
Submitted:
24 February 2023
Posted:
28 February 2023
Read the latest preprint version here
Abstract
White adipose tissue (WAT) has evolved to slowly mobilise energy stores in the form of lipids and a dynamic endocrine and immunologically active organ that regulates energy homeostasis. Breast WAT is involved not only in the biosynthesis of important molecular mediators but is also reported to be linked to the metastatic potential of breast tumours. The role of adiposity and consequently the role of systemic inflammation in immune responses and resistance to anti-cancer treatment in breast cancer (BC) patients is however still not clear. This review aims to evaluate the emerging evidence on the crosstalk between adiposity and the immune-tumour microenvironment in BC, its progression and treatment resistance. Adiposity, and by extension subclinical inflammation, are associated with metabolic dysfunction and changes in the immune-tumour microenvironment in BC. In oestrogen receptor positive (ER+) breast tumours, it is proposed that these changes are mediated via a paracrine interaction between macrophages and preadipocytes leading to elevated aromatase expression and secretion of pro-inflammatory cytokines and adipokines in the breast tissue in patients who are obese or overweight. In HER2+ breast tumours, WAT inflammation has been reported to induce trastuzumab resistance via activation of MAPK or PI3K signalling pathways. Furthermore, adipose tissue in patients with obesity is associated with upregulation of immune checkpoints on T-cells that is partially mediated via immunomodulatory effects of leptin and has been paradoxically associated with improved responses to immunotherapy in several cancers. In conclusion, evidence suggests that body composition and metabolic status are associated with patient outcomes. In order to optimise patient stratification and personalisation of treatment prospective studies are required to evaluate the role of body composition and metabolic parameters in metabolic immune reprogramming with and without immunotherapy in patients with BC.
Keywords:
breast cancer
; obesity
; inflammation
; metabolism
; treatment resistance
1. Introduction
BC is the most common type of malignancy worldwide in both sexes [1]. The worldwide average age-standardised incidence and mortality rates were estimated to be 47.8 and 13.6 per 100,000 persons per year, respectively in 2020 [1]. Therefore, BC is a major public health burden that needs to be addressed not only through primary prevention and early diagnosis but also via effective personalised treatments [2].
The increasing prevalence of obesity (body mass index [BMI] ≥25 kg/m2) has also become an important public health concern [3] [4]. The worldwide prevalence of obesity has increased dramatically by 27.5% for adults between 1980 and 2013 [3]. Overweight and obesity not only increase the risk of developing BC but also are associated with worse survival compared to patients with normal weight [3]. BMI is used as a surrogate of abnormal or excessive fat accumulation; however, it is not a reliable measure of body adiposity as BMI does not consider the percentages of lean and fat tissue mass [4]. Furthermore the same BMI may correspond to different lean and body fat mass that change with sex, ethnicity, and age [4].Therefore, further research is required to understand how body composition and BC interact.
Western lifestyle is associated with low-grade inflammation and chronic metabolic inflammatory diseases linked to decreased life expectancy [5]. Risk factors such as western diet, reduced physical activity, environmental or socioeconomic factors such as smoking, income, education and occupation, can result in chronic systemic inflammation that activates local immune cells, including macrophages, either directly or indirectly mediated via obesity [5,6,7]. In the context of nutrient excess, adipocytes increase in number and size that results in adipocyte disruption and cell death [8]. The innate immune cells, such as macrophages, respond to environmental danger signals that are released by the necrotic adipocytes that trigger chronic low-grade systematic inflammation via metabolic reprogramming [9]. During inflammation, there is an interplay between macrophages, that act as antigen-presenting cells, and adaptive immunity cells such as T-cells, by shaping T-cell responses and dysregulating the immunometabolic homeostasis towards a proinflammatory environment [10].
In this review, we evaluate the emerging evidence on the crosstalk between adiposity and tumour-immune microenvironment in BC progression and treatment resistance.
2. Immunometabolic changes in adipose tissue of patients with obesity
Adipose tissue represents an important component of human breast as it constitutes a dynamic endocrine and immunologically active organ that regulates energy homeostasis, as well as having important immunomodulatory properties [11]. White adipose tissue has evolved as a slowly mobilised energy store in the form of lipids [12]. Breast adipose tissue is involved not only in the biosynthesis of important molecular mediators but is also linked to the metastatic potential of breast tumours [13].
Under normal physiological conditions, in adipose tissue of individuals with healthy BMI, there is homeostasis between anti-inflammatory and proinflammatory molecules that maintains adipose tissue functions [14]. Adipose tissue is classified into white and brown adipose tissue (WAT and BAT respectively) that differ functionally and morphologically. WAT, distributed subcutaneously and viscerally, constitutes 20% of the body weight and 80% of total adipose tissue of a normal adult [15]. WAT is the largest store of energy, whilst BAT plays an important role in thermogenesis [15]. Under conditions of nutritional excess and during development of obesity, adipocytes undergo structural alterations, such as adipocyte hypertrophy, [16]. Then adipocytes become dysfunctional, undergo cell death, and secrete cytokines that contribute to adipose tissue inflammation and recruitment of pre-adipocytes leading to adipose tissue hyperplasia [17,18]. Apart from structural changes, adipose tissue also undergoes functional changes such as mitochondrial dysfunction and endoplasmic reticulum stress [11]. In obesity, there is a significant reduction in mitochondrial gene expression leading to downregulation of mitochondrial biogenesis in subcutaneous tissue associated with insulin resistance and inflammation in obese monozygotic twins compared with their leaner co-twins which was [19]. Furthermore, free fatty acid-mediated generation of reactive oxygen species is correlated to endoplasmic reticulum stress and up-regulation of pro-inflammatory gene signatures in adipose tissue [20,21]. Overall, these changes in the adipose tissue result in the activation of proinflammatory signalling pathways leading to chronic low-grade adipose tissue inflammation mediated by macrophage infiltration, neovascularization and increase of extracellular matrix [22,23] (Table 1).
3. Innate immunity in adipose tissue of patients with obesity
During weight gain, adipocytes increase the storage of lipids that results in structural changes, such as adipocyte hypertrophy, and adipocyte death. The mechanism of adipocyte death is still not clear, although it has been attributed to either inflammatory programmed cell death (pyroptosis) or necrosis [24,25]. Released intracellular contents of dead adipocytes such as free fatty acids, cytokines and damage-associated molecular patterns stimulate the recruitment of metabolically activated macrophages as well as in situ proliferation, that phagocytose the released cellular material forming crown-like structures (CLS) (Figure 1) [26].
ATM are metabolically activated, and their main function is to clear necrotic or apoptotic adipocytes via either phagocytosis or autophagic clearance mediated by lysosomal activation [27,28]. In obesity, free fatty acids released from ruptured adipocytes can initiate the intracellular NF-κB-mediated signalling pathway via Toll-like receptor-4, leading to activation of innate immune cells in the presence of interferon gamma (INF-γ) [29,30]. Proinflammatory macrophages are recruited around necrotic or pyroptotic hypertrophic adipocytes forming CLS that secrete proinflammatory mediators. In high-fat diet mice, obesity switched polarisation of ATM from an M2- to M1-like phenotype [31]. These pro-inflammatory ATM have been characterised as CD163+, an M2-like marker, and CD16+, an activating Fcγ receptor. ATM also express a range of other markers such as CD11c that plays an important role in T-cell activation in adipose tissue, and is associated with insulin resistance [32]. The CD11c+ CD163+ subset of ATM is associated with high BMI and accumulates in the adipose tissue of obese subjects [33]. CD11c+ CD206+ ATM confer pro-inflammatory properties that correlate with increased presence of CLS and insulin resistance in obese individuals [34].These suggest that CLS have a mixed phenotype, likely due to metabolic dysregulation, that are characterised by the concurrent expression of surface markers that describe M1- and M2-like macrophages [35]. Hence, ATM constitute a diverse population characterised by a mixed inflammatory phenotype that is dependent on the presence of adipose tissue in obese people [28].
4. Adaptive immunity in adipose tissue of patients with obesity
During the development of obesity, the composition of adaptive immunity cells resident in adipose tissue changes, with an increase in the CD8+ to CD4+ T-cell ratio, but a reduction in the number of regulatory T cells (Treg) within the adipose tissue [36]. Both CD4+ and CD8+ T-cells play an important role in the recruitment and activation of ATM via secretion of cytokines such as IFN-γ [37]. Flow-cytometric and immunohistochemical analyses demonstrated higher numbers of CD8+ effector T-cells and lower numbers of CD4+ helper T- cells in obese murine epididymal adipose tissue compared to lean mice on normal diet [37]. In addition, CD8+ T-cells were found within CLS in obese epididymal adipose tissue whereas there was no association between CD4+ T-cells and CLS [37]. A time course evaluation of immune cells in adipose tissue in C57BL/6 mice during high-fat diet , showed that CD8+ T-cell infiltration preceded the recruitment of macrophages [37]. In contrast, the number of CD4+CD8- helper T-cells and CD4+CD25+FoxP3+ Treg decreased suggesting that CD8+ T-cell infiltration is a crucial event during inflammation in adipose tissue [37]. This was further validated by depleting CD8+ T-cells in C57BL/6 mice using anti-CD8 antibody which resulted in reduction of M1-like macrophages and CLS without affecting M2-like macrophages [37]. In addition, high-fat diet did not increase levels of IL-6 and TNF-α mRNA in CD8-deficient mice, whereas adoptive transfer of CD8+ T-cells into CD8-deficient mice increased M1-like macrophage infiltration [37].
These findings suggest that Treg maintain immune homeostasis by suppressing inflammation induced by pro-inflammatory macrophages in adipose tissue under physiological conditions. CD8+ T-cell infiltration is required for adipose tissue inflammation in obesity as it precedes macrophage accumulation in adipose tissue and plays an important role in macrophage polarisation and infiltration. Metabolic dysregulation in the context of obesity may disrupt crosstalk between macrophages and T-cells [37]. Phenotypic differences in ATM in adipose tissue between lean and obese subjects may be attributed to their different immunometabolic functions influenced by metabolic stress and chronic inflammation that is promoted by enlarged or necrotic adipocytes.
5. Adipose tissue macrophages and breast cancer
CLS are correlated to a proinflammatory environment, as defined by elevated serum proinflammatory cytokines, and constitute not only a biomarker of WAT inflammation but also correlate with metabolic dysfunction[26]. WAT inflammation can occur concurrently not only in breasts but also in subcutaneous WAT located elsewhere as shown in women who underwent bilateral mastectomy and immediate autologous flap reconstruction suggesting that CLS are a marker of low-grade systemic inflammation [38]. Epidemiological studies have revealed that the presence of CLS in breast tumours is associated with raised inflammatory markers such as high sensitivity C-Reactive Protein (hsCRP) and IL-6 [39,40]. The effect of BMI in peripheral inflammation was shown in a cohort of BC patients with normal BMI where the correlation between CLS and high levels of serum hsCRP and IL-6 were either weak or non-significant [41]. These findings indicate that peripheral inflammation may be prominent in patients with high BMI and CLS which in turn may play a critical role in therapeutic responses.
In patients with primary BC of any subtype, CLS at any location, identified using the pan-macrophage marker CD68, were present in 36-65% of patients and have been correlated with postmenopausal status, high BMI, and larger mammary adipocyte size [40,42,43,44]. In addition, the presence of CLS is associated with dyslipidaemia as well as increased glucose and glycated haemoglobin (HbA1) levels compared to patients with no CLS [40,42]. Although there is no clear evidence of association between the presence of CD68+ CLS and clinicopathological features [40,43,45], the presence of macrophage scavenger receptor CD163+ CLS, is associated with the HER2+ and triple negative breast cancer (TNBC) subtypes [45]. Previous studies have shown varying degrees of association between the presence of CLS and poor clinical outcomes in patients with BC with three out of five studies demonstrating improved outcomes as shown in Table 2. [40,44,45,46]. This discrepancy in clinical outcomes in different cohorts can be attributed to patient and tumour heterogeneity, small sample size and different quantification methods and outcome assessments. Specifically, these cohorts included patients of any hormonal status and evaluated the presence of CLS within the whole observed adipose tissue area.
There is also strong evidence of correlation between elevated levels of serum leptin and adiponectin with CLS [40]. The role of leptin in trastuzumab resistance was evaluated by Griner et al. In vitro, differentiated adipocytes negatively affected trastuzumab-induced growth inhibition of HER2+ BT474 and SKBR-3 cell lines, an effect that was mediated via AKT phosphorylation [47]. Pharmacological blockade of PI3K via the PI3K inhibitor LY294002 or transfection with an inactive AKT1 kinase mutant suppressed the ability of differentiated adipocytes to promote resistance to trastuzumab [47].
In ER+ BC, aromatase expression and activity are associated with both high BMI and WAT inflammation [48]. Interestingly, WAT inflammation , as defined by the presence of CLS, is associated with raised aromatase expression and activity in women with normal BMI [41,43]. Furthermore, adipocyte size and markers of subclinical systemic inflammation are strongly associated with increased levels of aromatase in postmenopausal women [48]. In addition, menopause is associated with a reduction in oestrogen levels which are linked to the development of obesity [49]. Treatment with 17β-oestradiol protected ovariectomised mice against high fat diet-induced weight gain and was associated with a reduction in aromatase expression, WAT inflammation and the associated proinflammatory mediators in the mammary glands which is mediated via oestrogen receptor-α [49]. Hence, administration of oestrogen in obese mice may modulate WAT inflammation either through weight loss or due to its potential anti-inflammatory properties [49].
These findings suggest that hyperadiposity can induce WAT inflammation and metabolic dysfunction. In ER+ breast tumours, this is potentially mediated via a paracrine interaction between macrophages and preadipocytes leading to elevated aromatase expression and secretion of pro-inflammatory cytokines and adipokines in the breast tissue in patients with obesity or overweight [26]. Whereas in HER2+ breast tumours, WAT inflammation can induce trastuzumab resistance via activation of MAPK or PI3K signalling pathways [47].
6. BMI and clinical outcomes in different breast cancer subtypes and responses to treatment
Adiposity has been shown to affect the local immune environment of solid tumours in both pre-clinical and clinical studies where obesity was associated with enhanced response to immune checkpoint inhibitors (ICPI) [50,51]. Nevertheless, evidence for the influence of adiposity in therapeutic response in BC is conflicting which may be attributed to the heterogeneity of breast tumours.
7. HER2+ breast cancer
In HER2+ breast tumours, a multicentre retrospective cohort showed no evidence of association between BMI and clinical outcomes in patients with metastatic HER2+ BC [52]. In contrast, Krasniqi E et al., showed that BMI≥ 30 was correlated with poor overall survival (OS) in patients with HER2+ metastatic BC treated with pertuzumab and/or trastuzumab emtansine [53]. In early BC, Do Cosimo et al., demonstrated that patients with high BMI and HER2+ER+ early primary BC achieved lower complete pathological responses compared to patients with healthy or underweight BMI, after receiving neoadjuvant treatment with anti-HER2+ agents [54]. Mazzarella et al., reported that in non-metastatic HER2+ patients not treated with adjuvant trastuzumab, BMI≥30 was correlated with shorter OS and increased incidence of distant metastases in HER2+ER- BC whereas the outcome in HER2+ER+ tumours did not significantly differ between the different BMI groups [55].These findings indicate that adiposity and hormonal profile may explain the differential responses to anti-HER2 agents in patients with HER2+ primary BC.
Subclinical inflammation has also been correlated to therapeutic responses in patients with HER2+ BC. A cross-sectional study that included 175 patients with luminal B HER2-positive tumours, showed that these patients had a 2 times higher risk of having a waist circumference of ≥ 80 cm and a 3 times higher CRP compared to luminal A patients [56]. In a cohort of 66 patients with metastatic HER2+ BC, treated with trastuzumab alone or trastuzumab and chemotherapy, patients with elevated markers of systemic inflammation prior to treatment demonstrated worse progression-free survival and shorter OS [57]. Preclinical studies showed that PTEN deletion-induced resistance in HER2-amplified BC cell lines, was mediated by a significant increase in the secretion of proinflammatory cytokines including IL6 [58]. IL-6 induced a positive feedback loop that was dependent on NF-kB signalling and resulted in the generation of a cancer stem-like cell (CSC) population. Pharmacological inhibition of IL6 receptor signalling, alone or in combination with trastuzumab, blocked this inflammatory feedback loop and led to a decrease in the CSC population, tumour growth and metastasis in mouse xenografts [58]. Shou Liu et al, demonstrated that HER2+ overexpression promoted tumour-derived secretion of IL1a and IL6 that in turn induced the NF-kB and STAT3 pathways and led to the expansion of breast CSC [59]. Furthermore, in human breast tumour samples, there was strong evidence of association between high IL1a/IL6 expression and CSC-positive phenotype. In addition, in IL1a knock out mice, there was inhibition of HER2-induced tumorigenesis and reduction of inflammatory cytokine secretion. Chemical inhibition of IL1a signalling reduced the CSC population and improved chemotherapeutic responses both in vitro and in vivo [59]. Collectively, these demonstrate that inflammation mediates BC progression and metastatic spread as well as treatment resistance in pre-clinical models and clinical studies.
8. Oestrogen receptor negative breast cancer
The role of adiposity and inflammation was also demonstrated in ER- BC. In a study of 1779 patients with primary invasive BC, patients with triple-negative disease had a 3-fold risk of being overweight and of having raised CRP compared to luminal A subjects [56]. The associations between BMI and gene expression of both tumour and adjacent tissue were investigated in 519 postmenopausal women from the Nurse’s Health Study [60]. In ER- tumours and tumour-adjacent tissues, high BMI was associated with enhanced inflammation pathways with increased expression of genes associated with IFN-α and INF-γ response and activated mTORC1 complex. Tumour-adjacent tissues in ER- disease displayed activated inflammation pathways including IL-6 and IFN gamma with increasing BMI [60]. Hence, obesity is positively associated with inflammatory and aggressive molecular phenotypes in patients with BC.
9. Oestrogen receptor positive breast cancer
Gene set enrichment analysis (GSEA) of ER+ tumours from the Nurse’s Health Study, showed that high BMI was significantly correlated with upregulated cellular proliferation pathways in the primary tumours and epithelial mesenchymal transition and inflammatory pathways in the tumour-adjacent tissues [60]. In a study of 137 patients with ER+ BC, obesity was associated with shorter overall, and progression-free survival compared to patients without obesity [61]. Transcriptomic analysis of these tumours revealed that insulin signalling, and inflammation were the possible mechanisms that underly the prognostic effect of obesity on ER+ BC. GSEA showed that protein kinase B (AKT) target genes, as well as genes involved in glucose metabolism, in the generation of precursors of metabolites and energy, as well as in the epithelial–mesenchymal transition and metastasis were upregulated in patients with obesity [61]. To explore the causal relationship between obesity and tumour progression in ER+ BC, Fuentes-Mattei et al. generated oncogene-induced BC obese mouse and lean mouse models [61]. Transcriptomic analyses of these tumours suggested that obesity was associated with tumour metastasis, invasion, inflammation, and cell death resistance that were mediated by oestrogen signalling, hyperinsulinemia, IGF-1, and adipokine secretion [61]. The role of peripheral inflammation and BMI in ER+ breast tumours was also supported by a study of 216 BC patients by Madeddu et al. In this study, leptin levels in ER+ patients were significantly higher compared to ER- patients. Multivariate regression analysis revealed that BMI, leptin, IL-6 and reactive oxygen species were predictive factors for tumour size, lymph node stage and metastasis status in ER+ patients [62]. These findings indicate that an imbalance in the secretion of adipokines may play a role in the link between obesity and ER+ BC. This imbalance may result in a pro-inflammatory environment that promotes tumour progression and metastasis.
Quigley et al. compared gene expression profiles in 195 breast tumours of all subtypes to gene expression in matched adjacent normal tissue and tissue from women who underwent mammoplastic reduction surgery [63]. The expression of cytokines associated with acute inflammatory response such as IL-1B, TNF-α and suppressor of cytokine signalling 3 were significantly higher in the normal tissue adjacent to the breast tumour compared with mammoplastic reduction samples from healthy donors. BMI was also significantly correlated with the expression of leptin and macrophage scavenger receptor expression as well as with macrophage pathway expression levels in adjacent normal tissue but not in tumours. The change in macrophage pathways was inversely correlated with the change in oestrogen receptor-a expression in ER+ but not ER- patients. ER- tumours highly expressed macrophage pathways compared to matched adjacent normal tissue. On the contrary, there was an inverse correlation between macrophage pathway expression and oestrogen receptor-a expression in the tumour of ER+ cancers compared with adjacent normal tissue. The effect of oestrogen signalling pathway on inflammation in breast tumours was investigated by Qureshi et al who showed that oestrone, which is upregulated in post-menopausal women, promotes NF-kB mediated inflammation which results in the increase of tumour-initiating stem cells and ER+ cancer initiation and progression as well as poor outcomes [64]. Thus, high BMI is associated with inflamed normal tissue adjacent to the tumour that induces aggressive phenotypes that are mediated by oestrogen signalling in ER+ breast tumours.
In summary, adiposity and by extension subclinical inflammation are reported to be associated with changes in the immune-tumour microenvironment in BC that can be subtype specific. Obesity has also been correlated to aggressive clinical and molecular phenotype as well as enriched inflammation pathways.
10. Adiposity and response to cancer immunotherapy in breast cancer
Recent advances in cancer immunotherapy with immune checkpoint inhibitors (ICPI) have significantly improved clinical outcomes in patients with tumour types that were previously difficult to treat [65,66,67]. Nevertheless, only a limited proportion of patients demonstrate durable responses to immunotherapy [65,66,67]. This can be potentially explained by a number of parameters that affect immunotherapy responses such as the presence of TILs, the absence of immune checkpoints, systemic inflammation, hypoxic TME and low tumour mutational burden [68]. Breast tumours are considered immunologically quiescent compared to other tumour types. The heterogeneity in immunological profiles and mutational burden among the different subtypes of BC partially explains the differential responses to ICPI. For instance, TNBC and especially basal subtype as well as HER2+ breast carcinomas are associated with higher mutational burden and higher frequency of TILs compared to the ER+ tumours [69,70]. Higher rate of TILs, has been associated with improved clinical outcomes in patients with HER2+ and TNBC following neoadjuvant chemotherapy [69]. TILs can also identify a subset of patients with stage I TNBC who may not require adjuvant chemotherapy [71]. Chemotherapy may promote the release of tumour neoantigens and consequently tumour-specific immune responses. ICPI in combination to chemotherapy has been predominantly evaluated in patients with TNBC where pembrolizumab and atezolizumab have been approved at neoadjuvant and metastatic setting [72,73]. Currently, PD-L1 expressed by the tumour is used as a biomarker to select patients with metastatic BC who are most likely to benefit from ICPI. However, PD-L1 is not used in primary TNBC setting. Despite the positive results, it is not clear who can benefit from immunotherapy in this group of patients.
Randomised clinical trials investigating PD-1/PD-L1 blockade either as monotherapy or in combination with chemotherapy or targeted agents, in patients with metastatic BC, demonstrated objective responses and median OS ranging from 0% to 52% and from 8.1 to 17.2 months, respectively [69,72,74]. Higher responses were confined to patients with triple negative, HER2+, PD-L1+, BRCA1/2 deficient and treatment-naïve subgroups of BC patients underlying the fact that person stratification is important for the optimisation of immune responses in BC patients [69,72,74]. Nevertheless, none of these published RCT stratified the patients with BC by body composition parameters indicating the need to explore the potential impact of metabolic parameters on immune responses, disease progression and treatment outcomes in RCTs.
Currently, the evidence on body composition and immunotherapy responses are derived from other types of solid tumours with lack of data in BC. A systematic review and meta-analysis evaluated the association between BMI and the efficacy of ICPI in 5279 patients with cancer (renal cell carcinoma, malignant melanoma, and lung cancer) from 13 retrospective cohorts [75]. This study showed improved clinical outcomes in patients with high BMI who were treated with ICPI compared to patients with healthy BMI with no statistically significant difference in the frequency of immune-related adverse events [75]. This observation is supported by Maslov et. al., who demonstrated that high BMI in patients with metastatic cancer, across 20 different tumour types, is associated with a 48% lower risk of disease progression or death compared to patients with healthy BMI [76]. In addition, in a cohort of 250 patients diagnosed with different types of cancers who were treated with ICPI, individuals with a BMI ≥30 had significantly better progression-free survival and OS compared to patients without obesity [50]. Similar results were shown in a multi-cohort analysis that included two cohorts of patients with advanced melanoma who received ICPI [51]. In the first cohort, 207 patients received ipilimumab plus dacarbazine whereas in the second cohort 329 patients were treated with either pembrolizumab, nivolumab, or atezolizumab. The pooled analysis showed that patients with a BMI≥30 demonstrated longer progression-free and OS compared to non-obese individuals. These observations may be explained by the high levels of systemic inflammation in patients with high BMI. This is supported by small cohort of 26 healthy human volunteers where significantly elevated levels of serum leptin were observed in individuals with obesity obese compared to those without which was also associated with higher PD-1 expression on CD8+ T-cells [50].
Tumours evolve multiple mechanisms of immune escape and new therapeutic strategies to prevent this are of particular significance [77]. Western diet and obesity can promote systemic chronic inflammation with increased leptin that is associated with resistance to antibody treatment such as trastuzumab [47]. Leptin is an established pro-inflammatory cytokine that has effects both on innate and adaptive immunity. Immune cells express leptin receptor and leptin promotes proliferation of naïve T-cells, induces Th-1 responses and restores the function of impaired T-cells [78]. Leptin also contributes to the activation of monocytes and macrophages via the upregulation of proinflammatory cytokines, promotes survival of dendritic cells and neutrophil chemotaxis as well as acts as a negative regulator for the proliferation of Tregs via the activation of the mTOR pathway [78]. Wang et al investigated the effect of obesity and leptin levels on T-cell responses in multiple species and tumour models [50]. Upregulation of PD-1 in CD8+ T-cells together with reduced proliferative capacity, INF-γ and TNFα production were observed in diet-induced obese mice compared to controls [50]. Similar findings were reported in non-human primates and healthy human donors that were stratified by body weight and BMI, respectively [50]. In addition, obesity promotes tumour growth and T-cell exhaustion with PD-1 upregulation in tumour-infiltrating CD8+ T-cells in both B16F0 melanoma and 4T1 BC murine models which were shown to be partly mediated via leptin [50]. Zhang et al., showed that leptin in breast adipose tissue downregulates the effector functions of CD8+ T-cells by activating STAT3-Fatty Acid Oxidation (FAO) pathway and by inhibiting glycolysis and INF-γ secretion [79]. In addition, PD-1 increases FAO and inhibits IFN-γ and glycolysis via STAT3 activation in the tumour infiltrating CD8+ T-cells [79]. PD-1 signalling pathway reprograms T-cells metabolically by suppressing AKT and mTOR signalling which also leads to FAO rather than glycolysis [80]. Anti-PD-1 blockade resulted in higher responses, significant reduction of the tumour burden and improvement in survival in obese compared to lean B16 tumour bearing mice [50]. Moreover, higher CD8+ T-cell tumour infiltration and reduction in the metastatic burden in B16-bearing mice was reported [50]. These results are supported by Dyck et al., that showed a decreased number of tumour-infiltrating lymphocytes that were characterised by decreased proliferation and cytokine secretion, in high-fat diet compared to standard diet MC38 and B16-F10 tumour-bearing mice [81]. Overall, leptin is associated with a STAT3-mediated metabolic reprogramming of CD8+ T-cells that compromises their antitumour properties.
Collectively, obesity and systemic inflammation are associated with upregulation of immune checkpoints on T-cells that is partially mediated via the immunomodulatory effects of leptin in pre-clinical tumour models and clinical studies. In chronic inflammatory conditions such as obesity, T-cells become exhausted due to the persistent antigenic exposure that leads to the expression of inhibitory receptors and metabolic reprogramming of these immune cell populations [82,83]. In addition, tumour associated macrophages can directly suppress T-cell responses via overexpression of inhibitor receptors and by interfering in signalling pathways that are involved in inflammation, metabolism or proliferation and hypoxia [84,85]. This has been paradoxically associated with improved responses to ICPI in several cancers. Currently, there is lack of evidence in the role body composition and metabolic parameters in immune responses in BC. Hence, further research is needed in this field that can be potentially used to optimise patient stratification and personalisation of BC treatment.
11. Metabolic interventions in immunometabolic reprograming in breast cancer: the paradigm of metformin
Metformin, a dimethyl biguanide antidiabetic drug with pleiotropic effects, has demonstrated anti-tumourigenic properties both in pre-clinical and clinical studies [86]. In vitro and in vivo preclinical models of endometrial cancer showed that metformin reverses obesity-induced tumour aggressiveness by downregulating lipid and protein biosynthesis in obese compared to lean mice [87]. In addition, in a phase II randomised clinical trial (RCT), premenopausal women with obesity or overweight and characteristics of metabolic syndrome without history of BC were treated with metformin or placebo [88]. After 12 months of treatment, metformin was associated with a significant reduction in waist circumference and waist-to-hip ratio as well as a decrease in markers of systemic inflammation such as serum leptin and the leptin-to-adiponectin ratio and neutrophil-to-lymphocyte ratio compared to the placebo group [88]. Together, these findings suggest that metformin may have antitumour properties via the modulation of obesity-mediated systemic inflammation and tumour metabolism.
Previous studies demonstrated that metformin exerts pleiotropic antitumour effects that involve tumour metabolism, cell cycle, DNA repair mechanism, reactive oxygen species, angiogenesis and inflammatory pathways as shown in preclinical studies. In addition, metformin modulates the density of immune cell infiltrates in human and murine tumours [86,89,90]. Previous studies also showed that metformin polarized M2-like macrophages to M1-like phenotype within the tumour microenvironment which led to the recruitment of CD8+ T-cells into tumour and reduction of immunosuppressive infiltration of myeloid-derived suppressor cells and regulatory T-cells which may be mediated by the secretion of proinflammatory cytokines [91,92,93,94]. Specifically, metformin is associated with increased CD68+, F4/80+ and decreased CD11c+ and CD206+ macrophage densities in murine models [95,96]. In vitro and in vivo experimental models showed that metformin was correlated with reduced secretion of anti-inflammatory and increased production of pro-inflammatory cytokines by macrophages [97,98]. Regarding T-cells, metformin was associated with higher CD8+ T-cell density and reduced CD4+Foxp3+ regulatory T-cells, in vivo [99,100]. Interestingly, there was an increase in the density of regulatory T-cells derived from B16F10 murine melanoma tumours [101]. In addition, tumour infiltrating lymphocytes were associated with enhanced production of proinflammatory cytokines, granzyme B and perforin after the administration of metformin both in vitro and in mice [101,102]. Furthermore, tumours downregulated the expression of PD-L1 and restored the MHC-1 expression[99,103]. These findings suggest that metabolic reprogramming of macrophages with the use of metformin may repolarize tumour-associated macrophages towards an anti-M1-like phenotype that could then enhance CD8+ T-cell effector function and suppress the recruitment of regulatory T-cells. Thus, the combination of metformin and ICPI may further enhance immune responses. Nevertheless, further research is required to optimise the identification subgroups of BC patients that may benefit from metformin.
Pre-clinical studies evaluated the efficacy of metformin in combination with ICPI in several solid tumours including BC. In 4T1 murine BC experimental models, the use of nanodrug that contained metformin and the anticancer agent SN38 (7-ethyl-10-hydroxycamptothecin) resulted in improved outcomes compared to metformin or ICPI alone, an effect that was mediated by the downregulation of PD-L1 expressed by the tumour cells [104]. Cha et al., demonstrated that metformin-activated AMPK directly binds to and phosphorylates PD-L1 that induces abnormal glycosylation leading to endoplasmic reticulum-associated protein degradation [99]. In addition, in MYC-overexpressed breast tumour models, metformin combined with anti-apoptotic B-cell lymphoma-2 inhibitors, navitoclax or venetoclax, led to tumour growth inhibition, improved clinical outcomes and tumour infiltration by immune cells [105]. In another study, where 4T1 murine breast tumours were treated with metformin-loaded mannose-modified macrophage-derived microparticles (Man-MPs), reported that metformin polarized M2-like macrophages to M1-like phenotype within the TME [91]. This resulted in the recruitment of CD8+ T-cells into tumour tissues and reduction of myeloid-derived suppressor cells and regulatory T-cells. Similarly, in experimental lung cancer and malignant melanoma murine models, the combination of metformin and anti-PD1 ICPI was associated with CD8+ T-cell tumour infiltration, reduction of myeloid derived suppressive cells, inhibition of tumour growth and improved outcomes [92,93,94,106,107]. Hence, pre-clinical studies that evaluated the efficacy of metformin in combination with ICPI or other novel drugs in several solid tumours including BC showed promising results. Nonetheless, further experimental models need to be developed to evaluate immunometabolic reprogramming in BC that will improve personalisation of treatment and enable us to support clinical randomised controlled trial to investigate metabolic immune reprogramming with and without immunotherapy in patients with primary BC.
Despite promising pre-clinical data, randomised clinical trials RCT evaluating the impact of metformin on clinical outcomes did not show statistically significant results. Specifically, a recent phase III, double blind RCT that included high risk non-metastatic and non-diabetic BC patients showed no difference in disease-free and OS between patients randomised to adjuvant metformin or placebo in the ER/PR+ group [108]. However, in the same study, exploratory analysis in the HER2+ group revealed a statistically significant improvement in disease-free and OS in the metformin arm [108]. Similarly, a systematic review and metanalysis that included three cohort studies of diabetic patients with stage I-III BC, who received adjuvant metformin, showed no difference in recurrence-free, overall and cancer-specific survival between metformin and controls [109]. In addition, three phase II RCTs with non-diabetic patients diagnosed with metastatic BC failed to show any benefit in the metformin arm compared to the control group [110,111,112]. In contrast, in neoadjuvant setting there was a non-statistically significant trend of association between metformin and improved objective response rate in non-diabetic patients with locally advanced disease [113,114]. Furthermore, in a retrospective cohort of patients with metastatic malignant melanoma treated with ICPI with or without metformin, patients who received combination treatment demonstrated a trend for improved clinical outcomes, an association that was non-statistically significant likely due to the small sample size [115].
Although the clinical studies showed negative results, they are characterised by certain limitations. First, with the exception of the studies by Goodwin et al., and Lega et al., the remainder were characterised by small sample size that may have introduced random error. Secondly, none of the studies focused on a specific BC subtype. Hence, tumour heterogeneity might have confounded any possible clinical benefit related to metformin treatment. Furthermore, these studies did not stratify patients by metabolic or inflammatory parameters such as serum leptin or CRP which may help optimise the identification of patients who may have clinical responses to metformin. Other possible confounders include local adiposity and inflammation such as the presence of CLS, and body composition such as Fat Mass Index (FMI) and BMI which may affect response to treatment. Also, the higher pathological response in patients who received neoadjuvant metformin may indicate that metformin may have a direct tumour effect and that it may be best given in a neoadjuvant setting. However, these studies had a small sample size and might have been underpowered.
Two key preoperative window of opportunity clinical trials investigated the mechanism of action of metformin in non-diabetic patients with early primary BC [116,117]. The first trial by Hadad et al., recruited 47 non-diabetic patients who were randomly allocated to a two-week course of metformin or no medication [116,118]. Immunohistochemistry for metabolic and cell proliferation markers was performed on formalin-fixed paraffin embedded core biopsies that were taken at baseline and post-treatment [116]. RNA microarray analysis in metformin and control samples was also carried out [118]. In patients treated with metformin there was an increase in the expression of tumour pAMPK, reduction in pAkt, and suppression of insulin responses as well as there was a significant decrease in Ki67 and cleaved caspase-3 suggesting that metformin may exert a cytostatic anti-tumour effect [116]. Ingenuity pathway analysis demonstrated that metformin was associated with pathways involved in TNFR1, cell cycle regulation and metabolism [118]. The second window of opportunity study by Lord et al., integrated dynamic PET imaging, metabolomics, and transcriptomics pre- and post-metformin in a single arm cohort of 41 non-diabetic patients with primary BC. This study demonstrated evidence that metformin is associated with low levels of mitochondrial metabolites, with activation of multiple mitochondrial metabolic pathways, and increase in 18-FDG flux in tumours [117]. Also, two distinct metabolic responses were identified , the glycolytic and oxidative phosphorylation responders with the latter conferring resistance to metformin [117]. Overall, metformin appears to have an antitumour effect that is mediated by metabolic and cell cycle pathways.
In summary, metformin modulates the tumour microenvironment via the enhancement of anti-tumour immune responses which may be partially mediated by the secretion of cytokines by both T-cells and macrophages. Although RCT showed no evidence of association between metformin and improved clinical outcomes in patients with BC, preclinical studies showed that the combination of metformin and ICPI in breast, lung cancer and malignant melanoma murine models is correlated with enhanced tumour-infiltrating lymphocytes and improved outcomes. This discrepancy in the findings between clinical and preclinical studies may be explained by biological or methodological reasons. For instance, all tumour subtypes were included indicating that tumour heterogeneity might have masked any possible associations. In addition, these studies did not stratify patients by metabolic or inflammatory parameters which may help optimise the identification of patients who may respond to metformin. Also, the small sample size and confounding such as anthropometric, clinical, and metabolic parameters might have not been considered, whilst preclinical studies were characterised by a well-defined population and controlled conditions that minimised confounding and bias. There are a limited number of human studies that mainly studied small cell lung cancer and malignant melanoma and that showed weak evidence of better responses and survival outcomes, respectively [115,119]. However, these studies are characterised by their retrospective nature and small size samples size. Nevertheless, the immunomodulatory properties of metformin and potential underlying mechanism in patients with BC require further investigation.
12. Conclusions
There is mounting evidence that chronic inflammation in adipose tissue is associated with the promotion of protumourigenic macrophages and exhausted T-cell phenotypes that are linked to resistance to anti-cancer therapy [47,120]. Although the underlying biology is complex, a possible mechanism is that chronic inflammation enhances metabolic competition between tumour and immune cells for nutrients in favour of the tumour that leads to metabolic dysregulation of macrophage and T-cells [121]. In obesity, systemic inflammation is initiated, promoted, and maintained partly via the metabolic reprogramming of adipose tissue macrophages [38,39,40,41,43,45,122]. Obesity is also associated with the upregulation of immune checkpoints on T-cells which has been correlated to improved responses to ICPI in several cancers suggesting that body composition can be used to optimise patient personalisation of treatment. Metabolic interventions such as metformin, targeting this metabolic dysregulation may potentially restore anti-cancer function of the immune cells and improve clinical outcomes in patients with BC. This effect may be mediated by the modulation of obesity-mediated systemic inflammation, tumour metabolism and tumour-immune microenvironment. The combination of metformin and ICPI in murine models is associated with enhanced tumour-infiltrating lymphocytes and improved outcomes. However, prospective studies considering body composition and metabolic parameters are required to investigate metabolic immune reprogramming with and without immunotherapy in patients with BC.
Author Contributions
CS wrote the manuscript and prepared the figures and tables. EC, PWMJ, RIC and SAB critically reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
CS receives funding from National Institute for Health and Care Research (NIHR). CS and SAB received funding from Cancer Research UK. SAB gratefully acknowledge a programme grant from Against Breast Cancer (www.againstbreastcancer.org.uk; UK Charity 1121258).
Acknowledgements
We thank members of the wider Antibody and Vaccine Group and School of Cancer Sciences for useful discussions.
Conflicts of Interest
SAB has acted as a consultant for a number of biotech companies and has received institutional support for grants and patents from BioInvent International. Medical body composition analysers provided by SECA as part of an investigator-led Collaborative Research Agreement between Seca GmbH & Co. KG. (Hamburg, Germany), University Hospital Southampton NHS Foundation Trust and the University of Southampton have been utilised by RIC for research purposes. EC received honorarium: from Astra-Zeneca, Novartis, Pfizer, Roche, Lilly; Advisory boards: Pfizer, Nanostring, Lilly; Expert panel participation: World Cancer Research Fund. RIC and EC report Research funding from SECA and Astra-Zeneca. CS and PWMJ declare no competing interests.
References
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun (Lond) 2021, 41, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Jeibouei, S.; Akbari, M.E.; Kalbasi, A.; Aref, A.R.; Ajoudanian, M.; Rezvani, A.; Zali, H. Personalized medicine in breast cancer: pharmacogenomics approaches. Pharmgenomics Pers Med 2019, 12, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (London, England) 2014, 384, 766–781. [Google Scholar] [CrossRef]
- James, F.R.; Wootton, S.; Jackson, A.; Wiseman, M.; Copson, E.R.; Cutress, R.I. Obesity in breast cancer--what is the risk factor? Eur J Cancer 2015, 51, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Latz, E. The Western lifestyle has lasting effects on metaflammation. Nature Reviews Immunology 2019, 19, 267–268. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Frontiers in Immunology 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Matz, A.J.; Karlinsey, K.; Cao, Z.; Vella, A.T.; Zhou, B. Macrophages at the Crossroad of Meta-Inflammation and Inflammaging. Genes 2022, 13, 2074. [Google Scholar] [CrossRef]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nature Reviews Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e114. [Google Scholar] [CrossRef]
- Schmidt, V.; Hogan, A.E.; Fallon, P.G.; Schwartz, C. Obesity-Mediated Immune Modulation: One Step Forward, (Th)2 Steps Back. Frontiers in Immunology 2022, 13. [Google Scholar] [CrossRef]
- Schaffler, A.; Scholmerich, J. Innate immunity and adipose tissue biology. Trends in immunology 2010, 31, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Pond, C.M. The Evolution of Mammalian Adipose Tissues. In Adipose Tissue Biology, Symonds, M.E., Ed.; Springer International Publishing: Cham, 2017; pp. 1–59. [Google Scholar]
- Gesta, S.; Kahn, C.R. White Adipose Tissue. In Adipose Tissue Biology, Symonds, M.E., Ed.; Springer International Publishing: Cham, 2017; pp. 149–199. [Google Scholar]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annual review of pathology 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed]
- Torres, N.; Vargas-Castillo, A.E.; Tovar, A.R. Adipose Tissue: White Adipose Tissue Structure and Function. In Encyclopedia of Food and Health, Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Oxford, 2016; pp. 35–42. [Google Scholar]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, Adipose Tissue and Vascular Dysfunction. Circulation research 2021, 128, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Frontiers in Cardiovascular Medicine 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circulation research 2016, 118, 1786–1807. [Google Scholar] [CrossRef]
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Frontiers in Pharmacology 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. The Journal of biological chemistry 2014, 289, 7884–7896. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Chawla, A. Alternative macrophage activation and metabolism. Annual review of pathology 2011, 6, 275–297. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of Lipid Research 2005, 46, 2347–2355. [Google Scholar] [CrossRef]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res 2013, 54, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nature Reviews Endocrinology 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Grijalva, A.; Skowronski, A.; van Eijk, M.; Serlie, M.J.; Ferrante, A.W., Jr. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell metabolism 2013, 18, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Zuany-Amorim, C.; Hastewell, J.; Walker, C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nature reviews. Drug discovery 2002, 1, 797–807. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Wu, H.; Perrard, X.D.; Wang, Q.; Perrard, J.L.; Polsani, V.R.; Jones, P.H.; Smith, C.W.; Ballantyne, C.M. CD11c Expression in Adipose Tissue and Blood and Its Role in Diet-Induced Obesity. Arteriosclerosis, Thrombosis, and Vascular Biology 2010, 30, 186–192. [Google Scholar] [CrossRef]
- Nakajima, S.; Koh, V.; Kua, L.F.; So, J.; Davide, L.; Lim, K.S.; Petersen, S.H.; Yong, W.P.; Shabbir, A.; Kono, K. Accumulation of CD11c+CD163+ Adipose Tissue Macrophages through Upregulation of Intracellular 11beta-HSD1 in Human Obesity. Journal of immunology (Baltimore, Md. : 1950) 2016, 197, 3735–3745. [Google Scholar] [CrossRef]
- Wentworth, J.M.; Naselli, G.; Brown, W.A.; Doyle, L.; Phipson, B.; Smyth, G.K.; Wabitsch, M.; O'Brien, P.E.; Harrison, L.C. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 2010, 59, 1648–1656. [Google Scholar] [CrossRef]
- Bourlier, V.; Zakaroff-Girard, A.; Miranville, A.; De Barros, S.; Maumus, M.; Sengenes, C.; Galitzky, J.; Lafontan, M.; Karpe, F.; Frayn, K.N.; et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation 2008, 117, 806–815. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 2009, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, N.M.; Morris, P.G.; Zhou, X.K.; Gucalp, A.; Giri, D.; Harbus, M.D.; Falcone, D.J.; Krasne, M.D.; Vahdat, L.T.; Subbaramaiah, K.; et al. Menopause is a determinant of breast adipose inflammation. Cancer Prev Res (Phila) 2015, 8, 349–358. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Chen, I.-C.; Zhou, X.K.; Giri, D.D.; Falcone, D.J.; Winston, L.A.; Wang, H.; Williams, S.; Lu, Y.-S.; Hsueh, T.-H.; et al. Adiposity, Inflammation, and Breast Cancer Pathogenesis in Asian Women. Cancer Prevention Research 2018, 11, 227–236. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Morris, P.G.; Howe, L.R.; Giri, D.D.; Morrow, M.; Wang, H.; Pollak, M.; Jones, L.W.; et al. Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin Cancer Res 2016, 22, 2283–2289. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Brown, K.A.; Zhou, X.K.; Gucalp, A.; Subbaramaiah, K.; Giri, D.D.; Zahid, H.; Bhardwaj, P.; Wendel, N.K.; Falcone, D.J.; et al. Metabolic Obesity, Adipose Inflammation and Elevated Breast Aromatase in Women with Normal Body Mass Index. Cancer Prev Res (Phila) 2017, 10, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Vaysse, C.; Lomo, J.; Garred, O.; Fjeldheim, F.; Lofteroed, T.; Schlichting, E.; McTiernan, A.; Frydenberg, H.; Husoy, A.; Lundgren, S.; et al. Inflammation of mammary adipose tissue occurs in overweight and obese patients exhibiting early-stage breast cancer. NPJ Breast Cancer 2017, 3, 19. [Google Scholar] [CrossRef]
- Mullooly, M.; Yang, H.P.; Falk, R.T.; Nyante, S.J.; Cora, R.; Pfeiffer, R.M.; Radisky, D.C.; Visscher, D.W.; Hartmann, L.C.; Carter, J.M.; et al. Relationship between crown-like structures and sex-steroid hormones in breast adipose tissue and serum among postmenopausal breast cancer patients. Breast cancer research : BCR 2017, 19, 8. [Google Scholar] [CrossRef]
- Birts, C.N.; Savva, C.; Laversin, S.A.; Lefas, A.; Krishnan, J.; Schapira, A.; Ashton-Key, M.; Crispin, M.; Johnson, P.W.M.; Blaydes, J.P.; et al. Prognostic significance of crown-like structures to trastuzumab response in patients with primary invasive HER2 + breast carcinoma. Sci Rep 2022, 12, 7802. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Kim, E.S.; Koo, J.S. Tumor-associated macrophages and crown-like structures in adipose tissue in breast cancer. Breast cancer research and treatment 2018, 170, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Koru-Sengul, T.; Santander, A.M.; Miao, F.; Sanchez, L.G.; Jorda, M.; Gluck, S.; Ince, T.A.; Nadji, M.; Chen, Z.; Penichet, M.L.; et al. Breast cancers from black women exhibit higher numbers of immunosuppressive macrophages with proliferative activity and of crown-like structures associated with lower survival compared to non-black Latinas and Caucasians. Breast cancer research and treatment 2016, 158, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Griner, S.E.; Wang, K.J.; Joshi, J.P.; Nahta, R. Mechanisms of Adipocytokine-Mediated Trastuzumab Resistance in HER2-Positive Breast Cancer Cell Lines. Curr Pharmacogenomics Person Med 2013, 11, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Subbaramaiah, K.; Wang, H.; Giri, D.D.; Morrow, M.; Falcone, D.J.; Wendel, N.K.; et al. Menopause Is a Determinant of Breast Aromatase Expression and Its Associations With BMI, Inflammation, and Systemic Markers. The Journal of clinical endocrinology and metabolism 2017, 102, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, P.; Du, B.; Zhou, X.K.; Sue, E.; Giri, D.; Harbus, M.D.; Falcone, D.J.; Hudis, C.A.; Subbaramaiah, K.; Dannenberg, A.J. Estrogen Protects against Obesity-Induced Mammary Gland Inflammation in Mice. Cancer Prev Res (Phila) 2015, 8, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med 2019, 25, 141–151. [Google Scholar] [CrossRef] [PubMed]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. The Lancet Oncology 2018, 19, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Martel, S.; Poletto, E.; Ferreira, A.R.; Lambertini, M.; Sottotetti, F.; Bertolini, I.; Montemurro, F.; Bernardo, A.; Risi, E.; Zanardi, E.; et al. Impact of body mass index on the clinical outcomes of patients with HER2-positive metastatic breast cancer. Breast 2018, 37, 142–147. [Google Scholar] [CrossRef]
- Krasniqi, E.; Pizzuti, L.; Barchiesi, G.; Sergi, D.; Carpano, S.; Botti, C.; Kayal, R.; Sanguineti, G.; Marchetti, P.; Botticelli, A.; et al. Impact of BMI on HER2+ metastatic breast cancer patients treated with pertuzumab and/or trastuzumab emtansine. Real-world evidence. J Cell Physiol 2020, 235, 7900–7910. [Google Scholar] [CrossRef]
- Di Cosimo, S.; Porcu, L.; Agbor-Tarh, D.; Cinieri, S.; Franzoi, M.A.; De Santis, M.C.; Saura, C.; Huober, J.; Fumagalli, D.; Izquierdo, M.; et al. Effect of body mass index on response to neo-adjuvant therapy in HER2-positive breast cancer: an exploratory analysis of the NeoALTTO trial. Breast cancer research : BCR 2020, 22, 115. [Google Scholar] [CrossRef] [PubMed]
- Mazzarella, L.; Disalvatore, D.; Bagnardi, V.; Rotmensz, N.; Galbiati, D.; Caputo, S.; Curigliano, G.; Pelicci, P.G. Obesity increases the incidence of distant metastases in oestrogen receptor-negative human epidermal growth factor receptor 2-positive breast cancer patients. Eur J Cancer 2013, 49, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Agresti, R.; Meneghini, E.; Baili, P.; Minicozzi, P.; Turco, A.; Cavallo, I.; Funaro, F.; Amash, H.; Berrino, F.; Tagliabue, E.; et al. Association of adiposity, dysmetabolisms, and inflammation with aggressive breast cancer subtypes: a cross-sectional study. Breast cancer research and treatment 2016, 157, 179–189. [Google Scholar] [CrossRef]
- Alkhateeb, A.A.; Leitzel, K.; Ali, S.M.; Campbell-Baird, C.; Evans, M.; Fuchs, E.M.; Köstler, W.J.; Lipton, A.; Connor, J. Elevation in inflammatory serum biomarkers predicts response to trastuzumab-containing therapy. PLoS One 2012, 7, e51379. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D'Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell 2012, 47, 570–584. [Google Scholar] [CrossRef]
- Liu, S.; Lee, J.S.; Jie, C.; Park, M.H.; Iwakura, Y.; Patel, Y.; Soni, M.; Reisman, D.; Chen, H. HER2 Overexpression Triggers an IL1α Proinflammatory Circuit to Drive Tumorigenesis and Promote Chemotherapy Resistance. Cancer Research 2018, 78, 2040–2051. [Google Scholar] [CrossRef]
- Heng, Y.J.; Wang, J.; Ahearn, T.U.; Brown, S.B.; Zhang, X.; Ambrosone, C.B.; de Andrade, V.P.; Brufsky, A.M.; Couch, F.J.; King, T.A.; et al. Molecular mechanisms linking high body mass index to breast cancer etiology in post-menopausal breast tumor and tumor-adjacent tissues. Breast cancer research and treatment 2019, 173, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Mattei, E.; Velazquez-Torres, G.; Phan, L.; Zhang, F.; Chou, P.C.; Shin, J.H.; Choi, H.H.; Chen, J.S.; Zhao, R.; Chen, J.; et al. Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor-positive breast cancer. J Natl Cancer Inst 2014, 106. [Google Scholar] [CrossRef]
- Madeddu, C.; Gramignano, G.; Floris, C.; Murenu, G.; Sollai, G.; Macciò, A. Role of inflammation and oxidative stress in post-menopausal oestrogen-dependent breast cancer. J Cell Mol Med 2014, 18, 2519–2529. [Google Scholar] [CrossRef]
- Quigley, D.A.; Tahiri, A.; Lüders, T.; Riis, M.H.; Balmain, A.; Børresen-Dale, A.L.; Bukholm, I.; Kristensen, V. Age, estrogen, and immune response in breast adenocarcinoma and adjacent normal tissue. Oncoimmunology 2017, 6, e1356142. [Google Scholar] [CrossRef]
- Qureshi, R.; Picon-Ruiz, M.; Aurrekoetxea-Rodriguez, I.; Nunes de Paiva, V.; D'Amico, M.; Yoon, H.; Radhakrishnan, R.; Morata-Tarifa, C.; Ince, T.; Lippman, M.E.; et al. The Major Pre- and Postmenopausal Estrogens Play Opposing Roles in Obesity-Driven Mammary Inflammation and Breast Cancer Development. Cell metabolism 2020, 31, 1154–1172.e1159. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Arén Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol 2019, 20, 1370–1385. [Google Scholar] [CrossRef] [PubMed]
- Blank Christian, U.; Haanen John, B.; Ribas, A.; Schumacher Ton, N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gatti-Mays, M.E.; Kalinsky, K.; Korde, L.A.; Sharon, E.; Amiri-Kordestani, L.; Bear, H.; McArthur, H.L.; Frank, E.; Perlmutter, J.; et al. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncology 2019, 5, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jonas, S.F.; Bataillon, G.; Criscitiello, C.; Salgado, R.; Loi, S.; Viale, G.; Lee, H.J.; Dieci, M.V.; Kim, S.B.; et al. Prognostic value of tumor-infiltrating lymphocytes in patients with early-stage triple-negative breast cancers (TNBC) who did not receive adjuvant chemotherapy. Annals of Oncology 2019, 30, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. New England Journal of Medicine 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. New England Journal of Medicine 2022, 386, 556–567. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncology 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Wu, Z.; Wang, N.; Yang, Z.; Li, Y.; Xu, B.; Sun, M. Association between body mass index and survival outcomes for cancer patients treated with immune checkpoint inhibitors: a systematic review and meta-analysis. Journal of translational medicine 2020, 18, 235. [Google Scholar] [CrossRef] [PubMed]
- Maslov, D.; Tawagi, K.; Simenson, V.; Yuan, H.; Parent, C.; Bamnolker, A.; Goel, R.; Blake, Z.; KC, M.; Matrana, M.R.; et al. Impact of body mass index on survival rates in patients receiving immune checkpoint inhibitors. Journal of Clinical Oncology 2020, 38, e15108. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Procaccini, C.; Jirillo, E.; Matarese, G. Leptin as an immunomodulator. Mol Aspects Med 2012, 33, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell metabolism 2020, 31, 148–161.e145. [Google Scholar] [CrossRef]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front Immunol 2018, 9, 2569. [Google Scholar] [CrossRef] [PubMed]
- Dyck, L.; Prendeville, H.; Raverdeau, M.; Wilk, M.M.; Loftus, R.M.; Douglas, A.; McCormack, J.; Moran, B.; Wilkinson, M.; Mills, E.L.; et al. Suppressive effects of the obese tumor microenvironment on CD8 T cell infiltration and effector function. J Exp Med 2022, 219. [Google Scholar] [CrossRef] [PubMed]
- Moro-García, M.A.; Mayo, J.C.; Sainz, R.M.; Alonso-Arias, R. Influence of Inflammation in the Process of T Lymphocyte Differentiation: Proliferative, Metabolic, and Oxidative Changes. Frontiers in Immunology 2018, 9. [Google Scholar] [CrossRef]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Frontiers in Immunology 2019, 10. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nature reviews. Immunology 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Ding, J.; Chen, Y. Role of CD8+ T lymphocyte cells: Interplay with stromal cells in tumor microenvironment. Acta Pharmaceutica Sinica B 2021, 11, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Martin-Castillo, B.; Menendez, J.A. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. OncoImmunology 2019, 8, e1633235. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Kong, W.; Zhang, L.; Han, J.; Clark, L.H.; Yin, Y.; Fang, Z.; Sun, W.; Wang, J.; Gilliam, T.P.; et al. Reversal of obesity-driven aggressiveness of endometrial cancer by metformin. Am J Cancer Res 2019, 9, 2170–2193. [Google Scholar] [PubMed]
- Tapia, E. Reduction of Obesity Associated Breast Cancer Risk in a Phase II Clinical Trial of Metformin. 2020.
- Wu, Z.; Zhang, C.; Najafi, M. Targeting of the tumor immune microenvironment by metformin. Journal of Cell Communication and Signaling 2021. [Google Scholar] [CrossRef]
- Kristófi, R.; Eriksson, J.W. Metformin as an anti-inflammatory agent: a short review. Journal of Endocrinology 2021, 251, R11–R22. [Google Scholar] [CrossRef]
- Wei, Z.; Zhang, X.; Yong, T.; Bie, N.; Zhan, G.; Li, X.; Liang, Q.; Li, J.; Yu, J.; Huang, G.; et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nature Communications 2021, 12, 440. [Google Scholar] [CrossRef]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; Pasca di Magliano, M.; Swanson, K.D.; Zheng, B. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J Invest Dermatol 2017, 137, 1740–1748. [Google Scholar] [CrossRef]
- Nojima, I.; Eikawa, S.; Tomonobu, N.; Hada, Y.; Kajitani, N.; Teshigawara, S.; Miyamoto, S.; Tone, A.; Uchida, H.A.; Nakatsuka, A.; et al. Dysfunction of CD8 + PD-1 + T cells in type 2 diabetes caused by the impairment of metabolism-immune axis. Sci Rep 2020, 10, 14928. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res 2017, 5, 9–16. [Google Scholar] [CrossRef]
- Zhao, D.; Long, X.-D.; Lu, T.-F.; Wang, T.; Zhang, W.-W.; Liu, Y.-X.; Cui, X.-L.; Dai, H.-J.; Xue, F.; Xia, Q. Metformin decreases IL-22 secretion to suppress tumor growth in an orthotopic mouse model of hepatocellular carcinoma. International Journal of Cancer 2015, 136, 2556–2565. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-C.; Sun, X.; Ma, Q.; Fu, G.-F.; Cong, L.-L.; Zhang, H.; Fan, D.-F.; Feng, J.; Lu, S.-Y.; Liu, J.-L.; et al. Metformin's antitumour and anti-angiogenic activities are mediated by skewing macrophage polarization. Journal of Cellular and Molecular Medicine 2018, 22, 3825–3836. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin improves obesity-associated inflammation by altering macrophages polarization. Molecular and cellular endocrinology 2018, 461, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kwak, H.J.; Cha, J.Y.; Jeong, Y.S.; Rhee, S.D.; Kim, K.R.; Cheon, H.G. Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. The Journal of biological chemistry 2014, 289, 23246–23255. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Molecular Cell 2018, 71, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, Y.; Eikawa, S.; Tomonobu, N.; Domae, S.; Uehara, T.; Hori, S.; Furusawa, Y.; Hase, K.; Sasaki, A.; Udono, H. Attenuation of CD4(+)CD25(+) Regulatory T Cells in the Tumor Microenvironment by Metformin, a Type 2 Diabetes Drug. EBioMedicine 2017, 25, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.V.; Melo, A.C.L.; Siong Low, J.; Arantes de Castro, Í.; Braga, T.T.; Almeida, D.C.; Batista de Lima, A.G.U.; Hiyane, M.I.; Correa-Costa, M.; Andrade-Oliveira, V.; et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Research 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Cufí, S.; Vazquez-Martin, A.; Menendez, O.J.; Bosch-Barrera, J.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle 2012, 11, 865–870. [Google Scholar] [CrossRef]
- Cai, S.; Chen, Z.; Wang, Y.; Wang, M.; Wu, J.; Tong, Y.; Chen, L.; Lu, C.; Yang, H. Reducing PD-L1 expression with a self-assembled nanodrug: an alternative to PD-L1 antibody for enhanced chemo-immunotherapy. Theranostics 2021, 11, 1970–1981. [Google Scholar] [CrossRef]
- Haikala, H.M.; Anttila, J.M.; Marques, E.; Raatikainen, T.; Ilander, M.; Hakanen, H.; Ala-Hongisto, H.; Savelius, M.; Balboa, D.; Von Eyss, B.; et al. Pharmacological reactivation of MYC-dependent apoptosis induces susceptibility to anti-PD-1 immunotherapy. Nat Commun 2019, 10, 620. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhao, Y.; Liu, G.; Zhou, H.L.; Fan, J.; Zhang, L.; Li, Y.L.; Wang, Y.; Liang, J.; Xu, Z.X. Upregulation of programmed death ligand 1 by liver kinase B1 and its implication in programmed death 1 blockade therapy in non-small cell lung cancer. Life Sci 2020, 256, 117923. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Liu, Y.; Chen, C.; Chi, J.; Zhong, L.; Zhao, Y.; Zhao, Y. Metformin loaded porous particles with bio-microenvironment responsiveness for promoting tumor immunotherapy. Biomater Sci 2021. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Chen, B.E.; Gelmon, K.A.; Whelan, T.J.; Ennis, M.; Lemieux, J.; Ligibel, J.A.; Hershman, D.L.; Mayer, I.A.; Hobday, T.J.; et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. Jama 2022, 327, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: a systematic review and meta-analysis. Ann Oncol 2016, 27, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L.; et al. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast cancer research and treatment 2019, 174, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gong, C.; Wang, Z.; Zhang, J.; Wang, L.; Zhang, S.; Cao, J.; Tao, Z.; Li, T.; Wang, B.; et al. A randomized phase II study of aromatase inhibitors plus metformin in pre-treated postmenopausal patients with hormone receptor positive metastatic breast cancer. Oncotarget 2017, 8, 84224–84236. [Google Scholar] [CrossRef] [PubMed]
- Barakat, H.E.; Hussein, R.R.S.; Elberry, A.A.; Zaki, M.A.; Ramadan, M.E. The impact of metformin use on the outcomes of locally advanced breast cancer patients receiving neoadjuvant chemotherapy: an open-labelled randomized controlled trial. Sci Rep 2022, 12, 7656. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Pernas, S.; Dorca, J.; Álvarez, I.; Martínez, S.; Pérez-Garcia, J.M.; Batista-López, N.; Rodríguez-Sánchez, C.A.; Amillano, K.; Domínguez, S.; et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: the METTEN study. Oncotarget 2018, 9, 35687–35704. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J Immunother Cancer 2018, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Hadad, S.M.; Coates, P.; Jordan, L.B.; Dowling, R.J.; Chang, M.C.; Done, S.J.; Purdie, C.A.; Goodwin, P.J.; Stambolic, V.; Moulder-Thompson, S.; et al. Evidence for biological effects of metformin in operable breast cancer: biomarker analysis in a pre-operative window of opportunity randomized trial. Breast cancer research and treatment 2015, 150, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Lord, S.R.; Cheng, W.-C.; Liu, D.; Gaude, E.; Haider, S.; Metcalf, T.; Patel, N.; Teoh, E.J.; Gleeson, F.; Bradley, K.; et al. Integrated Pharmacodynamic Analysis Identifies Two Metabolic Adaption Pathways to Metformin in Breast Cancer. Cell metabolism 2018, 28, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Hadad, S.; Iwamoto, T.; Jordan, L.; Purdie, C.; Bray, S.; Baker, L.; Jellema, G.; Deharo, S.; Hardie, D.G.; Pusztai, L.; et al. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast cancer research and treatment 2011, 128, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Vagia, E.; Viveiros, P.; Kang, C.Y.; Lee, J.Y.; Gim, G.; Cho, S.; Choi, H.; Kim, L.; Park, I.; et al. Overcoming acquired resistance to PD-1 inhibitor with the addition of metformin in small cell lung cancer (SCLC). Cancer Immunol Immunother 2020. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nature Reviews Clinical Oncology 2019. [Google Scholar] [CrossRef]
- Maliniak, M.L.; Cheriyan, A.M.; Sherman, M.E.; Liu, Y.; Gogineni, K.; Liu, J.; He, J.; Krishnamurti, U.; Miller-Kleinhenz, J.; Ashiqueali, R.; et al. Detection of crown-like structures in breast adipose tissue and clinical outcomes among African-American and White women with breast cancer. Breast Cancer Research 2020, 22, 65. [Google Scholar] [CrossRef]
Figure 1.
Crown-like structures. A, Representative image showing CD68+ Crown-like structures within the adipose tissue adjacent to a human breast tumour; B, Hypertrophic adipocytes surrounded by macrophages forming crown-like structures.
Figure 1.
Crown-like structures. A, Representative image showing CD68+ Crown-like structures within the adipose tissue adjacent to a human breast tumour; B, Hypertrophic adipocytes surrounded by macrophages forming crown-like structures.
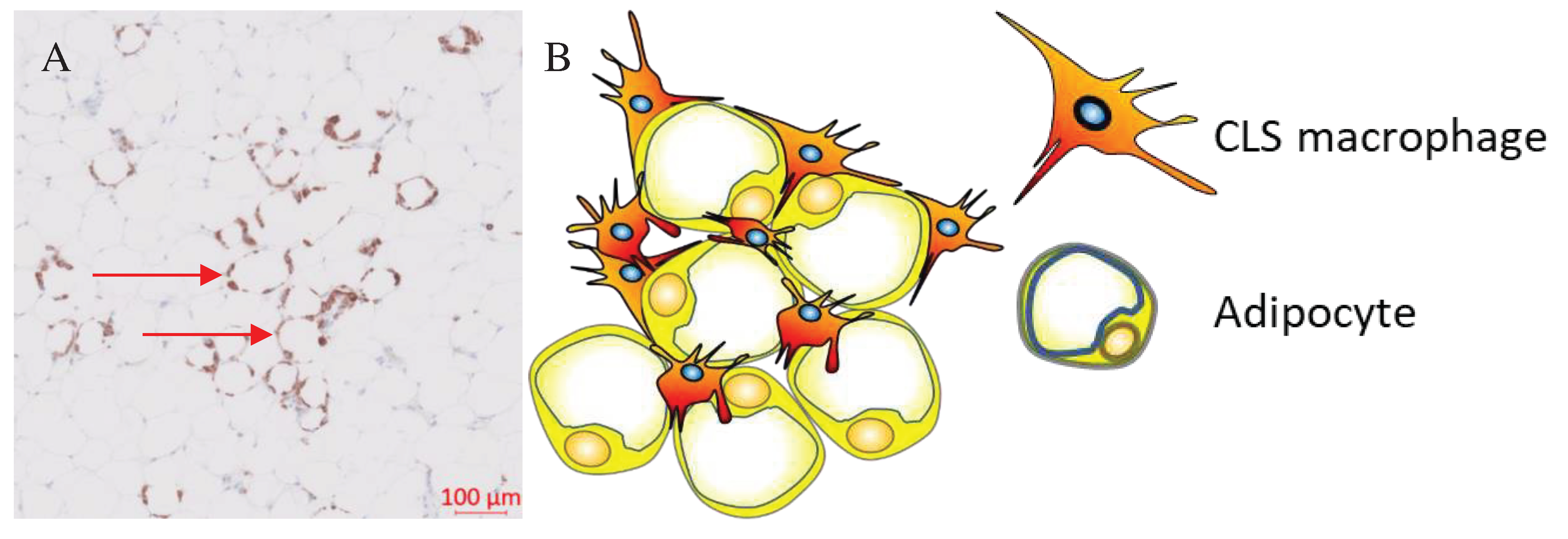

Table 1.
Obesity is associated with systemic immunometabolic changes.
| Immune changes | Metabolic changes |
|---|---|
| Upregulation of proinflammatory signalling pathways | Increased insulin and IGF levels |
| Increased immune cell infiltration | Insulin resistance |
| Upregulation of WNT signalling | Elevates leptin levels |
| Increased synthesis of arachidonic acid & PGE2 | Increases oestrogen and androgen levels |
| Downregulation of response to antigen and mitogen stimulation | Anti-apoptotic, promotes stemness |
Table 2.
Clinical outcomes of retrospective cohorts that evaluated the prognostic role of CLS in breast cancer.
Table 2.
Clinical outcomes of retrospective cohorts that evaluated the prognostic role of CLS in breast cancer.
| Study | Sample size | CLS- (n) | CLS+ (n) | CLS marker | RFS, CLS+ vs CLS- [HR (95%CI)] | OS, CLS+ vs CLS- [HR (95%CI)] |
|---|---|---|---|---|---|---|
| Iyengar N (cohort 2) (2016) | 127 | 75 | 52 | CD68 | 1.83 (1.07-3.13) a | not reported |
| Koru-Sengul T h (2016) | 134 | NR | NR | CD40 | 5.87 (0.73- 47.23) b | 13.59 (1.56-118.16) b |
| Koru-Sengul T h (2016) | 134 | NR | NR | CD163 | 2.21 (0.65-7.59) b | 2.42 (0.54-10.89) b |
| Koru-Sengul T h (2016) | 134 | NR | NR | CD206 | 1.17 (0.09-15.35) b | 0.74 (0.04-15.55) b |
| Cha YJ (2018)i | 140 | 122 | 18 | CD163 | 105 (94-116) vs 124 (118-131) c, d | 105 (94-116) vs 130 (124-136) c, d |
| Cha YJ (2018)i | 140 | 115 | 25 | CD68 | 106 (97-114) vs 124 (117-131) c, d |
106 (99-114) vs 130 (124-136) c, d |
| Cha YJ (2018)i | 56 | 49 | 7 | CD68 | 76 (56-96) vs 120 (108-132) c, d, e |
79 (63-96) vs 125 (114-136) c, d, e |
| Maliniak M (2020) | 319 | 223 | 96 | CD68 | 1.05 (0.64-1.72) f | 1.02 (0.55-1.87) f |
| Birts C (2022) | 117 | 47 | 61 | CD32B | 4.2 (1.01–17.4)g | not reported |
| Abbreviations: CLS, crown-like structures; NR, not reported; RFS, Recurrence Free Survival; OS, Overall Survival; a Multivariate model adjusted for age, race, BMI, breast cancer subtype, grade, stage, dyslipidaemia, hypertension, diabetes mellitus, and adjuvant therapy; b 90%CI, adjusted for HER2; c Univariate analysis; d Mean, month (95% CI); e node-positive patients; f multivariate model adjusted for age at diagnosis (years), BMI (kg/m2), and smoking status; g HER2+ breast cancer patients, multivariate model for time to metastatic disease, adjusted for peripheral white cell count; h,i same cohort; bold, statistically significant. | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



