
Submitted:
27 February 2023
Posted:
28 February 2023
You are already at the latest version
Abstract
Spinal muscular atrophy 5q (SMA) is the most common neuromuscular inherited disease in neonates which is associated with homozygous deletion of exon 7 in the SMN1 gene. Recently established drugs can improve the motor functions of SMA infants when treated in the pre-symptomatic stage. With aim of providing an early diagnosis, newborn screening (NBS) for SMA using real-time PCR assay with dried blood spots (DBS) was performed in Saint-Petersburg from January 2022 through November 2022. Here, 36,140 newborns were screened by GenomeX real-time PCR-based screening test, and three genotypes were determined. Homozygous deletion carriers (4 newborns), heterozygous carriers (772 newborns) and wild-type individuals (35,364 newborns) were identified. All four newborns screened positive for homozygous SMN1 deletion were confirmed by alternate methods. Two of the newborns had two copies of SMN2, and two of the newborns had three copies. We determined Saint-Petersburg spinal muscular atrophy incidence of 1 in 9,035 and SMA carrier frequency of 1 in 47. In conclusion, it can be summarized that providing both timely SMN1 information and confirmation along with SMN2 copy number as part of SMA newborn screening algorithm can significantly improve clinical follow-up, family members testing, and SMA patients' treatment.
Keywords:
spinal muscular atrophy
; newborn screening
; SMN1
; SMN2
; real-time PCR
; dried blood spots
; SMA incidence
; carrier frequency
1. Introduction
Spinal muscular atrophy is a severe autosomal recessive disorder characterized that results progressive muscular weakness caused by degeneration of motor neurons of the spinal cord. SMA is the most common genetic cause of infant mortality with the incidence of about 1 in 6,000-10,000 live births and the carrier frequency 1:40-1:60 [1,2]. SMA is classified into four main clinical groups (types I-IV) based on the age of onset and symptoms severity [3,4]. SMA Type I (Werdnig-Hoffmann disease) is the most acute form, characterized by onset in the first 6 months of life, no achievement of sitting and death within the first 2 years of life often caused by the respiratory distress. SMA type II patients demonstrate first symptoms after the age of 6 months and survive beyond 2 years, can sit unsupported, but never gain the ability to walk. Milder SMA type III (Kugelberg–Welander disease) develops after 18 months of life; patients achieve the ability to walk and have life expectancy close to a healthy population. Patients with SMA type IV usually present first symptoms in the second or third decade of life and have a normal life expectancy [3].
SMA is caused by homozygous mutations in the SMN1 gene [5,6]. About 97% of patients show homozygous deletions of the SMN1 gene, the rest exhibit small intragenic mutations [7]. The SMN1 gene has a nearly identical copy – the SMN2 gene – the result of duplication and inversion of the chromosomal segment of around 500 kb in chromosome 5q13. SMN1 differs from SMN2 by several single nucleotide exchanges [5]. Only one difference is functionally important - a translationally silent transition c.840C>T at +6 position in exon 7 that weakens the exonic splice site, causing exclusion of exon 7 from most of the SMN2 transcripts and production of a truncated non-functional SMN protein [8,9]. Still a little amount of full-length SMN protein produced by SMN2 gene makes this gene a principal disease severity modifier in SMA patients. Now there is growing evidence of additional factors contributing to SMA severity that can be found among multiple proteins interacting with SMN or affecting motor neuron survival, epigenetic modifications, transcriptional or splicing factors influencing SMN2 expression [10,11,12,13,14]. In rare cases protective modifiers may substantially ameliorate SMA progression up to almost complete absence of symptoms in patients [15,16]. But still only pathogenetic treatment may really change the course of the disease, being especially effective when given presymptomatically.
Antisense oligonucleotides technology was successfully used to modulate SMN2 gene exon 7 inclusion as possible SMA therapy [17]. Nusinersen was the first approved drug for treating spinal muscular atrophy [18]. It was registered in December 2016 as an orphan drug by the US Food and Drug Administration for SMA. Another SMN2 pre-mRNA splicing modifying drug is risdiplam [19]. It is an orally administered drug, which elevates SMN2 full-length mRNA production and spreads into the central nervous system (CNS) as well as peripheral tissues [20].
Gene replacement therapies are highly attractive therapeutic approaches to treat spinal muscular atrophy as it is classified as a monogenic disease. Many studies found that the injection of self-complementary scAAV9 vectors expressing full-length human SMN protein in different severe SMA mouse models significantly enhanced lifespan and disease severity [21,22]. The biotec company AveXis in Dallas has gained rapid track FDA approval for its scAAV9.CB.SMN vector, for which Foust et al. explained that it crosses blood-brain-barrier [23]. Onasemnogeneabeparvovec known as AVXS-101 that has the commercial name of Zolgensma is a new treatment for SMA as a biologic gene therapy drug. It consists of self-complementary AVV9 virus capsids which contain SMN1 transgene. A single dose of the drug might have a lasting influence throughout the lifetime of the patient as it is thought.
Importantly, the efficacy of any SMA therapy is highly dependent on period when treatment is initiated. Taking into account that the principle of existing treatment is to stop the disease progression rather than to reverse motor neuron death, the therapeutic intervention should occur before the onset of symptoms. The requirement of SMN as protein with housekeeping role and motoneuron-specific functions accounts for the narrow therapeutic window, especially in individuals with most severe SMA forms [24]. As most SMA cases develop in early postnatal period, the newborn screening for SMA is essential for giving patients the diagnosis and appropriate treatment in time. Evidences from in vivo studies in animal models and from clinical trials on children affected with SMA type I substantiate that early therapeutic intervention correlated with better motor performance and rescue of the pathological phenotype [25].
Currently, there was no newborn screening of spinal muscular atrophy in Saint-Petersburg. With a population of over 5,200,000 and birth rate 52,000 newborns per year, it represents a typical example of Russian megapolis. Our main aim is the establishment of the SMA newborn screening program in Saint-Petersburg. Second aim of this study is to determine the incidence of SMA and the carrier frequency in the population of Saint-Petersburg.
2. Materials and Methods
2.1. Study Recruitment
Twenty-one Saint-Petersburg maternity hospitals participated in pilot SMA screening program. Each hospital provided mothers with informed consent forms and informational brochures. Signed consent forms were further transported to Genetic Medical Center with dried blood spots (DBS) taken for mandated neonatal screening where only those DBS punches were taken for SMA analysis that had corresponding permit for this test. The SMA test was provided at no cost to participants.
2.2. Assay Validation
Validation of GenomeX technology proposed to use in SMA screening analysis was performed on DNA samples with known genotype extracted from whole blood by means of phenol-chloroform extraction. 250 samples were analyzed: SMN1 exon 7 homozygous deletion carriers (N=50), heterozygous carriers (N=58) and wild-type individuals (N=142). SMN1 deletions and copy number were determined before with TaqMan real-time PCR technology reported previously [26].
Trial screening test was performed on 400 DBS punches with unknown SMN1 copy number provided by the Genetic Medical Center by means of GenomeX assay kit (Genome-Mix LLC, Moscow, Russia).
2.3. Specimens
This study was performed using 250 DNA samples collected in large-scale research facilities #3076082 "Human Reproductive Health" in D.O. Ott Research Institute of Obstetrics, Gynecology and Reproductology.
Four hundred DBS on validation stage and 36,140 DBS on pilot screening stage were obtained from the Genetic Medical Center. Filter paper blood spots were obtained by a heel puncture on 4th day after birth (on 7th day in case of premature babies).
Capillary blood samples from SMA heterozygous carriers and SMA homozygous deletion carriers were obtained for verification from the Genetic Medical Center. DNA was extracted by means of salt (NaCl) extraction.
2.4. SMA Screening Assay
2.4.1. DNA extraction:
1. A 1X GenomeX elution reagent was prepared by diluting the 100X solution with ampouled or bidistilled water 1:99 to get 250 µl of 1X solution per sample.
2. A 3.2 mm diameter circle was knocked out from a DBS card into a 0.2 ml tube or a 96-well plate using a puncher.
3. 100 µl of 1X elution reagent was added to each tube.
4. Then tubes were incubated for 15 minutes at room temperature.
5. The liquid was stirred by pipetting twice and then removedfrom the tube.
6. Thesteps 3-5 were repeated one more time.
7. 50 ml of 1X elution reagent was added to the washed filter.
8. The tubes/plated were closed and centrifuged to remove drops.
9. The samples were heated for 15 minutes at +99 °C in a hot-lid thermal cycler Bio-Rad T100.
10. The samples were removed to +4°C and stored there up to 3 weeks.
2.4.2. Performing real-time PCR
1. GenomeX MasterMix was defrosted and joined with polymerase (17.6 µl of MasterMix + 0.4 µl of polymerase per sample, each DNA sample was analyzed twice).
2. 18 µl of prepared mix was added to each well of 96-well plate.
3. 2 µl of DNA sample was added to each well. No DNA was added in 2 wells (contamination control).
4. Plate was centrifuged to remove drops.
5. Plate was placed in a real-time PCR device under the following conditions: preincubation 95° - 5 min, then 40 cycles of 94° - 15 sec and 62° - 1 min.
Several real-time PCR machines were used: QuantStudio 5 (Applied Biosystems), LightCycler 96 (Roche), Applied Biosystems 7500, Rotor-Gene 3000 (Corbett).
The fluorescent signal was detected on FAM / Sybr Green and ROX / Texas Red channels.
2.4.3. Analysis of the results
The results were obtained based on ΔΔCt / relative quantitative analysis. When performing run on Rotor-Gene 3000 a calibration curve was prepared for calculation the results. A sample with 2 copies of SMN1 gene was used as calibrator (the value for this sample was equal to 1 and corresponded to the relative amount of the SMN1 gene according to the formula SMN1 = [SMN1] / [reference] (where [SMN1] is the concentration of the real-time PCR product from the SMN1 gene (FAM channel); [reference] is the concentration of the real-time PCR product from the reference gene (ROX channel). Then the 0 value for the sample corresponded to SMA patient, 0.5 – to SMA carrier (1 copy of SMN1), 1 – to 2 SMN1 copies, 1.5 – to 3, etc. Ct on the ROX detection channel should be < 33.
2.5. Screening, Notification of Results and Confirmatory Testing
In case of SMN1 deletions findings family members were invited to the Genetic Medical Center for medical genetic counseling and subsequent blood sampling (whole family in case of homozygous deletion and a newborn in case of heterozygous deletion). Family members got the printed results on SMA neonatal screening on DBS (only in case of SMN1 deletions findings). SMN1 deletions verification was performed in D.O.Ott Research Institute on DNA samples extracted from capillary blood. In case of homozygous deletion, MLPA analysis was performed according to manufacturer’s instructions (SALSA MLPA Probemix P021 SMA, MRC Holland). MLPA results visualization were obtained on Applied Biosystems 3130 Genetic Analyzer. After verification test family members got another printed results based on analysis performed on DNA extracted from whole blood and had genetic counseling.
3. Results and Discussion
3.1. Validation Study
We evaluated the Genome X real-time PCR-based assay using previously analyzed samples with different genotypes from homozygous SMN1 exon 7 deletion carriers, heterozygous carriers and wild-type individuals. We found clear non-overlapping ranges of values of the SMN1 copy number between the samples with 0, 1, and 2 or more copies of SMN1 exon 7, regardless of the SMN2 copy number. The significance of differences was confirmed by analysis of variance (p < 0.001).Based onthese data, ranges of SMN1 copy number values were assigned for each SMN1 genotype, e.g. range between 0.23–0.599 were considered heterozygous carriers, whereas range between 0.79–1.23 were considered SMN1 exon 7 deletion negative (2 SMN1 copies), and zero level were considered homozygous deletion carriers (Table 1). There was 100% concordance between the SMN1 copy number determined by the Genome X assay and known genotypes of 250 control samples. Specificity of the assay can be determined as 100%, the same for sensitivity if we do not take in account inability of this real-time PCR-based method to detect point mutations in SMN1 gene. Otherwise the sensitivity of the assay can be determined as 97% based of incidence of compound heterozygous carriers of SMN1 point mutations [7].
Additionally de-identified DBS of newborns (N=400) with unknown SMN1 copy number were provided by the Genetic Medical Center for the analysis. Total number of DBS samples roughly corresponds to number of babies who are born in Saint-Petersburg per two days. 48 hours were spent on screening of the 400 samples in the validation study. According to the results 8 individuals (2%) were SMA carriers, having one copy of SMN1 exon 7; 360 individuals (90%) have two copies of SMN1 and 32 (8%) have three copies. So the carrier frequency was estimated as of 1:50 that is in concordance with the reported data [27]. None had a homozygous SMN1 deletion and no samples failed.
3.2. Patients’ Flow
For this study, no additional biological material from newborns is required because the residual blood spots collected for mandated NBS are sufficient for the SMA testing. In fact the mandated DBS cards are collected under the order of the Health Committee of the Saint-Petersburg Government dated 05/29/2006 No. 220-r "On mass screening of newborn children for hereditary diseases in Saint-Petersburg." According to this order five inherited diseases (phenylketonuria, cystic fibrosis, galactosemia, congenital adrenal hyperplasia, primary congenital hypothyroidism) are being screened in Saint-Petersburg. The DBS collection is carrying out in maternity hospitals subordinate to the Saint-Petersburg Health Committee, as well as federal institutions that include maternity hospitals, perinatal centers and maternity wards. Final destination for DBS cards is Genetic Medical Center. Aliquots of samples (DBS punches) for SMA screening studies were taken out daily (on weekdays) at agreed hours in a specialized lab room of Genetic Medical Center by its personnel under control of D.O.Ott Research Institute research assistants. The mother’s informed consents and a paper referral for a SMA screening study were attached to the samples and transported to D.O.Ott Research Institute (Figure 1).
Analysis at the D.O.Ott Research Institute was carried out within 3 working days from the arrival of the sample to the Genetic Medical Center. It took 1-2 days to transfer DBS to the Genetic Medical Center. With positive screening results (presence of homo- or heterozygous mutations in the SMN1 gene) the independent aliquot of DBS was taken from the Genetic Medical Center, the analysis of the sample was repeated and in case of confirming the results the study coordinator sent a message to the Genetic Medical Center about the necessity of medical genetic consultation for the mother of a newborn with an identified risk of developing or carriage of SMA. In accordance to the screening algorthm, the family members (whole family in case of homozygous deletion and a newborn in case of heterozygous deletion) were invited to the Genetic Medical Center for medical genetic counseling and subsequent blood sampling for SMA diagnosis or carrier status verification in D.O.Ott Research Institute (Figure 2).
Verifying diagnostics of SMA was carried out within 1-2 working days from the receiving of the whole blood sample, while SMA carrier testing on blood samples was performed within 5-10 days. Confirmatory testing in case of homozygous SMN1 deletion always included determination of SMN2 gene copy number due to its importance as a main disease modifier [15]. If the results of verifying study confirmed positive detection of homo- or heterozygous mutations in the SMN1 gene, the mother of a newborn with an identified risk of developing or carriage of SMA were informed and genetic counseling was provided again by specialists of the Genetic Medical Center. If no mutations in the SMN1 gene were found in the newborn, the mother was informed that there is no risk of developing or carriage of SMA. Also, positive results for SMA were simultaneously reported to neurologists in Saint-Petersburg Consulting and Diagnostic Center for Children.
3.3. Screening for SMA
Within the time period of the SMA pilot study, 36,140 from 36,217 consent forms for participation in the NBS program were received. Therefore, 99.79% of these parents agreed for SMA testing of their newborn children. This is an outstanding result of the study awareness partially mediated by efforts of the Health Committee and general interest of people in SMA problem. Almost total agreement of parents to additional SMA testing differs markedly from numbers received by NBS programs in other countries [28,29].
Totally, 36,140 newborns were screened, three genotypes among them were determined. Our SMA NBS program resulted in finding of four homozygous SMN1deletion carriers, 772 heterozygous carriers and 35,364 wild-type individuals. No false positive SMA cases were detected; only two false positive heterozygous carriers were found. All four screen positive newborns were confirmed by alternate methods – different real-time PCR system and MLPA [26,30]. In all newborns with homozygous SMN1 deletion SMN2 copy number was determined – two of them had two copies of SMN2, and two had three copies. All the diagnosed individuals were found pre-symptomatic and already received gene therapy by onasemnogene abeparvovec.
3.4. Determination of SMA Carrier Frequency in Newborns
SMA is the leading genetic cause of infant mortality and it is of importance to carry out studies in its prevalence and incidence. Realization of SMA NBS program by means of GenomeX real-time PCR-based screening test gives us a unique opportunity to gain additional information on SMA incidence in Saint-Petersburg and to determine the carrier frequency. Information on possibility of SMA carrier status determination was included to the consent form and the parents were allowed not to be informed on the newborn status. However, none from the parents disagreed to know this information. Moreover, to date most of the parents of newborns with heterozygous SMN1 deletions received medical genetic counseling. Some parents used this information for preconception SMA screening and several dozens of SMA carriers were found between them and siblings. This work in underway and will be continued. It should be noted, that no families with SMA risk were found between parents of heterozygous newborns.
It can be concluded that in Saint-Petersburg newborn population we determined spinal muscular atrophy incidence of 1 in 9,035 and SMA carrier frequency of 1 in 47. Our data are in full agreement with previously reported SMA incidence and carrier frequency [27]. However, it differs from international summarized data of NBS SMA programs and study of the carrier frequency in three populations of Russian Federation [31,32].
5. Conclusions
In conclusion, it can be summarized that providing both timely SMN1 information and confirmation along with SMN2 copy number as part of SMA newborn screening algorithm can significantly improve clinical follow-up, family members testing, and SMA patients’ treatment. Presymptomatic SMA revealing is the basis of implementation of maximum therapy effectiveness especially for SMA type I patients. Information about the carrier status can be a part of advance preconceptional care.
Author Contributions
Conceptualization, A.K., A.G. and V.B.; methodology, A.K. and M.M.; formal analysis, A.K., A.G. and M.M.; investigation, M.M., S.S., H.A., N.M., M.P., E.S., M.F., N.S., N.K., A.I., S.F., N.O., I.S., and A.E.; resources, A.L., A.L.K., I.S. and Y.G.; writing—original draft preparation, A.K. and M.M.; writing—review and editing, A.K. and A.G.; supervision, V.B. and I.K.; project administration, A.K., A.G. and O.B.; funding acquisition, A.K., A.G. and V.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Novartis Farma LLC, grant number #1606-MD.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of D.O. Ott Research Institute of Obstetrics, Gynecology and Reproductology (protocol 112 was approved 23 September 2021).
Informed Consent Statement
Informed consent was obtained from all representatives of subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to restrictions of the subjects’ agreement.
Acknowledgments
The authors express their gratitude to Heads and medical personnel of Saint-Petersburg maternity hospitals participated in the study.
Conflicts of Interest
A.K. and A.G. received speaker honoraria from Novartis Farma LLC.
References
- Ogino, S.; Wilson, R.B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum. Genet. 2002, 111, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.A.; Singh, P.; Darras, B.T. Spinal muscular atrophy: A clinical and research update. Pediatr. Neurol. 2012, 46, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Russman, B.S. Spinal muscular atrophy: Clinical classification and disease heterogeneity. J. Child Neurol. 2007, 22, 946–951. [Google Scholar] [CrossRef]
- Zerres, K.; Wirth, B.; Rudnik-Schöneborn, S. Spinal muscular atrophy--clinical and genetic correlations. Neuromuscul. Disord. 1997, 7, 202–7. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Glotov, A.S.; Kiselev, A. V; Ivashchenko, T.E.; Baranov, V.S. [Analysis of deletional damage in SMN1, SMN2, and NAIP genes in patients with spinal muscular atrophy in the northwestern region of Russia]. Genetika 2001, 37, 1156–9. [Google Scholar]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.M.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Cuscó, I.; Barceló, M.J.; Rojas–García, R.; Illa, I.; Gámez, J.; Cervera, C.; Pou, A.; Izquierdo, G.; Baiget, M.; Tizzano, E.F. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J. Neurol. 2006, 253, 21–25. [Google Scholar] [CrossRef]
- Zheleznyakova, G.Y.; Nilsson, E.K.; Kiselev, A.V.; Maretina, M.A.; Tishchenko, L.I.; Fredriksson, R.; Baranov, V.S.; Schiöth, H.B. Methylation levels of SLC23A2 and NCOR2 genes correlate with spinal muscular atrophy severity. PLoS One 2015, 10. [Google Scholar] [CrossRef]
- Hosseinibarkooie, S.; Schneider, S.; Wirth, B. Advances in understanding the role of disease-associated proteins in spinal muscular atrophy. Expert Rev. Proteomics 2017, 14, 581–592. [Google Scholar] [CrossRef]
- Maretina, M.A.; Zheleznyakova, G.Y.; Lanko, K.M.; Egorova, A.A.; Baranov, V.S.; Kiselev, A. V. Molecular Factors Involved in Spinal Muscular Atrophy Pathways as Possible Disease-modifying Candidates. Curr. Genomics 2018, 19, 339–355. [Google Scholar] [CrossRef]
- Maretina, M.; Egorova, A.; Baranov, V.; Kiselev, A. DYNC1H1 gene methylation correlates with severity of spinal muscular atrophy. Ann. Hum. Genet. 2019, 83, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyakova, G.Y.; Kiselev, A. V.; Vakharlovsky, V.G.; Rask-Andersen, M.; Chavan, R.; Egorova, A.A.; Schiöth, H.B.; Baranov, V.S. Genetic and expression studies of SMN2 gene in Russian patients with spinal muscular atrophy type II and III. BMC Med. Genet. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.; Also-Rallo, E.; Martínez-Hernández, R.; Alías, L.; Rodríguez-Alvarez, F.J.; Millán, J.M.; Hernández-Chico, C.; Baiget, M.; Tizzano, E.F. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul. Disord. 2011, 21, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2 ) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [PubMed]
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam distributes and increases <scp>SMN</scp> protein in both the central nervous system and peripheral organs. Pharmacol. Res. Perspect. 2018, 6, e00447. [Google Scholar] [CrossRef]
- Glascock, J.J.; Osman, E.Y.; Wetz, M.J.; Krogman, M.M.; Shababi, M.; Lorson, C.L. Decreasing Disease Severity in Symptomatic, Smn −/−; SMN2 +/+, Spinal Muscular Atrophy Mice Following scAAV9-SMN Delivery. Hum. Gene Ther. 2012, 23, 330–335. [Google Scholar] [CrossRef]
- Benkhelifa-Ziyyat, S.; Besse, A.; Roda, M.; Duque, S.; Astord, S.; Carcenac, R.; Marais, T.; Barkats, M. Intramuscular scAAV9-SMN Injection Mediates Widespread Gene Delivery to the Spinal Cord and Decreases Disease Severity in SMA Mice. Mol. Ther. 2013, 21, 282–290. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, N.; et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J. Cell. Mol. Med. 2020, 24, 3034–3039. [Google Scholar] [CrossRef] [PubMed]
- Govoni, A.; Gagliardi, D.; Comi, G.P.; Corti, S. Time Is Motor Neuron: Therapeutic Window and Its Correlation with Pathogenetic Mechanisms in Spinal Muscular Atrophy. Mol. Neurobiol. 2018, 55, 6307–6318. [Google Scholar] [CrossRef] [PubMed]
- Anhuf, D.; Eggermann, T.; Rudnik-Schöneborn, S.; Zerres, K. Determination of SMN1 and SMN2 copy number using TaqManTM technology. Hum. Mutat. 2003, 22, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review. Orphanet J. Rare Dis. 2017, 12, 124. [Google Scholar] [CrossRef]
- Kraszewski, J.N.; Kay, D.M.; Stevens, C.F.; Koval, C.; Haser, B.; Ortiz, V.; Albertorio, A.; Cohen, L.L.; Jain, R.; Andrew, S.P.; et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet. Med. 2018, 20, 608–613. [Google Scholar] [CrossRef]
- Gailite, L.; Sterna, O.; Konika, M.; Isakovs, A.; Isakova, J.; Micule, I.; Setlere, S.; Diriks, M.; Auzenbaha, M. New-Born Screening for Spinal Muscular Atrophy: Results of a Latvian Pilot Study. Int. J. Neonatal Screen. 2022, 8, 15. [Google Scholar] [CrossRef]
- Scarciolla, O.; Stuppia, L.; De Angelis, M.V.; Murru, S.; Palka, C.; Giuliani, R.; Pace, M.; Di Muzio, A.; Torrente, I.; Morella, A.; et al. Spinal muscular atrophy genotyping by gene dosage using multiple ligation-dependent probe amplification. Neurogenetics 2006, 7, 269–276. [Google Scholar] [CrossRef]
- Dangouloff, T.; Vrščaj, E.; Servais, L.; Osredkar, D.; Adoukonou, T.; Aryani, O.; Barisic, N.; Bashiri, F.; Bastaki, L.; Benitto, A.; et al. Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go. Neuromuscul. Disord. 2021, 31, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Zabnenkova, V. V; Dadali, E.L.; Spiridonova, M.G.; Zinchenko, R.A.; Polyakov, A. V Heterozygous carrier rate for type I–IV proximal spinal muscular atrophy in Chuvashes, Udmurts, and residents of the Moscow region. Russ. J. Genet. 2012, 48, 838–845. [Google Scholar] [CrossRef]
Figure 1.
Scheme of DBS and documentation flow for realization of SMA newborn screening in Saint-Petersburg.
Figure 1.
Scheme of DBS and documentation flow for realization of SMA newborn screening in Saint-Petersburg.
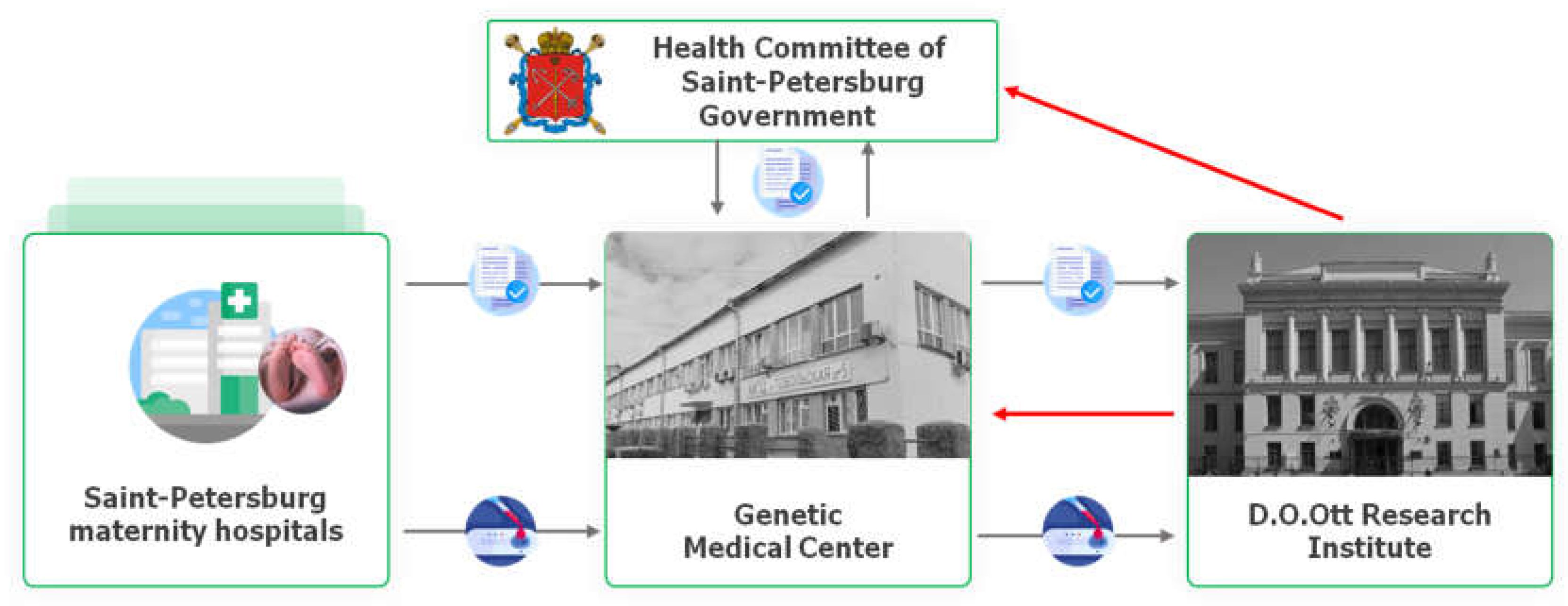
Figure 2.
Screening algorithm used in the SMA pilot study in Saint-Petersburg.
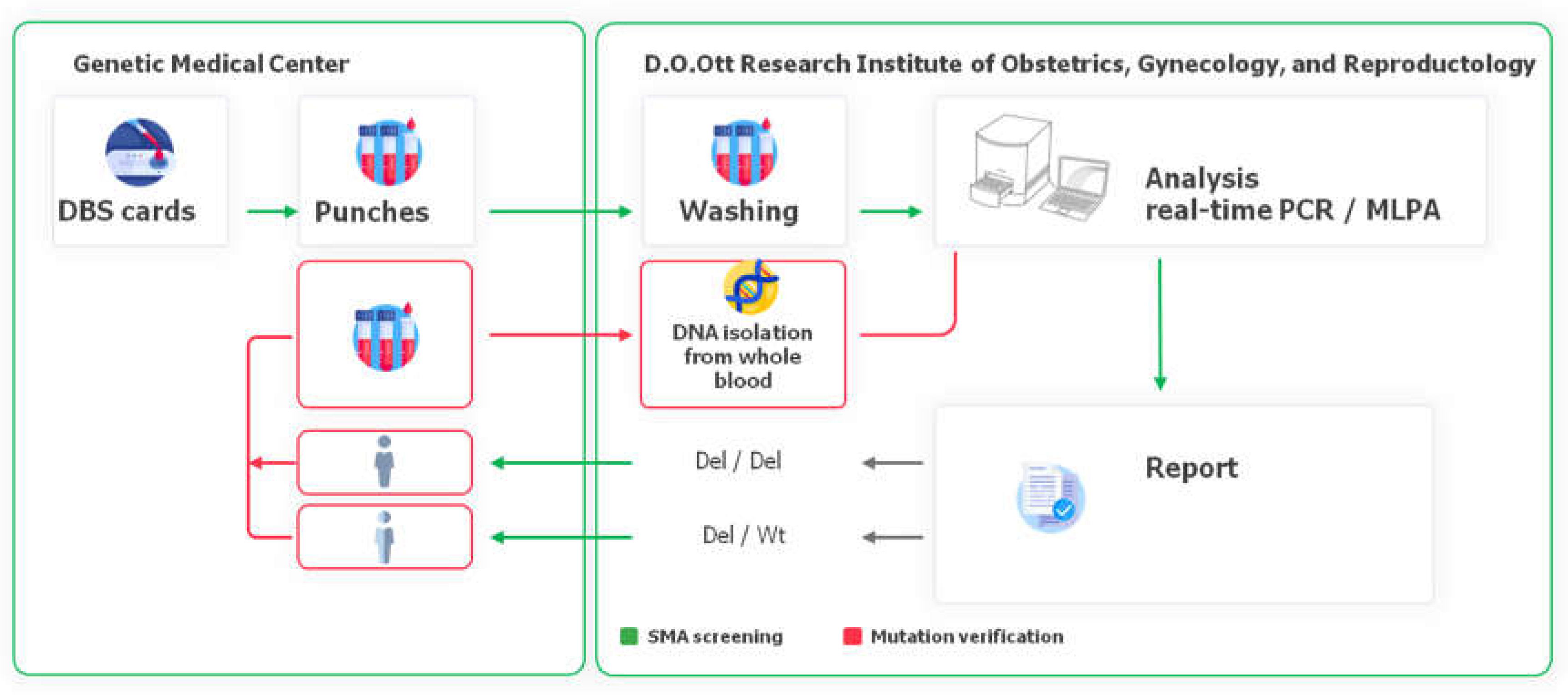

Table 1.
Results of validation study of SMA detection method.
| Genotype | Wt / Wt | Del 7 / Wt | Del 7 / Del 7 | Sensitivity* / Specificity |
|---|---|---|---|---|
| Del 7 / Del 7 | 0 | 0 | 50 | 100 % / 100% |
| Del 7 / Wt | 0 | 58 | 0 | 100 % / 100% |
| Wt / Wt | 142 | 0 | 0 | 100 % / 100% |
* Possible point mutations in SMN1 gene are not taken into account.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



