
Submitted:
14 February 2023
Posted:
16 February 2023
You are already at the latest version
Abstract
TGF-ꞵ signaling is a vital regulator for maintaining articular cartilage homeostasis. Runx transcription factors, downstream targets of TGF-ꞵ signaling, have been studied in the context of osteoarthritis (OA). Although Runx partner core binding factor β (Cbfβ) is known to play a pivotal role in chondrocyte and osteoblast differentiation, the role of Cbfβ in maintaining articular cartilage remains obscure. This study investigated Cbfβ as a novel anabolic modulator of TGF-ꞵsignaling and determined its role in articular cartilage homeostasis. Cbfβ significantly decreased in aged mouse articular cartilage and human OA cartilage. Articular chondrocyte-specific Cbfb-deficient mice (Cbfb△ac/△ac) exhibited early cartilage degeneration at 20 weeks old and developed OA at 12 months old. Cbfb△ac/△ac mice showed enhanced OA progression under the surgical-induced mice OA model. Mechanistically, forced expression of Cbfβ rescued Col2α1 and Runx1 expression in Cbfβ-deficient chondrocytes. TGF-ꞵ1-mediated Col2α1 expression failed despite the pSmad3 activation under TGF-ꞵ1 treatment in Cbfβ-deficient chondrocytes. Cbfβ protected Runx1 from proteasomal degradation through Cbfβ/Runx1 complex formation. These results indicate that Cbfβ is a novel anabolic regulator for cartilage homeostasis, suggesting that Cbfβ could protect OA development by maintaining the integrity of the TGF-ꞵ1 signaling pathway in articular cartilage.
Keywords:
articular cartilage
; Runx1/Cbfβ complex
; Osteoarthritis
; TGF-β signaling
; proteasomal degradation
1. Introduction
Osteoarthritis (OA), the most common and painful disease of the joints, affects nearly half of the world’s elderly population and represents an enormous socioeconomic challenge [1,2]. OA is a joint disease with structural and functional changes in the articular cartilage, subchondral bone, ligaments, capsule, synovium, sensory nerve endings, meniscus, and periarticular muscles. Among these structural components, many studies focus on cartilage or subchondral bone. Regarding OA initiation, both subchondral bone and articular cartilage changes are critical factors for OA initiation [1,3,4,5]. A dynamic equilibrium between synthesis and degradation of the extracellular matrix, such as Col2α1, proteoglycans, and Mmp13, controls articular cartilage homeostasis and integrity [4]. The inactivation of one copy of Col2α1 leads to articular cartilage degeneration and increased OA development [6]. In the early stage of OA, cartilage matrix degradation occurs in the superficial zone of the cartilage but later extends to deeper zones as OA progresses [7]. Mmp13 is a major enzyme that targets the breakdown of cartilage extracellular matrix such as type II collagen and proteoglycans [8]. Clinical investigation reveals a strong association between articular cartilage destruction with high Mmp13 expression [9].
Tight control of TGF-ꞵ signaling plays an essential role in articular cartilage homeostasis [10]. Injection of TGF-ꞵ into the knee joint increases proteoglycan, whereas injection of recombinant soluble TGF-ꞵ type II receptor, an endogenous inhibitor, markedly worsens OA phenotypes [11,12]. TGF-ꞵ Type I receptor ALK1 (Activin Receptor-like Kinase) correlated well with Mmp13 expression, whereas ALK5 correlates with aggrecan and collagen type II. In senescent and OA articular chondrocytes, ALK5 expression was significantly decreased compared to ALK1, increasing the ALK1/ALK5 ratio, which is associated with the upregulation of Mmp13 in OA [13]. Indeed, chondrocyte-specific loss of Smad3, ALK5, or the overexpression of dominant-negative TGF-ꞵ type II receptor in mice results in OA development at an early stage [14,15]. These findings indicate that abnormal TGF-ꞵ signaling increases OA development. Thus, the integrity of the TGF-ꞵ signaling pathway is essential for the healthy maintenance of articular cartilage and for preventing OA. Here, we explored the TGF-ꞵ signaling integrity regulated by Cbfβ.
Cbfβ is a partner protein of the runt-related transcription factor (Runx) family, which include Runx1, Runx2, and Runx3. It interacts with the Runt domain of Runx to enhance its DNA binding properties and transcription activity [16,17]. Runx1 is a pivotal transcription factor for chondrogenesis, chondrocyte proliferation, and survival. Runx1 in mouse embryonic mesenchymal cells results in a potent induction of early chondrocyte differentiation markers such as type II collagen but not the hypertrophy marker, type X collagen [18]. Runx1 also acts on the superficial zone of articular chondrocytes to stimulate chondrocyte proliferation and promote anabolic protein expression to maintain articular cartilage integrity [19]. Deletion or carboxyl terminus truncation of Runx2 inhibits endochondral bone formation with the arrest of chondrocyte maturation, resulting in the complete absence of mature osteoblast formation [20,21,22]. Runx2 and Runx3 are both expressed in cartilaginous condensations of the mouse limb. The cooperative roles of these two proteins in mediating chondrocyte maturation, hypertrophy, and cartilage formation have been established in vivo using double-knockout mice [23]. Homozygous deletion of Cbfb in mice causes embryonic lethality during pregnancy due to brain hemorrhage and reduces Runx1 stability and transcriptional activity to block fetal hepatic hematopoiesis [24,25]. Cbfβ plays an essential role in endochondral bone formation through skeletal tissue cell-type-specific Cbfb deletions, including mesenchymal cells [26,27], chondrocytes [28,29], and osteoblasts [30]. Since articular chondrocytes remain as non-hypertrophied hyaline cartilage during development, it is interesting to determine how the joint degenerates and progresses to OA as the articular chondrocytes die prematurely or undergo abnormal hypertrophy [7]. However, the role of Cbfβ during articular cartilage development and the evolution of OA is not well understood.
To determine the function of Cbfβ in articular cartilage and its effect on the development of OA, we analyzed the results of the deletion of Cbfb from articular chondrocytes using Gdf5-Cre and Cbfb-floxed mice together with a surgically induced OA model.
2. Materials and Methods
2.1. Antibodies and reagents
We purchased antibodies, including rabbit anti-HA, anti-Runx1, anti-Runx3, monoclonal anti-Actin, and goat anti-Lamin B antibodies from Santa Cruz Biotechnology (CA, USA), rabbit anti-Myc, anti-col2α1, mouse monoclonal anti-Mmp13, anti-Runx2, and anti-type II collagen from Abcam (MA, USA), rabbit anti-Smad3 and anti-phospho-Smad3 from Cell Signaling (MA, USA), monoclonal anti-Flag, an anti-HA-agarose antibody from Sigma-Aldrich (St. Louis, MO, USA). We obtained TGF- β 1, IL-1β, and recombinant TNF- α from R & D Systems (Minneapolis, USA), and hematoxylin, fast green, Safranin-O, human transferrin, and sodium selenite from Sigma-Aldrich.
2.2. Mice and experimental OA
Mice harboring an articular chondrocyte-specific deletion of Cbfb (CbfbΔac/Δac) obtained by crossing Gdf5-Cre transgenic mice (CreTg/+, kindly provided by Dr. David Kingsley from Stanford University, U.S.A) (38) with Cbfbfl/fl mice (39), both maintained on C57BL/6N background. The loxP sites of the Cbfbfl/fl mice encompass the exon 5 of Cbfβ, resulting in articular chondrocyte-specific deletion of exon 5 in Cbfb△ac/△ac mice. Cbfbfl/fl served as wild-type control (WT). Twelve-week-old male mice in both knees were conducted destabilization by the medial meniscus (DMM) OA surgery, and mice were sacrificed at 8 weeks after DMM surgery to assess OA progression (each group n=6) (40). We used twelve-month-old male mice for spontaneous OA progression analysis (WT=5, Cbfb△ac/△ac=5). All animal procedures followed the guidelines issued by the Institutional Animal Care and Use Committee of Kyungpook National University (KNU-201455).
2.3. Human subjects
Human OA joint cartilage tissues were obtained from OA surgery patients through total knee arthroplasty. The Institutional Review Board of Kyungpook National University Hospital approved using human OA cartilage. We obtained written informed consent from all patients prior to the surgical procedure (IRB File No of KNUH 2022-01-010-001).
2.4. β-galactosidase (β-gal) staining
Whole tissues were fixed for 30 min in 4% paraformaldehyde (PFA) in 0.1 M potassium phosphate buffer (pH 7.4) containing 2 mM MgCl2, 5 mM ethylene glycol tetra-acetic acid (EGTA), then washed for 30 min with 0.1 M potassium phosphate buffer. Tissues were kept in the X-gal staining solution overnight at 37℃ and then washed briefly in phosphate-buffered saline (PBS). We made cryosections of the stained samples, fixed the slides briefly in 4% PFA, washed them in PBS, and counterstained them with Nuclear Fast Red (Sigma). Samples were washed in PBS and analyzed with a Leica microscope [41].
2.5. Assessment of OA severity
Knee joints were fixed with 4% PFA and decalcified with 10% ethylenediaminetetraacetic acid (EDTA; pH 7.4) for 4 weeks. Decalcified tissues were dehydrated by ethanol, embedded in paraffin, and then sectioned with a thickness of 3 μm. We performed Safranin-O staining as in the previous study [5]. Briefly, sections were treated in the following order: deparaffinization, rehydration, and soaking in Weigert’s iron hematoxylin solution for 10 min each, followed by fast green solution and 0.1% Safranin-O solution for 5 min each. Assigning OA progression followed the Osteoarthritis Research Society International (OARSI) diagnosis criteria [5].
2.6. Immunohistochemistry
We quenched the sections with 3% H2O2 and retrieved antigens by boiling them in TEG buffer (1.211 g of Tris and 0.190 g of EGTA in 1L MilliQ-water, pH 9.0). After blocking with 1% bovine serum albumin for 1 hour at room temperature, sections were incubated with anti-Cbfβ, anti-Runx1, anti-type II collagen, and anti-Mmp13 antibodies overnight at 4°C and incubated with goat anti-rabbit IgG or goat anti-mouse IgG conjugated HRP for 1 hour. Signals developed with a DAB substrate-chromogen system (Dakocytomation, Denmark).
2.7. Cell culture
Primary articular chondrocytes derived from the hindlimb knee joint of wild-type and Cbfβ△ac/△acmice on postnatal day 5, as previously described (42). Chondrocytes were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Lonza, ME, USA) containing 10% fetal bovine serum (FBS) (Gibco-BRL, USA) and the appropriate penicillin/streptomycin. Chondrogenic ATDC5 cells were cultured in DMEM/F12 (Lonza) medium supplemented with 5% fetal bovine serum (FBS), 10 μg/ml human transferrin, and 3x10-8 M sodium selenite. Chondrocytes were plated at a density of 2 × 105 cells/well on 6 well plates for drug treatment. Protein or total RNA was isolated from chondrocytes after 24 h treatment with TGF-ꞵ1 (1, 10, or 100 ng/ml), IL-1β (10 ng/ml), or TNF-α (10 ng/ml).
2.8. Western blot analyses
Total protein lysates were isolated from human OA articular cartilage, mouse tibia articular cartilage of 12-month-old mice, and articular chondrocytes derived from articular cartilage by M-PER™ Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, Rockford) containing a cocktail of protease inhibitors and phosphatase inhibitors. Proteins were separated on 10% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes. The western blot was performed using the indicated antibodies.
2.9. qRT-PCR analyses
Total RNA from hind limbs joint cartilage of 12-month-old mice and articular chondrocyte cells were isolated using the easy-BLUE Total RNA Extraction Kit (iNtRON Biotechnology, Seongnam-si, Gyeonggi-do, Korea) and cDNA was synthesized from 1 μg of total RNA using SuperScript II Reverse Transcriptase (Invitrogen, CA, U.S.A). qRT-PCR was performed using the Power SYBR green master mixture (Applied Biosystems, Foster, CA). The qRT-PCR primers were designed using Primer Express software (Applied Biosystems). Sequences of qRT-PCR primers showed in Table 1.
| Target gene | Forward sequence (5’ - 3’) | Reverse sequence (5’ - 3’) |
| Gapdh | GCATCTCCCTCACAATTTCCA | GTGCAGCGAACTTTATTGATGG |
| Cbfb | TATGGGTTGCCTGGAGTT TG | AAGGCCTGTTGTGCTAATGC |
| Col2α1 | TTCCACTTCAGCTATGGCGA | GACGTTAGCGGTGTTGGGAG |
| Aggrecan | GAGAGAGGCGAATCGAACGA | CGTGAAGGGCAGCTGGTAAT |
| Mmp9 | AAACCAGACCCCAGACTCCTC | GAGGACACAGTCTGACCTGAA |
| Mmp13 | GCCAGAACTTCCCAACCATG | TCAGAGCCCAGAATTTTCTCC |
| Mmp14 | GGATGGACACAGAGAACTTCGTG | CGAGAGGTAGTTCTGGGTTGAG |
| Mmp15 | CTGAGCAGCTATGGCACAGACA | TGCTGTGTCTCCTCGTTGAAGC |
| IL-6 | TTGCCTTCTTGGGACTGATG | CTGAAGGACTCTGGCTTTGT |
| IL-17 | CTCAAAGCTCAGCGTGTCCAAACA | TATCAGGGTCTTCATTGCGGTGGA |
| IL-18 | CAGGCCTGACATCTTCTGCAA | TTTGATGTAAGTTAGTGAGAGTGA |
| IL-22 | GGTGACGACCAGAACATCCA | GACGTTAGCTTCTCACTTTCCTT |
2.10. Co-immunoprecipitation (Co-IP) assay
Primary articular chondrocytes were plated at a density of 2 × 106 cells/well in 100 mm plates for transfection experiments. Cells were transfected with 10μg DNA, including 5μg of the pcDNA3.1-myc-Cbfβ and 5μg of pCS4-2Flag-Runx1 by using Lipofectamine 2000 (Invitrogen) and grown for 24 h. Cell lysates (400 μg) were bound with anti-Myc at 40C overnight with gentle agitation, then incubated with 0.1g of Sepharose A beads (GE Healthcare) in lysis buffer for 2 h at 4 0C with gentle agitation. Mixtures were washed with lysis buffer five times, and proteins were eluted by boiling with 50μl protein loading buffer. Co-IP was performed by western blot with anti-Myc or anti-Flag antibody.
2.11. Poly-ubiquitination assay
ATDC5 chondrocytes were transfected with 12μg DNA, including 4μg pCS4-Flag-Runx1, 4μg pcDNA3.1-HA-Ubiquitin, and 4μg pCS4-Myc-Cbfβ by using Lipofectamine 2000 (Invitrogen) and grown for 24 h. After 24 h from transfection, cells were incubated with 20μm MG132 for 2 h. Cell lysates (400 μg) were allowed to bind with anti-Flag antibody at 4℃ overnight with gentle agitation, then incubated with 0.1g Sepharose A beads (GE Healthcare) in lysis buffer for 2 h at 4℃ with gentle agitation. Proteins were eluted by boiling with 50μl protein loading buffer. Ubiquitination levels were evaluated by western blot with anti-HA antibody.
2.11. Statistical analyses
We used GraphPad Prism 9 (GraphPad, San Diego) for statistical analysis. Paired or unpaired Student’s t-test was used to analyze the statistically significant between the two groups comparison. We considered a P <0.05 to be statistically significant (*P < 0.05, **P < 0.01), and ns represents no significance. Data are expressed as mean ± SD or mean ± SE.
3. Results
3.1. Cbfβ was enhanced by anabolism and suppressed by catabolism
To assess the involvement of Cbfβ in articular cartilage metabolism, we analyzed the expression of Cbfβ under TGF-ꞵ1, interleukin-1β (IL-1β), or TNF-α activation. Treatment of various concentrations (1, 10, and 100 ng/ml) of TGF-β1, one of the pivotal anabolic factors in the articular cartilage, increased pSmad3 and Cbfβ protein levels in the articular chondrocytes (Fig. 1a). TGF-β1 also increased the mRNA expression of Cbfβ, Type II collagen, and Aggrecan (Fig. 1b). The pro-inflammatory cytokines IL-1β or TNF-α decreased Cbfβ expression along with type II collagen and Aggrecan, while Mmp13 expression increased in primary cultured mouse articular chondrocytes (Fig. 1c). We confirmed that Cbfβ decreased under IL-1β or TNF-α treatment in the fluorescent staining (Fig. 1d). Moreover, Cbfꞵ silencing in chondrocytes upregulated the expression of Mmps such as Mmp9, Mmp13, Mmp14, and Mmp15, and inflammatory cytokines Il-6, IL-17, 1L-18, and IL-22 in the chondrocytes (Supplementary Fig. 1a and 1b). Taken together, Cbfβ was enhanced by anabolism and suppressed by catabolism in the articular chondrocytes.
3.2. Cbfβ loss is involved in articular cartilage degeneration
We then investigated whether there was a correlation between Cbfβ expression and articular cartilage degeneration in vivo. Aged articular cartilage reduced Cbfβ and its partner protein Runx1, but the expression of Runx2 and Runx3 was not significantly different compared to the young articular cartilage (Fig. 1E). Moreover, the expression of Cbfβ decreased in both mouse and human osteoarthritic cartilage (Fig. 1F-G). These results suggest that loss of Cbfβ may be involved in articular cartilage degeneration. Next, we investigated Cbfꞵ functions in joint development using articular cartilage-specific Cbfb deleted Gdf5-Cre; Cbfβfl/fl (Cbfb△ac/△ac) mice. With Rosa26 reporter (R26R) mice Gdf5-Cre activities were specifically observed in the articular cartilage, with around 75% deletion in articular cartilage by qRT- PCR (Supplementary Fig. 2A and 2B) and immunofluorescent staining in E16.5 (Supplementary Fig. 2C and 2D) and 20-week-old (Supplementary Figure 1e). Cbfb△ac/△ac mice displayed typical joint and cartilage formation at E14.5 and E16.5 embryonic stages (Fig. 2a and 2b). To further understand the effect of Cbfβ on articular cartilage homeostasis, we performed the articular cartilage integrity assessment with histochemical analysis at 12-month-old mice joints. Cbfb△ac/+ mice displayed higher OARSI scores than WT mice but without statistical significance. However, Cbfb△ac/△ac mice exhibited high OARSI scores with severe cartilage destruction (Fig. 2c and 2d). Cbfβ, Aggrecan, and Col2α1 mRNAs decreased, whereas Mmp13 expression increased in the articular cartilage of 12-month-old Cbfb△ac/△ac mice (Fig. 2e). These results indicate that Cbfβ physiologically protects articular cartilage during aging and that deletion of Cbfb causes age-dependent spontaneous OA progression.
3.3. Genetic deletion of Cbfb accelerated OA progression
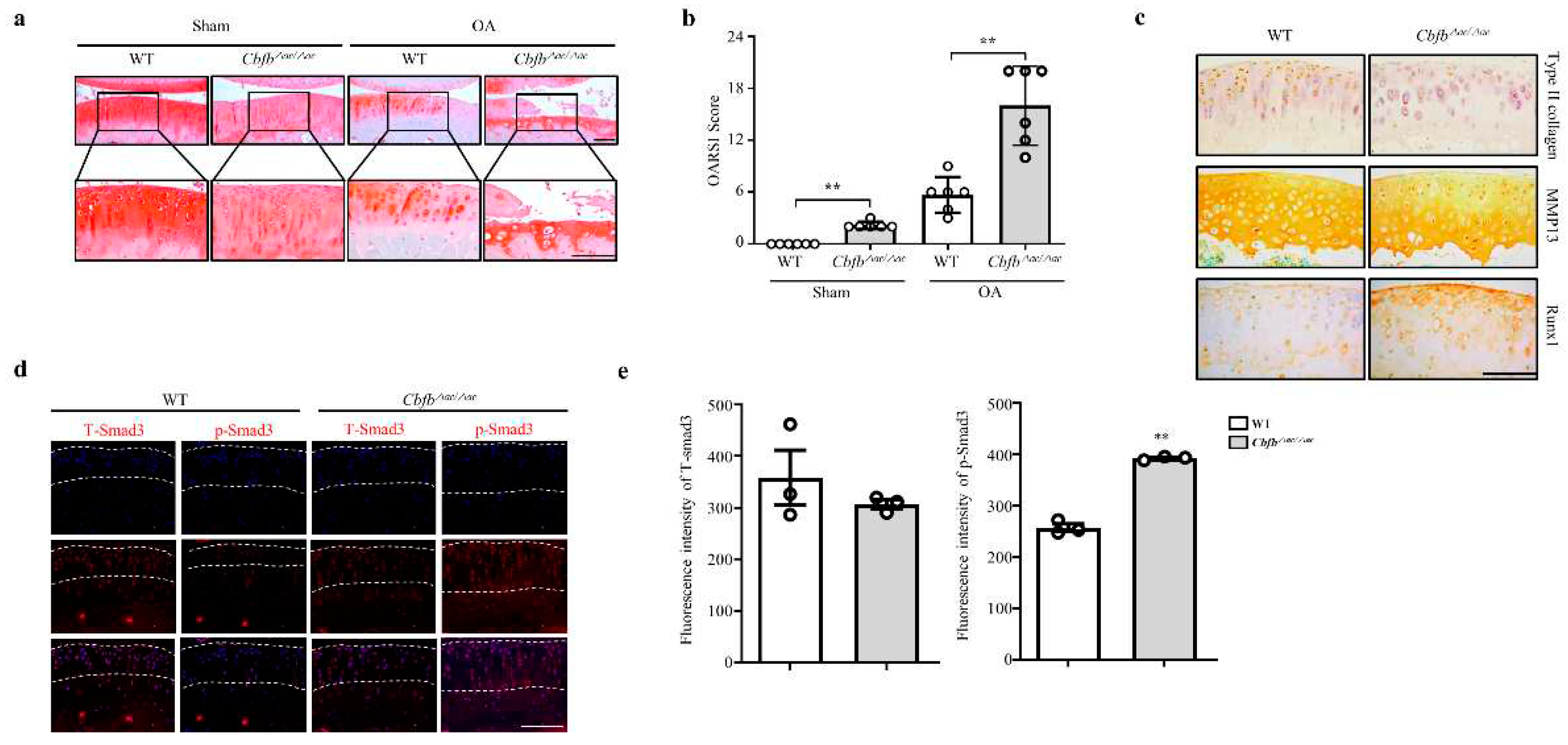
Cbfβ deficiency induced spontaneous OA development and presumed accelerated knee joint degradation in a surgically induced OA model. To explore this idea using a DMM surgery-induced mouse OA model, we confirmed the Cbfb△ac/△ac mice joints’ OA progression. Safranin O staining and scoring by the OARSI method revealed that Cbfb△ac/△ac mice joints induced severe damage to articular cartilage compared with controls. Indeed, Cbfb△ac/△ac mouse joints, even the sham group, showed more cartilage degeneration with a high OARSI score compared to WT mice at 20 weeks (Fig. 3a and 3b). Immunohistochemical staining decreased articular cartilage proteins that provide protection, such as Runx1 and Col2α, were decreased (Fig. 3c). Conversely, the matrix-degrading enzyme Mmp13 increased on the articular surface of Cbfb△ac/△ac mice compared to those of WT mice (Fig. 3c). TGF-ꞵ1 is one of the most critical anabolic regulators for articular cartilage integrity. Hence, we determined whether Cbfβ could modulate TGF-ꞵ1 signaling in the Cbfb△ac/△ac mouse articular cartilage. In fluorescence staining, the signal of pSmad3 unexpectedly increased in the Cbfb△ac/△ac mouse joint cartilage (Fig. 3d-e). These data raised another question as to why TGF-ꞵ1 signaling activation could not increase Type II collagen expression in Cbfb△ac/△ac mouse join cartilage.
Figure 3.
Deletion of Cbfb in the articular chondrocytes accelerated OA pathogenesis progression. (a) Articular cartilage destruction was determined by Safranin O staining 8 weeks after DMM surgery. Articular cartilage destructed severely in Cbfβ△ac/△ac mice. (b) The severity of OA was analyzed according to OARSI guidelines (n=6). Scale bars, 100 μm. (c) Immunohistochemical staining was performed on paraffin sections using anti-Runx1, -Col2α1, and -Mmp13 antibodies. (d) Expression of pSmad3 (left panel) and total Smad3 (T-Smad, right panel) in articular cartilage (AC) assessed by immunofluorescence staining in WT and Cbfβ△ac/△ac 20-week-old mice. (e) The pSmad3 and T-Smad3 fluorescence intensity of (e) were analyzed with the Leica fluorescence analysis program (n=3). Scale bars, 100 μm.
Figure 3.
Deletion of Cbfb in the articular chondrocytes accelerated OA pathogenesis progression. (a) Articular cartilage destruction was determined by Safranin O staining 8 weeks after DMM surgery. Articular cartilage destructed severely in Cbfβ△ac/△ac mice. (b) The severity of OA was analyzed according to OARSI guidelines (n=6). Scale bars, 100 μm. (c) Immunohistochemical staining was performed on paraffin sections using anti-Runx1, -Col2α1, and -Mmp13 antibodies. (d) Expression of pSmad3 (left panel) and total Smad3 (T-Smad, right panel) in articular cartilage (AC) assessed by immunofluorescence staining in WT and Cbfβ△ac/△ac 20-week-old mice. (e) The pSmad3 and T-Smad3 fluorescence intensity of (e) were analyzed with the Leica fluorescence analysis program (n=3). Scale bars, 100 μm.
3.4. Cbfβ modulates articular cartilage integrity by modulating TGF-ꞵ1 signaling
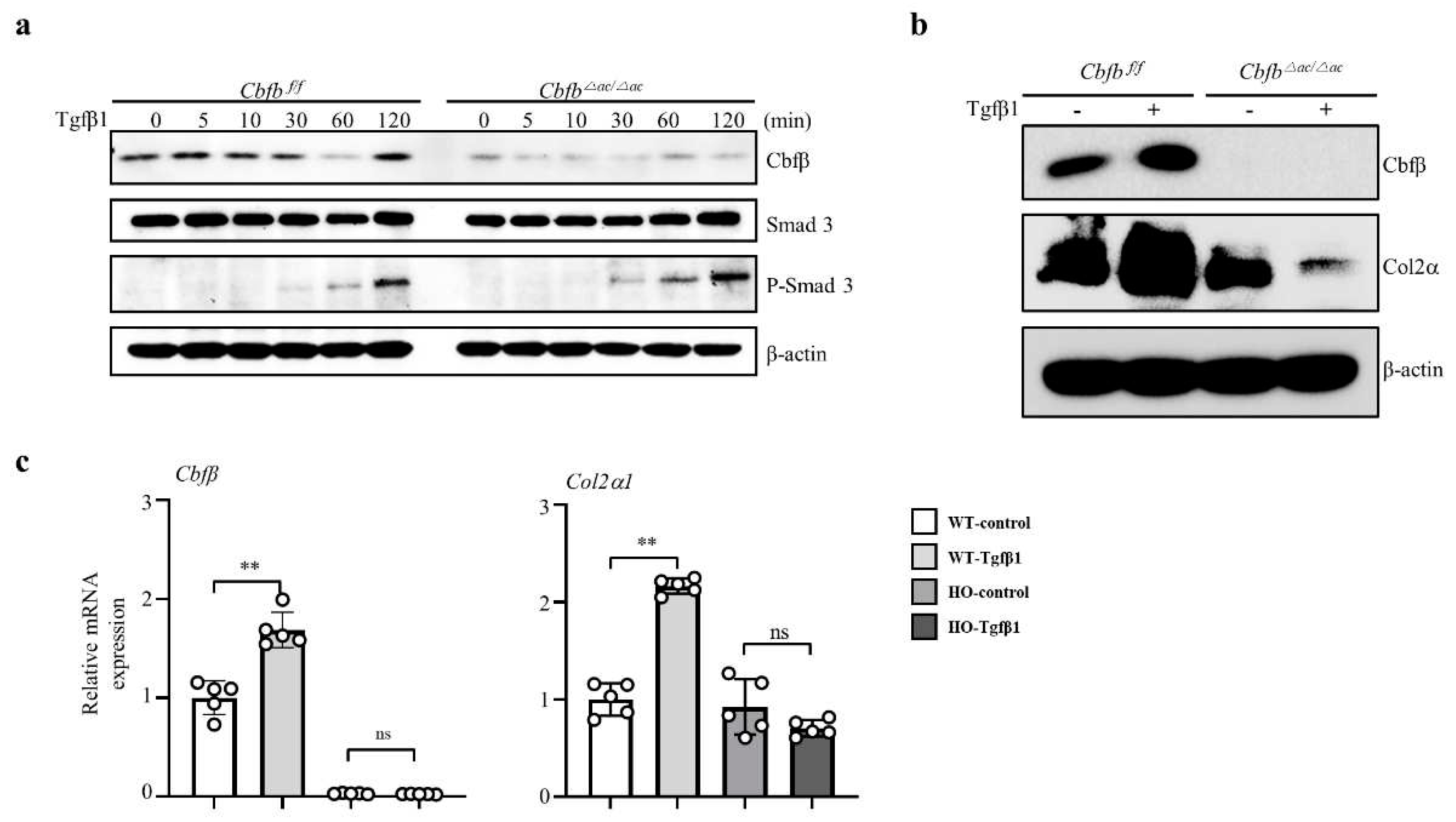
To clarify how Cbfβ is involved in the TGF-ꞵ1 mediated Type II collagen expression, we performed a TGF-ꞵ1 responsive assay in the Cbfβ deleted chondrocytes. Interestingly, TGF-ꞵ1 treatment increased p-Smad3 in the Cbfb△ac/△ac articular chondrocytes compared to WT cells under the (Fig. 4a). Consistently with in vivo data, TGF-ꞵ1-mediated Cbfβ and Col2α1 expression were reduced in Cbfb△ac/△ac articular chondrocytes compared with WT (Fig. 4b-c). These results indicate that Cbfβ plays an essential role in maintaining TGF-ꞵ signaling pathway integrity in the articular cartilage.
Figure 4.
Impaired TGF-β1-mediated Col2α1 transcription in Cbfβ-deficient articular chondrocytes. (a) Cbfβ, pSmad3, and T-smad3 expression were evaluated by western blotting in Cbfβ△ac/△ac articular chondrocyte. (b) Effects of TGF-ꞵ1 treatment for 24 h on Cbfβ and Col2α1 protein levels by western blotting. (c) The expression of Cbfβ, Runx1, and Col2α1 mRNA was measured by qRT-PCR.
Figure 4.
Impaired TGF-β1-mediated Col2α1 transcription in Cbfβ-deficient articular chondrocytes. (a) Cbfβ, pSmad3, and T-smad3 expression were evaluated by western blotting in Cbfβ△ac/△ac articular chondrocyte. (b) Effects of TGF-ꞵ1 treatment for 24 h on Cbfβ and Col2α1 protein levels by western blotting. (c) The expression of Cbfβ, Runx1, and Col2α1 mRNA was measured by qRT-PCR.
3.4. Cbfβ stabilizes Runx1 in articular chondrocytes
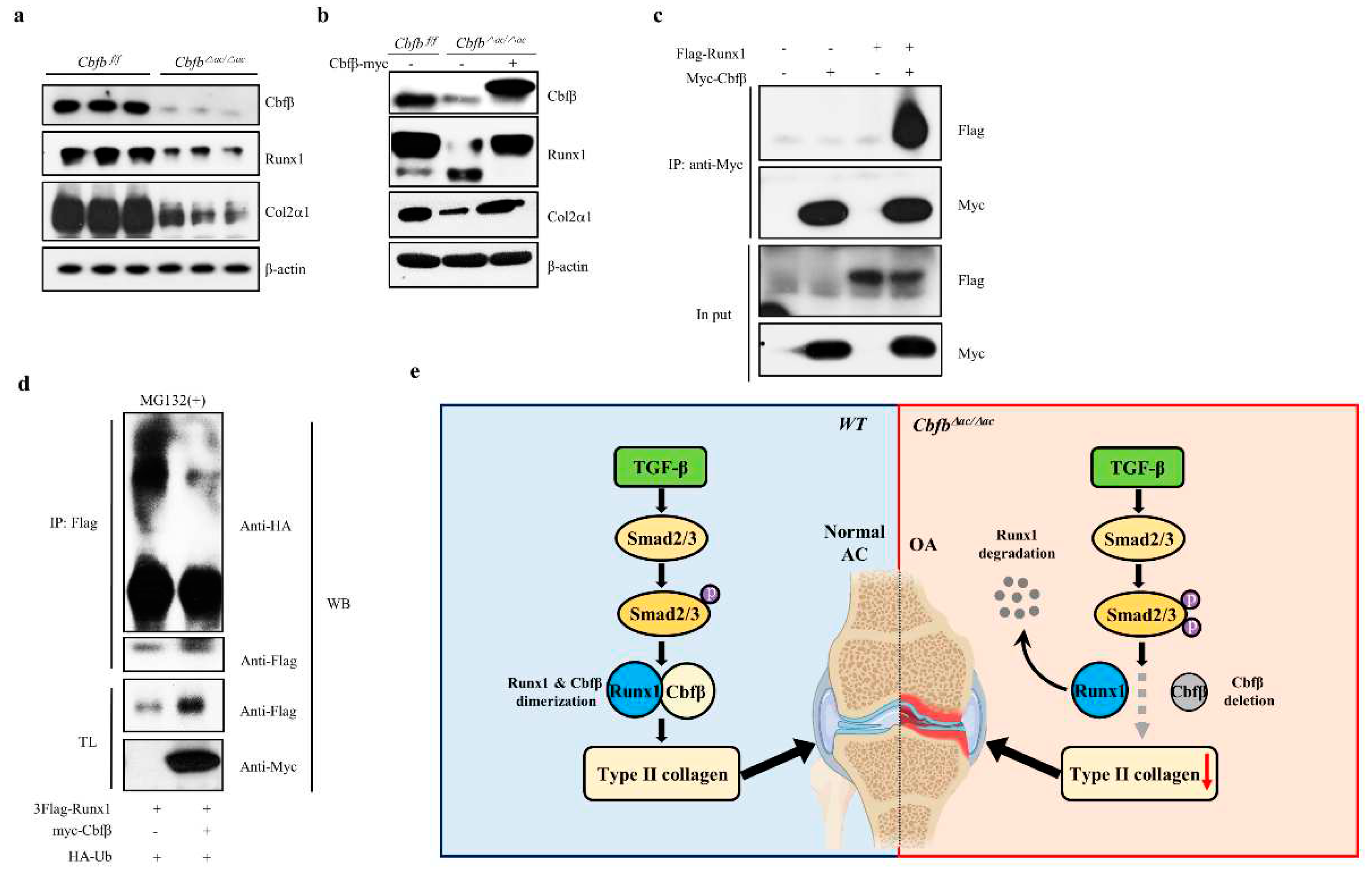
Runx1, Runx2, and Runx3 are crucial targets of TGF-ꞵ superfamily signaling [31]. Runx1 is a central regulator of articular cartilage integrity by coordinating YAP, TGF-β, and Wnt signaling in articular cartilage formation and maintenance by enhancing type II collagen and matrix production in collaboration with Sox trios [43]. We, therefore, hypothesized that Runx1 proteolysis resulted in accelerated cartilage degeneration in the Cbfβ deleted articular chondrocytes [32]. To test this hypothesis, we examined the expression of the Runx1 and Col2α1 in the Cbfβ deleted articular chondrocytes. The levels of Runx1 and Col2α1 were reduced in Cbfβ deleted articular chondrocytes from Cbfb△ac/△ac mice, whereas rescue of Cbfβ in Cbfb-deleted articular chondrocytes restored Runx1 and Col2α1 (Fig. 5a-b). To further investigate the effects of Cbfβ on Runx1stability in articular chondrocytes, we performed co-immunoprecipitation and poly-ubiquitination (Figure 5c). In articular chondrocytes, Cbfβ can endogenously bind Runx1 and protect Runx1 from polyubiquitination-mediated proteasomal degradation (Fig. 5c-d). These results suggest that Cbfβ makes the complex with Runx1, stabilizing Runx1 in articular chondrocytes.
4. Discussion
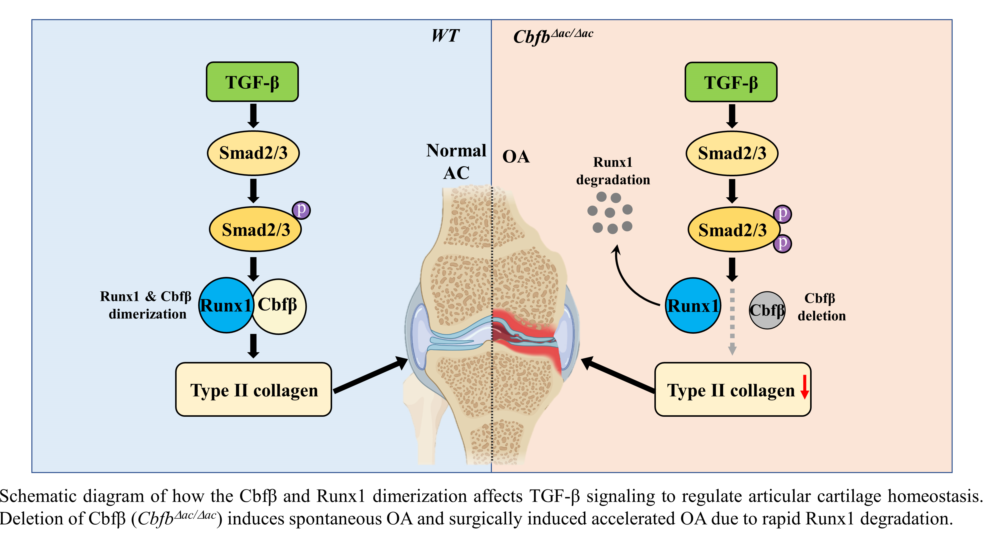
In this study, we found that the articular cartilage-specific deletion of Cbfβ resulted in spontaneous OA development and exacerbated OA progression in the surgically induced DMM model. Expression of Cbfβ decreased with aging and OA progression in articular cartilage, and only Runx1 among the Runx transcription factors downregulated in response to a decrease in Cbfβ (Fig. 1c and 1d). The absence or rescue of Cbfꞵ in Cbfb-deleted articular chondrocytes determined the levels of Runx1 and Cbfꞵ/Runx1 complex as well as Col2α1, indicating that Cbfβ/Runx1 complex formation plays a crucial role in maintaining articular cartilage homeostasis. Moreover, Cbfꞵ as a component of the Cbfꞵ/Runx1 complex was a key regulator of TGF-ꞵ signaling with modulation of pSmad3 in Cbfb-deleted articular chondrocytes. A proper Cbfβ/Runx1 complex formation via TGF-ꞵ signaling is vital for maintaining normal articular chondrocyte functional integrity and preventing degenerative cartilage diseases of joints such as OA.
Physiological maintenance of articular cartilage requires tight control of the TGF-ꞵ signaling pathway [10]. Our study demonstrates the critical role of Cbfβ and Runx1 as a mediator of TGF-ꞵ signaling (Fig. 5e). First, Cbfβ and Runx1, like various components of the TGF-ꞵ signaling pathway, such as the ALK1/ALK5 ratio [13], decreased with aging and OA progression. The activation of TGF-ꞵ signaling increased Cbfꞵ and Runx1 expression and Cbfꞵ/Runx1 complex formation. Deletion of Cbfꞵ attenuated the activation of the TGF-ꞵ signaling pathway; however, forced expression of Cbfβ recovered Runx1 and Col2α1 expression in Cbfꞵ-deficient chondrocytes. Moreover, the deletion of Cbfꞵ increased pSmad3 in the articular cartilage of Cbfb△ac/△ac mice. The increase of pSmad3 may result from the regulatory role of the negative feedback loop in the TGF-ꞵ signaling pathway. Unknown factors regulated by or involved in the Cbfꞵ/Runx1 in articular chondrocytes need further studies.
Several studies have suggested their involvement regarding the contribution of Runx transcription factors to OA. For example, Runx1 contributes to articular cartilage maintenance by increasing cartilage matrix production and inhibiting hypertrophic differentiation [32,33]. Another evidence is that the small molecule KGN binds to filamin A, a cytoplasmic sequestrant of Cbfꞵ, releasing Cbfꞵ from filamin A, which translocates Cbfꞵ to the nucleus and binds to Runx1 but not Runx2 of articular chondrocytes [34]. Runx1 is known to stimulate the differentiation of mesenchymal cells into chondrocytes by regulating the expression of type II collagen rather than type X collagen, a hypertrophy marker [18, 34). This study showed that Cbfꞵ levels correlated well with Runx1 but not with Runx2 and Runx3. In aged articular cartilage, Cbfꞵ and Runx1 decreased, but Runx2 and Runx3 were not, which also correlated well with the articular cartilage phenotype of Cbfb△ac/△ac mice. We also revealed that disruption of the TGF-ꞵ signaling integrity by deletion of Cbfꞵ in articular chondrocytes increases catabolic cytokines and enzymes like IL-6, IL-17, IL-18, IL-22, Mmp 9, Mmp 13, Mmp14, and Mmp15. Therefore, the positive relationship of the Cbfꞵ/Runx1 complex with Type II collagen is essential to the TGF-ꞵ signaling pathway in articular cartilage integrity.
Regarding Runx2 and Runx3, Runx2 expressed negligibly, while Runx3 was comparable to Runx1 in articular cartilage. Our study showed that diminished Runx1 correlated well with the level of Cbfꞵ, whereas increased Runx2 and Runx3 in the human OA samples did not. Our previous studies showed the protection of Runx2 by Cbfꞵ in both chondrocytes and osteoblasts [29,30], suggesting the presence of differential heterodimerization of Cbfꞵ with Runx transcription factors in skeletal tissues. Some cases of OA upregulate Runx2 with a transition from articular chondrocytes to hypertrophic chondrocytes (Kawaguchi, 2008). Runx2 haploinsufficiency mice show delayed OA progression after induction of knee joint instability [35]. Additionally, the deletion of Runx2 in articular chondrocytes slows the progression of surgery-induced OA [36]. Moreover, chondrocyte-specific Runx2 overexpression accelerates OA progression [37]. These works of literature indicate a positive relationship between OA progression and aberrant expression of Runx2 in articular chondrocytes. However, the Cbfꞵ study showed that Runx1, but not Runx2 or Runx3, was primarily involved in the aggravated OA phenotype in the DMM model and Cbfꞵ-deleted spontaneous OA and human OA samples.
5. Conclusions
In this study, deletion of Cbfβ in articular cartilage decreased Runx1 and type II collagen, which enhanced OA progression spontaneously in Cbfb△ac/△ac mice. These results indicate that Cbfβ regulates Runx1 protein stability and subsequently modulates Runx1 target genes such as type II collagen in the articular cartilage. Our findings indicate that Cbfβ prevents articular cartilage destruction by maintaining the integrity of the TGF-ꞵ signal pathway through the stabilization of Runx1.
Supplementary Materials
They will be provided when this paper is sent to review.
Author Contributions
Conceptualization, X.C., X.J. and J.Y.C.; methodology, X.C., X.J. and J.Y.C.; software, X.C. and X.J.; validation, X.C., X.J. and J.Y.C.; formal analysis, X.C. and X.J.; investigation, X.C. and X.J.; resources, H.J.K., and H.S.K.; data curation, X.C., X.J., H.J.K., and J.Y.C.; writing—original draft preparation, J.Y.C.; writing—review and editing, X.C., X.J., N.R.P., H.J.K., J.B.L., J.L.S., and G.S.S.; visualization, X.C., X.J.; supervision, H.J.K., J.B.L., J.L.S., and G.S.S.; project administration, X.C., X.J. and J.Y.C.; funding acquisition, X.C., and J.Y.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07043598 & NRF-2021R1I1A1A01047403), the Ministry of Science & ICT (NRF-2017R1A5A2015391, 2014M3A9D5A01073658). This study was also supported in part by R01DE029311 NIDCR and R01AR039588 NIAMS.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of Kyungpook National University (KNU-201455). Human OA joint cartilage tissues were obtained from OA surgery patients through total knee arthroplasty. The Institutional Review Board of Kyungpook National University Hospital approved the use of human OA cartilage and written informed consent was obtained from all patients prior to the surgical procedure (IRB File No of KNUH 2022-01-010-001).
Informed Consent Statement
The Institutional Review Board of Kyungpook National University Hospital approved the use of human OA cartilage, and written informed consent was obtained from all patients prior to the surgical procedure (IRB File No of KNUH 2022-01-010-001).
Data Availability Statement
The data that support the findings of this study are openly available in [repository name “Cells-GDF5-CBFB-Figures” and “Cells-GDF5-CBFB-Figures-Raw data”] at https://drive.google.com/drive/folders/15O7G0gX8ypbl9qFUUkizPEd6v3ycJyXC?usp=sharing and https://drive.google.com/drive/folders/18UjNmtxL8LjM9fcgU0ovSlN2WBioVYEA?usp=sharing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis. Nature reviews Disease primers. 2016;2:16072. [CrossRef]
- Hunter DJ, and Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393(10182):1745-1759. [CrossRef]
- Zhen G, Guo Q, Li Y, Wu C, Zhu S, Wang R, et al. Mechanical stress determines the configuration of TGFbeta activation in articular cartilage. Nat Commun. 2021;12(1):1706. [CrossRef]
- Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthritis. Lancet. 2015;386(9991):376-387. [CrossRef]
- Che X, Chi L, Park CY, Cho GH, Park N, Kim SG, et al. A novel method to detect articular chondrocyte death during early stages of osteoarthritis using a non-invasive ApoPep-1 probe. Arthritis Res Ther. 2015;17:309. [CrossRef]
- Hyttinen MM, Toyras J, Lapvetelainen T, Lindblom J, Prockop DJ, Li SW, et al. Inactivation of one allele of the type II collagen gene alters the collagen network in murine articular cartilage and makes cartilage softer. Ann Rheum Dis. 2001;60(3):262-268. [CrossRef]
- Goldring SR, and Goldring MB. Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage-bone crosstalk. Nat Rev Rheumatol. 2016;12(11):632-644. [CrossRef]
- Li H, Wang D, Yuan Y, and Min J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res Ther. 2017;19(1):248. [CrossRef]
- Wang M, Sampson ER, Jin H, Li J, Ke QH, Im HJ, et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther. 2013;15(1):R5. [CrossRef]
- Thielen N. G. M, Neefjes M, Vitters E. L, van Beuningen H. M, Blom A. B et al. Identification of Transcription Factors Responsible for a Transforming Growth Factor-β-Driven Hypertrophy-like Phenotype in Human Osteoarthritic Chondrocytes. Cells. 2022, 11(7), 1232-1249. [CrossRef]
- van Beuningen HM, van der Kraan PM, Arntz OJ, and van den Berg WB. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab Invest. 1994;71(2):279-290.
- Scharstuhl A, Glansbeek HL, van Beuningen HM, Vitters EL, van der Kraan PM, and van den Berg WB. Inhibition of endogenous TGF-beta during experimental osteoarthritis prevents osteophyte formation and impairs cartilage repair. J Immunol. 2002;169(1):507-514. [CrossRef]
- Blaney Davidson EN, Remst DF, Vitters EL, van Beuningen HM, Blom AB, Goumans MJ, et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J Immunol. 2009;182(12):7937-45. [CrossRef]
- Serra R, Johnson M, Filvaroff EH, LaBorde J, Sheehan DM, Derynck R, et al. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol. 1997;139(2):541-552. [CrossRef]
- Yang X, Chen L, Xu X, Li C, Huang C, and Deng CX. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153(1):35-46. [CrossRef]
- Speck NA, and Terryl S. A new transcription factor family associated with human leukemias. Crit Rev Eukaryot Gene Expr. 1995;5(3-4):337-64. [CrossRef]
- Bae SC, and Ito Y. Regulation mechanisms for the heterodimeric transcription factor, PEBP2/CBF. Histol Histopathol. 1999;14(4):1213-1221. [CrossRef]
- Wang Y, Belflower RM, Dong YF, Schwarz EM, O'Keefe RJ, and Drissi H. Runx1/AML1/Cbfa2 mediates onset of mesenchymal cell differentiation toward chondrogenesis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2005;20(9):1624-36. [CrossRef]
- LeBlanc KT, Walcott ME, Gaur T, O'Connell SL, Basil K, Tadiri CP, et al. Runx1 Activities in Superficial Zone Chondrocytes, Osteoarthritic Chondrocyte Clones and Response to Mechanical Loading. Journal of cellular physiology. 2015;230(2):440-8. [CrossRef]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765-71. [CrossRef]
- Choi JY, Pratap J, Javed A, Zaidi SK, Xing L, Balint E, et al. Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development. Proc Natl Acad Sci U S A. 2001;98(15):8650-5. [CrossRef]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755-64. [CrossRef]
- Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004;18(8):952-63. [CrossRef]
- Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, and Ito Y. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 2001;20(4):723-33. [CrossRef]
- Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL, Huang X, et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996;87(4):697-708. [CrossRef]
- Tian F, Wu M, Deng L, Zhu G, Ma J, Gao B, et al. Core binding factor beta (Cbfbeta) controls the balance of chondrocyte proliferation and differentiation by upregulating Indian hedgehog (Ihh) expression and inhibiting parathyroid hormone-related protein receptor (PPR) expression in postnatal cartilage and bone formation. J Bone Miner Res. 2014;29(7):1564-74. [CrossRef]
- Qin X, Jiang Q, Matsuo Y, Kawane T, Komori H, Moriishi T, et al. Cbfb regulates bone development by stabilizing Runx family proteins. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2015;30(4):706-14. [CrossRef]
- Wu M, Li YP, Zhu G, Lu Y, Wang Y, Jules J, et al. Chondrocyte-specific knockout of Cbfbeta reveals the indispensable function of Cbfbeta in chondrocyte maturation, growth plate development and trabecular bone formation in mice. Int J Biol Sci. 2014;10(8):861-72. [CrossRef]
- Park NR, Lim KE, Han MS, Che X, Park CY, Kim JE, et al. Core Binding Factor beta Plays a Critical Role During Chondrocyte Differentiation. J Cell Physiol. 2016;231(1):162-71. [CrossRef]
- Lim KE, Park NR, Che X, Han MS, Jeong JH, Kim SY, et al. Core binding factor beta of osteoblasts maintains cortical bone mass via stabilization of Runx2 in mice. J Bone Miner Res. 2015;30(4):715-22. [CrossRef]
- Ito Y, and Miyazono K. RUNX transcription factors as key targets of TGF-beta superfamily signaling. Curr Opin Genet Dev. 2003;13(1):43-7. [CrossRef]
- Yano F, Ohba S, Murahashi Y, Tanaka S, Saito T, and Chung UI. Runx1 contributes to articular cartilage maintenance by enhancement of cartilage matrix production and suppression of hypertrophic differentiation. Sci Rep. 2019;9(1):7666. [CrossRef]
- Zhou C, Cui Y, Yang Y, Guo D, Zhang D, Fan Y, et al. Runx1 protects against the pathological progression of osteoarthritis. Bone Res. 2021;9(1):50. [CrossRef]
- Johnson K, Zhu S, Tremblay MS, Payette JN, Wang J, Bouchez LC, et al. A stem cell-based approach to cartilage repair. Science. 2012;336(6082):717-21. [CrossRef]
- Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;54(8):2462-2470. [CrossRef]
- Liao L, Zhang S, Gu J, Takarada T, Yoneda Y, Huang J, et al. Deletion of Runx2 in Articular Chondrocytes Decelerates the Progression of DMM-Induced Osteoarthritis in Adult Mice. Sci Rep. 2017;7(1):2371. [CrossRef]
- Catheline SE, Hoak D, Chang M, Ketz JP, Hilton MJ, Zuscik MJ, et al. Chondrocyte-Specific RUNX2 Overexpression Accelerates Post-traumatic Osteoarthritis Progression in Adult Mice. J Bone Miner Res. 2019;34(9):1676-89. [CrossRef]
- Rountree RB, Schoor M, Chen H, Marks ME, Harley V, Mishina Y, et al. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004;2(11):e355. [CrossRef]
- Naoe Y, Setoguchi R, Akiyama K, Muroi S, Kuroda M, Hatam F, et al. Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med. 2007;204(8):1749-1755. [CrossRef]
- Glasson SS, Blanchet TJ, and Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15(9):1061-1069. [CrossRef]
- Jeong JH, Jin JS, Kim HN, Kang SM, Liu JC, Lengner CJ, et al. Expression of Runx2 transcription factor in non-skeletal tissues, sperm and brain. J Cell Physiol. 2008;217(2):511-517. [CrossRef]
- Gosset M, Berenbaum F, Thirion S, and Jacques C. Primary culture and phenotyping of murine chondrocytes. Nature protocols. 2008;3(8):1253-60. [CrossRef]
- Kim HJ, Lee DK, Jin X, Che X, Ryu SH, Choi JY. Phospholipase D2 controls bone homeostasis by modulating M-CSF-dependent osteoclastic cell migration and microtubule stability. Exp Mol Med. 2022;54(8):1146-1155. [CrossRef]
- Y. Zhang, T. Zuo, A. McVicar, H. L. Yang, Y. P. Li and W. Chen. Runx1 is a key regulator of articular cartilage homeostasis by orchestrating YAP, TGFβ, and Wnt signaling in articular cartilage formation and osteoarthritis. Bone Research, 2022; 10(1):63. [CrossRef]
Figure 1.
Cbfβ loss is involved in OA pathogenesis. (a) Cbfβ and pSmad 3 protein expression detected by Western blotting in articular chondrocytes under TGF-ꞵ1 treatment. NE, nuclear extracts; CE, cytoplasmic extracts (b) Cbfβ, Col2α1, and Aggrecan mRNA expression detected by qRT-PCR in articular chondrocytes under TGF-ꞵ1 treatment. (c) Cbfβ, Col2α1, and Mmp13 mRNA expression detected by qRT-PCR in articular chondrocytes under IL-1ꞵ or TNF-α treatment. (d) Cbfβ determined by immunofluorescence staining after treatment of IL-1ꞵ and TNF-α in articular chondrocytes. Scale bars, 100 μm. (e) Expression of Cbfβ, Runx1, Runx2, and Runx3 at 3 and 12 months of age evaluated by immunofluorescence staining (n=3). Scale bars, 100 μm. (f) Mouse joints OA development assessed by Safranin O staining, Cbfβ expression by immunohistochemistry. Scale bars, 100 μm. (g) Human OA development was assessed by Safranin O staining and Cbfβ expression by immunofluorescent (n=6). Scale bars, 100 μm.
Figure 1.
Cbfβ loss is involved in OA pathogenesis. (a) Cbfβ and pSmad 3 protein expression detected by Western blotting in articular chondrocytes under TGF-ꞵ1 treatment. NE, nuclear extracts; CE, cytoplasmic extracts (b) Cbfβ, Col2α1, and Aggrecan mRNA expression detected by qRT-PCR in articular chondrocytes under TGF-ꞵ1 treatment. (c) Cbfβ, Col2α1, and Mmp13 mRNA expression detected by qRT-PCR in articular chondrocytes under IL-1ꞵ or TNF-α treatment. (d) Cbfβ determined by immunofluorescence staining after treatment of IL-1ꞵ and TNF-α in articular chondrocytes. Scale bars, 100 μm. (e) Expression of Cbfβ, Runx1, Runx2, and Runx3 at 3 and 12 months of age evaluated by immunofluorescence staining (n=3). Scale bars, 100 μm. (f) Mouse joints OA development assessed by Safranin O staining, Cbfβ expression by immunohistochemistry. Scale bars, 100 μm. (g) Human OA development was assessed by Safranin O staining and Cbfβ expression by immunofluorescent (n=6). Scale bars, 100 μm.
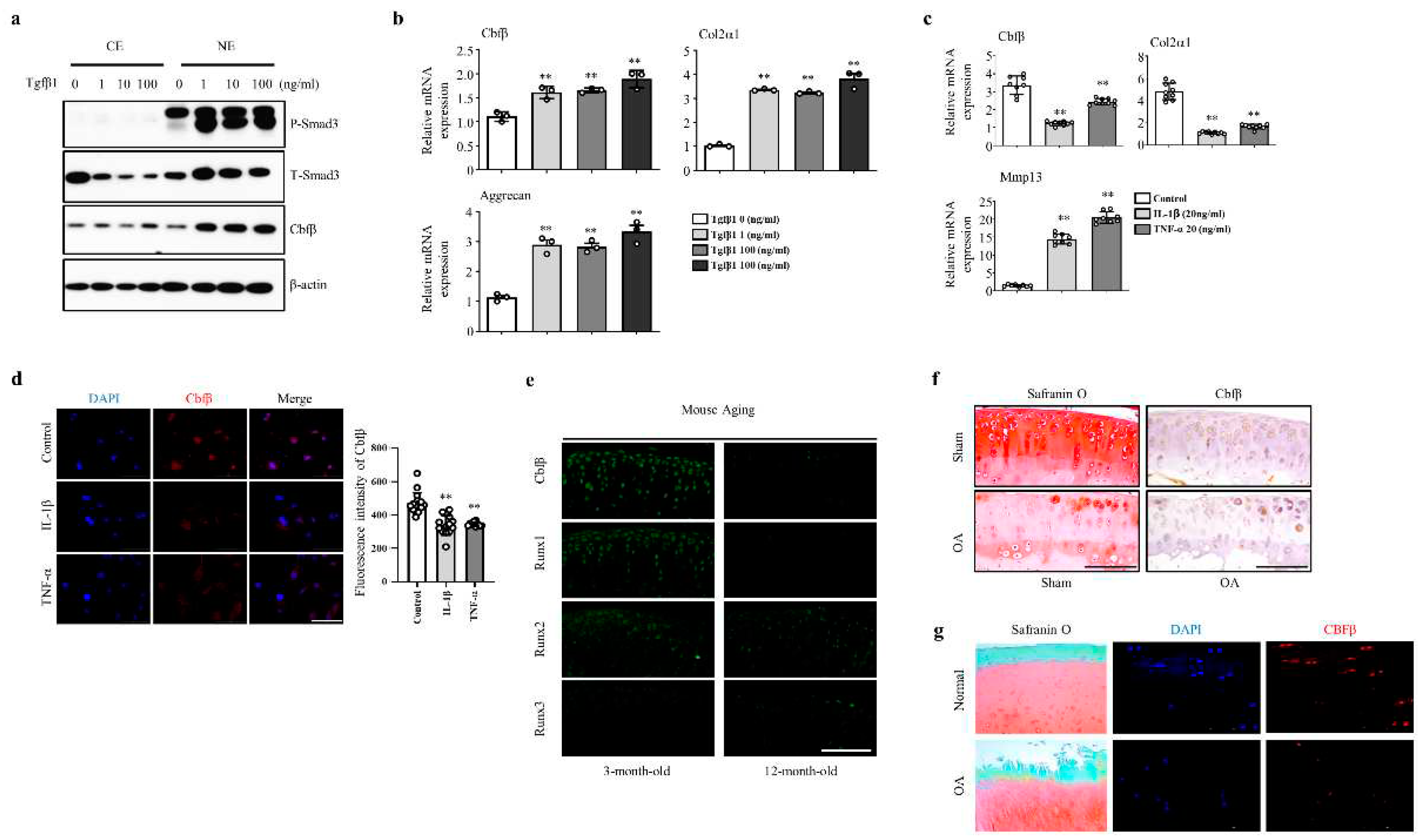
Figure 2.
Joint cartilage-specific Cbfb deleted mice exhibit spontaneous cartilage degeneration. (a, b) Evaluation of joint and limb formation by Safranin-O staining at E14.5 (WT=3, Cbfb∆ac/∆ac=4) and E16.5 (WT=7, Cbfb∆ac/∆ac=7) of Cbfb∆ac/∆ac mice. The humerus length was analyzed with the Leica program. Scale bars, 500 μm & 100 μm. (c) Spontaneous OA development assessed by Safranin O staining in 12-month-old Cbfb∆ac/∆ac mice. (d) OA severity was determined by histological analysis according to OARSI guidelines. Scale bars, 100 μm (WT=5, Cbfb∆ac/∆ac=4, Cbfb∆ac/∆ac=5). (e) Cbfβ, Col2α1, Aggrecan, and Mmp13 mRNA expression were evaluated by qRT-PCR in 12-month-old Cbfb∆ac/∆ac articular cartilage.
Figure 2.
Joint cartilage-specific Cbfb deleted mice exhibit spontaneous cartilage degeneration. (a, b) Evaluation of joint and limb formation by Safranin-O staining at E14.5 (WT=3, Cbfb∆ac/∆ac=4) and E16.5 (WT=7, Cbfb∆ac/∆ac=7) of Cbfb∆ac/∆ac mice. The humerus length was analyzed with the Leica program. Scale bars, 500 μm & 100 μm. (c) Spontaneous OA development assessed by Safranin O staining in 12-month-old Cbfb∆ac/∆ac mice. (d) OA severity was determined by histological analysis according to OARSI guidelines. Scale bars, 100 μm (WT=5, Cbfb∆ac/∆ac=4, Cbfb∆ac/∆ac=5). (e) Cbfβ, Col2α1, Aggrecan, and Mmp13 mRNA expression were evaluated by qRT-PCR in 12-month-old Cbfb∆ac/∆ac articular cartilage.
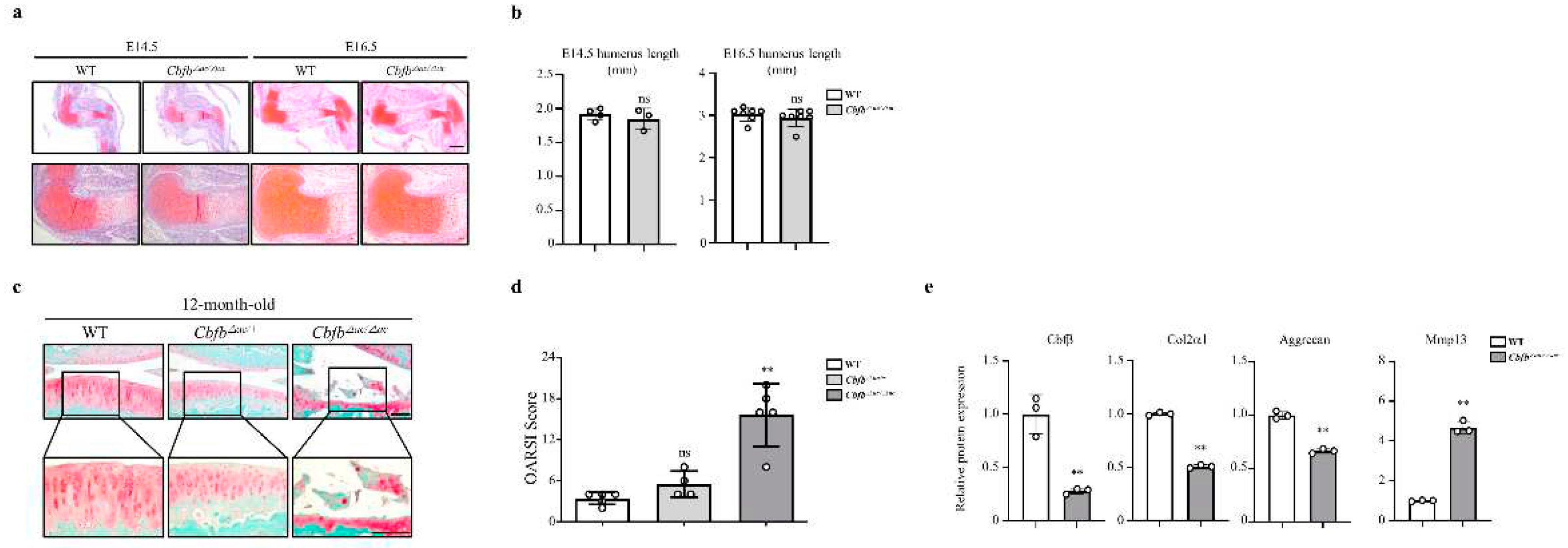
Figure 5.
Requirement of Cbfβ for Runx1 stabilization in articular chondrocytes. (a) Total protein was extracted from primary articular chondrocytes derived from WT or Cbfβ△ac/△ac mice and western blotting was performed with anti-Cbfβ, -Runx1, -Col2α1, and -β-actin antibodies. β-actin as the loading control. (b) Primary cultured articular chondrocytes derived from Cbfβ△ac/△ac mice were transiently transfected with Myc-Cbfβ for 24 h, and the expression levels of Cbfβ, Runx1, Col2α1, Mmp13, and β-actin analyzed by Western blotting. WT was relative control. (c) Primary cultures of articular chondrocytes were co-transfected with vectors expressing Flag-Runx1 and Myc-Cbfβ for 24 h. Total extracts were immunoprecipitated with anti-Myc antibody, and Runx1 was detected with anti-Flag antibody by Western blotting. (d) For polyubiquitination assay, ATDC5 chondrocytes were transfected with HA-Ub, Flag-Runx1, and Myc-Cbfβ for 24 h and treated with 10 μM MG132 for the last 2 h. After immunoprecipitation of Runx1 using anti-Flag antibody, Ub, Runx1, Cbfꞵ levels were determined by western blotting (WB) using anti-HA, anti-Flag, and anti-Myc antibodies. TL, total lysates. (e) Schematic diagram of how the Cbfβ/Runx1 complex affects TGF-ꞵ signaling to regulate articular cartilage homeostasis. Deletion of Cbfβ induces spontaneous OA and surgically induced accelerated OA due to rapid Runx1 degradation.
Figure 5.
Requirement of Cbfβ for Runx1 stabilization in articular chondrocytes. (a) Total protein was extracted from primary articular chondrocytes derived from WT or Cbfβ△ac/△ac mice and western blotting was performed with anti-Cbfβ, -Runx1, -Col2α1, and -β-actin antibodies. β-actin as the loading control. (b) Primary cultured articular chondrocytes derived from Cbfβ△ac/△ac mice were transiently transfected with Myc-Cbfβ for 24 h, and the expression levels of Cbfβ, Runx1, Col2α1, Mmp13, and β-actin analyzed by Western blotting. WT was relative control. (c) Primary cultures of articular chondrocytes were co-transfected with vectors expressing Flag-Runx1 and Myc-Cbfβ for 24 h. Total extracts were immunoprecipitated with anti-Myc antibody, and Runx1 was detected with anti-Flag antibody by Western blotting. (d) For polyubiquitination assay, ATDC5 chondrocytes were transfected with HA-Ub, Flag-Runx1, and Myc-Cbfβ for 24 h and treated with 10 μM MG132 for the last 2 h. After immunoprecipitation of Runx1 using anti-Flag antibody, Ub, Runx1, Cbfꞵ levels were determined by western blotting (WB) using anti-HA, anti-Flag, and anti-Myc antibodies. TL, total lysates. (e) Schematic diagram of how the Cbfβ/Runx1 complex affects TGF-ꞵ signaling to regulate articular cartilage homeostasis. Deletion of Cbfβ induces spontaneous OA and surgically induced accelerated OA due to rapid Runx1 degradation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



