
Submitted:
21 February 2023
Posted:
22 February 2023
You are already at the latest version
Abstract
Apoptosis, also known as the programmed death of cells, is responsible for maintaining the homeostasis of tissues and this function is carried out by caspases. The process of apoptosis is carried out via two distinct pathways: the extrinsic pathway, which is governed by death receptors, and the intrinsic pathway, also known as the mitochondrial pathway. The BCL-2 protein family encoded by the BCL-2 gene, located at the 18q21.33 chromosomal location, is in charge of regulating the intrinsic pathway, which is responsible for inducing cell death via the permeabilization of the mitochondrial membrane and the release of apoptosis - inducing components. The BCL-2 homology (BH1, BH2, BH3, BH4) domains of this family proteins are crucial for their functioning and their common BH domains allow interactions between members of the same family and can also serve as indications of pro- or anti-apoptotic activity. A direct correlation may be shown between the overexpression of BCL-2 and the postponement of cell death. It has been determined that a change in the expression of BCL-2 is the root cause of a variety of malignancies, including lung, breast, melanoma, and chronic lymphocytic leukemia, Multiple Sclerosis, Diabetes. In this review, we discuss the therapeutic potential of regulating BCL-2 family connections and their relevance to health and disease.
Keywords:
BCL-2 family protein
; BCL-2 Domains
; Apoptosis
; Cancer
; Biomarkers
; Therapeutic Agents
1. Introduction:
Apoptosis is a crucial biological process that induces cell death and maintains body homeostasis [1]. Apoptotic cell death is the most frequent consequence of infections [2] and it aids to remove the damaged or unnecessary cells without triggering an immune response or harming neighboring tissues [3]. The body-clearing process causes cells to round and shrink, chromatin to condense, and plasma membrane blebs to develop[4]. It is substantially different from other kinds of cell death depending on its frequency, appearance, and biochemistry [5]. Abnormalities in this process cause several disorders including autoimmune, neurological, cancer, coronary and infectious disease [6].
The apoptosis is regulated by two mechanisms, one is the extrinsic pathway and the other is the intrinsic pathway [7]. The extrinsic pathway activates apoptosis by integrating the extracellular signal into the intracellular signal transduction machinery [8]. Receptors including TNF-R1, CD95, TRAIL-R1, TRAIL-R2, DR3, and DR6 belonging to the death receptor family are transmembrane proteins that get activated when bound to their associated ligands (TNF, CD95L, TRAIL, and TL1A)[9]. Upon activation, the receptors are oligomerized and attract adaptor molecules that lead to the formation of the death-inducing signaling complex and caspase-8 activations. Once activated, caspase-8 has two possible modes of operation: caspase-8 can cleave caspase-3 or other effector caspases, or it can trigger the intrinsic (mitochondrial) apoptosis pathway by proteolytically cleaving Bid into tBid. Following this, tBid moves to mitochondrial membranes, where it interacts with other BCL-2 family members to open up the mitochondrial outer membrane [10][8]. Proteins like cytochrome c are released into the cytosol after the outer mitochondrial membrane becomes permeable. In this step, the cell makes the crucial decision of whether or not to commit suicide by activating this route. The BCL-2 family of proteins is crucially involved in these choices [11] [12]. The BCL-2 family of proteins contains both pro- and anti-apoptotic roles crucially involved in these choices [13] [14].
The BCL-2 (B cell lymphoma-2 ) family proteins are fundamental mediators of apoptosis [15] and it was first characterized as a proto-oncogene in the lymphoma cell. The BCL-2 gene was located at the breakpoint of the chromosomal translocation between chromosomes 18 and 14, t(14;18) [16]. In these chromosomal rearrangements, the BCL-2 gene, normally located at 18q21, is brought into close proximity to the IGH locus, 14q32, where strong enhancers linked with the IGH locus dysregulate BCL-2 transcription [17]. The proteins containing BCL-2 homology (BH) domains and playing a role in apoptosis regulation are collectively referred to as the BCL-2 family. Interactions between members of the same family are facilitated by their shared BH domains, which can also serve as indicators of pro- or anti-apoptotic activity [18]. The BCL-2 proteins family encoded by the BCL-2 gene is classified into three subfamilies based on the primary function of each member and their BH3 domain composition: (1) anti-apoptotic proteins (2) pro-apoptotic pore-formers and (3) pro-apoptotic BH3-only proteins [19].
Anti-apoptotic proteins, also known as pro-survival proteins, include BCL-2, BCLXL, BCLW, BCLB, MCL1, and BFL1 resist the apoptotic pathway [20]. The presence of the N-terminal BH4 domain defines the subset of survival proteins known as pro-survival proteins. In addition to their traditional function of suppressing apoptosis, BCL-2 and BCL-XL have been shown to have a role in the regulation of other critical cellular processes, including proliferation, autophagy, differentiation, DNA repair, tumor growth, and angiogenesis, via their BH4 domain [21]. The antiapoptotic and proapoptotic proteins contain four BH domains which take on a conserved functional group containing six or seven amphiphilic a-helices and two hydrophobic a-helices at the center to form a hydrophobic BH3 domain-binding cleft that works as a receptor for BH3 domains [17][22]. These four BH domain-containing (BH1–BH4) proteins inhibit apoptosis by interacting and trapping their pro-apoptotic counterparts [23].
The pro-apoptotic BH3-only proteins including BID, BIM, PUMA, BIK, BAD, BMF, NOXA, and HRK share only the BH3 domain which contains 9–16 amino acids. These proteins are necessary for the beginning stages of both developmentally programmed cell death and the apoptosis induced by stress [24][25]. This subgroup of BCL-2 can be divided into two categories: activator (BID, BIM, PUMA, NOXA) and sensitizer (BAD, BIK, BMF, HRK) [26]. The activator BH3-only proteins (BID, BIM) connect with the typical hydrophobic groove of BAX and BAK, which leads to their conformational change and activates them. On contrary, the sensitizer BH3 proteins (BIK, BMF, BAD) interact with the members of the ani-apoptotic family and induce the cells to apoptosis by preventing their binding to active effectors [27] [28] [29]. Certain BCL-2 family members don't follow traditional classification criteria due to their unclear roles and BH domain compositions presented here. BCL-2 family kin (BFK) is an uncharacterized member of the BCL-2 family that contains both a BH2 and a BH3 domain. The majority of human BFK expression occurs in the digestive system, and its forced expression modestly promotes apoptosis. Further, malignancies derived from different gastrointestinal organs show a marked downregulation of BFK expression [22].
In this study, we focus on the functions that BCL-2 family members play as both apoptotic and ani-apoptotic factors in several biological processes. In particular, we concentrated on how each of these members interacts with other members of the BCL-2 family. In addition, we cover multiple therapeutic drugs that target proteins belonging to the BCL-2 family in the context of breast cancer, Parkinson disease, Diabetes, lung cancer, Leukemia and Multiple Sclerosis.
2. Methods
In this review, we retrieved the relevant articles from online public resources like PubMed and Google Scholar using keywords like “BCL-2 family proteins”, “BCL-2 domains”, “apoptosis” “cancer” “mechanisms”, “biomarkers” and “therapeutic agents”. We have used relevant published manuscripts from 2000-2023 as references. The visual contents are processed via Microsoft Power Point.
3. Genomic-Proteomic Context
3.1. Genetic location of BCL-2 Family Member Gene
The BCL-2 family has crucial role in apoptosis. Since the discovery of BCL-2 in the 1980s in the context of B-cell lymphoma, a number of related proteins have been identified [18]. More than 20 proteins that control the intrinsic apoptosis pathway are encoded by the B cell lymphoma 2 (BCL-2) gene family. These proteins are essential for maintaining the proper ratio of cell survival to cell death [30]. BCL-2 gene is found in intracellular membranes such as the endoplasmic reticulum and mitochondria, and other members of the family are translocated from the cytoplasm to the mitochondria in response to a cell death trigger.
Figure 1.
BCL-2 translocation t(14;18) frequently found in human B-cell lymphoma. Human follicular center B-cell lymphomas typically have this chromosomal translocation. It relocates the BCL-2 gene from its usual location on chromosome 18q21 to a region of chromosome 14 called the 5' immunoglobulin heavy chain (IgH) intron enhancer E. Based on their key functions, the BCL-2 family is categorized into three groups: (1) anti-apoptotic proteins (BCL-2, BCL-XL, BCL-W, MCL-1, and BFL-1/A1), (2) pro-apoptotic proteins (BAX, BAK, and BOK), and (3) BH3-only proteins (BID, BAD, BIM, BIK, BMF, HRK, NOXA, PUMA, etc.). Each family member contains a BH3 domain, one of four BH domains involved in interactions between BCL-2 family proteins. The anti-apoptotic and pore-forming proteins are examples of multi-BH domain proteins because they have all four BH domains and adopt a highly conserved tertiary structure that creates a hydrophobic groove that binds BH3 domains from other family members.
Figure 1.
BCL-2 translocation t(14;18) frequently found in human B-cell lymphoma. Human follicular center B-cell lymphomas typically have this chromosomal translocation. It relocates the BCL-2 gene from its usual location on chromosome 18q21 to a region of chromosome 14 called the 5' immunoglobulin heavy chain (IgH) intron enhancer E. Based on their key functions, the BCL-2 family is categorized into three groups: (1) anti-apoptotic proteins (BCL-2, BCL-XL, BCL-W, MCL-1, and BFL-1/A1), (2) pro-apoptotic proteins (BAX, BAK, and BOK), and (3) BH3-only proteins (BID, BAD, BIM, BIK, BMF, HRK, NOXA, PUMA, etc.). Each family member contains a BH3 domain, one of four BH domains involved in interactions between BCL-2 family proteins. The anti-apoptotic and pore-forming proteins are examples of multi-BH domain proteins because they have all four BH domains and adopt a highly conserved tertiary structure that creates a hydrophobic groove that binds BH3 domains from other family members.
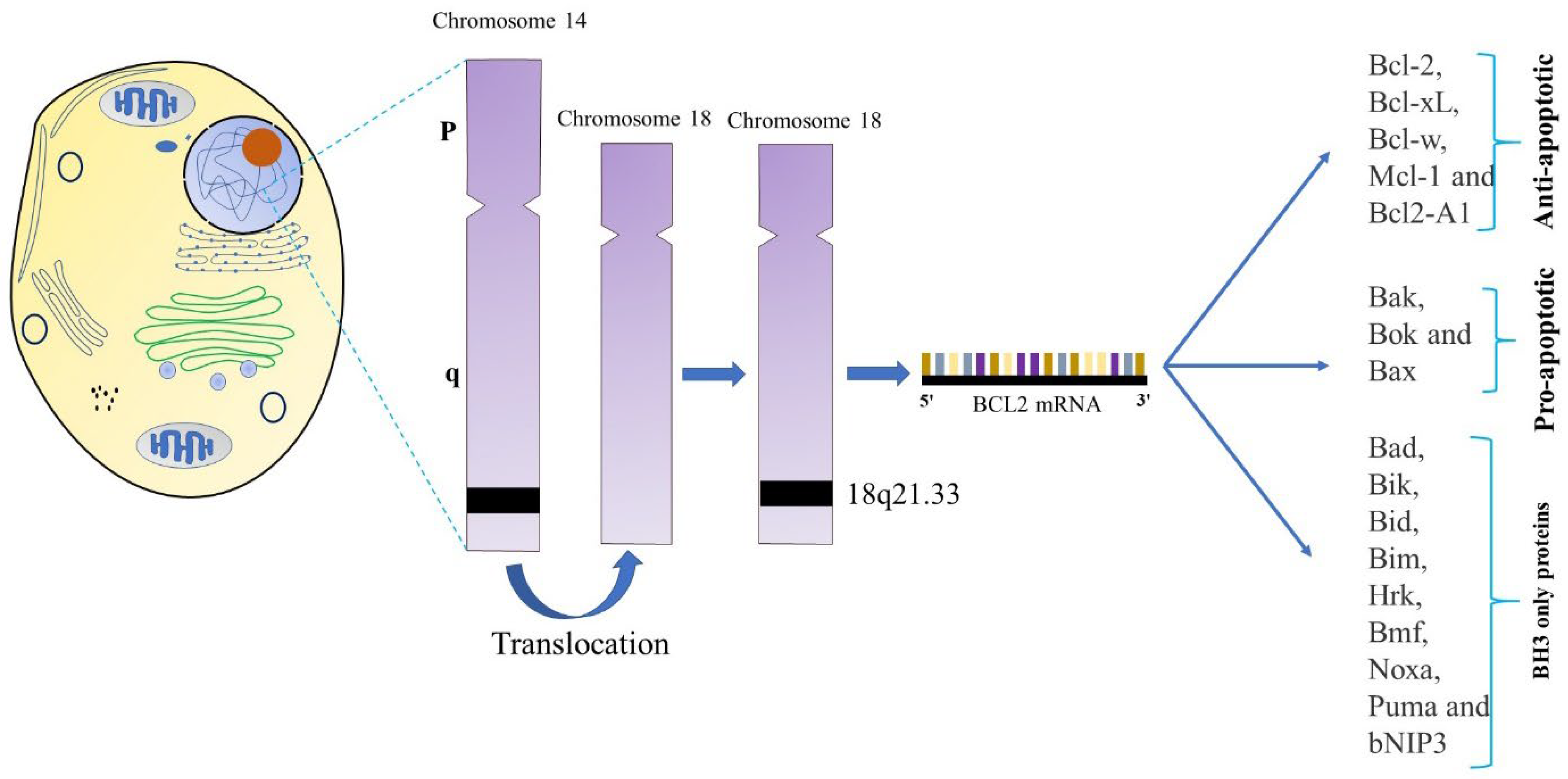
The prototype BCL-2 gene was discovered in humans at a The BCL-2 (B-cell lymphoma-2) gene was detected at the t(14;18) chromosomal translocation breakpoint in B-cell follicular lymphomas, where its transcription is severely promoted by the immunoglobulin heavy chain gene promoter and enhancer on chromosome 14 (Figure 1) [31] [32]. BCL-2's significant correlation with follicular lymphoma translocations indicated that it was an oncogene [33]. This translocation causes the BCL-2 gene on chromosome 18 to fuse with the immunoglobulin heavy chain promoter and enhancer on chromosome 14, which leads to excessive BCL-2 transcription.[34] About 30% of diffuse large B-cell lymphomas and 90% of follicular cell lymphomas exhibit this translocation to be present [35]. BCL-2 family proteins were developed to share BCL-2 homology (BH) domains. The encoded chromosomal translocation breakpoint and was eventually revealed to promote carcinogenesis by suppressing cell death rather than increasing cell-cycle progression [32].

Table 1.
Genetic Information of BCL-2 family proteins.
| Approved gene symbol | Approved Gene name | Aliases | Chromosomal Location | References |
|---|---|---|---|---|
| BCL-2 | BCL-2 apoptosis regulator | BCL-2, PPP1R50 |
18q21.33 | [31], [32] |
| BCL-2L1 | BCL-2 like 1 | BCLX, BCL-2L, BCL-X, BCL-XL, BCL-XS, PPP1R52 |
20q11.21 | [1] |
| BCL-2L2 | BCL-2 like 2 | KIAA0271, BCL-W, PPP1R51 |
14q11.2 | [36] |
| MCL1 | MCL1 apoptosis regulator, BCL-2 family member | BCL-2L3, Mcl-1 |
1q21.2 | [37] |
| BAX | BCL-2 associated X, apoptosis regulator | BCL-2L4 | 19q13.3 q13.4 | [38] |
| BAK1 | BCL-2 antagonist/killer 1 | BCL-2L7, BAK |
6p21.31 | [39,40] |
| BOK | BCL-2 family apoptosis regulator BOK | BCL-2L9, BOKL, MGC4631 |
2q37.3 | [39] |
| BCL-2L10 | BCL-2 like 10 | Diva, Boo, BCL-B |
15q21.2 | [39] |
| BIK | BCL-2-interacting killer (apoptosis-inducing) | NBK, BBC1 | 22q13.31 | [41] |
| PMAIP1 | Phorbol-12-myristate-13- acetate-induced protein 1 | NOXA, APR |
18q21 / (18q21.31) | [42,40] |
| BCL-2A1 | BCL-2-related protein A1 | GRS, BFL1, BCL-2L5, HBPA1 | 15q24.3 | [43] |
| BBC3 | BCL-2 binding component 3 | JFY1 , PUMA | 19q13.3-q13.4 | [44] |
| BNIP2 | BCL-2 interacting protein 2 | Nip2, BNIP-2 |
15q22.2 | [39] |
| HRK | Harakiri, BCL-2 interacting protein (contains only BH3 domain) | DP5 | 12q24.2 | [39] |
| BMF | BCL-2 modifying factor | 15q14 | [45] |
3.2. Structures of BCL-2 family members
Members of the BCL-2 family have three-dimensional structures that are composed of two core, mostly hydrophobic α-helices and six or seven amphipathic α-helices of different lengths. The first two α-helices are separated by an extended, unstructured loop. The overall fold of the pore-forming regions of bacterial toxins and the structures of the BCL-2 proteins are very similar [30][46]. BH domains are blocks of sequence homology found in proteins belonging to the BCL-2 family [47]. The BCL-2 family may be generally classified into the multi-motif BCL-2 proteins, which include numerous BH motifs and have anti- (BCL-2, BCL-XL, Bcl-w, Mcl-1, A1, Bcl-B) and pro-apoptotic (BAX, BAK) activities. The pro-apoptotic BH3-only proteins (Bim, Bad, tBid, Bmf, Bik, Noxa, Puma, and Hrk) have just one motif, a BH3. Almost all pro-apoptotic BCL-2 proteins have the BH3 motif, whereas anti-apoptotic proteins may or may not contain it (Figure 2) [1].
Figure 2.
BCL-2 homology domains (BH domains) and transmembrane domains (TM) are illustrated here. The BCL-2 protein family is divided into three groups such as multi-domain pro-apoptotic BCL-2 family proteins, multi-domain anti-apoptotic BCL-2 family proteins, and BH3-only proteins BCL-2 family proteins. The pro-apoptotic protein BAX, BAK, and BOK contain the BH1-3 domain. The anti-apoptotic BCL-2 family proteins, including BCL-2, BCL-XL, and BCL-W, contain the BH1-4 domain; MCL-1 and BFL-1/A1 contain the BH1-3 domain. The BH3-only proteins, including BID, BAD, BIM, BIK, BMF, HRK, NOXA, and PUMA, are composed of only the BH3 domain.
Figure 2.
BCL-2 homology domains (BH domains) and transmembrane domains (TM) are illustrated here. The BCL-2 protein family is divided into three groups such as multi-domain pro-apoptotic BCL-2 family proteins, multi-domain anti-apoptotic BCL-2 family proteins, and BH3-only proteins BCL-2 family proteins. The pro-apoptotic protein BAX, BAK, and BOK contain the BH1-3 domain. The anti-apoptotic BCL-2 family proteins, including BCL-2, BCL-XL, and BCL-W, contain the BH1-4 domain; MCL-1 and BFL-1/A1 contain the BH1-3 domain. The BH3-only proteins, including BID, BAD, BIM, BIK, BMF, HRK, NOXA, and PUMA, are composed of only the BH3 domain.
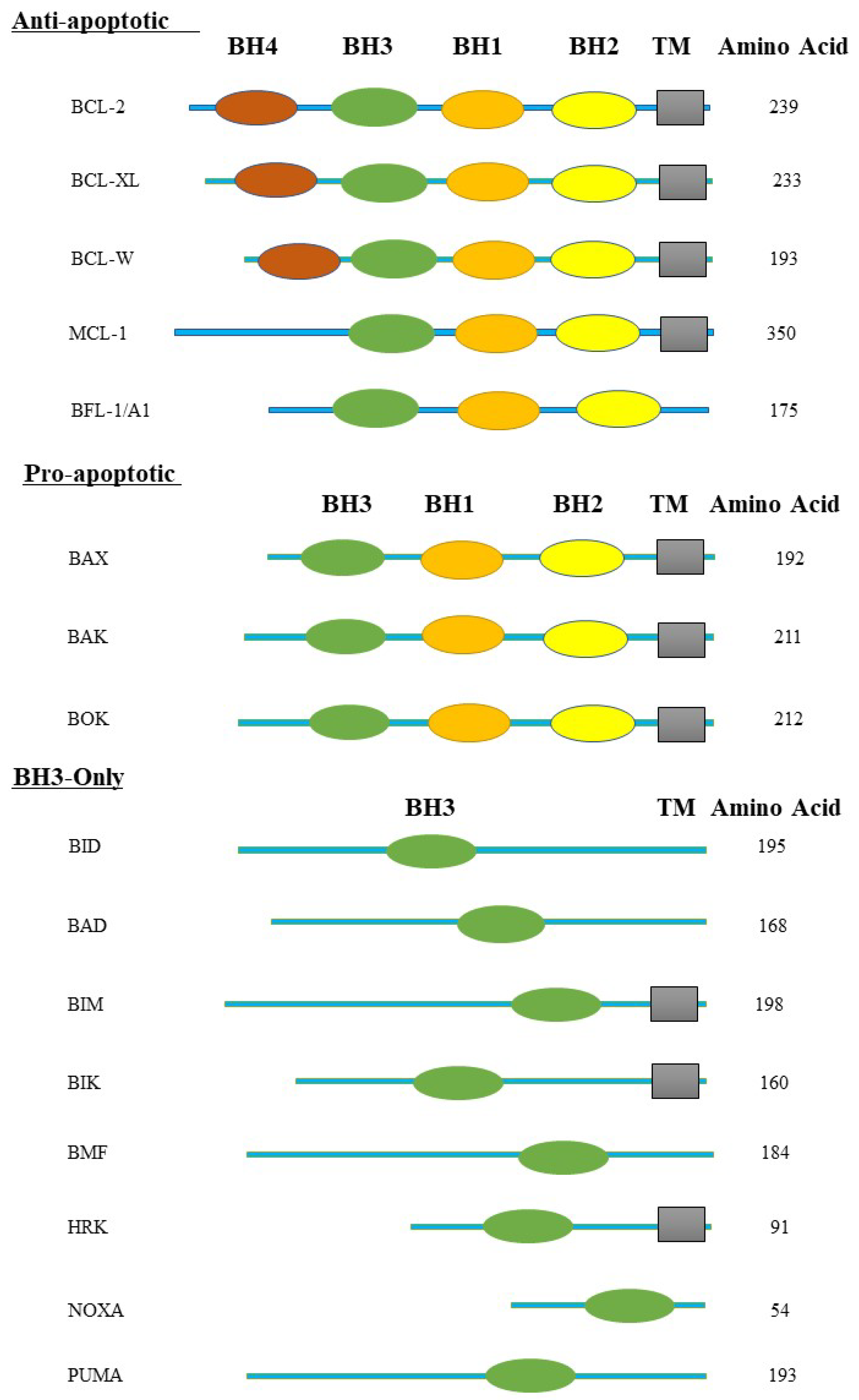
The term "structural homology" across family members refers to one to four areas that are highly homologous to BCL-2, known as the BH (BCL-2-homology region) domains (BH1, BH2, BH3, and BH4). The importance of BH domains for function and heterodimerization amongst members of the same family has been demonstrated via investigations on mutation and deletion [39]. The BCL-X gene, also known as BCL-2L1 (BCL-2-like 1), has a 44% homology to BCL-2. It has a variety of additional characterized isoforms in addition to the two well-known isoforms BCL-XL and BCL-XS [18]. The BH domain of the BCL-XL structure, which has eight helices (α1-α8), is crucial to the tertiary structure. In order to cause oligomerization, the pro-apoptotic protein's BH3 domain is placed into the hydrophobic pocket formed by the BH1-BH3 domain [48]. Although the BCL-XS protein's structure hasn't been fully defined, the hydrophobic binding cleft would be dramatically changed if both the BH1 and BH2 domains were lost [18]. Analysis on the structure of BAK and BAX monomers has shown that they are globular structures with an eight-helix encircling hydrophobic core in the center [34]. In the case of BAK, the carboxyl-terminal transmembrane helix, designated as α9, is permanently inserted into the MOM. But in the case of BAX, α9 is confined to the protein core within the hydrophobic BH3 domain-binding groove, which results in cytoplasmic localization [19]. Bid is composed of two core, predominantly hydrophobic α-helices, which are surrounded by six amphipathic helices. The amino-terminal region of the protein has the greatest change in total fold [46].
3.2. Gene Expression of BCL-2 Family Protein
BCL-2 expression can stop a variety of cytotoxic stimuli from killing cells. Acute myloid leukemia (AML), chronic lymphocytic leukemia (CLL), non-lymphoma Hodgkin's (NHL), myeloma, lung, prostate, and breast cancer, as well as melanoma, all have BCL-2 overexpression [49]. Chromosome translocations, gene amplification, and downregulation/deletion of genes producing microRNAs (miRNAs) involved in BCL-2. RNA degradation are just a few of the processes that might cause BCL-2 levels to rise.
BCL-2 overexpression in CLL, the most prevalent leukemia in humans, results from miRNA 15/16's lack of regulation [50]. The remarkable levels of BCL-XL expression are found in differentiating cells and they persist at higher levels throughout neuronal ontogeny. Ablation of the gene encoding BCL-XL, BCL-2l1, leads to embryonic mortality by E13.5 and significant apoptosis in the growing neurological and hematological systems [51]. BCLXL is a crucial element in the growth of tumors. Previous research has demonstrated that a number of solid tumors contain amplification of the BCL-2 L1 gene, which codes for BCLXL [23].
Mcl-1 expression has been linked to poor survival outcomes in ovarian cancer patients and in human cervical neoplasms. IL-6 controls Mcl-1 expression via a PI3K/Akt-dependent mechanism and promotes cervical cancer oncogenesis by suppressing cellular apoptosis [35]. Anti-apoptotic protein overexpression not only suppresses natural cell death but can also contribute to treatment resistance. BCL-XL and other proteins have been linked to therapeutic resistance because its overexpression disrupts the pathways that the therapies are supposed to target [52]. Anti-apoptotic protein overexpression, notably BCL-XL and Mcl-1, has been seen in a variety of cancers, including hepatocellular carcinoma, myeloma, CLL, and chronic myeloid leukemia (CML), where it is linked with resistance to therapy or a poor prognosis [35] [49].
BCLW facilitates tumor invasion by increasing the production of the transcriptional factor specificity protein 1, which then increases the expression of matrix metalloproteinase 2 [23]. The most notable maternally inherited mRNA is BCL-2L10 (also known as BCL-B or DIVA), which is greatest in the mature oocyte but diminishes during division, according to analysis of early mRNA expression from early embryos [51].
Table 2.
Localization and functions of BCL-2 family proteins.
| BCL-2 protein | Subcellular localization | Function | Reference |
| BCL-2 | Consistently linked to the MOM and/or ER membrane. | Both apoptosis effectors and BH3 only proteins are inhibited. | [34] [53] |
| BCL-XL | Located in the nucleus envelope, endoplasmic reticulum (ER), and mitochondrial membrane surfaces. | Bind to BAX or BAK to prevent apoptosis. | [19] [54] |
| MCL-1 | Placement to the outer mitochondrial membrane (OMM), inner mitochondrial membrane (IMM). | BH3-only proteins are inhibited as well as apoptotic effectors. Has an impact on cellular health and development in addition to its known pro-survival activities. | [55] |
| BCL-W | Loosely connected to the mitochondrial membrane and cytosol. | Inhibits BH3-only proteins as well as apoptotic effectors. | [19] [34] [36] |
| A1/Bfl-1 | Identified on the Golgi apparatus, cytosol, nuclear envelope, and ER membranes. | opposes the activation of caspase and prevents the release of proapoptotic cytochrome c from mitochondria. | [1] [56] |
| BAK | Always present on the OMM. | Oligomerizes to create holes in the OMM, which is considered to enhance MOMP. | [57] |
| BAX | Present on the OMM, often present in the cytoplasm as an inactive monomer. | Oligomerizes to create holes in the OMM, which is considered to enhance MOMP. | [57] |
| BOK | Detected on the ER membranes, nuclear outer membrane, and Golgi apparatus. | Depends on BAX and BAK to promote apoptosis. | [58] |
| BAD | Both the cytosol and the mitochondrial membrane | BAD is referred to as a "indirect" promoter of apoptosis because it causes cell death through inhibiting antiapoptotic proteins. | [59] |
| BIM | Nucleus envelope, endoplasmic reticulum (ER), cytoplasm, and mitochondrial outer membrane (MOM). | Apoptotic induction, it also controls mitochondrial activity and cellular metabolism. | [56] [60] |
| PUMA | Placement on the mitochondrial outer membrane (MOM), | Crucial p53-dependent apoptosis mediator. Activates BAK and BAX, resulting in MOMP. | [19] [61] |
| BIK | Mostly found in the ER membrane | Directly impacts BCL-2 and BCL-2-like 1 | [1] [62] |
Numerous regulatory mechanisms, as a result, reduce the production and function of BH3-only proteins in both healthy proliferating cells and malignancies. Some BH3-only proteins are continuously produced, whereas others are expressed in response to stressors like hypoxia or DNA damage [63].
BAX overexpression enhanced cell death, and the opposite effects supported a rheostat model in which cell fate is determined by the relative quantities of proapoptotic and antiapoptotic BCL-2 family members. Following the identification of BAK (BCL-2 antagonist/killer), a second proapoptotic protein that functions similarly to BAX while being more homologous to BCL-2 than BAX, it was shown that antiapoptotic action might be mediated by proteins other than BCL-2 [53]. The tumor suppressor TP53 has the ability to directly activate the promoter of BAX, which appears to be broadly expressed in both normal tissues and malignancies [34]. When initially obtained, human BCL-2A1 mRNA was shown to be overexpressed in stomach cancer relative to normal tissue, indicating that BCL-2A1 may also play a role in solid tumors [43]. Puma expression is generally kept to a minimum, but it can be increased by a number of transcription factors, including p53, p73, E2F1, and FOXO3a, which can cause an apoptotic response [64].
4. Apoptotic role of BCL-2 Family
Apoptosis is a strictly controlled form of cell death with unique biochemical and genetic processes that is essential for appropriate tissue growth and homeostasis. It helps to eliminate unneeded and undesired cells to keep the proper balance between cell death and survival in metazoan organisms [65] [66]. Signals at the cell surface and internal cell damage can both trigger apoptosis [67]. It has been found that the BCL-2 family is important for either activating or blocking the apoptotic pathway [68].
The BCL-2-regulated apoptotic pathway is activated in response to numerous upstream signaling events via transcriptional and/or posttranscriptional activation of the BH3-only proteins [69]. BH3-only proteins activate BAX and BAK. It can activate BAX and BAK through direct binding or indirectly through competition for pro-survival family members [70]. Activated BAX and BAK produce homodimers and heterodimers, which then come together to form pore-like structures in the outer mitochondrial membrane. This process, known as mitochondrial outer membrane permeabilization (MOMP), permits the cytosolic release of apoptogenic components, such as cytochrome c, which leads to caspase activation and apoptosis [71].
Additionally, MOMP promotes mitochondrial dysfunction, which depletes energy stores and results in cell death even when caspases are inactive. Therefore, only unusual circumstances will allow cells to recover from MOMP. Thus, MOMP is typically the key event that precedes cell death [72]. However, it is evident that certain of the BH3-only proteins (such as BIM, PUMA, and BID) are more effective apoptotic inducers than others (e.g., BAD, NOXA, BMF). Pro-apoptotic BH3-only proteins differ in how they interact with pro-survival proteins. BIM and PUMA are two examples of promiscuous proteins that have a high affinity for all pro-survival proteins, although some proteins only bind to subsets of these proteins, such as BAD, which only binds BCL-2, BCL-XL, and BCL-W but not MCL-1 or BFL-1, and NOXA, which binds mainly to MCL-1. This selectivity is also a characteristic of the drugs which have now been advanced to the goal of the apoptosis pathway [69] [73].
4.1. Models of BAX and BAK Activation
BAK is found in the mitochondria of healthy cells and has a transmembrane domain (α9) that spans the outer membrane. On the other hand, BAX is mainly found in the cytosol of healthy cells, where its transmembrane domain is tucked away in its conventional hydrophobic groove. However, cytotoxic signals encourage the buildup of BAX in the mitochondria [47]. The mechanism of BAX and BAK's activation has been highly debated and is regarded as the "holy grail" of apoptosis research. Both BAX and BAK undergo a noticeable change from innocuous proteins to deadly effectors during apoptosis [74].
There are three different models have been proposed to explain BAX and BAK activation. They are the direct activation model, the indirect activation model, and the membrane-mediated permissive Model [23] [28]. Direct binding of certain BH3-only proteins can activate BAX and BAK. While BH3-only family members easily attach to pro-survival BCL-2 proteins, it has been challenging to observe an earlier interaction with BAX and BAK [75]. The BH3-only proteins are divided into two groups in the direct activation model: sensitizers and activators. The sensitizer BH3-only proteins bind and inactivate the anti-apoptotic BCL-2 family members rather than binding and activating BAX and BAK [76]. BAK and BAX are directly bound by activators, changing their conformation, which causes BAK and BAX to oligomerize and become activated [23].
According to the indirect approach, the anti-apoptotic proteins are neutralized by BH3-only proteins, allowing BAK and BAX to be activated. This allows apoptosis to be mediated indirectly through BAK and BAX. Anti-apoptotic proteins constitutively bind with BAK and BAX to inhibit MOMP. The main purpose of BH3-only proteins is to attach to the anti-apoptotic BCL-2 proteins and displace BAK and BAX from anti-apoptotic proteins instead of binding BAK and BAX. This model is also known as the displacement model [77].
A mitochondrial outer membrane-mediated permissive model is another model of BAX and BAK activation. In healthy cells, BAX and BAK are typically inactive monomers or dimers. On the mitochondrial outer membrane, the anti-apoptotic BCL-2 family proteins bind to and are neutralized by activated BH3-only proteins during apoptosis. As a result, BAX and BAK are more likely to spontaneously activate, which leads to their homo-oligomerization and the formation of cytochrome c-releasing holes [78]. Both BAX and BAK associate with the membrane through helix 9, and this interaction causes BAX and BAK to spontaneously activate in the lipid bilayer of the MOM. The only purpose of the BH3-only proteins is to inactivate the anti-apoptotic BCL-2 proteins, which is similar to the indirect activation model. However, in the membrane-mediated permissive model, the MOM directly activates both BAX and BAK [74]. Although BH3-only proteins are the major activators of BAX and BAK, other proteins also seem to be capable of carrying out this role. It has been found that p53, Casp8p41, and retinoblastoma protein are also direct activators. Additionally, new research reveals that active BAK and BAX may operate as activators themselves [23].
4.2. Pore Formation by BAX and BAK
Biological membranes are essential information carriers and data storage structures for cell signaling and function. Membranes must allow data to flow in and out, so integral pore-forming proteins, active pumps, and cell adhesion molecules have all developed. These molecules all react to the flexibility of a lipid bilayer. All kingdoms of life have pore-forming proteins (PFPs), which alter cellular membranes by making them permeable to ions, tiny molecules, and occasionally bigger macromolecules. The BCL-2 protein family is capable of regulating pore-forming processes in mammalian apoptosis and inter-bacterial attack [79]. During apoptosis, the BAX and BAK proteins permeabilize the MOM, stimulating the release of a variety of proteins into the cytoplasm [80].
In healthy cells, BAX and BAK are globular monomers made up of helical bundles with a hydrophobic α-helix (α5) encircled by amphipathic helices. Their C-terminal helix (α9) is a hydrophobic transmembrane (TM) domain. It anchors them in the MOM. Probably the only way BAK is anchored there in healthy cells is by α9. BAX, on the other hand, is predominantly cytosolic and accumulates at the MOM following an apoptotic signal, where it inserts its α9. Other significant changes in the shape of BAK and BAX include BH3 (α2) being exposed and then being buried again in the surface groove of some other activated BAK or BAX molecule. The association of α 6 helices allows these new symmetric homo-dimers to multimerize [81][82]. BAX and BAK are pore-forming proteins that can sever the continuity of the lipid bilayer by opening toroidal pores. Toroidal pores are pore structures where the pore wall is made up of both lipids and proteins. As a result, the mechanical properties of the membrane as well as BAX and BAK affect the size and stability of these pores [83].
In model membranes, pores expand quickly after creation before relaxing to a smaller size that fluctuates between 3 and 8 nm depending on the protein content. There are two different types of BAX pores, one of which is voltage-gated, small, and weakly selective to cations but unable to permeabilize proteins. The other type of BAX pore is voltage-independent and is most likely large enough to allow the translocation of proteins, such as cytochrome c, across the membrane [84]. There are many structural explanations for how BAX and BAK are positioned at the mitochondria to form a toroidal pore [85]. To represent active BAX in the membrane, the umbrella model was created. The insertion of helices α5 and α6 into the lipid bilayer as a transmembrane hairpin is suggested by this mode [83] [86].
In the in-plane model, helices α4 and α5 form an aromatic planar surface on the membrane, which may raise membrane tension via local curvature stress and result in pores at the MOM. In this model, helices α9 are fully inserted into the lipid bilayer whereas helices α5 and α6 are only partially inserted [85]. According to the clamp model, an oligomer's BAX will generate dimer units. A dimerization domain with helices α2-α5 that lines the pore edge in a toroidal design in conjunction with the lipid head groups is responsible for protein interaction. Given that the multimerization method of PFPs does not include complete proteins crossing the bilayer, such organization may need monomer translocation to the inner side of the membrane. This model suggests that the most important reconfiguration of BAX occurs during the partial opening of the hairpin formed by helices α5 and α6, which come together to form a clamp-like structure. Helix 5 is situated on the pore surface and immediately interacts with the lipids, while helix 6 is at the membrane interface [87]. However, these pore structures play several important roles in the apoptosis pathway.
4.3. BH3-Only Proteins Stimulate Apoptotic Pathway
The expression of one or more conserved BCL-2 homology (BH) domains—BH1, BH2, BH3, and BH4—is a common characteristic of the structure of the BCL-2 protein family. The homology domain BH3 is expressed by the activator and sensitizer proteins, which are called BH3-only proteins. In contrast, effector and antiapoptotic proteins are multi-BH-domain relatives [88]. The sentinels of cellular stress are thought to be the BH3-only proteins. They can detect a wide range of stimuli, including developmental signals and growth factor deficiency, that can cause apoptosis, which kills off undesirable or diseased cells [89]. Eight BH3 proteins have been extensively studied in mammals: BIM, BAD, BMF, BID, NOXA, BIK, PUMA, and HRK [90]. Their fundamental individual roles as well as some of their overlapping functions have been explained via gene disruption [47]. They either directly activate BAX-like proteins or neutralize anti-apoptotic BCL-2 proteins to cause apoptosis. There are structural similarities between different anti-apoptotic compounds and viral homologs. A hydrophobic gap is formed by BH domains, to which BH3-only proteins bind. As a result, BH3-only proteins are promiscuous in their binding to antiapoptotic BCL-2 proteins, and signaling specificity results from a variety of regulatory mechanisms, including transcriptional and post-translational modifications [91].
The pro-apoptotic pore-forming proteins BAX and BAK are directly bound to and activated by BH3-only activators (BID, PUMA, and BIM). Activation and oligomerization of BAX and BAK form macropores in the mitochondrial outer membrane. The BH3-only pro-apoptotic sensitizing proteins (BAD, BIK, NOXA, HRK, BMF) cannot directly activate BAX and BAK. They counteract the effects of anti-apoptotic proteins by releasing the BH3-only activators and/or the constitutively activated BAX and BAK through competitive binding [92]. The six mammalian pro-survival BCL-2 families (BCL-2, BCL-XL, BCL-W, MCL-1, A1, and BCL-B) control BAX and BAK, and are inhibited by BH3-only proteins [93]. The primary event in mammalian apoptosis occurs upon oligomerization of BAX and BAK at the MOM and MOM permeabilization (MOMP), which causes the release of apoptogenic components from the mitochondrial intermembrane space. The apoptotic process signature proteolytic cascade of cysteine aspartyl proteases is triggered by the release of cytochrome c and other molecules from mitochondria [93] [94]. Increases in BH3-only protein expression frequently initiate MOM permeabilization [95].
PUMA and NOXA are two proteins that are direct transcriptional targets of p53. They play significant roles in the induction of apoptosis in response to DNA damage brought on by g-irradiation and chemotherapeutic medicines [94]. In the case of Bid, it has to be unfolded before caspase cleavage and activation. TBID could function as a membrane-targeting death ligand for BAK, causing the release of cytochrome c and apoptosis. BID, BIM, and even PUMA can directly activate BAX and BAK molecules, while all other BH3-only proteins exclusively function as de-repressors [89]. BIM and Puma appear to have an identical affinity for all members of the pro-survival family and are thus promiscuous binders, whereas the other members showed varying affinity (up to 1000-fold) for distinct pro-survival proteins [91]. There are small compounds known as BH3 mimetics, designed to prevent the interaction of pro- and anti-apoptotic proteins. They bind anti-apoptotic BCL-2 family proteins with high affinity and specificity through their hydrophobic groove [88] [92]. The first BH3 mimetic discovered, ABT-737 has a higher affinity for BCL-2, BCL-XL, and BCL-W and a lower affinity for MCL1 and A1/BFL1 [96]. ABT-263 (navitoclax) is another potent inhibitor that has effectiveness against chronic lymphocytic leukemia (CLL) [92]. As single drugs in xenograft models, ABT-737 and ABT-263 demonstrated proapoptotic effects in primary cancer cells [88].
5. Anti-apoptotic role of BCL-2 Family
Anti-apoptotic BCL-2 proteins’ primary function is to resist pro-apoptotic BCL-2 proteins, which prevents MOMP and blocks the mitochondrial pathway of apoptosis. Anti-apoptotic BCL-2 proteins include BCL-2, BCL-XL, BCL-W, MCL1, BFL1 (A1 in mouse), and BCL-B (BCL-2L10 in mouse) [97] [98]. At first, everything seems quite simple. Active BAX and BAK bind to the anti-apoptotic BCL-2 proteins. They specifically bind to the BH3 regions of BAX and BAK [11]. BCL-2, BCL-XL, MCL-1, BCL-W, and BCL-B all include C-terminal transmembrane (TM) domains that point them toward intracellular membranes, notably the MOM. Although BFL1 lacks a traditional TM, its C-terminal α-helix directs it toward mitochondria. All of these proteins have a very similar globular structure with a so-called BCL-2 core. The BCL-2 core consists of eight α-helices with helices 3, 4, and 5 forming a hydrophobic groove. The hydrophobic groove binds the BH3 domains of the pro-apoptotic family members [98]. It is also called the BC groove because it binds the BH3 region of binding partners [99]. The biology of anti-apoptotic BCL-2 family proteins depends on this BC groove [23]. However, there are variations in how well-suited some anti-apoptotic BCL-2 family members are to resist pro-apoptotic molecules.
The ability of specific anti-apoptotic compounds to bind and antagonize the BH3 domains of different pro-apoptotic molecules is determined by their hydrophobic BH3-domain binding pockets. Some members of the BH3-only family, such as BIM, BID, and PUMA, may bind all anti-apoptotic compounds with similar affinities. On the other hand, other BH3-only family members have a limited ability to connect with other antiapoptotic BCL-2 family members [100]. However, abundant evidence shows interactions between anti-apoptotic BCL-2 proteins and BAX or BAK to prevent apoptosis. The anti-apoptotic BCL-2 protein, which contains an intramolecular disulfide tether that prevents significant conformational changes, was discovered through studies of BAX variants. Indeed, intramolecular tethers can prevent important conformational changes in the BAX regulatory cycle, which store normal transient forms of BAX in mitochondria. The discovery of BAX retro-translocation was made possible by these results. The inactive conformation is continuously maintained by mitochondrial BAX being shuttled into the cytoplasm, which also shields healthy cells from BAX activity [101].
5.1. BCL-2
Due to chromosomal translocation, BCL-2 protein was first demonstrated to be overexpressed in B-cell lymphoma. Its level is additionally controlled by miRNAs. So far, 12 miRNAs have been linked to controlling BCL-2 expression [102]. The BCL-2 gene has three exons, the first two of which are responsible for encoding the four BH domains, and the third of which is responsible for encoding the transmembrane domain, which attaches the protein to intracellular membranes. The BCL-2 gene has two isoforms, BCL-2α and BCL-2ß. As shown in cell lines after exposure to cellular stress, BCL-2α binds to BAX via its BH1 and BH2 domains. This association is crucial to its function in regulating apoptosis. However, In numerous stress models, including gamma radiation, heat shock, glucocorticoid and cytotoxic drug therapy, and IL-3 deprivation of dependent cells, BCL-2 is upregulated and able to bind BAX, which reduces the ability of cells to undergo apoptosis [18]. When BCL-2 is overexpressed, it inhibits BAX-mediated cytochrome-c release, caspase activation, and cell death in nerve growth factor-deprived sympathetic neurons. Consequently, BCL-2 plays an important role in the development of the nervous system. BCL-2 overexpression increased cortical neuron survival after localized cerebral ischemia by preventing the transfer of apoptosis-inducing factor (AIF) from mitochondria to the nucleus [103]. In addition, BCL-2 protein helps malignant B-cells to resist apoptosis [104].
5.2. BCL-XL
The BCL-XL and BCL-XS protein isoforms are generated by alternative splicing of the BCL-X gene [103]. They have opposite functions in regulating apoptosis. BCL -XL is an anti-apoptotic protein that shares structural domains with BCL -2, BCL-XS promotes apoptosis since it lacks the area with the highest homology to BCL -2 [21]. The human BCL-XL protein was the first member of the BCL-2 family to have a three-dimensional structure. BCL-XL was determined via X-ray crystallography and nuclear magnetic resonance (NMR), with good agreement amongst the structures. There are eight alpha-helical regions in the structure of BCL-XL. A center hairpin configuration made up of helices α5 and α6 is surrounded on one side by helices α3 and α4 and on the other by helices α1, α2, and α8. The BH domains play an important role in the tertiary structure of the three-dimensional BCL-XL complex. In the BH1 and BH2 domains, the turn region connects the two helices in the BH1 and BH2 domains. The first is α4 to α5 for BH1, and the second is α7 to α8 for BH2. The BH3 domain is completely on α2, whereas the BH4 domain is on α1 and forms several stable hydrophobic interactions with α2, α5, and α6 [105][106]. Dual mechanisms are used by BCL-XL and BCL-2 to regulate apoptosis. First, BCL-XL and BCL-2 inhibit apoptosis by binding to the α-helical BH3 domain of pro-apoptotic regulators. Second, BCL-XL binds cytosolic p53 at a location distal to the hydrophobic groove, which prevents BAX activation and apoptosis from being p53-dependent [107]. Additionally, the anti-apoptotic action of BCL-XL is mediated via the BH4 domain. Along with preventing apoptosis, the BH4 domain of BCL-XL protein plays an important role in regulating cell migration and other important cellular processes. However, proteases from autophagosomes can degrade Bid, which affects BCL-XL-mediated apoptosis [48]. By attaching to the tumor suppressor Beclin-1 and subsequently blocking autophagy, BCL-XL has been associated with non-apoptotic cell death [18]. It has also been suggested that BCL-XL is a key factor in the emergence of medication resistance [108].
5.3. MCL-1
The human MCL-1 gene is found on chromosome 1q21, and MCL-1 proteins were first discovered in myeloid leukemia cells. MCL-1 is highly expressed in cardiac and skeletal muscles, sympathetic neurons, and neuroendocrine cells [103]. Although the structure of Mcl-1 is similar to other anti-apoptotic BCL-2 family members, it is considerably larger because it has an extended N-terminal regulatory domain. The abundance of proline [P], glutamic acid [E], serine [S], and threonine [T] residues is one of the most significant characteristics of this region. Commonly, PEST sequences frequently act as signals for rapid protein breakdown [109]. The MCL- gene has three distinct isoforms: MCL-1L, MCL-1S, and MCL-1ES. MCL-1L is anti-apoptotic, but MCL-1S and MCL-1ES are both pro-apoptotic, and these three proteins play distinct roles in the control of apoptosis [18]. The first isoform, Mcl-1L, binds to the outer mitochondrial membrane (OMM) by its transmembrane (TM) domain, where it inhibits the expression of cytochrome c. There are 350 amino acids encoded by three exons in MCL-1L. The MCL-1 gene skips the second exon, resulting in the 271 amino acid MCL-1S. Mcl-1S is mostly found in the cytosol and differs from Mcl-1L in length by lacking the BH1, BH2, and TM domains [110]. MCL-1ES results from alternative splicing at a non-canonical donor-acceptor site within the first exon. The resulting protein is 197 amino acids long and lacks the BH4 and PEST domains present in other MCL-1 isoforms [18].
MCL-1 is susceptible to several posttranslational, posttranslational, and transcriptional modifications. According to a number of studies, MCL-1 is regulated by a variety of cytokines and signaling pathways. Through autocrine signaling loops, VEGF and interleukin-6 (IL-6) can control MCL-1 expression. The synthesis of IL-6 is stimulated by the activation of the Notch-1 signaling pathway, which also increases the expression of MCL-1 [111]. By phosphorylating Thr-163, the ERK pathway protects the Mcl-1 protein from degradation [112]. Through the STAT3/MCL-1 pathway or the JAK/STAT and PI3K pathways, cytokines such as IL-15 and IL-22 can also regulate MCL-1 protein levels [113]. The pro-apoptotic proteins BAK/BAX are sequestered by MCL-1 through the BH3 domain, which contains the hydrophobic groove, to exert its antiapoptotic action [114]. Mcl-1 is essential for the survival of many different types of healthy cells, such as hepatocytes, neurons, cardiomyocytes, and others. Mcl-1 is often exploited by cancer cells to avoid apoptosis [109]. In addition to its anti-apoptotic action, MCL-1 is essential for embryogenesis, cell cycle regulation, and energy generation [115]. Selective MCL-1 inhibitors are needed as biochemical tools that can lead to new cancer chemotherapy [116].
5.4. BFL-1
A member of the BCL-2 family of proteins, BFL-1 inhibits the process of apoptosis and regulates the crucial equilibrium between cellular life and death in response to physiological stress [117]. The BCL-2A1 gene codes for the 175-residue BFL-1 protein [118]. Human BCL-2A1 (encoding BFL-1) was mapped to chromosome 15 in 1997, while mouse BCL-2A1 (encoding A1) was assigned to chromosome 9 [119]. Human BFL-1 and mouse A1 differ in a number of ways. Mice have four genes, three of which code for the functional paralogs BCL-2A1a, BCL-2A1b, and BCL-2A1d. These three have DNA and protein sequences that are more than 95% homologous, but BCL-2A1c encodes a pseudogene. Moreover, there is only one BCL-2A1 gene in human [120] [121]. BFL-1 shares structural similarities with other anti-apoptotic proteins. It contains BH1-4 hydrophobic plaques. BFL-1 binds to the BH3-only proteins BIM, PUMA, NOXA, BIK, BID, and HRK [119] [122]. BFL-1 inhibits the dimerization of BAX and BAK through a mechanism similar to other anti-apoptotic members. As a result, the release of apoptotic agents and depolarization of the mitochondrial membrane ceases. BFL-1 thus suppresses apoptosis by blocking the downstream caspase pathway. In some situations, BFL-1 promotes apoptosis. Full-length BFL-1 is cleaved at the C-terminal membrane, which turns BFL-1 into a pro-apoptotic protein. M-calpain mediates this mechanism. The α5 helix OF BFL-1 induces apoptosis through a BAX and BAK-dependent mechanism, whereas the α9 helix does so through a membrane instability mechanism [122].
Melanoma, lymphoma, and leukemia are all associated with the pathological expression of BFL-1 as an oncogenic driver [123]. Numerous human cancers, such as solid tumors and hematological malignancies, have been linked to the pathogenesis and chemoresistance of BFL-1 [124]. Many different B-cell lymphoma types have been shown to overexpress BFL-1. The increasing levels of BFL-1 seen in different types of cancers demonstrate the tremendous possibility of targeting BFL-1 for treating these diseases [119]. For the development of new classes of chemotherapeutic drugs that can combat the resistance of cancer cells to currently available chemotherapeutics, inhibiting the activity of BFL-1 represents an effective strategy. BFL-1 has several other physiological roles in post-allergic mast cells, cellular survival, B-cell maturation, and immune system modulation. Although little is known about the precise mechanisms involved, a role for BFL-1 in inflammation has been established [125].
5.5. BCL-W
BCL-W is an anti-apoptotic protein of the BCL-2 family. Compared to other anti-apoptotic molecules, BCL-W and BCL-XL have the highest sequence similarity (51%). BCL-W interacts with BAX, BAK, and several BH3-only proteins, including BAD, tBID, BIM, PUMA, BMF, and BIK, as demonstrated by co-immunoprecipitation [36]. BCL-W sometimes referred to as BCL-2-like 2 (BCL-2l2), is a highly conserved gene found in the q11.2-q1235 region of chromosome 14 in mice and humans. BCL-2L2 contains two non-coding exons in addition to two coding exons at the 5′ ends. The non-coding exon contains the minimal promoter region of the BCL-2L2 gene, which is highly conserved in humans, mice, and rats [36] [103]. BCL-W protein is typically located on the mitochondria's outer membrane. The BCL-W protein has nine helices, flanked by amphipathic helices α1, α2, α3, α4, α6, α7, α8, and α9, and a central hydrophobic groove created by a helix, α5 [126].
BCL-W is highly expressed, especially in the brain, colon, and testes. It is also connected to intracellular membranes [103]. The process of mitotic cell death is regulated by BCL-W. BCL-W used two additional siRNAs to accelerate mitotic cell death. The other two siRNAs were similarly able to accelerate mitotic cell death, although considerably less successful than the initial siRNA in depleting BCL-W [127]. In addition to its function as a pro-survival protein, BCL-W facilitates tumor invasion by increasing the production of transcription factor specificity proteins, which in turn increase the expression of matrix metalloproteinase-2 [23]. BCL-W also regulates the morphogenesis of mitochondria and has been linked to the development of dendrites. Additionally, increased levels of BCL-W cause neurological disorders such as Alzheimer's disease and Parkinson's disease [126]. The overexpression of BCL-W in diffuse large B cell lymphoma was associated with poor patient survival. BCL-W has significant effects on B cell lymphoma, and its expression can be used as a biomarker to diagnose and develop more precisely targeted treatments. The development of myeloid leukemia was accelerated by the interaction between MYC and BCL-W overexpression. Additionally, BCL-W overexpression has been observed in several human solid tumor cell lines as well as gastric and colorectal adenocarcinoma patient samples [128]. Numerous cancers are caused by genetic alterations in BCL-2L2, such as copy number variants in small and non-small lung cancer, high levels of BCL-w expression in gastric carcinomas, and low levels of BCL-w expression in colorectal cancer. Significantly higher BCL-W mRNA levels are found in breast cancer patients. Cancer of the urinary system has been profoundly impacted by BCL-W. Hepatocellular carcinoma (HCC), leiomyosarcomas, prostate cancer, cervical cancer, and prostate cancer are all linked to overexpression of BCL-W [126]. The BCL-W expression is abnormally high in samples from patients with Hodgkin lymphoma, Burkitt lymphoma (BL), and diffuse large B-cell lymphoma (DLBCL). However, Human BL and DLBCL cell line growth and survival are not always dependent on BCL-W. [129].
6. The Apoptosis Pathway
Apoptosis is an evolutionarily conserved cell death mechanism generally responsible for cell destruction throughout eukaryotic development and maintenance of organismal homeostasis. The BCL-2 protein family, which includes both pro- and anti-apoptotic members, regulates this pathway by balancing the choice between cellular life and death [130]. The mechanism of apoptosis is more complex and involves an energy-dependent cascade of molecular events [131]. Apoptosis can occur through two different pathways: the intrinsic pathway, which is activated downstream of cellular stress such as DNA damage, ER stress, or growth factor withdrawal, and the extrinsic pathway, which is initiated when death receptors on the cell surface are activated [132]. Additionally, a different pathway that involves T-cell-mediated and perforin/granzyme-dependent cell death is reported. This pathway is mediated by the granzyme types A and B [1]. These pathways converge when caspases (aspartate-specific cysteine proteases) are activated. These enzymes are responsible for cleaving essential proteins, which ultimately leads to cellular destruction [94].
The intrinsic pathway often referred to as the mitochondrial pathway, is triggered by signals within the cells, such as oncogenic stress, viral infection, or cytotoxic insults that damage DNA or protein molecules. Both pro- and anti-apoptotic BCL-2 family proteins control the entire mechanism [67]. The intrinsic apoptosis signaling cascade is initiated by BH3-only proteins, which are important sensors of cellular stress. They suppress the activity of anti-apoptotic BCL-2 family members by binding to the BH3 domain, thereby neutralizing their activity and promoting apoptosis. The anti-apoptotic BCL-2 protein is inhibited, which causes the apoptotic effectors BAX and BAK to become active. Some BH3-only proteins, including tBID (activated form of BID), BIM, and perhaps PUMA, may also directly bind and activate BAX and BAK [94]. Activated BAX and BAK oligomerize, which causes permeabilization of the Mitochondrial Outer Membrane (MOMP). The permeabilization enables the release of intermembrane proteins such as cytochrome c, a second mitochondria-derived activator of caspase (SMAC, also known as DIABLO), AIF (apoptosis-inducing factor), and Omi [66] [131]. Then the apoptosome is formed. The apoptosome is a complex made up of cytochrome c, procaspase-9, deoxyadenosine triphosphate (dATP), and apoptotic protease-activating factor-1 (APAF-1). Procaspase-9 becomes caspase-9 within the apoptosome [133]. The apoptosome then activates caspase-9, which in turn activates caspase-3 and thus induces apoptosis [1]. Intrinsic apoptosis involves additional processes that ensure cell death. The endogenous inhibitor of caspase function known as the X-linked inhibitor of apoptosis protein (XIAP) is inhibited by omi [134]. When apoptosome is formed, SMAC is released to block the inhibitor of apoptosis protein (IAP), which allows apoptosis to occur [66]. Direct nuclear translocation by AIF results in caspase-independent nuclear changes [131].
The extrinsic pathway is activated by the cell surface death receptors, tumor necrosis factor receptor (TNFR), FAS, and TNF-related apoptosis-inducing ligand receptor (TRAILR) upon stimulation by their respective ligands TNF, FASL, and TRAIL [132]. The intracellular death domain (DD) of death receptors serves as a docking site for the DD domain in the adapter protein FADD (Fas-associated death domain). By interacting with the DED domain in the pro-domain of caspase-8, a death effector domain (DED) in FADD can now draw procaspase-8 and concentrate it on the activated receptor. The formation of this complex, known as DISC (death-inducing signaling complex), results in the processing and activation of pro-caspase-8. Now that caspase-8 is activated, it can process and activate the effector caspases, caspases 3, 6, and 7, which finally leads to cell death [67]. In some cells, the BH3-only protein BID is cleaved by caspase-8 as a result of death receptor activation. BID cleavage produces a 15 kDa truncated active protein known as tBID, which translocates to mitochondria where it forms a heterodimer with the anti-apoptotic BCL-2 protein. As a result of this interaction, the anti-apoptotic BCL-2 proteins are inactivated, which in turn activates the pro-apoptotic proteins BAX and BAK. When BAX and BAK are activated, the mitochondrial membrane becomes permeable, cytochrome c is released, the apoptosome is formed, caspases are activated, and apoptosis occurs. Here, the extrinsic and intrinsic pathways converge, causing both to proceed normally and induce apoptosis [66] [94].
Figure 3.
There are three main types of apoptotic routes, which are known as the extrinsic pathway, intrinsic pathway, and perforin/granzyme pathway. Each pathway initiates with a specific death-inducing cell signal and is regulated by activating different caspases. The extrinsic route is activated by the interaction of death receptors (DRs) and death-inducing ligands via their adaptor domains. Subsequently, a multiprotein complex DISC and caspase-8 regulate this pathway. In the intrinsic apoptotic pathway, the stimuli first turn on BH3-only proteins. The apoptotic signals are then sent by turning on interactions between BCL-2 members, forming a MOMP complex, and then cytochrome c is released. This pathway is run by cytochrome c and other caspases activation. In the perforin/granzyme pathway, granzyme A and granzyme B are released by cytotoxic T cells. the granzyme A pathway functions caspase-independently method. Granzyme B activates caspase-10, which is coupled to the intrinsic pathway and induces apoptosis. Here, all pathways ultimately activate caspase-3. However, caspase-3 activation eventually induces apoptosis. Caspase-3 degrades essential parts of cells, like chromosomes and cytoskeletal proteins. This leads to the formation of apoptotic bodies, which are ingested and destroyed by phagocytic cells and finally removed by the organism.
Figure 3.
There are three main types of apoptotic routes, which are known as the extrinsic pathway, intrinsic pathway, and perforin/granzyme pathway. Each pathway initiates with a specific death-inducing cell signal and is regulated by activating different caspases. The extrinsic route is activated by the interaction of death receptors (DRs) and death-inducing ligands via their adaptor domains. Subsequently, a multiprotein complex DISC and caspase-8 regulate this pathway. In the intrinsic apoptotic pathway, the stimuli first turn on BH3-only proteins. The apoptotic signals are then sent by turning on interactions between BCL-2 members, forming a MOMP complex, and then cytochrome c is released. This pathway is run by cytochrome c and other caspases activation. In the perforin/granzyme pathway, granzyme A and granzyme B are released by cytotoxic T cells. the granzyme A pathway functions caspase-independently method. Granzyme B activates caspase-10, which is coupled to the intrinsic pathway and induces apoptosis. Here, all pathways ultimately activate caspase-3. However, caspase-3 activation eventually induces apoptosis. Caspase-3 degrades essential parts of cells, like chromosomes and cytoskeletal proteins. This leads to the formation of apoptotic bodies, which are ingested and destroyed by phagocytic cells and finally removed by the organism.
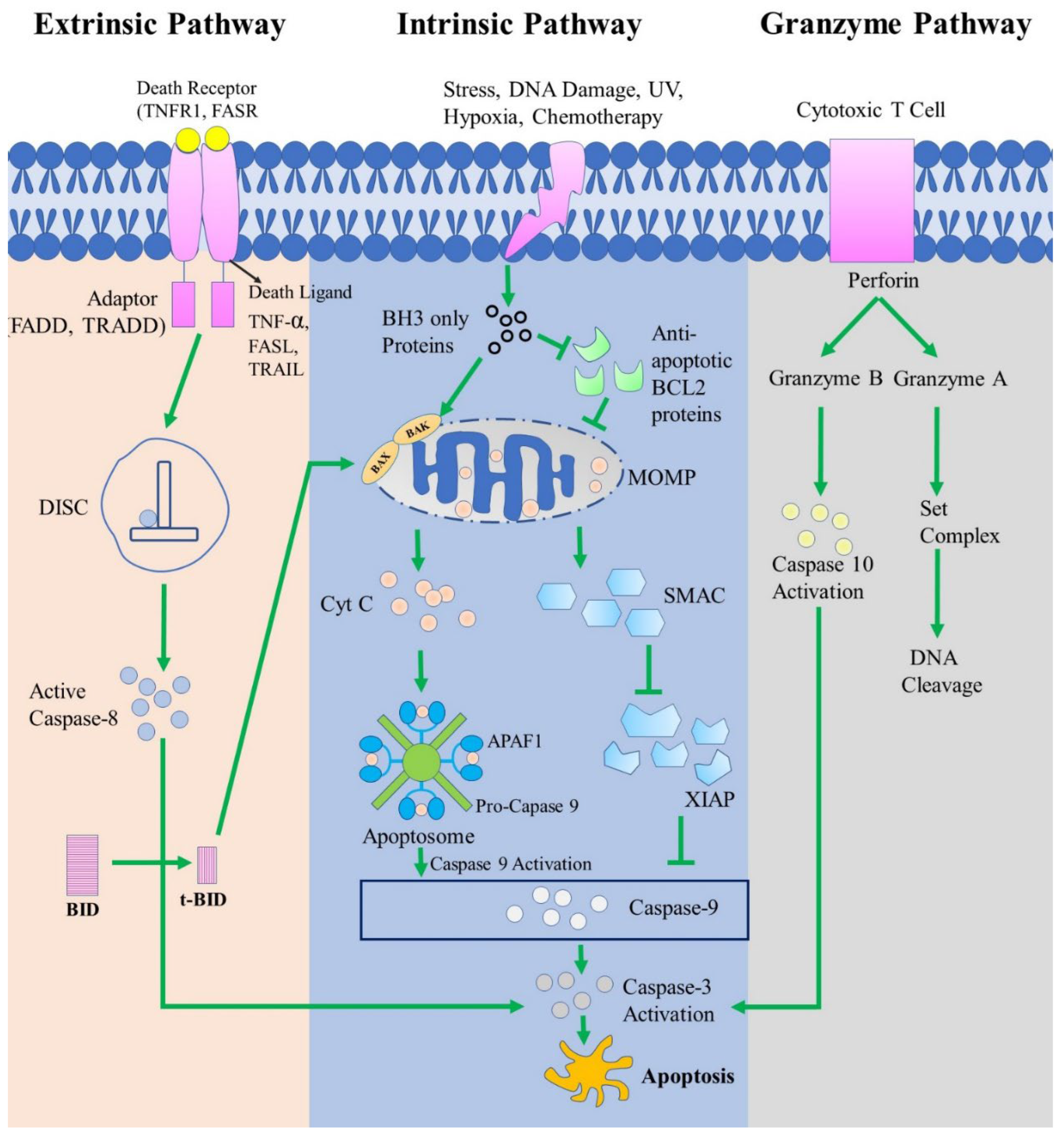
In the perforin/granzyme pathway, either granzyme A or granzyme B can be used to induce apoptosis. Granzymes are serine proteases that are released from granules found in the cytoplasm of NK and CTL cells [135][136]. Additionally, a glycoprotein called perforin helps granzymes enter target cells by creating pores in their cell membranes [135] [137]. Here, the granzyme A pathway is caspase-independent, whereas granzyme B is caspase-dependent [138]. Granzyme A has a vital role in cytotoxic T cell-induced apoptosis. Granzyme A protease breaks down the SET complex (protects the DNA and chromatin structure) and thereby releases inhibition of NM23-H1 (Nucleosome assembly protein SET, inhibits NM23-H1), which causes DNA to be degraded during apoptosis. These indicate that granzyme-mediated inactivation of this complex probably promotes apoptosis by preventing the maintenance of the integrity of DNA and chromatin structures [139]. On the other hand, Granzyme B activates caspase-3 either directly or indirectly through caspase-10. Granzyme B uses the mitochondrial pathway to release cytochrome c and amplifies the death signal by specifically cleaving the BID protein [131]. These three routes are visualized in Figure 3.
7. The Relationship Between BCL-2 Protein Family and P53
P53 is the gene that is mutated in most human cancers. It is estimated that p53 is mutated in about half of all cancers, and 20% of cancers involve p53 that is functionally inactive. The p53 gene is responsible for the development of a protein also known as tumor protein p53 [140]. The p53 protein is a regulator of many processes that cells need to function correctly, and it is involved in many processes related to their life and death [141]. The p53 protein inhibits the growth of tumors by controlling the expression of a vast number of genes involved in processes such as cell cycle arrest, DNA repair, senescence, apoptosis, cell motility, cell adhesion, and migration [142]. Besides, P53 has been shown to affect various physiological and pathological processes, including neurodegeneration, aging, autophagy, angiogenesis, maternal reproduction, and fertility [140]. P53 mediates apoptosis in both a transcription-independent and -dependent manner in response to different cellular stresses [143]. As a result of stress events, the p53 protein crosses into the mitochondria and promotes the expression of pro-apoptotic BCL-2 proteins such as PUMA, BAK, BAX, and NOXA, as well as inhibits the expression of anti-apoptotic BCL-2 proteins such as BCL-2 and BCL-XL [141]. In cells, p53 has been shown to interact with BCL-XL and BCL-2.
Through allosteric conformational changes in the BH3 peptide-binding region of BCL-XL, the p53 DNA-binding domain (p53DBD) promotes binding to pro-apoptotic BH3-only proteins. The transactivation domain of p53 (p53TAD) competitively inhibits BAK and BAX from binding to BCL-2 and BCL-XL, leading to the release of BAK and BAX. These results suggested that p53TAD may induce mitochondrial apoptosis by competitively inhibiting the interaction between pro-apoptotic and anti-apoptotic BCL-2 family proteins [143]. In addition, Granzyme B (GzmB) rapidly accumulates p53 in target cells. This p53 protein interacts with the BCL-2 protein and appears to be involved in the GzmB-induced apoptosis process. Therefore, inhibition of p53-BCL-2 interaction reduces GzmB-induced activation of BAX, the release of cytochrome c from mitochondria, and subsequent activation of effector caspases, thereby reducing the sensitivity of target cells to GzmB and CTL/NK-mediated death [144]. However, BCL-2 and p53 significantly determine tumor growth and may help identify high-risk patients more precisely [145]. High BCL-2 expression and low p53 expression were linked to indolent histopathological features of basal cell carcinoma (BCC). Based on these data, a thorough investigation of p53 and BCL-2 expression levels in BCC patients may yield helpful prognostic information. Some cases of BCC may develop and progress in response to these biomarkers [146].
8. Potential Biomarkers
8.1. P53
Pro-apoptotic molecules like BAK, BAX, PUMA, and Noxa are transcriptionally regulated by p53 and are crucial components of MOMP in response to death stimuli. BCL-2 and BCL-XL are also inhibited by p53, which enables pro-apoptotic components (BAK or BAX) to separate from heterodimeric complexes after oligomerization of BAX and BAK into the mitochondrial outer membrane (MOM), This produces lipid pores inside the MOM through which the apoptotic initiators are produced in response to death stimuli [147]. Additionally, p53 has the ability to directly activate BAK and/or BAX using a "hit and run" method to cause the permeabilization of the outer mitochondrial membrane. In various cellular compartments, P53 can have a dual function in apoptosis. In nucleus, its interaction with the fundamental elements of the transcriptional machinery, p53 functions as a transcriptional activator and stimulates the expression of the target gene. On the other hand, in mitochondria BCL-2 and BCL-XL interact with p53, leading transcription-independent apoptosis to develop [143]. In de novo diffuse large B-cell lymphoma patients, screening of P53 expression gives an important prognostic information, specially in subgroups with MYC-R, MYC expression, and MYC and BCL-2 double expression. P53 expression was demonstrated to significantly shorten survival in MYC/BCL-2 double-positive lymphomas[148]. BCL-XL inhibits p53, a direct BAX activator, characterizing it as a non-canonical prototype modulator of BCL-2 proteins-mediated MOMP [97].
The pro-apoptotic BCL-2 family protein PUMA level rises in response to p53 activation [149].
8.2. ABT-737
ABT-737 is a small-molecule BH3 mimetic that has an extremely high affinity for directly binding to BCL-2, BCL-XL, and Bcl-w. This action induces apoptosis by releasing BAX and BAK [150]. Synthetic small molecule cannot directly bind BAK or BAX, it can damage the complex protein unit that consists of BAX/BAK and BCL-2 [151].It has been shown that Mcl-1 overexpression makes some malignancies resistant to the BCL-2/BCL-XL inhibitors ABT-737 and ABT-263 [152]. In a xenograft mice model of PTLD, ABT-737 decreased tumor growth and improved overall survival while having no impact on BL xenograft mice [153]. ABT-737 therapy decreased bone marrow early and late apoptosis [154]. Both in vivo and in vitro studies of the BCL-2 inhibitor ABT-737 have revealed positive anticancer effectiveness. However, certain studies have shown that HCC cells are resistant to ABT-737, and the corresponding molecular mechanisms of this resistance are not clearly appreciated. ABT-737, a BH3 mimic, can selectively prevent the binding of BCL-2/xL and BAK/BAX by competing for the BH3 domain and then inducing apoptosis through the mitochondrial system [155]. When paclitaxel and ABT-737 were used together, PDA cell death was caused at a lower paclitaxel concentration than when paclitaxel was used alone [156].
8.3. BCR-ABL
The effectiveness of BCL-2 inhibition in TKI-resistant BCR-ABL1-positive cells generated from the blast crisis of CML patients who were previously in the chronic phase [157]. The BCR-ABL protein possesses a constitutive tyrosine kinase activity that contributes to cell apoptosis resistance and CML pathogenesis, yet the cellular and molecular mechanisms underlying BCR-ABL expression and apoptosis dysfunction in CML leukemic cells are still poorly understood [158]. Through a variety of methods, the oncogenic Bcr-Abl fusion protein encourages leukemic cell survival and proliferation. BCR-ABL1 may also control the amounts of other anti-apoptotic BCL-2 proteins, which could increase the threshold at which a death stimulus must be present in order for apoptosis to proceed [159]. The combined inhibition of BCL-2 and BCR-ABL tyrosine kinase has the potential to greatly increase depth of response and cure rates of chronic-phase and BC CML while BCL-2 is a crucial survival factor for CML stem/progenitor cells [160].
8.4. PUMA and NOXA
Bim, Noxa, Bim/Noxa, Bim/Puma, Bim/Bmf, BAX, BAK, or BAX/BAK-deficient cells are used to evaluate the significance of BCL-2, BCL-XL, and Mcl-1 using particular inhibitors [161]. Up-regulation of the tumor suppressor protein p53 and its transcriptional target PUMA suggests that they may be involved in inducing cell death [162]. BH3 is the only molecule that activate apoptosis. We provide biochemical and genetic proof that NOXA is an authentic BH3 activator. Stepwise, bimodal activation of BAX-BAK is directly induced by BID, BIM, PUMA, and NOXA [26]. It has been demonstrated that p53 can transcriptionally activate pro-apoptotic BCL-2 Homology domain-3 (BH3)-only genes, including p53 upregulated modulator of apoptosis (PUMA) and phorbol-12-myristate-13-acetate-induced protein 1. (PMAIP1 or NOXA) [163].
Apoptosis is caused by a number of well-known BCL-2 inhibitors, including ABT199 and HA14-1, that also bind to the hydrophobic groove of BCL-2. These inhibitors cause BCL-2 to become detached from or to compete with its proapoptotic partners BAK/BAX [164]. The most promising strategy for lowering a cell's overall radiosensitivity seemed to be apoptosis induction through an increase in the tumor suppressor protein p53 and its transcriptional target, the pro-apoptotic protein PUMA, which is regulated by ATM/ATR [149]. The presence of a MyoD responsive region in the PUMA gene will make it easier to test this theory in the future and to understand the mechanism by which MyoD promotes apoptosis rather than differentiation. Binding to two E-boxes or one E-box and a cooperative co-activator is necessary for MyoD-mediated transcription [165]. NOXA, in contrast, is regarded as a "weak" or indirect activator since it can only bind and block the pro-survival members MCL1 and BCL-2A1, preventing them from regulating BAX and BAK1 [166]. The anti-apoptotic member MCL-1's location and stability are controlled by Noxa, which modulates the sensitivity of ABT-737 [167]. BH3-only pro-apoptotic protein NOXA, which is encoded by the NOXA gene, serves as a sensitizer to prevent anti-apoptotic BCL-2 family proteins [168]. The histone deacetylase inhibitor panobinostat was used to pharmacologically induce NOXA, which led to decreased MCL1 protein abundance and improved lymphoma cell tolerance to BCL-2 inhibitors in vitro and in vivo [169].
9. Therapeutic Potential of BCL-2 Family Proteins
BCL-2 family proteins play an important role in modulating the apoptotic response in anticancer therapy. It is interesting to explore whether BCL-2 family proteins will be able to serve as biomarkers to predict treatment outcomes [170]. Targeted cancer therapy tries to stop the actions of proteins that are crucial for the development of cancer. Antiapoptotic proteins are an excellent target for cancer therapy because they are significantly elevated in many malignancies relative to normal cells [171]. A novel line of anti-cancer medications that precisely block the anti-apoptotic BCL-2 family proteins has undergone substantial advancement in recent years [172]. The clinical response of different malignancies and the chemotherapeutic sensitivity of lymphoma cell lines have been successfully predicted using BH3 profiling. The BH3 profiling appears useful for identifying highly primed tumors that are more likely to respond to conventional treatment as well as BH3 mimetics [170]. BH3-mimetics are a new class of anticancer drugs. They mimic the action of BH3-only proteins in that they bind to and inhibit anti-apoptotic BCL-2 family members and thereby activate apoptosis in cancer cells [69] [173]. When BH3-mimetics bind to such proteins in hydrophobic grooves, they displace sequestered BH3-only proteins, which can then activate BAK and BAX. Alternatively, BH3-mimetics can lower the apoptosis threshold by binding to the hydrophobic groove of unprimed cells and inhibiting any BH3-only proteins that are subsequently produced [174]. There are several compounds have been proposed to be BH3-mimetics. Such as ABT-737, ABT-263-navitoclax, ABT-199, GX15-070- obatoclax, AT-101, gossypol, BI-97D6, TW37, S1 derivative, BH3-M6, Marinopyrrole A, maritoclax, MIM1, BAM7, A-1155463, A-1210477 [175].
ABT-737 is considered a model of BH3 mimetics. It was a first-in-class molecule designed to mimic the action of BH3-only proteins. ABT-737 inhibits the activity of the anti-apoptotic proteins BCL-2, BCL-XL, and BCL-w by binding to them with a significantly higher affinity (1 nmol/L) than earlier drugs. It has a weak binding affinity for the anti-apoptotic BCL-2 family members BCL-B, MCL-1, and A1 [47] [50]. ABT-737 exhibits a wide range of anti-tumor activity, including activity against myeloma, chronic lymphocytic leukemia, acute myeloid leukemia, small-cell lung cancer, prostate cancer, and glioblastoma among others [175].
As a successor to ABT737, ABT 263 (Navitoclax) is an oral bioavailable relative that binds to and inhibits BCL-2 family proteins at nanomolar concentrations. Compared to MCL1 and BCL-W, ABT263 has a higher affinity for binding to BCL-2 and BCL-XL [171]. In preclinical studies, ABT-263 is used in combination with other conventional therapies. It has demonstrated efficacy in several additional cancers, including some forms of breast cancer [176]. In clinical trials, ABT 263 as a single agent significantly reduced tumor burden in the majority of chronic lymphocytic leukemia (CLL) patients [177]. However, as BCL-XL is essential for platelet survival, BAX and BAK-mediated thrombocytopenia is dose-limiting for ABT 263, and this is likely also true for BCL-XL selective BH3-mimetics [33]. The first highly specific BCL-2 inhibitor is ABT-199. ABT-199 has previously demonstrated significant antitumor activity in preliminary clinical tests in chronic lymphocytic leukemia (CLL) and non-Hodgkin's lymphoma with greater response rates than navitoclax and no thrombocytopenia [178].
A natural phenolic substance called gossypol, which is found in cotton plants, has been proven to inhibit apoptosis in mouse tumor models. Gossypol's synthetic derivative AT101 binds to BCL-2, BCL-XL, and MCL1 [171]. Another variant of the ABT-737 is the BM-957. Additionally, it triggers caspase 3-dependent apoptosis in tumor cell lines and tumor regression in xenograft models. BM-957 also binds to BCL-2 and BCL-XL with sub-nanomolar (nM) affinity. BM-1197 causes BAX and BAK-dependent apoptosis in small-cell lung cancer cell lines by binding to BCL-2 and BCL-XL with sub-nanomolar (nM) affinity, inhibiting their antiapoptotic actions. Additionally, in two separate small-cell lung cancer xenograft models, BM-1197 demonstrates better solubility and pharmacokinetic characteristics and causes complete and long-lasting tumor regression [178]. MCL-1 has some inhibitors because it is frequently elevated in a variety of malignancies. The first MCL-1 inhibitor was A-1210477. It has shown efficacy against multiple myeloma cells in vitro. S63845 was subsequently developed, a highly effective MCL-1-selective agent with significant single-agent activity in cell lines including multiple myeloma, lymphoma, chronic myeloid leukemia, and several solid organ cancers (such as non-small-cell lung cancer, breast cancer, melanoma cell lines). AMG 176 and AZD5991 are two other MCL-1 inhibitors under development with sub-nanomolar affinities. Both exhibit in vivo action in multiple myeloma and acute myeloid leukemia models [73].
A new anti-cancer drug could be produced from any substance that directly stimulates BAX or BAK [179]. Targeting the canonical grooves of BAX and BAK has the potential to develop therapeutic agents with reversible cell death properties. By inducing the conformational changes in the structure of BAX and BAK, substances that mimic BH3-peptide-mediated initiating events can promote apoptosis [180]. In more recent studies, small compounds that can directly bind to and activate proapoptotic BCL-2 family members have been designed using BH3 mimetic principles. The compound BAM-7 can induce apoptosis by forming a direct activator of BAX with high binding affinity. A BH3 peptide called PUMA induces the activation of BAK by binding with nanomolar (nM) affinity to its canonical binding pocket, which results in BAK-dependent MOMP and cell death. All of the anti-apoptotic BCL-2 family members can interact with and be inhibited by SAHB which is derived from the PUMA-BH3 helix. The pro-apoptotic BAX is also directly bound to and activated [178]. However, by focusing on the BCL-2 family of proteins, several of the significant future avenues in chemical and drug discovery are highlighted. In the meantime, the development of BH3-mimetics has reached a mature stage for the treatment of malignancies and possibly autoimmune diseases [180].
10. BCL-2 proteins as therapeutic target in diseases
10.1. Parkinson disease
Parkinson disease is a neurodegenerative disorder that affects the motor and non-motor neuron. It is defined by the progressive degeneration of dopaminergic neurons as well as the formation of Lewy bodies, consisting of α -synuclein [181]. a-synuclein is a tiny acidic protein that has a naturally unfolded structure; it is found mostly at presynaptic terminals and it could play role in vesicular transport and neurotransmitter release [182]. It has been revealed that abnormal aggregation of the protein may be harmful to dopaminergic neurons, which can contribute to neurodegeneration associated with Parkinson's disease [183]. The BCL-2 family protein[184] alongside with α -synuclein is used as a therapeutic target [185]. The activation of BAX enhanced the creation of -synuclein deposits and cell death, but the overexpression of the anti-apoptotic protein BCL-2 protect dopaminergic neurons [184].
According to the findings of many research, there are some chemicals that have the capability to trigger the overexpression of BCL-2 while simultaneously lowering the expression of BAX. B. Wang et al. demonstrated that Chitosan oligosaccharide (COSs) were capable of protecting the dopaminergic neurons by activating the PI3K/Akt/BCL-2 pathway and reducing the overexpression of α-synuclein [186].In PD treatment, the upregulation of BCL-2 and downregulation of BAX and caspase-3 are regulated by cannabidiol (CBD) as apoptosis inhibiting factor [187]. K. Guo et al. found that Paeoniflorin, active ingredient of a plant, downregulates proapoptotic proteins and upregulates ani-apoptotic proteins, suggesting a role for the BCL-2 family protein in PD therapy [188].
Citronellol, which is most often found in citrals, is an important chemotherapeutic drug that protects dopaminergic neurons and is used in the treatment of rotenone induced Parkinson's disease. By increasing the expression of the anti-apoptotic protein BCL-2 while simultaneously lowering the expression of the pro-apoptotic protein BAX, this chemical inhibits apoptosis. Additionally, it controls autophagy by preventing the formation of autophagy vacuoles and promoting the clearance of β-synuclein in lysosomes [189]. Citronellol's impact was also proven by J. Shao et al., who came to the conclusion that it may prevent apoptosis in PD by inhibiting the BCL-2/BAX pathway [190].
10.2. Breast Cancer
Breast cancer is the most prevalent cancer and the main cause of cancer-related mortality in women [191]. It may be inherited or linked to mutations in genes (BRCA1 and BRCA2, PTEN, TP53) involved in cellular proliferation, apoptosis, or other mitosis-related processes [192] [193] [194]. Apoptosis is particularly important for the healthy growth of the breast and the maintenance of homeostasis in the breast epithelial cell [195]. Disruptions in the apoptosis mechanism are strongly linked to the development of breast cancer [196].
The BCL-2 gene, which plays a role in the process of cell death, has been demonstrated to have a tight link to both the formation of breast cancer and the multiplication of breast cancer cells [196]. Furthermore, the expression of BCL-2 has been proven to be a feature that is related with a better prognosis in breast cancer patients [197]. In addition to preventing cells from committing suicide, the BCL-2 gene maintains the cells' ability to cycle for an extended period of time, which inhibits the progression of tumors. As a result, reduced BCL-2 expression may contribute to the development of breast cancer [198].
Usnic acid, a dibenzofuran derivative found in lichens, is employed as chemotherapeutic agent in treating the breast cancer as it has the ability to suppress cell growth and trigger cell death [199] [200]. It controls the up-regulation of miR-185-5p, which directly targets the 3'-untranslated region of BCL-2 and inhibits cellular growth by triggering apoptosis [201] [202]. In addition to lowering levels of the anti-apoptotic BCL-2, this agent also raises levels of the BAX, BOK protein [203].
In triple negative breast cancer cell (TNBC), Loperamide (LPR) is used as chemotherapeutic compound with doxorubicin (DXR) to sensitize the TNBC cell to DXR. The DXR and LPR combination decrease the BCL-2 and mTOR gene level and elevate the expression of the cell cycle inhibitor gene p21 [204] [205]. S.E. Simon et al. identified that SMS-IV-20 and SMS-IV-40, two bioactive phenylquinazoline derivatives, specifically target the pro-survival BCL-2 family members and promote apoptosis in MCF-7 and MCF-7-CR cells. These molecules induce the downregulation of of BCL-2, BCL-XL and simultaneously stimulate the release of mitochondrial cytochrome C that leads to the apoptosis of the cancer cell [206].
10.3. Lung cancer
Lung carcinoma has evolved over the past century from a rare and obscure condition to the most frequent cancer worldwide and the leading cause of cancer-related death[207]. Lung cancer with small cell content (SCLC) is the most dangerous type. A highly complex condition at the molecular level, with numerous mutations present in each tumor, when paired with frequent TP53 mutation [208]. Although new methods of diagnosis, chemotherapeutic agents, and therapeutic strategies have been developed for the treatment of cancer, the disease continues to present a significant challenge due to the development of drug resistance, the spread of metastases, and the low rates of long-term survival [209].
Apoptosis in lungs is strongly linked with both Small-Cell Lung Cancer (NSCLC) and Non-Small-Cell Lung Cancer (NSCLC) [210] [211]. The extrinsic and intrinsic pathways of apoptosis can be distinguished in lung carcinoma. Each pathway will eventually activate cell death proteases, which will then set off a cascade of proteolysis including effector caspases to carry out the apoptotic process's completion [212]. This process is regulated by the family members of BCL-2 and abnormalities BCL-2 family protein are reported in NSCLC, where overexpression of the anti-apoptotic BCL-2 and MCL-1 proteins is related with current clinical resistance [210] [213]. Antiapoptotic (BCL-2) and proapoptotic (BAX) proteins in healthy human airway epithelial cells and lung cancer cells are impacted by ATP-induced Ca2+ signaling[214]. Caspase-dependent and Caspase-independent signaling pathways that rely on mitochondria can be used to categorise the course of apoptosis. Cellular apoptosis was assisted by the caspase-dependent signaling pathway, which is linked to a balance between pro- and anti-apoptotic molecules (BAX, Bid) [215].
Long non-coding RNAs (lncRNAs) are being studied as a novel class of regulators of biological processes, including cellular proliferation, apoptosis, and carcinogenesis in lungs [216]. Reduced levels of miR-181 were linked to higher levels of BCL-2, and it was further hypothesized that miR-181 largely targets BCL-2 expression when conducting out its proapoptotic function in non small cell lung cancer [217].
Venetoclax, an anti-apoptotic BCL-2 inhibitor, induce BIM dependent apoptosis in Small-Cell Lung Cancers and for this inhibitory function it is used as a chemotherapeutic agent in lung cancer treatment [218].
Nur77, which is also known as TR3/NGFIB and is a member of the nuclear receptor superfamily, is utilized as a therapeutic target in the treatment of cancer due to its close association with both cell proliferation and apoptosis. [219]. It induces apoptosis through migrating from the nucleus to the mitochondria after which the remaining steps of the apoptosis mechanism are carried out. NuBCP-9, which simulates the functional and mechanistic actions of Nur77, convert the antiapoptotic BCL-2 to proapoptotic state and may serves as therapeutic target in the treatment of lung cancer patients [220].
ECPU-0001, a Carbazole-Piperazine Hybrid, greatly prevent the growth of lung tumors in a xenograft model without causing severe toxicity. In addition to this, it has been demonstrated to drastically reduce both the volume and burden of tumors. This chemical target the BCL-2 family of proteins and regulates the mitochondria-mediated apoptosis, which results in the over-expression of pro-apoptotic BAK and the down-expression of antiapoptotic mcl1. This event leads to the release of cyt c, apoptosome formation, caspase3,9 activation and finally cellular deaths [221].
Receptor tyrosine kinase-like orphan receptor 1 (ROR1), an oncofetal protein emerging as a therapeutic target that coexpresses with BCL-2 in various tumor types due to microRNA coregulation. Therapeutic BCL-2 inhibition works well with ROR1-targeted therapy to treat small-cell lung cancer effectively. ROR1 expression in SCLC patient samples and its potential for synergy with BCL-2 inhibition in SCLC cell lines were assessed [209].
BKA-o73 is a small molecule BAK activator with the highest affinity for the BH3 domain. Revealing that BAK protein is a desirable target for lung cancer therapy, increased BAK expression was associated with a poor prognosis in NSCLC patients. BAK binding to BKA-073 stimulates BAK oligomerization and mitochondrial priming, which activates BAK's proapoptotic activity. Venetoclax, a BCL-2 inhibitor, and BKA-073 work well together to inhibit lung cancer in vivo [222].
10.4. Multiple sclerosis (MS)
Multiple sclerosis (MS) is a central nervous system (CNS) inflammatory disease that causes demyelination and neurodegeneration [223] [224]. MS is a complex gene-environment interaction-driven disorder that is diverse, multifactorial, and immune-mediated [224]. MS, which now ranks as the most prevalent cause of neurological impairment in young adults, affects more than 2.5 million individuals globally. Immunoregulatory abnormalities, including loss of tolerance and a decline in the number of regulatory T cells, environmental variables, immunogenetic vulnerability, and environmental factors have all been connected to the etiology of MS [225]. Demyelinating lesions, which are seen in both the white and grey matter of the brain and spinal cord, are a pathological hallmark of multiple sclerosis [224]. Although the exact cause(s) of MS are unknown, a multifactorial model has been developed to explain its pathogenesis. This model is used to analyze the interactions between genetic, epigenetic, infectious, nutritional, climatic, and other environmental influences such as Epstein-Barr virus (EBV) infection, sun exposure, and smoking [226].
Degeneration of neurons and axonal myelin sheath degradation are hallmarks of multiple sclerosis (MS) [227]. In multiple sclerosis (MS), the significance of apoptosis is complicated by its two opposing effects. It causes the death of oligodendrocytes and neurons in the CNS, and it also eliminates autoreactive immune cells to put a stop to immunological reactions and maintain immune system homeostasis [228]. Apoptosis-inducing factor, which may cause DNA damage and apoptosis, is released from mitochondrial storage and translocated to the nucleus in multiple sclerosis lesions, demonstrating mitochondrial injury. This may explain why afflicted cells have an accumulation of fragmented DNA in their nucleus [229].
The etiology of Multiple Sclerosis(MS) may be influenced by defective apoptosis. Using BEAM or cyclophosphamide (CY)-based conditioning regimens, the authors assessed apoptosis-related markers in MS patients before and after autologous hematopoietic stem cell transplantation (AHSCT). When compared to their healthy counterparts, MS patients expressed more of the A1 gene and had lower levels of the proapoptotic genes BAD, BAX, and FASL. After AHSCT, the BAK, BIK, BIMEL, FAS, FASL, A1, BCL-2, BCLXL, CFLIPL, and CIAP2 genes were elevated in the BEAM-group. At D+720 post-transplantation, all genes, with the exception of BIK, BIMEL, and A1, attained levels comparable to controls. After transplanting, the CYgroup showed elevated BAX, BCLW, CFLIPL, and CIAP1 gene expressions and reduced BIK and BID gene expressions. increased BCL-2 expression prior to transplant. The early therapeutic benefits of AHSCT in MS patients are linked to normalization of molecules related to apoptosis [225].
Autophagy is the breakdown and recycling of dysfunctional cytoplasmic organelles, macromolecular aggregates, and long-lived proteins [230]. Autophagy is recognized to be involved in various neurodegenerative diseases and to be crucial for maintaining neuronal homeostasis. It is yet unknown, nevertheless, whether autophagy contributes to the processes of MS-related neuronal damage. Myelin oligodendrocyte glycoprotein p35-55 vaccination was used to generate experimental autoimmune encephalomyelitis (EAE), an in vivo model of MS, in female C57BL/6 mice. An autophagy-inducing drug called rapamycin reduced the aberrant clinical score, demyelination, inflammation, and neuronal damage brought on by EAE. Rapamycin reduced the BAX/BCL-2 ratio, inhibited caspase-3 production, and prevented cell death. contributed to the neuronal damage brought on by EAE, and pharmacological autophagy regulation may be a therapeutic approach for MS [231].
Solanesol (SNL) is a strong antioxidant and neuroprotective phytoconstituent that was successfully extracted from the Nicotiana tobaco plant. It is a member of the Solanaceae family and is a substantial biosynthetic precursor to CoQ10. Inducing MS-like neurobehavioral changes in Wistar rats was the neuroprotective potential of SNL (40–80 mg/kg) in intracerebropeduncular (ICP) ethidium bromide (EB). SNL therapy for a long period of time has neuroprotective potential by enhancing cognition, memory, grip strength, and motor function. SNL alters the amount of apoptotic markers (caspase-3, BAX, and BCL-2) in the rat brain along with mitochondrial ETC complex malfunction. SNL also reduced inflammatory cytokines (TNF-, IL-1), oxidative stress indicators, and restored levels of neurotransmitters. By reversing obvious pathogenic changes and lowering the amount of demyelination in rats, SNL has a neuroprotective impact [232].
10.5. Leukemia
Leukemias are a group of malignant disorders that present with increased numbers of leucocytes in the blood and bone marrow [233]. It can be categorized into several class including 1) Acute lymphoblastic leukemia (ALL); Acute Myeloblastic leukemia (AML); Chronic lymphocytic leukemia (CLL); and Chronic myeloid leukemia (CML) [233]. The BCL-2 family proteins play marked role in the formation and treatment of leukemia. BCL-2 is essential to the survival of AML cells; hence, blocking BCL-2 activity causes these cells to die [234]. Overexpression of anti-apoptotic BCL-2 proteins, such as BCL-2, BCL-Xl, and mcl-1, is linked with a poor prognosis and resistance to treatment in patients with acute myeloid leukemia (AML). So, the inhibitor of anti-apoptotic BCL-2 is targeted as therapeutic agent for leukemia patient [235].
The significance of microRNAs, also known as miRNAs, is demonstrated in a number of malignancies, including acute myeloid leukemia (AML). It was discovered that the levels of miR-182-5p expression were greater in AML tissues than it was in their normal controls. One example is that MiR-182-5p can operate as an oncogene or as a possible suppressive miRNA in malignancies. The authors showed that elevated levels of miR-182-5p in AML patients decrease cell apoptosis while simultaneously promoting cell proliferation. This is accomplished through miR-182-targeting 5p's of BCL-2L12 and BCL-2. Apoptosis was induced in AML cells through the targeting of BCL-2 or BCL-2L12 when miR-182-5p was inhibited and for this reason this miRNA may serve as therapeutic agent [236].
Venetoclax, a selective small-molecule inhibitor of BCL-2, induces apoptosis independently of p53 in chronic lymphocytic leukemia (CLL) cells. In vitro treatment of BCL-2-dependent cell lines induces rapid apoptosis, as demonstrated by detecting critical apoptotic markers such as cytochrome-c release and caspase activation. Since it has been demonstrated that CLL cells are predominantly dependent on BCL-2, this makes them obvious candidates for treatment with venetoclax [237].
10.6. Diabetes
Diabetes, which hampers the body's capability to convert food into energy, affects more than 300 million people around the world [238] [239]. The disease can be divided into two categories: Type 1 diabetes mellitus (T1DM) is an autoimmune disorder characterized by the destruction of beta cells in the pancreas. Type 2 diabetes mellitus (T2DM) occurs when the body's cells produce too little insulin to meet the body's growing needs. Numerous factors contribute to the condition, including genetics, lifestyle variables (high calorie diet, lack of exercise), and obesity [240].
It has been shown that diabetes is linked to programmed cell death, often known as apoptosis, and that apoptosis of beta cells may be the primary cause of the relative insulin deficit in DM [241]. Both type 1 and type 2 diabetes, as well as islet loss after transplantation, are characterized by the apoptosis of beta cells [242]. Type 2 diabetes mellitus (T2DM) has a complex etiology that includes obesity-related insulin resistance, poor insulin production, and loss of β-cell mass due to β-cell death [243]. The mitochondrial mechanism of β-cell apoptosis mediated by pro-inflammatory cytokines or lipotoxicity is tightly regulated by the BCL-2 family of proteins [239]. Placentas with both preeclampsia and gestational diabetes have a favorable response to the BAX protein. Sgarbosa et al. investigated BCL-2 expression in normoglycemic and diabetic placentas and found that hyperglycemic placentas had lower levels of the protein, which might indicate diseases that cause apoptosis [244].
Table 3.
Therapeutic agents/drugs and mechanism targeting BCL-2 family proteins.

MiRNAs have been identified as essential regulators of beta cell growth, glucose-stimulated insulin secretion (GSIS), and beta cell malfunction in several investigations [245][250][251]. As a biomarker for endothelial dysfunction in T2D and coronary artery disorders, miR-126 has been suggested. By directly lowering the expression of insulin receptor substrate1 (IRS1), miR-96 and miR-126 play a critical role in insulin resistance in the liver and hepatocytes [240]. miR-21 could be an important therapeutic target as inhibition of miR-21 increased levels of BCL-2 protein and decreased levels of cleaved caspase 3, whereas overexpression of miR-21 decreased levels of BCL-2 transcript and protein synthesis. It can degrade BCL-2 transcripts and prevents BCL-2 translation which leads to the apoptosis of beta cell [245].
Geraniol, a natural acyclic monoterpenoid, has several pharmacological activities. E. F. El Azab et al. found that it can improves HFD/STZ-induced type 2 diabetes mellitus in rats through dependently upregulating BCL-2 and downregulating Casp3 and BAX [246]. Piperine, produced from black pepper (Piper nigrum), is a significant plant alkaloid (Piper longum), has the ability to inhibit pancreatic β-cell apoptosis through decreasing BAX/BCL-2 ratio. It also decrease cytochrome c release. Thus, piperine improves pancreatic β-cell dysfunction in diabetes [247]. Another important therapeutic agent is Wogonin, a flavonoid from Scutellaria baicalensis Georgi root, which is discovered by X. qi Liu et al. The authors concluded that this compound reduce podocyte apoptosis by targetting BCL-2 protein henece it could be be a promising treatment for Diabetec Kidney Disease [248].
Melatonin is an antioxidant and free radical scavenger which is strongly connected with apoptosis. It is recognized as an important therapeutic target in diabetes patients because it prevents cardiac apoptosis by elevating the production of anti-apoptotic BCL-2 protein and blocking CD95 and caspases 9, 8, and 3 [249].
Conclusion
The present study describes BCL-2 family members and their apoptotic roles. BCL-2 family proteins consist of members that either activate or block apoptotic pathways. There are three types of BCL-2 protein family members. They are pro-apoptotic BCL-2 proteins, anti-apoptotic BCL-2 proteins, and BH3-only proteins. All these types have different biochemical roles. BH3-only proteins transduce signals to primary BCL-2 family members to induce apoptosis. They activate pro-apoptotic BCL-2 proteins such as BAX and BAK, which cause conformational changes and permeabilize the outer mitochondrial membrane. The anti-apoptotic BCL-2 protein inhibits MOMP and blocks the mitochondrial pathway of apoptosis. Apoptosis can occur through two pathways: the intrinsic and extrinsic pathways. A different pathway called the perforin/granzyme pathway has been reported. Through these pathways, the BCL-2 protein family regulates apoptosis in organisms. The BCL-2 protein family also has many therapeutic potentials. It plays a vital role in modulating the apoptotic response in anticancer therapy.
Multiple studies have shown that the BCL-2 family influences many other cellular processes in addition to apoptosis. These processes are generally independent of apoptosis regulation, suggesting that BCL-2 may have additional roles. Cancer, neurodegenerative disorders, ischemia, and autoimmune diseases are some conditions linked to abnormalities in the BCL-2 protein family's function. So, the BCL-2 family of proteins is the essential regulator of apoptosis [1]. The BCL-2 family of proteins has been shown to play a significant role in tumorigenesis, tumor maintenance, and the response of cancers to both traditional chemotherapies and targeted therapies [170]. BCL-2 anti-apoptotic protein function in solid tumor progression. Therefore, using of BCL-2 protein-targeting drugs as single agents or in combination with current standard-of-care therapies may offer a real opportunity to defeat therapy-resistant solid tumors and prolong the disease-free time in cancer patients [21]. Besides, the interactions between BH3-only proteins and anti-apoptotic BCL-2 family members can eliminate cancer cells by inhibiting all or some anti-apoptotic BCL-2 proteins concurrently to induce cell death [50]. Also, understanding BH3-only proteins’ role in disease has led to a new generation of cancer treatments, such as BH3 mimetics [89]. BH3-mimetic drugs specifically inhibit anti-apoptotic BCL-2 family members. Cancer, autoimmune, and infectious diseases can all be treated more effectively with BH3-mimetic drugs [33].
The BCL-2 family protein is already implicated in numerous beneficial biochemical and therapeutic processes. It also has tremendous potential for future development. Targeting the BCL-2 protein family offers crucial directions for the future of chemical and drug discovery. BH3 mimetics are currently in a mature stage of development to treat malignancies and possibly autoimmune illnesses [180]. There exist knowledge gaps concerning the anti-apoptotic BCL-2 family isoforms. To fully realize the potential of this pathway, further studies are needed on the many splice variants and isoforms and their biological involvement in apoptosis [18]. More research is needed to confirm that BCL-2 inhibitors can be used to treat chemotherapy-sensitive and chemotherapy-resistant cancers [108]. Additional research is needed to find drug-like inhibitors of BCL-XL and MCL-1, which will hopefully lead to better treatments [23]. In the near future, anti-apoptotic BCL-2 family members will be successfully targeted to improve therapy responses and patient survival for malignant diseases other than hematologic ones [21].
Author Contributions
SKP, MS, and MMUH conceptualized the framework and prepared the draft manuscript. SKP, MS, MAH, MAF, AM, SI and BP wrote the initial draft of the manuscript. MMUH was in charge of content supervision and was responsible for revising the final version of the article. All authors reviewed and approved the draft manuscript as the final version.
Funding
No fund was available for this project.
Data Availability Statement
Data will be made available on request.
Acknowledgment
Not Applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Additional Information
No additional information is available for this projecty.
Ethical Approval
This article does not contain any studies with human participants performed by any of the authors.
Consent for Publication
Not Applicable.
Abbreviations
| BCL-2 | B Cell Lymphoma Protein 2 |
| BCL-X | BCL-2-Like 1 |
| BCL-XL | B-Cell Lymphoma-Extra Large |
| BCL-XS | B-Cell Lymphoma-Extra Short |
| BCL-W | BCL-2-Like 2 Protein |
| BAG | BCL-2-Associated Athanogene |
| BCL-10 | B Cell Lymphoma Protein 10 |
| BAX | BCL-2-Associated X Protein |
| BAK | BCL-2 Antagonist Killer 1 |
| BID | BH3-Interacting Domain |
| BAD | BCL-2 Antagonist of Cell Death |
| BIM | BCL-2-Interacting Protein |
| BFL1 | BCL2-Related Protein A1 |
| BIK | BCL-2-Interacting Killer |
| BLK | BIK-Like Killer Protein |
| BH | BCL2 Homology Region |
| PUMA | P53 Upregulated Modulator of Apoptosis |
| BBC3 | BCL2 Binding Component 3 |
| APAF-1 | Apoptosis Protease-Activating Factor-1 |
| IAP | Inhibitor of Apoptosis Proteins |
| AIF | Apoptosis-Inducing Factor |
| CAD | Caspase-Activated Dnase |
| DR3 | Death Receptor 3 |
| DR6 | Death Receptor 6 |
| MOM | Mitochondrial Outer Membrane |
| MOMP | MOM Permeabilization |
| SMAC | Second Mitochondria-Derived Activator of Caspase |
| DISC | Death-Inducing Signaling Complex |
| TNFR | Tumor Necrosis Factor Receptor |
| TNFR1 | Tumor Necrosis Factor Receptor 1 |
| TRAILR | TNF-Related Apoptosis-Inducing Ligand Receptor |
| HRK | Harakiri |
| MCL1 | Myeloid Cell Leukemia 1 |
| MCL1L | MCL1 Long Form |
| MCL1S | MCL1 Short Form |
| BMF | BCL2-Modifying Factor |
| BNIP | BCL2 and the Nineteen Kda Interacting Protein |
| AML | Acute Myeloid Leukemia |
| BOO | BCL2 Homologue of the Ovary |
| CLL | Chronic Lymphocytic Leukemia |
| AIF | Apoptosis-Inducing Factor |
| IL | Interleukin |
| NMR | Nuclear Magnetic Resonance |
| TM | Transmembrane |
| PEST | Sequence Enriched in Proline/Glutamic Acid/Serine/Threonine |
| STAT3 | Signal Transducers and Activators of Transcription |
| HCC | Hepatocellular Carcinoma |
| DLBCL | Diffuse Large B-Cell Lymphoma |
| XIAP | X-linked Inhibitor of Apoptosis Protein |
| FADD | Fas-Associated Death Domain |
| DISC | Death-Inducing Signaling Complex |
| tBAD | Truncated BAD |
| BCC | Basal Cell Carcinoma |
| CLL | Chronic Lymphocytic Leukemia |
| AHSCT | Autologous Hematopoietic Stem Cell Transplantation |
| EAE | Autoimmune Encephalomyelitis |
| ALL | Acute Lymphoblastic Leukemia |
| AML | Acute Myeloblastic Leukemia |
| CLL | Chronic Lymphocytic Leukemia |
| CML | Chronic Myeloid Leukemia |
| CLL | Chronic Lymphocytic Leukemia |
| T1DM | Type 1 Diabetes Mellitus |
| T2DM | Type 2 Diabetes Mellitus |
References
- W. A. Siddiqui, A. Ahad, and H. Ahsan, “The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update,” Archives of Toxicology, vol. 89, no. 3. Springer Verlag, pp. 289–317, Feb. 2015. [CrossRef]
- K. Labbé and M. Saleh, “Cell death in the host response to infection,” Cell Death Differ., vol. 15, no. 9, pp. 1339–1349, 2008. [CrossRef]
- S. Arandjelovic and K. S. Ravichandran, “Phagocytosis of apoptotic cells in homeostasis,” Nat. Immunol., vol. 16, no. 9, pp. 907–917, 2015. [CrossRef]
- I. K. H. Poon, C. D. Lucas, A. G. Rossi, and K. S. Ravichandran, “Apoptotic cell clearance: basic biology and therapeutic potential,” Nat. Rev. Immunol., vol. 14, no. 3, pp. 166–180, Mar. 2014. [CrossRef]
- S. Goldar, M. S. Khaniani, S. M. Derakhshan, and B. Baradaran, “Molecular mechanisms of apoptosis and roles in cancer development and treatment,” Asian Pacific J. Cancer Prev., vol. 16, no. 6, pp. 2129–2144, 2015. [CrossRef]
- A. Hazafa et al., “Humanin: A mitochondrial-derived peptide in the treatment of apoptosis-related diseases,” Life Sci., vol. 264, p. 118679, 2021. [CrossRef]
- J. A. Glab, Z. Cao, and H. Puthalakath, Bcl-2 family proteins, beyond the veil, 1st ed., vol. 351. Elsevier Inc., 2020. [CrossRef]
- S. Fulda, “Targeting apoptosis for anticancer therapy,” Semin. Cancer Biol., vol. 31, pp. 84–88, 2015. [CrossRef]
- H. Walczak, “Death receptor-ligand systems in cancer, cell death, and inflammation,” Cold Spring Harb. Perspect. Biol., vol. 5, no. 5, 2013. [CrossRef]
- M. E. Guicciardi and G. J. Gores, “Life and death by death receptors,” FASEB J., vol. 23, no. 6, pp. 1625–1637, 2009. [CrossRef]
- D. R. Green, “The Mitochondrial Pathway of Apoptosis Part II: The BCL-2 Protein Family,” Cold Spring Harb. Perspect. Biol., vol. 14, no. 6, p. a041046, Jun. 2022. [CrossRef]
- X. Luo, K. L. O’Neill, and K. Huang, “The third model of Bax/Bak activation: a Bcl-2 family feud finally resolved?,” F1000Research, vol. 9, 2020. [CrossRef]
- H. M. Al-Aamri, H. R. Irving, C. Bradley, and T. Meehan-Andrews, “Intrinsic and extrinsic apoptosis responses in leukaemia cells following daunorubicin treatment,” BMC Cancer, vol. 21, no. 1, pp. 1–10, 2021. [CrossRef]
- I. Kapoor, J. Bodo, B. T. Hill, E. D. Hsi, and A. Almasan, “Targeting BCL-2 in B-cell malignancies and overcoming therapeutic resistance,” Cell Death Dis., vol. 11, no. 11, 2020. [CrossRef]
- J. T. Opferman and A. Kothari, “Anti-apoptotic BCL-2 family members in development,” Cell Death Differ. 2018 251, vol. 25, no. 1, pp. 37–45, Nov. 2017. [CrossRef]
- B. Antonsson and J. Martinou, “MINIREVIEW The Bcl-2 Protein Family,” vol. 57, p. 4839, 2000. [CrossRef]
- C. M. Croce and J. C. Reed, “Finally, an apoptosis-targeting therapeutic for cancer,” Cancer Res., vol. 76, no. 20, pp. 5914–5920, 2016. [CrossRef]
- C. F. A. Warren, M. W. Wong-Brown, and N. A. Bowden, “BCL-2 family isoforms in apoptosis and cancer,” Cell Death and Disease, vol. 10, no. 3. Nature Publishing Group, Mar. 2019. [CrossRef]
- J. Kale, E. J. Osterlund, and D. W. Andrews, “BCL-2 family proteins: Changing partners in the dance towards death,” Cell Death and Differentiation, vol. 25, no. 1. Nature Publishing Group, pp. 65–80, 2018. [CrossRef]
- E. L.Omonosova and G. C.Hinnadurai, “BH3-only proteins in apoptosis and beyond: An overview,” Oncogene, vol. 27, no. S1, pp. S2–S19, 2008. [CrossRef]
- S. D’Aguanno and D. Del Bufalo, “Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer,” Cells, vol. 9, no. 5. NLM (Medline), May 2020. [CrossRef]
- D. M. Jang, E. K. Oh, H. Hahn, H. S. Kim, and B. W. Han, “Structural insights into apoptotic regulation of human Bfk as a novel Bcl-2 family member,” Comput. Struct. Biotechnol. J., vol. 20, pp. 745–756, 2022. [CrossRef]
- H. Dai, W. Meng, and S. Kaufmann, “BCL2 Family, Mitochondrial Apoptosis, and Beyond,” Cancer Transl. Med., vol. 2, no. 1, p. 7, 2016. [CrossRef]
- A. Strasser, “The role of BH3-only proteins in the immune system,” Nat. Rev. Immunol., vol. 5, no. 3, pp. 189–200, 2005. [CrossRef]
- M. R. Schnorenberg, J. A. Bellairs, R. Samaeekia, H. Acar, M. V. Tirrell, and J. L. LaBelle, “Activating the intrinsic pathway of apoptosis using BIM BH3 peptides delivered by peptide amphiphiles with endosomal release,” Materials (Basel)., vol. 12, no. 16, 2019. [CrossRef]
- H. C. Chen et al., “An interconnected hierarchical model of cell death regulation by the BCL-2 family,” Nat. Cell Biol., vol. 17, no. 10, pp. 1270–1281, 2015. [CrossRef]
- A. Hantusch, M. Rehm, and T. Brunner, “Counting on Death – Quantitative aspects of Bcl-2 family regulation,” FEBS J., vol. 285, no. 22, pp. 4124–4138, 2018. [CrossRef]
- J. M. Adams, “BAX and BAK become killers without a BH3 trigger,” Cell Res., vol. 29, no. 12, pp. 967–968, 2019. [CrossRef]
- E. C. Donaldson, “Chapter 1 股関節 概念 Chapter 1 股関節,” An Autom. Irrig. Syst. Using Arduino Microcontroller, vol. 1908, no. January, pp. 2–6, 2011. [CrossRef]
- A. Ashkenazi, W. J. Fairbrother, J. D. Leverson, and A. J. Souers, “From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors,” Nature Reviews Drug Discovery, vol. 16, no. 4. Nature Publishing Group, pp. 273–284, Apr. 2017. [CrossRef]
- R. J. Youle and A. Strasser, “The BCL-2 protein family: Opposing activities that mediate cell death,” Nature Reviews Molecular Cell Biology, vol. 9, no. 1. pp. 47–59, Jan. 2008. [CrossRef]
- S. Choudhury, “A comparative analysis of BCL-2 family,” Bioinformation, vol. 15, no. 4, pp. 299–306, 2019. [CrossRef]
- A. R. D. Delbridge, S. Grabow, A. Strasser, and D. L. Vaux, “Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies,” Nature Reviews Cancer, vol. 16, no. 2. Nature Publishing Group, pp. 99–109, Feb. 2016. [CrossRef]
- H. Dai, W. Meng, and S. Kaufmann, “BCL2 Family, Mitochondrial Apoptosis, and Beyond,” undefined, vol. 2, no. 1, p. 7, 2016. [CrossRef]
- S. Thomas et al., “Targeting the Bcl-2 family for cancer therapy,” Expert Opinion on Therapeutic Targets, vol. 17, no. 1. pp. 61–75, Jan. 2013. [CrossRef]
- M. L. Hartman and M. Czyz, “BCL-w: apoptotic and non-apoptotic role in health and disease,” Cell Death and Disease, vol. 11, no. 4. Springer Nature, Apr. 2020. [CrossRef]
- C. Akgul, “Mcl-1 is a potential therapeutic target in multiple types of cancer,” Cellular and Molecular Life Sciences, vol. 66, no. 8. pp. 1326–1336, Apr. 2009. [CrossRef]
- D. Bhatt et al., “BCL-2 (-938C>A), BAX (-248G>A), and HER2 Ile655Val Polymorphisms and Breast Cancer Risk in Indian Population,” J. Oncol., vol. 2021, 2021. [CrossRef]
- H. Thomadaki and A. Scorilas, “BCL2 family of apoptosis-related genes: Functions and clinical implications in cancer,” Critical Reviews in Clinical Laboratory Sciences, vol. 43, no. 1. pp. 1–67, Jan. 2006. [CrossRef]
- S. Sethi, M. S. Benninger, M. Lu, S. Havard, and M. J. Worsham, “Noninvasive Molecular Detection of Head and Neck Squamous Cell Carcinoma An Exploratory Analysis,” 2009.
- S. Verma, M. L. Budarf, B. S. Emanuel, and G. Chinnadurai, “Structural analysis of the human pro-apoptotic gene Bik: Chromosomal localization, genomic organization and localization of promoter sequences,” 2000.
- Y. Sun and D. W. Leaman, “Involvement of noxa in cellular apoptotic responses to interferon, double-stranded RNA, and virus infection,” J. Biol. Chem., vol. 280, no. 16, pp. 15561–15568, Apr. 2005. [CrossRef]
- M. Vogler, “BCL2A1: The underdog in the BCL2 family,” Cell Death and Differentiation, vol. 19, no. 1. pp. 67–74, Jan. 2012. [CrossRef]
- F. K. Ahmad, S. Deris, and H. Othman, “Improved Bayesian Network Structure Learning for Breast Cancer Prognosis,” 2010.
- H. Puthalakath et al., “Bmf: A Proapoptotic BH3-Only Protein Regulated by Interaction with the Myosin V Actin Motor Complex, Activated by Anoikis.”.
- A. M. Petros, E. T. Olejniczak, and S. W. Fesik, “Structural biology of the Bcl-2 family of proteins,” Biochimica et Biophysica Acta - Molecular Cell Research, vol. 1644, no. 2–3. pp. 83–94, Mar. 2004. [CrossRef]
- P. E. Czabotar, G. Lessene, A. Strasser, and J. M. Adams, “Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy,” Nature Reviews Molecular Cell Biology, vol. 15, no. 1. pp. 49–63, Jan. 2014. [CrossRef]
- M. Li, D. Wang, J. He, L. Chen, and H. Li, “Bcl-XL: A multifunctional anti-apoptotic protein,” Pharmacological Research, vol. 151. Academic Press, Jan. 2020. [CrossRef]
- K. Papadopoulos, “Targeting the Bcl-2 Family in Cancer Therapy,” Semin. Oncol., vol. 33, no. 4, pp. 449–456, Aug. 2006. [CrossRef]
- G. F. Perini, G. N. Ribeiro, J. V. Pinto Neto, L. T. Campos, and N. Hamerschlak, “BCL-2 as therapeutic target for hematological malignancies,” J. Hematol. Oncol., vol. 11, no. 1, pp. 1–15, 2018. [CrossRef]
- J. T. Opferman and A. Kothari, “Anti-apoptotic BCL-2 family members in development,” Cell Death and Differentiation, vol. 25, no. 1. Nature Publishing Group, pp. 37–45, 2018. [CrossRef]
- B. Leibowitz and J. Yu, “Mitochondrial signaling in cell death via the Bcl-2 family,” Cancer Biology and Therapy, vol. 9, no. 6. Landes Bioscience, pp. 417–422, Mar. 2010. [CrossRef]
- A. Shamas-Din, J. Kale, B. Leber, and D. W. Andrews, “Mechanisms of action of Bcl-2 family proteins,” Cold Spring Harb. Perspect. Biol., vol. 5, no. 4, pp. 1–21, Apr. 2013. [CrossRef]
- F. Zhou, Y. Yang, and D. Xing, “Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis,” FEBS J., vol. 278, no. 3, pp. 403–413, Feb. 2011. [CrossRef]
- A. Bolomsky et al., “MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents,” Journal of Hematology and Oncology, vol. 13, no. 1. BioMed Central Ltd, Dec. 2020. [CrossRef]
- N. Popgeorgiev, L. Jabbour, and G. Gillet, “Subcellular localization and dynamics of the Bcl-2 family of proteins,” Frontiers in Cell and Developmental Biology, vol. 6, no. FEB. Frontiers Media S.A., Feb. 2018. [CrossRef]
- J. Lindsay, M. D. Esposti, and A. P. Gilmore, “Bcl-2 proteins and mitochondria-Specificity in membrane targeting for death,” Biochimica et Biophysica Acta - Molecular Cell Research, vol. 1813, no. 4. pp. 532–539, Apr. 2011. [CrossRef]
- N. Echeverry, D. Bachmann, F. Ke, A. Strasser, H. U. Simon, and T. Kaufmann, “Intracellular localization of the BCL-2 family member BOK and functional implications,” Cell Death Differ., vol. 20, no. 6, pp. 785–799, Jun. 2013. [CrossRef]
- A. C. Craik et al., “The BH3-only protein bad confers breast cancer taxane sensitivity through a nonapoptotic mechanism,” Oncogene, vol. 29, no. 39, pp. 5381–5391, Sep. 2010. [CrossRef]
- M. Miani, B. Elvira, and E. N. Gurzov, “Sweet Killing in Obesity and Diabetes: The Metabolic Role of the BH3-only Protein BIM,” Journal of Molecular Biology, vol. 430, no. 18. Academic Press, pp. 3041–3050, Sep. 2018. [CrossRef]
- P. Hikisz and Z. M. Kiliańska, “Puma, a critical mediator of cell death - one decade on from its discovery,” Cellular and Molecular Biology Letters, vol. 17, no. 4. BioMed Central Ltd., pp. 646–669, 2012. [CrossRef]
- J. Hatok and P. Racay, “Bcl-2 family proteins: Master regulators of cell survival,” Biomolecular Concepts, vol. 7, no. 4. Walter de Gruyter GmbH, pp. 259–270, Aug. 2016. [CrossRef]
- C. B. Karim et al., “Structural Mechanism for Regulation of Bcl-2 protein Noxa by phosphorylation,” Sci. Rep., vol. 5, Sep. 2015. [CrossRef]
- M. Fricker, J. O’Prey, A. M. Tolkovsky, and K. M. Ryan, “Phosphorylation of Puma modulates its apoptotic function by regulating protein stability,” Cell Death Dis., vol. 1, no. 7, Jul. 2010. [CrossRef]
- M. Hassan, H. Watari, A. Abualmaaty, Y. Ohba, and N. Sakuragi, “Apoptosis and molecular targeting therapy in cancer,” BioMed Research International, vol. 2014. Hindawi Limited, 2014. [CrossRef]
- C. M. Pfeffer and A. T. K. Singh, “Apoptosis: A target for anticancer therapy,” International Journal of Molecular Sciences, vol. 19, no. 2. MDPI AG, Feb. 2018. [CrossRef]
- R. Roufayel, “Regulation of stressed-induced cell death by the Bcl-2 family of apoptotic proteins,” Molecular Membrane Biology, vol. 33, no. 6–8. Taylor and Francis Ltd, pp. 89–99, Nov. 2016. [CrossRef]
- E. Khodapasand, N. Jafarzadeh, F. Farrokhi, B. Kamalidehghan, and M. Houshmand, “Is Bax/Bcl-2 ratio considered as a prognostic marker with age and tumor location in colorectal cancer?,” Iran. Biomed. J., vol. 19, no. 2, pp. 69–75, 2015. [CrossRef]
- A. R. D. Delbridge and A. Strasser, “The BCL-2 protein family, BH3-mimetics and cancer therapy,” Cell Death and Differentiation, vol. 22, no. 7. Nature Publishing Group, pp. 1071–1080, Jul. 2015. [CrossRef]
- R. W. Birkinshaw and P. E. Czabotar, “The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation,” Seminars in Cell and Developmental Biology, vol. 72. Elsevier Ltd, pp. 152–162, Dec. 2017. [CrossRef]
- R. Valentin, S. Grabow, and M. S. Davids, “The rise of apoptosis: targeting apoptosis in hematologic malignancies,” 2018.
- N. Volkmann, F. M. Marassi, D. D. Newmeyer, and D. Hanein, “The rheostat in the membrane: BCL-2 family proteins and apoptosis,” Cell Death and Differentiation, vol. 21, no. 2. pp. 206–215, Feb. 2014. [CrossRef]
- S. Arulananda, E. F. Lee, W. D. Fairlie, and T. John, “The role of BCL-2 family proteins and therapeutic potential of BH3-mimetics in malignant pleural mesothelioma,” Expert Review of Anticancer Therapy, vol. 21, no. 4. Taylor and Francis Ltd., pp. 413–424, 2021. [CrossRef]
- X. Luo, K. L. O’Neill, and K. Huang, “The third model of Bax/Bak activation: A Bcl-2 family feud finally resolved?,” F1000Research, vol. 9. F1000 Research Ltd, 2020. [CrossRef]
- D. Westphal, R. M. Kluck, and G. Dewson, “Building blocks of the apoptotic pore: How Bax and Bak are activated and oligomerize during apoptosis,” Cell Death and Differentiation, vol. 21, no. 2. pp. 196–205, Feb. 2014. [CrossRef]
- K. Huang, by Kai Huang, R. Singh, K. Johnson, and T. Bessho, “Mechanism of Bax/Bak Activation in Apoptotic Signaling Mechanism of Bax/Bak Activation in Apoptotic Signaling MECHANISM OF BAX/BAK ACTIVATION IN APOPTOTIC SIGNALING.”.
- Z. Liu, Y. Ding, N. Ye, C. Wild, H. Chen, and J. Zhou, “Direct Activation of Bax Protein for Cancer Therapy,” Med. Res. Rev., vol. 36, no. 2, pp. 313–341, Mar. 2016. [CrossRef]
- K. Huang et al., “BH3-only proteins target BCL-xL/MCL-1, not BAX/BAK, to initiate apoptosis,” Cell Res., vol. 29, no. 11, pp. 942–952, Nov. 2019. [CrossRef]
- R. J. C. Gilbert, M. D. Serra, C. J. Froelich, M. I. Wallace, and G. Anderluh, “Membrane pore formation at protein-lipid interfaces,” Trends in Biochemical Sciences, vol. 39, no. 11. Elsevier Ltd, pp. 510–516, 2014. [CrossRef]
- L. A. Gilliesa, H. Du, B. Peters, C. M. Knudson, D. D. Newmeyer, and T. Kuwana, “Visual and functional demonstration of growing Bax-induced pores in mitochondrial outer membranes,” Mol. Biol. Cell, vol. 26, no. 2, pp. 339–349, Jan. 2015. [CrossRef]
- D. Westphal et al., “Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane,” Proc. Natl. Acad. Sci. U. S. A., vol. 111, no. 39, pp. E4076–E4085, Sep. 2014. [CrossRef]
- R. T. Uren, S. Iyer, and R. M. Kluck, “Pore formation by dimeric Bak and Bax: an unusual pore?,” Philos. Trans. R. Soc. B Biol. Sci., vol. 372, no. 1726, 2017. [CrossRef]
- K. Cosentino and A. J. García-Sáez, “Bax and Bak Pores: Are We Closing the Circle?,” Trends in Cell Biology, vol. 27, no. 4. Elsevier Ltd, pp. 266–275, Apr. 2017. [CrossRef]
- K. Cosentino and A. J. García-Sáez, “Mitochondrial alterations in apoptosis,” Chemistry and Physics of Lipids, vol. 181. Elsevier Ireland Ltd, pp. 62–75, 2014. [CrossRef]
- A. Peña-Blanco and A. J. García-Sáez, “Bax, Bak and beyond — mitochondrial performance in apoptosis,” FEBS Journal, vol. 285, no. 3. Blackwell Publishing Ltd, pp. 416–431, Feb. 2018. [CrossRef]
- S. Bleicken, G. Jeschke, C. Stegmueller, R. Salvador-Gallego, A. J. García-Sáez, and E. Bordignon, “Structural Model of Active Bax at the Membrane,” Mol. Cell, vol. 56, no. 4, pp. 496–505, Nov. 2014. [CrossRef]
- U. Ros and A. J. García-Sáez, “More Than a Pore: The Interplay of Pore-Forming Proteins and Lipid Membranes,” J. Membr. Biol., vol. 248, no. 3, pp. 545–561, Jun. 2015. [CrossRef]
- T. Kuehl and D. Lagares, “BH3 mimetics as anti-fibrotic therapy: Unleashing the mitochondrial pathway of apoptosis in myofibroblasts,” Matrix Biology, vol. 68–69. Elsevier B.V., pp. 94–105, Aug. 2018. [CrossRef]
- J. A. Glab, G. W. Mbogo, and H. Puthalakath, “BH3-Only Proteins in Health and Disease,” in International Review of Cell and Molecular Biology, vol. 328, Elsevier Inc., 2017, pp. 163–196. [CrossRef]
- K. L. O’neill, K. Huang, J. Zhang, Y. Chen, and X. Luo, “Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane,” Genes Dev., vol. 30, no. 8, pp. 973–988, Apr. 2016. [CrossRef]
- M. Doerflinger, J. A. Glab, and H. Puthalakath, “BH3-only proteins: A 20-year stock-take,” FEBS Journal, vol. 282, no. 6. Blackwell Publishing Ltd, pp. 1006–1016, 2015. [CrossRef]
- M. Jullien, P. Gomez-Bougie, D. Chiron, and C. Touzeau, “Restoring Apoptosis with BH3 Mimetics in Mature B-Cell Malignancies,” Cells, vol. 9, no. 3. NLM (Medline), Mar. 2020. [CrossRef]
- M. Kvansakul and M. G. Hinds, “The structural biology of bh3-only proteins,” in Methods in Enzymology, vol. 544, Academic Press Inc., 2014, pp. 49–74. [CrossRef]
- K. J. Hutt, “The role of BH3-only proteins in apoptosis within the ovary,” Reproduction, vol. 149, no. 2. BioScientifica Ltd., pp. R81–R89, Feb. 2015. [CrossRef]
- H. Dai et al., “Measurement of BH3-only protein tolerance,” Cell Death Differ., vol. 25, no. 2, pp. 282–293, 2018. [CrossRef]
- H. Dai, X. W. Meng, and S. H. Kaufmann, “Mitochondrial apoptosis and BH3 mimetics,” F1000Research, vol. 5. Faculty of 1000 Ltd, 2016. [CrossRef]
- J. H. Zheng, A. Viacava Follis, R. W. Kriwacki, and T. Moldoveanu, “Discoveries and controversies in BCL-2 protein-mediated apoptosis,” FEBS Journal. Blackwell Publishing Ltd, pp. 2690–2700, Jul. 2016. [CrossRef]
- C. Correia et al., “Emerging understanding of Bcl-2 biology: Implications for neoplastic progression and treatment,” Biochimica et Biophysica Acta - Molecular Cell Research, vol. 1853, no. 7. Elsevier, pp. 1658–1671, Jul. 2015. [CrossRef]
- T. Moldoveanu, A. V. Follis, R. W. Kriwacki, and D. R. Green, “Many players in BCL-2 family affairs,” Trends in Biochemical Sciences, vol. 39, no. 3. pp. 101–111, Mar. 2014. [CrossRef]
- J. T. Opferman, “Attacking cancer’s Achilles heel: antagonism of anti-apoptotic BCL-2 family members,” FEBS Journal. Blackwell Publishing Ltd, pp. 2661–2675, Jul. 2016. [CrossRef]
- F. Edlich, “BCL-2 proteins and apoptosis: Recent insights and unknowns,” Biochem. Biophys. Res. Commun., vol. 500, no. 1, pp. 26–34, May 2018. [CrossRef]
- A. Basu, “The interplay between apoptosis and cellular senescence: Bcl-2 family proteins as targets for cancer therapy,” Pharmacology and Therapeutics, vol. 230. Elsevier Inc., Feb. 2022. [CrossRef]
- U. Anilkumar and J. H. M. Prehn, “Anti-apoptotic BCL-2 family proteins in acute neural injury,” Frontiers in Cellular Neuroscience, vol. 8, no. SEP. Frontiers Research Foundation, Sep. 2014. [CrossRef]
- A. Slomp and V. Peperzak, “Role and regulation of pro-survival BCL-2 proteins in multiple myeloma,” Frontiers in Oncology, vol. 8, no. NOV. Frontiers Media S.A., 2018. [CrossRef]
- E. F. Lee and W. Douglas Fairlie, “The structural biology of Bcl-xL,” International Journal of Molecular Sciences, vol. 20, no. 9. MDPI AG, May 2019. [CrossRef]
- C. Borrás et al., “BCL-xL, a mitochondrial protein involved in successful aging: From C. elegans to human centenarians,” Int. J. Mol. Sci., vol. 21, no. 2, 2020. [CrossRef]
- A. V. Follis et al., “Regulation of apoptosis by an intrinsically disordered region of Bcl-xL,” Nat. Chem. Biol., vol. 14, no. 5, pp. 458–465, May 2018. [CrossRef]
- S. Maji et al., “Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer,” in Advances in Cancer Research, vol. 137, Academic Press Inc., 2018, pp. 37–75. [CrossRef]
- V. V. Senichkin, A. Y. Streletskaia, A. S. Gorbunova, B. Zhivotovsky, and G. S. Kopeina, “Saga of Mcl-1: regulation from transcription to degradation,” Cell Death and Differentiation, vol. 27, no. 2. Springer Nature, pp. 405–419, Feb. 2020. [CrossRef]
- G. Morciano et al., “Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death,” Mol. Biol. Cell, vol. 27, no. 1, pp. 20–34, Jan. 2016. [CrossRef]
- H. Wang, M. Guo, H. Wei, and Y. Chen, “Targeting MCL-1 in cancer: current status and perspectives,” Journal of Hematology and Oncology, vol. 14, no. 1. BioMed Central Ltd, Dec. 2021. [CrossRef]
- R. Cherla, Y. Zhang, L. Ledbetter, and G. Zhang, “Coxiella burnetii inhibits neutrophil apoptosis by exploiting survival pathways and antiapoptotic protein Mcl-1,” Infect. Immun., vol. 86, no. 4, Apr. 2018. [CrossRef]
- A. R. Shenoy, S. Kirschnek, and G. Häcker, “IL-15 regulates Bcl-2 family members Bim and Mcl-1 through JAK/STAT and PI3K/AKT pathways in T cells,” Eur. J. Immunol., vol. 44, no. 8, pp. 2500–2507, 2014. [CrossRef]
- W. Xiang, C. Y. Yang, and L. Bai, “MCL-1 inhibition in cancer treatment,” OncoTargets and Therapy, vol. 11. Dove Medical Press Ltd., pp. 7301–7314, 2018. [CrossRef]
- Y. Fernández-Marrero, S. Spinner, T. Kaufmann, and P. J. Jost, “Survival control of malignant lymphocytes by anti-apoptotic MCL-1,” Leukemia, vol. 30, no. 11. Nature Publishing Group, pp. 2152–2159, Nov. 2016. [CrossRef]
- L. Chen and S. Fletcher, “Mcl-1 inhibitors: a patent review,” Expert Opinion on Therapeutic Patents, vol. 27, no. 2. Taylor and Francis Ltd, pp. 163–178, Feb. 2017. [CrossRef]
- E. P. Harvey, H. S. Seo, R. M. Guerra, G. H. Bird, S. Dhe-Paganon, and L. D. Walensky, “Crystal Structures of Anti-apoptotic BFL-1 and Its Complex with a Covalent Stapled Peptide Inhibitor,” Structure, vol. 26, no. 1, pp. 153-160.e4, 2018. [CrossRef]
- C. K. Hind, M. J. Carter, C. L. Harris, H. T. C. Chan, S. James, and M. S. Cragg, “Role of the pro-survival molecule Bfl-1 in melanoma,” Int. J. Biochem. Cell Biol., vol. 59, pp. 94–102, 2015. [CrossRef]
- G. Wang, S. T. Diepstraten, and M. J. Herold, “Last but not least: BFL-1 as an emerging target for anti-cancer therapies,” Biochem. Soc. Trans., vol. 50, no. 4, pp. 1119–1128, 2022. [CrossRef]
- L. Gangoda et al., “Absence of pro-survival A1 has no impact on inflammatory cell survival in vivo during acute lung inflammation and peritonitis,” Cell Death Differ., vol. 29, no. 1, pp. 96–104, 2022. [CrossRef]
- M. Sochalska, F. Schuler, J. G. Weiss, M. Prchal-Murphy, V. Sexl, and A. Villunger, “MYC selects against reduced BCL2A1/A1 protein expression during B cell lymphomagenesis,” Oncogene, vol. 36, no. 15, pp. 2066–2073, 2017. [CrossRef]
- X. Li, J. Dou, Q. You, and Z. Jiang, “Inhibitors of BCL2A1/Bfl-1 protein: Potential stock in cancer therapy,” European Journal of Medicinal Chemistry, vol. 220. Elsevier Masson s.r.l., Aug. 2021. [CrossRef]
- A. J. Huhn, R. M. Guerra, E. P. Harvey, G. H. Bird, and L. D. Walensky, “Selective Covalent Targeting of Anti-Apoptotic BFL-1 by Cysteine-Reactive Stapled Peptide Inhibitors,” Cell Chem. Biol., vol. 23, no. 9, pp. 1123–1134, 2016. [CrossRef]
- H. Flores-Romero et al., “BFL1 modulates apoptosis at the membrane level through a bifunctional and multimodal mechanism showing key differences with BCLXL,” Cell Death Differ., vol. 26, no. 10, pp. 1880–1894, 2019. [CrossRef]
- Z. O. Alabi, O. Tosin Olaoba, K. Sulaimon Ayinde, A. O. Akinyemi, and T. I. Adelusi, “Zaccheaus Oluwatayo Alabi, Olamide Tosin Olaoba , Kehinde Sulaimon Ayinde, Amos Olalekan Akinyemi and Temitope Isaac Adelusi. The Role of BFl-1 in Cancer Unravels Inhibition Mechanism,” Cancer Biol., vol. 9, no. 3, pp. 92–100, 2019. [CrossRef]
- P. Kumari and R. Rameshwari, “In silico mutational analysis to identify the role and pathogenicity of BCL-w missense variants,” J. Genet. Eng. Biotechnol., vol. 20, no. 1, 2022. [CrossRef]
- S. Huang, R. Tang, and R. Y. C. Poon, “BCL-W is a regulator of microtubule inhibitor-induced mitotic cell death,” Oncotarget, vol. 7, no. 25, pp. 38718–38730, 2016. [CrossRef]
- C. M. Adams, A. S. Kim, R. Mitra, J. K. Choi, J. Z. Gong, and C. M. Eischen, “BCL-W has a fundamental role in B cell survival and lymphomagenesis,” J. Clin. Invest., vol. 127, no. 2, pp. 635–650, Feb. 2017. [CrossRef]
- S. T. Diepstraten et al., “BCL-W is dispensable for the sustained survival of select Burkitt lymphoma and diffuse large B-cell lymphoma cell lines,” Blood Adv., vol. 4, no. 2, pp. 356–366, 2020. [CrossRef]
- R. Singh, A. Letai, and K. Sarosiek, “Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins,” Nature Reviews Molecular Cell Biology, vol. 20, no. 3. Nature Publishing Group, pp. 175–193, Mar. 2019. [CrossRef]
- T. K. Palai and S. R. Mishra, “Caspases: An apoptosis mediator,” Journal of Advanced Veterinary and Animal Research, vol. 2, no. 1. Network for the Veterinarians of Bangladesh, pp. 18–22, Mar. 2015. [CrossRef]
- D. Martinvalet, “Mitochondrial entry of cytotoxic proteases: A new insight into the granzyme B cell death pathway,” Oxidative Medicine and Cellular Longevity, vol. 2019. Hindawi Limited, 2019. [CrossRef]
- S. Zaman, R. Wang, and V. Gandhi, “Targeting the apoptosis pathway in hematologic malignancies,” Leukemia and Lymphoma, vol. 55, no. 9. Informa Healthcare, pp. 1980–1992, 2014. [CrossRef]
- M. S. Mohamed, M. K. Bishr, F. M. Almutairi, and A. G. Ali, “Inhibitors of apoptosis: clinical implications in cancer,” Apoptosis 2017 2212, vol. 22, no. 12, pp. 1487–1509, Oct. 2017. [CrossRef]
- D. Kashyap, V. K. Garg, and N. Goel, “Intrinsic and extrinsic pathways of apoptosis: Role in cancer development and prognosis,” in Advances in Protein Chemistry and Structural Biology, vol. 125, Academic Press Inc., 2021, pp. 73–120. [CrossRef]
- L. R. de Armas and E. R. Podack, “Natural killer cytolytic activity,” Nat. Kill. Cells Basic Sci. Clin. Appl., pp. 215–227, Jan. 2010. [CrossRef]
- I. Osińska, K. Popko, and U. Demkow, “Perforin: An important player in immune response,” Central European Journal of Immunology, vol. 39, no. 1. Termedia Publishing House Ltd., pp. 109–115, 2014. [CrossRef]
- J. G. Nirmala and M. Lopus, “Cell death mechanisms in eukaryotes,” Cell Biology and Toxicology, vol. 36, no. 2. Springer, pp. 145–164, Apr. 2020. [CrossRef]
- P. C. Kam, “Apoptosis : Mechanisms and clinical implications Apoptosis : mechanisms and clinical implications,” no. December 2000, 2018.
- C. Kunst, M. Haderer, S. Heckel, S. Schlosser, and M. Müller, “The p53 family in hepatocellular carcinoma,” Transl. Cancer Res., vol. 5, no. 6, pp. 632–638, 2016. [CrossRef]
- E. Wawryk-Gawda et al., “P53 protein in proliferation, repair and apoptosis of cells,” Protoplasma, vol. 251, no. 3, pp. 525–533, 2014. [CrossRef]
- L. Zhao and S. Sanyal, “p53 Isoforms as Cancer Biomarkers and Therapeutic Targets,” Cancers (Basel)., vol. 14, no. 13, p. 3145, Jun. 2022. [CrossRef]
- S. W. Chi, “Structural insights into the transcription-independent apoptotic pathway of p53,” BMB Rep., vol. 47, no. 3, pp. 167–172, 2014. [CrossRef]
- T. Ben Safta et al., “Granzyme B–Activated p53 Interacts with Bcl-2 To Promote Cytotoxic Lymphocyte–Mediated Apoptosis,” J. Immunol., vol. 194, no. 1, pp. 418–428, 2015. [CrossRef]
- H. Mansouri, L. F. Mnango, E. P. Magorosa, E. Sauli, and E. A. Mpolya, “Ki-67, p53 and BCL-2 Expressions and their Association with Clinical Histopathology of Breast Cancer among Women in Tanzania,” Sci. Rep., vol. 9, no. 1, pp. 1–11, 2019. [CrossRef]
- R. G. Mendez-Flores et al., “Role of Bcl-2, p53, and Ki-67 expression in basal cell carcinoma and their association with aggressive and non-aggressive histological phenotypes,” Postep. Dermatologii i Alergol., vol. 39, no. 3, pp. 517–523, 2022. [CrossRef]
- N. Dashzeveg and K. Yoshida, “Cell death decision by p53 via control of the mitochondrial membrane,” Cancer Lett., vol. 367, no. 2, pp. 108–112, 2015. [CrossRef]
- X. J. Wang et al., “P53 expression correlates with poorer survival and augments the negative prognostic effect of MYC rearrangement, expression or concurrent MYC/BCL2 expression in diffuse large B-cell lymphoma,” Mod. Pathol., vol. 30, no. 2, pp. 194–203, 2017. [CrossRef]
- J. Vávrová and M. Řezáčová, “Importance of proapoptotic protein PUMA in cell radioresistance,” Folia Biol. (Czech Republic), vol. 60, no. 2, pp. 53–56, 2014.
- J. E. Choi, S. M. Woo, K. J. Min, S. H. Kang, S. J. Lee, and T. K. Kwon, “Combined treatment with ABT-737 and VX-680 induces apoptosis in Bcl-2- and c-FLIP-overexpressing breast carcinoma cells,” Oncol. Rep., vol. 33, no. 3, pp. 1395–1401, 2015. [CrossRef]
- H. Wu et al., “Ionizing Radiation Sensitizes Breast Cancer Cells to Bcl-2 Inhibitor , ABT-737 , through Regulating Mcl-1 Ionizing Radiation Sensitizes Breast Cancer Cells to Bcl-2 Inhibitor , ABT-737 , through Regulating Mcl-1,” vol. 182, no. 6, pp. 618–625. [CrossRef]
- R. Pan et al., “Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia,” Blood, vol. 126, no. 3, pp. 363–372, 2015. [CrossRef]
- A. Robert, A. Pujals, L. Favre, J. Debernardi, and J. Wiels, “The BCL-2 family protein inhibitor ABT-737 as an additional tool for the treatment of EBV-associated post-transplant lymphoproliferative disorders,” Mol. Oncol., vol. 14, no. 10, pp. 2520–2532, 2020. [CrossRef]
- P. Gorombei et al., “Bcl-2 inhibitor abt-737 effectively targets leukemia-initiating cells with differential regulation of relevant genes leading to extended survival in a nras/bcl-2 mouse model of high risk-myelodysplastic syndrome,” Int. J. Mol. Sci., vol. 22, no. 19, pp. 1–21, 2021. [CrossRef]
- Z. Ni et al., “HCC cells with high levels of Bcl-2 are resistant to ABT-737 via activation of the ROS-JNK-autophagy pathway,” Free Radic. Biol. Med., vol. 70, pp. 194–203, 2014. [CrossRef]
- S. Kasai et al., “Bcl-2/Bcl-XL inhibitor ABT-737 sensitizes pancreatic ductal adenocarcinoma to paclitaxel-induced cell death,” Oncol. Lett., vol. 14, no. 1, pp. 903–908, 2017. [CrossRef]
- D. Hematology et al., “Targeting BCL-2 as a Therapeutic Strategy for Primary p210 BCR-ABL1 -positive B-ALL Cells,” vol. 516, pp. 511–516, 2020. [CrossRef]
- B. Z. Carter et al., “Synergistic effects of p53 activation via MDM2 inhibition in combination with inhibition of Bcl-2 or Bcr-Abl in CD34+ proliferating and quiescent chronic myeloid leukemia blast crisis cells,” Oncotarget, vol. 6, no. 31, pp. 30487–30499, 2015. [CrossRef]
- L. M. Brown, D. T. Hanna, S. L. Khaw, and P. G. Ekert, “Dysregulation of BCL-2 family proteins by leukemia fusion genes,” J. Biol. Chem., vol. 292, no. 35, pp. 14325–14333, 2017. [CrossRef]
- B. Z. Carter et al., “Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells,” Sci. Transl. Med., vol. 8, no. 355, 2016. [CrossRef]
- C. Kurschat, A. Metz, S. Kirschnek, and G. Häcker, “Importance of Bcl-2-family proteins in murine hematopoietic progenitor and early B cells,” Cell Death Dis., vol. 12, no. 8, pp. 1–8, 2021. [CrossRef]
- N. Singh, J. Sarkar, K. V. Sashidhara, S. Ali, and S. Sinha, “Anti-tumour activity of a novel coumarin-chalcone hybrid is mediated through intrinsic apoptotic pathway by inducing PUMA and altering Bax/Bcl-2 ratio,” Apoptosis, vol. 19, no. 6, pp. 1017–1028, 2014. [CrossRef]
- H. Kobeissy, R. Hage, S. Zeinab, D. Lina, and K. Ghassan, “Crosstalk between Noxa , Bcl - 2 , and ceramide in mediating p53 - dependent apoptosis in Molt - 4 human T - cell leukemia,” Mol. Cell. Biochem., no. 0123456789, 2020. [CrossRef]
- S. V. Vartak et al., “Novel BCL2 inhibitor, Disarib induces apoptosis by disruption of BCL2-BAK interaction,” Biochem. Pharmacol., vol. 131, pp. 16–28, 2017. [CrossRef]
- T. J. Harford, G. Kliment, G. C. Shukla, and C. M. Weyman, “The muscle regulatory transcription factor MyoD participates with p53 to directly increase the expression of the pro-apoptotic Bcl2 family member PUMA,” Apoptosis, vol. 22, no. 12, pp. 1532–1542, 2017. [CrossRef]
- J. E. Guikema, M. Amiot, and E. Eldering, “Exploiting the pro-apoptotic function of NOXA as a therapeutic modality in cancer,” Expert Opin. Ther. Targets, vol. 21, no. 8, pp. 767–779, 2017. [CrossRef]
- W. Nakajima, M. A. Hicks, N. Tanaka, G. W. Krystal, and H. Harada, “Noxa determines localization and stability of MCL-1 and consequently ABT-737 sensitivity in small cell lung cancer,” Cell Death Dis., vol. 5, no. 2, pp. 1–10, 2014. [CrossRef]
- Y. Yang et al., “SATB1 mediates long-range chromatin interactions: A dual regulator of anti-apoptotic BCL2 and pro-apoptotic NOXA genes,” PLoS One, vol. 10, no. 9, pp. 1–15, 2015. [CrossRef]
- Y. Liu et al., “NOXA genetic amplification or pharmacologic induction primes lymphoma cells to BCL2 inhibitor-induced cell death,” Proc. Natl. Acad. Sci. U. S. A., vol. 115, no. 47, pp. 12034–12039, 2018. [CrossRef]
- A. N. Hata, J. A. Engelman, and A. C. Faber, “The BCL2 family: Key mediators of the apoptotic response to targeted anticancer therapeutics,” Cancer Discov., vol. 5, no. 5, pp. 475–487, 2015. [CrossRef]
- G. Radha and S. C. Raghavan, “BCL2: A promising cancer therapeutic target,” Biochim. Biophys. Acta - Rev. Cancer, vol. 1868, no. 1, pp. 309–314, 2017. [CrossRef]
- C. Li, G. Zhang, L. Zhao, Z. Ma, and H. Chen, “Metabolic reprogramming in cancer cells: Glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer,” World J. Surg. Oncol., vol. 14, no. 1, pp. 1–7, 2016. [CrossRef]
- A. W. Roberts, “Therapeutic development and current uses of BCL-2 inhibition,” Hematol. (United States), vol. 20, no. 1, pp. 1–9, 2020. [CrossRef]
- T. Knight, D. Luedtke, H. Edwards, J. W. Taub, and Y. Ge, “A delicate balance – The BCL-2 family and its role in apoptosis, oncogenesis, and cancer therapeutics,” Biochem. Pharmacol., vol. 162, pp. 250–261, 2019. [CrossRef]
- L. Vela and I. Marzo, “Bcl-2 family of proteins as drug targets for cancer chemotherapy: the long way of BH3 mimetics from bench to bedside,” Curr. Opin. Pharmacol., vol. 23, no. Figure 1, pp. 74–81, 2015. [CrossRef]
- E. Y. Lee et al., “Human breast cancer cells display different sensitivities to ABT-263 based on the level of survivin,” Toxicol. In Vitro, vol. 46, pp. 229–236, Feb. 2018. [CrossRef]
- T. Song, G. Chai, Y. Liu, X. Yu, Z. Wang, and Z. Zhang, “Bcl-2 phosphorylation confers resistance on chronic lymphocytic leukaemia cells to the BH3 mimetics ABT-737, ABT-263 and ABT-199 by impeding direct binding,” Br. J. Pharmacol., vol. 173, no. 3, pp. 471–483, Feb. 2016. [CrossRef]
- S. Besbes, M. Mirshahi, M. Pocard, and C. Billard, “New dimension in therapeutic targeting of BCL-2 family proteins,” Oncotarget, vol. 6, no. 15, pp. 12862–12871, 2015. [CrossRef]
- S. Cory, A. W. Roberts, P. M. Colman, and J. M. Adams, “Targeting BCL-2-like Proteins to Kill Cancer Cells,” Trends in Cancer, vol. 2, no. 8, pp. 443–460, 2016. [CrossRef]
- M. J. Roy, A. Vom, P. E. Czabotar, and G. Lessene, “Cell death and the mitochondria: Therapeutic targeting of the BCL-2 family-driven pathway,” Br. J. Pharmacol., vol. 171, no. 8, pp. 1973–1987, 2014. [CrossRef]
- R. G. Micheletti, “An update on the diagnosis and treatment of hidradenitis suppurativa,” Cutis, vol. 96, no. 6, pp. 7–12, 2015.
- S. S. Chakrabarti et al., “Identifying the mechanisms of α-synuclein-mediated cytotoxicity in Parkinson’s disease: New insights from a bioinformatics-based approach,” Future Neurol., vol. 15, no. 3, pp. 13–15, 2020. [CrossRef]
- J. Jankovic and E. K. Tan, “Parkinson’s disease: Etiopathogenesis and treatment,” J. Neurol. Neurosurg. Psychiatry, vol. 91, no. 8, pp. 795–808, 2020. [CrossRef]
- J. Liu, W. Liu, and H. Yang, “Balancing apoptosis and autophagy for parkinson’s disease therapy: Targeting BCL-2,” ACS Chem. Neurosci., vol. 10, no. 2, pp. 792–802, 2019. [CrossRef]
- A. Jan, N. P. Gonçalves, C. B. Vaegter, P. H. Jensen, and N. Ferreira, “The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses,” Int. J. Mol. Sci. 2021, Vol. 22, Page 8338, vol. 22, no. 15, p. 8338, Aug. 2021. [CrossRef]
- B. Wang, L. Wang, Y. Qu, J. Lu, and W. Xia, “Chitosan oligosaccharides exert neuroprotective effects via modulating the PI3K/Akt/Bcl-2 pathway in a Parkinsonian model,” Food Funct., vol. 13, no. 10, pp. 5838–5853, May 2022. [CrossRef]
- L. Wang et al., “Cannabidiol Alleviates the Damage to Dopaminergic Neurons in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Parkinson’s Disease Mice Via Regulating Neuronal Apoptosis and Neuroinflammation,” Neuroscience, vol. 498, pp. 64–72, Aug. 2022. [CrossRef]
- K. Guo et al., “Neuroprotective effect of paeoniflorin in the mouse model of Parkinson’s disease through α-synuclein/protein kinase C δ subtype signaling pathway,” Neuroreport, vol. 32, no. 17, pp. 1379–1387, Dec. 2021. [CrossRef]
- R. L. Jayaraj, S. Azimullah, K. A. Parekh, S. K. Ojha, and R. Beiram, “Effect of citronellol on oxidative stress, neuroinflammation and autophagy pathways in an in vivo model of Parkinson’s disease,” Heliyon, vol. 8, no. 11, p. e11434, 2022. [CrossRef]
- J. Shao, X. Liu, M. Lian, and Y. Mao, “Citronellol Prevents 6-OHDA-Induced Oxidative Stress, Mitochondrial Dysfunction, and Apoptosis in Parkinson Disease Model of SH-SY5Y Cells via Modulating ROS-NO, MAPK/ERK, and PI3K/Akt Signaling Pathways,” Neurotox. Res., pp. 1–17, Sep. 2022. [CrossRef]
- T. Hong, J. Ding, and W. Li, “mir-7 reverses breast cancer resistance to chemotherapy by targeting MRP1 and BCL2,” Onco. Targets. Ther., vol. 12, pp. 11097–11105, 2019. [CrossRef]
- A. Srivastava and I. Jatoi, “Genetic Predisposition to Breast Cancer and Its Management,” Indian J. Surg. 2021 832, vol. 83, no. 2, pp. 273–274, May 2021. [CrossRef]
- M. Boujemaa et al., “Health influenced by genetics: A first comprehensive analysis of breast cancer high and moderate penetrance susceptibility genes in the Tunisian population,” PLoS One, vol. 17, no. 3, p. e0265638, Mar. 2022. [CrossRef]
- Y. Feng et al., “Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis,” Genes Dis., vol. 5, no. 2, pp. 77–106, 2018. [CrossRef]
- M. M. Williams and R. S. Cook, “Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance?,” Oncotarget, vol. 6, no. 6, pp. 3519–3530, 2015. [CrossRef]
- D. Merino, S. W. Lok, J. E. Visvader, and G. J. Lindeman, “Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer,” Oncogene 2016 3515, vol. 35, no. 15, pp. 1877–1887, Aug. 2015. [CrossRef]
- N. Honma et al., “Differences in clinical importance of Bcl-2 in breast cancer according to hormone receptors status or adjuvant endocrine therapy,” BMC Cancer, vol. 15, no. 1, 2015. [CrossRef]
- P. Raha, S. Thomas, K. T. Thurn, J. Park, and P. N. Munster, “Combined histone deacetylase inhibition and tamoxifen induces apoptosis in tamoxifen-resistant breast cancer models, by reversing Bcl-2 overexpression,” Breast Cancer Res., vol. 17, no. 1, pp. 1–16, 2015. [CrossRef]
- A. Pyrczak-Felczykowska et al., “Synthesis of Usnic Acid Derivatives and Evaluation of Their Antiproliferative Activity against Cancer Cells,” J. Nat. Prod., vol. 82, no. 7, pp. 1768–1778, 2019. [CrossRef]
- K. Kumar, J. P. Mishra, and R. P. Singh, “Anti-cancer efficacy and mechanisms of usnic acid,” Indian J. Pharm. Biol. Res., vol. 7, no. 03, pp. 1–4, 2019. [CrossRef]
- E. Deǧerli, V. Torun, and D. Cansaran-Duman, “MiR-185-5p response to usnic acid suppresses proliferation and regulating apoptosis in breast cancer cell by targeting Bcl2,” Biol. Res., vol. 53, no. 1, pp. 1–14, 2020. [CrossRef]
- N. Kiliç, Y. Ö. Islakoğlu, İ. Büyük, B. Gür-Dedeoğlu, and D. Cansaran-Duman, “Determination of Usnic Acid Responsive miRNAs in Breast Cancer Cell Lines,” Anticancer. Agents Med. Chem., vol. 19, no. 12, pp. 1463–1472, Nov. 2019. [CrossRef]
- R. Özben and D. Cansaran-Duman, “The expression profiles of apoptosis-related genes induced usnic acid in SK-BR-3 breast cancer cell,” Hum. Exp. Toxicol., vol. 39, no. 11, pp. 1497–1506, 2020. [CrossRef]
- “J Biochem Molecular Tox - 2021 - Elia - Loperamide potentiates doxorubicin sensitivity in triple-negative breast cancer.pdf.”.
- A. Ghanem et al., “Rumex Vesicarius L. extract improves the efficacy of doxorubicin in triple-negative breast cancer through inhibiting Bcl2, mTOR, JNK1 and augmenting p21 expression,” Informatics Med. Unlocked, vol. 29, no. December 2021, p. 100869, 2022. [CrossRef]
- S. Eugin Simon et al., “New synthetic phenylquinazoline derivatives induce apoptosis by targeting the pro-survival members of the BCL-2 family,” Bioorg. Med. Chem. Lett., vol. 67, p. 128731, Jul. 2022. [CrossRef]
- P. M. de Groot, C. C. Wu, B. W. Carter, and R. F. Munden, “The epidemiology of lung cancer,” Translational Lung Cancer Research, vol. 7, no. 3. AME Publishing Company, pp. 220–233, Jun. 2018. [CrossRef]
- L. A. Byers and C. M. Rudin, “Small cell lung cancer: Where do we go from here?,” Cancer, vol. 121, no. 5, pp. 664–672, 2015. [CrossRef]
- J. Fan et al., “Bruceine D induces lung cancer cell apoptosis and autophagy via the ROS/MAPK signaling pathway in vitro and in vivo,” Cell Death Dis., vol. 11, no. 2, 2020. [CrossRef]
- G. Liu et al., “Role of autophagy and apoptosis in non-small-cell lung cancer,” Int. J. Mol. Sci., vol. 18, no. 2, 2017. [CrossRef]
- X. Song, X. Xu, J. Lu, X. Chi, Y. Pang, and Q. Li, “Lamprey Immune Protein Mediates Apoptosis of Lung Cancer Cells Via the Endoplasmic Reticulum Stress Signaling Pathway,” Front. Oncol., vol. 11, no. July, pp. 1–14, 2021. [CrossRef]
- N. Othman and N. H. Nagoor, “The role of microRNAs in the regulation of apoptosis in lung cancer and its application in cancer treatment,” Biomed Res. Int., vol. 2014, 2014. [CrossRef]
- A. R. D. Delbridge and A. Strasser, “The BCL-2 protein family, BH3-mimetics and cancer therapy,” Cell Death Differ. 2015 227, vol. 22, no. 7, pp. 1071–1080, May 2015. [CrossRef]
- S. Song et al., “ATP promotes cell survival via regulation of cytosolic [Ca2+] and Bcl-2/Bax ratio in lung cancer cells,” Am. J. Physiol. - Cell Physiol., vol. 310, no. 2, pp. C99–C114, 2016. [CrossRef]
- S. Yu, L. sheng Gong, N. feng Li, Y. feng Pan, and L. Zhang, “Galangin (GG) combined with cisplatin (DDP) to suppress human lung cancer by inhibition of STAT3-regulated NF-κB and Bcl-2/Bax signaling pathways,” Biomed. Pharmacother., vol. 97, no. May 2017, pp. 213–224, 2018. [CrossRef]
- X. Shi et al., “A critical role for the long non-coding RNA GAS5 in proliferation and apoptosis in non-small-cell lung cancer,” Mol. Carcinog., vol. 54, no. S1, pp. E1–E12, 2015. [CrossRef]
- P. Huang, B. Ye, Y. Yang, J. Shi, and H. Zhao, “MicroRNA-181 functions as a tumor suppressor in non-small cell lung cancer (NSCLC) by targeting Bcl-2,” Tumor Biol., vol. 36, no. 5, pp. 3381–3387, 2015. [CrossRef]
- T. L. Lochmann et al., “Venetoclax is effective in small-cell lung cancers with high BCL-2 expression,” Clin. Cancer Res., vol. 24, no. 2, pp. 360–369, 2018. [CrossRef]
- L. Wu and L. Chen, “Characteristics of Nur77 and its ligands as potential anticancer compounds (Review),” Mol. Med. Rep., vol. 18, no. 6, pp. 4793–4801, 2018. [CrossRef]
- M. C. Pearce et al., “Induction of apoptosis and suppression of tumor growth by Nur77-derived Bcl-2 converting peptide in chemoresistant lung cancer cells,” Oncotarget, vol. 9, no. 40, pp. 26072–26085, 2018. [CrossRef]
- R. K. Mongre et al., “Novel carbazole-piperazine hybrid small molecule induces apoptosis by targeting BCL-2 and inhibits tumor progression in lung adenocarcinoma in vitro and xenograft mice model,” Cancers (Basel)., vol. 11, no. 9, pp. 1–25, 2019. [CrossRef]
- D. Park et al., “Discovery of Small Molecule Bak Activator for Lung Cancer Therapy,” Theranostics, vol. 11, no. 17, pp. 8500–8516, 2021. [CrossRef]
- M. Marrodan, M. F. Farez, M. E. Balbuena Aguirre, and J. Correale, “Obesity and the risk of Multiple Sclerosis. The role of Leptin,” Ann. Clin. Transl. Neurol., vol. 8, no. 2, pp. 406–424, 2021. [CrossRef]
- M. Filippi et al., “EM (Nature 2018),” pp. 1–27, 2018.
- A. F. Ferreira, “Aline fernanda ferreira,” vol. 55, no. 16, pp. 1–5, 2014.
- S. E. Baranzini and J. R. Oksenberg, “The Genetics of Multiple Sclerosis: From 0 to 200 in 50 Years,” Trends Genet., vol. 33, no. 12, pp. 960–970, 2017. [CrossRef]
- B. Macchi et al., “Role of inflammation and apoptosis in multiple sclerosis: Comparative analysis between the periphery and the central nervous system,” J. Neuroimmunol., vol. 287, pp. 80–87, 2015. [CrossRef]
- S. Hagman et al., “Analysis of apoptosis-related genes in patients with clinically isolated syndrome and their association with conversion to multiple sclerosis,” J. Neuroimmunol., vol. 280, pp. 43–48, 2015. [CrossRef]
- D. H. Mahad, B. D. Trapp, and H. Lassmann, “Pathological mechanisms in progressive multiple sclerosis,” Lancet Neurol., vol. 14, no. 2, pp. 183–193, 2015. [CrossRef]
- M. Igci et al., “Gene expression profiles of autophagy-related genes in multiple sclerosis,” Gene, vol. 588, no. 1, pp. 38–46, 2016. [CrossRef]
- X. Feng, H. Hou, Y. Zou, and L. Guo, “Defective autophagy is associated with neuronal injury in a mouse model of multiple sclerosis,” Bosn. J. Basic Med. Sci., vol. 17, no. 2, pp. 95–103, 2017. [CrossRef]
- N. Sharma et al., “Neuroprotection by solanesol against ethidium bromide-induced multiple sclerosis-like neurobehavioral, molecular, and neurochemical alterations in experimental rats,” Phytomedicine Plus, vol. 1, no. 4, p. 100051, 2021. [CrossRef]
- Y. Dong et al., “Leukemia incidence trends at the global, regional, and national level between 1990 and 2017,” Exp. Hematol. Oncol., vol. 9, no. 1, pp. 1–11, Jun. 2020. [CrossRef]
- S. Ribeiro, A. M. Eiring, and J. S. Khorashad, “Genomic abnormalities as biomarkers and therapeutic targets in acute myeloid leukemia,” Cancers (Basel)., vol. 13, no. 20, pp. 1–21, 2021. [CrossRef]
- Y. Wei et al., “Targeting Bcl-2 Proteins in Acute Myeloid Leukemia,” Front. Oncol., vol. 10, no. November, pp. 1–11, 2020. [CrossRef]
- S. Zhang, Q. Zhang, G. Shi, and J. Yin, “MiR-182-5p regulates BCL2L12 and BCL2 expression in acute myeloid leukemia as a potential therapeutic target,” Biomed. Pharmacother., vol. 97, no. November 2017, pp. 1189–1194, 2018. [CrossRef]
- M. Gentile et al., “Venetoclax for the treatment of chronic lymphocytic leukemia,” Expert Opin. Investig. Drugs, vol. 26, no. 11, pp. 1307–1316, 2017. [CrossRef]
- L. Ismail, H. Materwala, and J. Al Kaabi, “Association of risk factors with type 2 diabetes: A systematic review,” Comput. Struct. Biotechnol. J., vol. 19, pp. 1759–1785, 2021. [CrossRef]
- E. N. Gurzov and D. L. Eizirik, “Bcl-2 proteins in diabetes: Mitochondrial pathways of β-cell death and dysfunction,” Trends Cell Biol., vol. 21, no. 7, pp. 424–431, 2011. [CrossRef]
- E. Pishavar and J. Behravan, “miR-126 as a Therapeutic Agent for Diabetes Mellitus,” Curr. Pharm. Des., vol. 23, no. 22, pp. 3309–3314, 2017. [CrossRef]
- L. Demirtas et al., “Apoptosis, autophagy & endoplasmic reticulum stress in diabetes mellitus,” Indian J. Med. Res., vol. 144, no. OCTOBER, pp. 515–524, 2016. [CrossRef]
- H. E. Thomas, M. D. McKenzie, E. Angstetra, P. D. Campbell, and T. W. Kay, “Beta cell apoptosis in diabetes,” Apoptosis, vol. 14, no. 12, pp. 1389–1404, 2009. [CrossRef]
- T. Tomita, “Apoptosis in pancreatic β-islet cells in Type 2 diabetes,” Bosn. J. Basic Med. Sci., vol. 16, no. 3, pp. 162–179, 2016. [CrossRef]
- E. Gokalp-Ozkorkmaz et al., “Examination of Bcl-2 and Bax Protein Levels for Determining the Apoptotic Changes in Placentas with Gestational Diabetes and Preeclampsia,” p. 1548, 2018. [CrossRef]
- E. K. Sims, A. J. Lakhter, E. Anderson-Baucum, T. Kono, X. Tong, and C. Evans-Molina, “MicroRNA 21 targets BCL2 mRNA to increase apoptosis in rat and human beta cells,” Diabetologia, vol. 60, no. 6, pp. 1057–1065, 2017. [CrossRef]
- E. F. El Azab and H. S. Mostafa, “Geraniol ameliorates the progression of high fat-diet/streptozotocin-induced type 2 diabetes mellitus in rats via regulation of caspase-3, Bcl-2, and Bax expression,” J. Food Biochem., vol. 46, no. 7, p. e14142, Jul. 2022. [CrossRef]
- Q. He et al., “Piperine is capable of improving pancreatic β-cell apoptosis in high fat diet and streptozotocin induced diabetic mice,” J. Funct. Foods, vol. 88, p. 104890, Jan. 2022. [CrossRef]
- X. qi Liu et al., “Wogonin protects glomerular podocytes by targeting Bcl-2-mediated autophagy and apoptosis in diabetic kidney disease,” Acta Pharmacol. Sin., vol. 43, no. 1, pp. 96–110, 2022. [CrossRef]
- A. H. Amin, M. A. El-Missiry, and A. I. Othman, “Melatonin ameliorates metabolic risk factors, modulates apoptotic proteins, and protects the rat heart against diabetes-induced apoptosis,” Eur. J. Pharmacol., vol. 747, pp. 166–173, 2015. [CrossRef]
- S. R. Filios and A. Shalev, “β-Cell MicroRNAs: Small but Powerful,” Diabetes, vol. 64, no. 11, pp. 3631–3644, Nov. 2015. [CrossRef]
- K. Tugay et al., “Role of microRNAs in the age-associated decline of pancreatic beta cell function in rat islets,” Diabetologia, vol. 59, no. 1, pp. 161–169, Jan. 2016. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



