
Submitted:
28 February 2023
Posted:
01 March 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
COVID-19, the infectious disease caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), is frequently associated with pulmonary thrombotic events, especially in hospitalised patients. Severe SARS-CoV-2 infection is characterized by a proinflammatory state and an associated disbalance in hemostasis. Immune pathology analysis supports the inflammatory nature of pulmonary arterial thrombi composed by white blood cells, especially neutrophils, CD3+ and CD20+ lymphocytes, fibrin, red blood cells and platelets. Immune cells, cytokines, chemokines and the complement system are key drivers of immunothrombosis, as they induce the damage of endothelial cells and initiate pro-inflammatory and pro-coagulant positive feedback loops. Neutrophil extracellular traps induced by COVID-19-associated “cytokine storm”, platelets, red blood cells, and coagulation pathways close the inflammation-endotheliopathy-thrombosis axis, contributing to SARS-CoV-2 associated pulmonary thrombotic events. The hypothesis of immunothrombosis is also supported by the minor role of venous thromboembolism, chest CT imaging data showing peripheral blood clots associated with inflammatory lesions and the high incidence of thrombotic events despite routine thromboprophylaxis. Understanding the complex mechanisms behind COVID-19-induced pulmonary thrombosis will lead to future combination therapies for hospitalised patients with severe disease, that would target the crossroads of inflammatory and coagulation pathways.
Keywords:
COVID-19
; SARS-CoV-2 infection
; pulmonary in situ thrombosis
; embolism
; immunothrombosis
; inflammation
; coagulopathy
1. Introduction
SARS-CoV-2 associated micro- and macro-vascular thrombotic events frequently occur and they are associated with disease severity and worse clinical outcomes [1,2,3]. Severe hypercoagulability mainly develops in the lungs of COVID-19 patients [4], and the most common vascular thrombotic complication involves the pulmonary arteries [1,5,6]. The incidence of pulmonary artery thrombosis in patients with COVID-19 pneumonia varies between 18% and 57%, with a pooled determined incidence of 30.2 [7]. Post-mortem lungs from patients with SARS-CoV-2 infection showed severe coagulation abnormalities, especially fibrin- and platelet-rich thrombi, not observed in non-COVID-19 autopsy controls [4]. COVID-19 associated thrombotic complications are secondary to a synergistic interplay of endotheliopathy, coagulation pathways, platelet dysfunction and detrimental immune-mediated thrombosis [8]. However, it is not clear which of the pathobiological mechanisms, conventional risk factors and venous thromboembolic disease, in situ immunothrombosis, or additional thrombotic mechanisms contributes more to these COVID-19-associated pulmonary thrombotic events [2,9].
In this narrative review, we aimed to explore the pathophysiological pathways supporting the potential mechanisms responsible for COVID-19 associated pulmonary thrombosis, which could have implications for diagnostic and management strategies. We conducted free searches in PubMed database, for publications reporting on patients with COVID-19 and pulmonary thrombosis and/or associated mechanisms. Clinical, pathobiological and imaging data were extracted. In papers where the mechanism of thrombosis (embolism or local clot formation) could not be identified, we used the term pulmonary thrombosis covering both pulmonary embolism and in situ thrombosis. We searched only papers in English, using the following terms in various combinations: “SARS-CoV-2”, “COVID19”, “novel coronavirus”, “venous thromboembolism”, “thrombosis”, “pulmonary embolism”, “pulmonary thrombosis”, “immunothrombosis”, “in situ thrombosis”. We restricted our search to papers with full text or abstracts written in English and no other restrictions were applied. We considered relevant for this narrative review data from original human studies, preclinical/animal studies, case reports as well as previously published reviews.
2. COVID-19 associated pulmonary thrombosis is an in situ immunothrombosis
Pulmonary thrombosis in situ is a pathological condition non-related to embolism from deep vein thrombosis (DVT) in the lower extremities [10]. Non-pulmonary in situ thrombosis in the right ventricle was also described in COVID-19 patients [11,12]. COVID-19-associated pulmonary intravascular coagulopathy is a complex disease, “orchestrated” by a severe and dysregulated proinflammatory response that can lead to immunothrombosis [2,13]. The prothrombotic state in patients with COVID-19 is reminiscent of this immunothrombosis process, a result of the crosstalk between immune and haemostatic systems and characterised by the production of microthrombi in small capillaries, in which endothelial cells (ECs) adopt a pro-adhesive phenotype in contact with SARS-CoV-2 [14,15,16]. Pathological studies showed multiple microthrombi in pulmonary capillaries and larger primary thrombi in arterioles, considered to be primary in nature [12]. Autopsy lung samples from patients with COVID-19 showed important circulatory changes with inflammation-dependent intravascular thrombosis, a direct pathological evidence for immunothrombosis, which were not found in other organs like heart, brain, and kidneys [12]. There is a concept of a local lung associated coagulation system, the “bronchoalveolar hemostasis” and the formation of blood clots in the microvasculature of the lungs could be a part of the host immune defense against SARS-CoV-2 [17,18]. Normal host alveolar capillary associated immune defense system is presented in Figure 1.
In patients with in situ pulmonary thrombosis, in-depth immune pathology analysis by immunohistochemistry supports the inflammatory nature of arterial thrombi composed by white blood cells, especially neutrophils, CD3+ and CD20+ lymphocytes, fibrin, red blood cells and platelets, but not megakaryocytes [13]. Regional thrombosis of the lung microvasculature showed also compressed deformed red blood cells (RBCs), including polyhedrocytes and different morphological types of fibrin structures coated with sparse spherical microparticles, which could comprise virions or cellular ectosomes [12]. Different phenotypes of in situ thrombosis may exist and needs different intensity of anticoagulant therapy [19].
Severe COVID-19 is characterized by a proinflammatory state and an associated disbalance in hemostasis, which starts with the disruption of the alveolar epithelium, and involves intrinsic, extrinsic coagulation pathways, neutrophil extracellular traps (NETs) activation and release (NETosis), and impaired fibrinolysis secondary to high plasminogen activator inhibitor 1 (PAI-1) levels [15]. Acute respiratory failure in patients with severe COVID-19 is associated with diffuse alveolar damage, perialveolar microangiopathy, and obstructive neoangiogenesis [15,20,21,22,23,24,25,26,27]. In situ pulmonary thrombosis may appear in COVID-19 pneumonia patients, with peripheral distribution, either within the consolidation lesions of the infected lungs (due to active local inflammation), or in non-consolidation area (due to hypercoagulability caused by systemic inflammation) [19]. Systemic inflammatory markers like C-reactive protein (CRP) have higher values in patients with COVID-19 associated pulmonary artery thrombosis compared with those without pulmonary thrombi [28,29,30,31]. Inflammatory-mediated thrombosis is also supported by the similarity in terms of comorbidities and other classical risk factors for venous thromboembolic disease in patients with and without COVID-19 associated pulmonary thrombosis [28,29]. Elevated serum levels of CRP are associated with thrombotic disease and mortality in COVID-19 patients [32,33,34,35]. Moreover, CRP is an important link between inflammation and thrombosis, as it can activate the complement cascade, induces platelet adhesion to ECs, stimulates tissue factor (TF) expression by blood monocytes and it can alter the fibrinolytic balance of ECs [36]. Ferritin, another inflammatory biomarker, is also an independent predictive factor for both venous thrombotic events, especially pulmonary thrombosis, and also mortality, in COVID-19 patients [6,37,38]. Ferritin induces mitochondrial dysfunction in platelets and this could also contribute to inflammation and thrombosis [2].
3. Biological mechanisms for in situ pulmonary immunothrombosis
3.1. Inflammatory pathways
3.1.1. Macrophages (AMφs), monocytes, and T cells
Pathogen-associated molecular patterns (PAMPs), and endogenous damage-associated molecular pattern (DAMPs), release by cellular injury, when recognized by specialized receptors like toll-like receptors (TLRs), and C-type lectin receptors (CLRs), trigger intracellular signaling cascades and produce various inflammatory cytokines and chemokines that modulates coagulation abnormalities, especially mediated by nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway [39]. The SARS-CoV-2 spike (S) protein is a TLR-4 ligand and a NF-κB pathway activator in monocytes, but also in macrophages and its RNA can activate TLR-3 and TLR-7 cellular signaling pathways [40]. Synthesis of interferons (IFNs): IFN-α, IFN-β; interleukins (ILs): IL-1α, IL-1β, IL-2, IL-6, IL-8; tumor necrosis factor-alpha (TNF-α), can contribute to inflammation and thrombosis, through multiple pathways [40]. Comparing inflamed with normal alveoli areas from the same alveolar biopsies of COVID-19 lungs, in inflamed alveoli upregulated genes were enriched for innate immune and inflammatory pathways, including neutrophil degranulation, IFN-γ and interleukin signaling [41]. As SARS-CoV-2 replicates in the alveolar epithelial cells type II and interacts with pulmonary microvasculature at the pneumocyte-capillary interface, it locally activates the innate immunity leading to an exaggerated release of pro-inflammatory cytokines in severe COVID-19 cases, known as the “cytokine storm” [42]. Macrophages, monocytes, CD4+ T cells, and CD8+ T cells are increased and activated in COVID-19 patient lungs [4]. A cell census from the COVID-19 lungs autopsies showed also increased levels of dendritic cells (DCs) and natural killer (NK) cells [41]. In severe COVID-19, in the setting of inflammation, tissue resident AMφs in the bronchoalveolar space are diminished in number, a phenomenon described as the macrophage “disappearance reaction” [43]. Infected tissue resident AMφs express higher levels of proinflammatory chemokines, which leads to an excessive monocyte recruitment by a positive feedback loop [43]. Monocytes, in addition to their role against various pathogens, promote the activation of extrinsic coagulation pathway during inflammatory states and could be considered a bridge from inflammation to thrombosis [44]. A strong association of platelet-monocyte interaction with monocyte inflammatory activation and immune system dysfunction is seen in critically ill patients, contributing to thromboinflammation in COVID-19 [45]. Moreover, the axis NETs-CD14+/CD16+ inflammatory monocytes triggers an excessive amount of IL-6 and other pro-inflammatory molecules via a positive feedback mechanism, also contributing to the “cytokine storm” [42].
3.1.2. The NETs-thrombosis axis in COVID-19
SARS-CoV-2-induced “cytokine storm” primes neutrophils to release NETs, a distinct mechanism of innate immune response composed of web-like structures containing DNA filaments coated with histones and granule proteins, that can entrap and eliminate various pathogens [46]. The disbalance of vascular microenvironmental homeostasis and aberrant NETs formation leads to pulmonary thrombosis and it may augment SARS-CoV-2-induced “cytokine storm” and macrophage activation syndrome in patients with severe disease [47,48]. NETs, together with platelets, RBCs and procoagulant molecules (von Willebrand factor (vWF), fibronectin, fibrinogen, FXII, and TF), contribute to SARS-CoV-2 associated thrombotic events [8,16]. Pulmonary autopsies in patients with COVID-19 confirmed NETs-containing microthrombi with neutrophil-platelet infiltration [49]. NETs are responsible for the interplay between inflammation and thrombosis in the lungs of COVID-19 patients, promoting tissue injury and secondary immunothrombosis [46,50]. High levels of markers of NETs are found in the plasma of patients with severe COVID-19 [16,49,51,52]. NETs are implicated in SARS-CoV-2 associated acute respiratory distress syndrome (ARDS) pathogenesis, being present in high levels in the lower respiratory tract samples of these patients, but also in lung tissue from deceased patients with COVID-19 [53]. TLR2 and C-type lectin domain family 5 member A are critical in the induction of NETosis in lung tissues by the SARS-CoV-2 S protein, leading to COVID-19 associated pulmonary thromboinflammation [54].
3.1.3. Mast cells (MCs), cytokines and chemokines
MCs are specialized subendothelial innate sentinel cells characterized by antigen processing capabilities, being equipped with TLRs and other receptors involved in different inflammatory pathways, such as FcεRI, MAS-related G protein-coupled receptor-X2 (MRGPRX2), IgG receptors, Fc-gamma type 2 receptor A (FcγRIIA), dectin-1, IL-10 receptor, substance-P and complement receptors [55,56,57,58,59,60]. Viruses can activate MCs directly or by viral particles to release preformed inflammatory mediators, vasoactive autocoids such as histamine and catalytically active MCs-specific proteases, including β-tryptase, chymase, Granzyme B, carboxypeptidase-3 and β-hexosaminidase [59,60]. MCs also produce de novo lipid mediators such as prostaglandins (PGD2, PDE2), leukotrienes (LTB4, LTC4, LTD4), platelet-activating factor (PAF), cytokines (e.g., TNF, IL-6, IL-4, IL-5, IL-1β, IL-10, IL-13) and chemokines (e.g., CCL1, CCL2, CXCL1, CXCL8) [55,56,57,59], many of which now known to be associated with the “cytokine storm” observed in severe COVID-19.
Clusters of degranulating MCs expressing chymase and tryptase are seen in the lung areas with hemorrhagic phenomena and it could be connected to local histamine production, stored endogenously within the secretory granules of MCs, and released into the vessels after cell stimulation [61,62]. Resident phagocyte alveolar macrophages activated by SARS-CoV-2-TLR interaction produce IL-1 which further stimulates MCs to produce IL-6 [62]. IL-1 causes micro-thrombi and inflammation, by promoting ECs-leukocyte adhesion, endothelial dysfunction, and thromboxane A2, B2 (TxA2, TxB2) and TNF-α production [63].
Accumulation of MCs in the lungs can cause inflammation and thrombosis. In healthy lung tissue, MCs express low levels of critical cell-entry facilitators for SARS-CoV-2, such as angiotensin converting enzyme (ACE)-2 and its serine protease for S protein priming, transmembrane serine protease 2 (TMPRSS2) [64,65]. When analyzing mRNA expression of autoimmune-related genes in the lung tissue, MCs were the only type of all studied cells (MCs, neutrophils, macrophages, exhausted CD8+, CD8+ T cells, dendritic cells, cytotoxic cells, B-cells, CD45 cells and T-cells) that was highly expressed in patients with SARS-CoV-2 pneumonia compared to influenza [66]. A total of 43 genes were significantly differentially expressed between these patients, including chemokine C-X-C motif ligand 7 (CXCL7), which is present in platelets after thrombus formation followed by neutrophil attraction [66]. Microthrombi, present in more than half of the cases, as well as thrombi in small or large arteries, were more often seen in COVID-19 than influenza [66]. In SARS-CoV-2 infected patients, even without the replication of the coronavirus inside MCs, the interaction S-receptor binding domain (RBD)-ACE2 induces rapid MCs degranulation/activation [67]. Proteases released from MCs further enhance cell viral entry, as MCs-derived chymase interacts with SARS-CoV-2 S protein [68]. MCs degranulation degree rather than their total numbers is associated with the pro-thrombotic phenotype typical of COVID-19 [69]. Under IL-4 expression, early recruiting of CD117+ MCs progenitors in the alveolar septa will lead to MCs proliferation/differentiation and once activated, they will orchestrate the crosstalk between pro-inflammatory and procoagulative networks, such as the complement and the plasma kallikrein-kinin system [70]. Some of the cytokines associated with SARS-CoV-2 “cytokine release syndrome”, IL-2, IL-4, IL-6, IL-8, IL-1β, IL-13, IL-12, TNF-α, IL-7, especially IL-8 and TNF-α, further contribute to MCs chemotaxis [71,72,73]. MCs activation with subsequent degranulation in the respiratory tract submucosa will release high levels of IL-1, IL-6 and TNF-α, but also other proteins like matrix metalloproteinase 9 (MMP-9), PAF, substance P, transforming growth factor beta (TGF-β), TXB2, and vascular endothelial growth factor (VEGF), which will contribute to the pathogenesis as pro-inflammatory and pro-thrombotic molecular factors [74,75]. MCs and their associated proteases, chymase (CMA1), carboxypeptidase A3, and tryptase beta 2, also modulate indirectly the systemic thrombo-inflammatory response in patients with SARS-CoV-2 pneumonia, as they induce thrombosis through activation of clotting factors and platelets and this may affect the relatively high incidence of pulmonary thrombotic events in COVID-19 [66,69].
COVID-19 associated coagulopathy with microvascular thrombosis is secondary to inflammation and endotheliopathy that are “orchestrated” by IL-6, a pleiotropic proinflammatory cytokine [76]. The severity of SARS-CoV-2 infection is associated with increased blood levels but also with IL-6 expression on lung tissue [76,77]. IL-6 has a pro-inflammatory role in vascular endothelial cells, and it favors hypercoagulation by interfering with normal anticoagulant and profibrinolytic properties of ECs [78]. IL-1, another key proinflammatory cytokine, had the best correlation with COVID-19 associated thrombotic events compared to IL-6 and TNF-α in one study [79]. IL-1-mediated inflammation in COVID-19 associated acute lung injury (ALI) follows the biological path of NLRP3 inflammasome and caspase-1 activation, leading to production of major innate immune mediators, IL-1β and IL-18 [78]. A bi-directional relationship also exists between IL-1-mediated inflammation and coagulation [78]. IL-1 maintains thrombosis by increasing the time of clot lysis, upregulation of TF expression and activation of the endothelium via the IL-1β pathway, also promoting the recruitment of leukocytes [78]. In one study, in patients with COVID-19 associated thrombotic events no statistical significance was recorded between serum levels of inflammatory markers (CRP, ferritin), age and other clinical characteristics, except for IL-1β and soluble P-selectin [80].
3.1.4. Complement pathways
The complement system is a key mediator of the innate immune response and inflammation [81]. Lectin and alternative pathway mediates complement activation by SARS-CoV-2 S- and N-proteins [82,83,84], which will lead to ECs and MCs degranulation, enhanced phagocytic activity of neutrophils and monocytes, and “orchestration” of a proinflammatory environment [81]. Cleavage components C3a and C5a are potent inflammatory molecules capable of inducing the release of proinflammatory cytokines, as part of the COVID-19 associated “cytokine storm” [84]. Plasmin-mediated C5 activation is also involved in thrombosis by multiple mechanisms [85]. The intricate network between the complement system and the coagulation cascade could sustain an enhanced rate of COVID-19 associated coagulopathy [86]. Severe COVID-19 with respiratory failure is characterized by a diffuse lung microvascular injury mediated by complement activation indicated by both the presence of significant serum levels and deposits in lung microvasculature, of terminal complement components C5b-9 (membrane attack complex), C3b and C4d. [84,87,88,89]. Alternative Pathway Activator factor D and Lectin Pathway Activator Mannan-Binding Lectin-Associated Serine Protease 2, were also markedly increased in lungs from patients with COVID-19 [84,88]. Co-localization of S glycoproteins with C5b-9/C4d in the interalveolar septa in autopsy lung samples from patients with COVID-19 suggest a direct, local complement activation by SARS-CoV-2 [87]. Moreover, this cross talk between complement, immune and coagulation systems, amplifies the process of COVID-19 associated coagulopathty, by a positive feedback loop [16].
Immunothrombosis is an interplay between systemic and lung inflammatory pathways and coagulation/fibrinolysis systems (Figure 2).
3.2. Coagulation pathways
3.2.1. ECs and platelets
COVID-19 is an endothelial disease, triggering the “cytokine storm” and inflammation, oxidative stress, and coagulopathy [90]. SARS-CoV-2 S protein-ACE-2 interaction activates the endothelium, which will lead to stimulation of immune responses and ECs damage, activation of coagulation, reduction of fibrinolysis, and platelet adhesion and aggregation [91]. Damaged ECs lead to exposure of the prothrombogenic basal membrane, upregulation of TF and release of coagulation factor VIII, vWF and P-selectin from Weibel-Palade bodies (WPBs) [16]. Markers of ECs and platelet activation are significantly elevated especially in severe and critical SARS-CoV-2 infection, leading to diffuse endotheliitis and impaired microcirculatory function with an associated pro-coagulant state [92,93].
COVID-19 is also a disease of platelets pathology [94]. Platelets have the ability to interact with viruses, bacterial pathogens, and Plasmodium parasites [95]. SARS-CoV-2 S protein can interact with integrin α5β1, expressed on platelets [96]. Platelets are able to “orchestrate” monocyte responses to inflammatory activation, inflammatory cytokines secretion, and TF expression in COVID-19 patients [45]. Thrombocytopathy, as well as platelets associated abnormal hyper-reactivity phenotype in COVID-19, especially in the lungs, contributes to severe hypercoagulability and inflammation [4,97]. Key proinflammatory interleukins in COVID-19, IL-6, and IL-1, also interact with the platelet-thrombosis pathway. IL-6 induces platelet reactivity and high fibrinogen linking inflammation and thrombosis [26]. Platelet number and their degranulation activity are associated with IL-1β plasma concentration [98]. In COVID-19 patients, blood platelets showed upregulated receptors, contributing to aggregation and activation [4]. Upon activation, granules contained proteins with hemostatic functions are transfer to platelet membrane and release to extracellular space to further promote platelet adhesion and activation [99]. The lungs autopsy of COVID-19 patients showed upregulated platelet expression of PF4 (CXCL4), which promotes blood coagulation and activates the NF-κB signaling pathway in ECs, acting as a potential agent of both thrombosis and inflammation [4,100]. Anti-PF4 antibodies may be involved in the pathophysiology of severe clinical complications of COVID-19 [101]. Significant higher serum levels of other platelet activation markers involved in cell adhesion like LAMP-3, as well as transmembrane proteins from GPIIb/GPIIIa complex, vWF receptor units (GPIbα, GPIX), CD9, and CD40 (TNFRSF5, a transmembrane TNF superfamily receptor) were recorded in patients with SARS-CoV-2 infection [102]. However, the lungs autopsy of COVID-19 patients showed that platelet expression of some receptors like CD40L, CD42b (vWF receptor units, GPIbα), and CXCR4 were comparable to the control group [4]. CD40L and CXCR4 are involved in cell signaling in innate, adaptive immunity and thrombosis, mediating the acute lung injury (ALI) [99,103,104,105]. According to the same study, surface adhesion molecule expression like ICAM-1, VCAM-1, and E-selectin, was also reduced in lung ECs, suggesting that other mechanisms would be involved in the pulmonary infiltration of immune cells [4].
3.2.2. vWF
vWF is a multimeric glycoprotein procoagulant molecule synthesized by ECs and stored in lysosome related organelles like WPBs [106,107]. SARS-CoV-2-mediated ECs activation and damage in the pulmonary vascular bed by pro-inflammatory cytokines, complement activation, NETosis, and hypoxia increase local vWF release from WPBs [8]. ECs in the lungs of COVID-19 patients exhibit increased of both hypoxia-inducible factor 1-alpha (HIF-1α) and glucose transporter protein type 1 (GLUT1) expression, confirming local hypoxic stress as a potential trigger for this pathogenic process [4]. The lung’s autopsy of COVID-19 patients suggests increased vWF expression not only from ECs, but also by activated platelets, as it was detected in the outer layer of the thrombus and CD31-expressing vascular endothelium tissue samples [4]. vWF plays a critical role in thromboinflammation, with NFκB2 mediated vWF transcription as a potential direct biological link between immunity and thrombosis [8,108,109]. By using the TLR2-dependent activation of the NF-κB pathway in a MyD88-dependent manner, SARS-CoV-2 S protein induces inflammation and promotes vWF transcription, causing in situ thrombosis, as vWF binds to platelets, neutrophils, and monocytes [110,111,112]. ADAMTS13 functional metalloprotease cleaves newly released highly active ultra large vWF (UL-vWF) multimers into smaller, less thrombogenic and inflammatory fragments [8,106,108]. In COVID-19, the ADAMTS13/vWF ratio decreases, leading to an excess of overactive UL-vWF multimers as a key driver of micro-thromboses in the pulmonary vasculature and SARS-CoV-2-associated ARDS [8,92,108,113,114,115,116]. Plasma exchange could be a therapeutic option for critically ill COVID-19 patients in order to restore the disbalance in the ADAMTS13-vWF axis [113,114].
3.2.3. Thrombomodulin and P-selectin
Pulmonary vascular thrombosis is prevented by the ECs monolayer that expresses surface non-soluble thrombomodulin (nsTM), a type I transmembrane glycoprotein with important anticoagulant activity, secondary to a protein C-mediated pathway and a direct modulation of thrombin’s procoagulant effects [112,117]. Thrombomodulin has also cytoprotective and anti-inflammatory properties, as it blocks the TLR-4-specific ligands axis, inhibits nuclear translocation of NF-κB and induces direct mediated anti-inflammatory signals mediated by the endothelial protein C receptor (EPCR)-protease-activated receptor 1 (PAR1) system [117]. A dysfunctional prothrombotic phenotype of vascular ECs in the lungs of COVID-19 patients is characterized by a decreased expression of nsTM and other anticoagulants like EPCR [4]. Decreased nsTM expression in ECs is associated with increased immune cell infiltration in the lung of patients with SARS-CoV-2 infection [4]. Moreover, dysregulated pro-inflammatory cytokine generation by SARS-CoV-2 in the pulmonary microvasculature releases soluble thrombomodulin (sTM) from ECs surfaces that will further promote a procoagulant and pro-inflammatory local milieu [112]. Administration of a human recombinant thrombomodulin in sepsis-induced coagulopathy could have some favorable effects in terms of patient’s outcome [118]. Endothelial thrombomodulin expression could also be a potential therapeutic target for COVID-19-related immunothrombosis [4].
P-selectin, another biomarker of endothelial degranulation, blocks the initial attachment and rolling of platelets and leukocytes to inflammatory regions [14]. In COVID-19 patients, P-selectin could be a biomarker of severe disease and associated venous thrombosis [14,80,116,119,120,121]. P-selectin might also be detrimental in COVID-19 as it promotes NETosis through binding to P-selectin glycoprotein ligand-1 (PSGL-1), leading to the development of immuno-thrombosis and ALI/ARDS [48,122]. P-selectin could also be a marker for platelet and endothelial activation even weeks, up to one year after COVID-19 [123,124]. In one study, P-selectin level was increased in the hearts but not in the lungs of COVID-19 patient autopsies [4]. Crizanlizumab, a soluble P-selectin inhibitor, might increase endogenous thrombolysis in COVID-19 [119].
3.2.4. Intrinsic and extrinsic coagulation pathways
Coagulation pathways–inflammation–thrombosis axis could significantly contribute to the induction of COVID-19 coagulopathy. Human bronchial epithelial cells infected with SARS-CoV-2 showed upregulation of TF, the master regulator responsible for the initiation of the extrinsic coagulation pathway [125]. Cellular and humoral immune dysfunction, complement activation, IL6, and IL-1β-NETs axis also contributes to upregulation of TF and immunothrombotic events [126]. The disbalance between TF and endogenously encoded inhibitors, such as TF pathway inhibitor and protein S-protein C complex, further exacerbates the COVID-19–related thrombotic pathology [125]. Moreover, the genes for protein PAI-2 (plasminogen activator inhibitor-2), which inhibits urokinase and tissue plasminogen activator, are significantly expressed in human bronchial epithelial cells infected with SARS-CoV-2 and this contributes to pulmonary thromboses and distal coagulopathies [125]. Severe COVID-19 and pulmonary thrombosis are associated with elevated factor V and factor VIII activity [127,128]. Blood-clotting protein factor V is produced by circulating neutrophils, T-cells, and monocytes, being also expressed by lung-infiltrating leukocytes in patients with fatal SARS-CoV-2 infection [128]. The intrinsic pathway of coagulation is part of the innate immune system as inflammatory response mechanism against SARS-CoV-2; it is also involved in immunothrombosis, being activated especially by pro-inflammatory cytokines like IL-6, IL-1 and TNF-alpha [91,129].
3.2.5. Fibrinolytic disbalance and the central role of PAI-1
The altered fibrinolytic balance promotes the development of a hypercoagulable state that will lead to microvascular thrombosis [26]. In patients with COVID-19, impaired fibrinolysis is secondary to high plasminogen activator inhibitor 1 (PAI-1) levels, a member of the serine protease inhibitor superfamily that blocks the conversion of precursor plasminogen to active plasmin [15,130,131,132]. PAI-1 can also bind to TLR4 on macrophages and stimulates the secretion of proinflammatory cytokines and chemokines, being at the crossroads of hemostatic and inflammatory pathways [133,134]. SARS-CoV-2 S protein stimulates the production of PAI-1 by pulmonary microvascular ECs using a proposed mechanism involving the disbalance in zinc metallopeptidase STE24-ACE2 axis [130]. Inflammatory cytokines (IL-1, IL-6, IL-17A) and subsequent type II alveolar cells injury results in decreased surfactant and induction of the p53 pathway that also upregulates PAI-1 and changes the fibrinolytic balance in the lungs [135]. Higher PAI-1 levels are linked with severe COVID-19, as it is involved in ECs dysfunction and lung injury by pulmonary fibrin accumulation/hyaline membrane formation and promotes local hypoxia [130,131,132,136,137,138,139]. Diffuse alveolar damage and increased hyaluronan production leads to even higher levels of PAI-1 [133].
3.2.6. SARS-CoV-2-RBCs axis
SARS-CoV-2 can infect RBCs by attaching to Band-3 protein from the erythrocyte membrane [40]. This interaction is followed by alterations in CO2 uptake and oxygen release from hemoglobin, inducing thrombosis by tissue hypoxia [40]. Increased plasma levels of IL-1β, IL-6, and IL-8 in patients with COVID-19, has also a negative effect on the RBCs ultrastructure and induces signs of eryptosis, a form of suicidal death of RBCs, that exhibits an increased tendency of adhering to ECs as well as platelets, contributing to thrombosis [90]. Moreover, COVID-19 patients’ D-dimers are correlated with RBC surface phosphatidylserine (RBC-PS), another potential contribution of RBCs in the thrombotic diathesis [140]. Induced ECs dysfunction, and the presence of surface binding proteins for IL-8 and Duffy antigen receptor for chemokines on their surface [90], also link the RBCs with the immuno-stimulatory pathways and the crossroads of inflammation and thrombosis.
4. Chest CT imaging data supporting the role of pulmonary immunothrombosis
Diffuse, segmental/subsegmental thrombosis is predominant in COVID-19 patients [30,31,141,142,143,144]. As small peripheral lung thrombi are more prevalent in COVID-19, evaluating the subsegmental pulmonary arteries by computed tomography pulmonary angiogram is essential [30,145,146]. In their study, Cau et al. found that in 87% of cases, pulmonary thrombosis was in lung parenchyma affected by COVID-19 pneumonia [147]. Patients with COVID-19 pneumonia and associated pulmonary thrombosis had significantly worse segmental opacifications in CT imaging and all thrombi were located in segments with inflammation [5]. Percentage of ground-glass opacities, consolidation, and crazy paving in the lobes with thrombosis was higher than in the contralateral pulmonary lobes without pulmonary thrombosis [148]. In another paper, the distribution of thrombosis correlated with the pattern of consolidation and in 93% of cases it involved peripheral or subsegmental arteries [149]. Patients with pulmonary artery thrombosis have significantly higher inflammatory lesions on chest CT analysis compared to those without thrombosis, especially in the lower lobes of the lungs, where blood clots predominate [5,144,150]. When analysing quantitative chest CT data in patients with SARS-CoV-2 pneumonia and pulmonary thrombosis, more than half of them had more than 50% COVID-19-associated lung involvement, which is also associated with a higher mortality [144,151,152].
5. The role of venous thromboembolism
In non-ICU hospitalized COVID-19 patients who underwent systematic DVT screening, when pulmonary artery occlusion appeared, this was caused rather by in situ pulmonary thrombosis than by emboli from peripheral vein thrombi [153]. In a prospective multicenter cohort study including 1240 hospitalised COVID-19 patients who performed a CT pulmonary angiography, only 11.7% of 103 patients diagnosed with pulmonary thrombosis had DVT [154]. DVT was found in 59 patients (39.3%) when Doppler ultrasound was used systematically for evaluation of the lower extremities during hospital admission for COVID-19 patients [155]. In another study, none of the patients with pulmonary thrombosis had clinical evidence of DVT [149]. More than half of patients with pulmonary thrombosis lacked signs of DVT [6,7]. Not only clinical, but also histopathological data on COVID-19 patients showed the lack of evidence for venous occlusion of the lower limbs and vessels of the pelvis [12]. Pulmonary thrombosis cases occurred in the absence of a recognizable DVT, in two-thirds of cases, most of them involving the distal vessels [156]. Peripheral pulmonary blood clots, predominant in COVID-19 as showed earlier, reflect in situ thrombosis rather than DVT embolism, which is associated with a more centrally located clot burden [146,157]. The presence of DTV at a distal level (below knee) could be an indicator for in situ thrombosis (COVID-19 thrombotic phenotype) and at a proximal level (DTV above knee), especially if associate pulmonary central clots (in a main/lobar artery), could suggest an embolic origin [146]. Central pulmonary emboli are also related to lung cancer, and these patients usually have other associated risk factors for venous thromboembolic disease and worse outcomes [158]. Proximal asymptomatic DVT is also independently associated with active cancer [159]. Therefore, it is possible that some of the patients with DVT-associated pulmonary embolism, could also be more prone for venous thromboembolic events. In patients with known COVID-19 associated pulmonary thrombosis, there is a lower incidence of DVT compared to non-COVID-19 patients (6.9%-13.6%, vs. 45%-70%), as patients with SARS-CoV-2 infection often lack traditional risk factors and comorbidities for venous thromboembolic disease [145]. Venous thromboembolism could also play a role in pulmonary artery thrombosis in some cases since COVID-19 is a known risk factor for both DVT and associated pulmonary embolism [158,160,161]. In a study on 81 ICU patients with severe COVID-19 pneumonia and lower limb venous doppler ultrasound evaluation, venous thrombotic disease was reported in 25% of cases [162]. However, considering the hypercoagulable state of COVID-19 patients and the possibility of co-thrombotic events [163,164], it remains unclear if pulmonary artery thrombosis is a concurrent rather than a sequential event in patients with DVT, as specific data is missing. High rates of asymptomatic distal DVT reported in prolonged hospitalised patients with COVID-19 and no differences in prevalence of pulmonary thrombosis between patients with and without DVT could support this theory [165].
6. The failure of anticoagulation treatment in immunothrombosis
The failure of anticoagulation treatment in thrombotic diseases is very uncommon, but it is recognized as a problem especially among some patients with coexistent cancer [166,167,168]. The effect of anticoagulation in preventing pulmonary immunothombosis is less known in comparison with other kind of thrombotic events [169]. COVID-19 associated thrombotic complications could developed despite standard prophylactic anticoagulation, suggesting that a therapeutic anticoagulation or alternative anti-thrombotic agents could have much better results [8,16,170,171]. The failure of anticoagulation also for preventing pulmonary artery thrombosis was reported by some clinical studies on COVID-19 patients [30,144,165,172,173]. In patients with severe COVID-19, the poor response to systemic anticoagulation is supported not only by clinical studies, but also by histological examination of the lung tissue, which described a diffuse arterial thrombosis was described in the lungs secondary to vascular inflammatory, prothrombotic changes and hypoperfusion [174]. Moreover, the lungs autopsy of COVID-19 patients also showed no differences in the prevalence of microthombosis according to anticoagulation doses [169]. The use of intermediate instead of standard doses of prophylactic anticoagulants did not result in lower incidence of thrombosis or mortality in critically ill COVID-19 patients [175]. Pulmonary artery thrombosis and DVT reported in hospitalised patients with COVID-19 could develop despite prophylactic, intermediate and therapeutic doses of anticoagulation [30,144,165]. In most studies, LMWHs were used for prophylaxis of thrombosis. Because NETs, key molecules in immunothrombosis, are positively charged, unfractionated heparin may be a more effective option for SARS-CoV-2 associated pulmonary thrombosis [47]. Heparin also replaces molecules like histones from the chromatin backbone of NETs and destroys their stability [47]. According to current guidelines, therapeutic anticoagulation is now recommended for all non-ICU hospitalised COVID-19 patients with supplemental oxygen need regardless the presence of pulmonary artery thrombosis [176]. An extensive discussion about anticoagulation strategy and other pharmacological agents is beyond the scope of this review. A combination of therapeutic strategies to prevent thrombotic events in COVID-19, beyond traditional anticoagulation alone strategies, are needed [16].
7. Conclusions
COVID-19 associated pulmonary thrombosis is a complex pathology, being the consequence of a complex interplay between the inflammatory response, endothelium and coagulation system that leads to a systemic and also local, lung-associated, procoagulant state. Immunothrombosis is the main pathophysiological result, which significantly contributes to COVID-19 associated pulmonary thrombosis, being supported by clinical, molecular mechanisms, pathological studies and imaging data. The high incidence of thrombotic events despite routine thromboprophylaxis in severe disease, further supports this pathway. Venous thromboembolic disease could also play a role, especially in patients with other significant risk factor for thrombosis. However, considering the hypercoagulable state of COVID-19 patients and the possibility of co-thrombotic events, it remains unclear if pulmonary thrombosis is a concurrent or a sequential event in patients with DVT, as specific data is scarce and requires further studies. Understanding the mechanisms behind COVID-19-induced immunothrombosis will lead to future combination therapies for hospitalised patients with severe disease, that would target the crossroads of inflammatory and coagulation pathways.
Author Contributions
Conceptualization, C.M.N. and A.H; Methodology, C.M.N and A.H.; Writing-original draft preparation, C.M.N. and R.M.; Writing-review and editing, C.M.N., A.H. and R.M.; Image editing/visualization, R.M.; Supervision, A.H. All authors have read and agreed to the published version of the manuscript;
Funding
This research received no external founding.
Institutional Review Board Statement
Not applicable;
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Manolis, A.S., et al., COVID-19 Infection: Viral Macro- and Micro-Vascular Coagulopathy and Thromboembolism/Prophylactic and Therapeutic Management. J Cardiovasc Pharmacol Ther, 2021. 26(1): p. 12-24. [CrossRef]
- Loo, J., D.A. Spittle, and M. Newnham, COVID-19, immunothrombosis and venous thromboembolism: biological mechanisms. Thorax, 2021. 76(4): p. 412-420. [CrossRef]
- Tang, N., et al., Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost, 2020. 18(4): p. 844-847. [CrossRef]
- Won, T., et al., Endothelial thrombomodulin downregulation caused by hypoxia contributes to severe infiltration and coagulopathy in COVID-19 patient lungs. eBioMedicine, 2022. 75: p. 103812. [CrossRef]
- Mueller-Peltzer, K., et al., Pulmonary artery thrombi are co-located with opacifications in SARS-CoV2 induced ARDS. Respir Med, 2020. 172: p. 106135.
- Oba, S., et al., Arterial and Venous Thrombosis Complicated in COVID-19: A Retrospective Single Center Analysis in Japan. Front Cardiovasc Med, 2021. 8: p. 767074. [CrossRef]
- Suh, Y.J., et al., Pulmonary Embolism and Deep Vein Thrombosis in COVID-19: A Systematic Review and Meta-Analysis. Radiology, 2021. 298(2): p. E70-e80. [CrossRef]
- Portier, I., R.A. Campbell, and F. Denorme, Mechanisms of immunothrombosis in COVID-19. Curr Opin Hematol, 2021. 28(6): p. 445-453. [CrossRef]
- Conway, E.M., et al., Understanding COVID-19-associated coagulopathy. Nat Rev Immunol, 2022. 22(10): p. 639-649. [CrossRef]
- Payus, A.O., C.L.S. Lin, and A. Ibrahim, The poorly understood yet potent risk of pulmonary artery thrombosis in-situ in Post-Acute COVID-19 syndrome. J Cardiothorac Surg, 2023. 18(1): p. 42. [CrossRef]
- Cuevas Vilaplana, A., I. Roldan Torres, and J. Vizuete Del Rio, [Myocarditis and in situ thrombosis in the right ventricle in a COVID-19 patient]. Hipertens Riesgo Vasc, 2021. 38(3): p. 148-150. [CrossRef]
- Khismatullin, R.R., et al., Pathology of lung-specific thrombosis and inflammation in COVID-19. J Thromb Haemost, 2021. 19(12): p. 3062-3072. [CrossRef]
- Quartuccio, L., et al., Clinical, laboratory and immunohistochemical characterization of in situ pulmonary arterial thrombosis in fatal COVID-19. Thromb Res, 2022. 219: p. 95-101. [CrossRef]
- Agrati, C., et al., The Role of P-Selectin in COVID-19 Coagulopathy: An Updated Review. Int J Mol Sci, 2021. 22(15). [CrossRef]
- Lamers, M.M. and B.L. Haagmans, SARS-CoV-2 pathogenesis. Nature Reviews Microbiology, 2022. 20(5): p. 270-284. [CrossRef]
- Lim, M.S. and S. McRae, COVID-19 and immunothrombosis: Pathophysiology and therapeutic implications. Crit Rev Oncol Hematol, 2021. 168: p. 103529. [CrossRef]
- Thachil, J. and A. Srivastava, SARS-2 Coronavirus-Associated Hemostatic Lung Abnormality in COVID-19: Is It Pulmonary Thrombosis or Pulmonary Embolism? Semin Thromb Hemost, 2020. 46(7): p. 777-780. [CrossRef]
- Morrell, C.N., et al., Emerging roles for platelets as immune and inflammatory cells. Blood, 2014. 123(18): p. 2759-67. [CrossRef]
- Wang, L., et al., In situ pulmonary thrombosis in patients with COVID-19 pneumonia: different phenotypes may exist. Thromb Res, 2020. 196: p. 541-542. [CrossRef]
- Martín Giménez, V.M., et al., Lungs as target of COVID-19 infection: Protective common molecular mechanisms of vitamin D and melatonin as a new potential synergistic treatment. Life Sci, 2020. 254: p. 117808. [CrossRef]
- Calabrese, F., et al., Pulmonary pathology and COVID-19: lessons from autopsy. The experience of European Pulmonary Pathologists. Virchows Arch, 2020. 477(3): p. 359-372. [CrossRef]
- Ackermann, M., et al., Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med, 2020. 383(2): p. 120-128. [CrossRef]
- Menter, T., et al., Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology, 2020. 77(2): p. 198-209. [CrossRef]
- Carsana, L., et al., Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis, 2020. 20(10): p. 1135-1140. [CrossRef]
- Duarte-Neto, A.N., et al., Pulmonary and systemic involvement in COVID-19 patients assessed with ultrasound-guided minimally invasive autopsy. Histopathology, 2020. 77(2): p. 186-197. [CrossRef]
- Cacciola, R., et al., Cellular and molecular mechanisms in COVID-19 coagulopathy: role of inflammation and endotheliopathy. J Thromb Thrombolysis, 2022. 53(2): p. 282-290. [CrossRef]
- Golubeva, M.G., Role of P-Selectin in the Development of Hemostasis Disorders in COVID-19. Biology Bulletin Reviews, 2022. 12(4): p. 406-413. [CrossRef]
- Vivan, M.A., et al., Pulmonary embolism in patients with COVID-19 and D-dimer diagnostic value: A retrospective study. Braz J Infect Dis, 2022. 26(6): p. 102702. [CrossRef]
- Močibob, L., et al., COVID-19 and Pulmonary Thrombosis-An Unresolved Clinical Puzzle: A Single-Center Cohort Study. J Clin Med, 2022. 11(23). [CrossRef]
- Jalde, F.C., et al., Widespread Parenchymal Abnormalities and Pulmonary Embolism on Contrast-Enhanced CT Predict Disease Severity and Mortality in Hospitalized COVID-19 Patients. Front Med (Lausanne), 2021. 8: p. 666723. [CrossRef]
- Masselli, G., et al., Role of CT angiography in detecting acute pulmonary embolism associated with COVID-19 pneumonia. Radiol Med, 2021. 126(12): p. 1553-1560. [CrossRef]
- Smilowitz, N.R., et al., C-reactive protein and clinical outcomes in patients with COVID-19. Eur Heart J, 2021. 42(23): p. 2270-2279. [CrossRef]
- Sultana, G.N.N., et al., Studying C-reactive protein and D-dimer levels in blood may prevent severe complications: A study in Bangladeshi COVID-19 patients. Front Genet, 2022. 13: p. 966595. [CrossRef]
- Gorog, D.A., et al., Current and novel biomarkers of thrombotic risk in COVID-19: a Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nature Reviews Cardiology, 2022. 19(7): p. 475-495. [CrossRef]
- Lucijanić, M., et al., Clinical and prognostic significance of C-reactive protein to albumin ratio in hospitalized coronavirus disease 2019 (COVID-19) patients. Wiener klinische Wochenschrift, 2022. 134(9): p. 377-384. [CrossRef]
- Fay, W.P., Linking inflammation and thrombosis: Role of C-reactive protein. World J Cardiol, 2010. 2(11): p. 365-9. [CrossRef]
- Galland, J., et al., White blood count, D-dimers, and ferritin levels as predictive factors of pulmonary embolism suspected upon admission in noncritically ill COVID-19 patients: The French multicenter CLOTVID retrospective study. Eur J Haematol, 2021. 107(2): p. 190-201. [CrossRef]
- Kaushal, K., et al., Serum ferritin as a predictive biomarker in COVID-19. A systematic review, meta-analysis and meta-regression analysis. J Crit Care, 2022. 67: p. 172-181. [CrossRef]
- Wu, R., et al., Inflammasome-Dependent Coagulation Activation in Sepsis. Front Immunol, 2021. 12: p. 641750. [CrossRef]
- Tuculeanu, G., et al., Coagulation Disorders in Sepsis and COVID-19—Two Sides of the Same Coin? A Review of Inflammation–Coagulation Crosstalk in Bacterial Sepsis and COVID-19. Journal of Clinical Medicine, 2023. 12(2): p. 601. [CrossRef]
- Delorey, T.M., et al., COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature, 2021. 595(7865): p. 107-113. [CrossRef]
- Savla, S.R., K.S. Prabhavalkar, and L.K. Bhatt, Cytokine storm associated coagulation complications in COVID-19 patients: Pathogenesis and Management. Expert Rev Anti Infect Ther, 2021. 19(11): p. 1397-1413. [CrossRef]
- Bain, C.C., C.D. Lucas, and A.G. Rossi, Pulmonary macrophages and SARS-Cov2 infection. Int Rev Cell Mol Biol, 2022. 367: p. 1-28. [CrossRef]
- Faggioli, P.M., N. Mumoli, and A. Mazzone, Iloprost in COVID-19: The Rationale of Therapeutic Benefit. Front Cardiovasc Med, 2021. 8: p. 649499. [CrossRef]
- Hottz, E.D., et al., Platelet-monocyte interaction amplifies thromboinflammation through tissue factor signaling in COVID-19. Blood Adv, 2022. 6(17): p. 5085-5099. [CrossRef]
- Szturmowicz, M. and U. Demkow, Neutrophil Extracellular Traps (NETs) in Severe SARS-CoV-2 Lung Disease. Int J Mol Sci, 2021. 22(16). [CrossRef]
- Zhou, Y., et al., The Emerging Role of Neutrophil Extracellular Traps in Arterial, Venous and Cancer-Associated Thrombosis. Front Cardiovasc Med, 2021. 8: p. 786387. [CrossRef]
- Al-Kuraishy, H.M., et al., Neutrophil Extracellular Traps (NETs) and Covid-19: A new frontiers for therapeutic modality. Int Immunopharmacol, 2022. 104: p. 108516. [CrossRef]
- Middleton, E.A., et al., Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood, 2020. 136(10): p. 1169-1179. [CrossRef]
- Ackermann, M., et al., Patients with COVID-19: in the dark-NETs of neutrophils. Cell Death & Differentiation, 2021. 28(11): p. 3125-3139. [CrossRef]
- Pastorek, M., M. Dúbrava, and P. Celec, On the Origin of Neutrophil Extracellular Traps in COVID-19. Front Immunol, 2022. 13: p. 821007. [CrossRef]
- Teluguakula, N., Neutrophils Set Extracellular Traps to Injure Lungs in Coronavirus Disease 2019. J Infect Dis, 2021. 223(9): p. 1503-1505. [CrossRef]
- Ouwendijk, W.J.D., et al., High Levels of Neutrophil Extracellular Traps Persist in the Lower Respiratory Tract of Critically Ill Patients With Coronavirus Disease 2019. J Infect Dis, 2021. 223(9): p. 1512-1521. [CrossRef]
- Sung, P.-S., et al., CLEC5A and TLR2 are critical in SARS-CoV-2-induced NET formation and lung inflammation. Journal of Biomedical Science, 2022. 29(1): p. 52. [CrossRef]
- Lam, H.Y., et al., Mast cells: Therapeutic targets for COVID-19 and beyond. IUBMB Life, 2021. 73(11): p. 1278-1292. [CrossRef]
- Galli, S.J., N. Gaudenzio, and M. Tsai, Mast Cells in Inflammation and Disease: Recent Progress and Ongoing Concerns. Annu Rev Immunol, 2020. 38: p. 49-77. [CrossRef]
- Cildir, G., et al., The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J Exp Med, 2017. 214(9): p. 2491-2506. [CrossRef]
- Theoharides, T.C., P. Valent, and C. Akin, Mast Cells, Mastocytosis, and Related Disorders. New England Journal of Medicine, 2015. 373(2): p. 163-172. [CrossRef]
- Gebremeskel, S., et al., Mast Cell and Eosinophil Activation Are Associated With COVID-19 and TLR-Mediated Viral Inflammation: Implications for an Anti-Siglec-8 Antibody. Front Immunol, 2021. 12: p. 650331. [CrossRef]
- Marshall, J.S., L. Portales-Cervantes, and E. Leong, Mast Cell Responses to Viruses and Pathogen Products. Int J Mol Sci, 2019. 20(17). [CrossRef]
- Budnevsky, A.V., et al., Role of mast cells in the pathogenesis of severe lung damage in COVID-19 patients. Respir Res, 2022. 23(1): p. 371. [CrossRef]
- Conti, P., et al., Mast cells activated by SARS-CoV-2 release histamine which increases IL-1 levels causing cytokine storm and inflammatory reaction in COVID-19. J Biol Regul Homeost Agents, 2020. 34(5): p. 1629-1632. [CrossRef]
- Conti, P., et al., IL-1 induces throboxane-A2 (TxA2) in COVID-19 causing inflammation and micro-thrombi: inhibitory effect of the IL-1 receptor antagonist (IL-1Ra). J Biol Regul Homeost Agents, 2020. 34(5): p. 1623-1627. [CrossRef]
- Hoffmann, M., et al., SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell, 2020. 181(2): p. 271-280.e8. [CrossRef]
- Jackson, C.B., et al., Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol, 2022. 23(1): p. 3-20. [CrossRef]
- Wismans, L.V., et al., Increase of mast cells in COVID-19 pneumonia may contribute to pulmonary fibrosis and thrombosis. Histopathology, 2023. 82(3): p. 407-419. [CrossRef]
- Wu, M.L., et al., SARS-CoV-2-triggered mast cell rapid degranulation induces alveolar epithelial inflammation and lung injury. Signal Transduct Target Ther, 2021. 6(1): p. 428. [CrossRef]
- Liu, S., et al., Mast cells promote viral entry of SARS-CoV-2 via formation of chymase/spike protein complex. Eur J Pharmacol, 2022. 930: p. 175169. [CrossRef]
- Krysko, O., et al., Severity of SARS-CoV-2 infection is associated with high numbers of alveolar mast cells and their degranulation. Front Immunol, 2022. 13: p. 968981. [CrossRef]
- Motta Junior, J.D.S., et al., Mast Cells in Alveolar Septa of COVID-19 Patients: A Pathogenic Pathway That May Link Interstitial Edema to Immunothrombosis. Front Immunol, 2020. 11: p. 574862. [CrossRef]
- Gasparello, J., et al., In vitro induction of interleukin-8 by SARS-CoV-2 Spike protein is inhibited in bronchial epithelial IB3-1 cells by a miR-93-5p agomiR. Int Immunopharmacol, 2021. 101(Pt B): p. 108201. [CrossRef]
- Halova, I., L. Draberova, and P. Draber, Mast cell chemotaxis - chemoattractants and signaling pathways. Front Immunol, 2012. 3: p. 119. [CrossRef]
- Costela-Ruiz, V.J., et al., SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev, 2020. 54: p. 62-75. [CrossRef]
- Hafezi, B., et al., Cytokine Storm Syndrome in SARS-CoV-2 Infections: A Functional Role of Mast Cells. Cells, 2021. 10(7). [CrossRef]
- Theoharides, T.C., Potential association of mast cells with coronavirus disease 2019. Ann Allergy Asthma Immunol, 2021. 126(3): p. 217-218. [CrossRef]
- Setyo Nugroho, G.M., et al., Interleukin-6 (IL-6) expression of lung tissue in COVID-19 patient severity through core biopsy post mortem. Ann Med Surg (Lond), 2022. 82: p. 104648. [CrossRef]
- Harapan, H., et al., The prevalence, predictors and outcomes of acute liver injury among patients with COVID-19: A systematic review and meta-analysis. Rev Med Virol, 2022. 32(3): p. e2304. [CrossRef]
- Colafrancesco, S., et al., Targeting the Immune System for Pulmonary Inflammation and Cardiovascular Complications in COVID-19 Patients. Front Immunol, 2020. 11: p. 1439. [CrossRef]
- Rad, F., et al., The Relationship between Inflammatory Cytokines and Coagulopathy in Patients with COVID-19. J Clin Med, 2021. 10(9). [CrossRef]
- Abd El-Ghani, S.E.-S., et al., Serum interleukin 1β and sP-selectin as biomarkers of inflammation and thrombosis, could they be predictors of disease severity in COVID 19 Egyptian patients? (a cross-sectional study). Thrombosis Journal, 2022. 20(1): p. 77. [CrossRef]
- Gianni, P., et al., Complement-mediated microvascular injury and thrombosis in the pathogenesis of severe COVID-19: A review. World J Exp Med, 2022. 12(4): p. 53-67. [CrossRef]
- Ali, Y.M., et al., Lectin Pathway Mediates Complement Activation by SARS-CoV-2 Proteins. Front Immunol, 2021. 12: p. 714511. [CrossRef]
- Boussier, J., et al., Severe COVID-19 is associated with hyperactivation of the alternative complement pathway. J Allergy Clin Immunol, 2022. 149(2): p. 550-556.e2. [CrossRef]
- Niederreiter, J., et al., Complement Activation via the Lectin and Alternative Pathway in Patients With Severe COVID-19. Frontiers in Immunology, 2022. 13. [CrossRef]
- Foley, J.H., et al., Complement Activation in Arterial and Venous Thrombosis is Mediated by Plasmin. EBioMedicine, 2016. 5: p. 175-82. [CrossRef]
- Tomo, S., et al., Complement activation and coagulopathy - an ominous duo in COVID19. Expert Rev Hematol, 2021. 14(2): p. 155-173 . [CrossRef]
- Magro, C., et al., Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res, 2020. 220: p. 1-13. [CrossRef]
- Holter, J.C., et al., Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci U S A, 2020. 117(40): p. 25018-25025. [CrossRef]
- Peffault de Latour, R., et al., Complement C5 inhibition in patients with COVID-19 - a promising target? Haematologica, 2020. 105(12): p. 2847-2850. [CrossRef]
- Soma, P. and J. Bester, Pathophysiological Changes in Erythrocytes Contributing to Complications of Inflammation and Coagulation in COVID-19. Front Physiol, 2022. 13: p. 899629. [CrossRef]
- de Andrade, S.A., et al., Pathophysiology of COVID-19: Critical Role of Hemostasis. Front Cell Infect Microbiol, 2022. 12: p. 896972. [CrossRef]
- Goshua, G., et al., Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol, 2020. 7(8): p. e575-e582. [CrossRef]
- Varga, Z., et al., Endothelial cell infection and endotheliitis in COVID-19. Lancet, 2020. 395(10234): p. 1417-1418. [CrossRef]
- Tafazoli, A., S. Anil Kumar, and M. Othman, Thrombocytopathy vs Platelet hyper-reactivity in COVID-19: diverse pathologies, disease outcomes and therapeutic implications. Platelets, 2022. 33(1): p. 48-53. [CrossRef]
- Guo, L. and M.T. Rondina, The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells During Infectious Diseases. Front Immunol, 2019. 10: p. 2204. [CrossRef]
- Sharma, S., T. Tyagi, and S. Antoniak, Platelet in thrombo-inflammation: Unraveling new therapeutic targets. Front Immunol, 2022. 13: p. 1039843. [CrossRef]
- Comer, S.P., et al., COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol, 2021. 19(2): p. e3001109. [CrossRef]
- Tunjungputri, R.N., et al., The Inter-Relationship of Platelets with Interleukin-1β-Mediated Inflammation in Humans. Thromb Haemost, 2018. 118(12): p. 2112-2125. [CrossRef]
- Fard, M.B., et al., Thrombosis in COVID-19 infection: Role of platelet activation-mediated immunity. Thrombosis Journal, 2021. 19(1): p. 59. [CrossRef]
- Le, H.T., et al., Platelet factor 4 (CXCL4/PF4) upregulates matrix metalloproteinase-2 (MMP-2) in gingival fibroblasts. Scientific Reports, 2022. 12(1): p. 18636. [CrossRef]
- Liu, Q., et al., Anti-PF4 antibodies associated with disease severity in COVID-19. Proc Natl Acad Sci U S A, 2022. 119(47): p. e2213361119. [CrossRef]
- Bongiovanni, D., et al., SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis, 2021. 12(1): p. 50. [CrossRef]
- Aloui, C., et al., The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int J Mol Sci, 2014. 15(12): p. 22342-64. [CrossRef]
- Tian, X., et al., CXCR4 knockdown prevents inflammatory cytokine expression in macrophages by suppressing activation of MAPK and NF-κB signaling pathways. Cell & Bioscience, 2019. 9(1): p. 55. [CrossRef]
- Gavins, F.N., et al., Microvascular thrombosis and CD40/CD40L signaling. J Thromb Haemost, 2011. 9(3): p. 574-81. [CrossRef]
- Kremer Hovinga, J.A. and J.N. George, Hereditary Thrombotic Thrombocytopenic Purpura. New England Journal of Medicine, 2019. 381(17): p. 1653-1662. [CrossRef]
- Naß, J., J. Terglane, and V. Gerke, Weibel Palade Bodies: Unique Secretory Organelles of Endothelial Cells that Control Blood Vessel Homeostasis. Front Cell Dev Biol, 2021. 9: p. 813995. [CrossRef]
- Mei, Z.W., et al., Role of von Willebrand Factor in COVID-19 Associated Coagulopathy. J Appl Lab Med, 2021. 6(5): p. 1305-1315. [CrossRef]
- Manz, X.D., et al., Epigenetic Modification of the von Willebrand Factor Promoter Drives Platelet Aggregation on the Pulmonary Endothelium in Chronic Thromboembolic Pulmonary Hypertension. Am J Respir Crit Care Med, 2022. 205(7): p. 806-818. [CrossRef]
- Manz, X.D., H.J. Bogaard, and J. Aman, Regulation of VWF (Von Willebrand Factor) in Inflammatory Thrombosis. Arterioscler Thromb Vasc Biol, 2022. 42(11): p. 1307-1320. [CrossRef]
- Khan, S., et al., SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. Elife, 2021. 10.
- O’Sullivan, J.M., et al., Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol, 2020. 7(8): p. e553-e555. [CrossRef]
- Seibert, F.S., et al., Effect of plasma exchange on COVID-19 associated excess of von Willebrand factor and inflammation in critically ill patients. Sci Rep, 2022. 12(1): p. 4801. [CrossRef]
- Doevelaar, A.A.N., et al., von Willebrand Factor Multimer Formation Contributes to Immunothrombosis in Coronavirus Disease 2019. Crit Care Med, 2021. 49(5): p. e512-e520. [CrossRef]
- Mancini, I., et al., The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J Thromb Haemost, 2021. 19(2): p. 513-521. [CrossRef]
- Fenyves, B.G., et al., Plasma P-selectin is an early marker of thromboembolism in COVID-19. Am J Hematol, 2021. 96(12): p. E468-E471. [CrossRef]
- Ito, T., et al., Thrombomodulin in disseminated intravascular coagulation and other critical conditions—a multi-faceted anticoagulant protein with therapeutic potential. Critical Care, 2019. 23(1): p. 280. [CrossRef]
- Yamakawa, K., S. Murao, and M. Aihara, Recombinant Human Soluble Thrombomodulin in Sepsis-Induced Coagulopathy: An Updated Systematic Review and Meta-Analysis. Thromb Haemost, 2019. 119(1): p. 56-65. [CrossRef]
- Leucker, T.M., et al., Effect of Crizanlizumab, a P-Selectin Inhibitor, in COVID-19: A Placebo-Controlled, Randomized Trial. JACC Basic Transl Sci, 2021. 6(12): p. 935-945. [CrossRef]
- Neri, T., D. Nieri, and A. Celi, P-selectin blockade in COVID-19-related ARDS. Am J Physiol Lung Cell Mol Physiol, 2020. 318(6): p. L1237-L1238. [CrossRef]
- Karsli, E., et al., Soluble P-selectin as a potential diagnostic and prognostic biomarker for COVID-19 disease: A case-control study. Life Sci, 2021. 277: p. 119634. [CrossRef]
- Etulain, J., et al., P-selectin promotes neutrophil extracellular trap formation in mice. Blood, 2015. 126(2): p. 242-6. [CrossRef]
- Muller, R., et al., Increased plasma level of soluble P-selectin in non-hospitalized COVID-19 convalescent donors. Thromb Res, 2022. 216: p. 120-124. [CrossRef]
- Wang, S.S.Y., et al., Increased Platelet Activation demonstrated by Elevated CD36 and P-Selectin Expression in 1-Year Post-Recovered COVID-19 Patients. Semin Thromb Hemost, 2023. [CrossRef]
- FitzGerald, E.S., et al., Lung Epithelial Cell Transcriptional Regulation as a Factor in COVID-19-associated Coagulopathies. Am J Respir Cell Mol Biol, 2021. 64(6): p. 687-697. [CrossRef]
- Cañas, C.A., et al., Role of Tissue Factor in the Pathogenesis of COVID-19 and the Possible Ways to Inhibit It. Clin Appl Thromb Hemost, 2021. 27: p. 10760296211003983. [CrossRef]
- Stefely, J.A., et al., Marked factor V activity elevation in severe COVID-19 is associated with venous thromboembolism. Am J Hematol, 2020. 95(12): p. 1522-1530. [CrossRef]
- Wang, J., et al., Coagulation factor V is a T-cell inhibitor expressed by leukocytes in COVID-19. iScience, 2022. 25(3): p. 103971. [CrossRef]
- Antoniak, S., The coagulation system in host defense. Res Pract Thromb Haemost, 2018. 2(3): p. 549-557. [CrossRef]
- Han, M. and D. Pandey, ZMPSTE24 Regulates SARS-CoV-2 Spike Protein-enhanced Expression of Endothelial PAI-1. Am J Respir Cell Mol Biol, 2021. 65(3): p. 300-308. [CrossRef]
- Whyte, C.S., et al., The suboptimal fibrinolytic response in COVID-19 is dictated by high PAI-1. J Thromb Haemost, 2022. 20(10): p. 2394-2406. [CrossRef]
- Bielosludtseva, K., The diagnostic and prognostic role of plasminogen activator inhibitor-1 (PAI-1) in hospitalized patients with pneumonias of different etiologies. European Respiratory Journal, 2022. 60(suppl 66): p. 2495.
- Matsuyama, T., et al., An aberrant STAT pathway is central to COVID-19. Cell Death Differ, 2020. 27(12): p. 3209-3225. [CrossRef]
- Morrow, G.B., C.S. Whyte, and N.J. Mutch, A Serpin With a Finger in Many PAIs: PAI-1’s Central Function in Thromboinflammation and Cardiovascular Disease. Front Cardiovasc Med, 2021. 8: p. 653655. [CrossRef]
- Kwaan, H.C. and P.F. Lindholm, The Central Role of Fibrinolytic Response in COVID-19-A Hematologist’s Perspective. Int J Mol Sci, 2021. 22(3). [CrossRef]
- Kang, S., et al., IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A, 2020. 117(36): p. 22351-22356. [CrossRef]
- Al-Tamimi, A.O., et al., SARS-CoV-2 infection induces soluble platelet activation markers and PAI-1 in the early moderate stage of COVID-19. Int J Lab Hematol, 2022. 44(4): p. 712-721. [CrossRef]
- Poole, L.G., et al., Plasminogen Activator Inhibitor-1 Is Critical in Alcohol-Enhanced Acute Lung Injury in Mice. Am J Respir Cell Mol Biol, 2017. 57(3): p. 315-323. [CrossRef]
- Zuo, Y., et al., Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Scientific Reports, 2021. 11(1): p. 1580. [CrossRef]
- Bouchla, A., et al., Red Blood Cell Abnormalities as the Mirror of SARS-CoV-2 Disease Severity: A Pilot Study. Front Physiol, 2021. 12: p. 825055. [CrossRef]
- Espallargas, I., et al., CT imaging of pulmonary embolism in patients with COVID-19 pneumonia: a retrospective analysis. Eur Radiol, 2021. 31(4): p. 1915-1922. [CrossRef]
- Longchamp, G., et al., Proximal deep vein thrombosis and pulmonary embolism in COVID-19 patients: a systematic review and meta-analysis. Thrombosis Journal, 2021. 19(1): p. 15. [CrossRef]
- Valle, C., et al., Association between pulmonary embolism and COVID-19 severe pneumonia: Experience from two centers in the core of the infection Italian peak. Eur J Radiol, 2021. 137: p. 109613. [CrossRef]
- Niculae, C.M., et al., Acute Pulmonary Artery Thrombosis despite Anticoagulation in Patients with COVID-19 Pneumonia: A Single-Center Retrospective Cohort Study. J Clin Med, 2022. 11(9). [CrossRef]
- Trunz, L.M., et al., Imaging approach to COVID-19 associated pulmonary embolism. Int J Clin Pract, 2021. 75(10): p. e14340. [CrossRef]
- Barnett, N., et al., Prevalence of pulmonary embolism and deep venous thrombosis during the COVID-19 pandemic in an intensive care unit cohort: a service evaluation. Br J Anaesth, 2022. 129(5): p. e124-e126. [CrossRef]
- Cau, R., et al., Complications in COVID-19 patients: Characteristics of pulmonary embolism. Clin Imaging, 2021. 77: p. 244-249. [CrossRef]
- Scialpi, M., et al., Pulmonary embolism in COVID-19: Ancillary findings on chest CT angiography. Lung India, 2021. 38(Supplement): p. S123-S125. [CrossRef]
- Mandal, A.K.J., et al., Covid-19 and in situ pulmonary artery thrombosis. Respir Med, 2021. 176: p. 106176. [CrossRef]
- De Cobelli, F., et al., Pulmonary Vascular Thrombosis in COVID-19 Pneumonia. J Cardiothorac Vasc Anesth, 2021. 35(12): p. 3631-3641. [CrossRef]
- Bompard, F., et al., Pulmonary embolism in patients with COVID-19 pneumonia. Eur Respir J, 2020. 56(1). [CrossRef]
- Lazar, M., et al., Mortality Predictors in Severe SARS-CoV-2 Infection. Medicina (Kaunas), 2022. 58(7).
- Birocchi, S., et al., High rates of pulmonary artery occlusions in COVID-19. A meta-analysis. Eur J Clin Invest, 2021. 51(1): p. e13433. [CrossRef]
- Fauvel, C., et al., Pulmonary embolism in COVID-19 patients: a French multicentre cohort study. Eur Heart J, 2020. 41(32): p. 3058-3068. [CrossRef]
- Farouk, N., et al., Admission Levels of Serum P-Selectin and IL-6 Can Predict Development of Deep Venous Thrombosis in Hospitalized Covid-19 Patients. Int J Gen Med, 2022. 15: p. 5599-5607. [CrossRef]
- Marone, E.M., et al., Characteristics of Venous Thromboembolism in COVID-19 Patients: A Multicenter Experience from Northern Italy. Ann Vasc Surg, 2020. 68: p. 83-87. [CrossRef]
- Alonso Martinez, J.L., et al., Central Versus Peripheral Pulmonary Embolism: Analysis of the Impact on the Physiological Parameters and Long-term Survival. N Am J Med Sci, 2016. 8(3): p. 134-42. [CrossRef]
- Cha, S.-I., et al., Pulmonary embolism concurrent with lung cancer and central emboli predict mortality in patients with lung cancer and pulmonary embolism. Journal of Thoracic Disease, 2017. 10(1): p. 262-272. [CrossRef]
- Loffredo, L., et al., Asymptomatic and symptomatic deep venous thrombosis in hospitalized acutely ill medical patients: risk factors and therapeutic implications. Thrombosis Journal, 2022. 20(1): p. 72. [CrossRef]
- Katsoularis, I., et al., Risks of deep vein thrombosis, pulmonary embolism, and bleeding after covid-19: nationwide self-controlled cases series and matched cohort study. Bmj, 2022. 377: p. e069590. [CrossRef]
- Poor, H.D., Pulmonary Thrombosis and Thromboembolism in COVID-19. Chest, 2021. 160(4): p. 1471-1480. [CrossRef]
- Cui, S., et al., Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost, 2020. 18(6): p. 1421-1424. [CrossRef]
- Beretta, S., et al., Case Report: Concomitant Massive Cerebral Venous Thrombosis and Internal Iliac Vein Thrombosis Related to Paucisymptomatic COVID-19 Infection. Front Neurol, 2021. 12: p. 622130. [CrossRef]
- Malentacchi, M., et al., Concomitant brain arterial and venous thrombosis in a COVID-19 patient. Eur J Neurol, 2020. 27(9): p. e38-e39. [CrossRef]
- Lucijanic, M., et al., Asymptomatic deep vein thromboses in prolonged hospitalized COVID-19 patients. Wien Klin Wochenschr, 2021. 133(23-24): p. 1281-1288. [CrossRef]
- Horne, M.K., III, The dark side of deep venous thrombosis: the failure of anticoagulation. The American Journal of Medicine, 2001. 110(7): p. 589-590. [CrossRef]
- Rodger, M.A., et al., Management of suspected and confirmed recurrent venous thrombosis while on anticoagulant therapy. What next? Thrombosis Research, 2019. 180: p. 105-109. [CrossRef]
- Mosarla, R.C., et al., Anticoagulation Strategies in Patients With Cancer: JACC Review Topic of the Week. J Am Coll Cardiol, 2019. 73(11): p. 1336-1349. [CrossRef]
- Paez Vargas, J., et al., ANTICOAGULATION, BLEEDING, AND IMMUNOTHROMBOSIS IN CRITICALLY ILL PATIENTS WITH COVID-19. CHEST, 2021. 160(4): p. A994-A995. [CrossRef]
- Tan, B.K., et al., Arterial and venous thromboembolism in COVID-19: a study-level meta-analysis. Thorax, 2021. 76(10): p. 970-979. [CrossRef]
- Bikdeli, B., et al., COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J Am Coll Cardiol, 2020. 75(23): p. 2950-2973. [CrossRef]
- Klok, F.A., et al., Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res, 2020. 191: p. 145-147. [CrossRef]
- Llitjos, J.F., et al., High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost, 2020. 18(7): p. 1743-1746. [CrossRef]
- Porembskaya, O., et al., Thrombosis of pulmonary vasculature despite anticoagulation and thrombolysis: The findings from seven autopsies. Thrombosis Update, 2020. 1: p. 100017. [CrossRef]
- Sadeghipour, P., et al., Effect of Intermediate-Dose vs Standard-Dose Prophylactic Anticoagulation on Thrombotic Events, Extracorporeal Membrane Oxygenation Treatment, or Mortality Among Patients With COVID-19 Admitted to the Intensive Care Unit: The INSPIRATION Randomized Clinical Trial. Jama, 2021. 325(16): p. 1620-1630. [CrossRef]
- Lawler, P.R., et al., Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with Covid-19. N Engl J Med, 2021. 385(9): p. 790-802. [CrossRef]
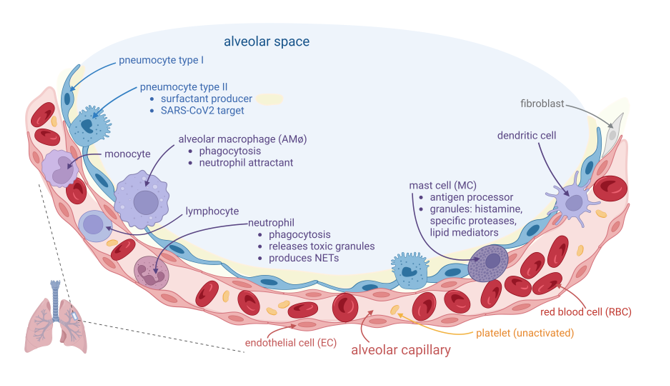
Figure 1.
Normal host alveolar capillary associated immune defense system. The alveolar space is lined with alveolar epithelium, consisting of two kinds of cells: type I and type II pneumocyte; type II cells are involved in surfactant secretion (light yellow in the figure) and in local innate immune defense. They are also a target for SARS-CoV2, which replicates inside these cells and can be further transferred into adjacent endothelial cells (ECs). The alveolar macrophages (AMφs) are placed into alveolar space, adjacent to the alveolar epithelium. AMφs are largely involved in immune defense, mainly by two processes: phagocytosis and chemoattraction of neutrophils. The alveolar-capillary layer is very thin over a large area, consisting of the adjacent membranes of both respiratory epithelium and vascular endothelium, and it is thicker where the body of these cells and other cells (fibroblasts in interstitium, mast cells (MCs) in subendothelial place) or other interstitial elements are placed. MCs are involved in COVID-19 pathogenicity. They contain granules with histamine and specific proteases. The capillary, lined with flat ECs, contains red blood cells (RBCs), platelets, and white blood cells. Neutrophils could pass into alveolar space if they are chemoattracted by activated AMφs. They are involved in the phagocytosis process. They contained granules with oxidants, that could be released outside the cell. They also can release extracellular traps (NETs) consisting of web-like structures containing DNA filaments coated with histones and granule proteins, that can entrap and eliminate various pathogens.
Figure 1.
Normal host alveolar capillary associated immune defense system. The alveolar space is lined with alveolar epithelium, consisting of two kinds of cells: type I and type II pneumocyte; type II cells are involved in surfactant secretion (light yellow in the figure) and in local innate immune defense. They are also a target for SARS-CoV2, which replicates inside these cells and can be further transferred into adjacent endothelial cells (ECs). The alveolar macrophages (AMφs) are placed into alveolar space, adjacent to the alveolar epithelium. AMφs are largely involved in immune defense, mainly by two processes: phagocytosis and chemoattraction of neutrophils. The alveolar-capillary layer is very thin over a large area, consisting of the adjacent membranes of both respiratory epithelium and vascular endothelium, and it is thicker where the body of these cells and other cells (fibroblasts in interstitium, mast cells (MCs) in subendothelial place) or other interstitial elements are placed. MCs are involved in COVID-19 pathogenicity. They contain granules with histamine and specific proteases. The capillary, lined with flat ECs, contains red blood cells (RBCs), platelets, and white blood cells. Neutrophils could pass into alveolar space if they are chemoattracted by activated AMφs. They are involved in the phagocytosis process. They contained granules with oxidants, that could be released outside the cell. They also can release extracellular traps (NETs) consisting of web-like structures containing DNA filaments coated with histones and granule proteins, that can entrap and eliminate various pathogens.
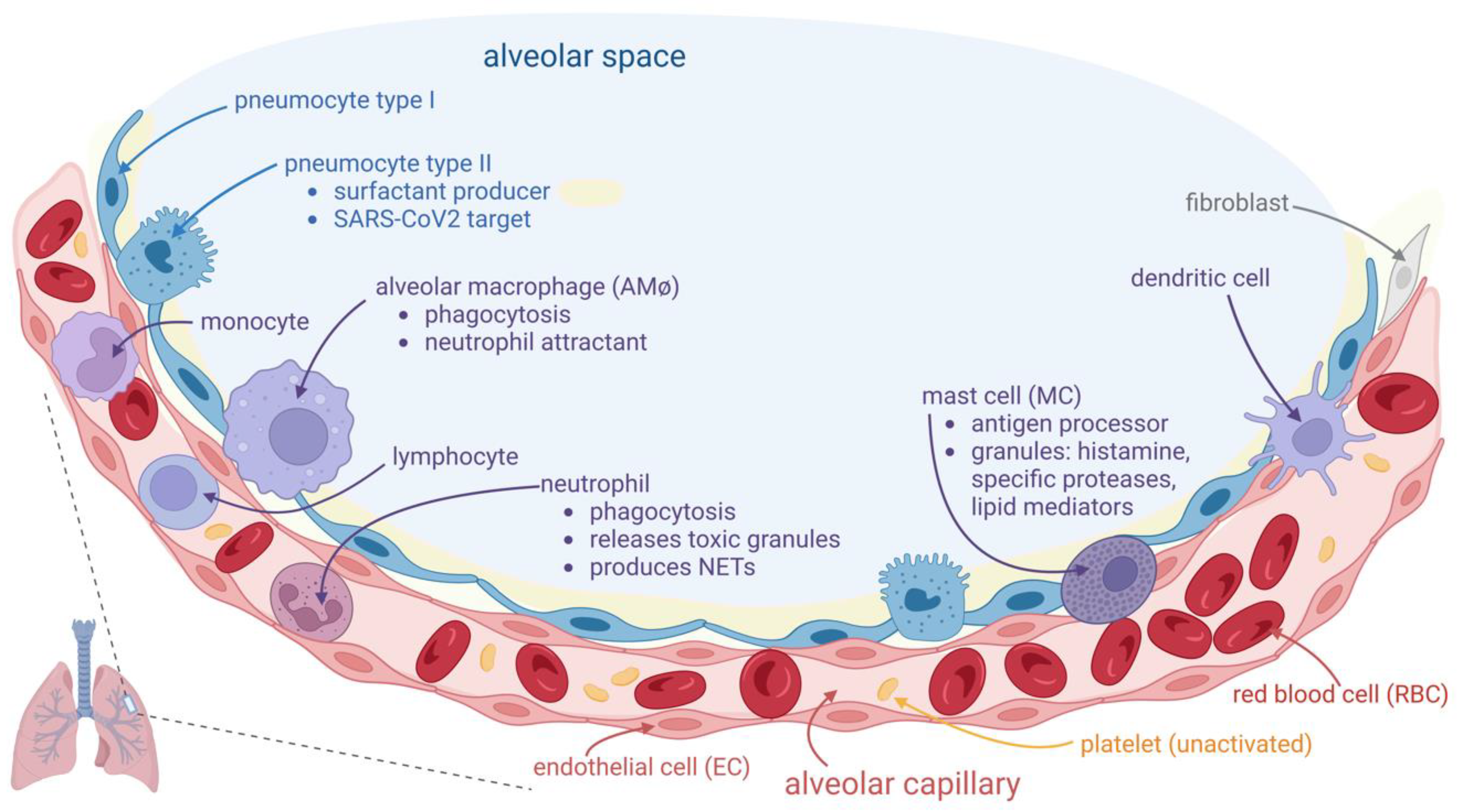
Figure 2.
The in situ immunothrombosis in COVID-19. SARS-CoV-2 replicates into the type II pneumocytes. Further, SARS-CoV-2 could be transferred to adjacent ECs, that has ACE-2 receptors and passes into the blood. AMφs interact with SARS-CoV-2 and activate themselves (1a). After activation, the major process in COVID-19 is the chemoattraction of neutrophils (1b). AMφs could also act like antigen presenters to T lymphocytes (1c). Neutrophils, attracted by AMφs, come into the area of the immune conflict, crawling, and squeezing (3a), and they activate and release neutrophil extracellular traps (NETosis) (3b); aberrant NETosis and oxidants release (3c) into the environment contribute to “cytokine storm” (4), tissue and vascular injuries and initiate immune-activated coagulation (6). SARS-CoV-2 activates MCs (2a) and they release histamine, other specific proteases, prostaglandins and leukotrienes, cytokines and chemokines (2b), contributing to capillary permeabilization and to the “cytokine storm” (4), ending with in situ immunothrombosis (6). Monocytes are excessively recruited in the area (5) and contribute to the “cytokine storm” (4) and to the activation of coagulation pathways (6). The blood clot (6) contains RBCs, activated platelets, fibrin, NETs, and lymphocytes, supporting the immune mechanism for thrombosis.
Figure 2.
The in situ immunothrombosis in COVID-19. SARS-CoV-2 replicates into the type II pneumocytes. Further, SARS-CoV-2 could be transferred to adjacent ECs, that has ACE-2 receptors and passes into the blood. AMφs interact with SARS-CoV-2 and activate themselves (1a). After activation, the major process in COVID-19 is the chemoattraction of neutrophils (1b). AMφs could also act like antigen presenters to T lymphocytes (1c). Neutrophils, attracted by AMφs, come into the area of the immune conflict, crawling, and squeezing (3a), and they activate and release neutrophil extracellular traps (NETosis) (3b); aberrant NETosis and oxidants release (3c) into the environment contribute to “cytokine storm” (4), tissue and vascular injuries and initiate immune-activated coagulation (6). SARS-CoV-2 activates MCs (2a) and they release histamine, other specific proteases, prostaglandins and leukotrienes, cytokines and chemokines (2b), contributing to capillary permeabilization and to the “cytokine storm” (4), ending with in situ immunothrombosis (6). Monocytes are excessively recruited in the area (5) and contribute to the “cytokine storm” (4) and to the activation of coagulation pathways (6). The blood clot (6) contains RBCs, activated platelets, fibrin, NETs, and lymphocytes, supporting the immune mechanism for thrombosis.
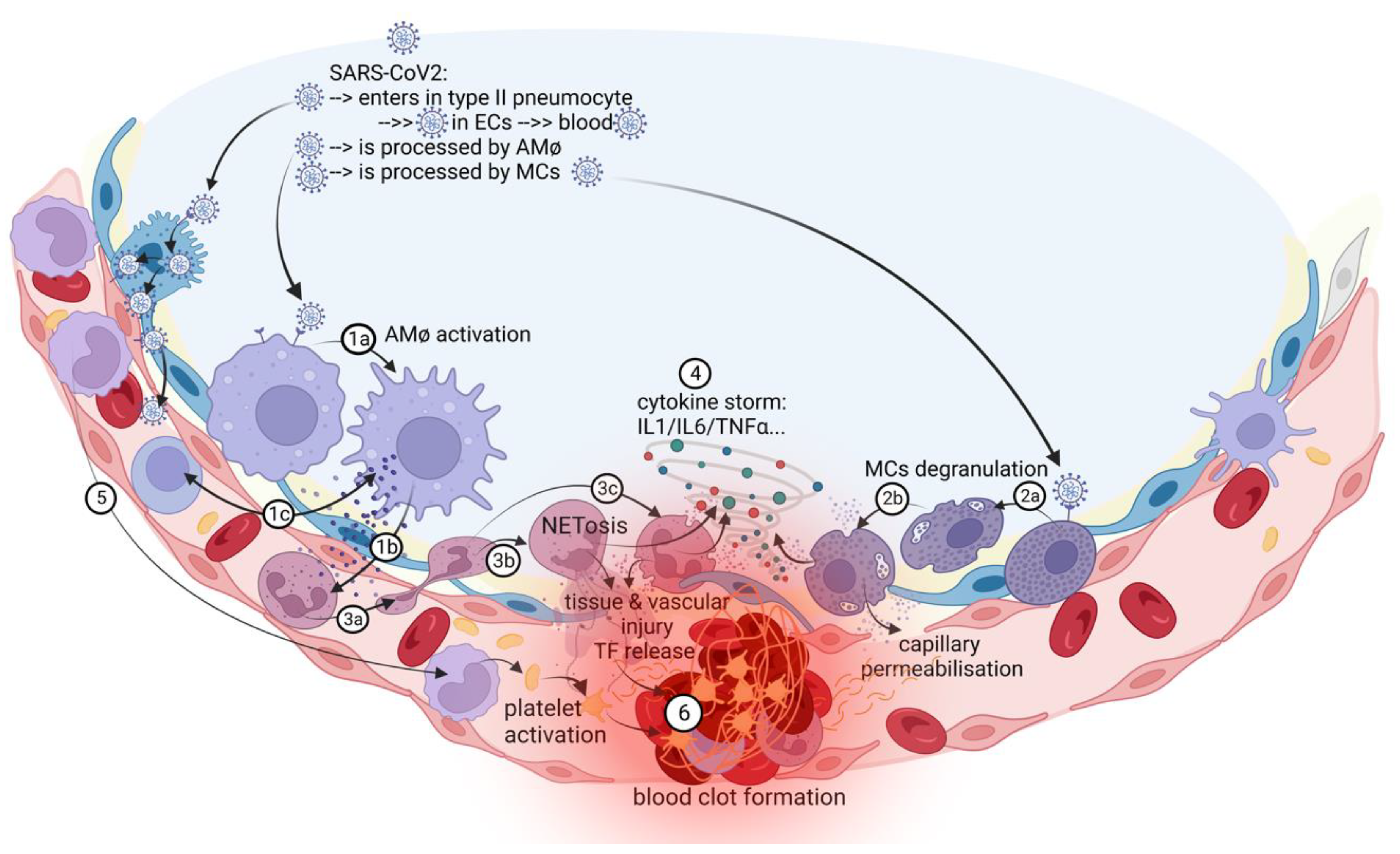
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



