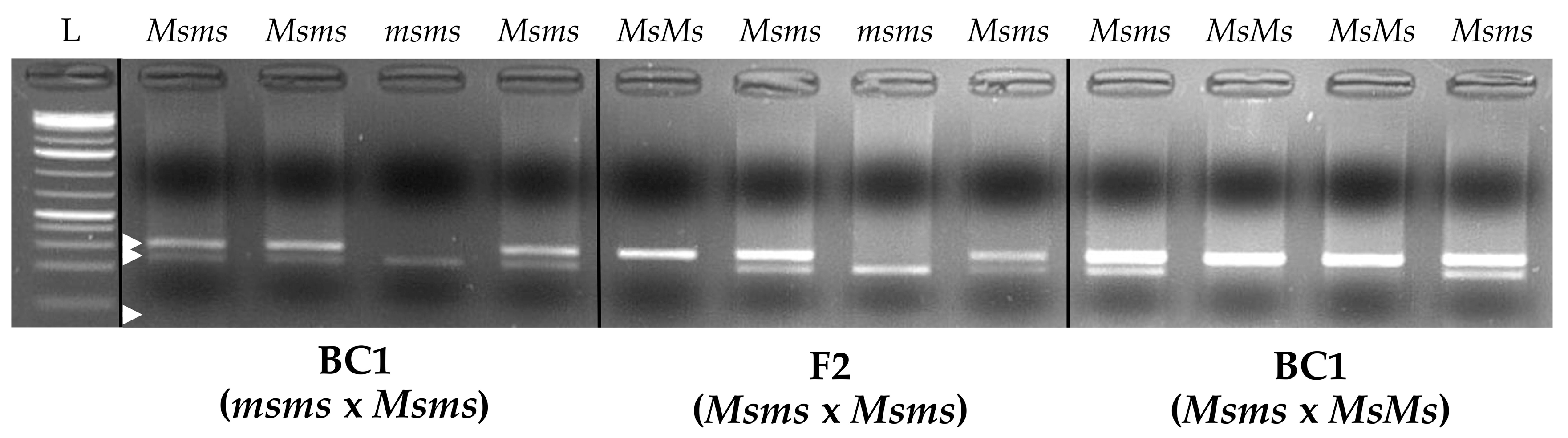
Figure 8.
Graphic representation of RADtag_2329 in the C. intybus genome with the closest AAP7-like and unpredicted genes. Starting and ending positions for each annotation and the RADtag are reported following the sequence direction. Genotype numbers for Myb80-like and RADtag_2329 are reported in the respective tables below the annotation.
Figure 8.
Graphic representation of RADtag_2329 in the C. intybus genome with the closest AAP7-like and unpredicted genes. Starting and ending positions for each annotation and the RADtag are reported following the sequence direction. Genotype numbers for Myb80-like and RADtag_2329 are reported in the respective tables below the annotation.

Figure 9.
Boxplot of the mean genetic similarity among the 40 selected suitable parental individuals calculated within populations (left side) and among populations (right side). Bubbles position in the chart indicates mean homozygosity, while their size reflects the respective standard deviation.
Figure 9.
Boxplot of the mean genetic similarity among the 40 selected suitable parental individuals calculated within populations (left side) and among populations (right side). Bubbles position in the chart indicates mean homozygosity, while their size reflects the respective standard deviation.
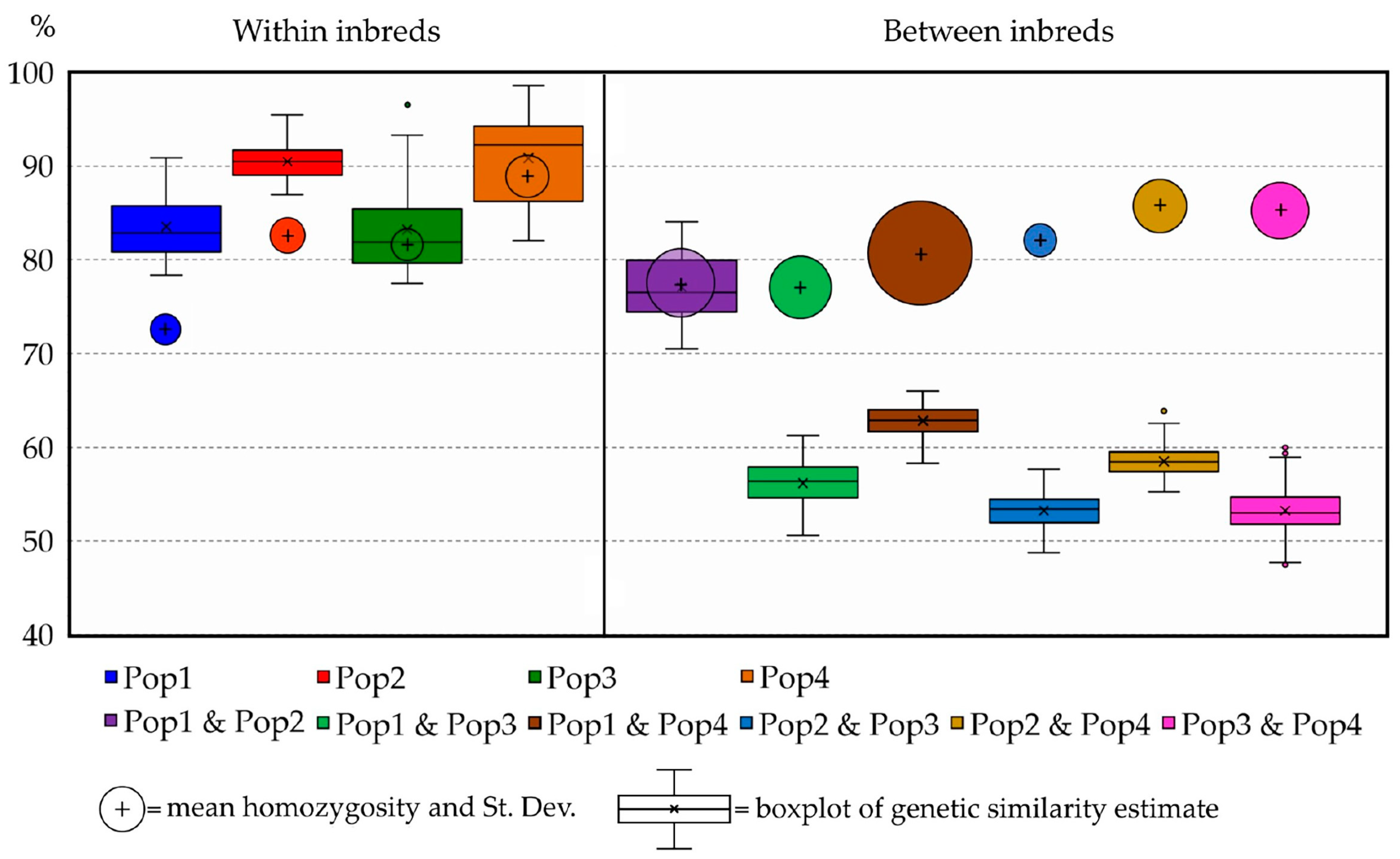
Figure 10.
Boxplot of the obtainable progenies’ heterozygosity (right scale) and homozygosity (left scale) in the case of selfing, full-sibling and pairwise crossing strategies. Maximum, minimum, and average computed values were considered in each grouping computation.
Figure 10.
Boxplot of the obtainable progenies’ heterozygosity (right scale) and homozygosity (left scale) in the case of selfing, full-sibling and pairwise crossing strategies. Maximum, minimum, and average computed values were considered in each grouping computation.
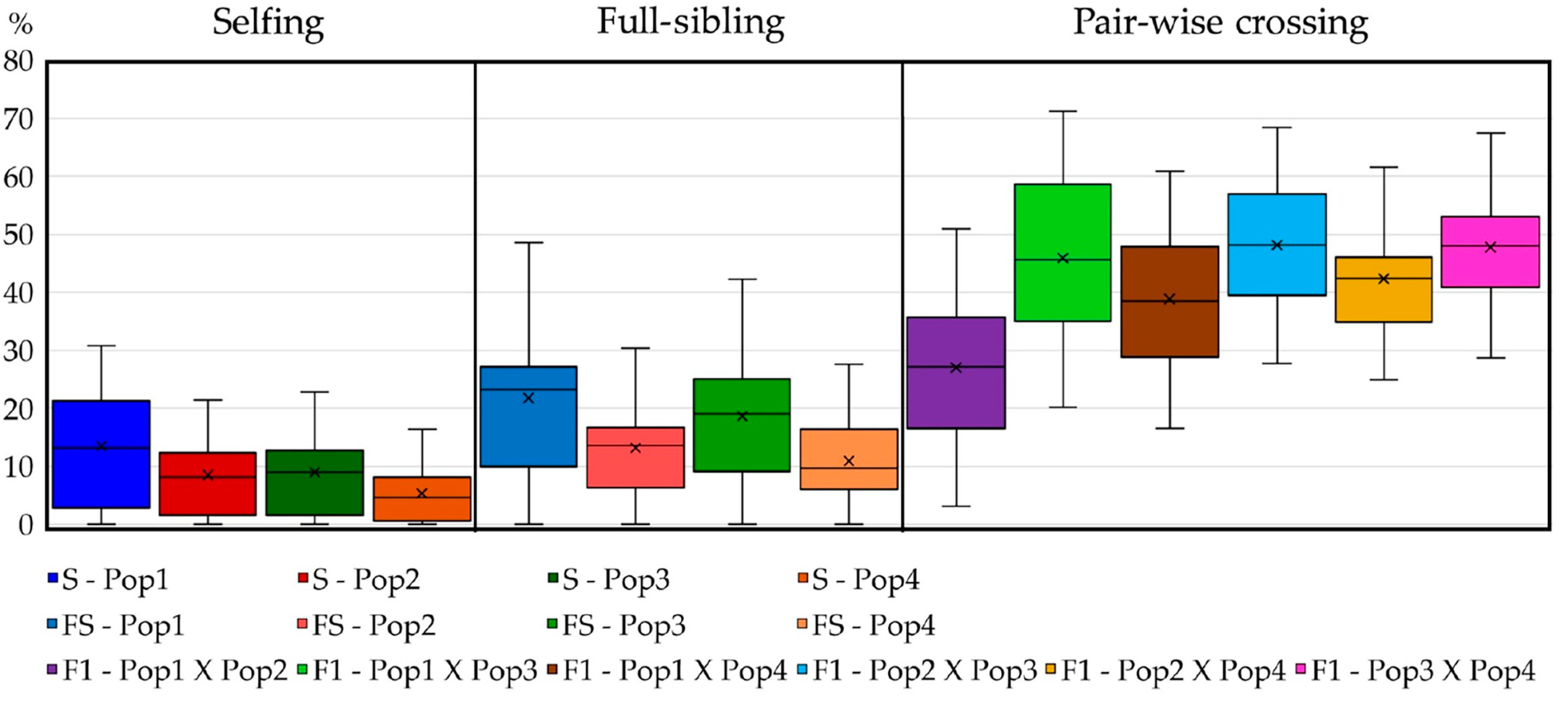

Table 1.
Genetic statistics calculated for each population and mean overall. Reported information is the number of individuals (N), number of observed (na) and expected (ne) alleles, observed (Ho) and expected (He) heterozygosity percentages, fixation index (F), and percentage of polymorphic loci (PL).
Table 1.
Genetic statistics calculated for each population and mean overall. Reported information is the number of individuals (N), number of observed (na) and expected (ne) alleles, observed (Ho) and expected (He) heterozygosity percentages, fixation index (F), and percentage of polymorphic loci (PL).
| Pop ID | N | na | ne | Ho (%) | He (%) | F | PL (%) |
|---|---|---|---|---|---|---|---|
| Pop1 | 15.00 | 1.56 | 1.43 | 25.96 | 23.43 | -0.10 | 56.48 |
| Pop2 | 17.00 | 1.36 | 1.27 | 16.33 | 14.02 | -0.14 | 36.23 |
| Pop3 | 37.00 | 1.54 | 1.38 | 17.17 | 20.77 | 0.17 | 54.01 |
| Pop4 | 25.00 | 1.37 | 1.21 | 12.09 | 11.93 | 0.07 | 37.45 |
| Mean | 23.50 | 1.46 | 1.32 | 17.89 | 17.54 | 0.01 | 46.05 |
Table 2.
Mean within-population expected heterozygosity (Hs), total heterozygosity (Ht), Wright’s F-statistics (Fis; Fit; Fst) and gene flow (Nm) calculated among the core collection. Standard errors (SEs) are also reported for each value.
Table 2.
Mean within-population expected heterozygosity (Hs), total heterozygosity (Ht), Wright’s F-statistics (Fis; Fit; Fst) and gene flow (Nm) calculated among the core collection. Standard errors (SEs) are also reported for each value.
| Hs | Ht | Fis | Fit | Fst | Nm | |
|---|---|---|---|---|---|---|
| Total | 0.18 | 0.36 | 0.03 | 0.48 | 0.48 | 0.63 |
| SE | 0.00 | 0.00 | 0.01 | 0.01 | 0.01 | 0.03 |
Table 3.
AMOVA results. Degrees of freedom (df), sum of squares (SS), means squares (MS), and estimated variance (Est. Var.) with the respective percentage (%) are reported.
Table 3.
AMOVA results. Degrees of freedom (df), sum of squares (SS), means squares (MS), and estimated variance (Est. Var.) with the respective percentage (%) are reported.
| Source | df | SS | MS | Est. Var. | % |
|---|---|---|---|---|---|
| Among Pops | 3 | 104,231 | 34,743 | 1,523 | 73.10 |
| Within Pops | 90 | 50,439 | 560 | 560 | 26.90 |
| Total | 93 | 154,671 | 2,083 | 100.00 |
Table 4.
Observed homozygosity (%) and mean pairwise genetic similarity matrix calculated within and among the four radicchio populations; standard errors are also reported.
Table 4.
Observed homozygosity (%) and mean pairwise genetic similarity matrix calculated within and among the four radicchio populations; standard errors are also reported.
| Obs. Homozygosity (%) | Population ID | Mean Genetic Similarity (%) | |||
|---|---|---|---|---|---|
| 74.04 ± 0.31 | Pop1 | 82.33 ± 0.04 | |||
| 83.67 ± 0.15 | Pop2 | 76.60 ± 0.01 | 90.12 ± 0.01 | ||
| 82.83 ± 0.13 | Pop3 | 56.46 ± 0.00 | 54.13 ± 0.00 | 82.50 ± 0.01 | |
| 87.91 ± 0.13 | Pop4 | 62.59 ± 0.01 | 58.46 ± 0.00 | 54.08 ± 0.00 | 91.05 ± 0.01 |
| Pop1 | Pop2 | Pop3 | Pop4 | ||
Table 5.
BLASTn analysis results of the no-missing data containing RADtags against the C. intybus and C. endivia exomes. Numbers of matching RADtags, CDSs, average mismatches (Avg. mis), average identical positions (Avg. ident) and exome-specific RADtags (Ex. Spec.) are reported alongside the percentages of average identity (Avg. ident), the average length of the alignments (Avg. length), exome-specific RADtags (Ex. Spec. %) and shared RADtags. The supporting statistics, such as E-value, bit score (Bs) and score (score), are also reported as average values for both analyses.
Table 5.
BLASTn analysis results of the no-missing data containing RADtags against the C. intybus and C. endivia exomes. Numbers of matching RADtags, CDSs, average mismatches (Avg. mis), average identical positions (Avg. ident) and exome-specific RADtags (Ex. Spec.) are reported alongside the percentages of average identity (Avg. ident), the average length of the alignments (Avg. length), exome-specific RADtags (Ex. Spec. %) and shared RADtags. The supporting statistics, such as E-value, bit score (Bs) and score (score), are also reported as average values for both analyses.
| BLASTn results |
RADtags (n) | CDS (n) | Avg. ident (%) |
Avg. ident (n) | Avg. length (bp) | Avg. mis (n) |
Avg. E-value |
Avg. Bs | Avg. score | Ex. Spec. (n) | Ex. Spec. (%) | Shared (n/%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
C. intybus exome |
1,308 | 1,131 | 98.00 | 62.22 | 63.50 | 1.26 | 9.04E-13 | 109.60 | 120.55 | 224 | 15.15 | 1,084 / 73.29% |
|
C. endive exome |
1,255 | 1,071 | 97.23 | 61.91 | 63.70 | 1.74 | 4.24E-13 | 107.73 | 118.43 | 171 | 11.56 |
Table 6.
Parental genotype × genotype crossing combination and relative expected average heterozygosity (He) in the progeny
Table 6.
Parental genotype × genotype crossing combination and relative expected average heterozygosity (He) in the progeny
| P1 genotype* | P2 genotype* | P1×P2 avg. He |
|---|---|---|
| 0 | 0 | 0 |
| 1 | 1 | 0 |
| 0 | 1 | 1 |
| 1 | 0 | 1 |
| 0/1 | 2 | 0.5 |
| 2 | 0/1 | 0.5 |
| 2 | 2 | 0.5 |
* 0: homozygous for allele A; 1: homozygous for allele B; 2: heterozygous.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



