
Submitted:
01 March 2023
Posted:
03 March 2023
You are already at the latest version
Abstract
Numerous studies have demonstrated that multiple intrinsic and extrinsic factors shape the structure and composition of gut microbiota in a host. The disorder of gut microbiota may trigger various host diseases. Here, we collected fecal samples from wild-caught Japanese geckos (Gekko japonicus) and captive conspecifics fed with mealworms (mealworm-fed geckos) and fruit flies (fly-fed geckos), aiming to examine dietary and sexual correlates of gut microbiota. We used the 16S rRNA gene sequencing technology to determine the composition of gut microbiota. The dominant phyla with a mean relative abundance higher than 10% were Verrucomicrobiota, Bacteroidota and Firmicutes. Gut microbial community richness was higher in mealworm-fed geckos than in fly-fed and wild geckos, and community diversity was higher in mealworm-fed geckos than in wild geckos. Neither alpha nor beta diversity of gut microbiota differed among wild, mealworm-fed and fly-fed geckos. The beta rather than alpha diversity of gut microbiota was sex-dependent. Based on the relative abundance of gut bacteria and its gene functions, we concluded that gut microbiota contributed more significantly to the host’s metabolic and immune functions. Higher diversity of gut microbiota in mealworm-fed geckos could result from higher chitin contents of insects of the order Coleoptera. This study not only provides basic information about the gut microbiota of G. japonicus, but also shows that gut microbiota correlates with dietary habit and sex in the species.
Keywords:
ASVs
; diet habit
; Gekko japonicus
; gut microbiota
; sex
1. Introduction
Gut microbiota is known as the second genome of the host [1], encoding the 10-100 times the number of genes of the host genome [2]. Gut microbiota plays a key role in host survival and adaptation, with its functions mainly manifested in a host’s life history [3], physiology [4], immune [5], growth [6], development [4] and behavior [7]. Gut microbiota can change rapidly in response to changes in the host’s environmental conditions and dietary habits [8], induce a host’s metabolic flexibility and phenotypic plasticity, and therefore enhance its ability to adapt to the environment [3]. For example, taxonomical shifts in gut bacterial communities in juvenile ostriches (Struthio camelus) coincide with the cessation of yolk absorption, co-occurring with their dietary switch [6]. These shifts may help ostriches adapt to dietary changes. For example, short chain fatty acids produced by the gut microbiota can maintain gut homeostasis [9]. Gut microbial dysbiosis can induce various host diseases and even threaten host survival [10].
The structure and diversity of gut microbiota are susceptible to numerous external and internal factors, including the host’s taxonomic category [11,12], sex [13], healthy status [14], age [6], dietary habit [15] and living environment [16]. These factors can substantially influence the composition, abundance and diversity of gut bacterial communities. In lizards, for example, captivity changes the gut microbial composition in Shinisaurus crocodilurus [17], Takydromus septentrionalis [18], and Tremarctos ornatus [19]. Toad-headed lizards (Phrynocephalus vlangalii) from the highest-altitude population have the lowest gut microbial diversity [16]. On the contrary, Glires mammals from high-latitude regions have a higher gut microbial diversity than their low-latitude conspecifics, because the increased energy demands in cold and hypoxic environments cannot be met without increasing gut microbial diversity [20].
The gut microbial composition is largely host-taxa specific [12]. In invertebrates, for example, the dominant gut microbial phyla are Tenericutes, Firmicutes, and Proteobacteria in snails [21,22], and Proteobacteria and Firmicute in insects [23]. In vertebrates, the dominant gut microbial phyla are Firmicutes and Bacteroidetes in amphibians [24], reptiles [25], and mammals [12], and Proteobacteria and Firmicutes in birds [26]. It is of great significance to explore the factors affecting gut microbiota and host-microbe symbiotic relationships. One widely accepted idea is that diet and host genetic status have a key role in shaping gut microbial structures [12,23].
Each microbial taxon has its functional roles in the host gut. Bacteria of the phylum Bacteroidetes are the homeostasis cornerstone in a health gut and involve in various functions, including the gut-brain-axis interactions, the immune system and metabolic homeostasis [27]. A large number of genes of the phylum Firmicutes are clustered to encode the ABC-type sugar transport systems, and bacteria of this phylum usually are active in carbohydrate metabolism [28]. Therefore, the ratio of Bacteroidetes to Firmicutes in relative abundance is correlated with the slim figure, with a higher ratio hinting a healthier host, which in turn correlates with host obesity [29]. Proteobacteria is regarded as a potential diagnostic signature of dysbiosis and risk of disease in human [28], but the ratio Proteobacteria to Firmicutes and Bacteroidetes in relative abundance is correlated with the bacterial stress tolerance under cold environments [30]. It is the role of gut microbiota that assists the host to adapt to a wide variety of diets and environments.
Reptiles are the first group of vertebrates that can truly live out of water on land and their gut bacterial variation has therefore attracted much attention. As in other animal taxa, gut microbiota is affected by many factors in reptiles, including host genetic status [25], captivity [18], environment [16], and diet [17]. Previous studies on reptiles have showed that a given factor may affect the gut microbiota in some species but not in others. For instance, diet shapes gut microbiota in S. crocodilurus [17] but not in Varanus salvator [31]. Compared with other reptile taxa, studies on gut microbiota in geckos have been limited, focusing only on the effects of fasting on gut microbiota in Eublepharis macularius [32,33] and the structure of gut microbiota in Hemidactylus frenatus [34]. Here, we used high-throughput sequencing to study dietary and sexual correlates of the gut microbiota in the Japanese gecko, Gekko japonicus. This gecko is a small-sized, oviparous species of the family Gekkonidae, occurring in the central and southeastern parts of China, Japan and Korea. The gecko is a comparatively well-known lizard species in China, with data collected over the past few years covering a wide range of topics such as genomics [35], temperature-dependent sex determination [36,37], molecular basis of character development [38], and microhabitat use [39].
2. Materials and Methods
2.1. Sample Collection
We used 49 adult geckos without any signs of disease (including ectoparasites) to conduct this study. All these geckos were collected in Xianlin Campus of Nanjing Normal University (NNU), 25 (14♀♀and 11♂♂) in June 2020 and 24 (11♀♀ and 13♂♂) in September 2020. Geckos collected in June were individually housed in 175 × 175 × 152 mm (length × width × height) plastic cages placed in a room where temperatures varied naturally. Of the 25 geckos, 13 (7♀♀and 6♂♂; hereafter mealworm-fed geckos) were fed with mealworms (larvae of Tenebrio molitor), and 12 (7♀♀ and 5♂♂; hereafter fly-fed geckos) with fruit flies (Drosophila melanogaster), both for three months, during which period distilled water was available ad libitum. All facilities were disinfected by wiping with 97% alcohol every other day. Mealworm- and fly-fed geckos always had free access to food sterilized with UV light for 1 h in advance. Geckos collected in mid-September (hereafter wild geckos) were individually housed in sterile 175 × 175 × 152 mm cages overnight and then collected fecal samples. In September, we used light traps to collect insects at the sites where we collected geckos, thereby assessing prey items potentially available to geckos in the wild. Insects of the orders Lepidoptera and Diptera were the most abundant prey items potentially available to Japanese geckos in Xianlin Campus of NNU (Table 1).
We put fecal samples collected from mealworm-fed, fly-fed and wild geckos into sterile tubes, labeled these tubes, and then stored them at -20 °C for late DNA extraction. We released all geckos at their point of capture soon after the collection of fecal samples in mid-September. Geckos of different groups did not differ from each other in mean values for body mass (H2,49 = 1.95, p = 0.38) and snout-vent length (H2,49 = 2.62, p = 0.27). Our experimental procedures complied with laws on animal welfare and research in China, and were approved by the Animal Research Ethics Committee of Nanjing Normal University (Permit No. IACUC 20200511).
2.2. DNA Extraction, PCR Amplification and Sequencing
We used the Mag-Bind Soil DNA Kit (Omega, Shanghai, China) to extract the microbial DNA from the fecal samples according to the manufacturer protocols. We used 2.0% agarose gel electrophoresis and Qubit 3.0 DNA detection kit (Thermo Fisher Scientific) to purify and quantify the DNA products, respectively. The bacterial V3-V4 region of the 16S rRNA gene were amplified using PCR with a 30 μL reaction system including 15 μL of 2× Hieff® Robust PCR Master Mix (2×), 1 μL of each primer (10 μM), 20 ng of genomic DNA, and ddH2O. The universal primers 341F (5’-CCTACGGGNGGCWGCAG-3’) and 805R (5’-GACTACHVGGGTATCTAATCC-3’) were selected to perform the PCR reaction. The first round of PCR thermal cycling conditions was performed as follows: initial denaturation at 94 °C for 3 min, followed by 5 cycles of denaturation at 94 °C for 30 s, annealing at 45 °C for 20 s and extension at 65 °C for 30 s. The other 20 cycles consisted of 94 °C for 20 s, 55 °C for 20 s, and 72 °C for 30 s, with a final extension at 72 °C for 5 min. In the second round, PCR products of the first round were used for amplification, and llumina bridge PCR compatible primers were introduced. The PCR reaction system was the same as the first round. The thermal cycling conditions were as follows: denaturation at 95 °C for 3 min, followed by 5 cycles of denaturation a 94 °C for 20 s, at 55 °C for 20 s and 72 °C for 30 s, and a final extension at 72 °C for 5 min. Sequencing of the PCR-amplified products was conducted on an Illumina MiSeq.
2.3. Quality Control and Data Standardization
We imported the raw paired-end sequence into Quantitative Insights into Microbial Ecology 2 (QIIME2) using the manifest file and trimmed the primers [40]. We used the DADA2 to filter and truncate low-quality reads and produce paired-end reads [41]. These reads after quality control were generated the raw amplicon sequence variants (ASV) with a minimum overlap of 12 bp. The raw paired-end sequences were submitted to the National Genomics Data Center (NGDC) GSA database (accession number CRA007161).
We used QIIME2 to classify ASVs into organisms based on pre-formatted SILVA 138 SUU NR99 ASVs full-length reference sequences following the q2-fragment-classifier method in QIIME2. The sequencing depth for each sample was calculated using QIIME2 and visualized using R 4.0 [42]. We removed ASVs with the number less than 10 in only one sample for further analysis to avoid large partial sample deviations. The abundance information was standardized based on the sample with the least ASVs number.
2.4. Estimation of Alpha and Beta Diversity
We used QIIME2 to calculate alpha diversity indexes, including community richness (observed species), community diversity (Shannon’s entropy index), and community evenness (Pielou’s evenness index). We used Kruskal-Wallis H and Mann-Whitney U test to examine whether alpha diversity indexes differed between (mealworm-fed, fly-fed and wild) gecko groups and between sexes, respectively. Pairwise comparisons using Wilcoxon rank sum test with continuity correction were performed when necessary. For beta diversity, we used principal coordinate analysis (PCoA) and permutational multivariate analysis of variance (Adonis) to show differences in microbial community structure among gecko groups. Adonis was performed based on the Bray-Curtis distance with 999 permutations. The linear discriminant analysis of effect sizes (LEfSe) and linear discriminant analysis (LDA) were conducted to compare the microbial abundances from the phylum to genus levels based on the relative abundance higher than 1% [43]. The unique bacterial taxa were determined based on log LDA score > 2 and p < 0.05. Kruskal-Wallis H test was used to verify whether the bacteria detected by LDA had a higher relative abundance among the different diet × sex combinations.
2.5. Gene Function Predication
PICRUST2 was used to explore gene functions of all ASVs in gut microbiota based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [44]. We allocated these gene functions to the corresponding KEGG pathways and obtained KEGG Orthology (KO) information for each gene function for three KEGG pathways [45]. The relative abundance of these gene functions for each sample was calculated to assess the functional differences in gut microbiota among different gecko groups. The LEfSe and LDA were performed to compare the relative abundance of KEGG gene functions from level 1 to level 3 based on the relative abundance higher than 1%. Only the gene functional category with a log LDA score > 2 and p < 0.05 was used in this analysis. Kruskal-Wallis H test was used to verify whether the gene function detected by LDA had a higher relative abundance among the different diet × sex combinations. The unique and shared gene functions were visualized using Venn diagram. All values were presented as mean ± standard error (SE), and the significance level was set at α < 0.05.
3. Results
We obtained 4,299,671 raw reads and 2473062 high-quality reads from the 49 fecal samples (Table A1). The number of observed bacterial ASVs firstly increased with the increase of the number of sequences and then leveled out in each sample (Figure A1). We identified 976 bacterial ASVs, with 114−214 ASVs per sample (Table A2). These ASVs could be allocated to 12 phyla, 19 classes, 49 orders, 83 families, and 168 genera.
The top four dominant bacterial phyla were Verrucomicrobiota (36.6 ± 3.5%), Bacteroidota (29.4 ± 2.2%), Firmicutes (18.9 ± 2.2%), and Proteobacteria (9.6± 2.3%) (Figure 1A). The dominant bacterial families with a relative abundance > 3% were Akkermansiaceae (35.3 ± 3.5%), Bacteroidaceae (18.0 ± 1.6%), Tannerellaceae (8.1 ± 1.0%), Enterobacteriaceae (6.2 ± 1.6%), Lachnospiraceae (4.4 ± 0.8%) and Clostridiaceae (4.3 ± 1.0%) (Figure 1B). The dominant genera with a relative abundance > 3% were Akkermansia (35.3 ± 3.5%), Bacteroides (18.0± 1.6%), Parabacteroides (5.7 ± 0.7%), and Clostridium_sensu_stricto_1 (4.3 ± 1.0%) (Figure 1C).
3.2. Dietary and Sexual Correlates of Gut Microbiota
Kruskal-Wallis showed that community diversity (H= 7.80, df=2, p = 0.02) and richness (H = 7.53, df=2, p = 0.02) rather than community evenness (H = 5.93, df=2, p = 0.05) differed among mealworm-fed, fly-fed and wild geckos. Specifically, gut microbial community richness and gut microbial community diversity were significantly higher in mealworm-fed geckos than in wild geckos (Figure 2). None of the above three diversity indexes differed between the sexes (all p > 0.05) (Figure 2).
The PCoA based on the Bray-Curtis distance showed a significant separation of gut microbiota among six diet × sex combinations (Adonis: r2 = 0.15, F5,43 = 1.46, p = 0.006), with the first and second axes respectively explaining 16.9% and 11.2% of the total variance (Figure 3A). However, the significant separation of gut microbiota was be found only between the sexes (Adonis: r2 = 0.06, F1,47 = 2.89, p = 0.001; Figure 3A), rather than in different diet groups (Adonis: r2 = 0.05, F1,46 = 1.21, p = 0.174; Figure 3B). In addition, neither in males (Adonis: r2 = 0.09, F2,21 = 1.10, p = 0.32; Figure 3C) nor in females (Adonis: r2 = 0.09, F2,24 = 1.11, p = 0.30; Figure 3D) did gut microbiota differed among mealworm-fed, fly-fed and wild geckos. LEfSe analysis showed significant differences in the unique gut microbiota among fly- and mealworm-fed females, mealworm-fed males, and wild males (Figure 4). Specifically, the unique bacteria family Desulfovibrioria and Marinifilaceae was found in mealworm-fed males, the families Eggerthellaceae and Caulobacteraceae was unique in wild males, the unique bacteria genera Eggerthella, Bacteroides and Odoribacter was found in fly-fed females, and the family Erysipelatoclostridiaceae and Tannerellaceae, and genera Desulfovibrio and Clostridium_sensu_stricto_1 was unique in mealworm-fed females (Figure 4). Kruskal-Wallis H test showed that the relative abundance of above bacterial taxon had significant differences among different groups except for the family Tannerellaceae (Table A3).
3.2. The Predicted Metagenomes
The predicted functions in gut microbiota were mainly involved in metabolism (80.8 ± 0.2%), genetic information processing (12.8 ± 0.2%), cellular processes (3.2 ± 0.1%), environmental information processing (2.4 ± 0.1%), organismal systems (0.4 ±0.01%), and human diseases (0.32 ± 0.02%) at the first level (Figure 5A). The second KEGG category level was composed of 31 functions, among which the most abundant categories with a relative abundance > 5% in gut microbiota had functions associated with carbohydrate metabolism (15.0 ± 0.1%), metabolism of cofactors and vitamins (13.5 ± 0.2%), amino acid metabolism (12.2 ± 0.1%), metabolism of terpenoids and polyketides (8.9 ± 0.1%), glycan biosynthesis and metabolism (6.9 ± 0.2%), metabolism of other amino acids (6.8 ± 0.1%), lipid metabolism (6.1 ± 0.1%), replication and repair (5.9 ± 0.1%) and energy metabolism (5.3 ± 0.05%) (Figure 5B). Among 157 KEGG functions at the third level, those with a relative abundance > 2% were biosynthesis of ansamycins (3.7 ± 0.1%), other glycan degradation (2.7 ± 0.1%), biosynthesis of vancomycin group antibiotics (2.6 ± 0.1%), valine, leucine and isoleucine biosynthesis (2.1 ± 0.02%) (Figure 5C).
A total of 157 known KO functional genes were identified. Geckos in six diet × sex combinations shared 136 genes (Figure 5D). LEfSe analysis based on KOs revealed that an unique gene function related to energy metabolism was found in fly-fed females (Figure 5E). In wild females, gene functions related to carbohydrate metabolism (Ko00010 and Ko00051) and environmental information processing and membrane transport were unique (Figure 5E). Gut microbial functions in fly-fed males had three unique functions related to metabolism (Ko00473, Ko01055, and biosynthesis of other secondary metabolites; Figure 5E). Gut microbial gene functions in mealworm-fed males were mainly associated with metabolism (Ko00340, Ko00720, Ko00790, and metabolism of cofactors and vitamins; Figure 5E). Kruskal-Wallis H test showed that the relative abundance of above unique gene functions had significant differences among different groups (Table A4).
4. Discussion
At the phylum level, the dominant gut microbes in G. japonicus were Verrucomicrobiota, Bacteroidota, Firmicutes, and Proteobacteria (Figure 1A), This is consistent with what has been observed in leopard geckos (Eublepharis macularius) [32], but differs from the results reported for other reptilian taxa. For example, the dominant gut microbial phyla are Proteobacteria, Bacteroidetes, and Firmicutes in lizards [16,31,46] and snakes [47,48], Bacteroidetes and Firmicutes in turtles [25,49], and Fusobacteria, Proteobacteria, Firmicutes, and Bacteroidetes in crocodiles [50,51]. This indicates that the dominant gut microbial phyla differ among animal taxa. In fact, even among animals of the same evolutionary clade, their gut microbiota may differ significantly. For example, the dominant gut microbial phyla differ significantly between two species of turtles [25] and among four species of snakes [47] reared under the same conditions. This inconsistency between species provides evidence for the genetic correlates of gut microbiota in reptiles.
Taxonomically, all gut dominant genera and families in G. japonicus belong to the four dominant phyla mentioned above. The members of the phylum Verrucomicrobiota are correlated with mucin-degrading, glucose homeostasis and inducing regulatory immunity [52], as well as reducing obesity risk [53]. Bacteria of the phylum Bacteroidota have functional roles in degrading the high molecular weight organic matter, activating T-cell mediated responses and producing butyrate to maintain a health gut [54]. Many studies have showed that bacteria of the phylum Firmicutes contribute to degrading complex carbohydrates of both plant and hosts [55]. Members of the phylum Proteobacteria are related to degrading and fermenting the complex sugars and producing the vitamins for their hosts [56].
There has been evidence that gut microbial compositions are closely correlated with food ingested by hosts [31] and with their sex [22]. In this study, mealworm-fed geckos had higher gut microbial community diversity and richness although diet diversity was higher in wild geckos (Figure 2). That food diversity is not associated with gut bacterial alpha diversity in G. japonicus is similar to the findings demonstrated in V. salvator [31], Anser anser [57], and Ochotona curzoniae [58]. However, there are some species such as Gasterosteus aculeatus and Perca fluviatilis [59] and Fejervarya limnocharis [60] where gut microbial alpha diversity is negatively correlated with diet diversity. Gut microbial alpha diversity did not differ between the sexes in G. japonicus, similar to the result reported for a wide range of vertebrates including fish [61], amphibians [62], birds [57], and mammals [63]. However, sexual differences in gut microbial diversity do exist in many animals, also including fish [64], birds [65], and mammals [66]. Taken together, available data show that dietary and/or sexual correlates of host gut microbial alpha diversity are species- or taxon-specific.
Sex rather and diet shaped beta diversity of the gut microbiota in G. japonicus (Figure 3 and Figure 4). However, PCoA showed that gut microbial structure differed only between sexes, but not among mealworm-fed, fly-fed and wild geckos (Figure 3). LEfSe showed that gut bacterial relative abundance differed not only between the sexes but also among three groups of geckos ingesting different prey items (Figure 4). Bacteria of the families Eggerthellaceae and Caulobacteraceae were enriched in wild males. Eggerthellaceae bacteria play an import in the transformation of bioactive secondary plant compounds in human feces [67], and Caulobacteraceae bacteria actively metabolize linear alkylbenzene sulfonates in soil [68]. The enrichment of bacteria of the genera Bacteroides, Eggerthella and Odoribacter in fly-fed females was correlated with metabolism [69], polysaccharide degradation [70] and immune [71], respectively. A higher relative abundance of Erysipelatoclostridiaceae at the family level, and Desulfovibrio and Clostridium_sensu_stricto_1 at the genus level in mealworm-fed females also was enriched. Bacteria of Bacteroidales [72], Desulfovibrio [55] and Clostridium_sensu_stricto_1 [73] could contribute to metabolism, and members of Erysipelatoclostridiaceae play a role in immunity in the host gut [74]. Therefore, the differences in relative abundance of gut microbiota may contribute more to metabolic and immune functions in the gecko.
The gut microbial functions in G. japonicus were mainly related to metabolism at the first function level with a relative abundance > 80%, the metabolism-related function, replication and repair at the second level, antibiotic and partial amino acid biosynthesis, other glycan degradation at the third level with higher relative abundances (Figure 5). Gut microbial functions in most animals are closely related to metabolism, including fish [61], amphibians [75], reptiles [18], birds [76] and mammals [19]. Therefore, the gut microbiota plays an important role in host energy metabolism. This is also evidenced by the enrichment of gene functions with high relative abundance in different diet × sex combinations in G. japonicus (Figure 5). For example, a higher relative abundance of gene functions related to metabolism were enriched in all male geckos, fly-fed female and wild female geckos.
Prey items potentially available for the Nanjing population of G. japonicus in September consisted of insects of the orders Lepidoptera and Diptera (Table 1). Insects mainly contain protein (30−70% of dry mass), fat (~35% of dry mass), minerals and vitamins [77], and can modulate the gut microbiota and improve host health status [78]. Fruit flies and mealworms used in this study belong to the orders Diptera and Coleoptera, respectively. This might be the main reason for why the gut microbiome of fly-fed geckos was closer to that of wild geckos. However, mealworm-fed geckos fed on mealworm containing more chitin. Chitin is one of the most abundant biopolymers in nature [79] and can restore the compositional balance of the bacterial community [78,80]. In this study, more diverse gut bacteria in mealworm-fed geckos might result from abundant chitin in diets. Therefore, the gut bacterial alpha diversity in G. japonicus might be correlated with the type of insect diets.
5. Conclusions
Gut microbial community diversity and richness rather than community evenness differed among mealworm-fed and wild geckos. Gut microbial community richness and diversity were significantly higher in mealworm-fed geckos than in wild geckos. None of the above three diversity indexes differed between the sexes. There was a significant separation of gut microbiota between the sexes. Such a separation of gut microbiota did not exist among geckos ingesting different prey items in both sexes. The relative abundance of unique gut bacteria and gene functions differed among different diet × sex combinations. Our study demonstrated dietary and sexual correlates of gut microbiota in a gecko species.
Author Contributions
Conceptualization, Y.-Y.D., P.L., X.J., and Y.-F.Q.; formal analysis, X.-R.J., Y.-R.W., K. G., J.-F.G., and Y.-F.Q.; Investigation, Y.-Y.D., L.-H.L., H.L., and Y.-F.Q.; Methodology, Y.-Y.D., Y. D., L.-H.L., P.L., H.L., X.J., and Y.-F.Q.; Supervision, Y.-F.Q. and X.J.; writing—original draft preparation, Y.-F.Q., X.-R.J. and X.J.; writing—review and editing, L.-H.L., J.-F.G., X.J., and Y.-F.Q.; funding acquisition, Y. D., and Y.-F.Q. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by grants from the National Natural Science Foundation of China (Grant no. 32171498) to Y-FQ, Finance science and Technology project of Hainan Province (ZDKJ2016009-1-2), and Hainan Key Laboratory of Herpetological Research (HKLHR202002) to YD.
Institutional Review Board Statement
The work was carried out in compliance with laws on animal welfare and research in China, and approved by the Animal Research Ethical Committees of Nanjing Normal University (Approval number: IACUC-20200511).
Informed Consent Statement
Not applicable.
Data Availability Statement
All 16S rRNA gene sequences obtained in this study have been deposited in the National Genomics Data Center (NGDC) GSA database (accession number CRA007161).
Acknowledgments
We thank Yi-Jin Jiang and Yu-Tian Zhao for help in sample collection during the research. We thank Jun-Qiong Chen for help in data analysis and comments on early versions of the manuscript.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Appendix A
Figure A1.
Rarefaction curves based on ASVs for individual fecal samples. Each color represents a sample.
Figure A1.
Rarefaction curves based on ASVs for individual fecal samples. Each color represents a sample.
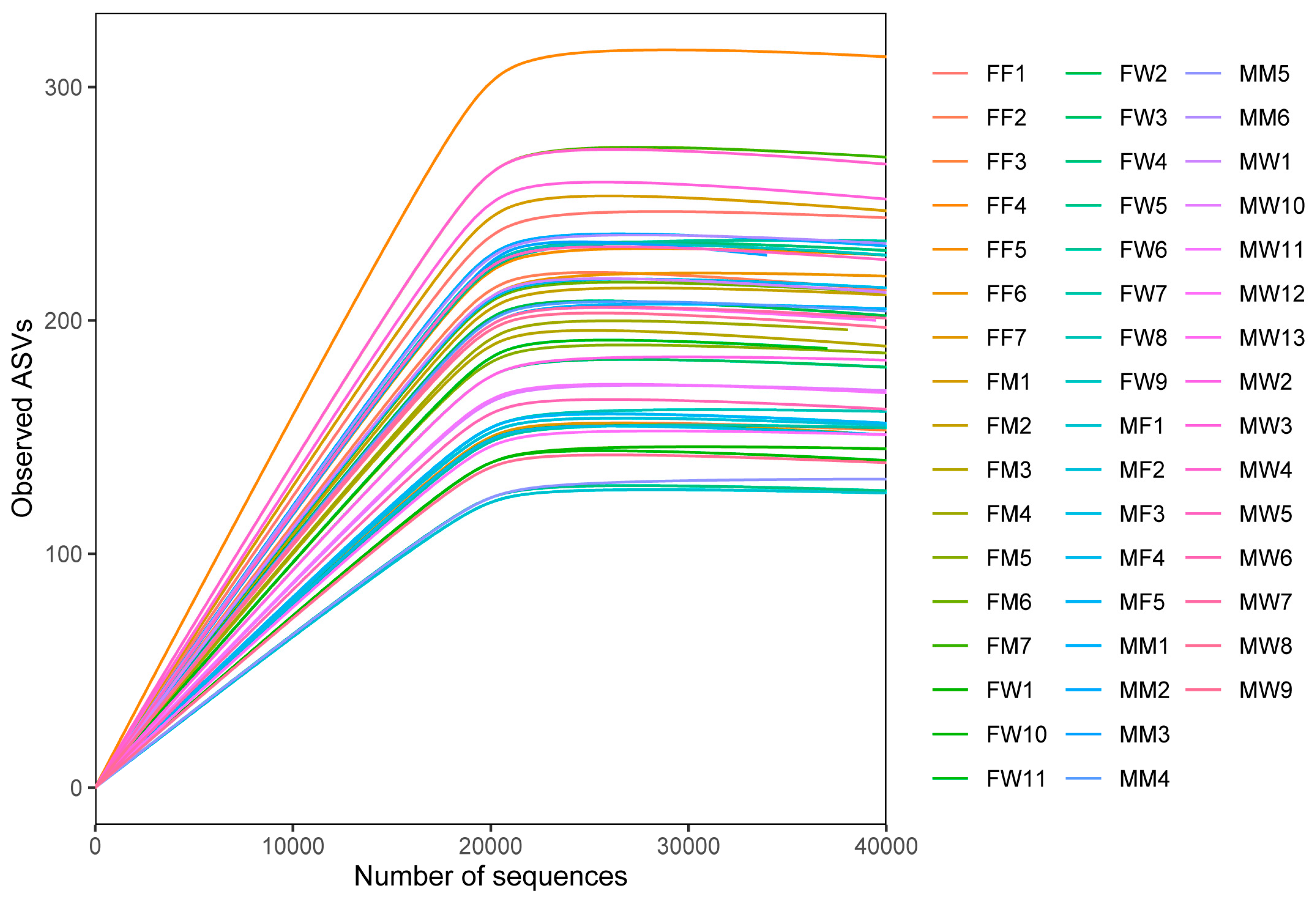

Table A1.
The number of valid reads and sequences information in each fecal sample.
| Sample ID | Group | Raw reads | High-quality reads | Average sequence length | Minimum sequence length | Maximum sequence length | Accession number |
|---|---|---|---|---|---|---|---|
| FF1 | FF | 73684 | 40053 | 406.59 | 258 | 422 | SAMC798099 |
| FF2 | FF | 88949 | 60703 | 406.61 | 257 | 422 | SAMC798104 |
| FF3 | FF | 90123 | 51799 | 408.68 | 395 | 422 | SAMC798108 |
| FF4 | FF | 94885 | 66593 | 407.10 | 395 | 422 | SAMC798112 |
| FF5 | FF | 88354 | 44352 | 406.84 | 259 | 422 | SAMC798130 |
| FF6 | FF | 87440 | 53660 | 407.19 | 260 | 422 | SAMC798131 |
| FF7 | FF | 73619 | 38059 | 411.72 | 395 | 422 | SAMC798132 |
| FM1 | FM | 75766 | 41882 | 405.6 | 258 | 422 | SAMC798096 |
| FM2 | FM | 102760 | 70196 | 408.12 | 395 | 422 | SAMC798098 |
| FM3 | FM | 72769 | 40632 | 409.31 | 395 | 422 | SAMC798101 |
| FM4 | FM | 67705 | 38056 | 408.60 | 260 | 422 | SAMC798103 |
| FM5 | FM | 74409 | 43685 | 403.84 | 261 | 422 | SAMC798105 |
| FM6 | FM | 72310 | 43249 | 409.58 | 395 | 422 | SAMC798107 |
| FM7 | FM | 81193 | 59389 | 407.66 | 259 | 422 | SAMC798110 |
| FW1 | FW | 112652 | 66225 | 406.46 | 259 | 422 | SAMC798095 |
| FW2 | FW | 86098 | 43304 | 409.95 | 395 | 422 | SAMC798097 |
| FW3 | FW | 76989 | 41571 | 411.82 | 395 | 422 | SAMC798100 |
| FW4 | FW | 84867 | 46340 | 404.12 | 261 | 422 | SAMC798102 |
| FW5 | FW | 69268 | 42933 | 409.83 | 259 | 422 | SAMC798106 |
| FW6 | FW | 150444 | 86689 | 401.75 | 260 | 422 | SAMC798109 |
| FW7 | FW | 92941 | 52980 | 408.74 | 395 | 422 | SAMC798111 |
| FW8 | FW | 86275 | 48301 | 409.68 | 395 | 422 | SAMC798137 |
| FW9 | FW | 82425 | 50606 | 407.51 | 395 | 422 | SAMC798138 |
| FW10 | FW | 81409 | 47539 | 411.59 | 260 | 422 | SAMC798139 |
| FW11 | FW | 72672 | 37028 | 408.33 | 257 | 422 | SAMC798140 |
| MF1 | MF | 77935 | 47389 | 408.57 | 395 | 421 | SAMC798115 |
| MF2 | MF | 82673 | 45365 | 410.01 | 307 | 422 | SAMC798122 |
| MF3 | MF | 93453 | 52888 | 406.79 | 257 | 422 | SAMC798123 |
| MF4 | MF | 92104 | 41266 | 401.99 | 259 | 422 | SAMC798124 |
| MF5 | MF | 90387 | 50096 | 411.24 | 395 | 422 | SAMC798129 |
| MM1 | MM | 82136 | 45335 | 408.02 | 395 | 422 | SAMC798114 |
| MM2 | MM | 104528 | 55091 | 410.22 | 395 | 422 | SAMC798117 |
| MM3 | MM | 92997 | 33970 | 408.92 | 258 | 422 | SAMC798119 |
| MM4 | MM | 96831 | 44585 | 411.18 | 395 | 422 | SAMC798121 |
| MM5 | MM | 94575 | 57840 | 407.83 | 395 | 421 | SAMC798126 |
| MM6 | MM | 88806 | 51696 | 408.94 | 395 | 422 | SAMC798128 |
| MW1 | MW | 98013 | 54728 | 407.02 | 395 | 422 | SAMC798113 |
| MW2 | MW | 86628 | 56998 | 403.80 | 283 | 422 | SAMC798116 |
| MW3 | MW | 97957 | 70420 | 405.41 | 260 | 422 | SAMC798118 |
| MW4 | MW | 90098 | 60973 | 406.60 | 260 | 423 | SAMC798120 |
| MW5 | MW | 96383 | 51194 | 405.50 | 258 | 422 | SAMC798125 |
| MW6 | MW | 90030 | 47425 | 413.33 | 395 | 422 | SAMC798127 |
| MW7 | MW | 95586 | 53061 | 408.23 | 258 | 422 | SAMC798133 |
| MW8 | MW | 71594 | 47456 | 408.50 | 395 | 422 | SAMC798134 |
| MW9 | MW | 89601 | 45778 | 406.39 | 258 | 422 | SAMC798135 |
| MW10 | MW | 90782 | 53788 | 406.60 | 257 | 422 | SAMC798136 |
| MW11 | MW | 89660 | 55005 | 407.86 | 260 | 422 | SAMC798141 |
| MW12 | MW | 93227 | 55424 | 406.95 | 281 | 422 | SAMC798142 |
| MW13 | MW | 73681 | 39467 | 410.10 | 395 | 422 | SAMC798143 |
Table A2.
The number of bacterial amplicon sequence variants (ASVs) at different taxonomic levels in each fecal sample.
Table A2.
The number of bacterial amplicon sequence variants (ASVs) at different taxonomic levels in each fecal sample.
| Sample ID | Group | ASVs | Genus | Family | Class | Order | Phylum |
|---|---|---|---|---|---|---|---|
| FF1 | FF | 211 | 68 | 46 | 14 | 29 | 8 |
| FF2 | FF | 189 | 78 | 52 | 15 | 34 | 10 |
| FF3 | FF | 146 | 43 | 34 | 13 | 25 | 8 |
| FF4 | FF | 197 | 84 | 52 | 15 | 35 | 10 |
| FF5 | FF | 211 | 59 | 35 | 12 | 21 | 9 |
| FF6 | FF | 173 | 61 | 41 | 14 | 29 | 8 |
| FF7 | FF | 132 | 41 | 31 | 12 | 19 | 8 |
| FM1 | FM | 213 | 71 | 43 | 11 | 24 | 7 |
| FM2 | FM | 189 | 75 | 51 | 14 | 32 | 10 |
| FM3 | FM | 179 | 52 | 36 | 12 | 23 | 7 |
| FM4 | FM | 188 | 60 | 40 | 12 | 27 | 8 |
| FM5 | FM | 162 | 49 | 38 | 12 | 23 | 7 |
| FM6 | FM | 178 | 72 | 45 | 12 | 27 | 8 |
| FM7 | FM | 197 | 80 | 49 | 14 | 31 | 10 |
| FW1 | FW | 126 | 55 | 34 | 10 | 21 | 7 |
| FW2 | FW | 179 | 50 | 37 | 12 | 24 | 8 |
| FW3 | FW | 167 | 51 | 37 | 10 | 21 | 7 |
| FW4 | FW | 191 | 59 | 37 | 12 | 25 | 8 |
| FW5 | FW | 115 | 41 | 29 | 10 | 19 | 7 |
| FW6 | FW | 206 | 56 | 38 | 13 | 26 | 8 |
| FW7 | FW | 145 | 42 | 28 | 10 | 19 | 7 |
| FW8 | FW | 147 | 41 | 28 | 10 | 18 | 7 |
| FW9 | FW | 173 | 56 | 40 | 11 | 23 | 6 |
| FW10 | FW | 114 | 35 | 32 | 12 | 22 | 7 |
| FW11 | FW | 171 | 48 | 31 | 12 | 21 | 9 |
| MF1 | MF | 122 | 35 | 30 | 12 | 22 | 8 |
| MF2 | MF | 150 | 43 | 32 | 13 | 21 | 8 |
| MF3 | MF | 142 | 45 | 33 | 12 | 22 | 8 |
| MF4 | MF | 187 | 53 | 34 | 12 | 22 | 7 |
| MF5 | MF | 149 | 43 | 32 | 11 | 19 | 8 |
| MM1 | MM | 197 | 58 | 38 | 11 | 22 | 8 |
| MM2 | MM | 212 | 56 | 37 | 11 | 22 | 6 |
| MM3 | MM | 214 | 60 | 39 | 13 | 25 | 9 |
| MM4 | MM | 186 | 46 | 31 | 13 | 21 | 8 |
| MM5 | MM | 114 | 34 | 33 | 11 | 23 | 6 |
| MM6 | MM | 210 | 58 | 44 | 12 | 27 | 7 |
| MW1 | MW | 204 | 54 | 33 | 13 | 23 | 8 |
| MW2 | MW | 134 | 52 | 40 | 11 | 28 | 7 |
| MW3 | MW | 128 | 59 | 44 | 14 | 28 | 9 |
| MW4 | MW | 183 | 77 | 47 | 13 | 30 | 9 |
| MW5 | MW | 204 | 50 | 33 | 11 | 20 | 7 |
| MW6 | MW | 147 | 36 | 24 | 11 | 16 | 7 |
| MW7 | MW | 190 | 48 | 29 | 12 | 19 | 8 |
| MW8 | MW | 117 | 39 | 28 | 12 | 19 | 8 |
| MW9 | MW | 172 | 53 | 35 | 14 | 23 | 9 |
| MW10 | MW | 161 | 48 | 31 | 12 | 20 | 8 |
| MW11 | MW | 152 | 58 | 40 | 14 | 27 | 9 |
| MW12 | MW | 135 | 44 | 26 | 10 | 18 | 7 |
| MW13 | MW | 173 | 50 | 35 | 12 | 22 | 7 |
Table A3.
The relative abundance of unique bacterial taxon among different groups based on Kruskal-Wallis H test. The letters “f” and “g” indicate family and genus, respectively.
Table A3.
The relative abundance of unique bacterial taxon among different groups based on Kruskal-Wallis H test. The letters “f” and “g” indicate family and genus, respectively.
| Taxonomy | df | H | p |
|---|---|---|---|
| f__Caulobacteraceae | 5 | 11.76 | 0.04 |
| f__Desulfovibrioria | 5 | 13.85 | 0.02 |
| f__Eggerthellaceae | 5 | 17.08 | 0.004 |
| f__Erysipelatoclostridiaceae | 5 | 11.23 | 0.05 |
| f__Marinifilaceae | 5 | 13.23 | 0.02 |
| f__Tannerellaceae | 5 | 10.51 | 0.06 |
| g__Bacteroides | 5 | 17.77 | 0.003 |
| g__Clostridium_sensu_stricto_1 | 5 | 14.60 | 0.01 |
| g__Desulfovibrio | 5 | 16.18 | 0.006 |
| g__Eggerthella | 5 | 15.56 | 0.008 |
| g__Odoribacter | 5 | 13.62 | 0.02 |
Table A4.
The relative abundance of unique predicated functions among different groups based on Kruskal-Wallis H test.
Table A4.
The relative abundance of unique predicated functions among different groups based on Kruskal-Wallis H test.
| Level name | df | H | p |
|---|---|---|---|
| Metabolism|Energy metabolism | 5 | 15.66 | 0.008 |
| Environmental Information Processing | 5 | 13.22 | 0.02 |
| Environmental Information Processing|Membrane transport | 5 | 13.54 | 0.02 |
| Metabolism|Biosynthesis of other secondary metabolites | 5 | 12.35 | 0.03 |
| Metabolism|Metabolism of cofactors and vitamins | 5 | 12.03 | 0.03 |
| ko00010 | 5 | 11.47 | 0.04 |
| ko00051 | 5 | 11.26 | 0.05 |
| ko00340 | 5 | 12.82 | 0.02 |
| ko00473 | 5 | 11.90 | 0.04 |
| ko00720 | 5 | 14.23 | 0.01 |
| ko00790 | 5 | 12.88 | 0.02 |
| ko01055 | 5 | 13.40 | 0.02 |
References
- Bruls, T.; Weissenbach, J. The human metagenome: our other genome? Hum. Mol. Genet. 2011, 20, R142–R148. [Google Scholar] [CrossRef]
- Qin, J.-J.; Li, R.-Q.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Macke, E.; Tasiemski, A.; Massol, F.; Callens, M.; Decaestecker, E. Life history and eco-evolutionary dynamics in light of the gut microbiota. Oikos 2017, 126, 508–531. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota — masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J. Y.; Groer, M.; Dutra, S.; Sarkar, A.; McSkimming, D. Gut microbiota and immune system interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef] [PubMed]
- Videvall, E.; Song, S.J.; Bensch, H.M.; Strandh, M.; Engelbrecht, A.; Serfontein, N.; Hellgren, O.; Olivier, A.; Cloete, S.; Knight, R.; et al. Major shifts in gut microbiota during development and its relationship to growth in ostriches. Mol. Ecol. 2019, 28, 2653–2667. [Google Scholar] [CrossRef]
- Parashar, A.; Udayabanu, M. Gut microbiota regulates key modulators of social behavior. Eur. Neuropsychopharmacol. 2016, 26, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Brun, A.; Magallanes, M.; Brinkerhoff, J.; Laspiur, A.; Acosta, J.C.; Caviedes-Vidal, E.; Bordenstein, S.R. Gut microbial ecology of lizards: insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol. Ecol. 2017, 26, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.; Chen, Y.; Ma, Z.; Zhang, X.; Shi, D.; Khan, J. A.; Liu, H. Gut microbiota-derived short chain fatty acids are potential mediators in gut inflammation. Anim. Nutr. 2022, 8, 350–360. [Google Scholar] [CrossRef]
- Sun, J.; Kato, I. Gut microbiota, inflammation and colorectal cancer. Genes Dis. 2016, 3, 130–143. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Kartzinel, T.R.; Hsing, J.C.; Musili, P.M.; Brown, B.R.P.; Pringle, R.M. Covariation of diet and gut microbiome in African megafauna. Proc. Natl. Acad. Sci. USA. 2019, 116, 23588–23593. [Google Scholar] [CrossRef]
- Org, E.; Mehrabian, M.; Parks, B. W.; Shipkova, P.; Liu, X.-Q.; Drake, T.A.; Lusis, A.J. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef]
- Zeevi, D.; Korem, T.; Godneva, A.; Bar, N.; Kurilshikov, A.; Lotan-Pompan, M.; Weinberger, A.; Fu, J.-Y.; Wijmenga, C.; Zhernakova, A.; et al. Structural variation in the gut microbiome associates with host health. Nature 2019, 568, 43–48. [Google Scholar] [CrossRef]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of diet on the gut microbiota: rethinking intervention duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef]
- Zhang, W.-Y.; Li, N.; Tang, X.-L.; Liu, N.-F.; Zhao, W. Changes in intestinal microbiota across an altitudinal gradient in the lizard Phrynocephalus vlangalii. Ecol. Evol. 2018, 8, 4695–4703. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.-S.; Liang, X.-X.; Yang, M.-Y.; Wang, T.-T.; Chen, J.-P.; Du, W.-G.; Li, H.; Sun, B.-J. Captivity influences gut microbiota in crocodile lizards (Shinisaurus crocodilurus). Front. Microbiol. 2020, 11, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhao, Y.-T.; Dai, Y.-Y.; Jiang, Y.-J.; Lin, L.-H.; Li, H.; Li, P.; Qu, Y.-F.; Ji, X. Captivity affects diversity, abundance and functional pathways of gut microbiota in the northern grass lizard Takydromus septentrionalis. MicrobiologyOpen 2020, 9, e1095. [Google Scholar] [CrossRef]
- Borbón-García, A.; Reyes, A.; Vives-Flórez, M.; Caballero, S. Captivity shapes the gut microbiota of Andean bears: insights into health surveillance. Front. Microbiol. 2017, 8, 1316. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qu, J.-P.; Li, T.-T.; Yao, M.-J.; Li, J.-Y.; Li, X.-Z. Gut microbiota may predict host divergence time during Glires evolution. FEMS Microbiol. Ecol. 2017, 93, fix009. [Google Scholar] [CrossRef]
- Hu, Z.-H.; Chen, X.; Chang, J.; Yu, J.-H.; Tong, Q.; Li, S.-G.; Niu, H.-X. Compositional and predicted functional analysis of the gut microbiota of Radix auricularia (Linnaeus) via high-throughput Illumina sequencing. PeerJ 2018, 6, e5537. [Google Scholar] [CrossRef]
- Chen, L.; Li, S.-X.; Xiao, Q.; Lin, Y.; Li, X.-X.; Qu, Y.-F.; Wu, G.-G.; Li, H. Composition and diversity of gut microbiota in Pomacea canaliculata in sexes and between developmental stages. BMC Microbiol. 2021, 21, 200. [Google Scholar] [CrossRef]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef]
- Vences, M.; Lyra, M.L.; Kueneman, J.G.; Bletz, M.C.; Archer, H.M.; Canitz, J.; Handreck, S.; Randrianiaina, R.-D.; Struck, U.; Bhuju, S.; et al. Gut bacterial communities across tadpole ecomorphs in two diverse tropical anuran faunas. Sci. Nat. 2016, 103, 25. [Google Scholar] [CrossRef]
- Qu, Y.-F.; Wu, Y.-Q.; Zhao, Y.-T.; Lin, L.-H.; Du, Y.; Li, P.; Li, H.; Ji, X. The invasive red-eared slider turtle is more successful than the native Chinese three-keeled pond turtle: evidence from the gut microbiota. PeerJ 2020, 8, e10271. [Google Scholar] [CrossRef]
- Dewar, M.L.; Arnould, J.P.Y.; Krause, L.; Dann, P.; Smith, S.C. Interspecific variations in the faecal microbiota of Procellariiform seabirds. FEMS Microbiol. Ecol. 2014, 89, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Gibiino, G.; Lopetuso, L.R.; Scaldaferri, F.; Rizzatti, G.; Binda, C.; Gasbarrini, A. Exploring Bacteroidetes: metabolic key points and immunological tricks of our gut commensals. Digest. Liver Dis. 2018, 50, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Ottman, N.; Smidt, H.; de Vos, W.M.; Belzer, C. The function of our microbiota: who is out there and what do they do? Front. Cell. Infect. Microbiol. 2012, 2, 104. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.; McAllister, T.A. Antimicrobial usage and resistance in beef production. J. Anim. Sci. Biotechnol. 2016, 7, 68. [Google Scholar] [CrossRef]
- Du, Y.; Chen, J.-Q.; Liu, Q.; Fu, J.-C.; Lin, C.-X.; Lin, L.-H.; L, H.; Qu, Y.-F.; Ji, X. Dietary correlates of oral and gut microbiota in the water monitor lizard, Varanus salvator (Laurenti, 1768). Front. Microbiol. 2022, 12, 771527. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Amaya, J.; Passement, C.A.; Dearing, M.D.; McCue, M.D. Unique and shared responses of the gut microbiota to prolonged fasting: a comparative study across five classes of vertebrate hosts. FEMS Microbiol. Ecol. 2014, 90, 883–894. [Google Scholar] [CrossRef] [PubMed]
- McCue, M.D.; Passement, C.A.; Meyerholz, D.K. Maintenance of distal intestinal structure in the face of prolonged fasting: a comparative examination of species from five vertebrate classes. Anat. Rec. 2017, 300, 2208–2219. [Google Scholar] [CrossRef]
- Naher, K.; Alam, A.S.; Rahman, S.; Kabir, M.M. Gut contents of common house gecko, Hemidactylus frenatus (Schlegel, 1836) in Jahangirnagar university campus, Savar, Bangladesh. Bangl. J. Zool. 2015, 41, 229–232. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Q.; Wang, Y.-J.; Luo, L.-H.; Yang, J.; Yang, L.-F.; Liu, M.; Li, Y.-R.; Qian, T.-M.; Zhng, Y.; et al. Gekko japonicus genome reveals evolution of adhesive toe pads and tail regeneration. Nat. Commun. 2015, 6, 10033. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.-H.; Yang, J.; Wang, J.; Ji, X. Offspring sex in a TSD gecko correlates with an interaction between incubation temperature and yolk steroid hormones. Naturwissenschaften 2012, 99, 999–1006. [Google Scholar] [CrossRef]
- Li, S.-R.; Xu, Z.-W.; Luo, L.-G.; Ping, J.; Zhou, H.-B.; Xie, L.; Zhang, Y.-P. Latitudinal variation in the pattern of temperature-dependent sex determination in the Japanese gecko, Gekko japonicus. Animals 2022, 12, 942. [Google Scholar] [CrossRef]
- Wang, F.-F.; Chen, M.-Y.; Cai, F.-N.; Li, P.; Yan, J.; Zhou, K.-Y. Expression of specific corneous beta proteins in the developing digits of the Japanese gecko (Gekko japonicus) reveals their role in the growth of adhesive setae. Comp. Biochem. Physiol. B. 2020, 240, 110370. [Google Scholar] [CrossRef]
- Kim, D.-I.; Choi, W.-J.; Park, I.-K.; Kim, J.-S.; Kim, I.-H.; Park, D. Comparisons of microhabitat use of Schlegel’s Japanese gecko (Gekko japonicus) among three populations and four land cover types. J. Ecol. Environ. 2018, 42, 24. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J; Arumugan, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- R Development Core Team. R: a language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. 2022. Available at: http://www.R-project.org.
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Ma, J.-E.; Li, J.; Zhang, X.-J.; Li, L.-M.; He, N.; Liu, H.-Y.; Luo, S.-Y.; Wu, Z.-J.; Han, R.-C.; et al. Diets alter the gut microbiome of crocodile lizards. Front. Microbiol. 2017, 8, 2073. [Google Scholar] [CrossRef]
- Zhang, B.; Ren, J.; Yang, D.-D.; Liu, S.-R.; Gong, X.-G. Comparative analysis and characterization of the gut microbiota of four farmed snakes from southern China. PeerJ 2019, 7, e6658. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.-F.; Wu, Y.-Q.; Jiang, Y.-J.; Ji, X. Diet rather than genetic status shapes the gut microbiota in two congeneric snakes. In Review 2022. [CrossRef]
- Campos, P.; Guivernau, M.; Prenafeta-Boldú, F.X.; Cardona, L. Fast acquisition of a polysaccharide fermenting gut microbiome by juvenile green turtles Chelonia mydas after settlement in coastal habitats. Microbiome 2018, 6, 69. [Google Scholar] [CrossRef]
- Keenan, S.W.; Elsey, R.M. The good, the bad, and the unknown: Microbial symbioses of the American alligator. Integr. Comp. Biol. 2015, 55, 972–985. [Google Scholar] [CrossRef]
- Tang, K.-Y.; Wang, Z.-W.; Wan, Q.-H.; Fang, S.-G. Metagenomics reveals seasonal functional adaptation of the gut microbiome to host feeding and fasting in the Chinese alligator. Front. Microbiol. 2019, 10, 2409. [Google Scholar] [CrossRef] [PubMed]
- Gryaznova, M.; Dvoretskaya, Y.; Burakova, I.; Syromyatnikov, M.; Popov, E.; Kokina, A.; Mikhaylov, E.; Popov, V. Dynamics of changes in the gut microbiota of healthy mice fed with lactic acid bacteria and bifidobacteria. Microorganisms 2022, 10, 1020. [Google Scholar] [CrossRef]
- Ulker, İ.; Yildiran, H. The effects of bariatric surgery on gut microbiota in patients with obesity: a review of the literature. Biosci. Microb. Food Health 2019, 38, 3–9. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut Bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef] [PubMed]
- Blaut, M. Composition and function of the gut microbiome. In The Gut Microbiome in Health and Disease; Haller, D., Ed.; Springer International Publishing: Cham, 2018; pp. 5–30. [Google Scholar]
- Colston, T.J.; Jackson, C.R. Microbiome evolution along divergent branches of the vertebrate tree of life: what is known and unknown. Mol. Ecol. 2016, 25, 3776–3800. [Google Scholar] [CrossRef] [PubMed]
- Ricaud, K.; Even, M.; Lavigne, F.; Davail, S.; Arroyo, J. Evolution of intestinal microbiota and body compartments during spontaneous hyperphagia in the Greylag goose. Poultry Sci. 2019, 98, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, T.-T.; Beasley, D.E.; Heděnec, P.; Xiao, Z.-S.; Zhang, S.-H.; Li, J.-B.; Lim, Q.; Li, X.-Z. Diet diversity is associated with beta but not alpha diversity of pika gut microbiota. Front. Microbiol. 2016, 7, 1169. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, D.I.; Snowberg, L.K.; Hirsch, P.E.; Lauber, C.L.; Knight, R.; Caporaso, J.G.; Svanbäck, R. Individuals’ diet diversity influences gut microbial diversity in two freshwater fish (threespine stickleback and Eurasian perch). Ecol. Lett. 2014, 17, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Liao, W.-B. Seasonal variation in gut microbiota related to diet in Fejervarya limnocharis. Animals 2021, 11, 1393. [Google Scholar] [CrossRef]
- Jiang, M.; Xu, M.-Y.; Ying, C.-P.; Yin, D.-H.; Dai, P.; Yang, Y.-P.; Ye, K.; Liu, K. The intestinal microbiota of lake anchovy varies according to sex, body size, and local habitat in Taihu Lake, China. MicrobiologyOpen 9, 2020, e00955. [CrossRef]
- Shu, Y.-L.; Hong, P.; Tang, D.; Qing, H.; Donde, O.O.; Wang, H.; Xiao, B.-D.; Wu, H.-L. Comparison of intestinal microbes in female and male Chinese concave-eared frogs (Odorrana tormota) and effect of nematode infection on gut bacterial communities. MicrobiologyOpen 2019, 8, e749. [Google Scholar] [CrossRef]
- Zhu, L.-F.; Clayton, J.B.; Suhr Van Haute, M. J.; Yang, Q.-N.; Hassenstab, H.R.; Mustoe, A.C.; Knights, D.; Benson, A.K.; French, J.A. Sex bias in gut microbiome transmission in newly paired marmosets (Callithrix jacchus). mSystems 2020, 5, e00910-19. [Google Scholar] [CrossRef]
- Méndez-Pérez, R.; García-López, R.; Bautista-López, J.S.B.-L.; Vázquez-Castellanos, J.; Alvarez-González, C.; Peña-Marín, E.; Rodríguez, V.I.D.; Melgar-Valdés, C.; Moya, A.; Alvarez-González, C.A.; et al. High-throughput sequencing of the 16S rRNA gene to analyze the gut microbiome in juvenile and adult tropical gar (Atractosteus tropicus). Lat. Am. J. Aquat. Res. 2020, 48, 456–479. [Google Scholar] [CrossRef]
- Zhu, C.-H.; Xu, W.-J.; Tao, Z.-Y.; Song, W.-T.; Liu, H.-X.; Zhang, S.-J.; Li, H.-F. Effects of rearing conditions and sex on cecal microbiota in ducks. Front. Microbiol. 2020, 11, 565367. [Google Scholar] [CrossRef]
- Pafčo, B.; Sharma, A.K.; Petrželková, K.J.; Vlčková, K.; Todd, A.; Yeoman, C.J.; Wilson, B.A.; Stumpf, R.; White, B.A.; Nelson, K.; et al. Gut microbiome composition of wild western lowland gorillas is associated with individual age and sex factors. Am. J. Phys. Anthropol. 2019, 169, 575–585. [Google Scholar] [CrossRef]
- Matthies, A.; Blaut, M.; Braune, A. Isolation of a human intestinal bacterium capable of daidzein and genistein conversion. Appl. Environ. Microbiol. 2009, 75, 1740–1744. [Google Scholar] [CrossRef]
- Cortés-Lorenzo, C.; Sánchez-Peinado, M. del M.; Oliver-Rodríguez, B.; Vílchez, J.L.; González-López, J.J.; Rodríguez-Díaz, M. Two novel strains within the family Caulobacteraceae capable of degradation of linear alkylbenzene sulfonates as pure cultures. Int. Biodeter. Biodegr. 2013, 85, 62–65. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Kaoutari, A.E.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.-X.; Liu, P.; Song, P.-X.; Chen, X.-Y.; Ma, X. Moderate dietary protein restriction alters the composition of gut microbiota and improves ileal barrier function in adult pig model. Sci. Rep. 2017, 7, 43412. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-L.; Wang, R.; Ji, G.-C.; Elmassry, M.M.; Zabet-Moghaddam, M.; Vellers, H.; Hamood, A.N.; Gong, X.X.; Mirzaei, P.; Sang, S.-M.; et al. Dietary supplementation of gingerols- and shogaols-enriched ginger root extract attenuate pain-associated behaviors while modulating gut microbiota and metabolites in rats with spinal nerve ligation. J. Nutr. Biochem. 2022, 100, 108904. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, J.-Q.; Liu, Y.; Zhang, J.; Chen, X.-H.; Qu, Y.-F. Comparative study on gut microbiota in three anura frogs from a mountain stream. Ecol. Evol. 2022, 12, e8854. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.-L.; Zhao, X.-Z.; Li, Q.; He, C.; Zhao, W.-J.; Liu, S.-Y.; Ding, J.-M.; Ye, W.-X.; Wang, J.; Chen, Y.; et al. Genome and metagenome analyses reveal adaptive evolution of the host and interaction with the gut microbiota in the goose. Sci. Rep. 2016, 6, 32961. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.; Pillai, V.V.; Goddard, J.M.; Park, H.G.; Kothapalli, K.S.; Ross, D.A.; Ketterings, Q.M.; Brenna, J.T.; Milstein, M.B.; Marquiset, H.; et al. Sustainable production of housefly (Musca domestica) larvae as a protein-rich feed ingredient by utilizing cattle manure. PLoS ONE 2017, 12, e0171708. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, L.; Coretti, L.; Dipineto, L.; Bovera, F.; Menna, F.; Chiariotti, L.; Nizza, A.; Lembo, F.; Fioretti, A. Insect-based diet, a promising nutritional source, modulates gut microbiota composition and SCFAs production in laying hens. Sci. Rep. 2017, 7, 16269. [Google Scholar] [CrossRef]
- Waśko, A.; Bulak, P.; Polak-Berecka, M.; Nowak, K.; Polakowski, C.; Bieganowski, C. The first report of the physicochemical structure of chitin isolated from Hermetia illucens. Int. J. Biol. Macromol. 2016, 92, 316–320. [Google Scholar] [CrossRef]
- Stull, V.J.; Finer, E.; Bergmans, R.S.; Febvre, H.P.; Longhurst, C.; Manter, D.K.; Patz, J.A.; Weir, T.L. Impact of edible cricket consumption on gut microbiota in healthy adults, a double-blind, randomized crossover trial. Sci. Rep. 2018, 8, 10762. [Google Scholar] [CrossRef]
Figure 1.
The relative abundance of the gut microbiota in each gecko group at the phylum (A), family (B), and genus (C) levels. Each color in a plot represents a taxonomic group, of which the name is shown on the right side of the plot. The color for ‘others’ indicates all other phyla (A), families (B), or genera (C) combined, of which the names are not listed in each plot. FF: fly-fed females; fly-fed males; FM; mealworm-fed females; MM: mealworm-fed males; FW: wild females; MW: wild males.
Figure 1.
The relative abundance of the gut microbiota in each gecko group at the phylum (A), family (B), and genus (C) levels. Each color in a plot represents a taxonomic group, of which the name is shown on the right side of the plot. The color for ‘others’ indicates all other phyla (A), families (B), or genera (C) combined, of which the names are not listed in each plot. FF: fly-fed females; fly-fed males; FM; mealworm-fed females; MM: mealworm-fed males; FW: wild females; MW: wild males.
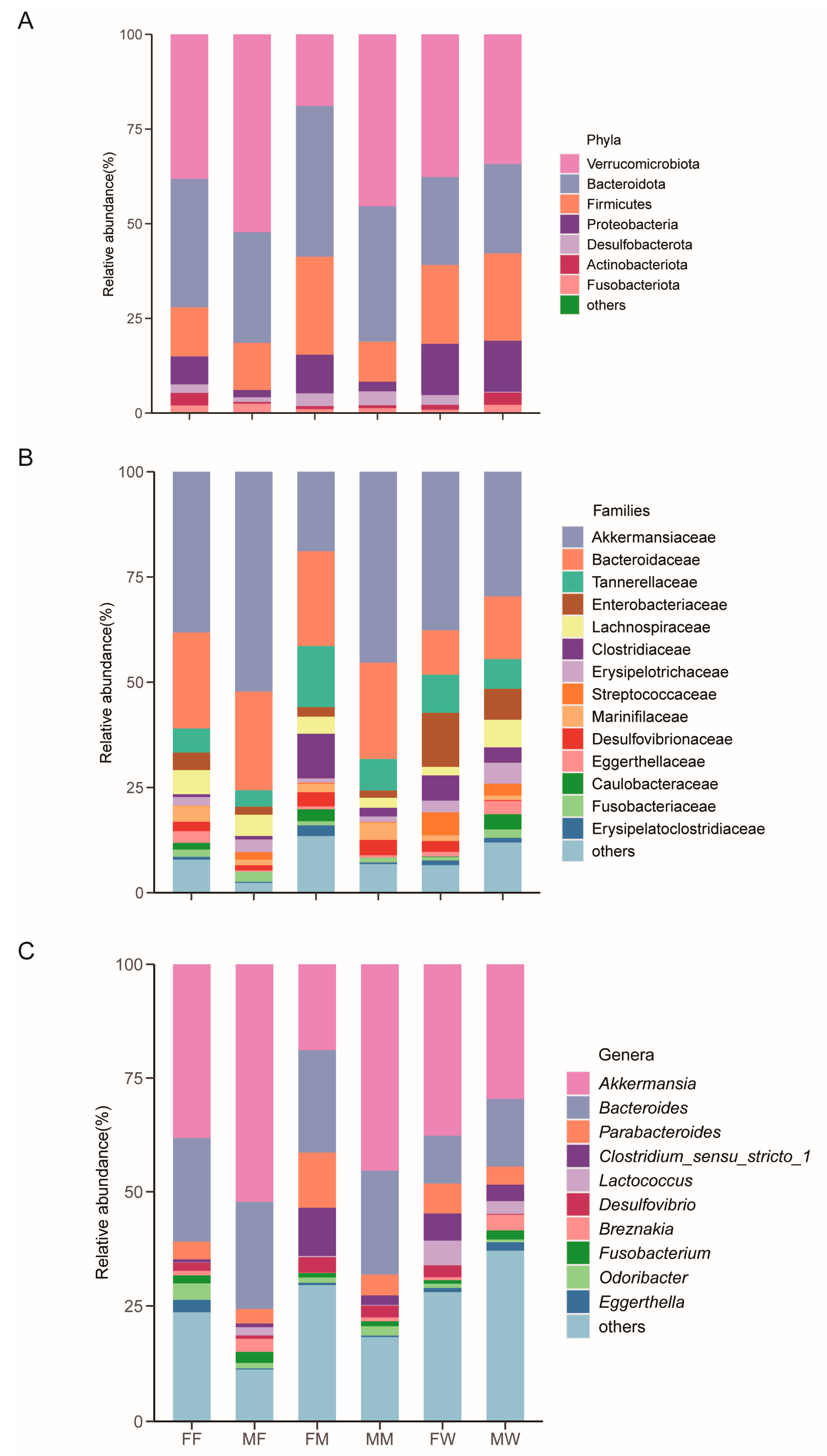
Figure 2.
The alpha diversity indexes of gut microbiota in six diet × sex combinations of fecal samples, including observed species (A), Shannon’s entropy index (B) and Pielou’s evenness index (C). F for fly-fed geckos, M for mealworm-fed geckos, and W for wild geckos.
Figure 2.
The alpha diversity indexes of gut microbiota in six diet × sex combinations of fecal samples, including observed species (A), Shannon’s entropy index (B) and Pielou’s evenness index (C). F for fly-fed geckos, M for mealworm-fed geckos, and W for wild geckos.
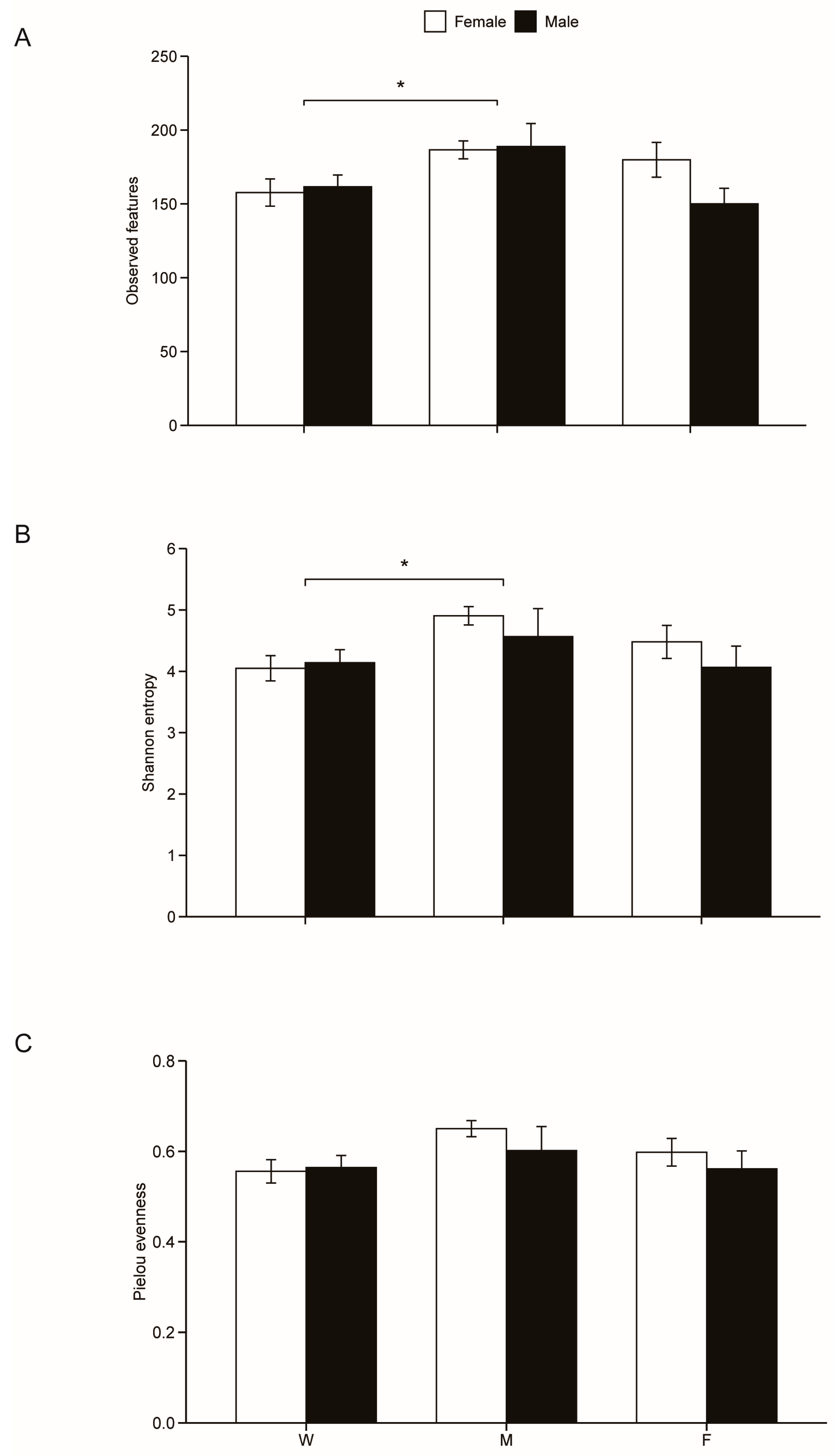
Figure 3.
Gut microbial diversity in six diet × sex combinations of fecal samples (A), fecal samples from three different diet group (B), male (C) and female (D) geckos ingesting different prey items. Principal coordinates analysis of Bray-Curtis distance matrix for bacterial community diversity. See Figure 1 for the definition of each group.
Figure 3.
Gut microbial diversity in six diet × sex combinations of fecal samples (A), fecal samples from three different diet group (B), male (C) and female (D) geckos ingesting different prey items. Principal coordinates analysis of Bray-Curtis distance matrix for bacterial community diversity. See Figure 1 for the definition of each group.
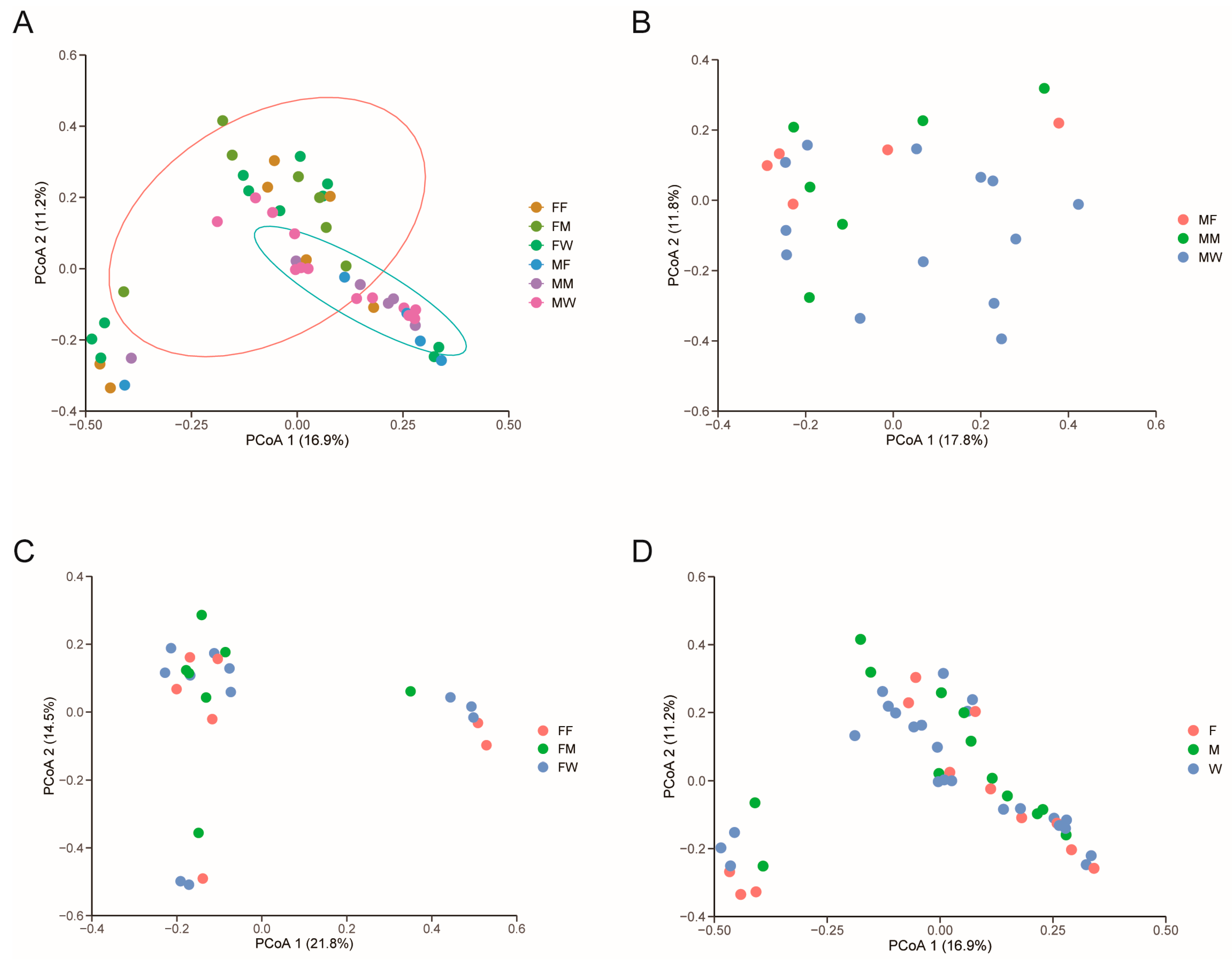
Figure 4.
Differences in gut microbiota among the four groups are determined by LEfSe (A). LDA scores reflect the differences in relative abundance among the four groups (B). See Figure 1 for the definition of each group. The letters “o”, “f” and “g” indicate order, family and genus, respectively.
Figure 4.
Differences in gut microbiota among the four groups are determined by LEfSe (A). LDA scores reflect the differences in relative abundance among the four groups (B). See Figure 1 for the definition of each group. The letters “o”, “f” and “g” indicate order, family and genus, respectively.
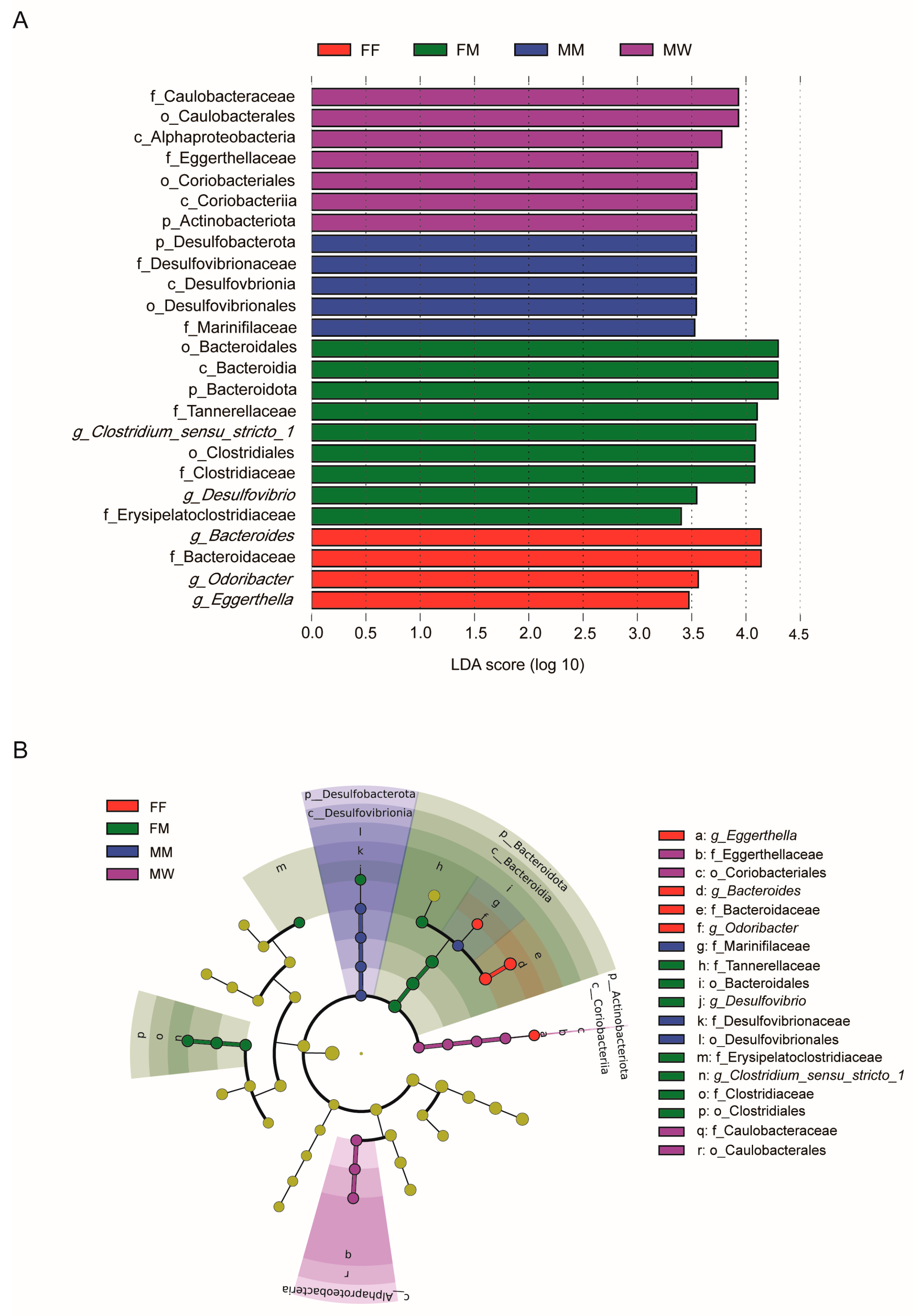
Figure 5.
The relative abundance of gene functional categories based on 16S RNA in the gut microbiota at top (A), second (B) and third (C) levels, and the Venn diagram of functional gene among the diet and gender combine groups (D). LDA scores reflect the differences in relative abundance among among the diet and gender combine groups (E). Each color in a plot indicates one gene function. Detailed descriptions are shown on the right side of each plot. The colors for others in plots B and C indicate all other gene functions not listed in these two plots. See Figure 1 for the definition of each group.
Figure 5.
The relative abundance of gene functional categories based on 16S RNA in the gut microbiota at top (A), second (B) and third (C) levels, and the Venn diagram of functional gene among the diet and gender combine groups (D). LDA scores reflect the differences in relative abundance among among the diet and gender combine groups (E). Each color in a plot indicates one gene function. Detailed descriptions are shown on the right side of each plot. The colors for others in plots B and C indicate all other gene functions not listed in these two plots. See Figure 1 for the definition of each group.
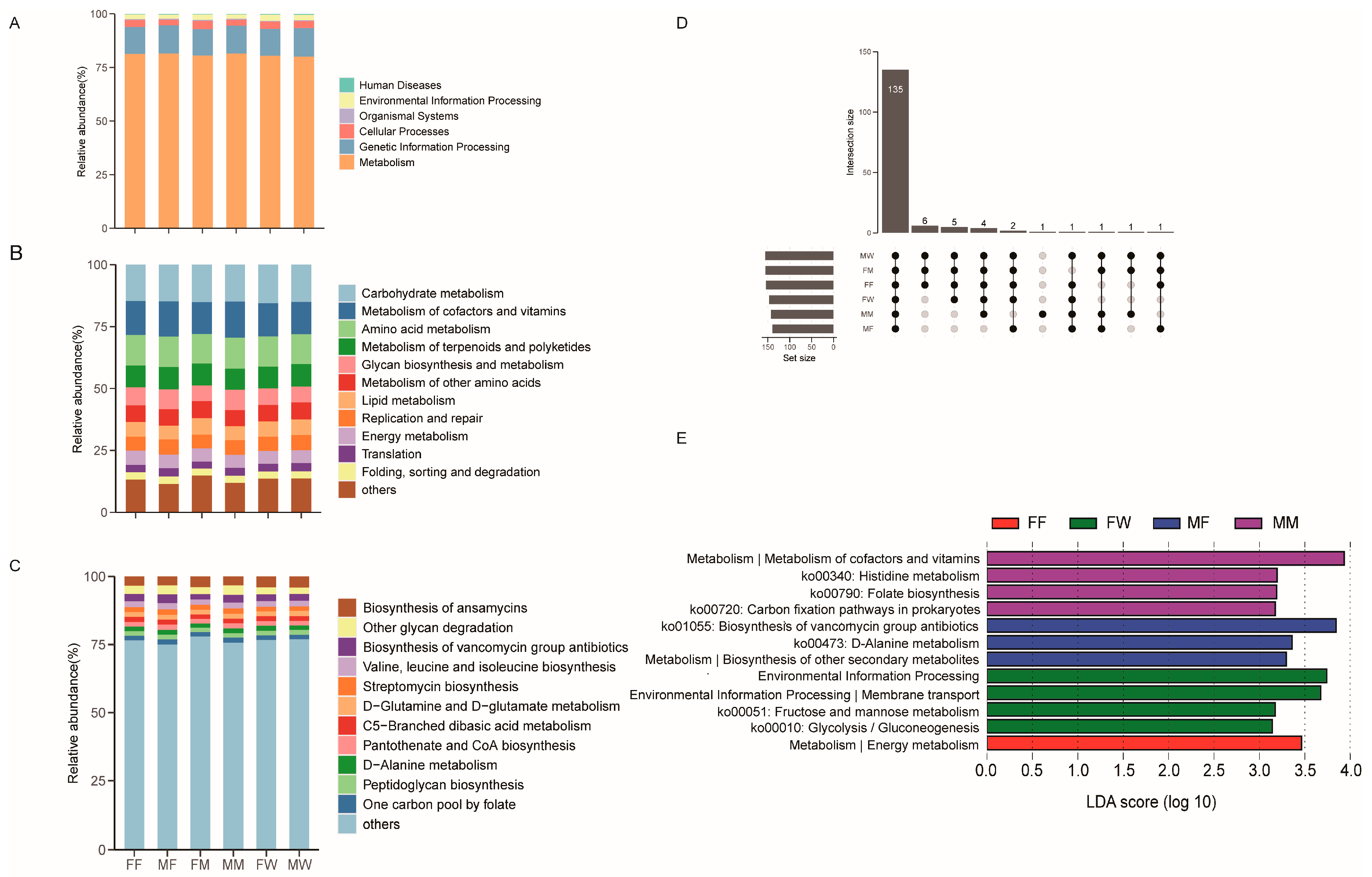
Table 1.
Prey items potentially available to Japanese geckos in the wild.
| Abundance of prey items | Order |
| Numerous (> 500) | Lepidoptera, Diptera |
| More (between 100 and 500) | Coleoptera, Hemiptera |
| Medium (between 50 and 100) | Hymenoptera, Ephemeroptera, Trichoptera |
| Fewer (between 10 and 50) | Orthoptera, Mantodea, Neuroptera, Megaloptera, Thysanoptera, Plecoptera, Blattodea |
| Least (< 10) | Dermaptera, Odonata, Corrodentia, Rhaphidioptera |
1 The abundance of prey items is sorted by the number of insects found in the light trap.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



