
Submitted:
01 March 2023
Posted:
01 March 2023
You are already at the latest version
Abstract
Hsp70.1 has dual functions as chaperone protein and lysosomal stabilizer. Previously, we reported that calpain-mediated cleavage of carbonylated Hsp70.1 causes ischemic neuronal death by inducing lysosomal rupture. Recently, we found that the consecutive injections of vegetable oil-peroxidation product ‘hydroxynonenal’ induces hepatocyte death via the similar cascade. As Hsp70.1 is related also to fatty acid β-oxidation in the liver, its deficiency is known to cause accumulation of fat. Genetic deletion of betaine-homocysteine S-methyltransferase (BHMT) was reported to perturb choline metabolism, inducing decrease of phosphatidylcholine with the resultant hepatic steatosis. Here, focusing on disorders of Hsp70.1 and BHMT, we studied the mechanism of hepatocyte degeneration and steatosis, using monkeys after the consecutive injections of synthetic hydroxynonenal. As these monkeys showed a significant impairment of liver functions, the liver tissues without and with hydroxynonenal injections were compared by proteomics, immunoblotting, immunohisto-chemical and electron microscopic analyses. Western blotting showed upregulation of neither Hsp70.1 nor BHMT, but an increased cleavage of both proteins. Proteomics showed downregulation of Hsp70.1, while 2-fold increments of carbonylated BHMT. Hsp70.1 carbonylation was negligible, showing a marked contrast to ischemic neurons which were associated with ~10-fold increments. The control liver histologically showed lipid droplets in Ito cells, but lipid depositions within hepatocytes were very little. In contrast, after hydroxynonenal injections, widespread fatty degeneration and focal coagulation necrosis were observed with accumulation of numerous tiny lipid droplets within and around the degenerating/dying hepatocytes. Electron microscopy showed lysosomal membrane permeabilization/rupture, remarkable dissolution of mitochondria and rough ER membrane, and proliferation of abnormal peroxisomes. It is probable that disruptions of rough ER caused impaired synthesis of Hsp70.1 and BHMT proteins, while impairments of mitochondria and peroxisomes presumably contributed to the sustained generation of reactive oxygen species. Since both Hsp70.1 and BHMT are vulnerable to the long-standing oxidative stressor, hydroxynonenal-induced disorders facilitated degeneration and steatosis of hepatocytes, respectively.
Keywords:
betaine-homocysteine S-methyltransferase
; calpain-cathepsin hypothesis
; carbonylation
; cell death
; lysosomal rupture
; phosphatidylcholine
Introduction
Currently, obesity and type 2 diabetes have reached epidemic proportions in the Western countries [1]. Both conditions are closely associated with nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) [2,3,4,5]. The clinicopathological picture of both diseases resembles that of alcohol-induced liver injury, however, they occur in persons who consume little or no alcohol [6]. NAFLD is defined by the excess fat accumulation, and is classified into simple steatosis and NASH. NASH is a progressive subtype of NAFLD, being first defined by analogy to alcoholic hepatitis. NASH is characterized by the accumulation of fat in the liver (steatosis) along with inflammation and different degrees of scarring or fibrosis [7,8]. It is a potentially serious condition, because about 10-25% of patients with NASH may progress to liver cirrhosis, and eventually to hepatocellular carcinoma [9,10]. The mechanism of progression from NAFLD to NASH is still not completely elucidated. Lipotoxicity is the harmful effect of lipid accumulation in the non-adipose tissue, and nowadays the occurrence of NASH is mainly viewed as a consequence of liver lipotoxicity. Using diverse experimental paradigms of cultured cells, mice, monkeys, and humans, Seike et al. recently reported that hydroxynonenal can cause hepatocyte injury by inducing lysosomal membrane permeabilization/rupture [11].
Until now, it was shown in the brain and pancreas that chronic exposure to hydroxynonenal induces degeneration and death of brain neurons and Langerhans β cells by lysosomal membrane permeabilization/rupture [12,13,14]. The most important and largest problem to be addressed here is how hydroxynonenal can induce lysosome-mediated cell degeneration and death. Lysosomal cell death has been widely accepted to occur in vivo not only under pathological circumstances but also under physiological conditions. An exquisite physiological example of lysosome-mediated programmed cell death is the post-lactational regression (involution) of the mammary gland to remove alveolar mammary epithelium and return the gland to its pre-pregnant state, which is one of the complex and highly-regulated cell death programmes occurring in the adult mammalian organism [15,16,17]. Milk fat globules during mammary gland involution upon cessation of lactation are known to be toxic to epithelial cells. Perturbation of lysosomal limiting membranes by high concentrations of free fatty acids being derived from milk triglycerides, results in the controlled, extra-lysosomal leakage of cathepsins culminating in physiological cell death [17]. It is likely that certain lysosomal membrane proteins are vulnerable to excessive fatty acids. The same cascade may occur also in pathological conditions. About the mechanism of ischemic neuronal death, Yamashima et al. have formulated the ‘calpain-cathepsin hypothesis’ in 1998 [18], and in 2009 modified it by adding the concept of ‘calpain-mediated cleavage of carbonylated heat-shock protein 70.1 (Hsp70.1)’ [19,20]. They first reported such a concept that Hsp70.1 (also called Hso70 or Hap72) with dual functions of molecular chaperone and lysosomal stabilizer, is vulnerable to the cleavage by activated μ-calpain, especially after carbonylation by hydroxynonenal. After the specific oxidative modification followed by μ-calpain-mediated cleavage, functional Hsp70.1 decreases, and this results in accumulation of garbage proteins and/or damaged organella, as well as permeabilization/rupture of the lysosomal membrane. Although best known for their chaperone functions, Hsp70.1 has such widely-accepted role as the regulation of metabolism. However, very little is known about the role of Hsp70.1 in the liver metabolism [21,22].
Liver is the major site of choline metabolism, where it is found primarily as phosphatidylcholine [23]. In the normal hepatocytes, a critical purpose of lipid droplet formation is to regulate the intracellular concentrations of unesterified fatty acids and cholesterol, because they are cytotoxic at the increased concentrations [24]. Phosphatidylcholine in the lipid droplet monolayer acts as a surfactant to prevent the coalescing of each lipid droplet that would yield larger lipid droplets which are less likely to undergo lipolysis [25]. Hepatic steatosis develops when fatty acid influx, de novo hepatic lipogenesis, or triglyceride (triacylglycerol) synthesis exceeds lipid efflux or oxidation. Alterations in choline and phosphatidylcholine metabolism have an impact on hepatic steatosis [26]. In the nonfatty liver, there is a homeostatic balance between triglyceride synthesis and efflux. Given that phosphatidylcholine is required for the efflux of very low-density lipoproteins (VLDL), reduction of phosphatidylcholine would cause the triglyceride accumulation in the liver of NAFLD [27].
Until now, the mechanisms that alter susceptibility for hepatic steatosis are poorly understood in both NAFLD and NASH. Betaine-homocysteine S-methyltransferase (BHMT) is an enzyme predominantly found in the liver as a major regulator of choline metabolism [28,29]. However, except for its roles in the ‘one carbon metabolism’, mechanisms of BHMT function and disorder are incompletely elucidated. It catalyzes formation of methionine from homocysteine, using the choline metabolite betaine as the methyl donor. So, BHMT deficiency leads to elevated betaine and homocysteine concentrations as well as reduced choline concentration [30]. Consequently, the choline deficiency influences hepatic lipid accumulation by reducing phosphatidylcholine concentration. BHMT overexpression increases phosphatidylcholine synthesis, leading to the reduced hepatic lipid accumulation [31], whereas BHMT deficiency leads to fatty liver [30]. Protein oxidation is currently thought to be an important player in various pathological conditions such as Alzheimer’s and Parkinson’s diseases, cardiovascular disease, type 2 diabetes, and stroke (ischemia/reperfusion injury)[32]. The most widely studied product of protein oxidation is the formation of carbonyl derivatives to side chains of certain amino acids, e.g., arginine, lysine, and proline [33,34,35]. For example, by the proteomics analysis of ischemic neuronal death, Oikawa et al. found more than 10-fold increase of carbonylated Hsp70.1 which was followed by calpain-mediated cleavage [35]. Accordingly, in this study we specifically focused on protein carbonylation of not only Hsp70.1 but also BHMT by the proteomics (2D-Oxyblot) analysis, because carbonylated BHMT was previously found in the rodent model of alcoholic steatosis and aging liver [32,36].
Here, we present evidence that normal chow-fed, young monkeys after the consecutive injections of the synthetic hydroxynonenal, showed both deficiency of the Hsp70.1 protein and increase of the carbonylated BHMT. By focusing disorders of both Hsp70.1 and BHMT, the mechanism of hepatocyte degeneration/death and steatosis was discussed.
Materials and Methods
Animal Experiments
After the referee of animal experimentation about the ethical or animal welfare, seven young (4-5 year-old; expected life span is 25-35 years) Japanese macaque monkeys (Macaca fuscata) were supplied by the National Bio-Resource Project (NBRP) for this study. These monkeys were reared in a wide cage with autofeeding and autocleaning machines as well as appropriate toys to play with for 1 year to facilitate acclimation. Room temperature was maintained at 22-24°C with a humidity of 40-50%. They were fed approximately 100 g x 2/day of a non-purified diet (CLEA Old World Monkey Diet CMK-2 containing 344.7 kcal/100 g, but only 4.05% crude fat, CLEA Japan, Inc., Tokyo). Apples, bananas, or sweet potatoes were given twice every week. Animal care staff monitored health and well-being of animals by checking the consumption of foods, the pupilar reflex to light, and the conditions of standing and jumping.
When 5 mg of hydoxynonenal (Cayman Chemical, Michigan, USA) was administered to monkeys, the serum hydoxynonenal concentration immediately after the intravenous injection was estimated to be 60 μM/L, as calculated from the blood volume converted from the monkey’s body weight of 6~7 Kg. On the basis of a report that the serum concentration of hydoxynonenal in the patients with Alzheimer’s disease is 20.65 μM/L at the median level (range: 6.02~25.20)[37] and considering the fact that hydoxynonenal is rapidly metabolized in the liver, we can assume that the concentration of hydoxynonenal in the monkey serum after 5mg/week injection is not too far from the pathological state in humans. So, seven monkeys were randomly divided into sham-operated controls (n=3) and those undergoing hydoxynonenal injections (n=4). In the latter monkeys, 5 mg of hydroxynonenal was intravenously injected every week for 24 weeks (total amount 120 mg for each monkey).
All experimental procedures were in accordance with the guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health, which met the ‘International Guiding Principles for Biomedical Research Involving Animals’, as issued by the council for the International Organizations of Medical Sciences. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Kanazawa University Graduate School of Medical Sciences (Protocol Number: AP-153613, 194062).
Blood Sampling
In the 4 monkeys with hydroxynonenal injections, venous blood was sampled once every month for 5 months prior to the hydroxynonenal injections (total sample number=20) and for 6 months during injections (total sample number=24), so the blood data were traced for 11 months in all 4 animals. The blood sampling was done also in the cotrol 3 monkeys. To speculate when liver injuries had occurred in the hydroxynonenal-treated monkeys, the average level at each time points were shown in time series of 11 months. The blood sampling was done at the time of each hydroxynonenal injection, but only the first blood sampling was done at the next week after the initial injection. The two–way ANOVA with Bonferroni’s post-test was used to compare the data before and after hydroxynonenal injections, and p < 0.05 was considered significant.
Tissue Collection
Both monkeys after hydroxynonenal injections (n=4) and sham-operated controls (n=3) were immobilized by the intramuscular injection of 10 mg/Kg body weight ketamine hydrochloride. In addition, to ameliorate animal suffering, the monkey was deeply anesthetized with 1~1.5% halothane plus 60% nitrous oxide. After the perfusion of 500 mL saline through the left ventricle, the liver and pancreas were removed. Half of the tissue was fixed in 4% paraformaldehyde for light microscopy, and 2.5% glutaraldehyde for electron microscopy. The remaining half was stocked in -80˚C deep freezer for the measurement of phosphatidylcholine concentration, as well as for Western blotting and proteomics analyses.
Analysis of Phosphatidylcholine Concentration in the Liver Tissues
The liver tissues without (n=3) and with (n=3) hydroxynonenal injections served for the measurement of phosphatidylcholine concentration. Phosphatidylcholine analysis was done according to Lipidome lab Phospholipid Enzymatic Fluorometric Assays package (Lipidome lab, Akita, Japan). First, total lipids were extracted from 100 mg of monkey liver samples by the Bligh-Dyer method [38]. An aliquot of the lower/organic phase was evaporated to dryness under N2 gas, and the residue was dissolved in methanol for phosphatidylcholine measurement. Details of the enzymatic fluorometric assays to quantify phospholipid classes were described previously [39]. Briefly, each sample (2.5 mg tissue/mL) was added to Reagent C1 (100 U/mL GPL-PLD, 1.5 mM CaCl2, 50 mM NaCl, and 50 mM Tris-HCl (pH 7.4)) and was incubated at 37°C for 30 min. After the incubation, Reagent C2 (4 U/mL choline oxidase, 5 U/mL peroxidase, 300 μM Amplex Red, 0.2% Triton X-100, 50 mM NaCl, and 50 mM Tris-HCl (pH 7.4)) was added to each well, and was incubated at room temperature for 30 min. Then, Amplex Red Stop Reagent was added. The fluorescence intensity was measured at excitation at 544 nm and emission at 590 nm using a microplate reader (SpectraMax iD3, Molecular device).
Western Blotting
For Western blotting of the liver tissues after hydroxynonenal injections, the pancreas tissue was utilized as a positive control of stress-induced upregulation of Hsp70.1 [14]. Total protein extraction was done for each samples, using protease inhibitor cocktail (Sigma-Aldrich, USA) and PhosSTOP phosphatase inhibitor cocktails tablets (Roche, Germany). After centrifugation at 12,000 rpm for 10 min, the supernatant proteins were determined by Bradford Assay (Thermo Fisher, USA). Twenty μg proteins were separated by SDS-PAGE in SuperSep (TM) Ace 5–20% gel (Wako, Japan) at 40 mA for 1h. The total proteins were transferred to PVDF membrane (Millipore, USA). Transferred protein quantities were detected with Ponseau S solution (Sigma-Aldrich, USA). Transferred proteins were blocked with 1% bovine serum albumin (KPL DetectorTM Block, USA) for 1h.
The blots were incubated with mouse monoclonal anti-human Hsp70 antibody (BD Bioscience, USA) at the dilution of 1:4,000, rabbit anti-human BHTM antibody (abcam, USA) at 1:1000, or mouse anti-human hydroxynonenal antibody (JaICA, Japan) at 1:500 overnight. GAPDH or β-actin were utilized as an internal control (Sigma-Aldrich, USA) at a dilution of 10,000. The immunoblots were subsequently incubated for 1h with secondary antibodies at 1:10,000 dilution of anti-mouse IgG (Santa Cruz, USA) or at 1:10,000 dilution of anti-rabbit IgG (Sigma, USA). An enhanced chemiluminescence (ECL) HRP substrate detection kit (Millipore, USA) was used to visualize the reactive protein bands with ImageQuant LAS 4000 mini (GE Life Science, USA).
Detection of Carbonyl Modified Proteins (2D-Oxyblot Analysis)
The liver tissues were directly transferred into a reaction tube containing 100 µl of lysis buffer (30 mM Tris-HCl, 7 M urea, 2M thiourea, 4 % w/v 3-[(3-cholamidopropyl) dimethylammonio] propanesulfonate, a protease inhibitor cocktail, pH 8.5). The hippocampus after transient ischemia was utilized as a positive control of Hsp70.1 carbonylation [35]. The tissue samples were homogenized using the Sample Grinding Kit (GE Healthcare UK Ltd., England) and incubated for 60 min on ice. All samples were centrifuged at 30,000 g for 30 min at 4 ˚C. The supernatant was collected and stored at -80 ˚C. Total protein of sample was quantified by the Bradford assay, each sample was prepared to 100 µg of protein. Derivatization with 2,4-dinitrophenylhydrazine (DNPH) was done according to the procedure of Nakamura and Goto [40]. Carbonylated proteins in each protein samples (100 µg protein) were labeled by derivatization of carbonyl group with 2,4-dinitrophenylhydrazone (DNP) by reaction with DNPH and separated by two-dimensional electrophoresis. Two-dimensional electrophoresis and subsequent immunoblotting for protein carbonyls, were done as described previously [41]. First dimension, isoelectric focusing, was performed on IPG strips (IPG, pH 3-10 NL strips, 24 cm, GE Healthcare) and Ettan IPGphor isoelectric focusing system (GE Healthcare). Second dimension, 12.5% SDS–polyacrylamide gel electrophoresis, was run on an Ettan DALT Six large electrophoresis system (GE Healthcare).
After the second-dimension, the proteins from the gels were transferred to polyvinylidene fluoride membrane (Immobilon-P transfer membrane; Millipore), using a TE77 semidry transfer unit (GE Healthcare) at 50 V for 30 min. The DNP adduct of the carbonyls of the proteins was detected on the PVDF membrane, using an immunoblot Kit (OxySelectTM Protein Carbonyl Immunoblot Kit, CELL BIOLABS, INC.). The chemiluminescence signal was detected on X-ray films. The spot intensities of carbonylated proteins were quantified using PDQuest ver. 8.0 (Bio-Rad, California, CA). We repeatedly confirmed reproducibility of 2D-Oxyblot analysis. The specific oxidation was estimated as relative carbonyl level (obtained from 2D-Oxyblot) per relative protein expression (obtained from 2D-DIGE). For the protein identification, spots were excised from 2D gels obtained with non-DNPH-treated samples and analyzed by the mass spectrometry. Mass analysis was performed with a matrix-assisted laser desorption ionization time-of-flight tandem mass spectrometry (MALDI-TOF/TOF MS; 4800 Plus MALDI-TOF/TOFTM Analyzer, AB SCIEX, Framingham, MA). Protein identification was performed with the MS/MS ion search tool in ProteinPilot software (AB SCIEX).
Two-Dimensional Differential in-Gel Electrophoretic (2D-DIGE) Analysis
Each soluble protein sample (25 µg protein) was minimally labeled with CyDye DIGE fluors according to the manufacturer's protocol (GE Healthcare). Each Cy3 labeled samples were combined with an equal amount of Cy5 labeled samples, and the pooled Cy2 labeled sample was added as an internal standard. After incubation on ice for 30 min in the dark, the samples were separated by 2D-electrophoresis [42]. The 2D-DIGE gels were scanned in a Typhoon FLA 9500 (GE Healthcare). Spot detection, gel matching, and statistical analysis were performed with DeCyder 2D software version 7.2 (GE Healthcare).
By considering 1) heterogeneity of hydroxynonenal-induced hepatic lesions in the given monkey, and 2) the small number of experimental animals because of high costs of both Japan macaque monkeys and the synthetic hydroxynonenal, the statistical analyses of phosphatidylcholine measurement, Western blotting, and proteomics data were not done.
Histological and Immunofluorescence Histochemical Analyses
The liver tissues after fixation with 4% paraformaldehyde for 2 weeks were embedded in paraffin, and 5μm sections were stained by hematoxylin-eosin (H-E). For the immunofluorescence histochemistry, the cryoprotected liver tissues embedded in the OCT medium (Sakura Finetek, Japan) were cut by cryotome (Tissue-Tek1Polar1, Sakura, Japan), and 5μm sections were immersed with heated 0.01% Citrate retrieval buffer to induce epitope retrieval. Non-specific staining was blocked with 1% bovine serum albumin (Nacalai tesque, Japan), and were incubated overnight at 4˚C with the primary antibodies at the dilution of 1:100. We used mouse monoclonal anti-human Hsp70 (BD Bioscience, USA), rabbit anti-human activated μ-calpain (recognizing only activated form of 76 kDa, order made by Peptide Institute, Japan), and rabbit anti-human cathepsin B (Cell Signaling, USA) antibodies. After washings, the sections were incubated for 30 min with secondary antibodies; Alexa FluorTM 594 goat anti-mouse IgG [H+L] (Invitrogen, USA), or Alexa FluorTM 488 goat anti-rabbit IgG (Invitrogen, USA) at the dilution of 1:500. To block autofluorescent staining, Autofluorescence Quenching Kit (Vector Laboratories, USA) was utilized. The immunoreactivity was observed with the laser confocal microscope (LSM5 PASCAL, Software ZEN 2009, Carls Zeiss, Germany).
Ultrastructural Analyses
For the electron microscopic analysis, small specimens of the liver tissue were fixed with 2.5% glutaraldehyde for 2h and 1% OsO4 for 1h. Subsequently, they were dehydrated with graded acetone, and embedded in resin (Quetol 812, Nisshin EM Co. Tokyo), then thin sections were made to stain with 0.5% toluidine blue (T-B). After trimming resin-embedded tissues, the ultrathin (70 nm) sections of selected area were stained with uranyl acetate (15 min) and lead citrate (3min), and were observed by the electron microscope (JEM-1400 Plus, JEOL Ltd., Tokyo).
Results
The comparison of blood data before (n=20 being collected for 5 months, Figure 1A, open circles) and after (n=24 for 6 months, Figure 1A, closed circles) the start (Figure 1A, arrows) of hydroxynonenal injections, showed a significant (P<0.05) increase of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and γ-glutamyl transferase (γ-GTP). This was observed as long as 24 weeks in all of the 4 monkeys which underwent 5 mg/week (total dose, 120mg for each monkey) of hydroxynonenal injections. The difference was almost the same, when comparing with the blood data of the control 3 monkeys without hydroxynonenal injections. Since increments of AST, ALT, and γ-GPT became prominent at the second month after the 5mg/week x 4 injections in 4 monkeys, the hepatic injury was thought to occur within a couple of weeks after the initial injection (Figure 1A, arrows). At autopsy 6 months after, the surface of the liver of non-injected, control monkeys looked reddish-brown (Figure 1B, Cont), whereas the surface of hydroxynonenal-treated liver showed a heterogenous, whitish-yellow discoloring (Figure 1B, HNE, arrows) being intermingled with the dark-brown area. The whitish-yellow portion histologically showed a severe degeneration of hepatocytes with accumulation of lipid droplets (Figures 4B and 5B). In contrast, the surrounding dark-brown area showed a mild degeneration.
During the intravenous injections of 5mg/week of hydroxynonenal (HNE) injections (total dose, 120 mg), the blood analysis showed increased levels of AST, ALT and γ-GTP (1A, closed circles), compared to the controls prior to hydroxynonenal injections (1A, white circles). The surface of the control liver looked diffusely reddish-brown at autopsy (1B, Cont), while at autopsy six months after the initial hydroxynonenal injections, the liver surface of hydroxynonenal-treated monkeys showed heterogenous, whitish-yellow discoloring (1B, HNE, black and green arrows) being intermingled with the dark-brown area. These whitish-yellow areas histologically showed a severe degeneration, while the dark-brown area showed a mild degeneration. Western blotting using anti-BHMT antibody showed not only decrease of the BHMT naïve proteins (1C, BHMT) but also approximately 1.5-fold increment of BHMT cleaved bands on both short and long exposures (1C, Cleaved 1 & 2), compared to the controls (1C).
The concentration of phosphatidylcholine within the liver tissues of 3 control monkeys was 19.39, 18.17 and 19.74 (average: 19.10) μg/mg tissue, while that of hydroxynonenal-treated 3 monkeys was 22.26, 17.46 and 14.85 (average: 18.19) μg/mg tissue. After hydroxynonenal injections, one monkey showed a higher level, while two monkeys showed a lower phosphatidylcholine level, compared to the controls. By considering the heterogeneity of hepatic legions in the given monkey (Figure 1B), we speculated that phosphatidylcholine concentration also showed a regional heterogeneity. Interestingly, however, the monkey with the lowest concentration (14.85 μg/mg tissue) showed the strongest hepatic injury among 3 monkeys studied.
Western blotting using anti-activated μ-calpain antibody showed a significant increase of activated μ-calpain in all of the 4 liver tissues after hydroxynonenal injections as reported previously [11] (data not shown here). As already demonstrated in the brain tissue [20], calpain-mediated cleavage of Hsp70.1 was shown to be increased also in the liver, compared to the controls (Figure 2A, Liver). However, surprisingly, upregulation of Hsp70.1, albeit the stress-inducible protein, was not observed in response to the chronic insult of hydroxynonenal injections. This was a marked contrast to the pancreas of the same monkey which showed increments of both Hsp70.1 main band and its cleavage at the simultaneous electrophoresis (Figure 2A, Pancreas). Using the whitish-yellow portion (Figure 1B, green arrow; Figure 2B, upper rectangle) of the liver associated with severe degeneration, SDS-PAGE followed by immunostaining using anti-DNP antibody, showed increments of a few adduct proteins which were modified by hydroxynonenal. However, the band indicating carbonylated Hsp70.1 was negligible around 70 kDa (Figure 2B, red arrow).
By the Western blotting after electrophoresis on the same gel, in both the liver and pancreas Hsp70.1 cleavage was increased after hydroxynonenal injections (2A, HNE), compared to the control (2A, Cont). Albeit a stress-inducible protein, Hsp70.1 showed no upregulation of naïve protein to the long-standing insult of consecutive hydroxynonenal injections in the liver (Figure 2A, Liver). This was a marked contrast to the pancreas which showed upregulation of Hsp70.1 naïve proteins after the same insult (Figure 2A, Pancreas). SDS-PAGE using whitish-yellow areas (1B HNE, green arrow, 2B, upper) and anti-DNP antibody showed negligible expression of carbonylated Hsp70.1 (2B, HNE). These were consistent with the data of 2D-Oxyblot analysis using anti-DNP antibody, which showed negligible expression of carbonylated Hsp70.1 (2C-b), although 2D-DIGE showed decreased expression of Hsp70.1 after hydroxynonenal injections (2C-a). Reduction of the carbonylated Hsp70.1 after the consecutive hydroxynonenal injections, showed a marked contrast to the significant increase of carbonylated Hsp70.1 in the hippocampal CA1 after transient ischemia (2C-c)[35].
2D-Oxyblot analysis using aniti-DNP antibody also showed negligible expression of carbonylated Hsp70.1 (Figure 2C-b), although 2D-DIGE analysis showed expression of the Hsp70.1 protein (Figure 2C-a). The 2D-DIGE showed down-regulation of Hsp70.1 protein on the corresponding spot. In our study using the same procedure of proteomics, we already reported that 2D-Oxyblot analysis of the hippocampus (CA1) of monkey brains showed a significant upregulation of carbonylated Hsp70.1 after transient ischemia, as shown in Figure 2C-c)[35]. In the previous study, the insult of brain was merely an acute insult of transient ischemia, and analysis was done as early as days 3-7 after the insult. In contrast, the insult of liver in the present experiment was chronic exposure to hydroxynonenal lasting for 24 weeks, and analysis was done as late as 6 months after the first insult of hydroxynonenal injection. Therefore, it is likely that due to both 1) impairments of synthesis due to rough ER damages as demonstrated by electron microscopy (Figure 8), and 2) long-standing cleavage of carbonylated Hsp70.1 (Figure 2A), reduction of both naïve and carbonylated Hsp70.1 proteins (Figure 2C-a,b), progressed step by step during 24 weeks of hydroxynonenal injections.
As BHMT carbonylation was previously reported in the rodent model of alcoholic steatosis [36], here we focused on spots of the similar MW and pI. Then, the spot 2406 with pI of 6.58 and MW of 44,998 (red circles) was identified as BHMT by the mass spectrometry analysis and the database matching. Compared to the controls (3A,B, Cont), the degenerating liver after hydroxynonenal injections (3A,B, HNE) showed two-fold increments of specific oxidation of BHMT (3B). In contrast, spots of carbonylated Hsp70.1 were not detected on 2D-Oxyblots (Figure 2C-b), which showed a marked contrast with ischemic brain [35](Figure 2C-c). As gel of 2D-electrophoresis is much larger than that of SDS-PAGE, molecular marker shows merely approximate positions.
By Western blotting using anti-BHMT antibody, the liver tissues after hydroxynonenal injections showed not only decrease of the BHMT naïve protein but also 1.5 fold-increase of BHMT cleaved bands on both short and long exposures, compared to the controls (Figure 1C). Carbonylation of BHMT was previously reported in the rodent model of alcoholic steatosis as a target protein of fatty liver development [36]. Accordingly, by the 2D-Oxyblot analysis, we focused on spots of similar MW and pI of BHMT. The spot 2406 with pI of 6.58 and MW of 44,998 was identified as BHMT by the mass spectrometry analysis and the database matching (Figure 3). The spots 2406 (Figure 3A, red circles) in both liver tissues without (Cont) and with (HNE) hydroxynonenal injections were enlarged in Figure 3B. Approximately 2-fold increase of the carbonylated BHMT (Figure 3B) was observed.
Since lysosomal cell death was thought to occur in the liver as shown in the ischemic brain [19,20], the following light microscopic, immunofluorescence histochemical, and electron microscopic analyses were focused on the lysosomal membrane integrity of the affected hepatocytes. By the microscopic observation, the whitish-yellow area showed widespread foamy degeneration which comprised of hepatocytes with advanced cytoplasmic degeneration around the portal triad (Figure 4B). The translucent cytoplasm indicated almost complete dissolution of lipid components during ethanol dehydration for the microscopic preparation. Many tiny granules were seen within the cytoplasm (Figure 4B, circle), which were thought to be degenerating mitochondria and peroxisomes as confirmed by the electron microscopy (Figures 8A,B, 9B,C). Focal necrotic area was detected (Figure 4B, star; Figure 5B, star), but apoptotic bodies were not observed. The necrotic area showed an infiltration of inflammatory cells. The degenerating hepatocytes (Figure 4B) did not show ballooning, being as large as the normal hepatocytes (Figure 4A). The cell boundary was distinct, which was compatible with thickened basement membrane as shown by the ultrastructural observation (Figure 8A, BM). In the degenerating hepatocytes, the nuclei often showed a dense chromatin, compared to the controls. These histological changes were never observed in the control liver, the cytoplasm of which was filled with eosinophilic substance (Figure 4A). The degenerating hepatocytes showed a marked contrast with the control hepatocytes.
By light microscopy, the whitish-yellow portion (Figures 1B, 2B) of the liver after the consecutive injections of hydroxynonenal, showed widespread fatty degeneration of hepatocytes and focal necrosis (4B, star). The cytoplasm of affected hepatocytes around the portal triad (4B, circle) was translucent, presumably due to the dissolution of lipid components during ethanol dehydration. This showed a marked contrast with the normal hepatocyte (4A). Star: focal necrotic area, H-E staining.
Procedures for the electron microscopic observation could preserve lipid components involved within tissues by double fixations using glutaraldehyde and OsO4. Accordingly, T-B staining of the semi-thin sections of resin-embedded tissues, revealed slightly-yellowish lipid droplets in both the control (Figure 5A) and the hydroxynonenal-treated, degenerating (Figure 5B) hepatocytes. In the control liver, lipid droplets were mainly seen within Ito cells which were localized at the subendothelial space of sinusoids (Figure 5A, black asterisks). The hepatocytes showed tiny lipid droplets, if present. In contrast, in the liver after hydroxynonenal injections, other than Ito cells (Figure 5B, black asterisks), many small lipid droplets were seen within and in the vicinity of degenerating/dying hepatocytes (Figure 5B, white asterisks, arrows). Lipid droplets measuring 2~15μm were formed within the degenerating and dying hepatocytes, the cytoplasm of which were translucent by the light microscopic observation (Figures 4B circle, 5B circle) or showed decreased electron-density by the electron microscopic observation (Figure 10A, circle). Abundant debris of cell organelle was seen as coarse granules within and around the dead cells. The hepatocytes without and with hydroxynonenal injections showed a remarkable contrast on both H-E and T-B stainings (Figure 4 and Figure 5).
In the control liver (5A), lipid droplets were seen mainly within Ito cells beside the sinusoid lumen (5A, black asteriskas). In contrast, after hydroxynonenal injections, many lipid droplets were seen within and outside the degenerating/necrotic hepatocytes (5B; white asterisks, arrows). The double fixation for the electron microscopic preparation could preserve lipid components against acetone dehydration. The translucent cytoplasm of degenerated hepatocytes (5B, circle) shows dissolution of cytoplasmic organelles, as confirmed by electron microscopy (Figure 10A, circle). Star: focal necrotic area, T-B staining.
Immunofluorescence histochemical staining showed that activated μ-calpain immunoreactivity was seen as tiny granules at the hepatocytes of control monkeys without hydroxynonenal injections (Figure 6A), whereas it was increased remarkably within hepatocytes after hydroxynonenal injections (Figure 6B). In the control liver, immunofluorescence histochemical co-staining of Hsp70.1 with activated μ-calpain was observed only in the small round cells in the vicinity of the sinusoidal endothels, but most of the hepatocytes showed negligible immunoreactivity (Figure 7B). In contrast, after hydroxynonenal injections, the hepatocytes showed a remarkable increase of merged immunoreactivity of Hsp70.1 and activated μ-calpain (Figure 7C, yellow). Interestingly, Western blotting using the same pair of monkey samples showed an increase of the Hsp70.1 cleaved band (Figure 7A). These results suggested interaction of activated μ-calpain upon Hsp70.1 in the hepatocytes after hydroxynonenal injections, which conceivably contributed to the calpain-mediated cleavage of carbonylated Hsp70.1 [20], although hardly possible to demonstrate in vivo. Presence of the Hsp70.1 protein on 2D-DIGE (Figure 2C-a) but absence of the carbonylated Hsp70.1 on 2D-Oxyblot (Figure 2C-b), were consistent with these data. Further, immunofluorescence histochemical staining of cathepsin B showed small immunoreactivity in the control hepatocytes (Figure 6C). In contrast, the cytoplasm of hepatocytes after hydroxynonenal injections showed a coarse granular staining with the diffuse cytoplasmic immunoreactivity (Figure 6D). This suggested that leakage of the lysosomal contents into the cytoplasm had occurred by the lysosomal membrane permeabilization/rupture presumably due to calpain-mediated cleavage of the carbonylated Hsp70.1 as previously demonstrated in neurons after ischemia [19,20] and β-cells after hydroxynonenal injections [14].
Electron microscopic analysis of the normal hepatocytes showed membrane-bound, electron-dense lysosomes in the hepatocytes of control monkeys (Figure 8C). In contrast, most of the hepatocytes after hydroxynonenal injections showed a remarkable decrease of membrane-bound, normal lysosomes (Figures 9B,C, 10), which was characteristic to the degenerating hepatocytes. Furthermore, the remaining lysosomes showed disintegrity of the limiting membrane with an irregular configuration (Figure 8A, arrows; 8D), which was associated with leakage of the lysosomal contents. In the hepatocytes of control monkeys without hydroxynonenal injections, numerous electron-dense glycogen granules, and rough ER filled the cytoplasm. Especially, the cytoplasm contained numerous mitochondria with crista (Figure 9A). In contrast, after hydroxynonenal injections, both mitochondria and rough ER showed a marked decrease or showed intense vacuolar degeneration with loss of cristae and/or accumulation of lipid debris (Figure 9B,C, m). Since mitochondrial function is critical to liver function such as degradation of fatty acids by β-oxidation, mitochondrial degeneration and reduction were thought to be closely related with increased lipid accumulation through membrane disruption (Figures 8B, 9C, arrows) [43,44]. Numerous fine-granular substance with fuzzy material was in continuity with disrupting membranes of mitochondria (Figure 9C, circles). Because of the impaired synthesis of ATP due to the damages of mitochondria and glycogen granules, Hsp70.1 was thought to be dysfunctional for achieving autophagic removal by autophagolysosome formation, because ATP is indispensable for its function. In addition, disruption of rough ER contributed to the reduction of Hsp70.1 synthesis. Electron-dense peroxisomes devoid of distinct limiting membrane and with irregular configuration (Figure 9B, circles) were increased (Figure 9B, arrows) around the degenerating mitochondria (Figure 9B, m). Electron-dense glycogen granules as seen in the normal hepatocytes (Figure 9A) disappeared, instead, the cytoplasm was filled with fine-granular substance (Figure 9B,C). Both the translucent cytoplasm as observed by the light microscopic observation (Figure 4B) and the cytoplasmic fine-granular material as observed by the electron microscopic observation (Figure 9B,C, Figure 10A, circle), indicated that the degenerating cytoplasm contained abundant fatty components which were mostly dissolved by ethanol during fixation for the microscopic preparation.
Compared to the membrane-bound, vivid lysosomes (8C) in the control hepatocytes, lysosomes of the degenerating hepatocytes after hydroxynonenal injections showed disintegrity of the limiting membrane (8A, arrows & D). In the liver after hyfroxynonenal injections, lsosomes often showed leakage of the content or formation of autophagolysosomes, and normal lysosomes like Figure 8C were extremely rare. Tiny lipid droplets (8A,B, asterisks) and membrane-derived lipid debris (8B, arrows) were observed in the vicinity of degenerating mitochondria and rough ER. The basement membrane (8A, BM) of degenerating hepatocytes was thickened, which is compatible with the distinct cell boundaries as seen by light microscopy (Figure 4B).
The cytoplasm of the control hepatocytes is filled with normal mitochondria and glycogen granules (9A, Cont). In contrast, most of the mitochondria (9B,C, m) in the degenerating hepatocytes showed a remarkable dissolution of cristae with the formation of membrane debris (9C, HNE, arrows). The outer membrane of degenerating mitochondria is in direct continuity with electron-dense particles (9C, HNE, circles). Compared to the control, abnormal peroxisomes show a marked increase after hydroxynonenal injections (9B, HNE, arrows). They are characterized by electron-dense deposits, irregular configuration, and absence of the limiting membrane. (9B, HNE, circles).
Low-magnification, electron microscopic observation of the hepatocytes after hydroxynonenal injections, showed formation of an electron-dense, small lipid droplet among the degenerating hepatocytes (Figure 10A,B, asterisks). This was consistent with the finding of T-B staining (Figure 5B, arrows). At one glance, it was ultrastructurally similar to red blood cell within the sinusoidal lumen, but the configuration and the localization were distinct. Red blood cell was bound by distinct membrane and was located within the sinusoidal lumen (Figure 10A, R, Sin), whereas lipid droplets showed irregular configuration and was localized among or within the degenerating hepatocytes (Figure 10A,B, asterisks). Lipid droplets were surrounded by degenerating mitochondria and peroxisomes which transferred to the cell periphery of degenerating hepatocytes (Figures 9B, 10B). High-magnification observation indicated generation of lipid-like material (Figure 10B, asterisk) in close vicinity of degenerating membranes of mitochondria (Figure 9C, arrows, circles) and rough ER (Figure 8B, arrows, asterisks). Such membrane debris was thought to be source of the lipid droplets which was often seen within and in the vicinity of degenerating hepatocytes (Figure 5B).
All hepatocytes showed diverse degree of degeneration after hydroxynonenal injections: the extent of degeneration is reversely parallel with electron-density of the cytoplasm. The electron-luscent hepatocyte (10A, circle) indicates a remarkable degeneration of the cytoplasm as seen by light microscopy (Figure 4B). A small lipid droplet as observed by T-B staining (Figure 5B, arrows) was formed among the degenerating hepatocytes (10A,B, white asterisks). The rectangle in Figure 10A is enlarged in Figure 10B. R: red blood cell, Sin: Sinusoidal lumen.
Discussion
Among the different oxidative modifications, protein carbonylation is the most common and irreversible, because of the permanent modification of target proteins [45]. The proteomics analyses of the present study showed paucity of carbonylated Hsp70.1 but an increase of carbonylated BHMT, which showed a marked contrast each other. Approximately 1.5-fold increase of carbonylated BHMT was observed (Figure 3), although both naïve Hsp70.1 and carbonylated Hsp70.1 were down-regulated (Figure 2C-a,b). This was thought to be reasonable, if considering 1) identification of carbonylated proteins in the normal liver tissue is not surprising, because basal level of oxidative stress exists in all cells due to the normal metabolism [45], and 2) the global levels of protein carbonyls in aging liver tissue of old mice were reported to be similar level; 2-fold of young mice [32]. On the contrary, induction of Hsp70 expression by stresses is known to decline significantly with age in a variety of tissues from rats to humans, presumably from a defect in the heat shock transcription factor. For example, using hepatocytes freshly isolated from young adult and old rats, Heydari et al. have shown that the induction of hsp70 expression by heat shock is reduced approximately 50% with age [46]. Accordingly, discrepancy of the dynamic changes between Hsp70.1 and BHMT in response to consecutive hydroxynonenal injections, is compatible with the previous reports. One possible explanation of such discrepancy is that the life-span (turn over) is different between the stress-inducible protein, Hsp70.1 and the house-keeping enzyme, BHMT. Anyway, disorders of Hsp70.1 and BHMT, although dynamic changes of each are distinct, must be critical for considering the mechanism of degeneration and steatosis of hepatocytes after hydroxynonenal injections. So, two issues were discussed separately, here.
Hydroxynonenal-Induced Hsp70.1 Deficiency Causes Lysosomal Membrane Disintegrity and Cell Degeneration
Lipotoxicity is harmful effects of lipid accumulation in the non-adipose tissue. Nowadays, the occurrence of NASH and its progression to cirrhosis or hepatocellular carcinoma, are thought to be closely related with lipotoxicity due to free fatty acids [47]. For example, Feldstein et al. previously reported that exposure of mouse and human hepatocytes to long-chain fatty acids induces lysosomal membrane permeabilization and release of cathepsin B into the cytosol [48,49]. Moreover, they demonstrated that leakage of cathepsin B into the cytosol occurs in human NAFLD, being correlated with disease severity, whereas inhibition of cathepsin B shows protection against diet-induced fatty liver disease in mice. This is quite similar to the monkey models of transient brain ischemia in which cathepsin release or inhibition revealed delayed neuronal death or neuroprotection, respectively [18,50]. Currently, the increment of dietary energy availability and sedentary lifestyle leads to obesity, and this causes not only an excessive accumulation of body fat but also an elevated level of free fatty acids in the blood. In both in vivo and in vitro, free fatty acids are known to undergo lipid peroxidation chain reactions, which, in turn, lead to the formation of highly-reactive electrophilic aldehydes. As linoleic acid is the most abundant ω-6 polyunsaturated fatty acid (PUFA) involved in Western diets, it’s end products are detectable in the human blood and diverse organs including liver. Mice with an oxidized linoleic acid-rich diet show increased levels of reactive aldehydes such as hydroxynonenal in the liver [51]. The same may occur in humans with daily intake of high-fat diets or deep-fried foods cooked by ω-6 PUFA-, especially linoleic acid- rich vegetable oils [13].
An initial event contributing to hepatocyte injury during NASH development is an enhanced oxidative stress which results in the short-term generation of reactive oxygen species. The latter in turn induce lipid peroxidation of linoleic acids at biomembranes and generate reactive aldehydes [52], which would be a long-lasting oxidative stressor. In human NASH as well as murine disease models, antibodies against reactive aldehydes show increased reactivity [53,54,55]. Although reactive aldehydes are recognized as a main cause of hepatocyte injury for the NASH development, the role of the most popular but toxic reactive aldehyde ‘hydroxynonenal’ as a cause of chronic liver injury, long remained unexplored. Until now, the cell toxicity of hydroxynonenal was studied mainly in cultured cells. Although studies of animal models were present, those focusing humans, especially lifestyle-related diseases were extremely rare. For example, Bruce-Keller et al. has demonstrated damage to neuronal perikarya after injection of hydroxynonenal directly into the basal forebrain of rats [56]. Vigh et al. demonstrated that repeated intrathecal injections of sublethal doses of hydroxynonenal caused motor neuron loss in the spinal cord of rats [57]. In addition, intraperitoneal injection of hydroxynonenal was found to exacerbate colonic inflammation through activation of Toll-like receptor 4 signaling in mice [58].
Interestingly, implication of lysosomal cell death in hydroxynonenal-induced hepatic injury was recently reported by Seike et al., using diverse experimental paradigms such as cultured cells, mice, monkeys, and human patients [11]. Although role of calpain and cathepsin was elucidated in detail, role of Hsp70.1 in hydroxynonenal-induced hepatocyte injury still remained unclear. Hsp70.1 becomes prone to calpain-mediated cleavage, especially after the specific oxidation, carbonylation by hydroxynonenal [20]. As demonstrated in hippocampal neurons after transient ischemia, calpain-mediated cleavage of the carbonylated Hsp70.1 was a main cause of lysosomal membrane permeabilization/rupture. This resulted in extra-lysosomal release of cathepsin enzymes, which is known as the ‘calpain-cathepsin hypothesis’ [18,19,20,59]. Hydroxynonenal can induce not only Hsp70.1 carbonylation but also calpain activation, and facilitates calpain-mediated cleavage of carbonylated Hsp70.1 [13]. Given the liver has high mitochondrial density and fast mitochondrial degradation or turnover [60], it is likely that Hsp70.1, as a molecular chaperone, plays an important role in maintaining mitochondrial homeostasis in the liver. By helping degradation of damaged mitochondria, Hsp70.1 must play a crucial role for maintaining function and health of the liver. However, due to both the down-regulation of Hsp70.1 in hydroxynonenal-induced liver injuries, and heterogeneity of affected lesions, immunohistochemical and immunoblot analyses could not precisely detect dynamic changes of Hsp70.1. So, the proteomics analyses by 2D-DIGE and 2D-Oxyblot were indispensable.
Although Hsp70.1 is a stress-inducible protein which is upregulated by the cell stress, decreased Hsp70.1 protein levels were found to be correlated with progression of NAFLD in the livers of patients with obesity [61]. Shearn et al. also found a decreased expression of Hsp70 in the NASH liver in comparison with the normal human liver by Western blotting, but an increased carbonylation of Hsp70 by LC-MS/MS analysis [52]. The present study showed something similar but something different results with these reports. Western blotting showed upregulation of Hsp70.1 protein in the pancreas (positive control of naïve Hsp70.1 protein) after hydroxynonenal injections, however, the liver of the same monkeys showed no up-regulation (Figure 2A). Instead, down-regulation of the Hsp70.1 protein was confirmed by 2D-DIGE analysis (Figure 2C-a). In addition, both SDS-PAGE (Figure 2B) and 2D-Oxyblot (Figure 2C-b) failed to detect carbonylated Hsp70.1. This showed a remarkable contrast to the hippocampus (positive control of carbonylated Hsp70.1 protein expression) after transient ischemia which showed more than 10-fold increase of carbonylated Hsp70.1 on postischemic day 5 (Figure 2C-c)[35], but was consistent with the results of NAFLD [61] and NASH [52]. However, immunohistochemical analysis showed an intense cross-immunoreactivity of activated μ-calpain with Hsp70.1 (Figure 7C), which indicated implication of activated μ-calpain for the cleavage of carbonylated Hsp70.1 (Figure 7A). This was very similar to the case of ischemic hippocampal neuronal death.
There should be certain difference in the response of Hsp70.1 between acute insult as transient brain ischemia and chronic insult as consecutive hydroxynonenal injections. We speculate that chronic exposure to hydroxynonenal (5 mg/week x 24 weeks) was such a long-standing, severe insult to the monkey liver that carbonylated Hsp70.1 might have been continuously and steadily cleaved by activated μ-calpain (Figure 7A) at each time of hydroxynonenal injections. Presumably, this lasted as long as 24 weeks until Hsp70.1 deficiency is completed at the time point of tissue resection. Since Hsp70.1 is a stress-inducible protein which is characterized by short life-span and fast turn-over, severe disruption of rough ER and mitochondria presumably caused both impairment of synthesis and dysfunction of Hsp70.1 in the hepatocytes after the repeated insults of hydroxynonenal injections. If considering elevation of AST, ALT, and γ-GTP indicating liver injury occurred as early as a few weeks after the initial hydroxynonenal injection, it is probable that both carbonylation and cleavage of Hsp70.1 had occurred at the early phase of hydroxynonenal injections and progressed as long as 24 weeks. It is likely that severe damages of rough ER and mitochondria as shown by electron microscopy (Figure 8, Figure 9 and Figure 10), has gradually facilitated deficiency of the Hsp70.1 protein (Figure 2C-a). Impairment of the protein synthesis, combined with the increased cleavage of Hsp70.1 (Figures 2A, 7A), conceivably contributed Hsp70.1 deficiency (Figure 2C-a).
Fatty acids are mainly oxidized in mitochondria through the subsequent reactions of β-oxidation. Acetyl-CoA is produced from fatty acids, and this enters the TCA cycle to generate energy by completely oxidizing it to CO2 [62]. Archer et al. recently found that Hsp70.1 is important in maintaining mitochondrial integrity and fatty acid oxidation in the liver, therefore being capable of both preserving hepatic homeostasis and preventing lipid storage [63]. Until the present study, there have been no studies concerning the direct effect of Hsp70.1 deficiency on hepatic lipid metabolism. As hydroxynonenal induced a remarkable damage such as loss of mitochondria and cleavage of Hsp70.1, both may have a crucial role not only for hepatocyte degeneration/death but also for lipid accumulation. In this study, along with mitochondrial degeneration, a remarkable proliferation of abnormal peroxisomes was observed in the degenerating hepatocytes. Peroxisomes are single membrane-bounded organelles that are well-known for their involvement in the lipid metabolism and the redox balance [64,65,66]. Peroxisomes contain several oxidases involved in the production of H2O2 as well as catalase involved in the decomposition of H2O2 to oxygen and water [67,68]. To absorb nutrients that the cell has acquired, peroxisomes digest long-chain fatty acids and break them down into smaller molecules by β-oxidation. Representative byproducts of β-oxidation are H2O2 and hydroxyradicals (OH•). The proliferating peroxisomes ultrastructurally showed no limiting membrane with an irregular configuration (Figure 9B), as shown in the pancreatic Langerhans β-cells in the same monkeys after hydroxynonenal injections [14]. Accordingly, we speculate that abnormal peroxisomes which were found in the degenerating hepatocytes are related to the sustained generation of reactive oxygen species and disorder of β-oxidation.
BHMT Carbonylation and Cleavage Cause Phosphatidylcholine Decrease and Hepatic Steatosis
There are three major dietary source of methyl groups in the ‘one carbon metabolism’; methionine, betaine, and choline. Methionine is an essential sulfur-containing gluconeogenic amino acid that is required for the normal development and cell growth. Using ATP as co-substrate, it is converted to S-adenosylmethionine by the enzyme methionine adenosyltransferase, Methyl group of S-adenosylmethionine is transferred to a large variety of substrates, such as DNA, RNA, proteins, phosphatidyl-ethanolamine, glycine, and guanidinoacetate for the cell homeostasis. These reactions receive the general name of ‘transmethylation reactions’, and are catalyzed by specific methyltransferases. Methionine is also regenerated from homocysteine via methionine synthase, using vitamin B12 as a cofactor and vitamin B9 as a methyl donor. In addition, BHMT catalyzes methionine synthesis from homocysteine, using betaine which is synthesized from choline by the enzyme choline oxidase, as a methyl donor [69]. Choline is a nutrient obtained through both dietary intake and endogenous synthesis. It is either oxidized to form betaine by the enzyme choline dehydrogenase, or used to generate phosphatidylcholine (CDP-choline pathway). Choline deficiency leads to the deficiency of phosphatidylcholine. Choline deficiency also perturbs protein kinase C signaling, which results in altered cell proliferation signals [70]. So, both humans and rats with choline deficiency develop not only fatty liver [70,71] but also hepatocyte death and liver damage [72]. In addition, the product of BHMT, methionine, is required for the synthesis of phosphatidylcholine from phosphatidylethanolamine by the enzyme, phosphatidyl-ethanolamine methyltransferase pathway (PEMT pathway). These two pathways generate phosphatidylcholine in the liver [26]. Studies of phosphatidylcholine anabolism in mice indicated that 70% of the hepatic synthesis of phosphatidylcholine is derived from the CDP-choline pathway, requiring dietary choline, while 30% is derived from the PEMT pathway [23]. Since BHMT influences hepatic lipid accumulation, deletion of the BHMT protein perturbs choline metabolism, and alter phosphatidylcholine concentration by disturbing the above two pathways.
Protein carbonyls are commonly used as a representative marker of protein oxidation in cells and tissues. Carbonylation of BHMT was previously reported by mass spectrometry in the rat model of alcoholic steatosis which was characterized by accumulation of fat in the liver at 3 and 6 weeks after ethanol exposure [36]. Furthermore, mice with the gene deletion encoding BHMT (Bhmt-/-) developed fatty liver, and showed elevation of ALT indicating hepatic damage at 5 weeks of age. The Bhmt-/- mice showed 21-fold increase of the liver betaine and ~27% decrease of phosphatidylcholine concentrations, possibly due to increased use of choline to form betaine. Consequently, 6-fold increase was observed in hepatic triglyceride concentrations, compared to the wildtype mice (Bhmt+/+), which was due to a decrease in the secretion of VLDL [30]. Since even ~27% decrease of phosphatidylcholine can cause 6-fold increase of hepatic triglyceride concentrations, it is probable that slight decrease of phosphatidylcholine concentration, as observed in this study, can more or less contribute to lipid depositions in the monkey liver. Phosphatidylcholine is a membrane constituent important for its structure and function, and in the liver it is crucial for the efflux of VLDL [27,73,74]. Phosphatidylcholine is required for the assembly/secretion of lipoproteins in the liver [75] and for solubilizing cholesterol in bile [76]. Accordingly, the link between choline/phosphatidylcholine deficiency and hepatic steatosis has been well recognized since more than half century ago [77]. The present experimental paradigm, albeit heterogeneity of hydroxynonenal-induced lesions (Figures 1B, 2B), could confirm tendency of phosphatidylcholine decrease in two of the three samples studied. This was compatible with decrease of the BHMT naïve protein (Figure 1C), increase of BHMT cleavage (Figure 1C), and increase of carbonylation of BHMT (Figure 3). These data altogether suggest that calpain-mediated cleavage of the carbonylated BHMT had occurred in response to hydroxynonenal.
Hepatic steatosis develops when fatty acid influx, de novo hepatic lipogenesis, or triglyceride synthesis exceeds lipid efflux or β-oxidation. Alterations in choline and phosphatidylcholine metabolism may have an impact of predisposing the subject to fatty liver. Concerning the mechanism of poor choline and phosphatidylcholine availability in the liver, there are three explanations which are widely accepted at present; 1) low dietary intake, 2) low estrogen status, and 3) genetic polymorphisms. All of these three affect the de novo phosphatidylcholine synthesis pathway [26]. Although exposure to alcohol [36] or vegetable oils [13] appears to be distinct with regard to the cellular toxicity, alcohol metabolite ‘acetaldehyde’ and lipid peroxidation product ‘hydroxynonenal’ have actually the same active group (-CHO). Regardless of the designation as ‘-nal’ or ‘-aldehyde’, either hydroxynonenal or acetaldehyde can cause protein carbonylation. One should note that consumption of linoleic acid-rich vegetable oils or alcohol has exactly the same adverse effect of generating -nal (-aldehyde) in the body. The concept of 4) acquired BHMT disorder due to ‘hydroxynonenal-mediated oxidation (carbonylation) followed by cleavage’ was the first concept which was unreported ever. Importantly, we should keep in mind that phosphatidylcholine deficiency due to BHMT disorder, may occur more drastically especially in the aged people with an increased serum concentration of hydroxynonenal due to the reduction of detoxification enzymes [78]. Especially, in those with aldehyde dehydrogenase 2 (ALDH2) gene mutation [79], we speculate that intake of vegetable oils for years may cause hepatic steatosis via accumulation of hydroxynonenal, by the similar BHMT disorder as seen in acetaldehyde-induced ALDH2 deficiency of rats after 3~6 week ethanol exposure [36].
In conclusion, especially in those with daily intake of ω-6 PUFA-rich, deep-fried foods containing abundant, exogenous hydroxynonenal, vegetable oil-dependent BHMT disorder had better be focused on as the fourth, but possibly the most common, explanation of phosphatidylcholine deficiency and hepatic steatosis. Alternatively, high-fat diets may generate excess reactive oxygen species in mitochondria, and this would oxidize phospholipids containing ω-6 PUFA of biomembranes to generate intrinsic hydroxynonenal. Since either exogenous or intrinsic hydroxynonenal may cause lysosomal membrane destabilization by Hsp70.1 deficiency [80] and the resultant hepatocyte degeneration/death with steatosis in long years, one should keep in mind that daily intake of ω-6 PUFA-rich vegetable oils and/or high-fat diets is detrimental for the liver, especially in the elderly people and those with ALDH2 deficiency [78,79].
Summary
Lipotoxicity implicates very complex cellular mechanisms in which the excess adiposity results in cell degeneration/death. However, the exact molecular cascade of lipotoxicity in the liver has not been elucidated in detail until now. The ‘calpain-cathepsin hypothesis’ has explained implication of lipid peroxidation product hydroxynonenal for ischemic neuronal death, because calpain-mediated cleavage of the carbonylated Hsp70 induced lysosomal membrane permeabilization/rupture in the brain and pancreas. Here, we studied whether the similar molecular cascade occurs in the liver. Intriguingly, we found that reduction of phosphatidylcholine, increments of lipid depositions, lysosomal membrane disintegrity, dissolution of mitochondria and rough ER, and hepatocyte degeneration/death, occurred by Hsp70.1 and BHMT disorders in the monkey liver after the consecutive hydroxynonenal injections. Accordingly, it is concluded from the present study that targeting ‘hydroxynonenal’ would be a viable strategy to prevent chronic liver diseases. In those with daily intake of linoleic acid-rich vegetable oils and/or high-fat diets, Hsp70.1 deficiency and acquired BHMT disorder had better be more and more focused on as a common cause of hepatic injury and steatosis.
Funding
This research was funded by Kiban-Kenkyu (B) (19H04029) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Institutional Review Board Statement
The protocol of monkey experiments done by the author was approved by the Committee on the Ethics of Animal Experiments of the Kanazawa University Graduate School of Medical Sciences (Protocol Number: AP-153613, AP-194062).
Informed Consent Statement
Informed consent utilizing human liver samples for the electron microscopic analysis was obtained at Kanazawa University Graduate School of Medical Sciences.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| ALDH2 | aldehyde dehydrogenase 2 |
| AST | aspartate aminotransferase |
| ALT | alanine aminotransferase |
| γ-GTP | γ-glutamyl transferase |
| BHMT | betaine-homocysteine S-methyltransferase |
| DNP | 2,4-dinitrophenylhydrazone |
| DNPH | 2,4-dinitrophenylhydrazine |
| Hsp70.1 | heat-shock protein 70.1 |
| MALDI-TOF/TOF MS | matrix-assisted laser desorption ionization time-of-flight tandem mass spectrometry |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| PEMT pathway | phosphatidyl-ethanolamine methyltransferase pathway |
| PUFA | polyunsaturated fatty acid |
| 2D-DIGE | two-dimensional differential in-gel electrophoretic |
| VLDL | very low-density lipoproteins |
References
- Friedman, J.M. Obesity in the new millennium. Nature 2000, 404, 632–634. [Google Scholar] [CrossRef]
- Diehl, A.M. Fatty liver, hypertension, and the metabolic syndrome. Gut 2004, 53, 923–924. [Google Scholar] [CrossRef]
- Wieckowska, A.; Feldstein, A.E. Nonalcoholic fatty liver disease in the pediatric population: a review. Curr. Opin. Pediatr. 2005, 17, 636–641. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. New Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Brancati, F.L.; Diehl, AM. Nonalcoholic fatty liver disease. 2002, 122, 1649–1657. [Google Scholar] [CrossRef]
- Brunt, E.M.; Tiniakos, DG. Pathology of steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 691–707. [Google Scholar] [CrossRef]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; Rizzetto, M. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neuschwander-Tetri, B.A. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Clin. Liver Dis. 2004, 8, 861–879. [Google Scholar] [CrossRef]
- Matteoni, C.A.; Younoss, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Seike, T.; Boontem, P.; Yanagi, M.; Li, S.; Kido, H.; Yamamiya, D.; Nakagawa, H.; Okada, H.; Yamashita, T.; Harada, K.; Kikuchi, M.; Shiraishi, Y.; Ozaki, N.; Kaneko, S.; Yamashima, T.; Mizukoshi, E. Hydroxynonenal causes hepatocyte death by disrupting lysosomal integrity in non-alcoholic steatohepatitis. Cell. Mol. Gastro. Hepatol. 2022, 14, 925–944. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T.; Ota, T.; Mizukoshi, E.; Nakamura, H.; Yamamoto, Y.; Kikuchi, M.; Yamashita, T.; Kaneko, S. Intake of ω-6 polyunsaturated fatty acid-rich vegetable oils and risk of lifestyle diseases. Adv. Nutr. 2020, 11, 1489–1509. [Google Scholar] [CrossRef] [PubMed]
- Boontem, P.; Yamashima, T. Hydroxynonenal causes Langerhans cell degeneration in the pancreas of Japanese macaque monkeys. PLoS ONE 2021, 16(11), e0245702. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J. Involution: apoptosis and tissue remodelling that convert the mammary gland from milk factory to a quiescent organ. Breast Cancer Res. 2006, 8(2), 203. [Google Scholar] [CrossRef] [PubMed]
- Kreuzaler, P.A.; Staniszewska, A.D.; Li, W.; Omidvar, N.; Kedjouar, B.; Turkson, J.; Poli, V.; Flavell, R.A. , Clarkson, R.W.; Watson, C.J. Stat3 controls lysosomal-mediated cell death in vivo. Nat. Cell Biol. 2011, 13, 303–309. [Google Scholar] [CrossRef]
- Sargeant, T.J.; Lloyd-Lewis, B.; Resemann, H.K.; Ramos-Montoya, A.; Skepper, J.; Watson, C.J. Stat3 controls cell death during mammary gland involution by regulating uptake of milk fat globules and lysosomal membrane permeabilization. Nature Cell Biol. 2014, 16, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T.; Kohda, Y.; Tsuchiya, K.; Ueno, T.; Yamashita, J.; Yoshioka, T.; Kominamin, E. Inhibition of ischaemic hippocampal neuronal death in primates with cathepsin B inhibitor CA-074: A novel strategy for neuroprotection based on ’calpain-cathepsin hypothesis’. Eur. J. Neurosci. 1998, 10, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T.; Oikawa, S. The role of lysosomal rupture in neuronal death. Prog. Neurobiol. 2009, 89, 343–358. [Google Scholar] [CrossRef]
- Sahara, S.; Yamashima, T. Calpain-mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem. Biophys. Res. Commun. 2010, 393, 806–811. [Google Scholar] [CrossRef]
- Chung, J.; Nguyen, A.K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; Horvath, I.; Mestril, R.; Watt, M.J.; Hooper, P.L.; Kingwell, B.A.; Vigh, L.; Hevener, A.; Febbraio, M.A. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA, 2008; 105, 1739–1744. [Google Scholar] [CrossRef]
- Henstridge, D.C.; Bruce, C.R.; Drew, B.G.; Tory, K.; Kolonics, A.; Estevez, E.; Chung, J.; Watson, N.; Gardner, T.; Lee-Young, R.S.; Connor, T.; Watt, M.J.; Carpenter, K.; Hargreaves, M.; McGee, S.L.; Hevener, A.L.; Febbraio, M.A. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 2014, 63, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Vance, D.E. Phosphatidylcholine and choline homeostasis. Lipid Res. J. 2008, 49, 1187–1194. [Google Scholar] [CrossRef]
- 24. Suzuki, M.; Shinohara, Y.; Ohsaki, Y.; Fujimoto, T. Lipid droplets: Size matters. J. Electron Microsc. (Tokyo) 2011. 60 Supplement 1:S101–16. [CrossRef]
- Krahmer, N.; Guo, Y.; Wilfling, F.; Hilger, M.; Lingrell, S.; Heger, K.; Newman, H.W.; Schmidt-Supprian, M.; Vance, D.E.; Mann, M.; Farese, Jr. R.V; Walther, T.C. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 2011, 14, 504–515. [Google Scholar] [CrossRef]
- Sherriff, J.L.; O’Sullivan, T.A.; Properzi, C.; Oddo, J.L.; Adams, LA. Choline, its potential role in nonalcoholic fatty liver disease, and the case for human and bacterial genes. Adv. Nutr. 2016, 7, 5–13. [Google Scholar] [CrossRef]
- Noga, A.A.; Zhao, Y.; Vance, D.E. An unexpected requirement for phosphatidylethanolamine N-methyltransferase in the secretion of very low density lipoproteins. J. Biol. Chem. 2002, 277, 42358–42365. [Google Scholar] [CrossRef]
- Pajares, M.A.; Pérez-Sala, D. Betaine-homocysteine S-methyltransferase: just a regulator of homocysteine metabolism? Cell. Mol. Life Sci. 2006, 63, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
- Szegedi, S.S.; Castro, C.C.; Koutmos, M.; Garrow, T.A. Betaine homocysteine S-methyltransferase-2 is an S-methylmethionine-homocysteine methyltransferase. J. Biol. Chem. 2008, 283, 8939–8945. [Google Scholar] [CrossRef]
- Teng, Y.W.; Mehedint, M.G.; Garrow, T.A.; Zeisel, S.H. Deletion of betaine-homocysteine S-methyltransferase in mice perturbs choline and 1-carbon metabolism, resulting in fatty liver and hepatocellular carcinomas. J. Biol. Chem. 2011, 286, 36258–36267. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Shinohara, M.; Vance, D.; Than, T. A.; Ookhtens, M.; Chan, C.; Kaplowitz, N. Effect of transgenic extrahepatic expression of betaine-homocysteine methyltransferase on alcohol or homocysteine-induced fatty liver. Alcohol. Clin Exp. Res. 2008, 32, 1049–1058. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; de Waal, E.M.; Pierce, A.; Van Remmen, H.; Ward, W.F.; Richardson, A. Detection of protein carbonyls in aging liver tissue: A fluorescence-based proteomic approach. Mech. Ageing Dev. 2006, 127, 849–861. [Google Scholar] [CrossRef]
- Doorn, J.A.; Petersen, D.R. ; Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem. Res. Toxicol. 2002, 15, 1445–1450. [Google Scholar] [CrossRef]
- Uchida, K. Histidine and lysine as targets of oxidative modification. Amino Acids 2003, 25, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S., Yamada, T., Minohata, T., Kobayashi, H., Furukawa, A., Tada-Oikawa, S., Hiraku, Y.; Murata, M.; Kikuchi, M.; Yamashima, T. Proteomic identification of carbonylated proteins in the monkey hippocampus after ischemia-reperfusion. Free Radic. Biol. Med. 2009. 46, 1472–1477. [CrossRef] [PubMed]
- Newton, B.W.; Russell, W.K.; Russell, D.H.; Ramaiah, S.; Jayaraman, A. Liver proteome analysis in a rodent model of alcoholic steatosis. J. Proteome Res. 2009, 8, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- McGrath, L.T.; McGleenon, B.M.; Brennan, S.; McColl, D.; McIlroy, S. Passmore, A.P. Increased oxidative stress in Alzheimer’s disease as assessed with 4-hydroxynonenal but not malondialdehyde. Q. J. Med. 2001, 94, 485–490. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Morita, S.; Tsuji, T.; Terada, T. Protocols for enzymatic fluorometric assays to quantify phospholipid classes. Int. J. Mol. Sci. 2020, 21, 1032. [Google Scholar] [CrossRef]
- Nakamura, A.; Goto, S. Analysis of protein carbonyls with 2,4-dinitrophenyl hydrazine and its antibodies by immunoblot in two-dimensional gel electrophoresis. J. Biochem. 1996, 119, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Nabeshi, H.; Oikawa, S.; Inoue, S.; Nishino, K.; Kawanishi, S. Proteomic analysis for protein carbonyl as an indicator of oxidative damage in senescence-accelerated mice. Free Radic. Res. 2006, 40, 1173–1181. [Google Scholar] [CrossRef]
- Mori, Y.; Oikawa, S.; Kurimoto, S.; Kitamura, Y.; Tada-Oikawa, S.; Kobayashi, H.; Yamashima, T.; Murata, M. Proteomic analysis of the monkey hippocampus for elucidating ischemic resistance. J. Clin. Biochem. Nutr. 2020, 67, 167–173. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J, N. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Song, K.X.; Qu, S. Nonalcoholic Fatty liver disease: pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid. Med. Cell Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Scaloni, A.; Giustarini, D.; Cavarra, E.; Tell, G.; Lungarella, G.; Colombo, R.; Rossi, R.; Milzani, A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: the contribution of redox proteomics. Mass Spectrom. Rev. 2005, 24, 55–99. [Google Scholar] [CrossRef] [PubMed]
- Heydari, A.R.; Takahashi, R.; Gutsmann, A.; You, S.; Richardson, A. Hsp70 and aging. Experientia. 1994, 50, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Li, Z.; Bronk, S.F.; Gores, G.J. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am. J. Physiol. Gastrointest. Liver Physiol. 2006. 290, G1339–1346, 2006. [CrossRef] [PubMed]
- Yamashima, T.; Saido, T.C.; Takita, M.; Miyazawa, A.; Yamano, J.; Miyakawa, A.; Nishijyo, H.; Yamashita, J.; Kawashima, S.; Ono, T.; Yoshioka, T. Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. Eur. J. Neurosci. 1996, 8, 1932–1944. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Johnson, C.D.; Hennebelle, M.; Holtmann, T.; Taha, A.Y.; Kirpich, I.A.; Eguchi, A.; Ramsden, C.E.; Papouchado, B.G.; McClain, C.J.; Feldstein, A.E. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J. Lipid Res. 2018, 59, 1597–1609. [Google Scholar] [CrossRef]
- Shearn, C.T.; Saba, L.M.; Roede, J.R.; Orlicky, D.J.; Shearn, A.H.; Peterson, D.R. Differential carbonylation of proteins in end-stage human fatty and nonfatty NASH. Free Rad. Biol. Med. 2017, 113, 280–290. [Google Scholar] [CrossRef]
- Albano, E.; Mottaran, E.; Vidali, M.; Reale, E.; Saksena, S.; Occhino, G.; Burt, A.D.; Day, C.P. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. 2005, 54, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Parola, M.; Alisi, A.; Marra, F.; Piemonte, F.; Mombello, C.; Sutti, S.; Povero, D.; Maina, V.; Novo, E.; Albano, E. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int. J. Mol. Med. 26, 471–476. [CrossRef] [PubMed]
- Sutti, S.; Jindal A, Locatelli, I.; Vacchiano, M.; Gigliotti, .L; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014, 59, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Bruce-Keller, A.J.; Li, Y.J.; Lovell, M.A.; Kraemer, P.J.; Gary, D.S.; Brown, R.R.; Markesbery, W.R.; Mattson, M.P. 4-Hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. J. Neuropathol. Exp. Neurol. 1998, 57, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Vigh L, Smith RG, Soós J, Engelhardt JI, Appel SH, Siklós L. Sublethal dose of 4-hydroxynonenal reduces intracellular calcium in surviving motor neurons in vivo. Acta Neuropathol. 2005. 109, 567-575. [CrossRef]
- Wang, Y.; Wang, W.; Yang, H.; Shao, D.; Zhao, X.; Zhang, G. Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of Toll-like receptor 4 signaling. Free Radic. Biol. Med. 2019, 131, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog. Neurobiol. 2000, 62, 273–295. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Lawless, C.; von Zglinicki, T. Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model. Aging Cell. 2008, 7, 920–923. [Google Scholar] [CrossRef] [PubMed]
- Di Naso, F.C.; Porto, R.R.; Fillmann, H.S.; Maggioni, L.; Padoin, A.V.; Ramos, R.J.; Mottin, C.C.; Bittencourt, A.; Marroni, N.A.P.; de Bittencourt, P.I.H., Jr. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity (Silver Spring) 2015, 23, 120–129. [Google Scholar] [CrossRef]
- Eaton, S. Control of mitochondrial β-oxidation flux. Prog. Lipid Res. 2002, 41, 197–239. [Google Scholar] [CrossRef]
- Archer, A.E.; Rogers, R.S.; Von Schulze, A.T.; Wheatley, J.L.; Morris, E.M.; McCoin, C.S.; Thyfault, J.P.; Geiger, P.C. Heat shock protein 72 regulates hepatic lipid accumulation. Heat shock protein 72 regulates hepatic lipid accumulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R696–R707. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The peroxisome: a new cytoplasmic organelle. Proc. R. Soc. Lond. B. Biol. Sci. 1969, 173, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M.; Martínez-Chantar, M.L.; Lu, S.C. S-adenosylmethionine metabolism and liver disease. Ann. Hepatol. 2013, 12, 183–189. [Google Scholar] [CrossRef] [PubMed]
- da Costa, K.A.; Cochary, E.F.; Blusztajn, J.K.; Garner, S.C.; Zeisel, S.H. Accumulation of 1,2-sn-diradylglycerol with increased membrane-associated protein kinase C may be the mechanism for spontaneous hepatocarcinogenesis in choline-deficient rats. J. Biol. Chem. 1993, 268, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.M.; da Costa, K.A.; Kwock, L.; Stewart, P.W.; Lu, T.S.; Stabler, S.P.; Allen, R.H.; Zeisel, S.H. Sex and menopausal status influence human dietary requirements for the nutrient choline. Am. J. Clin. Nutr. 2007, 85, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- da Costa, K.A.; Badea, M.; Fischer, L.M.; Zeisel, S.H. Elevated serum creatine phosphokinase in choline-deficient humans: mechanistic studies in C2C12 mouse myoblasts. Am. J. Clin. Nutr. 2004, 80, 163–70. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.M.; Vance, D.E. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J. Biol. Chem. 1988, 263, 2998–3004. [Google Scholar] [CrossRef]
- Yao, Z.M.; Vance, D.E. 1989. Head group specificity in the requirement of phosphatidylcholine biosynthesis for very low density lipoprotein secretion from cultured hepatocytes. J. Biol. Chem. 264, 11373–11380. https://www.jbc.org/article/S0021-9258(18)60474-0/pdf.
- Zeisel, S.H.; da Costa, K.A. Choline: an essential nutrient for public health. Nutr. Rev. 2009, 67, 615–23. [Google Scholar] [CrossRef]
- Jüngst, D.; Lang, T.; Huber, P.; Lange, V.; Paumgartner, G. Effect of phospholipids and bile acids on cholesterol nucleation time and vesicular/micellar cholesterol in gallbladder bile of patients with cholesterol stones. J. Lipid Res. 1993, 34, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nakamura, S.; Karoji, N.; Aikawa, T.; Suzuki, O.; Onodera, A.; Ono, Y. Hepatic function tests in heavy drinkers among workmen. Tohoku J. Exp. Med. 1967, 93, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Schaur, R.J.; Siems, W.; Bresgen, N.; Eckl, P.M. 4-Hydroxy-nonenal – A bioactive lipid peroxidation product. Biomolecules 2015, 5, 2247–2337. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chang, B.; Li, X.; Zou, Z. Role of ALDH2 in hepatic disorders: Gene polymorphism and disease pathogenesis. J. Clin. Transl.. Hepatol. 2021, 9, 90–98. [Google Scholar] [CrossRef]
- Yamashima, T. Implication of vegetable oil-derived hydroxynonenal in the lysosomal cell death for lifestyle-related diseases. nutrients 2023, 15, 609. [Google Scholar] [CrossRef]
Figure 1.
Blood data, inspection of the liver at autopsy, and Western blotting of BHMT.
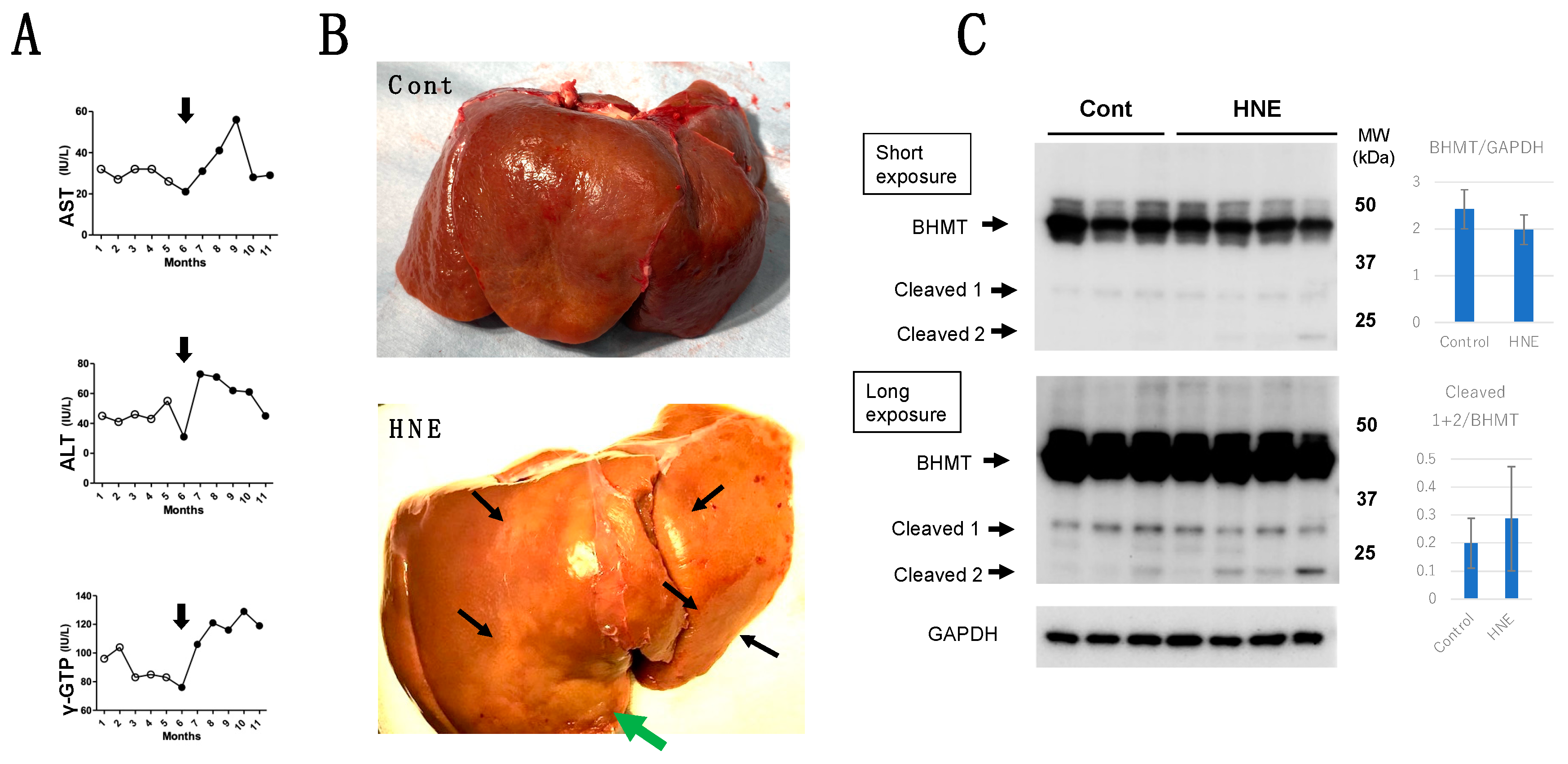
Figure 2.
Western blotting, SDS-PAGE, and proteomics analyses of Hsp70.1.
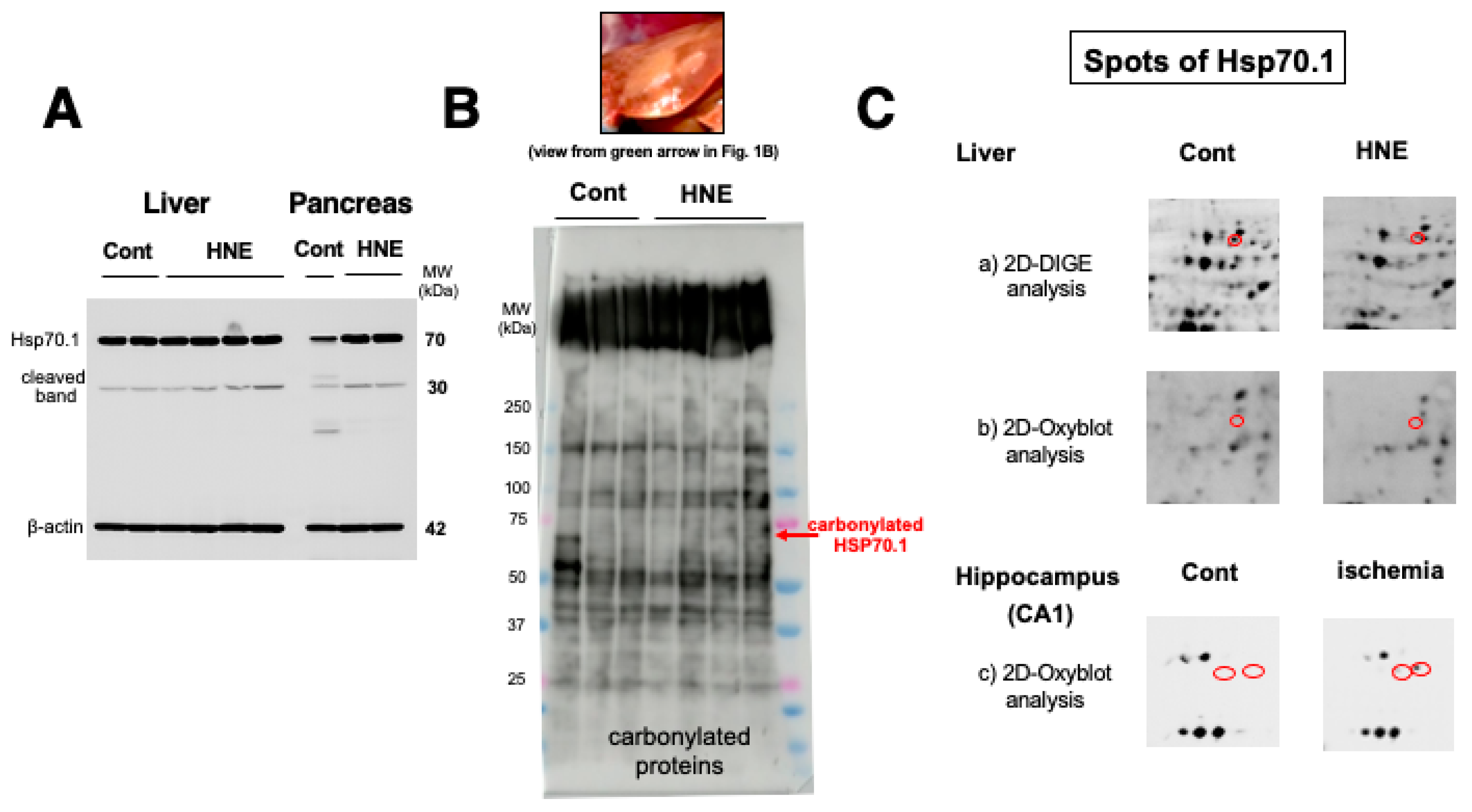
Figure 3.
2D-Oxyblot analysis of BHMT in the control and hydroxynonenal (HNE)-treated livers.
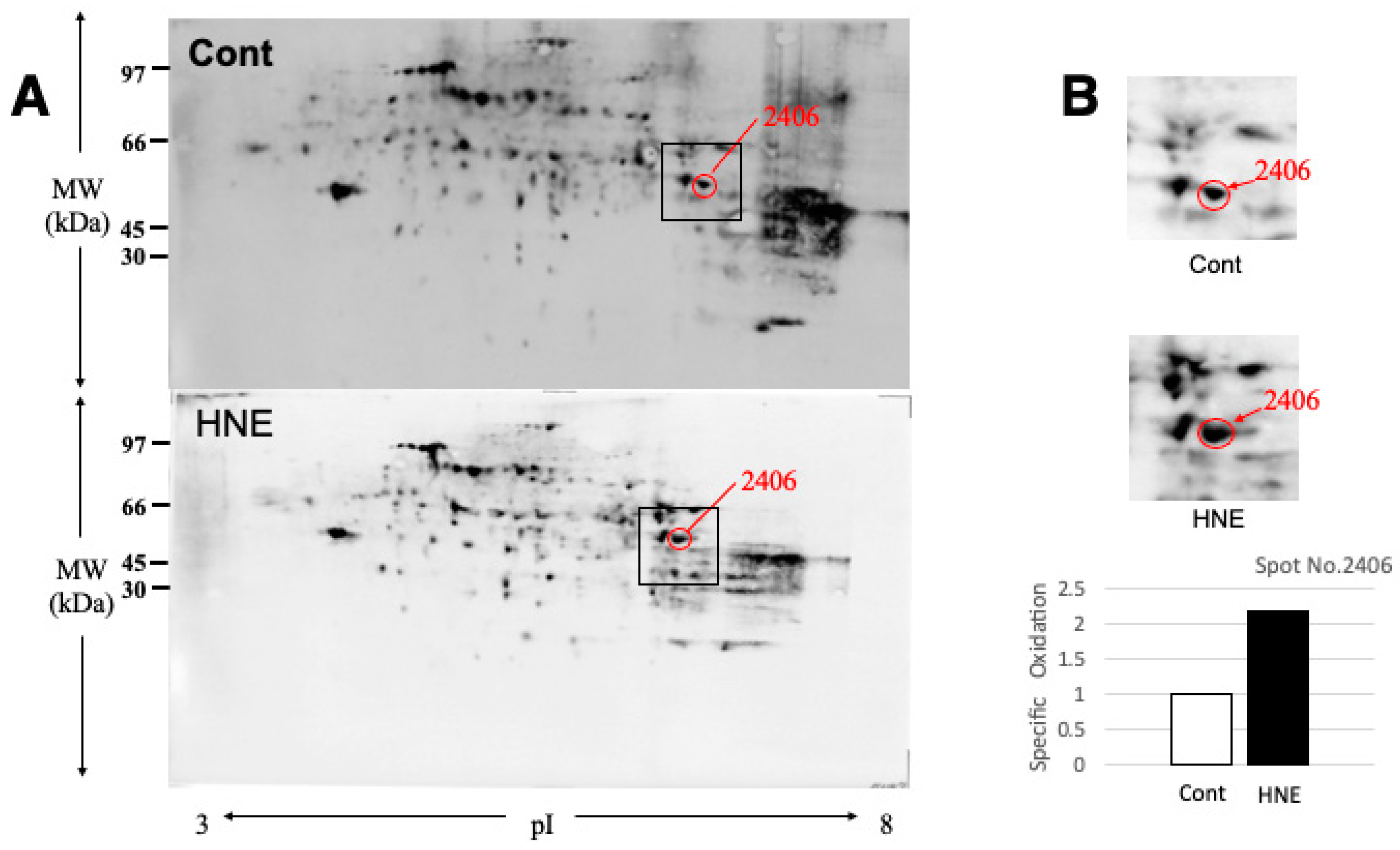
Figure 4.
Histological finding of the hydroxynonenal-treated liver.
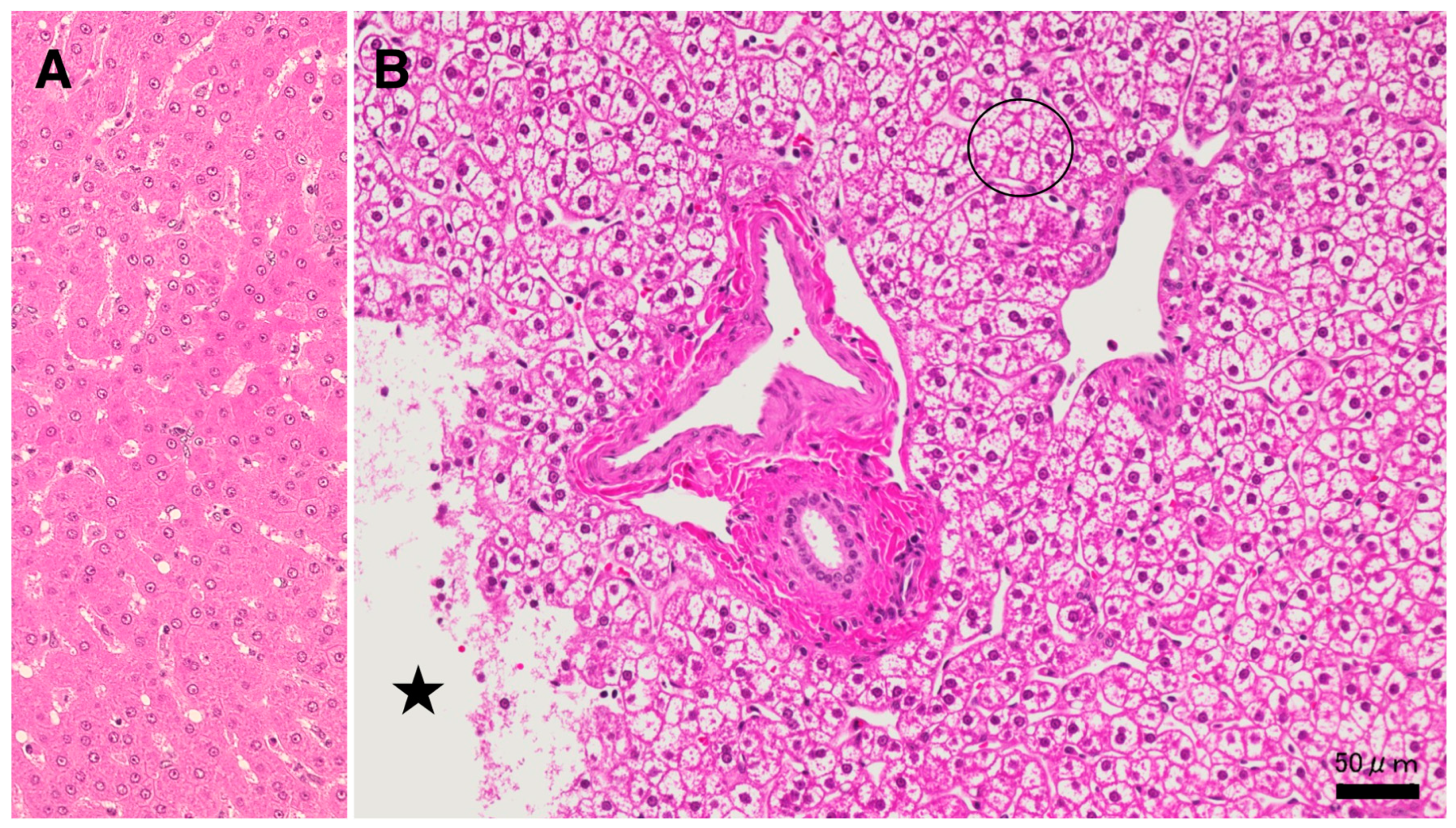
Figure 5.
Accumulation of lipid droplets after hydroxynonenal injections.
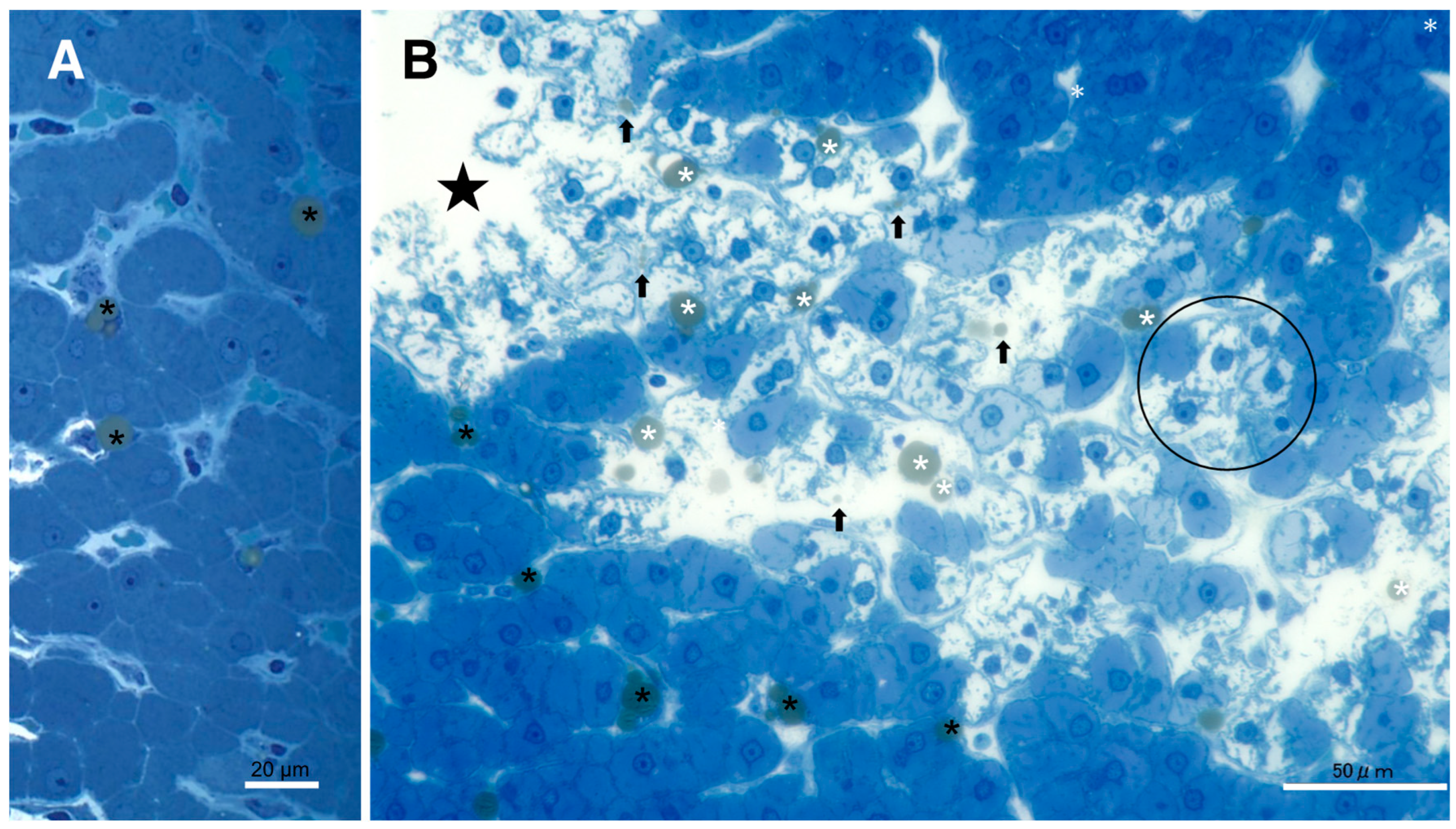
Figure 6.
Immunofluorescence histochemical stainings of activated µ-calpain and cathepsin B. Compared to the control liver without hydroxynonenal treatment (6A, Cont), the liver after hydroxynonenal injections (6B, HNE) showed an increased immunoreactivity of activated µ-calpain within hepatocytes. Concomitantly, coarse granular and diffuse cytoplasmic cathepsin B immunoreactivity was seen after hydroxynonenal injections (6D, HNE), showing a marked contrast to the control (6C, Cont). This indicates extra-lysosomal leakage of cathepsin B which was localized within lysosomes prior to hydroxynonenal injections (6C, Cont).
Figure 6.
Immunofluorescence histochemical stainings of activated µ-calpain and cathepsin B. Compared to the control liver without hydroxynonenal treatment (6A, Cont), the liver after hydroxynonenal injections (6B, HNE) showed an increased immunoreactivity of activated µ-calpain within hepatocytes. Concomitantly, coarse granular and diffuse cytoplasmic cathepsin B immunoreactivity was seen after hydroxynonenal injections (6D, HNE), showing a marked contrast to the control (6C, Cont). This indicates extra-lysosomal leakage of cathepsin B which was localized within lysosomes prior to hydroxynonenal injections (6C, Cont).
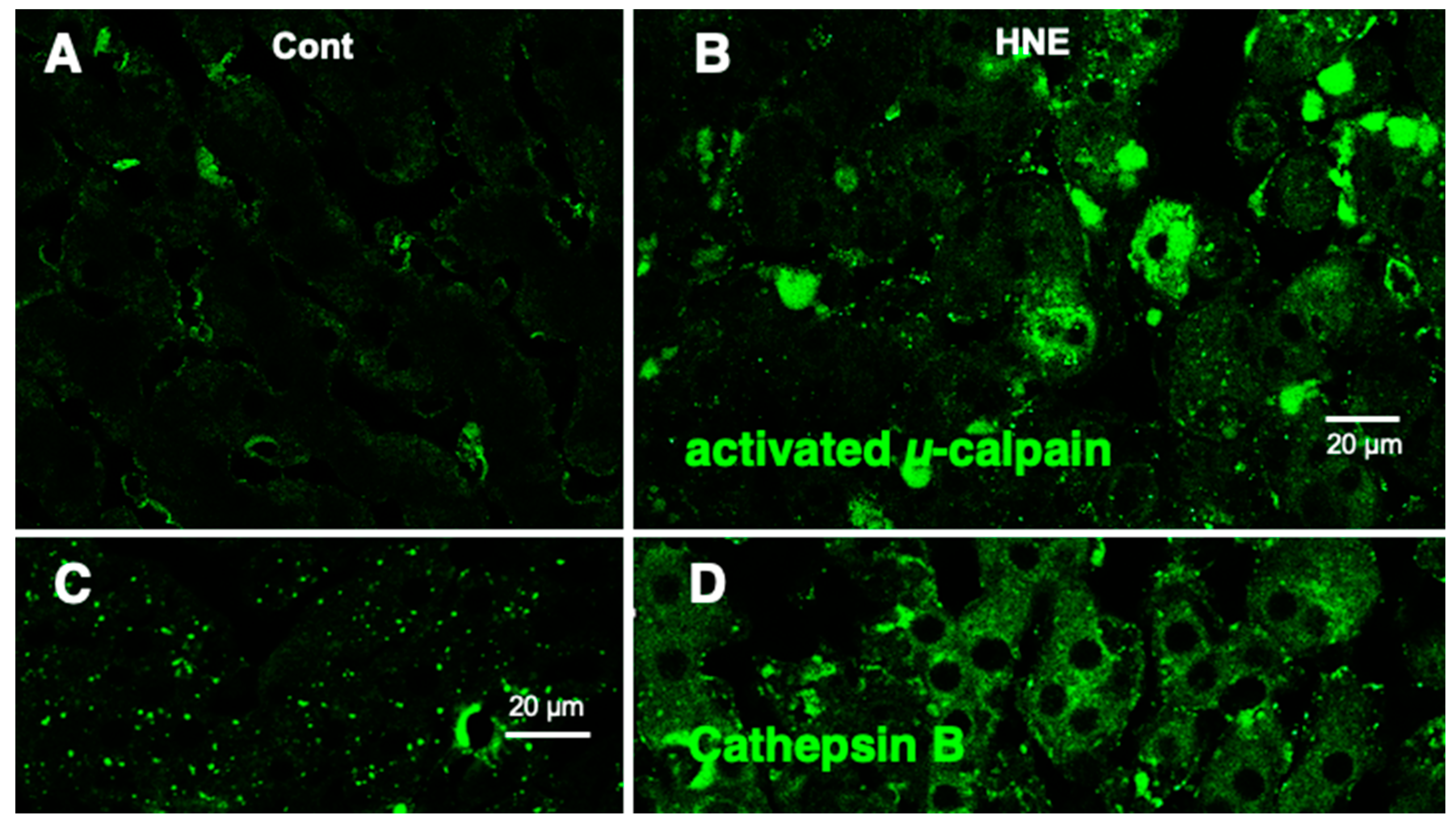
Figure 7.
Western blotting and immunohistochemical data without (Cont) and with (HNE) hydroxynonenal injections. Western blotting indicates calpain-mediated cleavage of Hsp70.1 was increased after hydroxynonenal injections (HNE), compared to the control (Cont). Immunohistochemical analysis indicates interaction of activated µ-calpain (green) upon Hsp70.1 (red) especially after hydroxynonenal injections (HNE).
Figure 7.
Western blotting and immunohistochemical data without (Cont) and with (HNE) hydroxynonenal injections. Western blotting indicates calpain-mediated cleavage of Hsp70.1 was increased after hydroxynonenal injections (HNE), compared to the control (Cont). Immunohistochemical analysis indicates interaction of activated µ-calpain (green) upon Hsp70.1 (red) especially after hydroxynonenal injections (HNE).
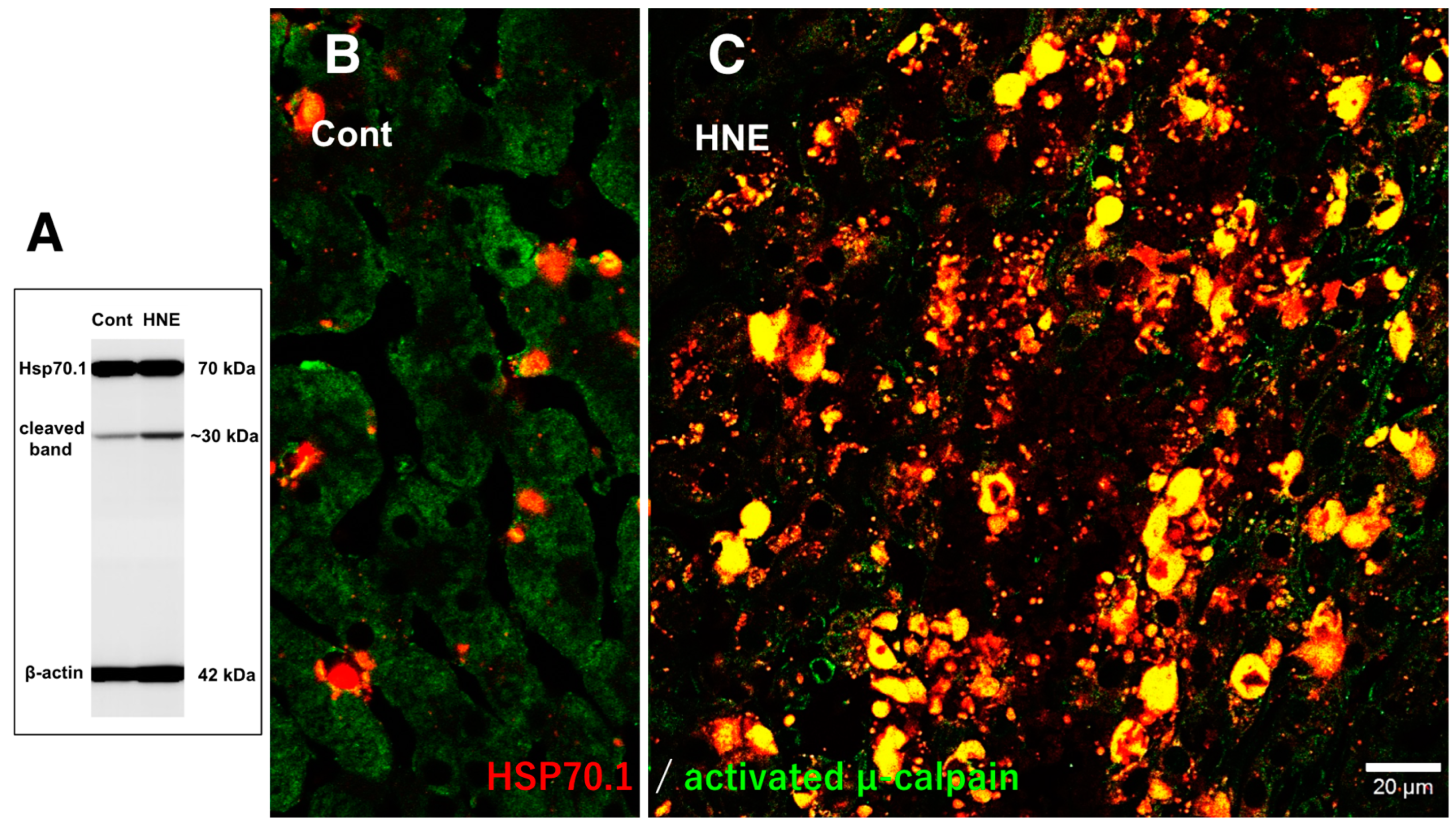
Figure 8.
Electron-microphotographs of the degenerating hepatocytes after hydroxynonenal injections.
Figure 8.
Electron-microphotographs of the degenerating hepatocytes after hydroxynonenal injections.
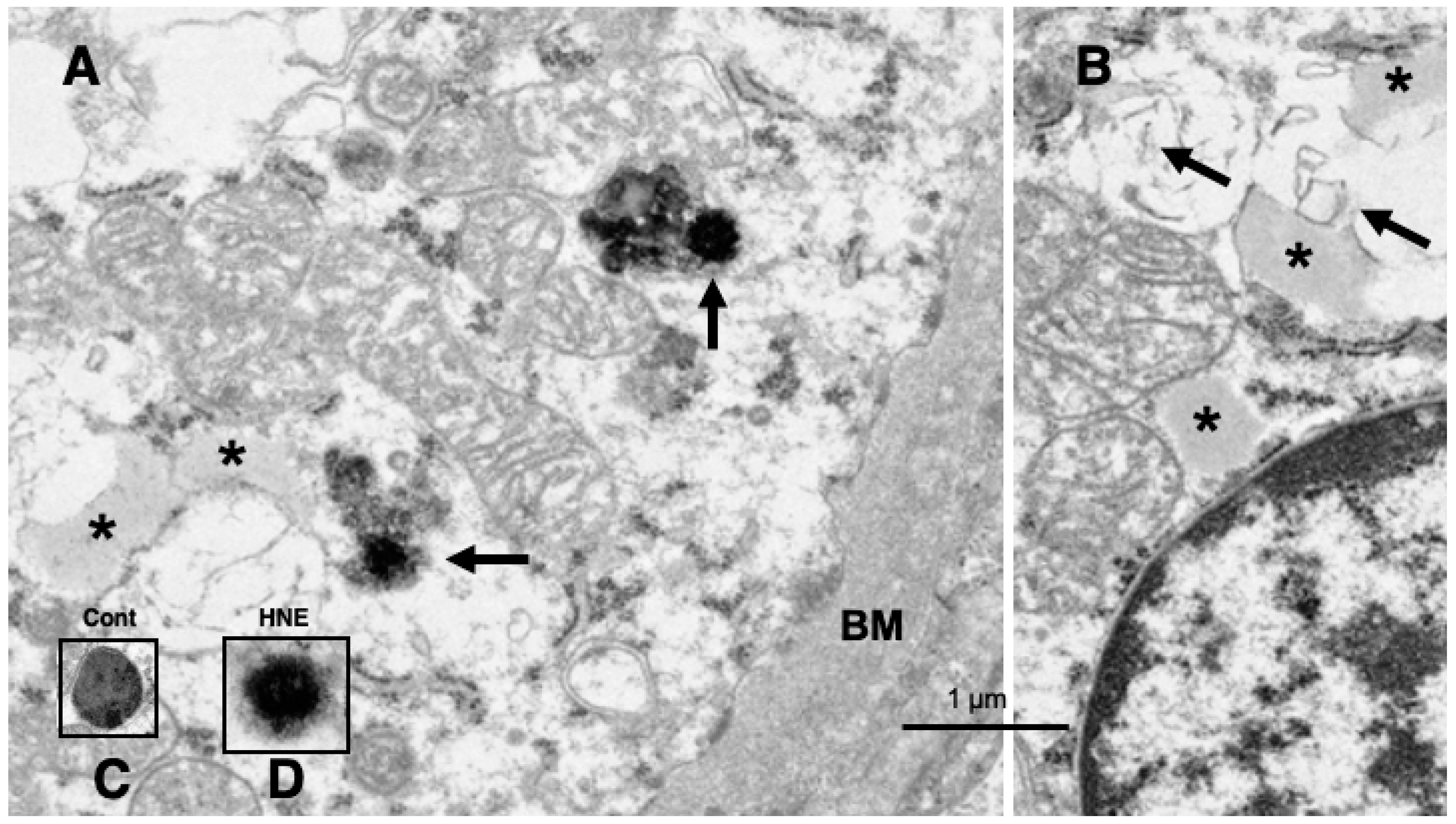
Figure 9.
Ultrastructural changes of mitochondria and peroxisomes in the degenerating hepatocytes after hydroxynonenal injections, compared to the control.
Figure 9.
Ultrastructural changes of mitochondria and peroxisomes in the degenerating hepatocytes after hydroxynonenal injections, compared to the control.
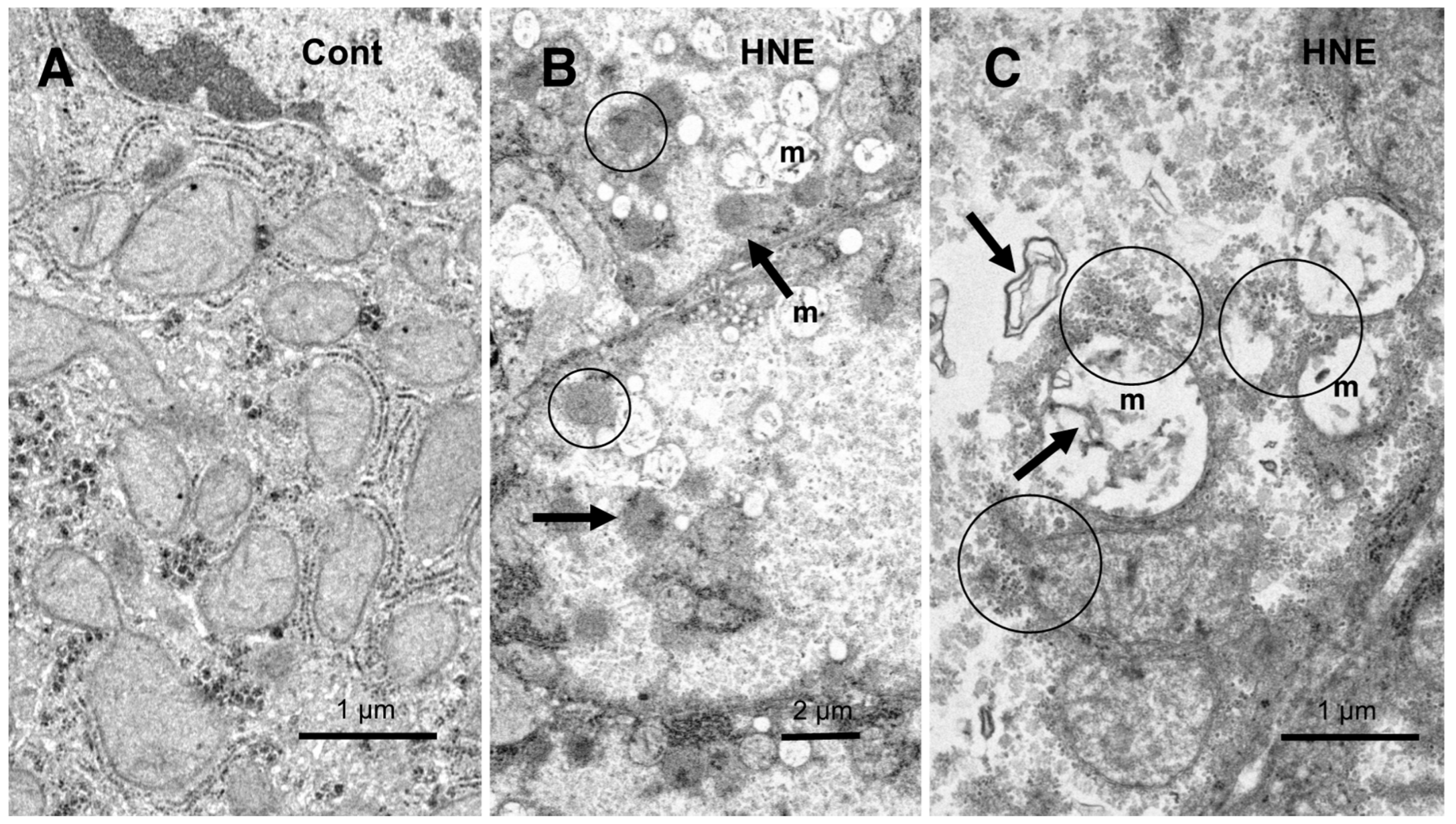
Figure 10.
Low-magnification electron-microphotographs of the degenerating hepatocyte after hydroxynonenal injections.
Figure 10.
Low-magnification electron-microphotographs of the degenerating hepatocyte after hydroxynonenal injections.
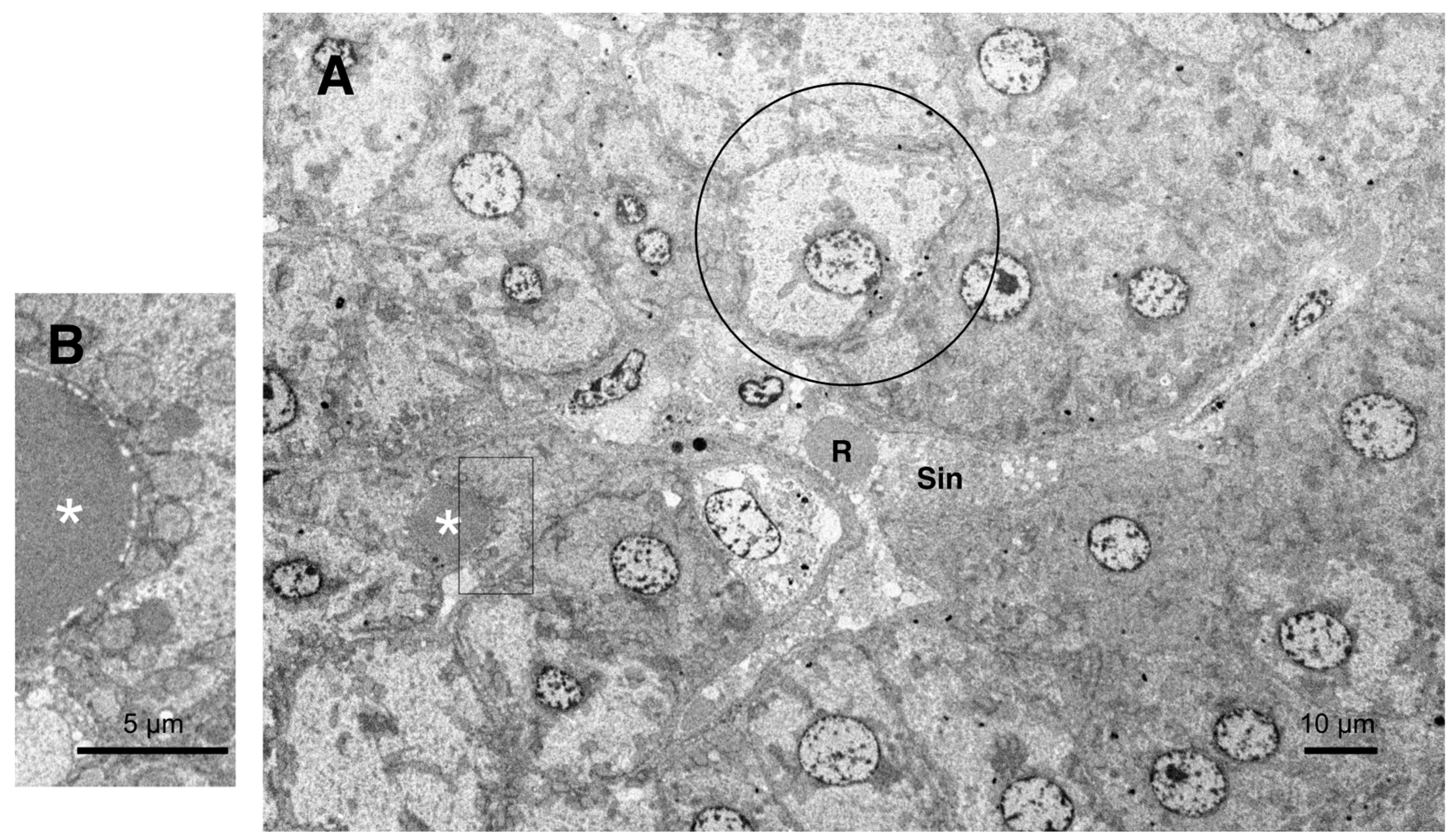
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



