
Submitted:
17 February 2023
Posted:
21 February 2023
You are already at the latest version
Abstract
About 50% of colorectal cancer patients develop liver metastases. Patients with metastatic colorectal cancer have 5-year survival rates below 20% despite new therapeutic regimens. Tumor heterogeneity has been linked with poor treatment response and clinical outcome, but was so far mainly studied via bulk genomic analyses. In this study we performed spatial proteomics via MALDI mass spectrometry imaging on six patient-matched CRC primary tumor and liver metastases to characterize interpatient, intertumor and intratumor hetereogeneity. We found several peptide features that were enriched in vital tumor areas of primary tumors and liver metastasis and tentatively derived from tumor cell specific proteins such as annexin A4 and prelamin A/C. Liver metastases of colorectal cancer showed higher heterogeneity between patients than primary tumors while within patients both entities show similar intratumor heterogeneity sometimes organized in zonal pattern. Together our findings give new insights into the spatial proteomic heterogeneity of primary CRC and patient-matched liver metastases.
Keywords:
colorectal cancer
; metastasis
; tumor heterogeneity
; spatial proteomics
; mass spectrometry imaging
; MALDI imaging
; formalin-fixed paraffin-embedded tissues
1. Introduction
Colorectal cancer (CRC) is among the most deadly and prevalent cancers worldwide 1,2. About half of all CRC patients develop liver metastases 3,4. Synchronous metastases are present at the time of diagnosis in two-third of the patients, while metachronous metastases develop after the primary tumor surgery. Paradoxically, despite progress in therapy, survival rates for metastasized colorectal cancer are still poor. 5-year survival rates for metastatic CRC are below 20% compared to about 90% for localized CRC. New combinatorial and even personalized therapeutic regimens extend the median survival time only by 2 – 8 months 5–8.
Tumor heterogeneity is an important, complex and multifactorial property of CRC, which contributes to the poor clinical outcome of metastasized CRC. Tumor heterogeneity refers to cellular, genetic and molecular differences between individual patients (interpatient heterogeneity), between tumors of the same patient (intertumor heterogeneity) as well as inside an individual tumor (intratumor heterogeneity) 9. In CRC these levels of heterogeneity are well described on a genetic level by using genomic approaches 10. Genomic differences are evident between different patients, histological tumor subtypes, colon localization, multiple primary or metastatic lesions even within tumors of the same patient and different regions of the same tumor 11,12. About 70% of CRC occur sporadically caused by somatic mutations leading to heterogeneity on genetic and molecular level 3,13. The genetic and molecular differences between the same histological tumor subtypes in different patients require personalized treatment regimens that take the individual tumor composition into account. For CRC targeted therapy approaches are available and especially KRAS and BRAF mutation status have been taken into account for personalized treatment regimens, however they only increased median survival rates by 2-4 months compared to chemotherapy alone 5,6,8. Tumor heterogeneity remains one of the main reasons for overall poor survival in metastasized cancers because it has been associated with progression and poor prognosis in CRC but is also considered to be one important factor for therapy resistance and treatment failure in CRC 9,14,15.
Intratumor heterogeneity in CRC is characterized by its complex tissue architecture with molecular differences within the tumor itself and the tumor periphery called tumor microenvironment. The tumor microenvironment consists of a variety of components such as extracellular matrix and secreted factors; mesenchymal, immune and endothelial cells; structures such as blood vessels, glands, and debris like necrosis and hemorrhage. The tumor microenvironment has been recognized to influence tumor progression, differentiation, metastasis formation, therapy response and heterogeneity within a single tumor area 16,17. Tumor cells within the same tumor display distinct genetic, phenotypic and functional profiles. Variations between the tumor cells are based on processes such as clonal development and adaptation to the microenvironment.
Genomic aberrations manifest in proteins, which are associated with changes in disease and common therapeutic targets. The limited correlation between the genetic and proteomic level was shown in a cohort of 146 patients with metastatic CRC. Primary tumors and metastases showed a high similarity on genetic but not on proteomic level, highlighting that genomics cannot cover all variations of the phenotype and the need for proteomic investigation of CRC heterogeneity 18. Proteomic differences at different CRC tumor sides (left vs. right) could be confirmed in several studies, including tissue biopsies 19 as well as plasma samples 20. Similarly proteomic differences between primary tumors and liver metastasis of the same patient as well as between recurrent liver metastases of the same patient were discovered 21,22. Proteomic intratumor heterogeneity was studied by Sugihara et al. using laser microdissection of ulcer floor, central area, and invasive front of CRC tumors to identify their different proteins and biological functions by shotgun proteomics 23. However, laser microdissection of tumor tissues and tumor subareas is a very laborious step resulting in limited spatial information. Therefore, the majority of proteomic studies analyze homogenized bulk tissue samples, which prevents detailed analysis of the different tissues in the tumor microenvironment and intratumor heterogeneity.
Matrix assisted laser desorption/ionization mass spectrometry imaging (MALDI imaging) enables measuring spatial protein distributions across different tissue and tumor compartment directly from thin tissue sections, without laborious preparation steps. Typically proteins are digested with trypsin to generate tryptic peptides, which are imaged at a spatial resolution between about 10 and 200 µm. Thus MALDI imaging is a predestined technique for the proteomic investigation of the tumor microenvironment and intratumor heterogeneity in complex tumor tissues 24. Tryptic peptide imaging was applied to CRC for the discovery of protein profiles associated with clinical marker 25, diagnosis 26, prognosis 27 and presence of lymph node metastases 28. Turtoi et al. applied MALDI imaging to analyze spatial proteomic heterogeneity in liver metastasis of CRC. They identified proteins that are zonally organized within CRC liver metastasis 29.
Here, we aim to characterize different levels of spatial proteomic heterogeneity in the complex tissue architecture of primary colorectal cancer and patient-matched liver metastasis tissues. In addition to the unique chance to study proteomic composition between primary CRC and liver metastasis of the same patients, we investigate interpatient and intratumor heterogeneity, which has been mainly studied on genome levels and rarely on proteome level. We apply MALDI imaging to study tryptic peptide distributions at 150 µm spatial resolution directly from formalin-fixed and paraffin-embedded tissue specimens from six patients. We found higher interpatient heterogeneity between liver metastasis than primary tumors and organized clusters with different proteomic content inside all tumor tissues.
2. Materials and Methods
Patient cohort
Tissues were obtained from 6 metastasized CRC patients whose tumors were resected at the University Medical Center in Freiburg. The study was approved by the Ethics Committee of the University Medical Center Freiburg (N° 504/17). All patients gave written informed consent. Before study inclusion, all patient data were pseudonymized. Only therapy naive patient samples were included. Tissue specimens were formalin-fixed directly after surgical removal and paraffin-embedded (FFPE) as previously described 30. All tissue specimens were reviewed by an experienced pathologist to confirm diagnosis of colorectal adenocarcinoma and ensure the presence of vital tumor tissue. 6 µm thick sections of the FFPE tissue blocks were sliced with a microtome and mounted onto indium tin oxide (ITO) coated glass slides (Bruker Daltonik, Bremen, Germany).
Sample preparation
The sliced and mounted tissue slides were deparaffinized according to a standard procedure 30 and prepared for MALDI imaging measurement as previously described 31. Samples were rinsed 10 mM ammonium bicarbonate, heated at 100°C for 1 hour in citrate buffer for antigen retrieval, twice rinsed in 10 mM ammonium bicarbonate and dried at room temperature. 1 mg/ml Trypsin (Worthington, Lakewood, NJ, USA) was sprayed over the whole slide using the iMatrixSpray (Tardo GmbH, Subingen, Switzerland, parameters: height: 60 mm, line distance: 1 mm, speed: 180 mm/s, density: 0.5 μL/cm3, cycles: 10, delay: 15 s, pressure: 1.6 bar). The specimen was placed in a digestion chamber containing a saturated potassium sulfate solution according to Ly et al. 32 and incubated at 50°C for 2 hours. 10 mg/ml alpha-cyano-4-hydroxycinnamic acid (CHCA, Sigma-Aldrich, Munich, Germany) matrix was prepared in 50% (v/v) acetonitrile and 1% (v/v) trifluoroacetic acid. A custom internal calibration mix including 0.08 µg/ml Angiotensin I (Anaspec, Seraing, Belgium), 0.04 µg/ml Substance P (Anaspec, Seraing, Belgium), 0.15 µg/µl [Glu]-Fibrinopeptide B (Sigma-Aldrich, Munich, Germany), and 0.30 µg/µl ACTH fragment (18-39) (Abcam, Cambridge, UK) was prepared 33 and mixed with the matrix solution with a ratio of 1:12 (v/v). The matrix/calibrant mix was sprayed across the slide with the iMatrixSpray (parameters: height: 60 mm, line distance: 1 mm, speed: 180 mm/s, density: 0.5 μL/cm3, cycles: 20, delay: 5 s, pressure: 1.6 bar). Teach marks were added manually in all 4 tissue corners by scratching a small cross into the slide surface and painting over it with a xylene resistant pen (LabID Technologies, BH 's- Hertogenbosch, Germany).
Data acquisition
Samples from the same patient were prepared together and measured consecutively on a 4800 MALDI-TOF/TOF Analyzer (Applied Biosystems, Waltham, MA, USA) using the 4000 Series Explorer software (Novartis and Applied Biosystems). Before measurement, internal calibrants outside the tissue regions were used for external mass calibration. A squared region containing tissue and teachmarks was measured with a laser diameter of about 100µm and a raster step size of 150 μm. Mass spectra were acquired averaging 500 laser shots each in a m/z range of 800-2500.
Tissue staining and annotation
The slides were rinsed with 70 % ethanol to remove the matrix after MALDI imaging. Then the slides were incubated for 4 min in hematoxylin solution modified according to Gill III, washed with acetic acid in aqueous solution and rinsed with water for about 5 min. Afterwards the tissues were incubated in eosin for 1 min and rinsed again with water. The stained tissues were scanned at x20 magnification with the Axio Scan.Z1 (ZEISS, Göttingen, Germany). A pathologist (AF) roughly annotated different tissue types in MS Powerpoint and another pathologist (GM) annotated only the vital tumor tissue in a detailed manner in Photoshop CS5 (Adobe, San Jose, USA).
Quality control and preprocessing
The Analyze7.5 MALDI imaging files were uploaded to the European Galaxy cloud (https://usegalaxy.eu), where the large majority of MALDI imaging data analysis was performed using previously developed mass spectrometry imaging (MSI) Galaxy tools and workflows 34,35. To obtain information about the tissue outline and annotated tumor areas in the H&E image, image co-registration of the H&E image and MALDI image was performed, according to a previously published workflow based on teachmarks and affine transformation in the Galaxy framework 31,35. Raw MALDI imaging data was assessed with the MSI qualitycontrol tool using the internal calibrant peaks as reference peaks with a mass range of 200 ppm. The samples were assessed for imaging artifacts, mass spectra quality including m/z accuracy and intensity variation. The findings were used to confirm sufficient data quality and find suitable preprocessing parameters. Before preprocessing only spectra from the tissue areas were kept while spectra from the slide background were removed. The detailed preprocessing parameters are listed in Table S1, including resampling, smoothing, baseline removal, m/z recalibration using the internal calibrants, peak picking, alignment and filtering, contaminants removal. Exemplary Galaxy workflow for preprocessing is displayed in Figure S1. As samples were separately measured, a common peak list was extracted by combining all preprocessed samples, removing empty m/z bins and exporting the peptide mass features. This list was used for peak integration of the raw data of the single, total ion current (TIC) normalized files. Between each preprocessing step, the MSI qualitycontrol tool was applied to monitor and optimize preprocessing parameters.
MSI analysis
- Tissue level proteomic differences
Unsupervised segmentation was performed to investigate tissue heterogeneity on sample level. Cardinal’s “Spatial Shrunken Centroids” (SSC) algorithm 36,37 was used in the MSI segmentation tool. The settings were spatial radius r=2, k=5, s=2. The initial number of clusters parameter k was set to 5, because on average, 4 major tissue types were expected to be in each sample, leaving room for an additional one. The discriminative m/z features were extracted from the SSC features output, results were The results were downloaded and further analyzed in R (version 4.0.4) in Jupyter notebook. The top 10 discriminative peptide mass features for the tumor clusters were selected according to their t-statistic values and their frequency calculated across all samples. Mean intensities of the top peptide mass features in tumor and non-tumor areas were calculated and significance tested using Wilcoxon rank sum test, corrected for multiple testing using "Benjamin Hochberg“ method. Ion images of these m/z were plotted in Galaxy with the MSI mz images tool (plusminus m/z: 0.25, contrast enhancement ‘histogram’) on the binned, and TIC normalized (using the MSI preprocessing tool) raw data.
- 2.
- Spatial intratumor heterogeneity
To investigate intratumor heterogeneity, primary and metastatic tumor regions were extracted from each sample based on the detailed tumor annotation of the pathologist in the H&E image. Unsupervised segmentation was performed with the SSC algorithm using the MSI segmentation tool with the setting r=2. The other parameters were varied to determine the optimal number of clusters using a method proposed by Bemis et al. 36 . Two starting cluster numbers k (3 and 6) were applied while increasing the shrinkage parameter s from 2 to 25. The segmentation results were projected back onto the whole sample for better visual evaluation.
- 3.
- Comparison of primary and liver metastases
Matched primary tumors and metastases were combined for each patient, the annotated tumor areas extracted and peak integration as well as TIC normalization performed as described above. Unsupervised segmentation was performed with the settings r = 2; k = 3, 6; s = 2, 5, 10, 15, 20, 25. Results were projected back onto the whole samples for better comparison and evaluated whether a) intratumor heterogeneity in each sample can be observed, b) resulting clusters are unique to one entity and c) if not, whether common clusters form specific patterns. A linear mixed model 38 was applied on log2 transformed, preprocessed tumor areas to find differentially abundant mass features between primary tumors and liver metastases.
- 4.
- Inter-patient tumor heterogeneity
To evaluate interpatient heterogeneity the discriminative mass features from the single sample tissue level segmentation were compared and a frequency distribution calculated and plotted in R (version 4.0.4) in Jupyter notebook.
- 5.
- Comparison with literature and tentative feature identification
We performed a literature review for MALDI imaging studies that compare solid tumors with normal tissues and extracted the m/z values and if available the information about the protein they derive from. We matched our m/z values with a 300 ppm window to the m/z values obtained from literature.
3. Results
3.1. Overview of the CRC patient cohort
The metastasized colorectal cancer (CRC) cohort contained 12 samples from six patients: six primary cancer tissues and six patient-matched liver metastases. MALDI imaging of tryptic peptides was performed separately on all samples using 150 µm raster step size. Across all samples about 56,000 tissue mass spectra were measured. In two samples, parts of the measured spectra were removed because they were part of an artificial stripe pattern, which would interfere with downstream analysis (PRIM_3, PRIM_6). Pre-processing resulted in a common peak list of 665 peptide mass features, for which the intensity values were obtained via the peak height within a 200 ppm m/z window from all mass spectra. After the measurement, the tissue sections were H&E stained and annotated by pathologists for i) tissue outline, ii) tissue types, and iii) detailed tumor tissue areas. Patient characteristics and numbers of spectra in the tissue of each sample are listed in Table 1.
3.2. Spatial proteome represents tissue morphologies
We performed MALDI imaging on whole CRC tissues, each between 1-3 cm2 in size to allow for the investigation of the whole tumor in its native tissue architecture. The different tissue types were annotated in the H&E images by a pathologist (Figure 1). Seven different tissue types are present in our CRC tissue cohort. Vital tumor tissue and peritumoral tissue consisting of stroma and necrosis were found in both primary tumor and liver metastasis tissues. In contrast, non-malignant colon tissue, smooth muscle tissue, connective / fatty tissue were only present in the primary tumor tissues, while liver parenchyma and duct structures were specific to the liver metastasis tissues. This depicts the heterogeneity of the spatial tissue morphologies in both entities. We hypothesized that the different proteomic composition of these tissue morphologies is also represented in the mass spectra of the corresponding tissue regions. To investigate this, we performed unsupervised segmentation with the spatial shrunken centroids algorithm 34 of each tissue specimen individually. The spatial shrunken centroids method is specifically designed for spatial data, agnostic to known tissue morphologies, and clusters similar mass spectra into the same segment based only on their molecular content. Side-by-side comparison of the obtained tissue segments with the tissue annotations shows that the segments resemble distinct morphological tissue areas (Figure 1).
The vital tumor regions were overall well represented in the segmentation results. To allow for a more detailed investigation a pathologist annotated the vital tumor areas in the H&E images with a high level of detail. Visual comparison of the tumor annotation and the spatial proteomic segmentation confirmed an overall good similarity regarding tumor border and level of detail even for highly segmented and multifocal regions (Figure S2). For primary tumors the segments resembled the detailed tumor annotation very well, with the exception for two primary tumors in which non-malignant colon epithelium could not be reliably separated from primary tumor areas. Tumor segments in the liver metastasis showed more deviations. Some liver tumor segments included small non-tumor areas and the tumor segment of one liver metastasis was missing a significant part of the tumor tissue (Figure S2).
Next, we aimed to find near-canonical m/z values for vital tumor segments. As previously explained, these segments were obtained by applying the spatial shrunken centroids method for spatial segmentation on each individual MALDI imaging dataset. As part of the segmentation, the spatial shrunken centroids method performs feature regularization. This means that only the most informative peptide features are used for the segmentation, while non-informative features are removed. This is reflected in the t-statistic value, which is calculated for each peptide feature. Higher t-statistics values mean higher contributions, while a value of zero means the peptide feature was not informative and not included in the segmentation. To find peptide features that are characteristic for a vital tumor, we extracted the ten peptide features with the highest t-statistics value for each sample and counted for each of these m/z values in how many samples it occurred in the top 10 tumor peptide features. Ten peptide features were present in at least four out of six samples of either primary tumor or metastasis and are listed in Table 2. For these tumor enriched peptide features, the average intensities were higher in the vital tumor clusters than in the non-tumor tissue clusters and five of them were significantly different (adj. p-value <0.05 in Wilcoxon Rank Sum Test): 929.51, 930.63, 943.37, 957.44, 1028.5 (Table 2, Figure S3). We plotted the spatial distribution of m/z 943.37, which correlated well with the detailed tumor annotation (Figure S2 iii). These findings underline the validity of our approach to define vital tumor enriched peptide features by their t-statistics value in the spatial shrunken centroids method.
Interestingly, 9 of our 10 tumor enriched peptide features matched within a 300 ppm window to peptide mass features that have been described as tumor specific in three other solid tumor MALDI imaging studies (Table 2). These studies compared vulvar squamous cell carcinomas to precursor lesions, head and neck cancer tumor epithelial regions with non-tumor tissue regions, and neuroblastoma cell rich tumor regions with surrounding non-tumor tissue 39–41. MALDI imaging measures majorly the most abundant proteins 42 and thus it is likely that universal solid tumor protein markers can be found even across very different cancer types. Two of the three mentioned studies run additional LC-MS/MS experiments to obtain tentative identifications for their peptide mass features. Often multiple identifications are obtained for one peptide mass feature and cannot be further specified, thus multiple identifications are reported (Table 2). Despite this uncertainty, the large majority of the tentatively identified proteins are known to be associated with tumor cells. Annexin A4, for which we have potentially found two tumor enriched tryptic peptides, is known to promote CRC tumorigenesis and epithelial-mesenchymal transition, which fosters metastasis formation 43. Prelamin A/C was a potential identification for peptide mass feature 1028.5 in both studies, which performed identification via LC-MS/MS. The role of lamin A/C, the products of prelamin A/C, in CRC cancer is not yet established. One study found lamin A/C in 70% of CRC tumor tissues and the expression was associated with higher mortality while another study reported increased recurrence with decreased lamin A/C 44,45.

Table 2.
Overview of the peptide mass features enriched in vital tumor areas. The frequency of the top 10 peptide mass features of each sample’s vital tumor segment were calculated across all samples, primary tumors and in liver metastases, respectively. Only peptide mass features that occurred in four or more samples of at least one entity (primary tumor or metastasis) are listed here (black). Complementary numbers are provided in gray. The list might include isotopes. Peptide mass features with significant different average intensities between vital tumor and non-tumor tissues are marked with * (adj. p-value <0.05 in Wilcoxon Rank Sum Test). Within a 300 ppm window, the 9 peptide mass features could be matched to tumor specific peptide mass features of other solid tumor MALDI imaging studies out of which two provided tentative protein identifications (shown as gene names).
Table 2.
Overview of the peptide mass features enriched in vital tumor areas. The frequency of the top 10 peptide mass features of each sample’s vital tumor segment were calculated across all samples, primary tumors and in liver metastases, respectively. Only peptide mass features that occurred in four or more samples of at least one entity (primary tumor or metastasis) are listed here (black). Complementary numbers are provided in gray. The list might include isotopes. Peptide mass features with significant different average intensities between vital tumor and non-tumor tissues are marked with * (adj. p-value <0.05 in Wilcoxon Rank Sum Test). Within a 300 ppm window, the 9 peptide mass features could be matched to tumor specific peptide mass features of other solid tumor MALDI imaging studies out of which two provided tentative protein identifications (shown as gene names).
| m/z | CRC all samples | CRC primary tumor | CRC liver metastasis | Vulvar cancer | Head and neck cancer | Neuroblastoma |
|---|---|---|---|---|---|---|
| 857.36 | 5/12 | 1/6 | 4/6 | ANXA4FDXR | - | 857.4 |
| 901.32 | 4/12 | 4/6 | 0/6 | - | HSP90 | 901.5 |
| 901.68 | 7/12 | 6/6 | 1/6 | - | - | - |
| 929.51* | 10/12 | 6/6 | 4/6 | - | - | 929.4 |
| 930.63* | 7/12 | 5/6 | 2/6 | - | - | 930.6 |
| 943.37* | 9/12 | 5/6 | 4/6 | HNRNPC | - | 943.5 |
| 944.5 | 7/12 | 2/6 | 5/6 | H2AC18 H2AC7 H2BC1 H2AX H2AZ2 |
VIM KRT78 |
944.4 |
| 957.44* | 5/6 | 1/6 | 4/6 | AHNAK COL6A1 FUBP1 PRPF8 |
NCL | 957.6 |
| 958.59 | 8/12 | 6/6 | 2/6 | ANXA4 HNRNPAB NAPRT TFRC |
- | 958.5 |
3.3. CRC tissues show organized intratumor heterogeneity
Due to the clonal evolution of tumors, different cell clones may proliferate in parallel causing different tumor subregions. This intratumor heterogeneity is a major factor in therapy resistance as only some cell populations might be affected by a treatment while others may continue growing. To probe intratumor heterogeneity, we assessed the annotated tumor tissue by spatial segmentation. This approach differs from the previously presented segmentation that also included large areas of non-tumor tissue. The strong differences between tumor- and non-tumor areas are likely to overshadow more subtle differences arising from intratumor heterogeneity. Hence, to assess intratumor heterogeneity, we focused the unsupervised segmentation on mass spectra that were annotated as vital tumor areas based on detailed histopathological annotation in the H&E image. Inside the tumor areas of each sample multiple segments were obtained, which indicates proteomic intratumor heterogeneity (Figure 2). Many tissues with larger tumor areas showed segments at their borders (PRIM_2, PRIM_3, PRIM_5 and PRIM_6), potentially representing the transition zone between tumor and peritumoral tissue. Three liver metastases (MET_2, MET_4 MET_5) show organized pattern formation as zonal structures forming differing layers from inwards to outwards.
Comparison of the segments with the H&E images showed that they did not correspond to visible histologic patterns. Most segments contained a mixture of tumor cells, fibrocytes, immune cells and some segments contained small amounts of necrosis or extracellular matrix. Unfortunately, with the used mass spectrometer, we were restricted to 150 µm spatial resolution and therefore not able to focus our proteomic analysis on individual tumor cells.
3.4. Spatial proteomic similarity of patient-matched primary tumors and liver metastases
Next, we were interested to compare the m/z profiles between patient-matched primary tumors and corresponding liver metastases. For this, we virtually combined the MALDI imaging data of primary tumor samples and corresponding liver metastasis for each patient. Then we repeated the spatial shrunken centroids based unsupervised segmentation on the vital tumor areas from these six combined datasets, hence enabling the direct delineation of shared segments. The resulting segments of the patient-matched tumor entities showed high similarity to the segments obtained by segmentation of the individual tumor tissues. The preservation of the intratumor segments even in the presence of a different entity underlines the robustness of the molecular segmentation. All patient-matched tumor tissue pairs show multiple clusters that span across both entities (Figure 3). This indicates a substantial proteomic concordance across primary tumor and liver metastasis of the same patient and the absence of batch effects between the samples of the same patient. We then investigated if metastasis forming segments can be found. These would be represented by a segment that covers a part of the primary tumor and a larger area in the matching liver metastasis. However, we could not find such segments. To follow up on the proteomic differences between patient-matched vital tumor areas of primary tumors and liver metastasis, we compared the averaged vital tumor intensities between them using a linear mixed model. The analysis resulted in very high adjusted p-values for all peptide features, indicating that none of the peptide features had a significantly different mean intensity between the vital tumor areas of primary tumor and liver metastases.
3.5. Interpatient proteomic differences are higher between liver metastases than in primary tumors
After having analyzed the spatial tumor proteome sample and patient wise, we now aimed to compare it between all six patients. For this, we re-inspected the tissue level unsupervised segmentation results shown in Figure 1. Here we focus on the proteomic comparison between the vital tumor segments of all patients. We had previously extracted a list of peptide mass features that were enriched in the vital tumor segments (Table S2). In total these were 248 peptide features in primary tumors and 363 peptide features in liver metastasis. We counted for each of these peptide features in how many patients they were part of the tumor enriched peptide feature list. We did this separately for primary tumors and liver metastases. For primary tumors 100 out of the 248 primary tumor enriched peptide features were found as tumor enriched in all six patients (Figure 4a). In contrast, for liver metastasis not a single peptide feature was found to be tumor enriched in all six patients and only 34 out of 363 peptide features occurred in more than 3 liver metastases (Figure 4b). This indicates a higher interpatient heterogeneity in CRC liver metastases compared to CRC primary tumors.
4. Discussion
We performed MALDI imaging on 12 whole-slide tissues from six patient-matched primary CRC and liver metastases. We obtained distribution heatmaps representing the complex tissue composition of CRC on a molecular level and could characterize interpatient, intertumor and intratumor hetereogeneity. In comparison to most proteomic CRC studies, our approach with a spatial resolution of 150µm allowed for the spatial separation of vital tumor tissue from non-tumor tissue and the investigation of intratumor heterogeneity without laborious and time consuming sample preparation. We found several peptide features that were enriched in vital tumor areas and found in other MALDI imaging studies of solid tumors. This suggests that high abundant tumor peptides may serve as general solid tumor markers in MALDI imaging, but more investigations are needed to confirm this hypothesis. Amongst the peptide features most enriched across our 12 vital tumors are tentatively peptides from several characteristic tumor proteins, including annexin A4 and prelamin A/C, which both were described in CRC before.
The peptide features that characterized the vital tumor areas in the primary tumors were more similar across patients than in the metastasized tumors. This higher interpatient heterogeneity in CRC metastases implicates patient specific differences during metastasis formation. Metastasis formation requires many different steps, including EMT, MET and plasticity through genetic alterations to allow dissemination and seeding into other organs. A genomic analysis in CRC revealed that rare subclones seed many metastases 46. This implicates a high number of differing genetic alterations in each rare subclone, which could explain the higher heterogeneity observed in metastasis. The same study also found that potential metastasis forming clones do not necessarily represent the most abundant subpopulations in the primary tumor. This is in concordance with our findings in terms of spatial comparison of primary tumors and metastasis, as we did not find potentially metastasis forming subclusters in the major tumor mass of primary tumors.
We found subclusters that were spanning across both entities and no significant differences between both entities could be confirmed. This is in contrast to two recent shotgun proteomic studies in which proteomic differences between primary CRC and liver metastasis were found 18,21. However, these studies included some patients that had already obtained treatment before surgery, while all our patients had no previous treatment and thus no selective pressures were present that might lead to a different adaptation of primary tumor and liver metastasis. Furthermore, the shotgun proteomic studies were performed on macrodissected tumor tissues and the obtained protein intensities are average values over the full tumor tissue, neglecting intratumor heterogeneity. In contrast, our study is the first that reveals spatially resolved proteomic compositions of both the primary tumor and liver metastases of the same patients, but might have missed differences between both entities in less abundant proteins due to the limited sensitivity of the MALDI mass spectrometer.
The separate assessment of intratumor heterogeneity in each vital tumor tissue found areas with distinct peptide profiles, sometimes organized in distinct zones between tumor and peritumoral tissue. Intratumor heterogeneity in CRC stems from mutational events during cancer formation and adaptation to the microenvironment that leads to a multitude of subclones. Importantly, these alterations are not evenly distributed across a tumor 47. Some proteomic studies have also shown that protein expression varies in different CRC tumor zones 48. The only other MALDI imaging study of CRC liver metastasis was able to show functional and therapeutic biomarker heterogeneity in peritumoral, rim and center zones of metastatic tumor tissues 27. Data from different in-vivo cancer models show functionally different zones in several tumors: proliferation at the outer zone, differentiation in the middle zone, and central cell death 49. This intratumor heterogeneity has direct clinical implications for example for therapy response and progression. Unfortunately, we had no access to such clinical data to correlate it with our intratumor segments, which should be part of future studies.
Although we studied only a limited number of patients, we were able to unravel their proteomic heterogeneity on patient, tumor and intratumor level. This aspect is especially important as therapeutic treatment is often chosen according to the properties of the primary tumors. Our data suggests that treatment of metastasis CRC needs to consider their diverse and distinct tumor phenotypes, which should be investigated in future MALDI imaging studies on a larger patient cohort and with data about therapy response.
5. Conclusions
MALDI imaging of six patient-matched CRC primary tumor and liver metastases allowed for the spatial proteomic characterization of interpatient, intertumor and intratumor heterogeneity. We found several peptide features that were enriched in vital tumor areas of primary tumors and liver metastasis and tentatively derived from tumor cell specific proteins such as annexin A4 and prelamin A/C, which were also found in other MALDI imaging studies of solid tumors. Liver metastases of colorectal cancer show higher proteomic heterogeneity between patients than primary tumors while within patients both entities show similar intratumor clusters and proteomes. The vital tumor areas of primary tumors as well as liver metastasis showed spatial segments with distinct proteomic profiles, sometimes organized in zonal patterns. Together our findings give new insights into the spatial proteome of primary CRC and patient-matched liver metastases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Final pre-processing workflow in Galaxy; Figure S2: Mean intensities of the top tumor peptide features per sample; Table S1: Galaxy tools and parameters applied for spectra pre-processing. Table S2: List of vital tumor area enriched peptide mass features.
Author Contributions
Conceptualization, S.F., A.J., O.S. and M.F.; methodology, L.M., O.S., and M.F.; software, L.M., M.S., and M.F.; validation, L.M., M.S., M.W., A.J., O.S., and M.F.; formal analysis, L.M., F.T., A.J., and M.F.; investigation, L.M., M.S., F.T. , and H.F.; resources, S.F., H.F., M.W., and O.S.; data curation, L.M., M.S., and M.F.; writing—original draft preparation, L.M., and M.F.; writing—review and editing, M.S., O.S., and M.F.; visualization, L.M.; supervision, M.W., S.F., O.S., and M.F.; project administration, O.S, and M.F.; funding acquisition O.S.. All authors have read and agreed to the published version of the manuscript.
Funding
O.S. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, projects 446058856, 466359513, 444936968, 405351425, 431336276, 43198400 (SFB 1453 “NephGen”), 441891347 (SFB 1479 “OncoEscape”), 423813989 (GRK 2606 “ProtPath”), 322977937 (GRK 2344 “MeInBio”)), the ERA PerMed program (BMBF, 01KU1916, 01KU1915A), the ERA TRANSCAN program (BMBF 01KT2201), the German Consortium for Translational Cancer Research (project Impro-Rec), the MatrixCode research group of FRIAS, Freiburg, the Fördergesellschaft Forschung Tumorbiologie, Freiburg im Breisgau, the invest BW program (BW1_1198/03), and the Centers for Personalized Medicine Baden-Württemberg. F.T. acknowledges funding by the Hungarian National Research, Development and Innovation Office (NFKIH) under grant: Development of scientific workshops of medical, health sciences and pharmaceutical educations, project identification number: EFOP-3.6.3-VEKOP-16-2017-00009.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by Ethics Committee of the University Medical Center Freiburg (N° 504/17).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The raw data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository.
Acknowledgments
The authors acknowledge the help of Anna Frey and Gulnara Mirzayeva for annotations of the H&E images and Larissa Meyer for performing literature review for tumor MALDI imaging studies. M.C.F. was supported by the Medical-Scientist-Programme, Faculty of Medicine, University of Freiburg. The authors acknowledge the support of the Freiburg Galaxy Team: Dr. Björn Grüning and Prof. Rolf Backofen, Bioinformatics, University of Freiburg, Germany funded by the SFB1425, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - Project #422681845 and German Federal Ministry of Education and Research [BMBF Grant 031 A538A RBC (de.NBI)]. We acknowledge support by the Open Access Publication Fund of the University of Freiburg.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- De Rosa, M.; Pace, U.; Rega, D.; Costabile, V.; Duraturo, F.; Izzo, P.; Delrio, P. Genetics, Diagnosis and Management of Colorectal Cancer (Review). Oncol. Rep. 2015, 34, 1087–1096. [Google Scholar] [CrossRef]
- Millikan, K.W.; Staren, E.D.; Doolas, A. INVASIVE THERAPY OF METASTATIC COLORECTAL CANCER TO THE LIVER. Surg. Clin. North Am. 1997, 77, 27–48. [Google Scholar] [CrossRef]
- Manfredi, S.; Lepage, C.; Hatem, C.; Coatmeur, O.; Faivre, J.; Bouvier, A.-M. Epidemiology and Management of Liver Metastases From Colorectal Cancer: Ann. Surg. 2006, 244, 254–259. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; Tonini, G.; Carlomagno, C.; Allegrini, G.; Chiara, S.; D’Amico, M.; Granetto, C.; Cazzaniga, M.; Boni, L.; Fontanini, G.; Falcone, A. FOLFOXIRI plus Bevacizumab versus FOLFIRI plus Bevacizumab as First-Line Treatment of Patients with Metastatic Colorectal Cancer: Updated Overall Survival and Molecular Subgroup Analyses of the Open-Label, Phase 3 TRIBE Study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; Steeghs, N.; Guren, T.K.; Arkenau, H.-T.; Garcia-Alfonso, P.; Pfeiffer, P.; Orlov, S.; Lonardi, S.; Elez, E.; Kim, T.-W.; Schellens, J.H.M.; Guo, C.; Krishnan, A.; Dekervel, J.; Morris, V.; Calvo Ferrandiz, A.; Tarpgaard, L.S.; Braun, M.; Gollerkeri, A.; Keir, C.; Maharry, K.; Pickard, M.; Christy-Bittel, J.; Anderson, L.; Sandor, V.; Tabernero, J. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Goldberg, R.M.; Sargent, D.J.; Morton, R.F.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Pitot, H.C.; Alberts, S.R. A Randomized Controlled Trial of Fluorouracil Plus Leucovorin, Irinotecan, and Oxaliplatin Combinations in Patients With Previously Untreated Metastatic Colorectal Cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar] [CrossRef]
- Venook, A.P.; Niedzwiecki, D.; Lenz, H.-J.; Innocenti, F.; Fruth, B.; Meyerhardt, J.A.; Schrag, D.; Greene, C.; O’Neil, B.H.; Atkins, J.N.; Berry, S.; Polite, B.N.; O’Reilly, E.M.; Goldberg, R.M.; Hochster, H.S.; Schilsky, R.L.; Bertagnolli, M.M.; El-Khoueiry, A.B.; Watson, P.; Benson, A.B.; Mulkerin, D.L.; Mayer, R.J.; Blanke, C. Effect of First-Line Chemotherapy Combined With Cetuximab or Bevacizumab on Overall Survival in Patients With KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2017, 317, 2392. [Google Scholar] [CrossRef] [PubMed]
- Molinari, C.; Marisi, G.; Passardi, A.; Matteucci, L.; De Maio, G.; Ulivi, P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? Int. J. Mol. Sci. 2018, 19, 3733. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.C.; Lim, Y.M. Proteome Heterogeneity in Colorectal Cancer. PROTEOMICS 2018, 18, 1700169. [Google Scholar] [CrossRef] [PubMed]
- Di, J.; Yang, H.; Jiang, B.; Wang, Z.; Ji, J.; Su, X. Whole Exome Sequencing Reveals Intertumor Heterogeneity and Distinct Genetic Origins of Sporadic Synchronous Colorectal Cancer. Int. J. Cancer 2018, 142, 927–939. [Google Scholar] [CrossRef]
- Lu, Y.-W.; Zhang, H.-F.; Liang, R.; Xie, Z.-R.; Luo, H.-Y.; Zeng, Y.-J.; Xu, Y.; Wang, L.-M.; Kong, X.-Y.; Wang, K.-H. Colorectal Cancer Genetic Heterogeneity Delineated by Multi-Region Sequencing. PLOS ONE 2016, 11, e0152673. [Google Scholar] [CrossRef]
- de la Chapelle, A. Genetic Predisposition to Colorectal Cancer. Nat. Rev. Cancer 2004, 4, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Ramón y Cajal, S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical Implications of Intratumor Heterogeneity: Challenges and Opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Quezada, S.A.; Larkin, J.; Swanton, C. Translational Implications of Tumor Heterogeneity. Clin. Cancer Res. 2015, 21, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia Promotes Invasive Growth by Transcriptional Activation of the Met Protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Mesker, W.E.; Junggeburt, J.M.C.; Szuhai, K.; de Heer, P.; Morreau, H.; Tanke, H.J.; Tollenaar, R.A.E.M. The Carcinoma–Stromal Ratio of Colon Carcinoma Is an Independent Factor for Survival Compared to Lymph Node Status and Tumor Stage. Anal. Cell. Pathol. 2007, 29, 387–398. [Google Scholar] [CrossRef]
- Li, C.; Sun, Y.-D.; Yu, G.-Y.; Cui, J.-R.; Lou, Z.; Zhang, H.; Huang, Y.; Bai, C.-G.; Deng, L.-L.; Liu, P.; Zheng, K.; Wang, Y.-H.; Wang, Q.-Q.; Li, Q.-R.; Wu, Q.-Q.; Liu, Q.; Shyr, Y.; Li, Y.-X.; Chen, L.-N.; Wu, J.-R.; Zhang, W.; Zeng, R. Integrated Omics of Metastatic Colorectal Cancer. Cancer Cell 2020, 38, 734–747.e9. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Huang, J.; Pei, H.; Zeng, S.; Tao, Y.; Shen, L.; Zeng, L.; Zhu, H. Comparative Proteomic Study for Profiling Differentially Expressed Proteins between Chinese Left- and Right-Sided Colon Cancers. Cancer Sci. 2013, 104, 135–141. [Google Scholar] [CrossRef]
- Surinova, S.; Radová, L.; Choi, M.; Srovnal, J.; Brenner, H.; Vitek, O.; Hajdúch, M.; Aebersold, R. Non-invasive Prognostic Protein Biomarker Signatures Associated with Colorectal Cancer. EMBO Mol. Med. 2015, 7, 1153–1165. [Google Scholar] [CrossRef]
- Fahrner, M.; Bronsert, P.; Fichtner-Feigl, S.; Jud, A.; Schilling, O. Proteome Biology of Primary Colorectal Carcinoma and Corresponding Liver Metastases. Neoplasia 2021, 23, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Voß, H.; Wurlitzer, M.; Smit, D.J.; Ewald, F.; Alawi, M.; Spohn, M.; Indenbirken, D.; Omidi, M.; David, K.; Juhl, H.; Simon, R.; Sauter, G.; Fischer, L.; Izbicki, J.R.; Molloy, M.P.; Nashan, B.; Schlüter, H.; Jücker, M. Differential Regulation of Extracellular Matrix Proteins in Three Recurrent Liver Metastases of a Single Patient with Colorectal Cancer. Clin. Exp. Metastasis 2020, 37, 649–656. [Google Scholar] [CrossRef]
- Sugihara, Y.; Taniguchi, H.; Kushima, R.; Tsuda, H.; Kubota, D.; Ichikawa, H.; Fujita, S.; Kondo, T. Laser Microdissection and Two-Dimensional Difference Gel Electrophoresis Reveal Proteomic Intra-Tumor Heterogeneity in Colorectal Cancer. J. Proteomics 2013, 78, 134–147. [Google Scholar] [CrossRef]
- Vaysse, P.M.; Heeren, R.M.A.; Porta, T.; Balluff, B. Mass Spectrometry Imaging for Clinical Research-Latest Developments, Applications, and Current Limitations. Analyst 2017, 142, 2690–2712. [Google Scholar] [CrossRef]
- Hinsch, A.; Buchholz, M.; Odinga, S.; Borkowski, C.; Koop, C.; Izbicki, J.R.; Wurlitzer, M.; Krech, T.; Wilczak, W.; Steurer, S.; Jacobsen, F.; Burandt, E.C.; Stahl, P.; Simon, R.; Sauter, G.; Schl??ter, H. MALDI Imaging Mass Spectrometry Reveals Multiple Clinically Relevant Masses in Colorectal Cancer Using Large-Scale Tissue Microarrays. J. Mass Spectrom. 2017, 52, 165–173. [Google Scholar] [CrossRef]
- Mittal, P.; Condina, M.R.; Klingler-Hoffmann, M.; Kaur, G.; Oehler, M.K.; Sieber, O.M.; Palmieri, M.; Kommoss, S.; Brucker, S.; McDonnell, M.D.; Hoffmann, P. Cancer Tissue Classification Using Supervised Machine Learning Applied to MALDI Mass Spectrometry Imaging. Cancers 2021, 13, 5388. [Google Scholar] [CrossRef]
- Martin, B.; Gonçalves, J.P.L.; Bollwein, C.; Sommer, F.; Schenkirsch, G.; Jacob, A.; Seibert, A.; Weichert, W.; Märkl, B.; Schwamborn, K. A Mass Spectrometry Imaging Based Approach for Prognosis Prediction in UICC Stage I/II Colon Cancer. Cancers 2021, 13, 5371. [Google Scholar] [CrossRef]
- Meding, S.; Balluff, B.; Elsner, M.; Schöne, C.; Rauser, S.; Nitsche, U.; Maak, M.; Schäfer, A.; Hauck, S.M.; Ueffing, M.; Langer, R.; Höfler, H.; Friess, H.; Rosenberg, R.; Walch, A. Tissue-Based Proteomics Reveals FXYD3, S100A11 and GSTM3 as Novel Markers for Regional Lymph Node Metastasis in Colon Cancer: Proteomic Markers for Regional Node Metastasis. J. Pathol. 2012, 228, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Turtoi, A.; Blomme, A.; Debois, D.; Somja, J.; Delvaux, D.; Patsos, G.; Di Valentin, E.; Peulen, O.; Mutijima, E.N.; De Pauw, E.; Delvenne, P.; Detry, O.; Castronovo, V. Organized Proteomic Heterogeneity in Colorectal Cancer Liver Metastases and Implications for Therapies. Hepatology 2014, 59, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Bronsert, P.; Weißer, J.; Biniossek, M.L.; Kuehs, M.; Mayer, B.; Drendel, V.; Timme, S.; Shahinian, H.; Küsters, S.; Wellner, U.F.; Lassmann, S.; Werner, M.; Schilling, O. Impact of Routinely Employed Procedures for Tissue Processing on the Proteomic Analysis of Formalin-Fixed Paraffin-Embedded Tissue. PROTEOMICS - Clin. Appl. 2014, 8, (9–10). [Google Scholar] [CrossRef] [PubMed]
- Föll, M.C.; Volkmann, V.; Enderle-Ammour, K.; Timme, S.; Wilhelm, K.; Guo, D.; Vitek, O.; Bronsert, P.; Schilling, O. Moving Translational Mass Spectrometry Imaging towards Transparent and Reproducible Data Analyses: A Case Study of an Urothelial Cancer Cohort Analyzed in the Galaxy Framework. Clin. Proteomics 2022, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Longuespée, R.; Casadonte, R.; Wandernoth, P.; Schwamborn, K.; Bollwein, C.; Marsching, C.; Kriegsmann, K.; Hopf, C.; Weichert, W.; Kriegsmann, J.; Schirmacher, P.; Kriegsmann, M.; Deininger, S. Site-to-Site Reproducibility and Spatial Resolution in MALDI–MSI of Peptides from Formalin-Fixed Paraffin-Embedded Samples. PROTEOMICS – Clin. Appl. 2019, 13, 1800029. [Google Scholar] [CrossRef]
- Gustafsson, J.O.R.; Eddes, J.S.; Meding, S.; Koudelka, T.; Oehler, M.K.; McColl, S.R.; Hoffmann, P. Internal Calibrants Allow High Accuracy Peptide Matching between MALDI Imaging MS and LC-MS/MS. J. Proteomics 2012, 75, 5093–5105. [Google Scholar] [CrossRef]
- The Galaxy Community; Afgan, E.; Nekrutenko, A.; Grüning, B.A.; Blankenberg, D.; Goecks, J.; Schatz, M.C.; Ostrovsky, A.E.; Mahmoud, A.; Lonie, A.J.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2022 Update. Nucleic Acids Res. 2022, gkac247. [Google Scholar] [CrossRef]
- Föll, M.C.; Moritz, L.; Wollmann, T.; Stillger, M.N.; Vockert, N.; Werner, M.; Bronsert, P.; Rohr, K.; Grüning, B.A.; Schilling, O. Accessible and Reproducible Mass Spectrometry Imaging Data Analysis in Galaxy. GigaScience 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Bemis, K.D.; Harry, A.; Eberlin, L.S.; Ferreira, C.R.; van de Ven, S.M.; Mallick, P.; Stolowitz, M.; Vitek, O. Probabilistic Segmentation of Mass Spectrometry (MS) Images Helps Select Important Ions and Characterize Confidence in the Resulting Segments. Mol. Cell. Proteomics 2016, 15, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Bemis, K.D.; Harry, A.; Eberlin, L.S.; Ferreira, C.; Van De Ven, S.M.; Mallick, P.; Stolowitz, M.; Vitek, O. Cardinal: An R Package for Statistical Analysis of Mass Spectrometry-Based Imaging Experiments. Bioinformatics 2015, 31, 2418–2420. [Google Scholar] [CrossRef] [PubMed]
- Bemis, K.A.; Guo, D.; Harry, A.J.; Thomas, M.; Lanekoff, I.; Stenzel-Poore, M.P.; Stevens, S.L.; Laskin, J.; Vitek, O. Statistical Detection of Differentially Abundant Ions in Mass Spectrometry-Based Imaging Experiments with Complex Designs. Int. J. Mass Spectrom. 2019, 437, 49–57. [Google Scholar] [CrossRef]
- Zhang, C.; Arentz, G.; Winderbaum, L.; Lokman, N.A.; Klingler-Hoffmann, M.; Mittal, P.; Carter, C.; Oehler, M.K.; Hoffmann, P. MALDI Mass Spectrometry Imaging Reveals Decreased CK5 Levels in Vulvar Squamous Cell Carcinomas Compared to the Precursor Lesion Differentiated Vulvar Intraepithelial Neoplasia. Int. J. Mol. Sci. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Hoffmann, F.; Umbreit, C.; Krüger, T.; Pelzel, D.; Ernst, G.; Kniemeyer, O.; Guntinas-Lichius, O.; Berndt, A.; von Eggeling, F. Identification of Proteomic Markers in Head and Neck Cancer Using MALDI–MS Imaging, LC–MS/MS, and Immunohistochemistry. Proteomics - Clin. Appl. 2019, 13, 1–10. [Google Scholar] [CrossRef]
- Wu, Z.; Hundsdoerfer, P.; Schulte, J.H.; Astrahantseff, K.; Boral, S.; Schmelz, K.; Eggert, A.; Klein, O. Discovery of Spatial Peptide Signatures for Neuroblastoma Risk Assessment by MALDI Mass Spectrometry Imaging. Cancers 2021, 13, 3184. [Google Scholar] [CrossRef]
- van de Ven, S.M.W.Y.; Bemis, K.D.; Lau, K.; Adusumilli, R.; Kota, U.; Stolowitz, M.; Vitek, O.; Mallick, P.; Gambhir, S.S. Protein Biomarkers on Tissue as Imaged via MALDI Mass Spectrometry: A Systematic Approach to Study the Limits of Detection. Proteomics 2016, 16, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhang, Z.; Zhang, A.; Liu, C.; Sun, Y.; Peng, Z.; Liu, Y. Membrane-Cytoplasm Translocation of Annexin A4 Is Involved in the Metastasis of Colorectal Carcinoma. Aging 2021, 13, 10312–10325. [Google Scholar] [CrossRef] [PubMed]
- Belt, E.J.T.; Fijneman, R.J.A.; van den Berg, E.G.; Bril, H.; Delis-van Diemen, P.M.; Tijssen, M.; van Essen, H.F.; de Lange-de Klerk, E.S.M.; Beliën, J.A.M.; Stockmann, H.B.A.C.; Meijer, S.; Meijer, G.A. Loss of Lamin A/C Expression in Stage II and III Colon Cancer Is Associated with Disease Recurrence. Eur. J. Cancer 2011, 47, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Willis, N.D.; Cox, T.R.; Rahman-Casañs, S.F.; Smits, K.; Przyborski, S.A.; van den Brandt, P.; van Engeland, M.; Weijenberg, M.; Wilson, R.G.; de Bruïne, A.; Hutchison, C.J. Lamin A/C Is a Risk Biomarker in Colorectal Cancer. PLoS ONE 2008, 3, e2988. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.X.; Krasnick, B.A.; White, B.S.; Grossman, J.G.; Strand, M.S.; Zhang, J.; Cabanski, C.R.; Miller, C.A.; Fulton, R.S.; Goedegebuure, S.P.; Fronick, C.C.; Griffith, M.; Larson, D.E.; Goetz, B.D.; Walker, J.R.; Hawkins, W.G.; Strasberg, S.M.; Linehan, D.C.; Lim, K.H.; Lockhart, A.C.; Mardis, E.R.; Wilson, R.K.; Ley, T.J.; Maher, C.A.; Fields, R.C. The Clonal Evolution of Metastatic Colorectal Cancer. Sci. Adv. 2020, 6, eaay9691. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; Curtis, C. A Big Bang Model of Human Colorectal Tumor Growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Shi, H.; Hood, K.A.; Hayes, M.T.; Stubbs, R.S. Proteomic Analysis of Advanced Colorectal Cancer by Laser Capture Microdissection and Two-Dimensional Difference Gel Electrophoresis. J. Proteomics 2011, 75, 339–351. [Google Scholar] [CrossRef]
- Mehrabi, M.; Amini, F.; Mehrabi, S. Active Role of the Necrotic Zone in Desensitization of Hypoxic Macrophages and Regulation of CSC-Fate: A Hypothesis. Front. Oncol. 2018, 8, 235. [Google Scholar] [CrossRef]
Figure 1.
Tissue morphologies of CRC primary tumor and liver metastasis tissues differ in their proteomic composition. a) Primary tumor and b) liver metastasis tissues for all patients are shown. i. Tissue annotations in H&E images. For primary tumors (a), vital tumor tissue (blue), peritumoral tissue (stroma, necrosis) (green), non-malignant colon tissue (black) smooth muscle tissue (red), connective / fatty tissue (pink) were annotated. For liver metastases (b), vital tumor tissue (blue), liver parenchyma (orange), peritumoral tissue (stroma, necrosis) (green), duct structure (gray). ii. MALDI imaging segments with different proteomic composition show organized tissue architecture. These segments resemble annotated tissue types shown in i: vital tumor (light and dark blue), peritumoral area (green), connective / fatty tissue (pink), smooth muscle tissue (red), liver parenchyma (yellow, orange). Tissue regions with artifacts were removed from PRIM_3 and PRIM_6 MALDI imaging data.
Figure 1.
Tissue morphologies of CRC primary tumor and liver metastasis tissues differ in their proteomic composition. a) Primary tumor and b) liver metastasis tissues for all patients are shown. i. Tissue annotations in H&E images. For primary tumors (a), vital tumor tissue (blue), peritumoral tissue (stroma, necrosis) (green), non-malignant colon tissue (black) smooth muscle tissue (red), connective / fatty tissue (pink) were annotated. For liver metastases (b), vital tumor tissue (blue), liver parenchyma (orange), peritumoral tissue (stroma, necrosis) (green), duct structure (gray). ii. MALDI imaging segments with different proteomic composition show organized tissue architecture. These segments resemble annotated tissue types shown in i: vital tumor (light and dark blue), peritumoral area (green), connective / fatty tissue (pink), smooth muscle tissue (red), liver parenchyma (yellow, orange). Tissue regions with artifacts were removed from PRIM_3 and PRIM_6 MALDI imaging data.
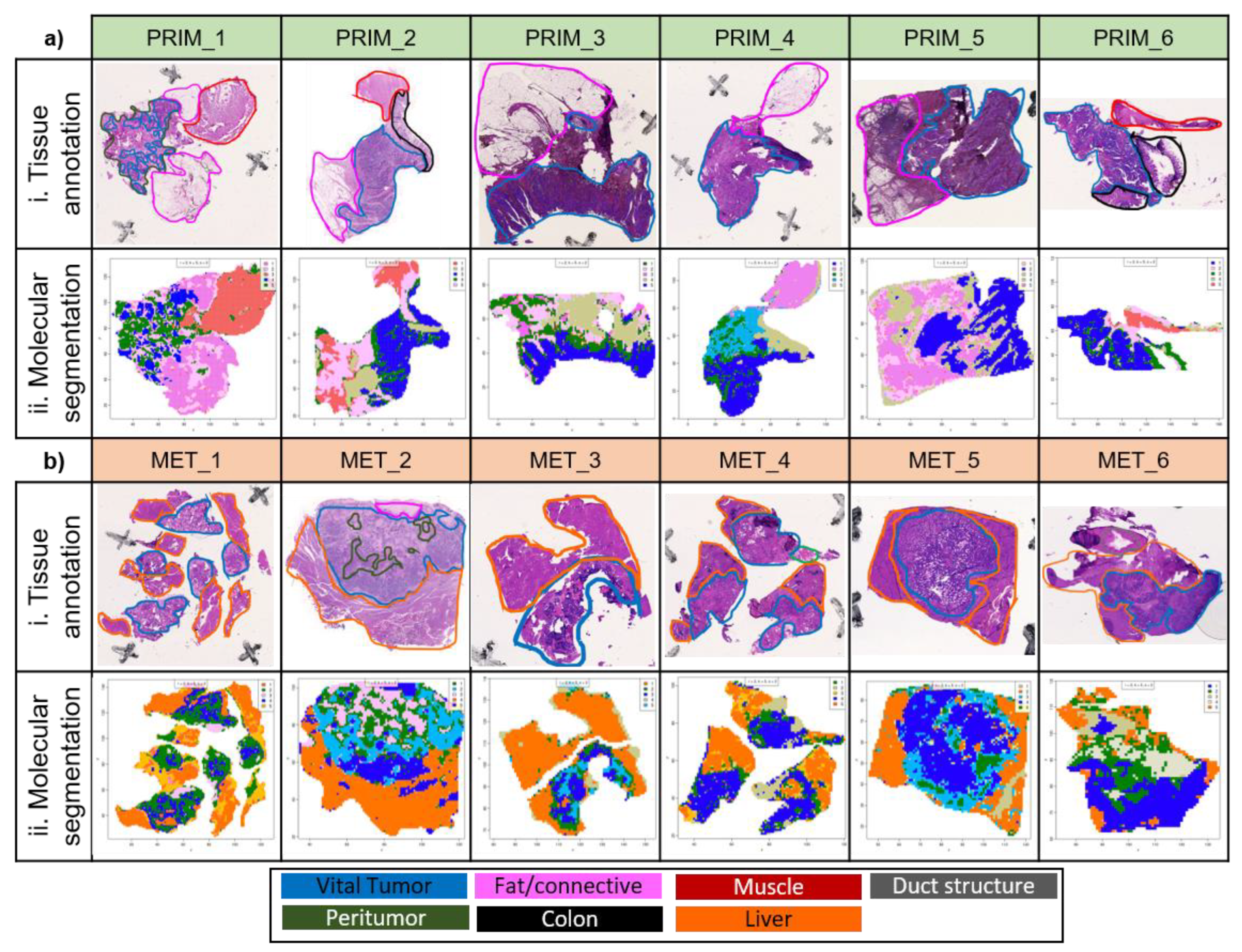
Figure 2.
Unsupervised segmentation of the vital tumor regions shows areas with distinct proteomic composition indicating intratumor heterogeneity. a) i. tumor annotation and ii. spatial proteomic segments for primary tumors. B) i. tumor annotation and ii. spatial proteomic segments for liver metastases. For better comparability, the tumor segments are projected onto the whole sample (beige).
Figure 2.
Unsupervised segmentation of the vital tumor regions shows areas with distinct proteomic composition indicating intratumor heterogeneity. a) i. tumor annotation and ii. spatial proteomic segments for primary tumors. B) i. tumor annotation and ii. spatial proteomic segments for liver metastases. For better comparability, the tumor segments are projected onto the whole sample (beige).
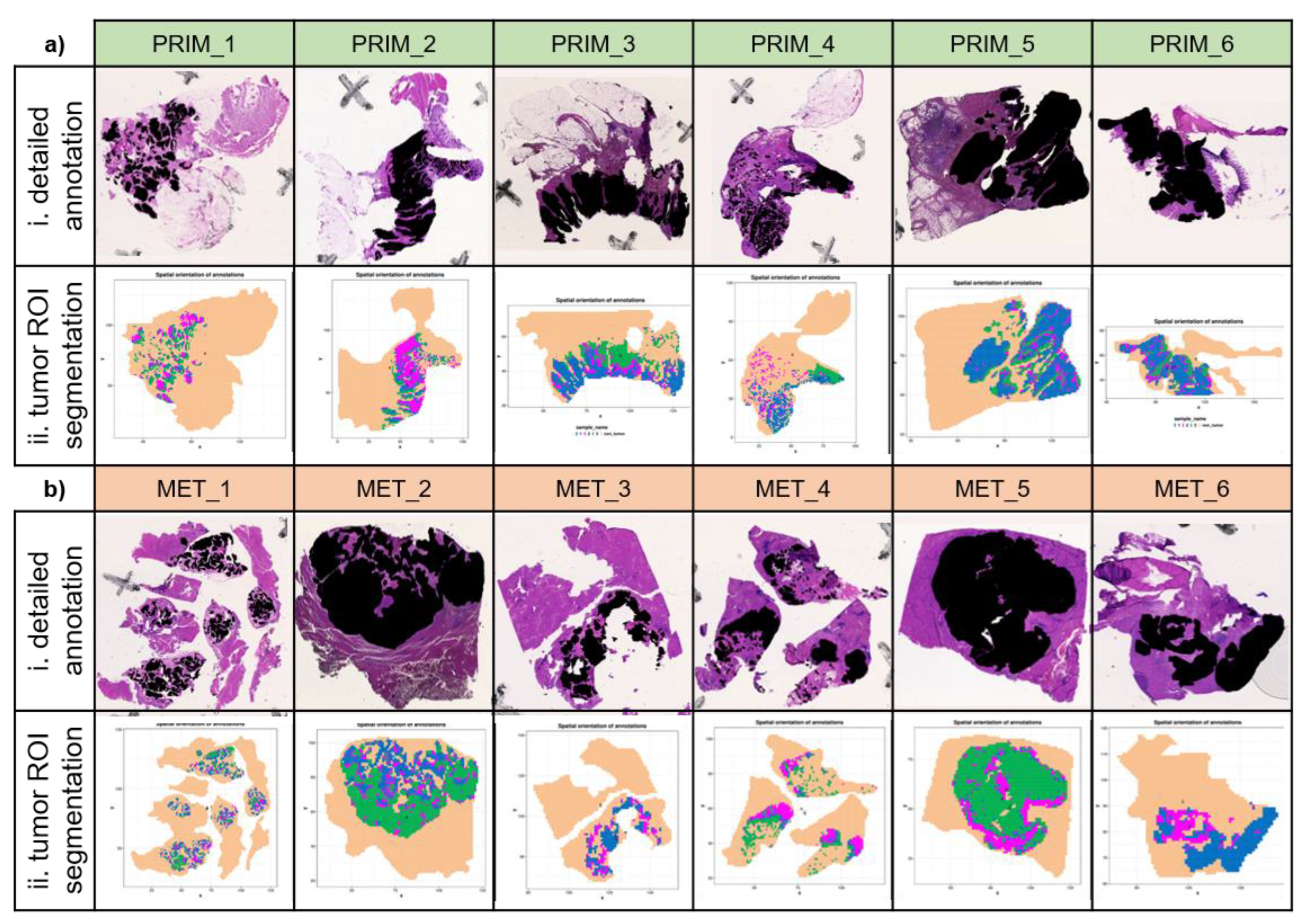
Figure 3.
Unsupervised segmentation of the tumor areas of matched primary tumors and liver metastases. The segmentation results are projected onto the whole sample (beige) for better comparability. All pairs show intratumor heterogeneity which spans both entities.
Figure 3.
Unsupervised segmentation of the tumor areas of matched primary tumors and liver metastases. The segmentation results are projected onto the whole sample (beige) for better comparability. All pairs show intratumor heterogeneity which spans both entities.
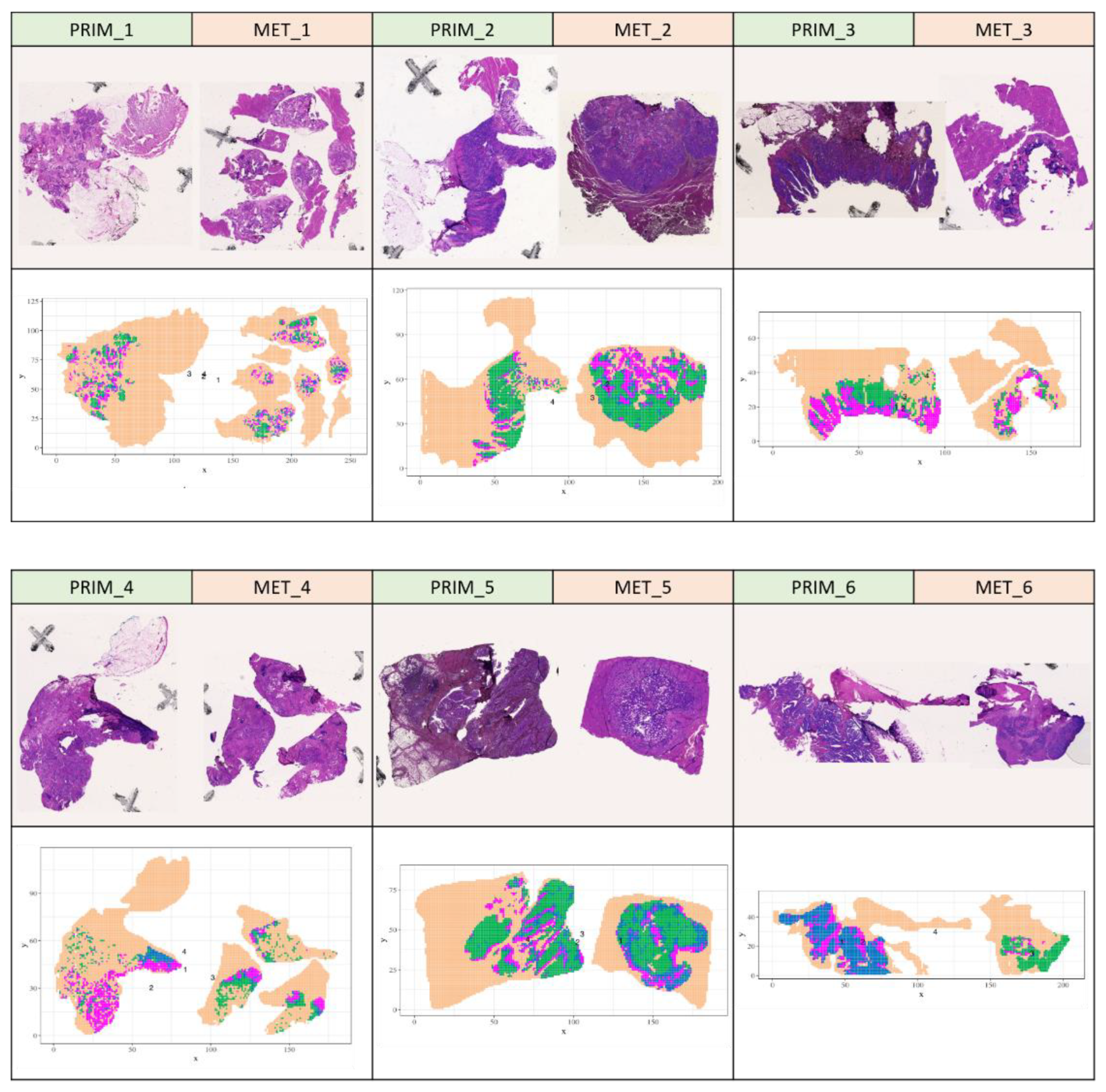
Figure 4.
Occurrence of the vital tumor enriched peptide features across primary tumors (a) and liver metastases (b). The different colors represent the six patients. In general, primary tumors show a higher overlap of tumor peptide features between samples in contrast to liver metastases, where only a small number of features occurs in more than 3 samples. Isotopes may be included in these plots and inflate the numbers because the data quality did not allow for reliable monoisotopic peak picking.
Figure 4.
Occurrence of the vital tumor enriched peptide features across primary tumors (a) and liver metastases (b). The different colors represent the six patients. In general, primary tumors show a higher overlap of tumor peptide features between samples in contrast to liver metastases, where only a small number of features occurs in more than 3 samples. Isotopes may be included in these plots and inflate the numbers because the data quality did not allow for reliable monoisotopic peak picking.
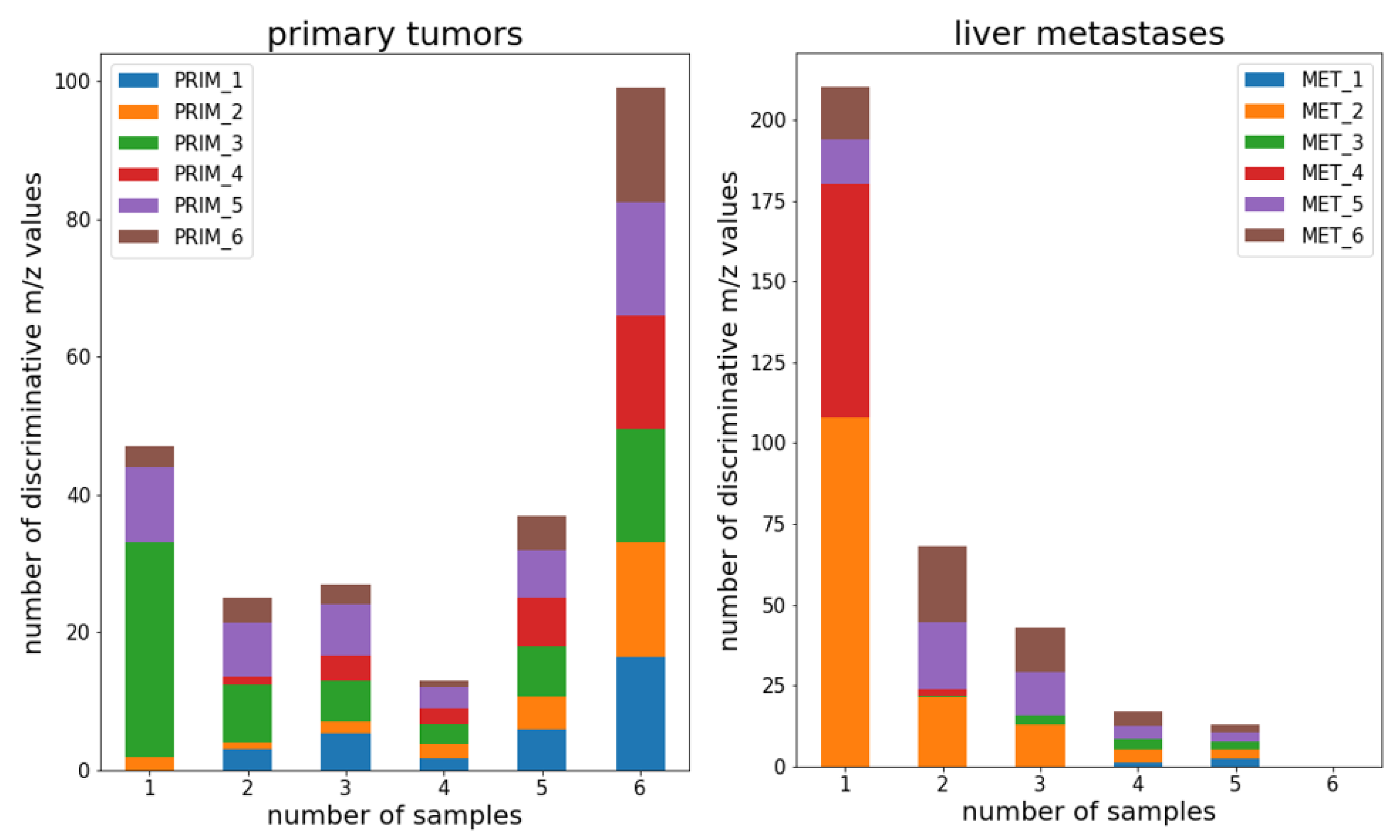
Table 1.
Overview of patient samples: Primary tumor cancer / metastases location, metastasis diagnosis, UICC Tumor Classification, and numbers of tissue spectra per sample. PRIM = primary tumor, MET = liver metastasis.
Table 1.
Overview of patient samples: Primary tumor cancer / metastases location, metastasis diagnosis, UICC Tumor Classification, and numbers of tissue spectra per sample. PRIM = primary tumor, MET = liver metastasis.
| Patient | Localization | Metastases | Metastasis diagnosis | UICC Tumor classification | Tissue spectra PRIM | Tissue spectra MET |
|---|---|---|---|---|---|---|
| P1 | Sigmoid | Liver, Peritoneal | Synchronous | pT4b | 8832 | 7222 |
| P2 | Rectosigmoid | Liver | Synchronous | pT3 | 5374 | 5708 |
| P3 | Sigmoid | Liver, Peritoneal | Metachronous | pT3 | 3353 | 2223 |
| P4 | Rectosigmoid | Liver | Synchronous | pT4a | 4319 | 3359 |
| P5 | Ascending | Liver | Synchronous | pT3 | 6273 | 3837 |
| P6 | Sigmoid | Liver | Synchronous | pT3 | 3429 | 1979 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



