Submitted:
20 June 2023
Posted:
20 June 2023
You are already at the latest version
Abstract
AB toxins have historically been associated with significant morbidity, mortality through infections such as botulinum, anthrax, cholera, and diphtheria. These AB toxin-mediated diseases remain prevalent in low and middle income countries, with intermittent outbreaks of Shiga toxin-producing Escherichia coli (STEC) or whooping cough by Bordetella pertussis in high-income countries. These reports warrant an investigation to better understand the distinct characteristics of AB toxins derived from different pathogens. As toxigenic pathogens broaden their scope and diversity, it amplifies the complexity of the problems posed by their AB toxins. Here, we discuss the history, structure and characteristics of key AB toxins, and report on historical and ongoing research on these toxins. We also explore research avenues that hold great promise in potentially improving clinical management of toxin-mediated diseases in the future.
Keywords:
AB toxin
; diphtheria
; botulinum
; tetanus
; anthrax
; cholera
; pertussis
; shiga toxin
; vaccines
1. Introduction
The origin of the word ‘toxin’ dates back to 1885 when it was first coined by a chemist named Ludwig Brieger [1]. The term comprises several groups of toxins which can include chemical, radioactive and biological agents. In the modern era, people have become increasingly concerned about biological toxins originating mainly from a bacterial source. There are two main categories for bacterial toxins: exotoxins and endotoxins. Within these two groups there are further sub-groups such as enterotoxins. There is some cross-over between exotoxins and endotoxins with some toxins categorised as both.
1.1. Exotoxins and endotoxins
Exotoxins are harmful proteins secreted by certain Gram-positive and Gram-negative bacteria [2]. They are secreted from the bacterial host, with the host cell staying intact. Exotoxins are highly potent and are the key virulence factors associated with diphtheria, anthrax, botulism, pertussis, and tetanus [3]. For some of these, toxoid vaccines and anti-toxins are available as prevention and treatment. These pharmaceuticals are purified exotoxins that are inactivated using either heat or formaldehyde to form toxoids, without compromising their immunogenicity [4].
Endotoxins are constituents of the outer membranes of the cell walls, making them exclusive to Gram-negative bacteria. They are released into the host when a pathogen undergoes cell lysis [5]. Endotoxins consist of lipopolysaccharide (LPS) and lipid complexes, which initiate host inflammatory responses to the infection [3]. All of the toxins covered in this review are exotoxins that fall under a sub-category known as AB toxins.
1.2. AB toxins
The first AB toxin was Diphtheria toxin (DT), isolated from Corynebacterium diphtheriae in 1883. The AB-toxin family has since expanded to include numerous other examples, including tetanus toxin, produced by Clostridium tetani, and botulinum toxin, of Clostridium botulinum, among others [6,7]. AB toxins are made up of two components: the ‘binding subunit’ B first binds to target receptors, facilitating the transport of the ‘active subunit’ A across the cellular membrane, which causes the toxigenic effect [8]. Figure 1 describes a simplified diagram of the process, from production to release by a bacterial cell, through to its binding to a target cell surface, followed by active toxicity. Furthermore, there is a subgroup of AB toxins known as AB5 toxins, where the B subunit is a pentamer i.e., composed of five chains [9]. The cholera toxin (CTX), produced by Vibrio cholerae, and Shiga (Stx) and Shiga-like toxins, produced by Shigella dysenteriae & some serogroups of Escherichia coli, are prominent examples of AB5 toxins [10]. AB toxins can also be found outside of microbes. An example of this is ricin, a highly potent toxin produced in seeds of Ricinus communis, the castor bean plant [10]. Table 1 lists some of the major bacterial AB toxins and the species they are commonly linked with.
2. Diphtheria Toxin
Diphtheria toxin (DT) is the main potent tool of C. diphtheriae, the cause of diphtheria. Diphtheria is a disease that affects thousands worldwide and is still a major cause of mortality, especially in children [11]. Harboured on a corynephage beta and encoded by the tox gene, DT is one of the most well-understood toxins, as it was the first AB toxin discovered [6]. Traditionally linked with C. diphtheriae, DT is also produced by both C. ulcerans and C. pseudotuberculosis. C. diphtheriae was first described as the causative agent of diphtheria in 1883 by Edwin Klebs, and cultured a year later by Friedrich Loeffler [7,12]. Evidence of products produced by the bacterium C. diphtheriae being the cause of diphtheria was presented by Roux and Yersin in 1888, after injecting animals with sterile filtrates of liquid C. diphtheriae culture [13]. The first Nobel Prize for medicine was awarded in 1901 to Emil von Behring, who with the help of Shibasaburō Kitasato discovered that the serum of immunized animals offered protection against the disease [14]. A toxoid vaccine developed during the 1920s was specifically designed to combat DT. Even before the diphtheria toxoid vaccine was fully implemented in the 1940s, the United States of America alone recorded 100,000-200,000 cases and 13,000–15,000 deaths [15]. With widespread vaccination, the number of cases had fallen dramatically, reaching a record low of 4,333 cases reported to the World Health Organization in 2006 [16].
Despite this success, several major outbreaks have occurred since the vaccine’s introduction, the largest of which was during the dissolution of the Soviet Union in the early-to-mid 1990s, resulting in over 157,000 cases and ~5,000 deaths over an eight year period [17]. This outbreak was likely the result of a variety of factors, but largely attributed to gaps in vaccine coverage, especially in children, due to regional changes in healthcare procedure and recommendations [17]. More recently, outbreaks in Yemen and among Rohingya refugee camps have demonstrated diphtheria’s ability to quickly re-emerge in favourable conditions [18,19]. Globally, the year 2019 saw the highest reported number of cases (22,986) to the WHO since 1996. This highlights the importance of this historic disease even today.
DT’s crystal structure was reported by Choe et al in 1992 to 2.5 Å resolution, as a Y-shaped molecule of three domains [20]. These were, the catalytic (C) domain, the constituent part of subunit A; and the transmembrane (T) and the receptor binding (R) domains, which together make up subunit B. The subunits are linked together by a disulfide bridge and peptide bond, both of which must be broken in order for the subunit A to activate. The B subunit’s function is to bind to heparin-binding epidermal growth factor (HB-EGF) and allow translocation of the toxin across the phospholipid bilayer via endocytosis [20]. The cleavage of the bonds between the DT subunits occurs by endosome-associated proteases and causes the acidification of the endosome, allowing subunit A to be translocated across the endosomal membrane into the cytosol [21]. Once in the cytosol, the A subunit catalyses the transfer of ADP-ribose of nicotinamide adenine dinucleotide (NAD) onto the elongation factor 2 (EF-2). The EF2 is an essential part of the protein synthesis process within cells and the ADP-ribosylation, which prevents protein synthesis inside the cell, resulting in apoptosis-driven cell death as seen in Figure 2 [20].
Diphtheria typically begins with angina, tonsillitis-like symptoms, and mild fever. If the symptoms are not treated, a white-grey pseudomembrane forms over the pharynx, larynx, and tonsils. This is considered a typical presentation of symptomatic diphtheria, as is a swollen neck, also known as ‘bull neck’, found in the majority of cases. If left untreated, infection can result in death. However, antibiotics coupled with diphtheria antitoxin (DAT) will typically lead to a full recovery. The main antibiotics prescribed for diphtheria treatment are penicillin and erythromycin, however resistance to both had been reported in some parts of the world, both individually and in combination [22,23,24]. DAT is an equine immunoglobulin preparation made by extracting blood plasma from horses immunized against DT. While part of the World Health Organization’s Essential Medicines list, its recommended use varies between countries. In the United Kingdom, guidance on the use of DAT indicates it should be used for the treatment of suspected and confirmed diphtheria cases, but not for prophylaxis [25]. In the USA, the Food and Drug Administration has not licenced DAT for use, but the Centers for Disease Control and Prevention (CDC) are authorised to distribute to treating clinicians for patients with suspected or confirmed respiratory diphtheria after discussions between the clinician and the CDC diphtheria duty officer [26].
The DT tox gene is suppressed in high iron environments with the assistance of the diphtheria toxin repressor (dtxR) gene repressor. dtxR was long believed to be the sole regulator of toxin production, but recent studies have demonstrated that other mechanisms may be involved, and that dtxR may play other roles besides toxin regulation [27,28,29]. Utilising genomics, advances have been made in identifying how the diphtheria tox gene, and by extension the DT protein, has changed and adapted [30]. After bringing together 502 genomes from 16 countries and territories over 122 years, 291 (58%) were identified to carry the tox gene using in-silico PCR [30]. Of these extracted genes, 18 variants were found, eight of which were found to contain non-synonymous SNP changes from the variant carried by wild strains used to produce the toxoid vaccine [30]. While two of these non-synonymous variations were in the signal sequence of the gene, six were plotted onto the protein’s 3D structure, and the impact of these mutations were estimated using computational tools [30]. One was estimated to be a mutation that would have a low impact on the protein structure, while four were estimated to have a moderate impact, and one to have a high impact [30]. These variations were not linked to variations within the corynephage, although it was only a preliminary analysis [30]. While there is no concern of vaccine efficacy being affected, the results have clear implications for future vaccine and antitoxin development, as other variants may already be present in strains circulating today, and the tox gene will continue to mutate and produce new variants into the future [30]. On a positive note DT has been utilised effectively as a transport agent in both medical and biochemical procedures. Attenuated DT can mediate the delivery of bound and conjugated elements, including siRNA and antibodies [31,32]. This mechanism has been described as a potential way of expanding targeted therapies, including cancer therapy, with a ‘detoxified’ DT acting as the delivery mechanism, moving a potentially wide range of passenger proteins across the cellular membrane and provide treatment to intra-cellular conditions [33].
2.1. Anthrax Toxin
First reported in 1954 by H. Smith and J. Keppie, anthrax toxin (AT) is a bacterial AB toxin produced by Bacillus anthracis [34]. AT is made up of proteins in a three-part combination; the B subunit anthrax protective antigen (PA), and the A subunits edema factor (EF) and lethal factor (LF) [35]. PA binds to the AT receptors ANTXR1 (TEM8) or ANTXR2 (CMG2) and is cleaved by the cell surface protease furin prior to EF and LF endocytosis into the cytosol [36,37]. Once inside, EF, a calcium-calmodulin (CaM)-dependent adenylate cyclase can convert 2000 molecules of ATP to cAMP per second, greatly increasing the production of cAMP, causing local inflammation and edema [37,38]. LF is a protease that cleaves mitogen-activated protein kinase (MAPK) kinases 1 and 2 (MAPKK1 and 2), inactivating the kinases and inhibiting the signal transduction pathway, leading to macrophage lysis as seen in Figure 3 [38,39,40,41]. Both EF and LF are toxigenic in isolation, but in combination cause major host damage [35]. The toxin components are encoded on separate genes, pagA (the protective antigen), cya (the edema factor) and lef (the lethal factor) [42]. These genes were discribed by Mikesell et al harboured on a large plasmid known as pXO1 and sequenced by Okinaka et al in 1999 [43,44]. The toxin expression peaks in the late log phase of bacterial growth, and is believed to be regulated by orthologs of the transition state regulator abrB [42], with similar genes found within both the chromosome and pXO1. Alongside toxin production, a capsule produced by genes carried on the pXO2 plasmid is required for B. anthracis virulence [45]. Without both major factors the bacterium is much less virulent [45].
B. anthracis is a disease that primarily affects animals, and can prove fatal to most wildlife, from deer and livestock to chimpanzees [35,46]. At least 109 bacteria per millilitre of blood, the release of spores from carcasses which can spread to contaminate large areas and can germinate into full infections when taken up by nonimmune hosts [35,47]. These spores can persist in the environment for long periods of time [47]. There are 3 modes of uptake: skin, respiratory or intestinal epithelia, the mode of uptake affects severity and mortality rates [48]. In the case of inhalation, first stage symptoms can take between hours and days to manifest, and can include fever, fatigue, headaches, spasms, coughing and vomiting [49,50]. If left untreated, the second stage can develop quickly with symptoms worsening, progressing to death in a very short time (2-48 hrs) [49]. Antibiotics are the primary treatment, often coupled with antitoxin [51]. While most isolates are still susceptible to antibiotics, antimicrobial resistance in environmental and engineered strains have previously been reported. This includes the potential for gaining quinolone, ciprofloxacin, tetracycline, doxycycline, penicillin and amoxicillin resistance through horizontal gene transfer and mutations across the genome [52].
Evolutionary changes play a major role in the effectiveness of bacterial toxins, and AT is no exception. Previous studies have highlighted the impact variations within ANTXR2 in humans have had on AT sensitivity [53]. A 2016 study identified multiple SNP variations in ANTXR2 and demonstrated a statistically significant correlation between AT sensitivity and variations within this host gene [54]. Also, variation within AT itself has been widely researched as a potential delivery system for tumour treatment, utilising the toxin in much the same way as DT [55,56,57,58]. AT has been widely examined as a potential high risk bioweapon and major concern for bioterrorism, owing to how deadly the toxin can be and how the spores can be found in environments for extended periods of time. Research into the viability of AT as a bioweapon occurred during both world wars with covert programs intended to infect livestock and animal feed, expanding into bioweapon programs experimenting on prisoners of war such as Unit 731 of the Imperial Japanese army [59]. Contrast to that in 1942, the UK demonstrated the effect of anthrax spore bombs on Gruinard Island (Scotland). The spores were highly lethal to the flocks of sheep and the island remained heavily contaminated for decades, up till the 1990s [59,60]. The first instance of AT bioterrorism was in Japan in 1993. This was a liquid suspension of anthrax that was aerosolised from a roof of an eight-story building in Tokyo, by the cult group Aum Shinrikyo, with no casualties [61]. They had used a vaccine strain of B. anthracis [61]. To understand where we are in preparedness, in the UK, there is a strategy for biological risks known as “Understand, Prevent, Detect, Respond”. In the USA, after recognising any unusual pattern of illness, doctors who suspect anthrax can order lab tests and contact the CDC. With enough evidence they can respond before the lab results come back by deploying field staff, continuous testing and shipping of anti-toxins and guidance to clinicians and public [62].
2.2. Botulinum Toxin
Botulinum neurotoxin (BoNT) is one of the most potent exotoxins known. It is produced by the bacterium Clostridium botulinum. BoNT has seven serologically distinct forms, denoted by letters A-G. These exotoxins produced by C. botulinum prevent acetylcholine from being released. However, types A, B, E and F of BoNT cause human botulism [63]. While, types C and D cause disease in other animals such as fish and birds [63]. The first accurate description of clinical botulism was in 1820 by Justinus Kerner. These reports of botulism were originally referred to as “sausage poisoning” as he made the association between the citizens of Germany falling ill and consumption of sausages [64]. Before the end of the century it was discovered by Émile van Ermengem that the toxin was produced by C. botulinum, after the isolation of C. botulinum from a piece of ham that poisoned 34 people in 1895 [64,65]. This Gram-positive spore-forming anaerobe is commonly found in soil, dust and sediments. It can cause foodborne botulism from canned or vacuum-packed food [66,67]. The characteristic symptoms are vomiting, stomach cramps, diarrhoea and descending paralysis [67]. A modified form of this toxin is used in cosmetic Botox, a purified and highly diluted form of BoNT-A [68]. Originally approved by the FDA in 1989 to treat strabismus, blepharospasm and hemifacial spasm, since 2002, Botox cosmetics were introduced to reduce skin wrinkles [69,70].
The exotoxin produced by C. botulinum is a 150 kDa protein composed of two subunits linked by a disulfide bond. Subunit B (100 kDa), also known as the heavy chain of the toxin, binds to GD1a receptors on presynaptic membranes of peripheral and cranial nerves [69]. Subunit A (50 kDa), also referred to as the light chain, cleaves a targeted member of the SNARE protein family, resulting in the inhibitation of the release of acetylcholine and the disease botulism [69]. The target cleaved for both BoNT/A and BoNT/E is the SNARE protein SNAP-25. For BoNT/B, BoNT/D, BoNT/G, the target to cleave is synaptobrevin [71]. BoNT/C is an exception with two targets, SNAP-25 and syntaxin [72]. The migration of BoNT occurs by the retrograde axonal transport system towards the spinal cord where it can migrate between postsynaptic and presynaptic neurons [73,74]. The toxin-receptor complex is then taken into the cell by endocytosis, cleavage of the disulfide bond occurs by its own proteases which activates the toxin and the subunit A escapes into the cytoplasm [69]. Subunit A (light chain) of the toxin acts as a zinc endopeptidase, selectively cleaving the targeted SNARE proteins including SNAP-25, synaptobrevin and syntaxin, which inhibits the release of acetylcholine at the synapse as seen in Figure 4 [75]. This prevents the release of acetylcholine, which causes flaccid paralysis in all seven serotypes of botulism [76]. The variant BoNT/A is the most potent, followed by BoNT/B and BoNT/F.
After exposure to the toxin, symptoms of botulism appear on average 12 to 72 hours (ranging from 2 hours to 8 days) later [77]. In general, botulism causes descending flaccid paralysis, which can result in respiratory failure. The early symptoms of foodborne and wound botulism are weakness, vertigo, blurred vision, fatigue, dry mouth with difficulty in speaking and swallowing. It may progress to weakness in the head, neck and arms, continuing to affect the respiratory muscles and the lower body [78]. In the case of infant botulism, the early symptoms may include difficulty in feeding, unfocused eyes, lack of facial expressions, and a weak cry. This can progress further to weakened muscles and difficulty in breathing [79].
Treatment involves two types of antitoxins, depending on the type of botulism. The heptavalent botulinum antitoxin (HBAT) is required for both foodborne and wound type botulism [80]. In 2010, the HBAT anti-toxin was produced and it is capable of neutralising all seven serologically distinct forms of botulinum toxin (types A, B, C, D, E, F and G) [80]. The HBAT functions by binding to the free botulinum toxin, preventing further internalisation at the acetylcholine receptor [81]. This replaced the first anti-toxin produced for botulinum in the 1970s by the US Army Medical Research Institute of Infectious Diseases. This 1970s, serum was developed from a single horse and was the only source for the anti-toxin till the 1990s [81]. Wound botulism may also require antibiotics to remove the bacterial source of the toxin from the body [81]. In the case of infant botulism a second type of antitoxin is used instead of HBAT, known as botulism immune globulin [82].
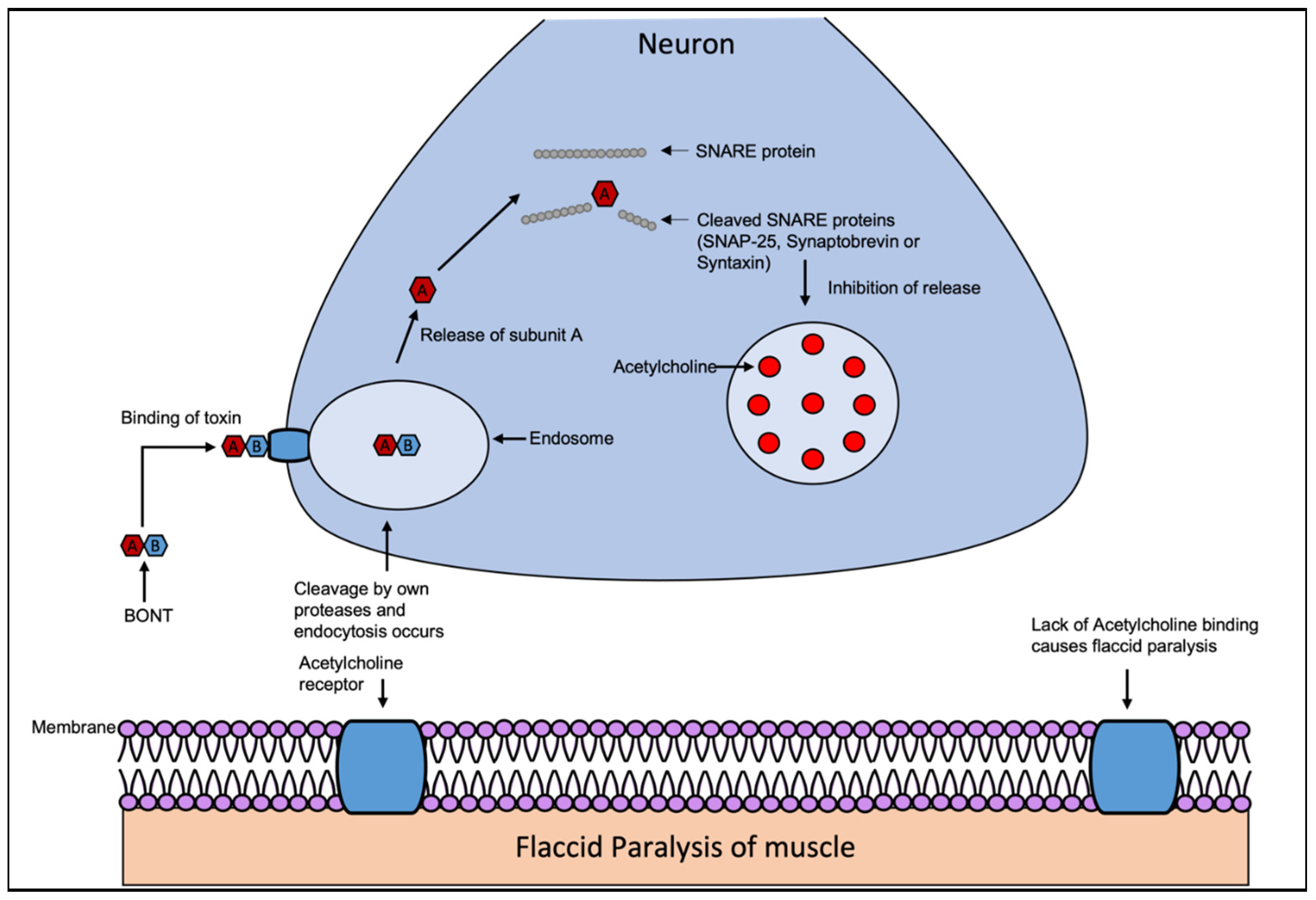
Figure 4.
The BoNT mechanism of action. BoNT are composed of two subunits linked by a disulfide bond. The exotoxins BoNT/A, BoNT/B and BoNT/F, BoNT/E cause human disease. All BoNT types target different members of the same SNARE protein family. The migration of BoNT occurs by the retrograde axonal transport system towards the spinal cord where it can migrate between postsynaptic and presynaptic neurons. The toxin-receptor complex is then taken into the cell by endocytosis, cleavage of the disulfide bond occurs by its own proteases activating the toxin and the subunit A escapes into the cytoplasm. Subunit A of the toxin, acting as a zinc endopeptidase, selectively cleaves the targeted SNARE protein including SNAP-25, synaptobrevin or syntaxin (depending on type of BoNT), which inhibits the release of acetylcholine at the synapse. Due to this the release of acetylcholine is prevented. This occurs in all 7 serotypes of botulism, resulting in flaccid paralysis. Adapted from Multani et all [83].
Figure 4.
The BoNT mechanism of action. BoNT are composed of two subunits linked by a disulfide bond. The exotoxins BoNT/A, BoNT/B and BoNT/F, BoNT/E cause human disease. All BoNT types target different members of the same SNARE protein family. The migration of BoNT occurs by the retrograde axonal transport system towards the spinal cord where it can migrate between postsynaptic and presynaptic neurons. The toxin-receptor complex is then taken into the cell by endocytosis, cleavage of the disulfide bond occurs by its own proteases activating the toxin and the subunit A escapes into the cytoplasm. Subunit A of the toxin, acting as a zinc endopeptidase, selectively cleaves the targeted SNARE protein including SNAP-25, synaptobrevin or syntaxin (depending on type of BoNT), which inhibits the release of acetylcholine at the synapse. Due to this the release of acetylcholine is prevented. This occurs in all 7 serotypes of botulism, resulting in flaccid paralysis. Adapted from Multani et all [83].
2.3. Tetanus Toxin
Tetanus Neurotoxin (TeNT) was first reported in 1884 by Carle and Rattone, when they produced tetanus in rabbits via injection using pus from a deceased tetanus case [82]. This exotoxin is produced by the anaerobic, Gram-positive spore-forming bacterium Clostridium tetani [84]. The spores of TeNT are found naturally in the environment; particularly in soil, intestinal tracts/faeces of both animals and humans and on the surfaces of rusty tools like nails and needles [85]. Tetanospasmin and tetanolysin are two exotoxins produced from C. tetani. Tetanospasmin is the main toxin that is produced and causes disease, and it may be aided further by tetanolysin, but the mechanism is currently unknown [86]. In the year 1889, Kitasato isolated the organism from a fatal human case and provided evidence that tetanospasmin could be neutralised using specific antibodies [87]. In 1897 the effect of passively transferred antitoxin was shown, leading to passive immunisation later being used in treatment as well as prophylaxis during World War I [88]. Since the toxoid vaccine introduction in 1924, tetanus in countries with high vaccine coverage has decreased significantly [89]. Almost half of tetanus deaths are in children under the age of 5, primarily due to low vaccination rates, unsanitary delivery, and poor neonatal care [90].
Same as DT, tetanospasmin is made up of three fragments that combine to form a 150 kDa toxin. One making up subunit A (50-kDa) also known as the light chain (Lc) and the other two fragments that form the B subunit (100-kDa total) also known as the heavy chains (Hc and Hn) [91]. The Hc fragment binds to the axonal membrane, while Hn is responsible for the translocation of the subunit A across the layer in an endosome [91]. This toxin is translated from the tetX gene as one protein, located on the pE88 plasmid [92]. The other toxin produced from C. tetani (tetanolysin), is believed to assist the main toxin (tetanospasmin) but the mechanism is currently unknown [86]. These channels have been previously described as bearing similar properties to bilayer channels used by DT. Tetanospasmin binds to the GD2/GD1b receptors on the presynaptic membranes of peripheral nerves, before migrating via the retrograde axonal transport system to the spinal cord and towards the brainstem [85]. From the brainstem, tetanospasmin reaches the terminal of the glycinergic interneurons and y-aminobutyric acid (GABA) secreting neurons [85,86]. Once reaching the targeted area, chemical reduction of the disulfide bond occurs to separate the Lc [93]. This enables the Lc to degrade synaptobrevin, a SNARE protein, that is also targeted by BoNT, that is needed to dock synaptic vesicles on the presynaptic membrane. This cleavage results in the prevention of both glycine and GABA from being secreted. Without glycine and GABA, the release of acetylcholine is unrestricted causing the paralysis as seen in Figure 5 [91].
The symptoms of TeNT occur in a descending order of muscle spasms, beginning with the head such as lockjaw before gradually worsening, fever, changes in blood pressure and heart rate and eventually spasms plus seizures [94]. The other types of tetanus are neonatal tetanus, a generalised tetanus in new-borns, while localised tetanus and cephalic tetanus are rarer manifestations [94]. Treatments include Human Tetanus Immunoglobulin antibody therapy (HTIG), coupled with the sterilisation and removal of necrotic tissue. This removes free tetanospasmin but the toxin that is already bound to the host is unaffected. This treatment shortens the length of illness and reduces the severity of the disease. Metronidazole is paired too as it has been linked to a decrease in mortality [94].
2.4. Cholera Toxin
Cholera toxin (CTX) is produced by the highly motile, Gram-negative, comma-shaped bacterium Vibrio cholerae. This bacterium causes the disease known as cholera, that originated from the Indian subcontinent [95]. The disease eventually spread outwards to most of Asia in 1817 leading to seven world-wide pandemics [95,96]. There are several different serogroups (>200 known); many containing non-pathogenic cholera, and two serogroups that are pathogenic. These two pathogenic serogroups produce the AB5 Cholera toxin, an enterotoxin that causes illness by inducing the expression of ion channels [97,98]. These two major serological groups are O1 consisting of two biotypes, classical or El Tor and the O139 serogroup. These are also referred to as toxigenic O1 and O139 [95,99]. The genes encoding CTX are carried on the CTXφ bacteriophage, with the ctxA and ctxB (ctxAB) genes encoding the A and B subunits. Other genes involved in increasing virulence are part of a cluster known as the CTX genetic element [98].
After ingestion of the bacterium via the consumption of contaminated water or food, V. cholerae colonise the intestinal lumen and adheres to the mucus layer [100]. The bacterium penetrates the mucus layer and attaches to the gut epithelial cells and begins to proliferate. Under certain host environmental stimuli the production of toxin coregulated pilus (TCP) occurs, resulting in the formation of microcolonies and thereby concentrated secreted toxin [101]. The structure of the 84kDa enterotoxin produced is composed of two subunits A+B linked by a disulfide bond [102,103]. Within the A subunit there are two domains A1 and A2. The A2 fragment, an extended alpha helix, functions by bonding the A and B subunits. Subunit B (56 kDa) consists of five identical B chains (11kDa each), which bind to five ganglioside GM1 receptors on the plasma membrane and triggers endocytosis of the toxin [97]. The A subunit dissociates and is transported into the cytoplasm. The A1 fragment is the pathogenic part, which carries out ADP-ribosylation of the ⍺ subunit of the G-protein (GS). This locks the G protein in its GTP-bound form, stimulating adenylate cyclase to constantly produce cAMP [97,103]. The high cAMP levels activate cystic fibrosis transmembrane conductance regulator (CFTR), increasing intestinal permability and leading to the rapid loss of water and ions from infected enterocytes as seen in Figure 6 [97].
The characteristic symptom of CTX is the rice water diarhoea, causing a rapid loss of fluids and electrolytes, which must be replaced immediately with intravenous fluid such as Ringers lactate solution, saline or oral rehydration salts (ORS) [104,105]. Antibiotics treatments are available for patients with severe dehydration, and the first-line choice is doxycycline followed by tetracycline. For children and pregnant women, other antibiotics such as erythromycin, azithromycin and ciprofloxacin should be used instead [100].
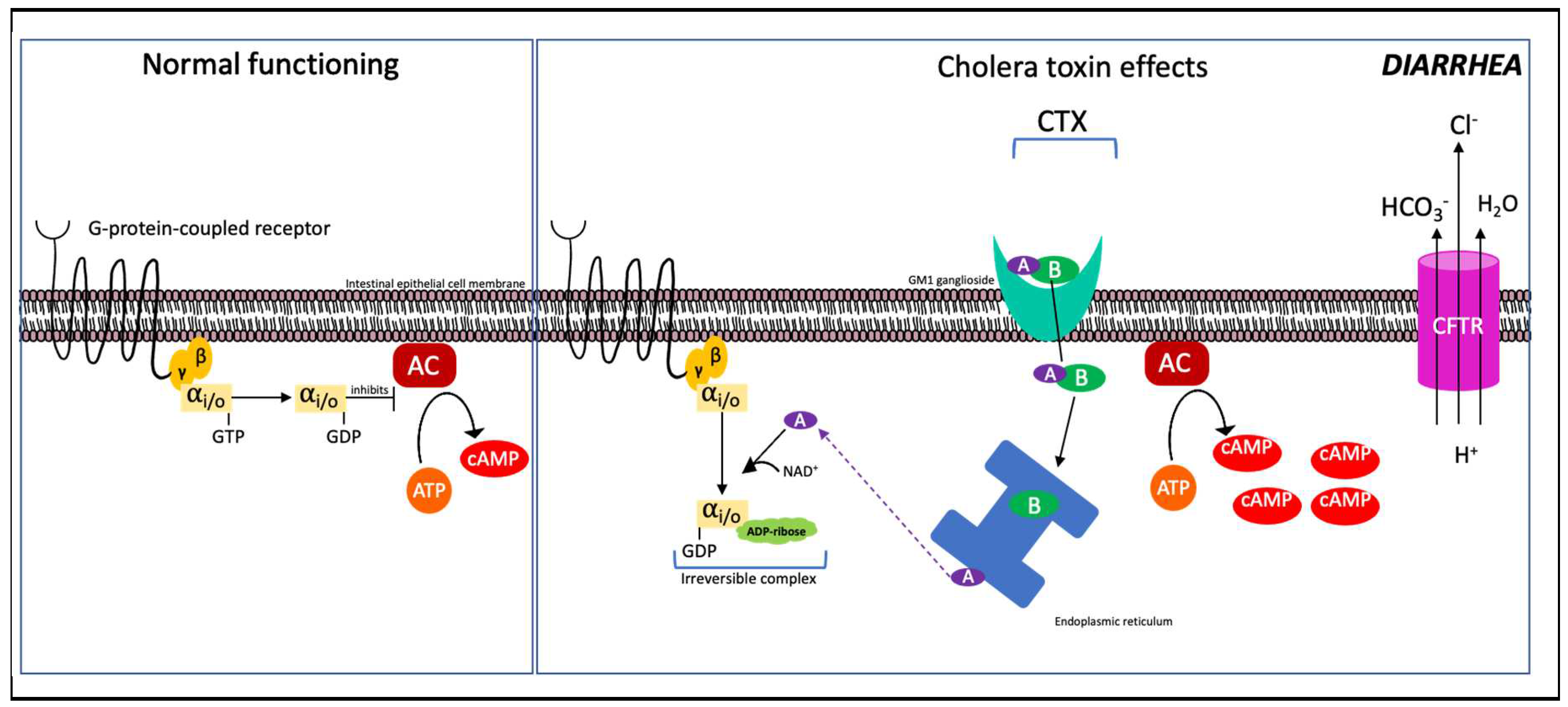
Figure 6.
Mechanism of CTX function. (left) Normal function of G-protein-coupled receptor in inhibiting production of cAMP; (Right) Cholera AB toxin enters the cell via GM1 receptors on intestinal epithelia via endocytosis. The A subunit dissociates from the AB complex in the endoplasmic reticulum and is transported out into the cytoplasm, ADP-ribosylating the ⍺ subunit of the G-protein. ATP is constitutively converted into cAMP, increasing intestinal permeability (also called “leaky gut”) which drives out H2O, HCO3- and Cl-, resulting in watery diarrhoea. Adapted from Ramamurthy et al., 2020 and Gabutti et al., 2020 [106,107].
Figure 6.
Mechanism of CTX function. (left) Normal function of G-protein-coupled receptor in inhibiting production of cAMP; (Right) Cholera AB toxin enters the cell via GM1 receptors on intestinal epithelia via endocytosis. The A subunit dissociates from the AB complex in the endoplasmic reticulum and is transported out into the cytoplasm, ADP-ribosylating the ⍺ subunit of the G-protein. ATP is constitutively converted into cAMP, increasing intestinal permeability (also called “leaky gut”) which drives out H2O, HCO3- and Cl-, resulting in watery diarrhoea. Adapted from Ramamurthy et al., 2020 and Gabutti et al., 2020 [106,107].
2.5. Pertussis Toxin
Pertussis toxin (PTX) is also an AB5 family member, produced by the Gram-negative aerobic pathogen Bordetella pertussis. PTX plays a role in the pathogenesis of whooping cough, a respiratory disease that causes severe coughing and can lead to fractures of the ribcage, hernias, and unconsciousness before eventually leading to death if left untreated [108]. Antibiotics are the recommended treatment, including macrolides erythromycin, clarithromycin, and azithromycin.
PTX acts in a similar way to cholera toxin, preventing the cell from inhibiting the conversion of ATP to cAMP via adenylate cyclase, but with distinct mechanistic differences. Structurally, PTX is made up of six subunits; the A-promoter (S1 subunit) and the B-oligomer (S2-S5 subunit), with two copies of S4. This was determined by Stein et al in 1994, when they reported the crystal structure to a resolution of 2.9 å [109]. Moreover, despite its comparable structure, the pentamer of PTX subunit B displays relatively low sequence homology compared to other AB5 toxins [109].
The A-promoter (s1 subunit) of the toxin enters the cell and induces ADP-ribosylation of the heterotrimeric Gi/o protein’s α-subunit (Gαi/o) in an irreversible reaction as seen in Figure 7 [110]. In the absence of an inhibitory signal from the Gαi/o, adenylate cyclase is constitutively converted to cyclic adenosine monophosphate (cAMP). These molecules are involved in stimuli signalling, thus breaking down communication between cells [110]. cAMP serves as a secondary intracellular messenger which activates protein kinases for downstream signalling and has been linked with developing inflammatory and cancer-related pathologies [111].
Other effects linked to PTX include impaired macrophage function, a rise in white blood cell levels (leukocytosis), and an increase in insulin levels, shown to lead to hypoglycemia [112]. The links between PTX and the characteristic coughing symptom have not been fully elucidated. Earlier research using rat model testing revealed that wild type B. pertussis strains caused coughing, while rats infected with B. pertussis strains lacking PTX did not [112,113]. Despite this, when rat subjects were injected with purified PTX, no coughing was observed, while in rhesus macaques leukocytosis similar to the pathology of normal B. pertussis infection was observed after injections with 25 µg/kg PTX, but not at 12.5 µg/kg [112,114,115,116]. At higher levels the toxin is fully lethal in mice and rats [114].
2.6. Shiga And Shiga-Like Toxins
Shiga and Shiga-like toxins (also known as Stxs and Vero toxins) are a well studied toxin group, first discovered in 1898 by Dr. Kiyoshi Shiga when they reported a dysentery-causing bacillus [117]. In honour of his contribution, this bacterium was given the name Shigella dysenteriae. Five years later, Shiga and Neisser published details on the dysentery-causing toxin produced by the bacteria, as did Conradi when they demonstrated the bacillus paralysing and killing rabbits in the same year [118,119]. 80 years later, Shiga-like toxin production was reported in O157:H7 E. coli strains in the United States of America [120]. Since then, Stxs have been extensively studied, in no small part due to their affinity with E. coli. Similarly to DT, the stx operon can be found harboured in an inducible, lysogenic, lambda-like bacteriophage [121]. Based on their antigens, Stxs are classified into two main groups: Stx1 and Stx2. There are further subdivisions within these groups, three in group 1, and seven in group 2 [122]. Not all toxins are equally potent, with Stx2a demonstrated to be more so than the other Stx2 variants [123]. Meanwhile, Stx1 has been rarely linked to serious human infections [122].
Stx is another member of the AB5 toxin family, consisting of an A subunit (A1 + A2) and five identical B subunits. Subunit B of Stx binds at the glycosphingolipid Gb3 site and the function of Gb3 is currently unknown [121,124]. The mechanism begins with Stx binding to the Gb3 receptors and endocytosis occurs as seen in Figure 8. Cleavage of the bond between the A and B subunits occur by furin during this transport. The A1 and A2 fragments remain together due to the disulfide bond between them. Stx migrates in a retrograde manner inside of the cell, towards the golgi body (apparatus) and further to the endoplasmic reticulum [121,124] where the disulfide bond is reduced and A1 is released into the cytosol. The A1 fragment then targets an adenine in the 28S rRNA of the 60S ribosomal subunit for removal/depurination. As a result, protein synthesis is halted and the ribosome no longer associates with elongation factor 1 leading to cell death [121,124]. Symptomatically, infection typically causes bloody diarrhoea, with some patients developing hemolytic uremic syndrome (HUS). This condition results in blood platelet degradation, anaemia, and eventually kidney failure [125]. It has been found that Gb3 is in higher concentrations in kidney tissue, as well as throughout the central nervous system in neurons, explaining the toxin’s links to kidney diseases and nerve conditions such as paralysis [125,126]. Toxin gene expression had been linked to the late promoter pR’ carried on the Shiga toxin prophage in E. coli, while in S. dysenteriae a B subunit promoter in the Shiga toxin operon was identified as well [127,128,129].
Current treatment recommendations focus on non-specific supportive therapy focussing on hydration [130]. Antibiotics are not recommended as it may enhance production of Stx and increase propagation of Stx Phages, with studies suggesting quinolones and trimethoprim may intensify the risk of HUS [130,131]. Despite years of study, no specific therapy for HUS has passed clinical trials, given the fact that everything from antisera to camelid single chain antibodies, and even a manganese-based treatment, having been through various stages of development [132,133,134]. Some of the main challenges in Stx and HUS treatment development are the sporadic and geographically diverse nature of outbreaks, as well as the limited amount of financial gains available from production.
3. Notable comparisons between DT and other AB Toxins.
The two bacterial AB toxins that are typically compared are BoNT and TeNT, as both use similar mechanisms but with opposite effects. Both of these toxins have a very similar basic structure; the difference being, tetanus has two fragments that form the B subunit (heavy chain) rather than one. These two fragments are the Hc and Hn [91]. In the same way, DT has two fragments in its B component, known as the transmembrane (T) domain and the receptor binding (R) domain [20]. Comparing the mechanism between these three toxins, both bind to the receptors of the presynaptic membranes and then migrate backwards via the axonal transport system towards the cell body [73,91]. Both BoNT and TeNT travel to the spinal cord and brainstem to negatively affect the host. In contrast, DT binds to the HB-EGF and translocates across the plasma membrane to target the EEF2 [20]. The main difference between DT, BoNT and TeNT is the method of the toxin entry. Botulinum is most often caused via ingestion of contaminated food, tetanus is through wound infection/penetration, while DT is normally transmitted through respiratory droplets carrying the toxigenic bacteria in air. Another example of similar mechanisms is CTX and PTX as both affect cAMP production. In CTX the 5 B fragments bind to five ganglioside GM1 receptors on the plasma membrane and trigger endocytosis of the toxin [97]. The A1 fragment activates the G protein GS, locking in its GTP-bound form and stimulating adenylate cyclase to produce cAMP [97]. The increased cAMP levels activate the CFTR, resulting in rapid loss of water and ions. In contrast to CTX, PTX carries out this same process with the exception that it ADP-ribosylates the Ga subunit rather than the Gαs, rendering it unable to inhibit cAMP production, thus increasing the intracellular levels of cAMP [110].
AT has a rather unusual structure, with one B fragment known as protective antigen (PA) and two A fragments subunits edema factor (EF) and lethal factor (LF), which are polar opposites for most bacterial AB toxins other than CTX. The combination of PA (B) and LF (A) has a lethal toxigenic effect, due to LF induced macrophage apoptosis [35]. There are a few bacterial AB toxins, that have similar effect of disrupting cellular processes leading to apoptosis. This includes AT, DT and Stx. In the case of anthrax, macrophage apoptosis occurs due to the LF cleavage of protein kinases (MAPKK1 and 2) inhibiting the signal transduction pathway [40]. In contrast, both DT and Stx prevent protein synthesis leading to apoptosis. DT does this by catalysing the transfer of ADP-ribose from NAD onto the EF-2 of the ribosomal unit and Stx with the cleavage of an adenine residue on the 28S rRNA of a 60S ribosomal subunit. As previously mentioned, the structure of CTX contains two fragments that form subunit A, which is uncommon in bacterial AB toxins. Interestingly, rather than providing a pathogenic function, the A2 fragment in CTX holds together the 5 subunit B fragments [97]. This in contrast to AT, where both A fragments EF and LF act with pathogenic functions.
4. Toxin therapeutics and variant concerns
One of the best preventative measures are vaccinations against the pathogens producing these toxins. However, vaccines are not available for all of these AB toxin producers. As with Shiga, there are no specific therapeutics licensed for use in humans yet. Even with developed therapeutics, in the case of DT, shortages have occurred in many countries. This can be put down to the limited financial incentive to the development and the rarity of outbreaks in many parts of the world. As countries no longer buy the serum regularly, many pharmaceutical companies have scaled down their production lines [135]. Even among the available stock, there are issues with quality control and batches often fail to meet WHO recommended concentration requirements [136]. This is a concern for future vaccine and antitoxin development as with a rise in diphtheria cases, variants of DT are already seen. These isolates will continue to gain mutations producing more distant variants in the future [30]. In the case of DT, there is a effective vaccine, antitoxin (DAT) is available for use on suspected and confirmed diphtheria cases, but not for prophylaxis in the UK. Other avenues such as human antibody therapies, including monoclonals have been explored, but none have yet passed clinical trials [136,137].
For tetanus, since 1924, cases in countries with a high vaccine coverage had decreased massively [89]. This was followed up with a more effective version in 1938. Eventually combined into the DTP vaccine for diphtheria, tetanus and pertussis were introduced in 1948 [138]. Since the introduction of DTP, cases decreased rapidly, although for whooping cough 24 million cases and 160,700 deaths were estimated globally in 2014 among children under 5 years old [139]. A newer acellular DTP vaccine was developed and introduced in the 1990s, although long term coverage has been questioned, leading to both vaccines remaining in use [138]. In the UK a 5-in-1 (Pentavalent) vaccine was used in new-borns until 2017, covering diphtheria, tetanus, pertussis, polio and Hib disease. This was replaced with a 6-in-1 vaccine that also included protection against hepatitis B. Despite the high vaccine coverage in many high-income countries whooping cough remains prevalent, primarily due to waning immunity years after vaccination, alongside adaptation of the bacteria itself [140]. Additional research is required to further elucidate the role of PTX in disease and vaccine-mediated protection, in order to develop more effective treatments and vaccines [112].
Similarly, there was also a vaccine developed for botulism called formalin-inactivated penta-serotype-BoNT/A-E toxoid, however this vaccine was discontinued before it could be approved [81]. This was the decision from the CDC based on their assessment of the available data, which indicated a decline in immunogenicity of some of the toxin serotypes. For anthrax prevention, a killed vaccine is available against B. anthracis and is given to at risk populations, with further vaccines in development [141,142,143,144]. Currently, the only cholera vaccine in the UK is an orally administered inactivated V. cholerae whole-cell vaccine with recombinant CT subunit B (WC/rBS). In countries with more frequent outbreaks such as India, other types of vaccines are used. This vaccine is based on a bivalent killed whole-cell oral cholera (BivWC) formulation, as it uses both whole cells from serogroup 01 and 0139 covering all toxigenic cholera. It is inexpensive to use and effective for up to five years [145].
All of the bacterial AB toxins mentioned in this review necessitate further research into treatment and prevention. In the case of DT, the need is for further research into the toxin evolution, and then onwards to the antitoxin development/usage. The need to understand DT evolution is due to the moderate to high impact of mutations on its protein structure. The implications of this on future vaccine and antitoxin development should be thoroughly investigated, as other variants may already be circulating [30]. AT requires more research into the antitoxin treatment due to evolutionary changes in the toxin. The toxin potency, like that of several others on this list, raises the possibility of its use as a bioweapon, emphasising the importance of effective treatment options. The current research on BoNT focuses on the recent toxins discovered such as BoNT/X. Even with research focusing on new variants and treatments, there is a lack of research in the prevention of infant botulism. There is currently no way to protect an infant below the age of one, if exposed to botulinum spores. In the same light, future research into tetanus toxin should focus on tackling neonatal tetanus, which makes up almost 50% of cases each year [90]. Areas of focus include ways of improving vaccine coverage as well as sanitation. Tetanolysin produced by C. tetani should be a focus of ongoing research, as the mechanism of if or how it does so, are yet to be determined [146]. Recent publications have highlighted the importance of an improved vaccine for pertussis, as well as producing new tools to study this disease [138,147,148]. The links between PTX and whooping cough are still not entirely understood, and will continue to be an area for future study [148]. For shiga toxin further research into the toxin in general is required across its different hosts and the effective treatment of HUS in severe cases, as there is no specific therapy to treat this. The development and testing of cholera rapid diagnostics have been a growing area of cholera research in recent years. The goal of these tools is to bring testing equipment to cholera-endemic areas, allowing for rapid diagnosis and treatment of cholera cases as soon as possible.
4.1. Conclusions
The potential for pathogens carrying this group of toxins to re-emerge and cause havoc as a result of few adaptations or a decrease of vaccinations within communities should be a matter of concern. The year 2019 saw the highest reported number of diphtheria cases since 1996. With reported case numbers rising again, it is more important than ever to understand DT and these other bacterial AB toxins. We continue to live in the seventh pandemic of cholera and its end is not in sight. Tetanus, anthrax, whooping cough and botulism are also still a living reality. It is critical to monitor these toxin mediated diseases and effectiveness of vaccines where available. Only then we would be able to stay one step ahead of these continuously evolving pathogens and their toxins.
Author Contributions
Conceptualization, B.P. and A.M.; writing—original draft preparation, B.P. and A.Ma. and V.S.; writing—review and editing, B.P. and A.Ma. and V.S. and A.M.; visualization, B.P. and A.Ma and V.S.; supervision, B.P. and A.M.; project administration, B.P. V.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable.,
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Law, S.R. From the Stench of Death to an Antidote for Plant Aluminium Toxicity. Physiologia Plantarum 2019, 167, 469–470. [Google Scholar] [CrossRef]
- Barbieri, J.T. Exotoxins. In Encyclopedia of Microbiology (Third Edition); Schaechter, M., Ed.; Academic Press: Oxford, 2009; pp. 355–364. ISBN 978-0-12-373944-5. [Google Scholar]
- Ramaiah, S.K.; Rose, R.E. 9.08 - Endotoxin-Induced Hepatotoxicity. In Comprehensive Toxicology (Second Edition); McQueen, C.A., Ed.; Elsevier: Oxford, 2010; pp. 613–625. ISBN 978-0-08-046884-6. [Google Scholar]
- Yadav, D.K.; Yadav, N.; Khurana, S.M.P. Chapter 26 - Vaccines: Present Status and Applications. In Animal Biotechnology; Verma, A.S., Singh, A., Eds.; Academic Press: San Diego, 2014; pp. 491–508. ISBN 978-0-12-416002-6. [Google Scholar]
- Stetzenbach, L.D. Airborne Infectious Microorganisms. In Encyclopedia of Microbiology (Third Edition); Schaechter, M., Ed.; Academic Press: Oxford, 2009; pp. 175–182. ISBN 978-0-12-373944-5. [Google Scholar]
- Collier, R.J. Understanding the Mode of Action of Diphtheria Toxin: A Perspective on Progress during the 20th Century. Toxicon 2001, 39, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.R. Corynebacterium Diphtheriae. In Medical Microbiology; University of Texas Medical Branch at Galveston, 1996 ISBN 0963117211.
- Piot, N.; van der Goot, F.G.; Sergeeva, O.A. Harnessing the Membrane Translocation Properties of AB Toxins for Therapeutic Applications. Toxins 2021, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Beddoe, T.; Paton, A.W.; Le Nours, J.; Rossjohn, J.; Paton, J.C. Structure, Biological Functions and Applications of the AB5 Toxins. Trends Biochem Sci 2010, 35, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Odumosu, O.; Nicholas, D.; Yano, H.; Langridge, W. AB Toxins: A Paradigm Switch from Deadly to Desirable. Toxins (Basel) 2010, 2, 1612–1645. [Google Scholar] [CrossRef]
- WHO World Health Organization: Immunization, Vaccines And Biologicals. Vaccine Preventable Diseases Vaccines Monitoring System 2020 Global Summary Reference Time Series: DIPHTHERIA. Available online: https://apps.who.int/immunization_monitoring/globalsummary/timeseries/tsincidencediphtheria.html (accessed on 16 October 2021).
- Loeffler, F. Untersuchungen Über Die Bedeutung Der Mikroorganismen Für Die Entstehung Der Diphtherie Beim Menschen, Bei Der Traube Und Beim Kalbe. Mitt KJin Gesundh 1884, 2, 421–499. [Google Scholar]
- Roux, E.; Yersin, A. Contribution a l’etude de La Diphtherie. Annales de l’Institut Pasteur 1888, 2, 620–629. [Google Scholar]
- Behring; Kitasato Ueber Das Zustandekommen Der Diphtherie-Immunitüt Und Der Tetanus-Immunitüt Bei Thieren. Deutsche Medizinische Wochenschrift 1890, 16, 1113–1114. [CrossRef]
- Centers for Disease Control and Prevention (CDC) Diphtheria. In Pink Book; Centers for Disease Control and Prevention: Atlanta, GA, 2012; pp. 75–86.
- World Health Organisation Diphtheria Reported Cases. Available online: http://apps.who.int/immunization_monitoring/globalsummary/timeseries/tsincidencediphtheria.html (accessed on 25 February 2019).
- Dittmann, S.; Wharton, M.; Vitek, C.; Ciotti, M.; Galazka, A.; Guichard, S.; Hardy, I.; Kartoglu, U.; Koyama, S.; Kreysler, J.; et al. Successful Control of Epidemic Diphtheria in the States of the Former Union of Soviet Socialist Republics: Lessons Learned. The Journal of Infectious Diseases 2000, 181, S10–S22. [Google Scholar] [CrossRef]
- Rahman, M.R.; Islam, K. Massive Diphtheria Outbreak among Rohingya Refugees: Lessons Learnt. Journal of Travel Medicine 2019, 26. [Google Scholar] [CrossRef]
- Dureab, F.; Müller, O.; Jahn, A. Resurgence of Diphtheria in Yemen Due to Population Movement. Journal of Travel Medicine 2018, 25. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.G.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The Crystal Structure of Diphtheria Toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.C.; Efstratiou, A.; Mokrousov, I.; Mutreja, A.; Das, B.; Ramamurthy, T. Diphtheria. Nat Rev Dis Primers 2019, 5, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Floros, P.; Gunaratne, D.A.; Chang, A.; Coman, W.B. A Rare Case of Toxigenic Diphtheria Tonsillitis Resistant to Penicillin Causing Sepsis and Death. Australian Journal of Otolaryngology 2018, 1. [Google Scholar] [CrossRef]
- Martini, H.; Soetens, O.; Litt, D.; Fry, N.K.; Detemmerman, L.; Wybo, I.; Desombere, I.; Efstratiou, A.; Piérard, D. Diphtheria in Belgium: 2010-2017. Journal of medical microbiology 2019, 68, 1517–1525. [Google Scholar] [CrossRef]
- Forde, B.M.; Henderson, A.; Playford, E.G.; Looke, D.; Henderson, B.C.; Watson, C.; Steen, J.A.; Sidjabat, H.E.; Laurie, G.; Muttaiyah, S.; et al. Fatal Respiratory Diphtheria Caused by β-Lactam-Resistant Corynebacterium Diphtheriae. Clinical Infectious Diseases 2020. [Google Scholar] [CrossRef]
- Gayatri Amirthalingam; Charlotte Gower; Meera Chand; Mary Ramsay Guidance on the Use of Diphtheria Anti-Toxin (DAT); London, 2018.
- CDC Diphtheria Antitoxin (DAT). Available online: https://www.cdc.gov/diphtheria/dat.html (accessed on 19 August 2020).
- White, A.; Ding, X.; vanderSpek, J.C.; Murphy, J.R.; Ringe, D. Structure of the Metal-Ion-Activated Diphtheria Toxin Repressor/Tox Operator Complex. Nature 1998, 394, 502–506. [Google Scholar] [CrossRef]
- Wang, Z.; Schmitt, M.P.; Holmes, R.K. Characterization of Mutations That Inactivate the Diphtheria Toxin Repressor Gene (DtxR). Infection and Immunity 1994, 62, 1600–1608. [Google Scholar] [CrossRef]
- Wittchen, M.; Busche, T.; Gaspar, A.H.; Lee, J.H.; Ton-That, H.; Kalinowski, J.; Tauch, A. Transcriptome Sequencing of the Human Pathogen Corynebacterium Diphtheriae NCTC 13129 Provides Detailed Insights into Its Transcriptional Landscape and into DtxR-Mediated Transcriptional Regulation. BMC Genomics 2018, 19, 82. [Google Scholar] [CrossRef]
- Will, R.C.; Ramamurthy, T.; Sharma, N.C.; Veeraraghavan, B.; Sangal, L.; Haldar, P.; Pragasam, A.K.; Vasudevan, K.; Kumar, D.; Das, B.; et al. Spatiotemporal Persistence of Multiple, Diverse Clades and Toxins of Corynebacterium Diphtheriae. Nat Commun 2021, 12, 1500. [Google Scholar] [CrossRef]
- Shafiee, F.; Aucoin, M.G.; Jahanian-Najafabadi, A. Targeted Diphtheria Toxin-Based Therapy: A Review Article. Front Microbiol 2019, 10, 2340. [Google Scholar] [CrossRef]
- Woo, J.H.; Lee, Y.-J.; Neville, D.M.; Frankel, A.E. Pharmacology of Anti-CD3 Diphtheria Immunotoxin in CD3 Positive T-Cell Lymphoma Trials. In Immunotherapy of Cancer: Methods and Protocols; Yotnda, P., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2010; pp. 157–175. ISBN 978-1-60761-786-0. [Google Scholar]
- Auger, A.; Park, M.; Nitschke, F.; Minassian, L.M.; Beilhartz, G.L.; Minassian, B.A.; Melnyk, R.A. Efficient Delivery of Structurally Diverse Protein Cargo into Mammalian Cells by a Bacterial Toxin. Mol. Pharmaceutics 2015, 12, 2962–2971. [Google Scholar] [CrossRef]
- Smith, H.; Keppie, J. Observations on Experimental Anthrax: Demonstration of a Specific Lethal Factor Produced in Vivo by Bacillus Anthracis [3]. Nature 1954, 173, 869–870. [Google Scholar] [CrossRef]
- Collier, R.J.; Young, J.A.T. Anthrax Toxin. Annual Review of Cell and Developmental Biology 2003, 19, 45–70. [Google Scholar] [CrossRef]
- van der Goot, G.; Young, J.A.T. Receptors of Anthrax Toxin and Cell Entry. Molecular Aspects of Medicine 2009, 30, 406–412. [Google Scholar] [CrossRef]
- Bhatnagar, R.; Batra, S. Anthrax Toxin. Crit Rev Microbiol 2001, 27, 167–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Moayeri, M.; Zhao, H.; Crown, D.; Leppla, S.H.; Purcell, R.H. Potent Neutralization of Anthrax Edema Toxin by a Humanized Monoclonal Antibody That Competes with Calmodulin for Edema Factor Binding. PNAS 2009, 106, 13487–13492. [Google Scholar] [CrossRef] [PubMed]
- Friebe, S.; van der Goot, F.G.; Bürgi, J. The Ins and Outs of Anthrax Toxin. Toxins 2016, 8, 69. [Google Scholar] [CrossRef]
- Park, J.M.; Greten, F.R.; Li, Z.W.; Karin, M. Macrophage Apoptosis by Anthrax Lethal Factor through P38 MAP Kinase Inhibition. Science 2002, 297, 2048–2051. [Google Scholar] [CrossRef]
- Guichard, A.; Jain, P.; Moayeri, M.; Schwartz, R.; Chin, S.; Zhu, L.; Cruz-Moreno, B.; Liu, J.Z.; Aguilar, B.; Hollands, A.; et al. Anthrax Edema Toxin Disrupts Distinct Steps in Rab11-Dependent Junctional Transport. PLOS Pathogens 2017, 13, e1006603. [Google Scholar] [CrossRef]
- Saile, E.; Koehler, T.M. Control of Anthrax Toxin Gene Expression by the Transition State Regulator AbrB. J Bacteriol 2002, 184, 370–380. [Google Scholar] [CrossRef]
- Mikesell, P.; Ivins, B.E.; Ristroph, J.D.; Dreier, T.M. Evidence for Plasmid-Mediated Toxin Production in Bacillus Anthracis. Infection and Immunity 1983, 39. [Google Scholar] [CrossRef]
- Okinaka, R.T.; Cloud, K.; Hampton, † O; Hoffmaster, A.R.; Hill, K.K.; Keim, P.; Koehler, T.M.; Lamke, G.; Kumano, ‡ S; Mahillon, § J; et al. Sequence and Organization of PXO1, the Large Bacillus Anthracis Plasmid Harboring the Anthrax Toxin Genes. Journal of Bacteriology 1999, 181, 6509–6515. [Google Scholar] [CrossRef]
- Green, B.D.; Battisti, L.; Koehler,’, T.M.; Thorne,’, C.B.; Ivins2, B.E. Demonstration of a Capsule Plasmid in Bacillus Anthracis. Infection and Immunity 1985, 49, 291–297. [Google Scholar] [CrossRef]
- Hoffmann, C.; Zimmermann, F.; Biek, R.; Kuehl, H.; Nowak, K.; Mundry, R.; Agbor, A.; Angedakin, S.; Arandjelovic, M.; Blankenburg, A.; et al. Persistent Anthrax as a Major Driver of Wildlife Mortality in a Tropical Rainforest. Nature 2017, 548, 82–85. [Google Scholar] [CrossRef]
- Dragon, D.C.; Rennie, R.P. The Ecology of Anthrax Spores: Tough but Not Invincible. The Canadian Veterinary Journal 1995, 36, 295–301. [Google Scholar] [PubMed]
- Chambers, J.; Yarrarapu, S.N.S.; Mathai, J.K. Anthrax Infection. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2021. [Google Scholar]
- Goldenberg, A.; Shmueli, G.; Caruana, R.A.; Fienberg, S.E. Early Statistical Detection of Anthrax Outbreaks by Tracking Over-the-Counter Medication Sales. Proceedings of the National Academy of Sciences of the United States of America 2002, 99, 5237–5240. [Google Scholar] [CrossRef] [PubMed]
- Mahan, C.M.; Kang, H.K.; Dalager, N.A.; Heller, J.M. Anthrax Vaccination and Self-Reported Symptoms, Functional Status, and Medical Conditions in the National Health Survey of Gulf War Era Veterans and Their Families. Annals of Epidemiology 2004, 14, 81–88. [Google Scholar] [CrossRef] [PubMed]
- CDC Treatment | Anthrax. Available online: https://www.cdc.gov/anthrax/medical-care/treatment.html (accessed on 4 September 2020).
- McLaughlin, H.P.; Bugrysheva, J. V.; Conley, A.B.; Gulvik, C.A.; Cherney, B.; Kolton, C.B.; Marston, C.K.; Saile, E.; Swaney, E.; Lonsway, D.; et al. Rapid Nanopore Whole-Genome Sequencing for Anthrax Emergency Preparedness. Emerging Infectious Diseases 2020, 26, 358–361. [Google Scholar] [CrossRef]
- Martchenko, M.; Candille, S.I.; Tang, H.; Cohen, S.N. Human Genetic Variation Altering Anthrax Toxin Sensitivity. Proc Natl Acad Sci U S A 2012, 109, 2972–2977. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Shi, M.; Ye, B.; Shen, W.; Li, P.; Xing, L.; Zhang, X.; Hou, L.; Xu, J.; et al. Anthrax Susceptibility: Human Genetic Polymorphisms Modulating ANTXR2 Expression. Toxins (Basel) 2015, 8, E1. [Google Scholar] [CrossRef]
- Bachran, C.; Leppla, S.H. Tumor Targeting and Drug Delivery by Anthrax Toxin. Toxins 2016, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Wein, A.N.; Peters, D.E.; Valivullah, Z.; Hoover, B.J.; Tatineni, A.; Ma, Q.; Fattah, R.; Bugge, T.H.; Leppla, S.H.; Liu, S. An Anthrax Toxin Variant with an Improved Activity in Tumor Targeting. Sci Rep 2015, 5, 16267. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ma, Q.; Fattah, R.; Bugge, T.H.; Leppla, S.H. Anti-Tumor Activity of Anthrax Toxin Variants That Form a Functional Translocation Pore by Intermolecular Complementation. Oncotarget 2017, 8, 65123–65131. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.D.; Fattah, R.J.; Crown, D.; Zhang, Y.; Liu, S.; Moayeri, M.; Fischer, E.R.; Hansen, B.T.; Ghirlando, R.; Nestorovich, E.M.; et al. Engineering Anthrax Toxin Variants That Exclusively Form Octamers and Their Application to Targeting Tumors. J Biol Chem 2013, 288, 9058–9065. [Google Scholar] [CrossRef] [PubMed]
- Hilmas, C.J.; Anderson, J. Chapter 29 - Anthrax. In Handbook of Toxicology of Chemical Warfare Agents (Second Edition); Gupta, R.C., Ed.; Academic Press: Boston, 2015; pp. 387–410. ISBN 978-0-12-800159-2. [Google Scholar]
- Martin, G.J.; Friedlander, A.M. 209 - Bacillus anthracis (Anthrax). In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases (Eighth Edition); Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; W.B. Saunders: Philadelphia, 2015; pp. 2391–2409.e2. ISBN 978-1-4557-4801-3. [Google Scholar]
- Takahashi, H.; Keim, P.; Kaufmann, A.F.; Keys, C.; Smith, K.L.; Taniguchi, K.; Inouye, S.; Kurata, T. Bacillus Anthracis Bioterrorism Incident, Kameido, Tokyo, 1993. Emerg Infect Dis 2004, 10, 117–120. [Google Scholar] [CrossRef]
- The Threat of an Anthrax Attack | CDC. Available online: https://www.cdc.gov/anthrax/bioterrorism/threat.html (accessed on 31 August 2021).
- Peng Chen, Z.; Morris, J.G.; Rodriguez, R.L.; Wagle Shukla, A.; Tapia-Núñez, J.; Okun, M.S. Emerging Opportunities for Serotypes of Botulinum Neurotoxins. Toxins (Basel) 2012, 4, 1196–1222. [Google Scholar] [CrossRef]
- Erbguth, F.J. From Poison to Remedy: The Chequered History of Botulinum Toxin. J Neural Transm (Vienna) 2008, 115, 559–565. [Google Scholar] [CrossRef]
- Erbguth, F.J. Historical Notes on Botulism, Clostridium Botulinum, Botulinum Toxin, and the Idea of the Therapeutic Use of the Toxin. Mov Disord 2004, 19 Suppl 8, S2–6. [Google Scholar] [CrossRef]
- Dhaked, R.K.; Singh, M.K.; Singh, P.; Gupta, P. Botulinum Toxin: Bioweapon & Magic Drug. Indian J Med Res 2010, 132, 489–503. [Google Scholar]
- Aureli, P. Botulism. In International Encyclopedia of Public Health; Elsevier, 2017; pp. 254–262 ISBN 978-0-12-803708-9.
- Nayyar, P.; Kumar, P.; Nayyar, P.V.; Singh, A. BOTOX: Broadening the Horizon of Dentistry. J Clin Diagn Res 2014, 8, ZE25–29. [Google Scholar] [CrossRef] [PubMed]
- Nigam, P.K.; Nigam, A. Botulinum Toxin. Indian J Dermatol 2010, 55, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Sheen, D.; Clarkson, E. Botox and Dermal Fillers: Review and Its Role in the Dental Office. Dent Clin North Am 2020, 64, 325–339. [Google Scholar] [CrossRef]
- Poulain, B.; Molgó, J.; Popoff, M.R. 11 - Clostridial neurotoxins: from the cellular and molecular mode of action to their therapeutic use. In The Comprehensive Sourcebook of Bacterial Protein Toxins (Fourth Edition); Alouf, J., Ladant, D., Popoff, M.R., Eds.; Academic Press: Boston, 2015; pp. 287–336. ISBN 978-0-12-800188-2. [Google Scholar]
- Zanetti, G.; Sikorra, S.; Rummel, A.; Krez, N.; Duregotti, E.; Negro, S.; Henke, T.; Rossetto, O.; Binz, T.; Pirazzini, M. Botulinum Neurotoxin C Mutants Reveal Different Effects of Syntaxin or SNAP-25 Proteolysis on Neuromuscular Transmission. PLoS Pathog 2017, 13, e1006567. [Google Scholar] [CrossRef] [PubMed]
- von Berg, L.; Stern, D.; Pauly, D.; Mahrhold, S.; Weisemann, J.; Jentsch, L.; Hansbauer, E.-M.; Müller, C.; Avondet, M.A.; Rummel, A.; et al. Functional Detection of Botulinum Neurotoxin Serotypes A to F by Monoclonal Neoepitope-Specific Antibodies and Suspension Array Technology. Sci Rep 2019, 9, 5531. [Google Scholar] [CrossRef]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol Rev 2017, 69, 200–235. [Google Scholar] [CrossRef]
- Lu, B. The Destructive Effect of Botulinum Neurotoxins on the SNARE Protein: SNAP-25 and Synaptic Membrane Fusion. PeerJ 2015, 3, e1065. [Google Scholar] [CrossRef]
- Pellett, S.; Yaksh, T.L.; Ramachandran, R. Current Status and Future Directions of Botulinum Neurotoxins for Targeting Pain Processing. Toxins (Basel) 2015, 7, 4519–4563. [Google Scholar] [CrossRef]
- Jeffery, L.A.; Karim, S. Botulism. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2021. [Google Scholar]
- Rao, A.K.; Lin, N.H.; Griese, S.E.; Chatham-Stephens, K.; Badell, M.L.; Sobel, J. Clinical Criteria to Trigger Suspicion for Botulism: An Evidence-Based Tool to Facilitate Timely Recognition of Suspected Cases During Sporadic Events and Outbreaks. Clinical Infectious Diseases 2018, 66, S38–S42. [Google Scholar] [CrossRef]
- Lindström, M.; Korkeala, H. Laboratory Diagnostics of Botulism. Clin Microbiol Rev 2006, 19, 298–314. [Google Scholar] [CrossRef]
- Yu, P.A.; Lin, N.H.; Mahon, B.E.; Sobel, J.; Yu, Y.; Mody, R.K.; Gu, W.; Clements, J.; Kim, H.-J.; Rao, A.K. Safety and Improved Clinical Outcomes in Patients Treated With New Equine-Derived Heptavalent Botulinum Antitoxin. Clin Infect Dis 2017, 66, S57–S64. [Google Scholar] [CrossRef] [PubMed]
- Sundeen, G.; Barbieri, J.T. Vaccines against Botulism. Toxins (Basel) 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Vanella de Cuetos, E.E.; Fernandez, R.A.; Bianco, M.I.; Sartori, O.J.; Piovano, M.L.; Lúquez, C.; de Jong, L.I.T. Equine Botulinum Antitoxin for the Treatment of Infant Botulism. Clin Vaccine Immunol 2011, 18, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- Multani, I.; Manji, J.; Hastings-Ison, T.; Khot, A.; Graham, K. Botulinum Toxin in the Management of Childrenwith Cerebral Palsy. Pediatr Drugs 2019, 21, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Stock, I. [Tetanus and Clostridium tetani--a brief review]. Med Monatsschr Pharm 2015, 38, 57–60. [Google Scholar]
- Hassel, B. Tetanus: Pathophysiology, Treatment, and the Possibility of Using Botulinum Toxin against Tetanus-Induced Rigidity and Spasms. Toxins (Basel) 2013, 5, 73–83. [Google Scholar] [CrossRef]
- Nathan, B.R.; Bleck, T.P. CHAPTER 42 - Tetanus. In Tropical Infectious Diseases: Principles, Pathogens and Practice (Third Edition); Guerrant, R.L., Walker, D.H., Weller, P.F., Eds.; W.B. Saunders: Edinburgh, 2011; pp. 284–288. ISBN 978-0-7020-3935-5. [Google Scholar]
- Aubert, N.; Brachet-Botineau, M.; de Olivera Preto, G.E.; Benz-de Bretagne, I.; Watier, H.; Brachet, G. History, Extensive Characterization and Challenge of Anti-Tetanus Serum from World War I: Exciting Remnants and Deceived Hopes. Immunol Res 2020, 68, 7–12. [Google Scholar] [CrossRef]
- Pinkbook: Tetanus | CDC. Available online: https://www.cdc.gov/vaccines/pubs/pinkbook/tetanus.html (accessed on 27 June 2021).
- Callison, C.; Nguyen, H. Tetanus Prophylaxis. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2021. [Google Scholar]
- Behrens, H.; Ochmann, S.; Dadonaite, B.; Roser, M. Tetanus. Our World in Data 2019. [Google Scholar]
- Pellizzari, R.; Rossetto, O.; Schiavo, G.; Montecucco, C. Tetanus and Botulinum Neurotoxins: Mechanism of Action and Therapeutic Uses. Philos Trans R Soc Lond B Biol Sci 1999, 354, 259–268. [Google Scholar] [CrossRef]
- Eisel, U.; Jarausch, W.; Goretzki, K.; Henschen, A.; Engels, J.; Weller, U.; Hudel, M.; Habermann, E.; Niemann, H. Tetanus Toxin: Primary Structure, Expression in E. Coli, and Homology with Botulinum Toxins. EMBO J 1986, 5, 2495–2502. [Google Scholar] [CrossRef]
- Bleck, T.P.; Reddy, P. Chapter 16 - Toxin-mediated syndromes of the nervous system. In Handbook of Clinical Neurology; Bacterial Infections of the Central Nervous System; Elsevier, 2010; Vol. 96, pp. 257–272.
- Bae, C.; Bourget, D. Tetanus. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2021. [Google Scholar]
- Harris, J.B.; LaRocque, R.C.; Qadri, F.; Ryan, E.T.; Calderwood, S.B. Cholera. The Lancet 2012, 379, 2466–2476. [Google Scholar] [CrossRef]
- Lippi, D.; Gotuzzo, E. The Greatest Steps towards the Discovery of Vibrio Cholerae. Clin Microbiol Infect 2014, 20, 191–195. [Google Scholar] [CrossRef]
- Bharati, K.; Ganguly, N.K. Cholera Toxin: A Paradigm of a Multifunctional Protein. Indian J Med Res 2011, 133, 179–187. [Google Scholar]
- Faruque, S.M.; Albert, M.J.; Mekalanos, J.J. Epidemiology, Genetics, and Ecology of Toxigenic Vibrio Cholerae. Microbiol Mol Biol Rev 1998, 62, 1301–1314. [Google Scholar] [CrossRef]
- Nelson, E.J.; Harris, J.B.; Morris, J.G.; Calderwood, S.B.; Camilli, A. Cholera Transmission: The Host, Pathogen and Bacteriophage Dynamic. Nat Rev Microbiol 2009, 7, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Ojeda Rodriguez, J.A.; Kahwaji, C.I. Vibrio Cholerae. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2021. [Google Scholar]
- Almagro-Moreno, S.; Pruss, K.; Taylor, R.K. Intestinal Colonization Dynamics of Vibrio Cholerae. PLoS Pathog 2015, 11, e1004787. [Google Scholar] [CrossRef]
- Moss, J.; Haun, R.S.; Tsai, S.-C.; Welsh, C.F.; Scott Lee, F.-J.; Russ Price, S.; Vaughan, M. [5] Activation of cholera toxin by ADP-ribosylation factors: 20-kDa guanine nucleotide-binding proteins. In Methods in Enzymology; Iyengar, R., Ed.; Heterotrimeric G Proteins; Academic Press, 1994; Vol. 237, pp. 44–63.
- Sanchez, J.; Holmgren, J. Cholera Toxin - a Foe & a Friend. Indian J Med Res 2011, 133, 153–163. [Google Scholar] [PubMed]
- Information, N.C. for B.; Pike, U.S.N.L. of M. 8600 R.; MD, B.; Usa, 20894 Diarrhoea; World Health Organization, 2013.
- Dan-Nwafor, C.C.; Ogbonna, U.; Onyiah, P.; Gidado, S.; Adebobola, B.; Nguku, P.; Nsubuga, P. A Cholera Outbreak in a Rural North Central Nigerian Community: An Unmatched Case-Control Study. BMC Public Health 2019, 19, 112. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, T.; Nandy, R.K.; Mukhopadhyay, A.K.; Dutta, S.; Mutreja, A.; Okamoto, K.; Miyoshi, S.-I.; Nair, G.B.; Ghosh, A. Virulence Regulation and Innate Host Response in the Pathogenicity of Vibrio Cholerae. Frontiers in Cellular and Infection Microbiology 2020, 10, 520. [Google Scholar] [CrossRef]
- Gabutti, G.; Rossanese, A.; Tomasi, A.; Giuffrida, S.; Nicosia, V.; Barriga, J.; Florescu, C.; Sandri, F.; Stefanati, A. Cholera, the Current Status of Cholera Vaccines and Recommendations for Travellers. Vaccines 2020, 8, 606. [Google Scholar] [CrossRef]
- Kilgore, P.E.; Salim, A.M.; Zervos, M.J.; Schmitt, H.J. Pertussis: Microbiology, Disease, Treatment, and Prevention. Clinical Microbiology Reviews 2016, 29, 449–486. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Cockle, S.A.; Klein, M.H.; Read, R.J. The Crystal Structure of Pertussis Toxin. Structure 1994, 2, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Teter, K. Intracellular Trafficking and Translocation of Pertussis Toxin. Toxins 2019, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Gao, L.-N.; Cui, Y.-L.; Zhang, Y.; Zhou, X. The Cyclic AMP Signaling Pathway: Exploring Targets for Successful Drug Discovery (Review). Molecular Medicine Reports 2016, 13, 3715–3723. [Google Scholar] [CrossRef]
- Gregg, K.A.; Merkel, T.J. Pertussis Toxin: A Key Component in Pertussis Vaccines? Toxins 2019, 11, 557. [Google Scholar] [CrossRef]
- Parton, R.; Hall, E.; Wardlaw, A.C.Y. 1994 Responses to Bordetella Pertussis Mutant Strains and to Vaccination in the Coughing Rat Model of Pertussis. Journal of Medical Microbiology 40, 307–312. [CrossRef]
- Toyota, T.; Kai, Y.; Kakizaki, M.; Sakai, A.; Goto, Y.; Yajima, M.; Ui, M. Effects of Islet-Activating Protein (IAP) on Blood Glucose and Plasma Insulin in Healthy Volunteers (Phase 1 Studies). The Tohoku Journal of Experimental Medicine 1980, 130, 105–116. [Google Scholar] [CrossRef]
- Hinds, P.W.; Yin, C.; Salvato, M.S.; Pauza, C.D. Pertussis Toxin Induces Lymphocytosis in Rhesus Macaques. Journal of Medical Primatology 1996, 25, 375–381. [Google Scholar] [CrossRef]
- Warfel, J.M.; Beren, J.; Kelly, V.K.; Lee, G.; Merkel, T.J. Nonhuman Primate Model of Pertussis. Infection and Immunity 2012, 80, 1530–1536. [Google Scholar] [CrossRef]
- K Shiga Ueber Den Disenteriebacillus (Bacillus Dysenteriae). Shiga, K. (1898). Ueber den Disenteriebacillus (Bacillus dysenteriae). Zentralblat fuer Bakteriologie, Parasitenkunde und Infektionskrankheiten Erste Abteilung 1898, 24, 913–918.
- Neisser, M.; Shiga, K. Ueber Freie Receptoren von Typhus-Und Dysenteriebazillen Und Über Das Dysenterietoxin. DMW-Deutsche Medizinische Wochenschrift 1903, 29, 61–62. [Google Scholar] [CrossRef]
- H Conradi Ueber Lösliche, Durch Aseptische Autolyse Erhaltene Giftstoffe von Ruhr-Und Typhusbazillen. DMW-Deutsche Medizinische Wochenschrift 1903, 29, 26–28. [CrossRef]
- O’Brien, A.D.; Lively, T.A.; Chen, M.E.; Rothman, S.W.; Formal, S.B. Escherichia Coli O157:H7 Strains Associated with Haemorrhagic Colitis in the United States Produce a Shigella Dysenteriae 1 (SHIGA) like Cytotoxin. The Lancet 1983, 321, 702. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiology Spectrum 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Baranzoni, G.M.; Fratamico, P.M.; Gangiredla, J.; Patel, I.; Bagi, L.K.; Delannoy, S.; Fach, P.; Boccia, F.; Anastasio, A.; Pepe, T. Characterization of Shiga Toxin Subtypes and Virulence Genes in Porcine Shiga Toxin-Producing Escherichia Coli. Front Microbiol 2016, 7, 574. [Google Scholar] [CrossRef]
- Fuller, C.A.; Pellino, C.A.; Flagler, M.J.; Strasser, J.E.; Weiss, A.A. Shiga Toxin Subtypes Display Dramatic Differences in Potency. Infect Immun 2011, 79, 1329–1337. [Google Scholar] [CrossRef]
- Puente, J.L.; Finlay, B.B. CHAPTER 9 - Pathogenic Escherichia coli. In Principles of Bacterial Pathogenesis; Groisman, E.A., Ed.; Academic Press: San Diego, 2001; pp. 387–456. ISBN 978-0-12-304220-0. [Google Scholar]
- Obrig, T.G. Escherichia Coli Shiga Toxin Mechanisms of Action in Renal Disease. Toxins 2010, 2, 2769–2794. [Google Scholar] [CrossRef]
- Obata, F.; Tohyama, K.; Bonev, A.D.; Kolling, G.L.; Keepers, T.R.; Gross, L.K.; Nelson, M.T.; Sato, S.; Obrig, T.G. Shiga Toxin 2 Affects the Central Nervous System through Receptor Globotriaosylceramide Localized to Neurons. J Infect Dis 2008, 198, 1398–1406. [Google Scholar] [CrossRef]
- Habib, N.F.; Jackson, M.P. Identification of a B Subunit Gene Promoter in the Shiga Toxin Operon of Shigella Dysenteriae 1. Journal of Bacteriology 1992, 174, 6498–6507. [Google Scholar] [CrossRef]
- Wagner, P.L.; Neely, M.N.; Zhang, X.; Acheson, D.W.K.; Waldor, M.K.; Friedman, D.I. Role for a Phage Promoter in Shiga Toxin 2 Expression from a Pathogenic Escherichia ColiStrain. Journal of Bacteriology 2001, 183, 2081–2085. [Google Scholar] [CrossRef]
- Zhang, L.X.; Simpson, D.J.; McMullen, L.M.; Gänzle, M.G. Comparative Genomics and Characterization of the Late Promoter PR’ from Shiga Toxin Prophages in Escherichia Coli. Viruses 2018, 10, 595. [Google Scholar] [CrossRef]
- CDC Questions and Answers | E. Coli | CDC. Available online: https://www.cdc.gov/ecoli/general/index.html (accessed on 3 September 2020).
- Mühlen, S.; Dersch, P. Treatment Strategies for Infections With Shiga Toxin-Producing Escherichia Coli. Frontiers in Cellular and Infection Microbiology 2020, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Mejías, M.P.; Hiriart, Y.; Lauché, C.; Fernández-Brando, R.J.; Pardo, R.; Bruballa, A.; Ramos, M. V.; Goldbaum, F.A.; Palermo, M.S.; Zylberman, V. Development of Camelid Single Chain Antibodies against Shiga Toxin Type 2 (Stx2) with Therapeutic Potential against Hemolytic Uremic Syndrome (HUS). Scientific Reports 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga Toxin Therapeutics: Beyond Neutralization. Toxins 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Linstedt, A.D. Manganese Blocks Intracellular Trafficking of Shiga Toxin and Protects against Shiga Toxicosis. Science 2012, 335, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.S.; Stickings, P.; White, J.M.; Neal, S.; Crowcroft, N.S.; Sesardic, D.; Efstratiou, A. A Review of the International Issues Surrounding the Availability and Demand for Diphtheria Antitoxin for Therapeutic Use. Vaccine 2009, 28, 14–20. [Google Scholar] [CrossRef]
- Huygen, K. Development of Human Monoclonal Antibodies to Diphtheria Toxin: A Solution for the Increasing Lack of Equine DAT for Therapeutic Use? Virulence 2016, 7, 613–615. [Google Scholar] [CrossRef]
- Wenzel, E.V.; Bosnak, M.; Tierney, R.; Schubert, M.; Brown, J.; Dübel, S.; Efstratiou, A.; Sesardic, D.; Stickings, P.; Hust, M. Human Antibodies Neutralizing Diphtheria Toxin in Vitro and in Vivo. Scientific Reports 2020, 10, 1–21. [Google Scholar] [CrossRef]
- Kuchar, E.; Karlikowska-Skwarnik, M.; Han, S.; Nitsch-Osuch, A. Pertussis: History of the disease and current prevention failure. In Pulmonary Dysfunction and Disease; Springer New York LLC, 2016; pp. 77–82.
- Yeung, K.H.T.; Duclos, P.; Nelson, E.A.S.; Hutubessy, R.C.W. An Update of the Global Burden of Pertussis in Children Younger than 5 Years: A Modelling Study. The Lancet Infectious Diseases 2017, 17, 974–980. [Google Scholar] [CrossRef]
- Mooi, F.R.; Van Der Maas, N.A.T.; De Melker, H.E. Pertussis Resurgence: Waning Immunity and Pathogen Adaptation - Two Sides of the Same Coin. Epidemiology and Infection 2014, 142, 685–694. [Google Scholar] [CrossRef]
- Hopkins, R.J.; Howard, C.; Hunter-Stitt, E.; Kaptur, P.E.; Pleune, B.; Muse, D.; Sheldon, E.; Davis, M.; Strout, C.; Vert-Wong, K. Phase 3 Trial Evaluating the Immunogenicity and Safety of a Three-Dose BioThrax® Regimen for Post-Exposure Prophylaxis in Healthy Adults. Vaccine 2014, 32, 2217–2224. [Google Scholar] [CrossRef]
- Gu, M.L.; Leppla, S.H.; Klinman, D.M. Protection against Anthrax Toxin by Vaccination with a DNA Plasmid Encoding Anthrax Protective Antigen. Vaccine 1999, 17, 340–344. [Google Scholar] [CrossRef]
- Verma, A.; Burns, D.L. Improving the Stability of Recombinant Anthrax Protective Antigen Vaccine. Vaccine 2018, 36, 6379–6382. [Google Scholar] [CrossRef] [PubMed]
- Bower, W.A.; Schiffer, J.; Atmar, R.L.; Keitel, W.A.; Friedlander, A.M.; Liu, L.; Yu, Y.; Stephens, D.S.; Quinn, C.P.; Hendricks, K. Use of Anthrax Vaccine in the United States: Recommendations of the Advisory Committee on Immunization Practices, 2019. MMWR Recommendations and Reports 2019, 68, 1–20. [Google Scholar] [CrossRef]
- Desai, S.N.; Cravioto, A.; Sur, D.; Kanungo, S. Maximizing Protection from Use of Oral Cholera Vaccines in Developing Country Settings. Hum Vaccin Immunother 2014, 10, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, N.; Mitsui, K.; Hase, J. Purification and Some Properties of Tetanolysin. Microbiol Immunol 1980, 24, 575–584. [Google Scholar] [CrossRef]
- Choi, Y.H.; Campbell, H.; Amirthalingam, G.; van Hoek, A.J.; Miller, E. Investigating the Pertussis Resurgence in England and Wales, and Options for Future Control. BMC Medicine 2016, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, K.M.; Snyder, Y.G.; Skerry, C.; Carbonetti, N.H. Fatal Pertussis in the Neonatal Mouse Model Is Associated with Pertussis Toxin-Mediated Pathology beyond the Airways. Infection and Immunity 2017, 85. [Google Scholar] [CrossRef]
Figure 1.
Simplified graphical description of AB toxin mode of action. 1) Bacterial cell secretes AB toxin into the environment. 2) The B subunit binds to the target cell’s membrane. 3) The AB toxin permeates across and into the cell as a vacuole (excluding CTX and few other AB toxins where subunit B does not enter). 4) The bond connecting the A and B subunits breaks, and the A subunit enters the cytoplasm as an active toxin.
Figure 1.
Simplified graphical description of AB toxin mode of action. 1) Bacterial cell secretes AB toxin into the environment. 2) The B subunit binds to the target cell’s membrane. 3) The AB toxin permeates across and into the cell as a vacuole (excluding CTX and few other AB toxins where subunit B does not enter). 4) The bond connecting the A and B subunits breaks, and the A subunit enters the cytoplasm as an active toxin.
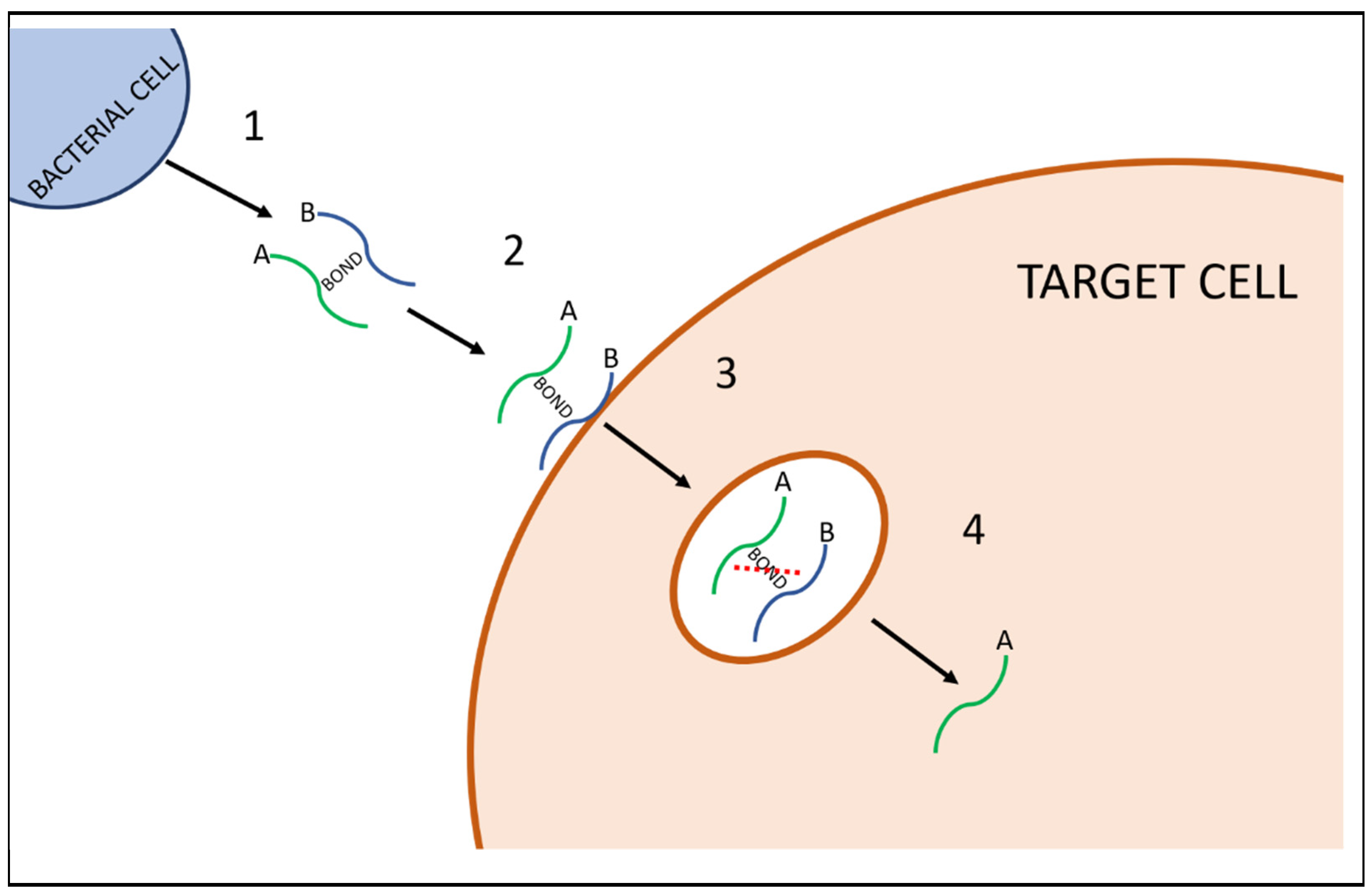
Figure 2.
Graphical description of the mechanism of working DT. DT has three parts the catalytic domain (C) aka subunit A, the transmembrane (T) domain, and the receptor binding (R) domain, which both make up subunit B. The B subunit binds to HB-EGF and translocation of the toxin across the phospholipid bilayer takes place via endocytosis. Cleavage of the bond between DT subunits by endosome-associated proteases and acidification of the endosome occurs allowing subunit A to be translocated across the endosomal membrane into the cytosol. Once in the cytosol, the A subunit catalyses the transfer of ADP-ribose of NAD onto the elongation factor 2 (EF-2). This ADP-ribosylation, prevents protein synthesis inside the cell, leading to apoptosis-driven cell death. Adapted from Sharma et al [21].
Figure 2.
Graphical description of the mechanism of working DT. DT has three parts the catalytic domain (C) aka subunit A, the transmembrane (T) domain, and the receptor binding (R) domain, which both make up subunit B. The B subunit binds to HB-EGF and translocation of the toxin across the phospholipid bilayer takes place via endocytosis. Cleavage of the bond between DT subunits by endosome-associated proteases and acidification of the endosome occurs allowing subunit A to be translocated across the endosomal membrane into the cytosol. Once in the cytosol, the A subunit catalyses the transfer of ADP-ribose of NAD onto the elongation factor 2 (EF-2). This ADP-ribosylation, prevents protein synthesis inside the cell, leading to apoptosis-driven cell death. Adapted from Sharma et al [21].
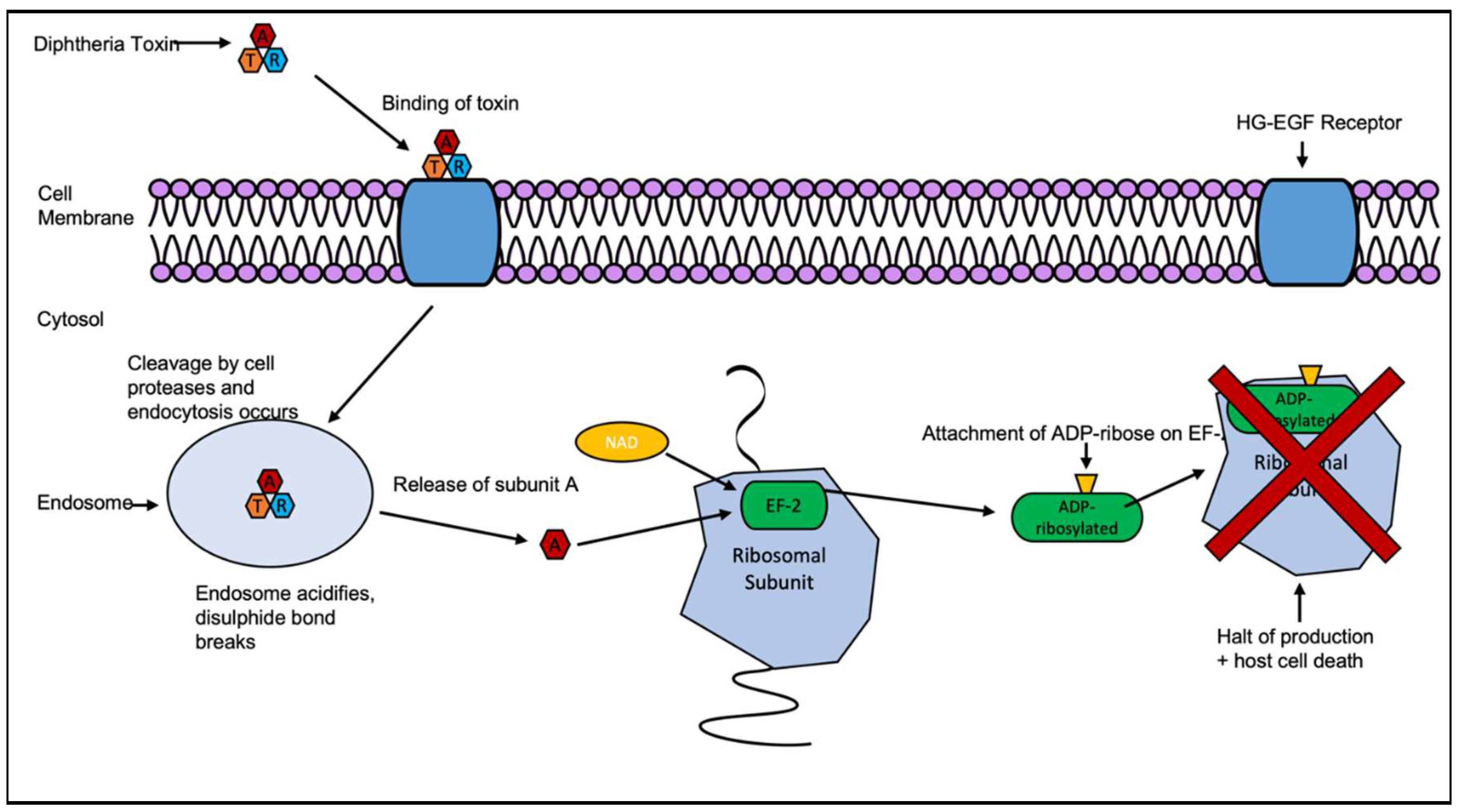
Figure 3.
An overview of the working of anthrax toxin. AT is made up of three proteins: the B subunit anthrax protective antigen (PA), and the A subunits edema factor (EF) and lethal factor (LF). PA binds to the anthrax toxin receptors ANTXR1 (TEM8) or ANTXR2 (CMG2) and cleaved by cell surface protease furin, before endocytosis of the EF and LF into the cytosol. Once inside, EF a CaM-dependent adenylate cyclase converts molecules of ATP to cAMP leading to an increased production of cAMP causing local inflammation and edema. LF is a protease that cleaves MAPKK1 and MAPKK2 and this cleavage inactivates the kinases inhibiting the signal transduction pathway, leading to macrophage lysis. Adapted from Friebe et all [39].
Figure 3.
An overview of the working of anthrax toxin. AT is made up of three proteins: the B subunit anthrax protective antigen (PA), and the A subunits edema factor (EF) and lethal factor (LF). PA binds to the anthrax toxin receptors ANTXR1 (TEM8) or ANTXR2 (CMG2) and cleaved by cell surface protease furin, before endocytosis of the EF and LF into the cytosol. Once inside, EF a CaM-dependent adenylate cyclase converts molecules of ATP to cAMP leading to an increased production of cAMP causing local inflammation and edema. LF is a protease that cleaves MAPKK1 and MAPKK2 and this cleavage inactivates the kinases inhibiting the signal transduction pathway, leading to macrophage lysis. Adapted from Friebe et all [39].
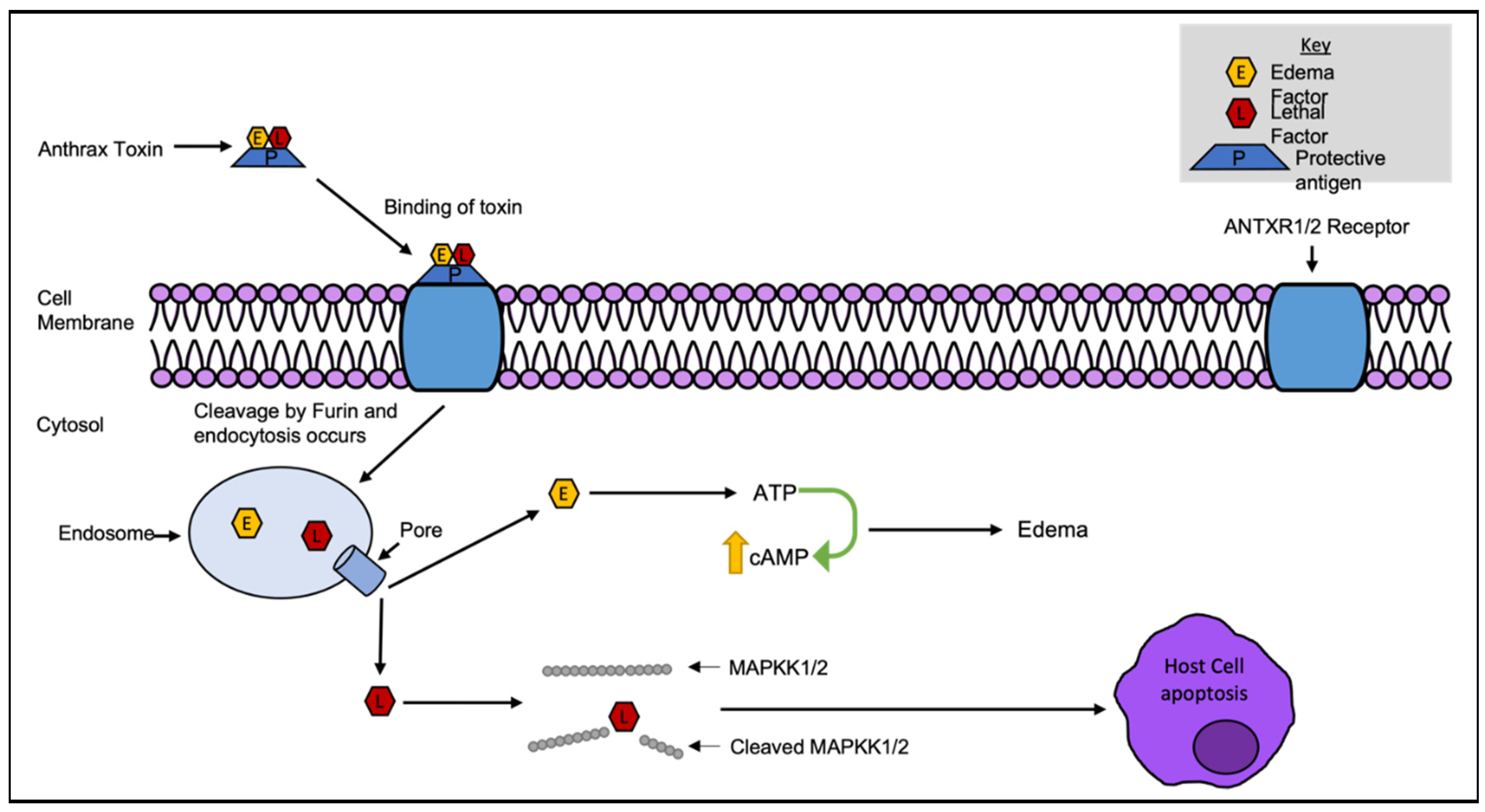
Figure 5.
Simplified graphical description of the working of Tetanus toxin. 1) The B subunit binds to the target neuronal membrane. 2) The tetanospasmin toxin crosses across the neuronal membrane. 3) The toxin travels backwards towards to the spinal cord and brainstem. 4) Once reaching the targeted area, chemical reduction of the disulfide bond occurs to separate the Lc. 5) The A subunit cleaves the SNARE protein (synaptobrevin) preventing the release of GABA + Glycin. 6) This causes acetylcholine to be left uninhibited causing the characteristics of tetanus.
Figure 5.
Simplified graphical description of the working of Tetanus toxin. 1) The B subunit binds to the target neuronal membrane. 2) The tetanospasmin toxin crosses across the neuronal membrane. 3) The toxin travels backwards towards to the spinal cord and brainstem. 4) Once reaching the targeted area, chemical reduction of the disulfide bond occurs to separate the Lc. 5) The A subunit cleaves the SNARE protein (synaptobrevin) preventing the release of GABA + Glycin. 6) This causes acetylcholine to be left uninhibited causing the characteristics of tetanus.
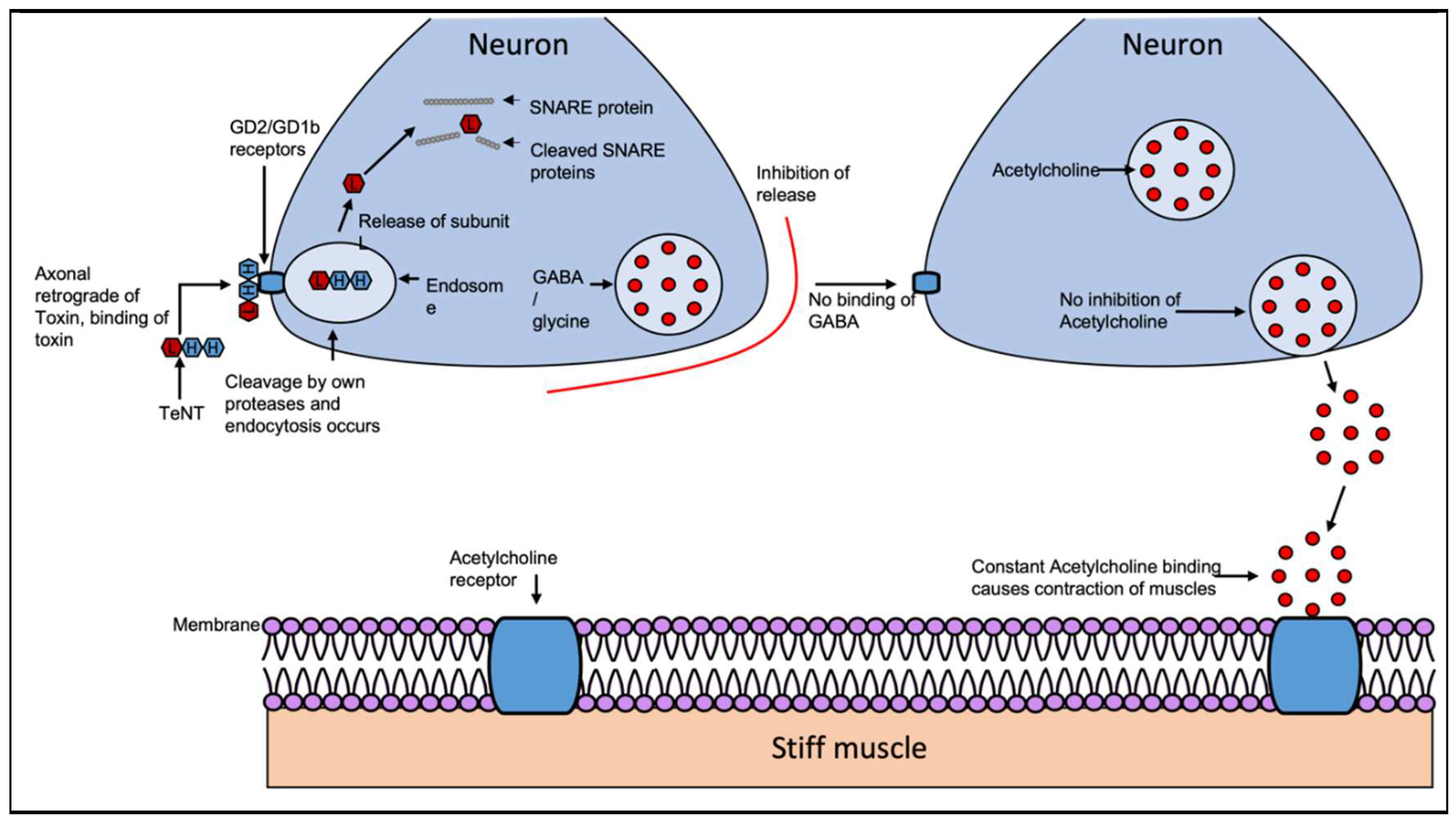
Figure 7.
PTX mechanism. Normal function of G-protein-coupled receptor in inhibiting production of cAMP compared to dysregulated constitutive production of cAMP due to A subunit of PTX and G-protein-independent effects mediated by B subunit of PTX. Adapted from Mangmool and Kurose, 2011 [111].
Figure 7.
PTX mechanism. Normal function of G-protein-coupled receptor in inhibiting production of cAMP compared to dysregulated constitutive production of cAMP due to A subunit of PTX and G-protein-independent effects mediated by B subunit of PTX. Adapted from Mangmool and Kurose, 2011 [111].
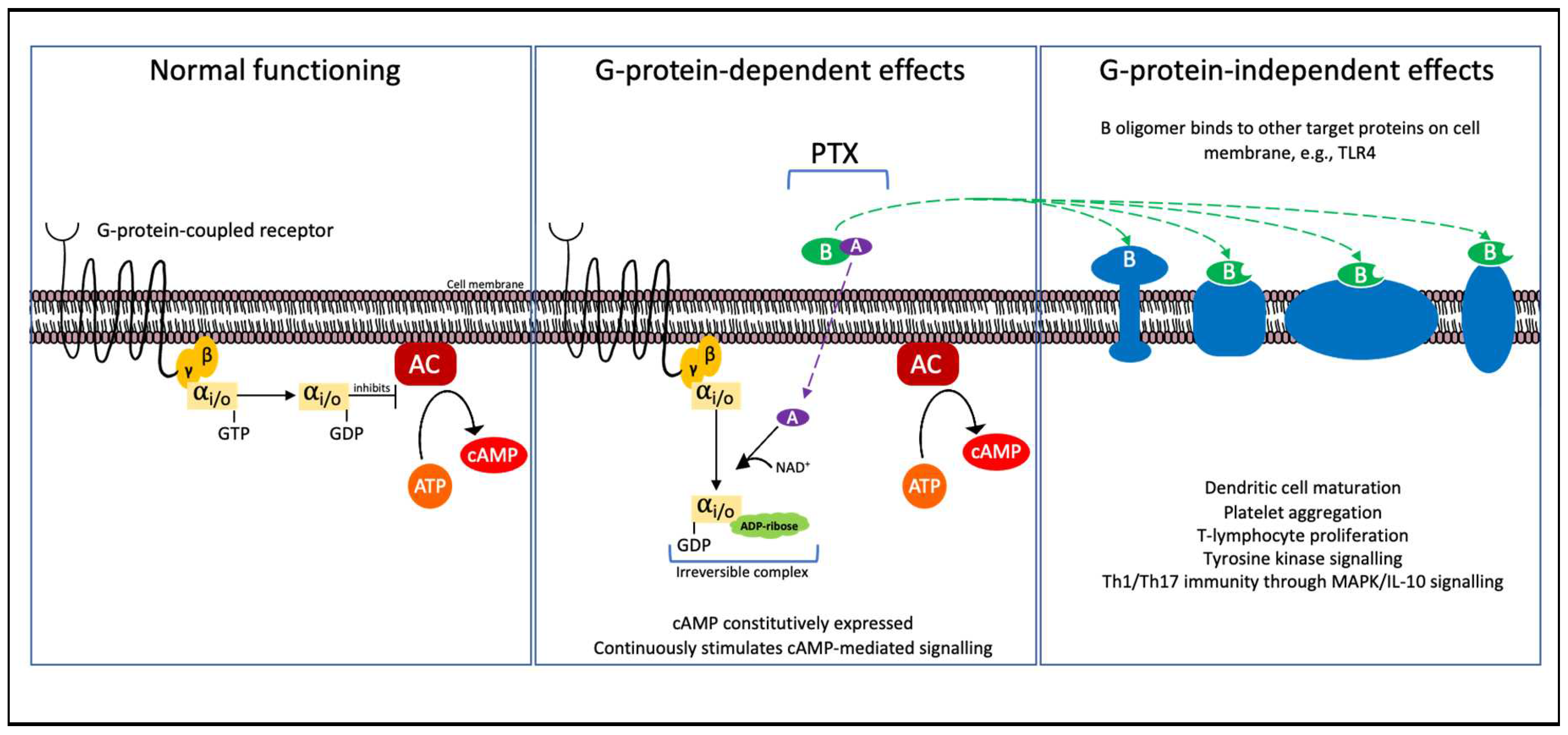
Figure 8.
Simplified mechanism for Shiga toxin. Stx is composed of subunit A (A1 + A2) and five identical B subunits. Subunit B binds at Gb3 and endocytosis occurs. Cleavage by furin occurs during the transport, still bound together due to the disulfide bond. The toxin migrates further inside of the cell, towards the golgi body and towards the endoplasmic reticulum (ER). The disulfide bond is reduced and A1 is released into the cytosol. The A1 fragment targets an adenine in the 28S rRNA of 60S ribosomal subunit for depurination. Causing protein synthesis to be halted as the ribosome no longer associates with elongation factor 1 and cell death occurs. Adapted from Puente and Finlay [124].
Figure 8.
Simplified mechanism for Shiga toxin. Stx is composed of subunit A (A1 + A2) and five identical B subunits. Subunit B binds at Gb3 and endocytosis occurs. Cleavage by furin occurs during the transport, still bound together due to the disulfide bond. The toxin migrates further inside of the cell, towards the golgi body and towards the endoplasmic reticulum (ER). The disulfide bond is reduced and A1 is released into the cytosol. The A1 fragment targets an adenine in the 28S rRNA of 60S ribosomal subunit for depurination. Causing protein synthesis to be halted as the ribosome no longer associates with elongation factor 1 and cell death occurs. Adapted from Puente and Finlay [124].
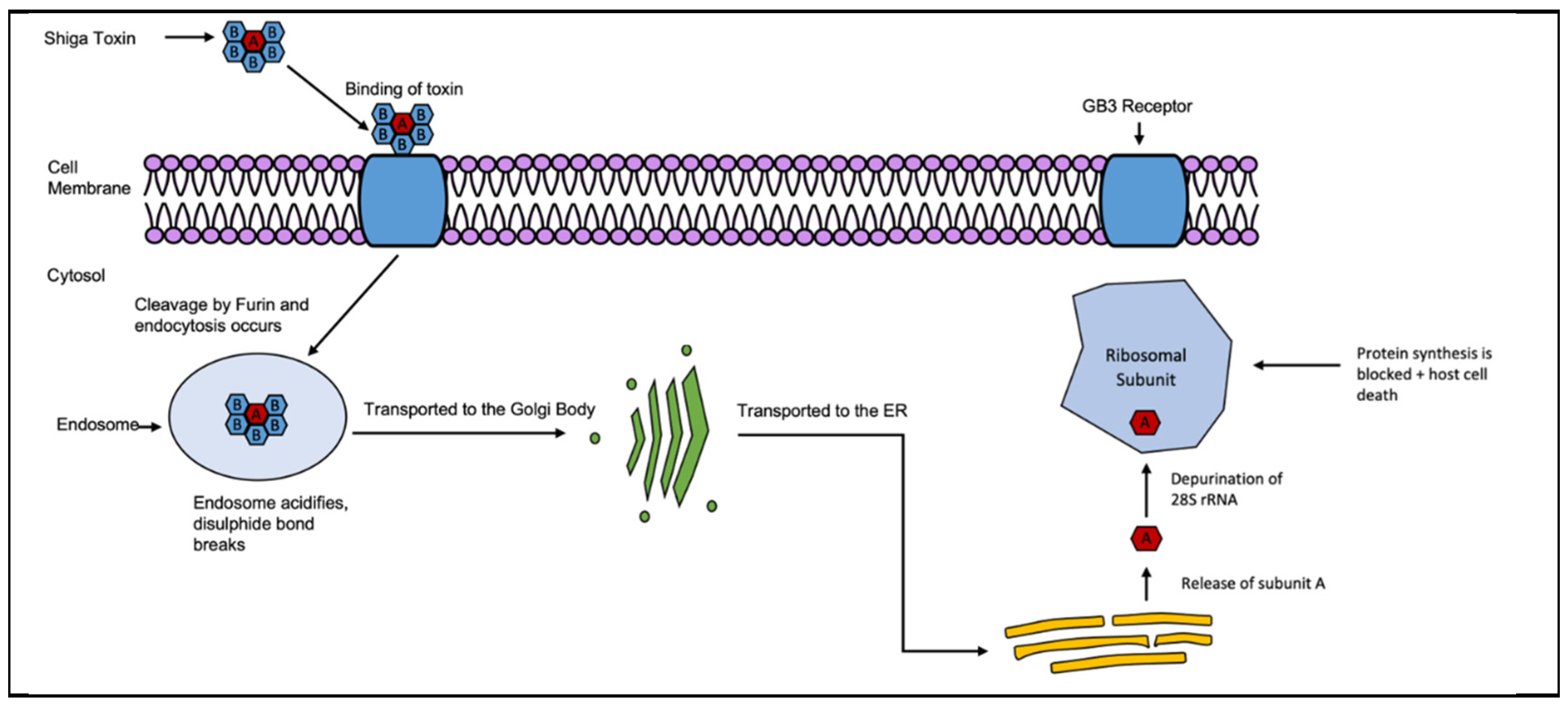

Table 1.
Major bacterial AB toxins and their associated species.
| Toxin | Bacterial Species | Targets |
|---|---|---|
| Anthrax Toxin, AB | Bacillus anthracis | MAPKK1 and 2 |
| Botulinum Toxin, AB | Clostridium botulinum | SNARE protein family |
| Cholera Toxin, AB5 | Vibrio cholerae | α-subunit of G-protein (GS) |
| Diphtheria Toxin, AB | Corynebacterium diphtheriae | Elongation factor 2 EF2 |
| Pertussis Toxin, AB5 | Bordetella pertussis | Gi/o protein’s α-subunit (Gαi/o) |
| Shiga and Shiga-like Toxins (Stxs/Vero toxins), AB5 | Shigella dysenteriae & Escherichia coli | 28S rRNA |
| Tetanus Toxin, AB | Clostridium tetani | Synaptobrevin |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



