
Submitted:
20 February 2023
Posted:
22 February 2023
You are already at the latest version
Abstract
The detection and quantification of protein–protein interactions (PPIs) is a crucial technique that often involves the use of recombinant proteins with fusion-protein tags, such as maltose-binding protein (MBP) and glutathione-S-transferase (GST). In this study, we improved the cohesive and sticky properties of gelatinized starch by supplementing it with agarose, resulting in a harder gel that could coat the bottom of a microtiter plate. The resulting gelatinized starch/agarose mixture allowed for the efficient immobilization of MBP-tagged proteins on the coated plates, enabling the use of indirect ELISA-like PPI assays. By using the enzymatic activity of GST as an indicator, we succeeded in determining the dissociation constants between MBP-tagged and GST-tagged proteins on 96-well microtiter plates and a microplate reader without any expensive specialized equipment.
Keywords:
gelatinized starch
; maltose-binding protein
; microplate based assay
; protein-protein interaction
; dissociation constant determination
1. Introduction
Detection and quantitative analysis of protein–protein interactions (PPIs) is a key technique for fundamental biochemical experiments. For this purpose, purified recombinant proteins are often employed because of their convenience. Most of these recombinant proteins are expressed and purified according to various fusion-protein-based strategies with popular affinity tags, such as hexahistidine-tag, maltose binding protein (MBP), glutathione S-transferase (GST), His-Patch thioredoxin, Halo-tag, and PA-tag [1]. These affinity tags are used with their specific ligand-immobilized medium. Among these, MBP-fusion proteins are frequently purified using chemically cross-linked starch, known as amylose-linked agarose bead [2,3]. Although amylose-linked agarose beads are commercially available, their relatively high cost (33,000 JPY/15 mL) may hamper large-scale or highly parallel experiments. Immobilized amylose on beads can also be degraded by amylase derived from Escherichia coli, decreasing the binding capacity when reusing the medium [4]. By contrast, starch is a polysaccharide containing 20%–25% amylose and 75%–80% amylopectin by weight [5]. The surface area of starch is increased by heat gelatinized treatment of raw corn starch, providing an increased binding capacity to capture MBP-fusion proteins. Here, we describe a protocol that expands our previous gelatinized starch technique by adopting it for the detection and quantification of PPIs in microtiter plate-based assays [6].
2. Materials and Methods
2.1. Preparation of MBP and GST Fusion Proteins
The constructs of MBP-fusion and GST-fusion proteins used in this study are illustrated in Figure 1a,b. To rapidly construct MBP-fusion protein expression vectors, we employed our “unidirectional” TA-cloning technology (PRESAT-vector technology) and constructed a pET-MBP-HRV3C-PRESAT vector from pET-21b by inserting PRESAT-linker [7]. The vector was subsequently used to clone the genes of interest and express them as MBP-fusion proteins under the T7 promoter [7]. The DNA fragment coding mouse ARHGEF11 fragment (residues 1334–1406 except for deleted residues 1352–1394 by alternative splicing) was amplified by PCR with the primers 5’-GCT GAA GAG GCT TCA AGC TC-3’ and 5’-ATG ATT AAG GTT CTG CTG GCA TGC TG-3’ from plasmid DNA subcloned into the coding region of the C-terminal ARHGEF11 (1247–1552) originating from mouse kidney QUICK-CloneTM cDNA (Becton, Dickinson and Company, Franklin Lakes, NJ, USA). The fragment was subcloned into a pET-MBP vector with an In-Fusion HD cloning kit (Takara Bio Inc., Shiga, Japan) according to the manufacturer’s instructions. The DNA fragment coding mouse ZO-1 ZU5 domain (residues 1624–1745) was amplified by PCR with the primers5’- GAG GAT GGT CAT ACT GTA GTG-3’ and 5’-ATG CTC GAG ATT AAA AGT GGT CAA TCA GGA CAG AAA C-3’ from plasmid DNA kindly gifted by Dr. Mikio Fruse (NIPS, Japan).
All fusion proteins were expressed in E. coli BL21(DE3) grown in an LB medium containing ampicillin (50 μg/mL) under isopropyl-β-D-1-thiogalactopyranoside (IPTG) induction. The supernatants of the sonicated cells of the MBP-fusion proteins were directly used for subsequent immobilization experiments without additional purification. The supernatants of the sonicated cells of GST-fusion proteins were purified using DEAE Sepharose (Cytiva, Tokyo, Japan) and a GST-Accept affinity column (Nacalai Tesque, Kyoto, Japan). The fusion proteins were eluted from glutathione (GSH) beads with a buffer containing 10 mM GSH (reduced form), 150 mM NaCl, and 50 mM Tris-HCl buffer (pH 7.4). The purified proteins were stored at 4°C until use.
2.2. Preparation of Gelatinized Corn Starch-Agarose Mixed Gel for Affinity Column Chromatography
We suspended 0.2 g of agarose (for ≥ 1 kbp fragment, agarose fine powder; 02468-66, Nacalai Tesque) in 5 mL double distilled water (DDW) and pre-incubated the mixture for more than 10 min at 95°C. We suspended 0.2 g of corn starch (193-09925, FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) in 5 mL DDW and added it to the pre-incubated agarose solution. The mixture was gently mixed and further incubated for 10 min at 95°C. This gelatinized starch-agarose (GSA) mixture was cooled and allowed to solidify in a 10 mL disposable syringe. This gel was squeezed out through a 22G syringe needle (Terumo) to obtain the GSA beads. The beads were washed three times using DDW and equilibrated with GSA buffer (150 mM NaCl and 50 mM Tris-HCl, pH 7.4) before use.
2.3. Immobilization of MBP Fusion Protein in a 96-Well Plate
A melted starch-agarose mixture solution was poured over the bottom of the 96-well plate and allowed to solidify by cooling to room temperature for 30 min. Aliquots of supernatants containing MBP-fusion proteins were overlaid and allowed to absorb onto the GSA-coated 96-well plate. The solution was discarded, and the well was washed twice or thrice using the GSA buffer before use.
2.4. Interaction Assay
Supernatants containing GST-fusion proteins of interest were overlaid onto the immobilized MBP-tagged proteins and incubated at 4°C for 30 min. The solutions were discarded, and the wells were washed thrice with the GSA buffer. The MBP-fusion and GST-fusion proteins were co-eluted using the same GSA buffer supplemented with 10 mM maltose. The eluents were analyzed by either SDS-PAGE or colorimetric enzymatic assay using a 1-chloro-2,4-dinitro-benzen (CDNB) assay (see below). The SDS-PAGE gels were stained with Coomassie brilliant blue.
2.5. CDNB Assay
Quantification of GST-fusion proteins was done by a colorimetric assay that measured the changes in absorbance at 340 nm. A reaction solution containing 2 mM of CDNB and 2 mM of GSH (reduced form) was added to the eluents, and the absorbance at 340 nm was monitored every 1 min over 5 min. The slope of the absorbance change was calculated according to the manufacturer’s instructions for the pGEX fusion expression system (Cytiva). Absorbance was measured with an EnSpire multimode plate reader (Perkin-Elmer, Norwalk, CT, USA).
2.6. Determination of Dissociation Constant (KD) Value
The KD of ARHGEF11/ZO1-ZU5 complex was obtained using a GSA-coated microtiter plate. MBP-ARHGEF11 was immobilized onto 96-well plates, overlaid with 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 8, and 16 μM of the affinity purified GST-ZO1-ZU5, and allowed to bind at 4°C for 30 min. The complex was eluted with a maltose solution, as described above. The backgrounds of the non-specific binding of GST-fusion proteins to GSA gel were also measured in the same manner using the MBP-only construct instead of MBP-ARHGEF11. Blank wells (coated with MBP) were used as a negative control, and their values were subtracted from those with corresponding actual samples. The amounts of ZO1-ZU5 bound to ARHGEF11 were quantified using a CDNB assay. The data were subjected to non-linear fitting to calculate KD. The experiments were performed in triplicate, and the standard deviations were calculated. An overview of the experimental processes of the PPI assay are illustrated in Figure 1c and Figure 2a–e.
3. Results and Discussion
The muddled and sticky properties of gelatinized starch could be improved by adding agarose. In preliminary experiments, we crushed the solidified GSA gel and filled the beads into a small polystyrene column filter. We demonstrated that the GSA beads were easily handled by the gravity-flow column filter more conveniently than the original gelatinized starch. These house-made GSA beads were useful for the purification of MBP-fusion mARHGEF11, with an absorption capacity of approximately half that of commercially available amylose-resin (data not shown). Considering the low cost of the GSA beads, this performance was acceptable. Accordingly, we examined whether the GSA gel was able to absorb MBP-tagged ARHGEF11 in PPI experiments (Figure 2a–e). Indeed, the GSA gel could capture MBP-ARHGEF11 from the crude extract of E. coli, and was capable of purifying the fusion protein in a single step (Figure 3a). The relatively low capacity of the GSA gel could be improved by repeating the absorption-washing step three times (Figure 3b). We then confirmed that MBP-ARHGEF11 captured on the GSA gel surface was able to bind its partner GST-ZO1-ZU5, as expected, demonstrating the success of this assay system in detecting the specific PPI between ARHGEF11 and ZO1-ZU5 (Figure 3c). Thus, a 96-well microtiter plate can be used to detect PPIs between MBP- and GST-fusion proteins of interest in a high throughput manner using our GSA gel immobilization method.
To detect these PPIs more sensitively, we employed an enzymatic colorimetric CDNB assay for GST-fusion proteins. We verified that the protein-binding reaction rates in the PPI pair were higher than those of the controls, proportional to the amount of GST-fusion protein (Figure 4a,b). With a completion time of 5 min, which is faster than detection by SDS-PAGE, our method is useful as a high-throughput initial screening for detecting PPIs. This could be beneficial in high-throughput screening for developing PPI inhibitors against potential drug targets.
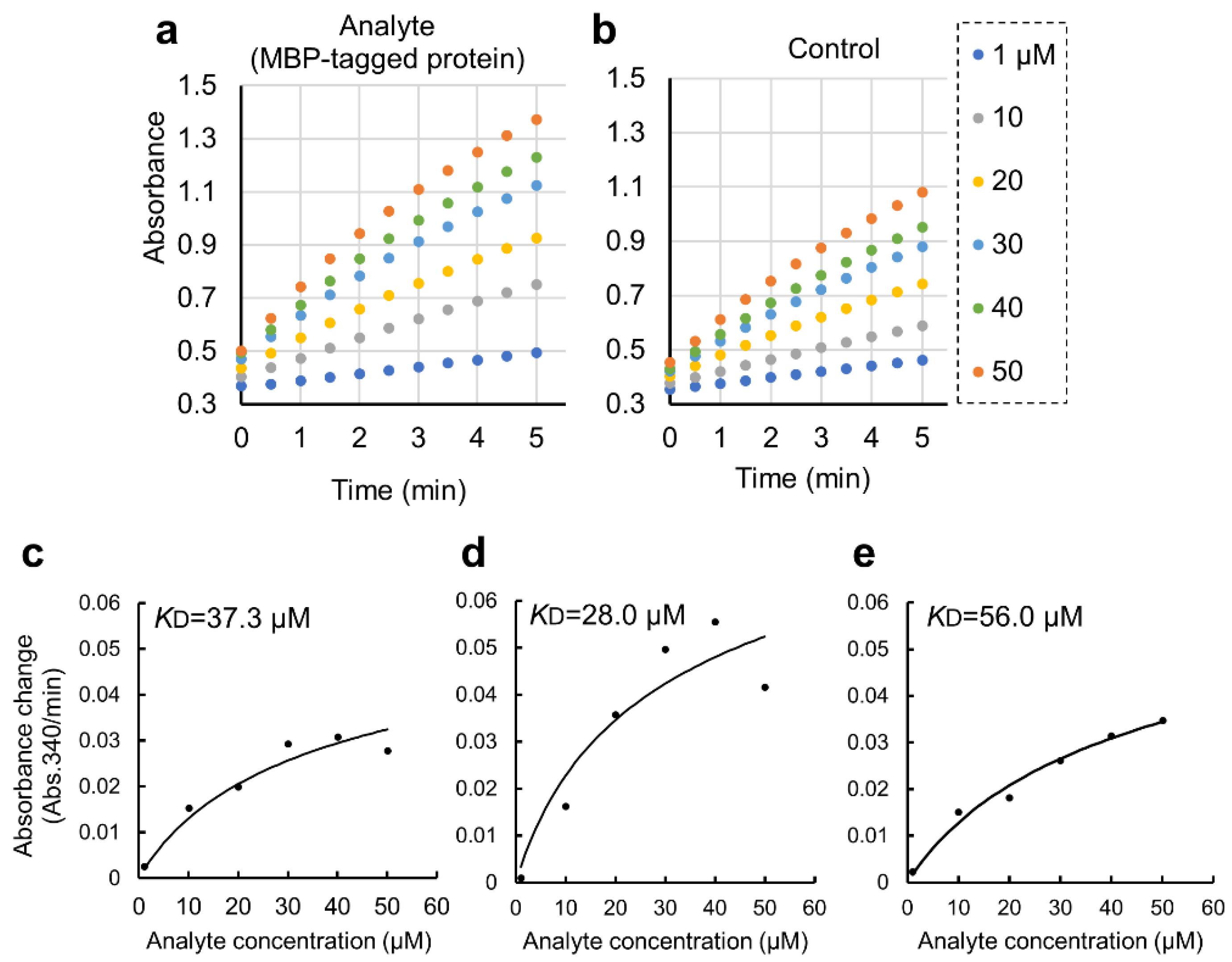
We further applied the GSA gel immobilization method to determine the KD value of the binding of ARHGEF11 and ZO1-ZU5 (Figure 4c–e). The KD value estimated from the three independent experiments performed across different dates was 41.7 ± 14.0 μM. The time required for each experiment was approximately 3 to 4 h. The obtained KD value was confirmed as reasonable based on an NMR titration assay (data not shown).
Although our GSA gel immobilization method of MBP-fusion proteins for PPI experiments is convenient, scalable, reproducible, and cost-effective, we are aware of one major demerit. Since the GSA gel is opaque (Figure 1b), the wells where the protein of interest was immobilized could not be used for the calorimetric assay of the plate reader. The opacity of the GSA gel obscured the absorbance measurement of CDNB at 340 nm. However, we could not reduce the gel volume further to improve the translucence of the media. As a result, bound GST-fusion proteins must be transferred to new wells for colorimetric quantification (Figure 2e). To address this limitation, future studies may consider using fluorescent GST substrates, such as DNs-Rh 9, to quantify GST-fusion protein levels in wells with GSA gel [8].
Figure 3.
(a) Binding and single-step purification of MBP-tagged protein using a GSA-coated 96-well microtiter plate. lysate: a crude extract of MBP-ARHGEF11-expressing E. coli; eluates: MBP-ARHGEF11 captured by GSA-gel was washed twice and then eluted with the maltose-containing buffer. Co-purification of MBP-tagged and GST-tagged proteins by GSA-gel. Eluates from the GSA-gel from the well containing MBP-ARHGEF11 only, MBP-ARHGEF11 and GST-ZO1-ZU5 (analyte), or GST-ZO1-ZU5 only (control) were analyzed by SDS-PAGE. (c) Effect of the number of repeating absorption-washing steps on MBP-tagged protein immobilization, comparing one, two, or three repeats of absorption, by comparing the amount of MBP-ARHGEF11 after elution.
Figure 3.
(a) Binding and single-step purification of MBP-tagged protein using a GSA-coated 96-well microtiter plate. lysate: a crude extract of MBP-ARHGEF11-expressing E. coli; eluates: MBP-ARHGEF11 captured by GSA-gel was washed twice and then eluted with the maltose-containing buffer. Co-purification of MBP-tagged and GST-tagged proteins by GSA-gel. Eluates from the GSA-gel from the well containing MBP-ARHGEF11 only, MBP-ARHGEF11 and GST-ZO1-ZU5 (analyte), or GST-ZO1-ZU5 only (control) were analyzed by SDS-PAGE. (c) Effect of the number of repeating absorption-washing steps on MBP-tagged protein immobilization, comparing one, two, or three repeats of absorption, by comparing the amount of MBP-ARHGEF11 after elution.
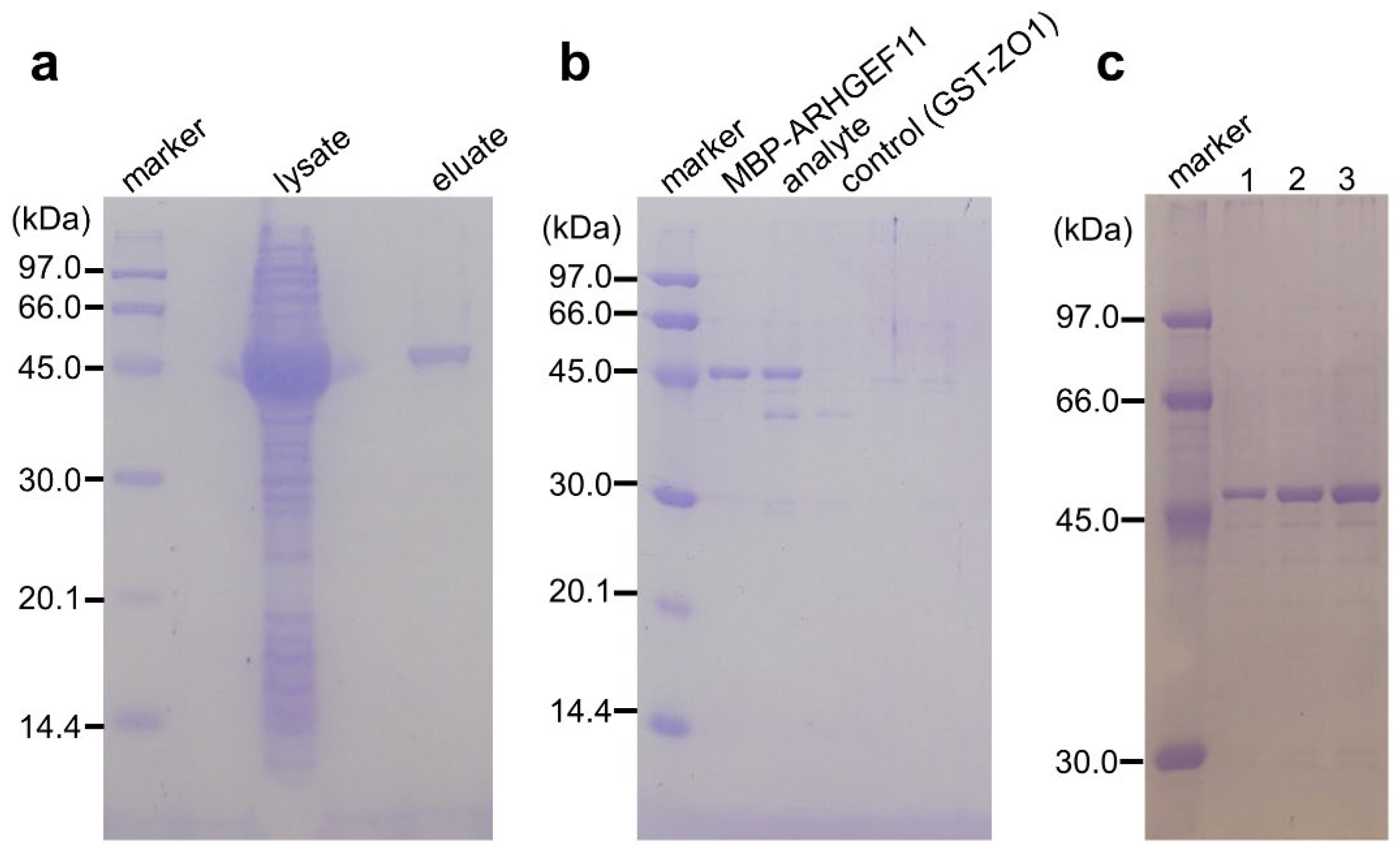
Figure 4.
Quantitative binding experiments of GST-ZO1-ZU5 bound to GSA-immobilized MBP-ARHGEF11 and the estimation of KD. (a,b) Representative results of CDNB assay of increasing concentration of GST-ZO1-ZU5 (1, 10, 20, 30, 40, and 50 mM) captured by GSA-immobilized MBP-ARHGEF11 (a) or MBP-only (control, b). (c–e) Estimations of KD from the triplicate measurements are represented. Solid lines are calculated values fitted by the theoretical equation. Estimated KD were shown in the graphs.
Figure 4.
Quantitative binding experiments of GST-ZO1-ZU5 bound to GSA-immobilized MBP-ARHGEF11 and the estimation of KD. (a,b) Representative results of CDNB assay of increasing concentration of GST-ZO1-ZU5 (1, 10, 20, 30, 40, and 50 mM) captured by GSA-immobilized MBP-ARHGEF11 (a) or MBP-only (control, b). (c–e) Estimations of KD from the triplicate measurements are represented. Solid lines are calculated values fitted by the theoretical equation. Estimated KD were shown in the graphs.
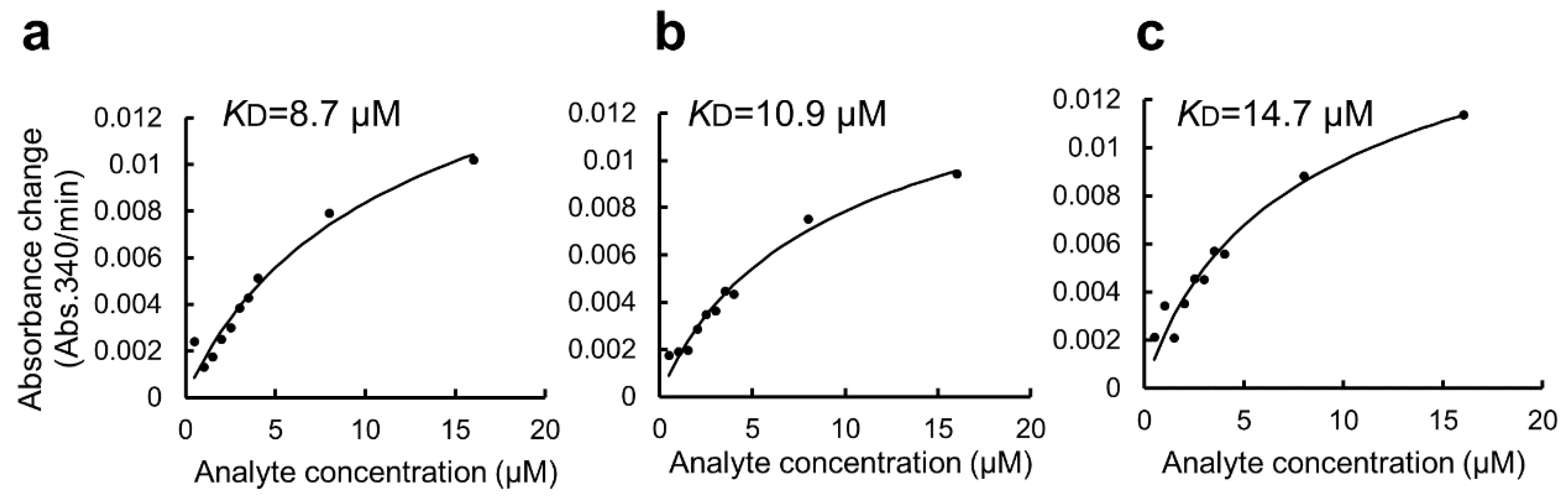
In this study, we used BSA as a blocking agent for the GSA-captured MBP-fusion protein to suppress non-specific interactions. However, BSA has a high capacity to promiscuously bind many small organic molecules. This might be an additional disadvantage of the present protocol when applied to a high-throughput drug screening experiment. Thus, we attempted to modify the protocol by omitting the BSA blocking step. The modified protocol was compared with the original protocol and is summarized in Supplementary Figure S1 (right). Due to the non-specific binding of GST-ZO1-ZU5 to GSA gel, the apparent KD value was 6.7 ± 4.4 μM (Figure 5a–c). Thus, the step of BSA blocking seemed necessary for accurate KD determination, and only omittable for limited purposes.
4. Conclusions
In this study, we developed a simple and cost-effective method for immobilizing MBP-tagged proteins on the bottom of 96-well microtiter plates. We showed that the PPI assay in 96-well plates using our GSA gel was sufficiently accurate and quantitative in determining KD values without the need for specialized or expensive equipment. Furthermore, the use of the GSA gel in 96-well plates was found to be an effective method for interaction screening with a large number of samples, which could potentially accelerate PPI studies.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Comparison of a GSA-based protein–protein interaction assay using MBP- and GST-fusion proteins with and without the BSA blocking step.
Author Contributions
Conceptualization, H.M. and H.H.; methodology, Y.K.; investigation, Y.E., R.K., E.H. and S.I.; resources, T.T.; data curation, H.H.; writing—original draft preparation, H.M. and H.H.; writing—review and editing, H.M. and H.H.; visualization, Y.E.; supervision, E.H. and T.T.; project administration, H.H.; funding acquisition, H.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Aichi Cancer Research Foundation and the Uehara Memorial Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The evidence data analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The authors would like to thank Scribendi Editing Services (https://www.scribendi.com/) for the English language review.
Conflicts of Interest
H.H. and T.T. are the founders of a Nagoya University-based spinoff startup company, BeCellBar LLC. R.K., R.H., S.I., N.G., E.H., Y.K. and H.M. declare that they have no conflict of interest regarding the content of this article.
References
- Ki, M.-R.; Pack, S.P. Fusion Tags to Enhance Heterologous Protein Expression. Appl Microbiol Biotechnol 2020, 104, 2411–2425. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, T.; Klotz, U. Affinity Chromatographic Isolation of the Periplasmic Maltose Binding Protein of Escherichia Coli. FEBS Lett 1978, 94, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Kellermann, O.K.; Ferenci, T. Maltose-Binding Protein from Escherichia Coli. Methods Enzymol 1982, 90 Pt E, 459–463. [Google Scholar] [CrossRef]
- Routzahn, K.M.; Waugh, D.S. Differential Effects of Supplementary Affinity Tags on the Solubility of MBP Fusion Proteins. J Struct Funct Genomics 2002, 2, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-N.; Lai, Y.-T.; Chou, W.-I.; Chang, M.D.-T.; Lyu, P.-C. Solution Structure of Family 21 Carbohydrate-Binding Module from Rhizopus Oryzae Glucoamylase. Biochem J 2007, 403, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, Y.; Namikawa, M.; Sekiguchi, M.; Inada, Y.; Yamauchi, S.; Kimoto, Y.; Okazaki, K.; Toyota, Y.; Sato, T.; Morioka, H. Expression, Purification and Characterization of CAR/NCOA-1 Tethered Protein in E. Coli Using Maltose-Binding Protein Fusion Tag and Gelatinized Corn Starch. Biol Pharm Bull 2021, 44, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Goda, N.; Tenno, T.; Takasu, H.; Hiroaki, H.; Shirakawa, M. The PRESAT-Vector: Asymmetric T-Vector for High-Throughput Screening of Soluble Protein Domains for Structural Proteomics. Protein Sci 2004, 13, 652–658. [Google Scholar] [CrossRef]
- Zhang, J.; Shibata, A.; Ito, M.; Shuto, S.; Ito, Y.; Mannervik, B.; Abe, H.; Morgenstern, R. Synthesis and Characterization of a Series of Highly Fluorogenic Substrates for Glutathione Transferases, a General Strategy. J Am Chem Soc 2011, 133, 14109–14119. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The concept framework of gelatinized starch-agarose (GSA)-based protein–protein interaction assay experiments. (a) Overview of the preparation of GSA-coated microtiter plates (right upper panel). The well coated with gelatinized starch without agarose was also shown as a control (right lower panel). (b) Constructs of fusion proteins used in this study with their residue numbers, MBP-ARHGEF11 and GST-ZO1-ZU5. (c) Scheme for 1-chloro-2,4-dinitro-benzen (CDNB)-based colorimetric detection of glutathione S-transferase (GST) with glutathione (GSH) as a substrate.
Figure 1.
The concept framework of gelatinized starch-agarose (GSA)-based protein–protein interaction assay experiments. (a) Overview of the preparation of GSA-coated microtiter plates (right upper panel). The well coated with gelatinized starch without agarose was also shown as a control (right lower panel). (b) Constructs of fusion proteins used in this study with their residue numbers, MBP-ARHGEF11 and GST-ZO1-ZU5. (c) Scheme for 1-chloro-2,4-dinitro-benzen (CDNB)-based colorimetric detection of glutathione S-transferase (GST) with glutathione (GSH) as a substrate.
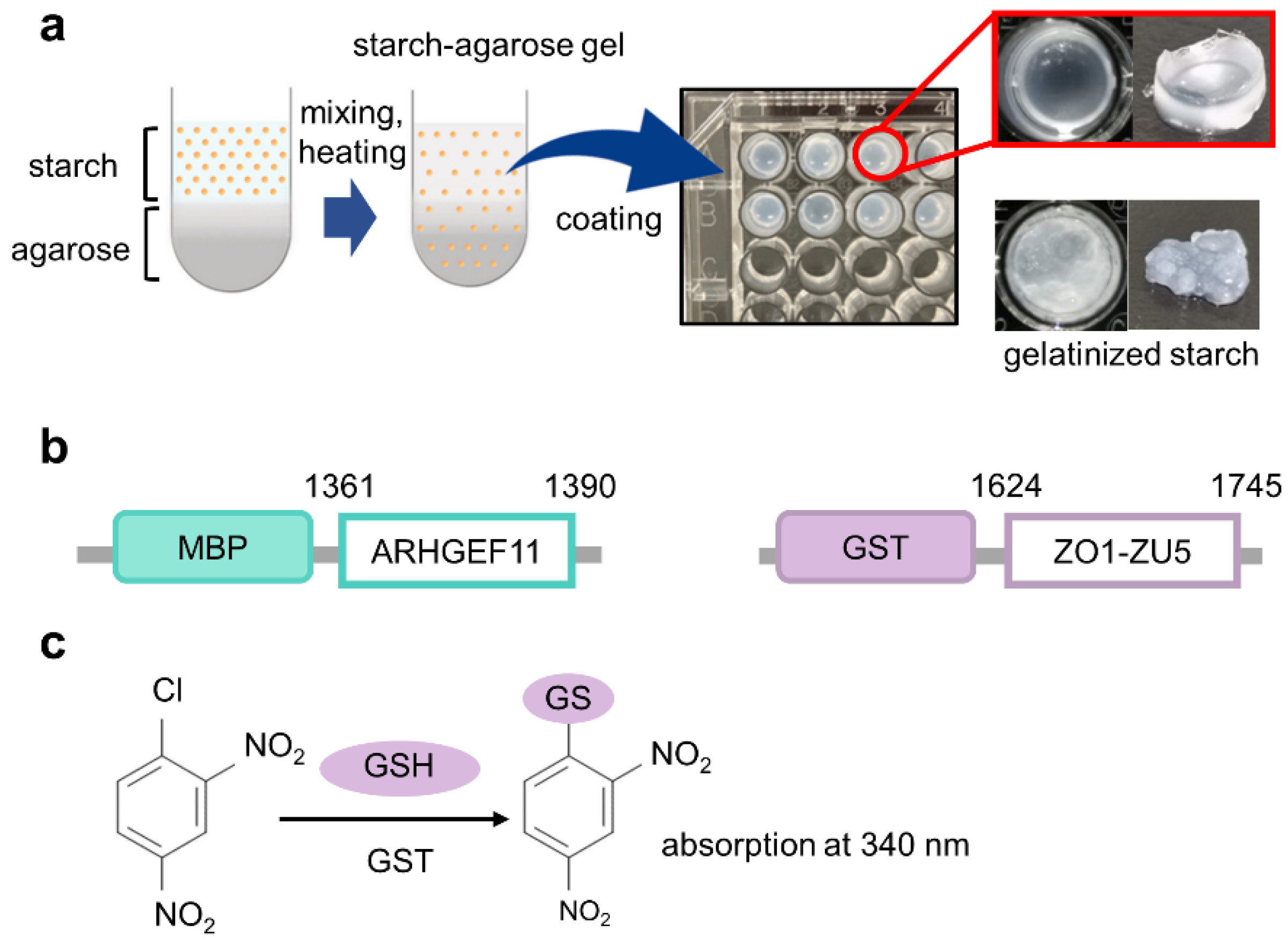
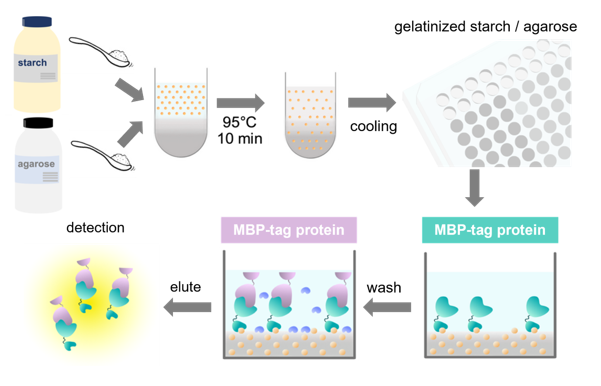
Figure 2.
Overview of a GSA-based protein–protein interaction assay using MBP- and GST-fusion proteins. (a) Schematic representation of fusion proteins used in this study: MBP-ARHGEF11, MBP (as a negative control), and GST-ZO1-ZU5. (b) Immobilization step. MBP-tagged protein (MBP-ARHGEF11) and MBP (control) were immobilized on a GSA-gel at the bottom surface of a 96-well microtiter plate. Yellow balls represent the amylose in gelatinized corn starch. (c) Protein–protein interaction step with bovine serum albumin blocking. GST-tagged protein (GST-ZO1-ZU5) specifically binds MBP-tagged protein (MBP-ARHGEF11) (left), whereas GST-tagged protein merely binds to the bottom of the control well. (d) Elution step. MBP-tagged protein and GST-tagged protein are eluted by maltose-containing buffer (left), whereas the control well does not contain GST-tagged protein. (e) Quantification step. Both eluates were subjected to CDNB assay to quantify the concentration of GST-tagged protein.
Figure 2.
Overview of a GSA-based protein–protein interaction assay using MBP- and GST-fusion proteins. (a) Schematic representation of fusion proteins used in this study: MBP-ARHGEF11, MBP (as a negative control), and GST-ZO1-ZU5. (b) Immobilization step. MBP-tagged protein (MBP-ARHGEF11) and MBP (control) were immobilized on a GSA-gel at the bottom surface of a 96-well microtiter plate. Yellow balls represent the amylose in gelatinized corn starch. (c) Protein–protein interaction step with bovine serum albumin blocking. GST-tagged protein (GST-ZO1-ZU5) specifically binds MBP-tagged protein (MBP-ARHGEF11) (left), whereas GST-tagged protein merely binds to the bottom of the control well. (d) Elution step. MBP-tagged protein and GST-tagged protein are eluted by maltose-containing buffer (left), whereas the control well does not contain GST-tagged protein. (e) Quantification step. Both eluates were subjected to CDNB assay to quantify the concentration of GST-tagged protein.
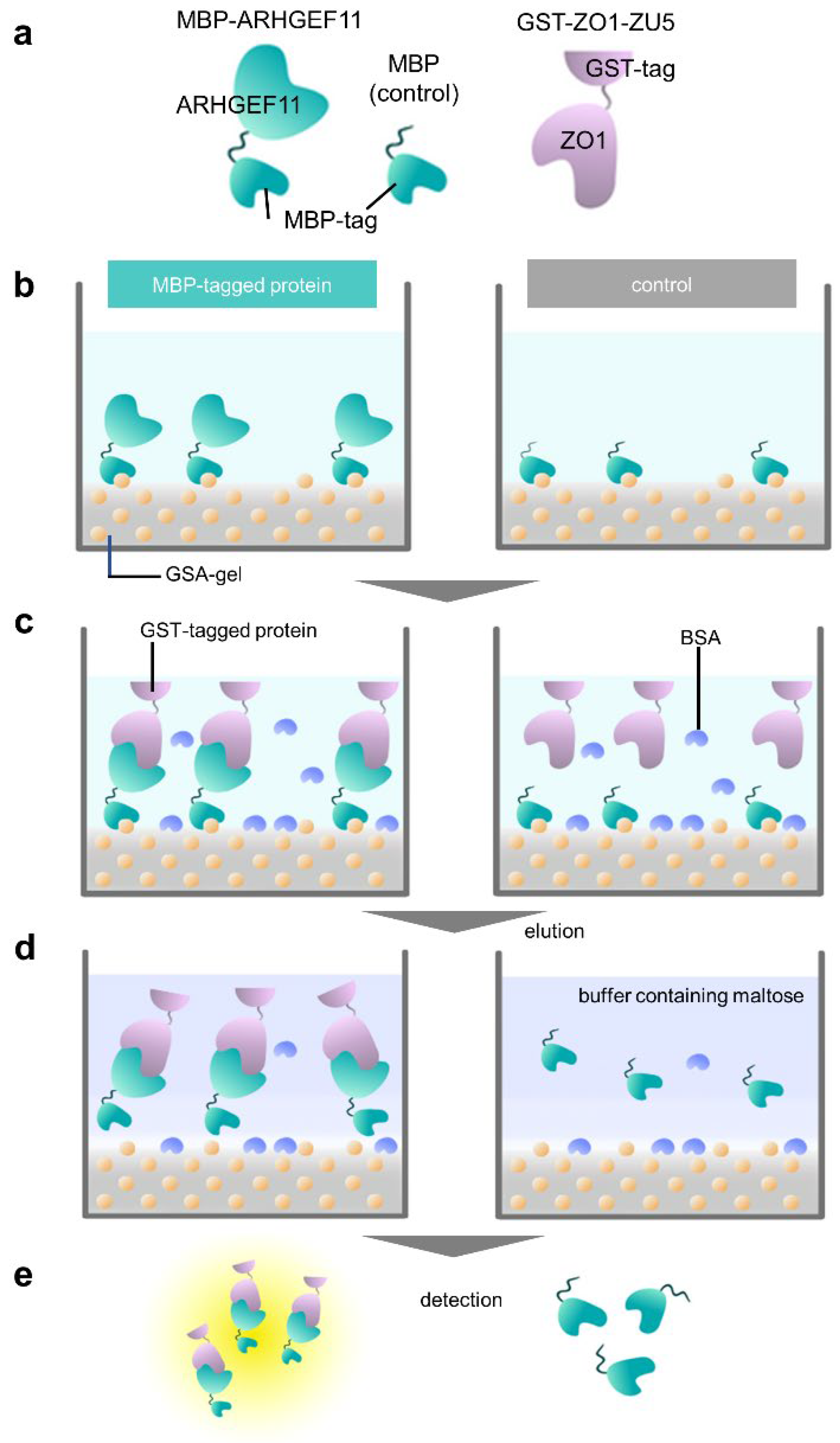
Figure 5.
Estimation of KD of the binding of GST-ZO1-ZU5 to GSA-immobilized MBP-ARHGEF11 using a GSA-coated 96-well microtiter plate without BSA blocking. (a–c) Estimations of KD from the triplicate measurements are represented. Solid lines are calculated values fitted by the theoretical equation. Estimated KD were shown in the graphs.
Figure 5.
Estimation of KD of the binding of GST-ZO1-ZU5 to GSA-immobilized MBP-ARHGEF11 using a GSA-coated 96-well microtiter plate without BSA blocking. (a–c) Estimations of KD from the triplicate measurements are represented. Solid lines are calculated values fitted by the theoretical equation. Estimated KD were shown in the graphs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



