
Submitted:
02 March 2023
Posted:
03 March 2023
You are already at the latest version
Preprints.org 2023 Most Popular Preprints Award Winner Collection
Abstract
Over recent decades, therapeutic proteins have had widespread success in treating a myriad of diseases. Glycosylation, a near universal feature of this class of drugs, is a critical quality attribute that significantly influences the physical properties, safety profile and biological activity of therapeutic proteins. Optimizing protein glycosylation, therefore, offers an important avenue to developing more efficacious therapies. In this review, we discuss specific examples of how variations in glycan structure and glycoengineering impacts the stability, safety, and clinical efficacy of protein-based drugs that are already in the market as well as those that are still in preclinical development. We also highlight the impact of glycosylation on next generation biologics such as T cell-based cancer therapy and gene therapy.
Keywords:
glycosylation
; glycoengineering
; biologic
; therapeutic protein
; gene therapy
; cell-based therapy
; monoclonal antibody
1. Glycosylation of Therapeutic Proteins
Since the commercial release of human insulin as the first therapeutic protein in the early 1980s, biologics have been the fastest-growing class of therapeutic molecules. In 2019 alone, the market share for biopharmaceuticals amounted to over 200 billion dollars in the United States(Feng et al., 2022, Moorkens et al., 2017), with over 350 new products approved for clinical use by the Food and Drug Administration by 2021(Feng et al., 2022). Therapeutic proteins make up the biggest fraction of the biologics sector, encompassing a plethora of antibodies, vaccines, immune factors, hormones, blood factors, and enzymes and are used to treat both communicable and non-communicable diseases such as cancer, diabetes, multiple sclerosis, and SARS-CoV-2, to name a few (Figure 1).
Most therapeutic proteins such as vaccines, monoclonal antibodies, hormones, enzymes and immune factors undergo N-linked or O-linked glycosylation. N-linked glycans consist of carbohydrate molecules that are attached to the nitrogen atom on Asparagine (Asn) residues in the protein, while O-linked glycans consist of carbohydrates linked to the oxygen atom on Serine (Ser) or Threonine (Thr) residues in the protein.
The dominance of protein-based therapeutics in the market speaks to their immense positive impact in the clinic. Compared to small molecule drugs, proteins demonstrate high target specificity, which can result in lower toxicity from fewer off-target effects and improved pharmacological potency. Additionally, one analysis suggests that development times for protein-based drugs is typically shorter than that for small molecules(Beall et al., 2019). However, these biopharmaceuticals are not without issues, such as intrinsic limitations in their physicochemical and pharmacological characteristics. Thus, a focal point of biologic development has been to generate more efficacious formulations of therapeutic proteins through protein and cellular engineering.
Many protein-based drugs are engineered glycoproteins that are recombinantly expressed in animal cell-lines, and almost all such biopharmaceuticals undergo post-translational modification (PTM). Perhaps the most important class of PTM for many biologics is glycosylation, a process that occurs on most eukaryotic secreted and membrane proteins. Glycosylation involves the covalent addition of carbohydrates (glycans) to a protein through two major linkages: (a) the amide nitrogen atom on an asparagine (Asn) residue (N-linked glycosylation), and (b) the hydroxyl oxygen on serine (Ser), threonine (Thr) and tyrosine (Tyr) residues (O-linked glycosylation). These carbohydrate groups can be a single monosaccharide or chains of branched or linear oligosaccharides(Reily et al., 2019, Varki et al., 2022) (Figure 1). Furthermore, a glycoprotein can have many different “glycoforms”, with variations pertaining to either glycosylation site occupancy (macroheterogeneity) or differences in glycan structure (microheterogeneity). Not only does glycosylation increase protein structural diversity, glycan heterogeneity is also a crucial contributor in determining biophysical, and pharmacological properties of glycoproteins. Glycoengineering—the manipulation of glycan composition—has therefore been an invaluable tool in generating products that demonstrate optimal therapeutic efficacies(Sola et al., 2007, Sola and Griebenow, 2009, Ma et al., 2020, Sinclair and Elliott, 2005, Chen et al., 2022, Dammen-Brower et al., 2022). As such, because of the impact of glycans on protein structure, function, and dynamics, we view glycoengineering as an essential protein engineering method that complements and amplifies changes introduced by mutagenesis. In this review, we describe how glycosylation significantly impacts key characteristics of protein-based drugs such as their stability, transport and uptake, half-life, therapeutic efficacy, and immunogenicity. We also discuss how glycoengineering can be applied to improve newer classes of biologics such as T cell- and oligonucleotide-based therapies.
1.1. Stability
Proteins are innately prone to degradation due to physical and chemical processes like denaturation, proteolysis, aggregation, oxidation and hydrolysis. Overcoming the inherent instability of glycoproteins is key to therapeutic protein development. Preservation of glycoprotein conformation ensures that they remain intact and functionally active during storage and after they have been administered to patients. Degradation of therapeutic proteins can result in reduced or complete loss of efficacy and compromised safety.
The presence of large, hydrophilic groups such as glycans on a protein can improve their stability by preventing aggregation, contributing to increased thermal and chemical stability, and making them more resistant to enzymatic degradation(Lis and Sharon, 1993, Mitra et al., 2006, Shental-Bechor and Levy, 2008, Sola and Griebenow, 2009, Sola et al., 2007, Zheng et al., 2011, Zhou and Qiu, 2019, Wang et al., 1996, Lee et al., 2015). Glycosylation can lead to an increase in internal electrostatic interactions, and strengthen hydrogen bonds and hydrophobic interactions within the protein, making it more resistant to denaturation(Sola and Griebenow, 2009, Sola and Griebenow, 2006, Sola et al., 2007, Lee et al., 2015). Glycans around the peptide backbone also make the protein less accessible to proteases through steric hindrance, thereby making them less susceptible to proteolysis(Nishiyama et al., 2000, Sola and Griebenow, 2009). One mechanism through which glycosylation improves solubility is to increase the solvent-accessible surface area of the glycoprotein facilitated by the presence of the glycans(Tams et al., 1999, Paul et al., 2021, Sola and Griebenow, 2006). This can prevent protein aggregation, which often leads to an increased likelihood of adverse immune reactions and accelerated clearance from the bloodstream(Lundahl et al., 2021, Pham and Meng, 2020, Wang, 2005). Resistance against aggregation is therefore important in preserving both the safety and efficacy of the molecule.
Most biologic drugs are typically more stable and less prone to aggregation, and denaturation than their non-glycosylated counterparts (Supplementary Table S1). Biologics such as interferon-β (IFN-β)(Runkel et al., 1998, Karpusas et al., 1998, Farrell et al., 2012), alpha-1 antitrypsin (AAT)(Jeppsson et al., 1975, Travis et al., 1985, Kwon and Yu, 1997), granulocyte stimulating factor (GCSF)(Oh-eda et al., 1990), erythropoietin (EPO)(Narhi et al., 1991) become more susceptible to aggregation and thermal degradation upon removal of glycan moieties, to name a few examples. Protection against protease digestion can also be imparted just by the presence of glycosylation, such as in the case of interferon-γ(Sareneva et al., 1995), interferon-α(Ceaglio et al., 2008, Ceaglio et al., 2010) and GCSF(Carter et al., 2004). Furthermore, the abundance and type of attached carbohydrates can also be tweaked to improve physical characteristics of the protein. Site specific addition of glucosyl moieties on human insulin improved physical stability in solution(Baudys et al., 1995, Uchio et al., 1999). Indeed, a glycoengineered variant of insulin that is O-mannosylated at Thr27 (B-chain) has a comparable activity to the naturally occurring protein while being more resistant to enzymatic degradation and oligomerization(Guan et al., 2018).
1.2. Half-life
The therapeutic efficacy of a biopharmaceutical is largely impacted by how long it remains functional in circulation. Glomerular filtration by the kidneys eliminates small proteins and peptides, and depends mostly on size and charge, with molecules under ~30 kDa typically being eliminated through this route(Tryggvason and Wartiovaara, 2005, Maack et al., 1979, Mahmood and Green, 2005, Dammen-Brower et al., 2022). Since the glomerular filter is negatively charged, anionic peptides are typically repelled by the charge similarity and therefore less likely to be eliminated(Rennke et al., 1975, Bocci, 1989, Tryggvason and Wartiovaara, 2005, Mahmood and Green, 2005). Apart from renal filtration, protein clearance is also mediated by receptors that recognize and bind specific types of glycans. Two avenues of serum clearance are through the mannose receptor (MR)(Stahl, 1992, Ashwell and Harford, 1982) and the asialoglycoprotein receptor (ASGPR)(Stockert, 1995, Ashwell and Harford, 1982, Ashwell and Morell, 1974) (Figure 2A). Both are C-type lectin receptors that recognize and bind specific carbohydrates decorating glycoproteins, thereby facilitating their elimination from the blood. Mannose receptors are found primarily in liver Kupffer and endothelial cells(Hubbard and Stukenbrok, 1979, Schlesinger et al., 1978, Linehan et al., 1999, Takahashi et al., 1998) and immune cells, such as macrophages(Shepherd et al., 1982, Stahl et al., 1978, Stahl and Gordon, 1982, Wileman et al., 1986, Largent et al., 1984, Martinez-Pomares, 2012) and immature dendritic cells(Sallusto and Lanzavecchia, 1994, Martinez-Pomares, 2012). They bind glycans bearing a terminal mannose, fucose, or N-acetylglucosamine (GlcNac)(Stahl, 1990, Taylor and Drickamer, 1993). ASGPRs, on the other hand—while also abundantly found in liver cells—recognize terminal β-linked galactose or N-acetylgalactosamine (GalNac) that have been desialylated; hence the prolonged serum longevity of highly sialylated glycoproteins(Stockert, 1995, Ashwell and Harford, 1982, Morell et al., 1968, Morell et al., 1971, Ashwell and Morell, 1974, Van Den Hamer et al., 1970, Park et al., 2003). Additionally, ASGPRs can bind and clear proteins bearing a terminal Sia2,6GalNac and Sia2,6Gal(Park et al., 2005), suggesting that in addition to sialic acid capping, engineering the terminal linkage to be α-2,3 instead of α-2,6 could prolong the half-life of a therapeutic protein(Andre et al., 2004, Andre et al., 1997, Unverzagt et al., 2002, Tian et al., 2019).
Because a protein’s circulatory half-life can be significantly impacted by glycosylation, evading and bypassing the different serum clearance mechanisms through glycoengineering is important aspect in biopharmaceutical design (Supplementary Table S2). Adding or modifying sugar groups to increase the size, hydrodynamic radius and net negative charge of the protein can decrease the rate of removal via kidney filtration. Incomplete glycosylation or glycan removal has resulted in recombinant human proteins with much shorter in vivo half-lives compared to their fully glycosylated counterparts—such as in the case of AAT(Travis et al., 1985, Ross et al., 2012, Yu and Gan, 1978, Weber et al., 1985), EPO(Wasley et al., 1991, Yamaguchi et al., 1991, Fukuda et al., 1989) and Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF)(Okamoto et al., 1991). Conversely, simply adding more attached carbohydrates to proteins—such as EPO(Su et al., 2010, Elliott et al., 2004), follicle-stimulating hormone (FSH)(Perlman et al., 2003, Weenen et al., 2004) and interferon-α(Ceaglio et al., 2008)—can prolong circulation and activity in vivo. Glycoengineering to increase in levels of sialylation or sialic acid capping of terminal galactose or GalNac is also utilized to inhibit ASGPR binding and limit hepatic clearance. This approach has been applied to improve serum longevity for therapeutic proteins such as EPO(Egrie et al., 2003, Egrie and Browne, 2001), human growth hormone(Flintegaard et al., 2010), α-galactosidase(Sohn et al., 2013a), iduronate sulfatase(Muenzer et al., 2007, Muenzer et al., 2006), and β-glucuronidase(Cadaoas et al., 2020).
1.3. Transport and Uptake
In addition to eliminating molecules from circulation, glycan-binding cell-surface receptors also participate in the cellular targeting and tissue distribution of therapeutic glycoproteins, particularly those used for enzyme replacement therapy (ERT). Lysosomal storage diseases (LSDs) are inherited metabolic disorders with a deficiency of lysosomal enzymes and an accumulation of unwanted metabolites, ultimately resulting in clinical dysfunction in peripheral organs and the central nervous system(Futerman and van Meer, 2004, Parenti et al., 2013, Bonam et al., 2019). Many LSDs are treated by ERT, whereby patients receive intravenous supplementation of the deficient enzyme—which is itself a glycoprotein. Trafficking and uptake of these lysosomal enzymes are mediated largely by the mannose-6-phosphate dependent pathway, which involves recognition and binding by mannose-6-phosphate receptors (MPRs)(Sly, 1985, Parenti et al., 2013, Varki and Kornfeld, 1980, Achord et al., 1978, Stahl et al., 1978) (Figure 2B). Like ASGPRs and MRs, MPRs are lectins that recognize and bind specific glycosylation features on glycoproteins, particularly those that bear mannose-6-phosphate residues(Kaplan et al., 1977, Seo and Oh, 2022). Glycan manipulation ERT enzymes for better lysosomal targeting has improved cellular uptake and increase therapeutic efficacy.
Gaucher’s Disease is one of the very first LSDs for which ERT was developed(Barton et al., 1991). This disorder is typified by a deficiency in β-glucocerebrosidase(Brady et al., 1966, Brady et al., 1965), a lysosomal hydrolase internalized by macrophages through an MR-dependent pathway(Sato and Beutler, 1993, Shaaltiel et al., 2007). Intravenous supplementation of the β-glucocerebrosidase is the most prevalent treatment modality among individuals affected with Gaucher’s disease. Enriching for exposed terminal mannose residues on this enzyme leads to better engagement of macrophages through MRs and increased uptake by affected cells(Doebber et al., 1982, Furbish et al., 1981, Friedman et al., 1999). Thus, the three commercially available versions of β-glucocerebrosidase (Imiglucerase(Grabowski et al., 1995), Velaglucerase alfa(Zimran et al., 2007) and Taliglucerase alfa(Shaaltiel et al., 2007)) were developed to contain high mannose structures(Tekoah et al., 2013).
Additional ERTs developed for the management of other LSDs include α-galactosidase for Fabry Disease(Desnick, 2001, Schiffmann et al., 2000, Schiffmann et al., 2001), α-glucosidase for Pompe Disease(Kishnani et al., 2006, Rossi et al., 2007, Van den Hout et al., 2004, Chen et al., 2009), α-L-Iduronidase for Mucopolysaccharidosis (MPS) Type I(Kakkis et al., 2001, Clarke et al., 2009, Parini and Deodato, 2020, Wraith et al., 2004, Dornelles et al., 2017), iduronate sulfatase for MPS Type II(Muenzer et al., 2002, Garcia et al., 2007, Muenzer et al., 2007, Muenzer et al., 2006, Sohn et al., 2013b, Wraith, 2008), GalNac-6-Sulfatase for MPS Type IVA(Hendriksz et al., 2014, Hendriksz et al., 2018), GalNac-4-Sulfatase for MPS Type IV(Harmatz et al., 2006, Harmatz et al., 2004), and β-glucuronidase for MPS Type VII(Fox et al., 2015, Wang et al., 2020, Harmatz et al., 2018); all of these rely on MPR signaling to facilitate lysosomal targeting and delivery (Seo and Oh, 2022, Oh, 2015). Similar to β-glucocerebrosidase, these therapeutic protein products have been glycoengineered to improve cellular uptake and biodistribution by enriching for glycoforms bearing mannose-6-phosphate residues (Oh, 2015, Seo and Oh, 2022, Lee et al., 2003, Sakuraba et al., 2006, Zhu et al., 2004, Zhu et al., 2009, Park et al., 2018, Lachmann, 2011, Tiels et al., 2012, Kakkis et al., 1994, Muenzer et al., 2007, Togawa et al., 2014, Parini and Deodato, 2020, Tomatsu et al., 2007) (Supplementary Table S3).
1.3. Immunogenicity
Even more important than biological potency, product safety is paramount in biologics. Adverse immunological responses to therapeutic proteins undermine both the safety of the subject and the molecule’s therapeutic efficacy. In addition to the development of unwanted acute allergic or inflammatory reactions against a recombinant protein, unwanted immune engagement can also lead to a partial or complete loss of pharmacological activity due to binding by neutralizing antibodies(Porter, 2001, Wadhwa et al., 2015, Wang et al., 2008) or accelerated serum clearance(Ehrenpreis, 2017, Lundahl et al., 2021, Filipe et al., 2014). The development of anti-drug antibodies has been documented among patients treated with recombinant EPO(Mayeux and Casadevall, 2003, Casadevall et al., 2002), interferon-α(Bonetti et al., 1994, Douglas et al., 1993, Fossa et al., 1992), interferon-β(Zang et al., 2000, Konrad et al., 1987, Larocca et al., 1989), insulin(Di Mario et al., 1986, Fineberg et al., 1983), thrombopoietin(Li et al., 2001), and factor VIII(Pratt and Thompson, 2009)—to name a few examples. Stimulation of the immune response against therapeutic proteins is driven by several factors, including aggregate formation(Pham and Meng, 2020, Lundahl et al., 2021, Wang, 2005)—which in turn, can be controlled through glycan manipulation (see Section 1.1). Numerous glycosylated therapeutic proteins, such as interferon-β(Runkel et al., 1998, Kivisakk et al., 2000) and EPO(Elliott et al., 1996), are less immunogenic compared to their deglycosylated counterparts due to increased stability and a decreased propensity for aggregation. In some cases, increasing protein sialylation could also be applied to minimize the antigenicity of the drug. Desialylated EPO for example, has demonstrated higher immunoreactivity(Wide et al., 2003), and decreased antigenicity was observed with increased sialylation for asparaginase(Fernandes and Gregoriadis, 2001) (Supplementary Table S4).
The presence of non-human glycan structures in recombinant biotherapeutics can also trigger adverse immune responses. Although host expression systems in bacteria, yeast and plants have been utilized in the past, mammalian systems such as Chinese Hamster Ovary (CHO) cells are predominantly used to manufacture recombinant glycoproteins with the aim of producing glycoforms that most closely resemble their human-derived counterparts. Despite this, inherent genetic differences between the glycosylation machineries of nonhuman mammals, such as murine myeloma cell lines (i.e. NS0 and Sp2/0), relative to humans can lead to the addition of glycan moieties that are normally absent in humans, and thus trigger adverse immunological reactions. The most commonly reported immunogenic glycan epitopes are the ɑ-Gal epitope (Galɑ1,3-Gal) and N-glycolylneuraminic acid (Neu5GC)(Butler and Spearman, 2014, Goh and Ng, 2018) (Figure 2C).
Antibodies against ɑ-Gal are naturally abundant in human serum (approximately 1% of circulating immunoglobulins), due to continuous antigenic stimulation from normal gut flora(Galili et al., 1988). This can lead to hypersensitivity against proteins terminating in ɑ-Gal residues. Unfortunately, many monoclonal antibodies generated from mouse-derived cell lines contain ɑ-Gal epitopes(Sheeley et al., 1997, Yoo et al., 2002, Macher and Galili, 2008), which undermines their clinical safety. Such was the case with cetuximab, a monoclonal antibody for treating colorectal and head neck cancers. A subset of patients treated with cetuximab experienced anaphylaxis due to the pre-existing population of ɑ-Gal IgE antibodies in their bloodstream(O'Neil et al., 2007, Chung et al., 2008). Roughly 30% of the 21 glycoforms present in cetuximab were capped by ɑ-Gal residues(Qian et al., 2007). Neu5GC is a modified sialic acid that is also potentially immunogenic glycan structure common to many nonhuman mammalian cell lines. It can be assimilated from exogenous sources into newly synthesized glycans and presented on human cells, but as with ɑ-Gal epitopes, humans produce circulating antibodies against Neu5GC(Altman and Gagneux, 2019, Padler-Karavani et al., 2008, Tangvoranuntakul et al., 2003, Nguyen et al., 2005). This co-existence of anti-Neu5GC antibodies and epitope incorporation correlates well with chronic inflammation-mediated diseases(Okerblom and Varki, 2017, Varki, 2017, Dhar et al., 2019). Adverse reactions have been observed among patients treated with rabbit-derived anti-thymocyte globulin, likely due to the immune response triggered by Neu5GC(Salama et al., 2017, Amon et al., 2017, Yehuda and Padler-Karavani, 2020). A comparative study of murine myeloma-derived cetuximab, bearing terminal Neu5GC residues, and CHO-derived panitumumab, containing negligible Neu5GC residues, also revealed that when exposed to human serum containing high levels of anti-Neu5GC antibodies, immune complex formation was only observed against cetuximab(Ghaderi et al., 2010); this suggests that patients who have higher levels of anti-Neu5GC antibodies could be prone to adverse reactions when treated with cetuximab (Supplementary Table S4).
Given the importance of glycosylation to modulating the glycoprotein immunogenicity, tweaking its composition can help elevate the immune response when necessary—as in the case of vaccines. Many viruses, such as SARS, influenza, and HIV, have surface proteins that are extensively coated by host-derived glycans. In addition to being essential to binding and entry into the host cells, these attached sugars form a glycan shield, masking the viral epitopes that can be neutralized by circulating antibodies and allowing them to evade the host immune system(Wanzeck et al., 2011, Fenouillet et al., 1994, Watanabe et al., 2020, Tate et al., 2014). In some cases, the presence of the glycan structures is integral to the immunogenic epitope and are necessary for engagement by neutralizing antibodies(Behrens et al., 2016). Modifying glycosylation on subunit viral vaccine candidates, such has hemagglutinin (influenza), S protein (SARS) and ENV protein (HIV), can significantly alter immune response and improve clinical efficacy. By altering glycans on hemagglutinin (HA) to trim heterogenous complex-type glycans down to only N-linked GlcNacs, better binding and neutralization was obtained, along with broader cross-strain activity in mice (Chen et al., 2014, Wang et al., 2009). Likewise, an S protein vaccine against SARS-CoV-2 that was enzymatically modified to be mono-GlcNac-decorated induced a stronger immune response compared to normally glycosylated S protein, and protected vaccinated animals against wild-type virus and other variants of concern(Huang et al., 2022). Furthermore, an mRNA vaccine candidate with specific glycosites removed in the S2 domain of the S protein resulted in higher antibody neutralization and CD8+ T cell activity against variants of concern relative to the wild-type SARS-CoV-2 mRNA vaccine(Wu et al., 2022). One caveat, however, of mRNA vaccines is that while genetic engineering of glycan occupancy can be achieved, optimizing glycan microheterogeneity at specific sites is not possible on this platform, due to the fact that the protein glycosylation machinery is completely dependent on the host cell. The stronger and broader immune engagement observed in vaccine candidates after glycan shield removal is likely due to the exposure of conserved previously hidden epitopes on the protein surface. Alternatively, the addition/removal of glycan structures can also lead to the creation of neoepitopes not found in the native pathogen, which can subsequently be targeted by the immune response. Using this approach, immune response to HIV vaccine candidates can be redirected. By knocking in a novel glycosite on an HIV Env trimer vaccine candidate, they masked a previously immunodominant epitope and instead created new epitopes on the protein that could be recognized by neutralizing antibodies by knocking out multiple N-linked glycosites(Ringe et al., 2019). It is important to note, however, that full removal of glycans could decrease vaccine efficacy for sites where the glycans impact protein conformation(Chen et al., 2014, Huang et al., 2022, Wu et al., 2022). Moreover, eliminating glycans entirely runs the risk of creating neoepitopes that are not recognized by the immune system upon infection by the native virus, as seen with deglycosylated HIV env (Zhou et al., 2017). Nevertheless, these examples demonstrate that by leveraging informed glycan design, viral vaccines could provide stronger and longer lasting protection, and also be effective against a broader spectrum of strains and variants, reducing the frequency of needed updates to formulations in response to new mutants.
2. Monoclonal Antibodies
Monoclonal antibodies (mAb), such as recombinant immunoglobulins of the IgG1 subtype, are monospecific in terms of the epitope recognized. Therapeutic mAbs account for 80% of total antibody sales in the United States(2020). Furthermore, the global therapeutic mAb market is expected to generate more than $300 billion globally per year (Lu et al., 2020). Therefore, understanding their critical quality attributes, specifically glycosylation, ensures their safety and similarity as effective therapies.
IgG antibodies contain two heavy chains and two light chains to form three major domains: two identical Fab domains for antigen binding and an Fc domain (dimeric base of the antibody)(Schroeder and Cavacini, 2010). Asparagine(N) 297-linked glycosylation occurs in the Fc region and in the form of biantennary complex structures(Mimura et al., 2018, Varki et al., 2022) (Figure 3). Glycosylation at the Fc region of IgG proteins can greatly impact antibody structure and effector functions (Jefferis, 2009a, Jefferis, 2012).
The constant region of antibodies contains binding sites for immune effector molecules such as the complement system or Fc receptors (Vidarsson et al., 2014). These receptors help recruit immune mediators, generally via Fc receptor binding. Antibody Fc receptors mediate the cell killing effects of mAbs by complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), or antibody-mediated phagocytosis by monocytes/macrophages(Weiner et al., 2010, Ludwig et al., 2003, Jefferis, 2009b). In ADCC, immune complexes are engaged with FcγRIIIa on natural killer (NK) cells or tumor cell apoptosis can be induced through the suppression of pro-survival ligands or inhibition of signal receptor dimerization. CDC is activated by the binding of complement component C1q to the mAb Fc region to initiate the complement cascade (Raju, 2008, Dekkers et al., 2017, Zhou et al., 2008b, Pereira et al., 2018). These mechanisms are particularly important for cancer immunotherapy; indeed, Fc-mediated effector cell recruitment and functions, such as ADCC, are crucial to tumor-targeting antibodies. For instance, rituximab or trastuzumab activity was abolished in genetically modified mice that lacked FcγR expression or had defective FcγR signaling. In contrast, FcγRIIb knockout mice showed enhanced efficacy(Clynes et al., 2000, de Haij et al., 2010). Although glycosylation at the N297 region accounts for only 2-3% of antibody mass, IgG-Fc glycosylation and structure are critical for Fc effector functions, such as ADCC and CDC (Chan and Carter, 2010, Jefferis, 2009b). Aglycosylated Fc-IgGs reduce binding affinity to FcγRI and eliminate binding to FcγRII and FcγRIII receptors, and C1q-mediated processes such as phagocytosis, ADCC, and CDC are abated or severely impaired in aglycosylated IgG. (Sazinsky et al., 2008, Raju et al., 2000, Nose and Wigzell, 1983, Pound et al., 1993, Sarmay et al., 1992, Tao and Morrison, 1989, Woof and Burton, 2004).
2.1. Fc Glycosylation and mAb Effector Function: Fucosylation
Core fucosylation of Fc N-glycans on mAbs occurs when ɑ-1,6-linked fucose is attached to the innermost GlcNAc moiety (Garcia-Garcia et al., 2021). The removal of the core fucose increases Fc affinity for FcγRIIIa in all IgG subclasses, thereby inclusively augmenting ADCC activity and improving therapeutic efficacy(Shields et al., 2002, Shinkawa et al., 2003). Due to the impact of Fc fucosylation on FcRγIIIa binding, many monoclonal antibodies and antibody-drug conjugates being developed for the clinic are being engineered or “glyco-optimized” to decrease fucose(Golay et al., 2022, Tong et al., 2021, Pereira et al., 2018), with a few already commercially available or in clinical trials, such as obinutuzumab (anti-CD20)(Tobinai et al., 2017), mogamulizumab (anti-CCR4)(Watson and Marx, 2019), belantamab mafodotin (anti-BCMA)(Lassiter et al., 2021), benralizumab(anti-IL5Rα)(Pelaia et al., 2018), imgatuzimab (anti-EGFR)(Temam et al., 2017) and tomuzotuximab (anti-EGFR)(Fiedler et al., 2018) (Supplementary Table S5). Furthermore, an increase or decrease in ADCC activity produced by other glycan features always occurs in the context of core fucosylation(Li et al., 2017, Shinkawa et al., 2003).
2.2. Fc Glycosylation and mAb Effector Function: Sialylation
Whether or not sialic acids are well-suited in ADCC- and CDC-related therapies, sialylated IgGs have recently garnered substantial interest as immunosuppressants for autoimmune and inflammatory diseases, as demonstrated by intravenous immunoglobulin (IVIG) therapy (Nimmerjahn and Ravetch, 2008, Kaneko et al., 2006). IVIG therapy involves administering concentrated IgG derived from pooled plasma to patients (Seite et al., 2008). As a highly effective biologic in treating several autoimmune diseases, including idiopathic thrombocytopenic purpura (ITP), chronic inflammatory demyelinating polyneuropathy, and myasthenia gravis, IVIG consumption has increased approximately 400-fold since 1980, and approximately 100 tons are consumed per year(Orange et al., 2006, 2019). Sialylated IgGs initiate anti-inflammatory responses through murine C-type lectin-like receptor-specific intracellular adhesion molecule-grabbing non-integrin R1 (SIGN-RI) (DCSIGN in humans), expressed by macrophages and dendritic cells. As a result, FcγRIIb is upregulated, increasing Treg cell populations and suppressing inflammatory responses (Anthony and Ravetch, 2010, Anthony et al., 2008, Kaneko et al., 2006, Sondermann et al., 2013). Although IgG sialylation is not the main determinant of anti-inflammatory effect of IVIG therapy, IgG sialylation does enhance the efficacy(Schwab and Nimmerjahn, 2013). Increased endogenous IgG sialylation improved treatment of Kawasaki disease during IVIG therapy(Ogata et al., 2013), and sialylated human IgG reduced the severity of rheumatoid arthritis in mouse models, an effect that was not observed using desialylated human IgG(Kaneko et al., 2006). Similarly, a 10-fold increase in anti-inflammatory activity was seen with tetra-Fc sialylation compared to asialylated IVIG across several animal models(Washburn et al., 2015).
2.3. Fc Glycosylation and mAb Effector Function: Galactosylation
The effector functions of galactosylation, particularly ADCC, should be addressed on a case-by-case basis. In some cases, the terminal galactose residue content had no effect on ADCC activity. For example, the degalactosylation of rituximab and other recombinant mAbs with variable galactose contents confirmed that ADCC activity was unaffected(Hodoniczky et al., 2005). To complicate matters, increased galactosylation can promote ADCC activity or inhibit ADCC activity(Houde et al., 2010, Nimmerjahn et al., 2007, Kumpel et al., 1994, Pereira et al., 2018, Zhang et al., 2020b, Aoyama et al., 2019). Interestingly, site-specific galactose attachment on the N-glycan structure in afucosylated palivizumab influences FcγRIIIA binding and ADCC activity, as shown with enzymatic transglycosylation using chemically defined N-glycans(Hatfield et al., 2022). Although terminal galactose may only play a minor role in ADCC activity, it is critical for CDC activity(Hodoniczky et al., 2005). Galactosylated rituximab had higher CDC than degalactosylated glycoforms due to its higher affinity to C1q receptors; when Campath-1H was deglycosylated, CDC activity was reduced by 50%(Peschke et al., 2017, Hodoniczky et al., 2005, Boyd et al., 1995). Based on these findings, additional research on the effects of galactosylation on ADCC activity and its subclasses is required, and different glycoforms should be considered whenever developing new drugs.
2.4. Fc Glycosylation and mAb Effector Function: Bisecting GlcNac
Human serum contains ~10% IgGs by protein mass, and many of their N-glycans contain bisecting GlcNAc structures(van de Bovenkamp et al., 2016, Gudelj et al., 2018). These features can only be produced in human or murine cells (e.g., S20); CHO cells are unable to produce N-glycans with bisecting GlcNAc because of a lack of active N-acetylglucosaminyltransferase-III (GnT-III) required for its synthesis(Campbell and Stanley, 1984, Sallustio and Stanley, 1989, Umana et al., 1999), although the enzyme can be genetically activated(Karottki et al., 2020, Shamie et al., 2021). Overexpression of GnT-III was applied to increase bisecting GlcNAc attachment to N-glycans in therapeutic mAbs, thereby improving FcR-binding(Davies et al., 2001). Specifically, ADCC activities were 10-30-fold higher when mAbs containing bisecting GlcNAc N-glycans bound to FcRIIIa receptors (Davies et al., 2001, Shinkawa et al., 2003). For example, adding bisecting GlcNAc without removing core fucosylation improved ADCC by approximately 10-fold, indicating that N-glycans with bisecting GlcNAc structures can enhance ADCC activity. Furthermore, trastuzumab-bearing N-glycans modified with bisecting GlcNAc increased ADCC 10-fold, comparable to that observed for similarly modified rituximab(Hodoniczky et al., 2005). However, these finding have been contradicted. Since incorporation of bisecting GlcNAc is not a suitable substrate for 1,6-fucosyltransferase, N-glycans containing such structures are always associated with loss of core fucosylation. As a result, removing the core fucose rather than bisecting GlcNAc may still have the greatest impact on the therapeutic antibody ADCC activity(Shinkawa et al., 2003, Schachter, 2000).
2.5. Fc Glycosylation and mAb Effector Function: Mannosylation
The prevalence of high-mannose N-glycans on recombinant mAbs can vary substantially (more than 1-20% in both CHO and murine cells), but endogenous human IgG contains only trace amounts (<0.1%) of these glycovariants(Flynn et al., 2010, Goetze et al., 2011). ADCC activity is enhanced in mAbs with high-mannose glycoforms(Liu, 2015, Yu et al., 2012, Shi and Goudar, 2014, Brady et al., 2015, Liu et al., 2017). However, similar to the other glycoforms mentioned previously, changes in ADCC are more directly associated with the loss of core fucosylation(Zhou et al., 2008a, Brady et al., 2015). Furthermore, mAbs with high mannose structures can have a negative impact on CDC activity by lowering the binding affinity with C1q(Walsh, 2018). Other studies have reported similar results for high-mannose mAbs that reduce CDC activity by lowering binding to C1q. Thus, Fc mannosylated mAbs have a positive effect on ADCC but a negative effect on CDC activity(Yu et al., 2012, Zhou et al., 2008a, Hiatt et al., 2014).
3. Future Perspectives: Next Generation Biologics
Glycoengineering of therapeutics has been applied to a variety of therapeutic glycoproteins(Walsh, 2018, Majewska et al., 2020). Beyond protein-based drugs, however, next-generation biologics, such as cell therapy- and nucleic acid-based therapeutics, offer opportunities for treating cancer, infectious diseases, immune disorders, and inherited genetic diseases(Kulkarni et al., 2021, Weber et al., 2020). Here we describe how glycosylation can impact cell therapy, gene therapy, and drug delivery platforms, including specific examples of glycoengineering to improve biological activity and clinical efficacy.
3.1. T-Cell Therapy
T-cell based immunotherapies, in particular chimeric antigen receptor (CAR) T-cells, are gaining traction especially for cancers of the blood and bone marrow(June et al., 2018, Feins et al., 2019). For these, T-cells are collected and transfected to express surface receptors that bind to and eliminate tumor cells. T-cell therapies rely on interactions between tumor cell ligands or antigen presenting cells (APCs) and co-stimulatory receptors on T-cells, such as CD28, inducible costimulatory (ICOS), and 4-1BB, which promote T-cell proliferation and cytotoxicity (Chen and Flies, 2013) (Figure 4A). Receptors on immune cells—including T-cell receptors (TCRs)—are glycosylated, and specific glycosylation patterns are pivotal in immune function, including communication between immune cells and the modulation of their anti-tumor activity(Sun et al., 2021, Mereiter et al., 2019). For example, inhibiting N-glycosylation of CD28 increased interaction with its CD80 ligand, thereby enhancing its co-stimulatory signaling activity(Ma et al., 2004). Sialidase treatment of T-cells and APCs also enhanced CD28-mediated activation of T-cells and reactivation of exhausted T-cells, possibly by eliminating sialic acid-containing glycans that compete for CD28-CD80 interactions(Edgar et al., 2021). Removing specific glycosites in ICOS led to intracellular sequestration of the receptor(Kamei et al., 2010) and altered its ligand binding affinity(Kamei et al., 2010, Rujas et al., 2020). Galectin-9 also binds 4-1BB, a co-stimulatory signaling receptor on T-cells, thus controlling T-cell function by facilitating 4-1BB surface expression(Madireddi et al., 2014). Deglycosylation of 4-1BB results in reduced galectin-9 binding(Madireddi et al., 2014). Furthermore, the mutation of N-glycosylation sites reduces membrane expression of 4-1BB and decreases polyubiquitination, thereby inducing multimerization of 4-1BB, which may hamper 4-1BB receptor signaling(Sun et al., 2022).
Co-inhibitory signaling pathways mediated by receptors on T-cells (e.g., PD-1, CTLA-4, and TIM-3) induce exhaustion and apoptosis and inhibit cytotoxic function(Chen and Flies, 2013) (Figure 4A). The PD-1/PD-L1 axis has been a target of cancer immunotherapy, and its inhibitory function depends on PD-1 glycosylation. PD-1 depends on core fucosylation, as the inhibition of FUT8 attenuated PD-1 cell surface expression and promoted PD-1 degradation, resulting in augmented T-cell activity and anti-tumor responses(Okada et al., 2017, Zhang et al., 2020a). Among the PD-1 N-glycosylation sites, N116 mediates the interaction between galectin-9 and PD-1, which induces TIM-3/PD-1 dimerization and attenuates galectin-9/TIM-3-mediated cell death(Yang et al., 2021). Furthermore, PD1 N-glycosylation maintains its expression and its interaction with PD-L1 (Sun et al., 2020). PD-1 N-glycosylation varies when produced by different host systems, leading to different the binding affinities of camrelizumab, an anti-PD-1 monoclonal antibody(Liu et al., 2020). Another co-inhibitory receptor expressed on T-cells, CTLA-4, interacts with CD80/86 proteins to transmit the inhibitory signal. N-glycosylation (N78/110) contributes to CTLA-4 dimerization and T-cell activity(Darlington et al.). CTLA-4 surface expression was decreased in Mgat5-negative T-cells, and increased upon hexosamine treatment, implying N-glycan branching enhances the CTLA-4 surface retention and suppresses its endocytosis(Grigorian et al., 2007, Lau et al., 2007). Furthermore, defective N-glycan branching reduced CTLA-4 surface expression and promoted multiple sclerosis(Mkhikian et al., 2011). Finally, TIM-3 N-glycosylation is critical its interaction with galectin-9, which inhibits T-cell effector function(Zhu et al., 2005a).
Manipulation of glycosylation patterns can also improve immunotherapeutic outcomes. The mutation of PD-1 N-glycosylation at site N74 decreased PD-1 surface expression in CAR T-cells, thereby enhancing their cytotoxicity and cytokine secretion(Shi et al., 2019). Exofucosylation can increase surface sialyl-Lewis X, a glycan that enhances E-selectin binding, thereby improving CAR T-cell targeting efficiency(Mondal et al., 2019). Furthermore, N-glycosylation on the CAR hinge domain derived from CD28 contributes to CAR surface expression and CAR-T cytotoxicity(Hirobe et al., 2022). Drugs, including small molecules, sugar analogues, and mAb, can also be utilized to target glycosylation on various proteins to support immunotherapy(Zheng et al., 2022). BMS1166 inhibits PD-L1 glycosylation and blocks its endomembrane transport, resulting in T-cell activation(Chen et al., 2020). Another small molecule, N-linked glycosylation inhibitor-1, targets oligosaccharyltransferase, thus inhibiting N-glycosylation of epidermal growth factor receptor, cyclooxygenase-2, B7-H4 and other proteins to impede tumor cell proliferation(Lopez-Sambrooks et al., 2016, Rinis et al., 2018, Song et al., 2020). Similarly, inhibition of N-glycosylation with 2-deoxy-D-glucose (2DG) enhances T-cell cytotoxicity and promotes memory T-cell differentiation (Sasawatari et al., 2020). 2DG hindered the N-glycosylation of target proteins, including MICA/B and PD-L1, and enhanced the anti-tumor activity combined with other treatments (Andresen et al., 2012, Shao et al., 2018, Kim et al., 2020). 2-fluoro-L-fucose has also inhibited fucosylation of PD-1 and B7-H3, resulting in T-cell proliferation and activation(Okada et al., 2017, Huang et al., 2021). Sialic acid mimetics can block sialylation to suppress tumor growth and increase the proportion of NK cells, CD8+ T-cells, and CD4+ T-cells with reduced regulatory T-cells(Bull et al., 2018). Enhanced T-cell activity is achieved by mAbs with superior binding affinity to the N-glycosylation at site N58 of PD-1(Liu et al., 2020, Wang et al., 2019b, Liu et al., 2019, Lu et al., 2022). Fusion proteins also show promise, such as a sialidase-conjugated mAb that targets human epidermal growth factor receptor 2 and enhances the NK cell antitumor activity by desialylating HER2-positive tumor cells(Xiao et al., 2016). Understanding and ultimately manipulating the glycoproteome can therefore be invaluable to immunotherapies.
3.2. Viral Drug Delivery
Successful delivery of therapeutic nucleic acids, such as plasmid DNA, mRNA, antisense oligonucleotide (ASO), and small interfering RNA (siRNA) remains challenging since their negative charges inhibit transfer across the plasma and nuclear membranes(Molle et al., 2022). However, delivery platforms such as viral vectors, including adeno-associated viruses (AAV), adenoviruses, and lentiviruses overcome this challenge and are used in gene therapy(Bulcha et al., 2021, Sharon and Kamen, 2018). However, the target specificity, vector yield and transduction efficiency of viral vectors, such as AAV, is determined by capsid serotypes and capsid protein PTMs, such as ubiquitination, SUMOylation, and O-glycosylation (Wang et al., 2019a, Mary et al., 2019a) .
Glycans have been detected on capsid proteins in several AAV serotypes, including AAV2, 3, 5, 7, 8, 9, rh10 (Mary et al., 2019a). N-glycosylation of viral proteins can affect infectivity and the immune response by assisting in protein folding and trafficking and modulating the interaction between virus and host immune system(Vigerust and Shepherd, 2007). Inhibition of N-glycosylation increased AAV2 transduction efficiency and decreased vector yield(Mary et al., 2019a, Mary et al., 2019b), and the site-specific mutation of glycosylation sites revealed that modulation of glycosylation increased the hepatic and ocular gene transfer of AAV2(Mary et al., 2019a, Mary et al., 2019b). Proteomic analysis of AAV5-producing HEK293 showed an upregulation of MAN2A2, an alpha-mannosidase which trims high-mannose structures resulting in complex N-glycans in Golgi, suggesting that the glycosylation pathway can be targeted to improve AAV production(Strasser et al., 2021). Furthermore, bioconjugation of GalNAc on capsid proteins can increase AAV2 transduction efficiency and reduce neutralizing antibody production against AAV2 and AAV8 (Mevel et al., 2019).
Glycans and glycan-binding proteins (lectins) are also important to virus and host interactions, including viral entry and tissue tropism(Raman et al., 2016) (Figure 4B). Cell-surface glycans, such as heparan sulfate proteoglycans (HSPG), sialic acids, and terminal galactose, aid in attachment and infection of AAV serotypes(Stroh and Stehle, 2014, Meyer and Chapman, 2022). Membrane-associated HSPG are receptors for AAV2 and AAV3, so disrupting HSPG synthesis genes and/or treatment with heparin inhibits attachment and infection (Summerford and Samulski, 1998, Handa et al., 2000, Rabinowitz et al., 2002). Terminal sialylation is also critical for AAV infectivity; heparan sulfate and sialic acids are necessary for binding and transduction of AAV6 (Halbert et al., 2001, Wu et al., 2006, Ng et al., 2010), while AAV4 and AAV5 use O-linked and N-linked sialic acids as a primary receptor, respectively(Chen et al., 2005, Walters et al., 2001, Kaludov et al., 2001, Walters et al., 2002). Inhibiting sialylation also decreases binding and transduction efficiency of AAV1 (Wu et al., 2006, Chen et al., 2005). For AAV9, however, enzymatic digestion of sialic acids increased the surface binding and transduction, suggesting that N-linked galactose facilitates its binding and transduction (Shen et al., 2011, Bell et al., 2011). Understanding the glycan binding specificity of different AAV serotypes can help improve gene therapy vectors (Mietzsch et al., 2014), and engineering capsid protein affinity to glycans is being attempted (Madigan and Asokan, 2016).
3.3. Non-Viral Drug Delivery
Nucleic acids and other therapeutic molecules can also be administered as naked molecules or with non-viral delivery platforms. Many cell types recognize and bind specific glycan structures, such as terminal mannose, fucose, galactose, GalNac and GlcNac; conjugation of carbohydrate moieties to oligonucleotide-based drugs can improve their stability, cell specificity, and delivery (Bakowski and Vogel, 2022) (Figure 4C). This can impact cancer therapy, where lectin receptors are often overexpressed in tumors versus healthy tissue(Berthe et al., 2003, Ishiwata et al., 1997, Pavelic et al., 2003, Laube, 2009, Hebert, 2006), and glycoengineering can be exploited to reduce drug toxicity in healthy cells and improve clinical efficacy.
Galactosylated polyethylene glycol and mannose 6-phosphate polyethylene glycol can be covalently linked to siRNA to successfully inhibit gene expression in hepatocytes in vitro (Zhu and Mahato, 2010), and conjugation of siRNA to GalNAc (a ligand for ASGP receptors), facilitated targeted delivery and robust gene silencing both in vitro and in vivo (Nair et al., 2014). Different designs of GalNAc conjugation have been tested to optimize synthesis and targeting efficiency(Matsuda et al., 2015, Rajeev et al., 2015). Furthermore, conjugation of multivalent GalNAc to antisense oligonucleotides and siRNAs using GalNAc phosphoramidite monomer has extended the structural flexibility of the number of GalNAc units for effective silencing(Yamamoto et al., 2016, Sharma et al., 2018).
Glycoengineering of delivery platforms can also improve uptake or alter biodistribution of nanoparticles (NP) and extracellular vesicles (EV) by leveraging glycan receptors(Bakowski and Vogel, 2022, Bost et al., 2021) (Figure 4C). NPs coated with fucose can transfer liposomes into pancreatic cancer cells(Yoshida et al., 2012), and galactosylated liposomes and lipid nanoparticles (LNP) can deliver therapeutic molecules such as azidothymidine, doxorubicin, paclitaxel, and siRNA (Garg and Jain, 2006, Wang et al., 2010, Jain et al., 2015, Wang et al., 2016, Yang et al., 2018). Mannosylated NPs demonstrated improved delivery of both DNA and RNA into dendritic cells(Kim et al., 2006, Markov et al., 2015, Goswami et al., 2019), and mannose-based NPs containing dasatinib showed efficient uptake into macrophages(Rushworth et al., 2020). Coating liposomes with mannose-6-phosphate also increased cellular uptake of a cytotoxic molecule C6Cer, and selectively induced apoptosis in cancer cells(Minnelli et al., 2018). The surface of EVs are naturally enriched with glycoproteins, and glycosidase treatment impacts EV binding affinity, showing that surface glycans are important in target cell uptake(Williams et al., 2019). Indeed, the inhibition of EV surface N- and O-glycosylation enhanced uptake, and O-glycan removal significantly increased the EV accumulation into lung tissues in vivo (Nishida-Aoki et al., 2020). Similarly, removal of sialic acid on EV surfaces shifted their biodistribution from the liver to the lungs (Royo et al., 2019). As with nanoparticles, adding glycans to EVs can target cells with specific lectin receptors. For example, compared to unconjugated EVs, mannose-conjugated carriers containing an immune stimulant MPLA, demonstrated higher cellular uptake and elevated cytokine secretion in dendritic cells, which primarily express mannose receptors (Choi et al., 2019). These findings underscore the immense potential impact of glycosylation on improving the efficacy and safety profiles of new and current platforms for drug delivery.
4. Conclusion and Outlook
There is no denying that optimizing glycosylation in the drug discovery and development workflow can immensely improve protein-based, oligonucleotide-based and cell-based therapeutic products. The last two decades have seen significant advancements with regard to the creation of glycoengineering platforms that involve genetic modification of plant(Grabowski et al., 2014), yeast(Jacobs et al., 2009, Beck et al., 2010, Arico et al., 2013), human(Hart et al., 2017, Meuris et al., 2014), and hamster(Shitara, 2009, Pereira et al., 2018) producer cell lines to generate recombinant products with homogenous and tailored glycan structures for improved safety and efficacy. By combining these technologies with systems biology approaches that aim to better understand the cell’s highly complex glycosylation machinery(Spahn et al., 2017, Spahn et al., 2016, Liang et al., 2020, Krambeck et al., 2017), we may be better able to predict and design optimal glycoprofiles and build the capacity to produce glycoengineered biologics more reliably and cost-effectively at scale.
Author Contributions
FR, AGP, SS, JS, MW, TF, EAT and NEL all contributed to the writing of the manuscript. FR, AGP and SS created and formatted the figures. FR, EAT, TF and NEL participated in the review and editing of the manuscript.
ACKNOWLEDGMENTS
This work was supported with funding from the Novo Nordisk Foundation (NNF20SA0066621) and NIGMS (R35 GM119850).
References
- Intravenous Immunoglobulin (IVIG) Market Global Opportunity Analysis and Industry Forecast, 2018 - 2025. Available: https://www.alliedmarketresearch.com/intravenous-immunoglobulin-IVIG-market.
- Expanding access to monoclonal antibody-based products: A global call to action. Available: https://www.iavi.org/phocadownload/expanding/Expanding access to monoclonal antibody-based products.pdf.
- ACHORD, D. T. , BROT, F. E., BELL, C. E. & SLY, W. S. 1978. Human beta-glucuronidase: in vivo clearance and in vitro uptake by a glycoprotein recognition system on reticuloendothelial cells. Cell, 15, 269-78.
- Altman, M.O.; Gagneux, P. Absence of Neu5Gc and Presence of Anti-Neu5Gc Antibodies in Humans—An Evolutionary Perspective. Front. Immunol. 2019, 10, 789. [Google Scholar] [CrossRef] [PubMed]
- Amon, R.; Ben-Arye, S.L.; Engler, L.; Yu, H.; Lim, N.; Le Berre, L.; Harris, K.M.; Ehlers, M.R.; Gitelman, S.E.; Chen, X.; et al. Glycan microarray reveal induced IgGs repertoire shift against a dietary carbohydrate in response to rabbit anti-human thymocyte therapy. Oncotarget 2017, 8, 112236–112244. [Google Scholar] [CrossRef] [PubMed]
- ANDRE, S. , UNVERZAGT, C., KOJIMA, S., DONG, X., FINK, C., KAYSER, K. & GABIUS, H. J. 1997. Neoglycoproteins with the synthetic complex biantennary nonasaccharide or its alpha 2,3/alpha 2,6-sialylated derivatives: their preparation, assessment of their ligand properties for purified lectins, for tumor cells in vitro, and in tissue sections, and their biodistribution in tumor-bearing mice. Bioconjug Chem, 8, 845-55.
- ANDRE, S. , UNVERZAGT, C., KOJIMA, S., FRANK, M., SEIFERT, J., FINK, C., KAYSER, K., VON DER LIETH, C. W. & GABIUS, H. J. 2004. Determination of modulation of ligand properties of synthetic complex-type biantennary N-glycans by introduction of bisecting GlcNAc in silico, in vitro and in vivo. Eur J Biochem, 271, 118-34.
- Andresen, L.; Skovbakke, S.L.; Persson, G.; Hagemann-Jensen, M.; Hansen, K.A.; Jensen, H.; Skov, S. 2-Deoxy d-Glucose Prevents Cell Surface Expression of NKG2D Ligands through Inhibition of N-Linked Glycosylation. J. Immunol. 2012, 188, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Anobile, C.J.; A Talbot, J.; McCann, S.J.; Padmanabhan, V.; Robertson, W.R. Glycoform composition of serum gonadotrophins through the normal menstrual cycle and in the post-menopausal state. Mol. Hum. Reprod. 1998, 4, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Nimmerjahn, F.; Ashline, D.J.; Reinhold, V.N.; Paulson, J.C.; Ravetch, J.V. Recapitulation of IVIG Anti-Inflammatory Activity with a Recombinant IgG Fc. Science 2008, 320, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Ravetch, J.V. A Novel Role for the IgG Fc Glycan: The Anti-inflammatory Activity of Sialylated IgG Fcs. J. Clin. Immunol. 2010, 30, 9–14. [Google Scholar] [CrossRef]
- Aoyama, M.; Hashii, N.; Tsukimura, W.; Osumi, K.; Harazono, A.; Tada, M.; Kiyoshi, M.; Matsuda, A.; Ishii-Watabe, A. Effects of terminal galactose residues in mannose α1-6 arm of Fc-glycan on the effector functions of therapeutic monoclonal antibodies. mAbs 2019, 11, 826–836. [Google Scholar] [CrossRef] [PubMed]
- ARICO, C. , BONNET, C. & JAVAUD, C. 2013. N-glycosylation humanization for production of therapeutic recombinant glycoproteins in Saccharomyces cerevisiae. Glycosylation Engineering of Biopharmaceuticals: Methods and Protocols (2013): 45-57.
- Ashwell, G.; Harford, J. Carbohydrate-Specific Receptors of the Liver. Annu. Rev. Biochem. 1982, 51, 531–554. [Google Scholar] [CrossRef]
- ASHWELL, G. & MORELL, A. G. 1974. The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins. Adv Enzymol Relat Areas Mol Biol, 41, 99-128.
- Bakowski, K.; Vogel, S. Evolution of complexity in non-viral oligonucleotide delivery systems: from gymnotic delivery through bioconjugates to biomimetic nanoparticles. RNA Biol. 2022, 19, 1256–1275. [Google Scholar] [CrossRef]
- BARTON, N. W. , BRADY, R. O., DAMBROSIA, J. M., DI BISCEGLIE, A. M., DOPPELT, S. H., HILL, S. C., MANKIN, H. J., MURRAY, G. J., PARKER, R. I., ARGOFF, C. E. & ET AL. 1991. Replacement therapy for inherited enzyme deficiency--macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med, 324, 1464-70.
- BASILE, I. , DA SILVA, A., EL CHEIKH, K., GODEFROY, A., DAURAT, M., HARMOIS, A., PEREZ, M., CAILLAUD, C., CHARBONNE, H. V., PAU, B., GARY-BOBO, M., MORERE, A., GARCIA, M. & MAYNADIER, M. 2018. Efficient therapy for refractory Pompe disease by mannose 6-phosphate analogue grafting on acid alpha-glucosidase. J Control Release, 269, 15-23.
- Baudyš, M.; Uchio, T.; Mix, D.; Kim, S.W.; Wilson, D. Physical Stabilization of Insulin by Glycosylation. J. Pharm. Sci. 1995, 84, 28–33. [Google Scholar] [CrossRef]
- Beall, R.F.; Hwang, T.J.; Kesselheim, A.S. Pre-market development times for biologic versus small-molecule drugs. Nat. Biotechnol. 2019, 37, 708–711. [Google Scholar] [CrossRef]
- BECK, A. , COCHET, O. & WURCH, T. 2010. GlycoFi's technology to control the glycosylation of recombinant therapeutic proteins. Expert Opin Drug Discov, 5, 95-111.
- Behrens, A.-J.; Vasiljevic, S.; Pritchard, L.K.; Harvey, D.J.; Andev, R.S.; Krumm, S.A.; Struwe, W.B.; Cupo, A.; Kumar, A.; Zitzmann, N.; et al. Composition and Antigenic Effects of Individual Glycan Sites of a Trimeric HIV-1 Envelope Glycoprotein. Cell Rep. 2016, 14, 2695–2706. [Google Scholar] [CrossRef]
- Bell, C.L.; Vandenberghe, L.H.; Bell, P.; Limberis, M.P.; Gao, G.-P.; Van Vliet, K.; Agbandje-McKenna, M.; Wilson, J.M. The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. J. Clin. Investig. 2011, 121, 2427–2435. [Google Scholar] [CrossRef]
- BERTHE, M. L. , ESSLIMANI SAHLA, M., ROGER, P., GLEIZES, M., LEMAMY, G. J., BROUILLET, J. P. & ROCHEFORT, H. 2003. Mannose-6-phosphate/insulin-like growth factor-II receptor expression levels during the progression from normal human mammary tissue to invasive breast carcinomas. Eur J Cancer, 39, 635-42.
- Bocci, V. Catabolism of therapeutic proteins and peptides with implications for drug delivery. Adv. Drug Deliv. Rev. 1989, 4, 149–169. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Bonetti, P.; Diodati, G.; Drago, C.; Casarin, C.; Scaccabarozzi, S.; Realdi, G.; Ruol, A.; Alberti, A. Interferon antibodies in patients with chronic hepatitic C virus infection treated with recombinant interferon alpha-2α. J. Hepatol. 1994, 20, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Bost, J.P.; Barriga, H.; Holme, M.N.; Gallud, A.; Maugeri, M.; Gupta, D.; Lehto, T.; Valadi, H.; Esbjörner, E.K.; Stevens, M.M.; et al. Delivery of Oligonucleotide Therapeutics: Chemical Modifications, Lipid Nanoparticles, and Extracellular Vesicles. ACS Nano 2021, 15, 13993–14021. [Google Scholar] [CrossRef]
- Boyd, P.; Lines, A.; Patel, A. The effect of the removal of sialic acid, galactose and total carbohydrate on the functional activity of Campath-1H. Mol. Immunol. 1995, 32, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Brady, L.J.; Velayudhan, J.; Visone, D.B.; Daugherty, K.C.; Bartron, J.L.; Coon, M.; Cornwall, C.; Hinckley, P.J.; Connell-Crowley, L. The criticality of high-resolution N-linked carbohydrate assays and detailed characterization of antibody effector function in the context of biosimilar development. mAbs 2015, 7, 562–570. [Google Scholar] [CrossRef]
- BRADY, R. O. , KANFER, J. N., BRADLEY, R. M. & SHAPIRO, D. 1966. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher's disease. J Clin Invest, 45, 1112-5.
- BRADY, R. O. , KANFER, J. N. & SHAPIRO, D. 1965. Metabolism of Glucocerebrosides. Ii. Evidence of an Enzymatic Deficiency in Gaucher's Disease. Biochem Biophys Res Commun, 18, 221-5.
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- BULL, C. , BOLTJE, T. J., BALNEGER, N., WEISCHER, S. M., WASSINK, M., VAN GEMST, J. J., BLOEMENDAL, V. R., BOON, L., VAN DER VLAG, J., HEISE, T., DEN BROK, M. H. & ADEMA, G. J. 2018. Sialic Acid Blockade Suppresses Tumor Growth by Enhancing T-cell-Mediated Tumor Immunity. Cancer Res, 78, 3574-3588.
- Butler, M.; Spearman, M. The choice of mammalian cell host and possibilities for glycosylation engineering. Curr. Opin. Biotechnol. 2014, 30, 107–112. [Google Scholar] [CrossRef] [PubMed]
- CADAOAS, J. , BOYLE, G., JUNGLES, S., CULLEN, S., VELLARD, M., GRUBB, J. H., JURECKA, A., SLY, W. & KAKKIS, E. 2020. Vestronidase alfa: Recombinant human beta-glucuronidase as an enzyme replacement therapy for MPS VII. Mol Genet Metab, 130, 65-76.
- CAMPBELL, C. & STANLEY, P. 1984. A dominant mutation to ricin resistance in Chinese hamster ovary cells induces UDP-GlcNAc:glycopeptide beta-4-N-acetylglucosaminyltransferase III activity. J Biol Chem, 259, 13370-8.
- Carter, C.R.D.; Whitmore, K.M.; Thorpe, R. The significance of carbohydrates on G-CSF: differential sensitivity of G-CSFs to human neutrophil elastase degradation. J. Leukoc. Biol. 2003, 75, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, N.; Nataf, J.; Viron, B.; Kolta, A.; Kiladjian, J.-J.; Martin-Dupont, P.; Michaud, P.; Papo, T.; Ugo, V.; Teyssandier, I.; et al. Pure Red-Cell Aplasia and Antierythropoietin Antibodies in Patients Treated with Recombinant Erythropoietin. New Engl. J. Med. 2002, 346, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Ceaglio, N.; Etcheverrigaray, M.; Conradt, H.S.; Grammel, N.; Kratje, R.; Oggero, M. Highly glycosylated human alpha interferon: An insight into a new therapeutic candidate. J. Biotechnol. 2010, 146, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Ceaglio, N.; Etcheverrigaray, M.; Kratje, R.; Oggero, M. Novel long-lasting interferon alpha derivatives designed by glycoengineering. Biochimie 2008, 90, 437–449. [Google Scholar] [CrossRef]
- Chan, A.C.; Carter, P.J. Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 2010, 10, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, W.; Li, Y.; Ma, B.; Shang, S.; Tan, Z. Impact of N-Linked Glycosylation on Therapeutic Proteins. Molecules 2022, 27, 8859. [Google Scholar] [CrossRef]
- Chen, F.-F.; Li, Z.; Ma, D.; Yu, Q. Small-molecule PD-L1 inhibitor BMS1166 abrogates the function of PD-L1 by blocking its ER export. OncoImmunology 2020, 9, 1831153. [Google Scholar] [CrossRef]
- Chen, J.-R.; Yu, Y.-H.; Tseng, Y.-C.; Chiang, W.-L.; Chiang, M.-F.; Ko, Y.-A.; Chiu, Y.-K.; Ma, H.-H.; Wu, C.-Y.; Jan, J.-T.; et al. Vaccination of monoglycosylated hemagglutinin induces cross-strain protection against influenza virus infections. Proc. Natl. Acad. Sci. 2014, 111, 2476–2481. [Google Scholar] [CrossRef]
- CHEN, L. & FLIES, D. B. 2013. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol, 13, 227-42.
- Chen, L.-R.; Chen, C.-A.; Chiu, S.-N.; Chien, Y.-H.; Lee, N.-C.; Lin, M.-T.; Hwu, W.-L.; Wang, J.-K.; Wu, M.-H. Reversal of Cardiac Dysfunction after Enzyme Replacement in Patients with Infantile-Onset Pompe Disease. J. Pediatr. 2009, 155, 271–275. [Google Scholar] [CrossRef]
- Chen, S.; Kapturczak, M.; Loiler, S.A.; Zolotukhin, S.; Glushakova, O.Y.; Madsen, K.M.; Samulski, R.J.; Hauswirth, W.W.; Campbell-Thompson, M.; Berns, K.I.; et al. Efficient Transduction of Vascular Endothelial Cells with Recombinant Adeno-Associated Virus Serotype 1 and 5 Vectors. Hum. Gene Ther. 2005, 16, 235–247. [Google Scholar] [CrossRef]
- Choi, E.S.; Song, J.; Kang, Y.Y.; Mok, H. Mannose-Modified Serum Exosomes for the Elevated Uptake to Murine Dendritic Cells and Lymphatic Accumulation. Macromol. Biosci. 2019, 19, e1900042. [Google Scholar] [CrossRef]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.-T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-Induced Anaphylaxis and IgE Specific for Galactose-α-1,3-Galactose. New Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef]
- Clarke, L.A.; Wraith, J.E.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Sidman, M.; Kakkis, E.D.; et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 2009, 123, 229–240. [Google Scholar] [CrossRef]
- CLYNES, R. A. , TOWERS, T. L., PRESTA, L. G. & RAVETCH, J. V. 2000. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med, 6, 443-6.
- Dammen-Brower, K.; Epler, P.; Zhu, S.; Bernstein, Z.J.; Stabach, P.R.; Braddock, D.T.; Spangler, J.B.; Yarema, K.J. Strategies for Glycoengineering Therapeutic Proteins. Front. Chem. 2022, 10, 863118. [Google Scholar] [CrossRef]
- Darlington, P.J.; Kirchhof, M.G.; Criado, G.; Sondhi, J.; Madrenas, J. Hierarchical Regulation of CTLA-4 Dimer-Based Lattice Formation and Its Biological Relevance for T Cell Inactivation. J. Immunol. 2005, 175, 996–1004. [Google Scholar] [CrossRef]
- Davies, J.; Jiang, L.; Pan, L.Z.; LaBarre, M.J.; Anderson, D.; Reff, M. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. . 2001, 74, 288–94. [Google Scholar] [CrossRef]
- de Haij, S.; Jansen, J.M.; Boross, P.; Beurskens, F.J.; Bakema, J.E.; Bos, D.L.; Martens, A.; Verbeek, J.S.; Parren, P.W.; van de Winkel, J.G.; et al. In vivo Cytotoxicity of Type I CD20 Antibodies Critically Depends on Fc Receptor ITAM Signaling. Cancer Res 2010, 70, 3209–3217. [Google Scholar] [CrossRef]
- Dekkers, G.; Treffers, L.; Plomp, R.; Bentlage, A.E.H.; de Boer, M.; Koeleman, C.A.M.; Lissenberg-Thunnissen, S.N.; Visser, R.; Brouwer, M.; Mok, J.Y.; et al. Decoding the Human Immunoglobulin G-Glycan Repertoire Reveals a Spectrum of Fc-Receptor- and Complement-Mediated-Effector Activities. Front. Immunol. 2017, 8, 877. [Google Scholar] [CrossRef] [PubMed]
- DESNICK, R. J. 2001. α-Galactosidase A Deficiency : Fabry Disease.
- Dhar, C.; Sasmal, A.; Varki, A. From “Serum Sickness” to “Xenosialitis”: Past, Present, and Future Significance of the Non-human Sialic Acid Neu5Gc. Front. Immunol. 2019, 10, 807. [Google Scholar] [CrossRef] [PubMed]
- DI MARIO, U. , ARDUINI, P., TIBERTI, C., LOMBARDI, G., PIETRAVALLE, P. & ANDREANI, D. 1986. Immunogenicity of biosynthetic human insulin. Humoral immune response in diabetic patients beginning insulin treatment and in patients previously treated with other insulins. Diabetes Res Clin Pract, 2, 317-24.
- Doebber, T.W.; Wu, M.S.; Bugianesi, R.L.; Ponpipom, M.M.; Furbish, F.S.; A Barranger, J.; O Brady, R.; Shen, T.Y. Enhanced macrophage uptake of synthetically glycosylated human placental beta-glucocerebrosidase. J. Biol. Chem. 1982, 257, 2193–2199. [Google Scholar] [CrossRef]
- Dornelles, A.D.; Artigalás, O.; da Silva, A.A.; Ardila, D.L.V.; Alegra, T.; Pereira, T.V.; Vairo, F.P.E.; Schwartz, I.V.D. Efficacy and safety of intravenous laronidase for mucopolysaccharidosis type I: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0184065. [Google Scholar] [CrossRef]
- DOUGLAS, D. D. , RAKELA, J., LIN, H. J., HOLLINGER, F. B., TASWELL, H. F., CZAJA, A. J., GROSS, J. B., ANDERSON, M. L., PARENT, K., FLEMING, C. R. & ET AL. 1993. Randomized controlled trial of recombinant alpha-2a-interferon for chronic hepatitis C. Comparison of alanine aminotransferase normalization versus loss of HCV RNA and anti-HCV IgM. Dig Dis Sci, 38, 601-7.
- Edgar, L.J.; Thompson, A.J.; Vartabedian, V.F.; Kikuchi, C.; Woehl, J.L.; Teijaro, J.R.; Paulson, J.C. Sialic Acid Ligands of CD28 Suppress Costimulation of T Cells. ACS Central Sci. 2021, 7, 1508–1515. [Google Scholar] [CrossRef]
- EGRIE, J. C. & BROWNE, J. K. 2001. Development and characterization of novel erythropoiesis stimulating protein (NESP). Br J Cancer, 84 Suppl 1, 3-10.
- Egrie, J.C.; Dwyer, E.; Browne, J.K.; Hitz, A.; A Lykos, M. Darbepoetin alfa has a longer circulating half-life and greater in vivo potency than recombinant human erythropoietin. Exp. Hematol. 2003, 31, 290–299. [Google Scholar] [CrossRef]
- Ehrenpreis, E.D. Pharmacokinetic Effects of Antidrug Antibodies Occurring in Healthy Subjects After a Single Dose of Intravenous Infliximab. Drugs R&D 2017, 17, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Chang, D.; Delorme, E.; Dunn, C.; Egrie, J.; Giffin, J.; Lorenzini, T.; Talbot, C.; Hesterberg, L. Isolation and characterization of conformation sensitive antierythropoietin monoclonal antibodies: effect of disulfide bonds and carbohydrate on recombinant human erythropoietin structure. Blood 1996, 87, 2714–2722. [Google Scholar] [CrossRef]
- Elliott, S.; Egrie, J.; Browne, J.; Lorenzini, T.; Busse, L.; Rogers, N.; Ponting, I. Control of rHuEPO biological activity: The role of carbohydrate. Exp. Hematol. 2004, 32, 1146–1155. [Google Scholar] [CrossRef]
- FARRELL, R. A. , MARTA, M., GAEGUTA, A. J., SOUSLOVA, V., GIOVANNONI, G. & CREEKE, P. I. 2012. Development of resistance to biologic therapies with reference to IFN-beta. Rheumatology (Oxford), 51, 590-9.
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef]
- FENG, X. , XIE, H.-G., MALHOTRA, A. & YANG, C. F. 2022. Biologics and biosimilars : drug development and clinical affairs, Boca Raton, Taylor and Francis.
- Fenouillet, E.; Gluckman, J.C.; Jones, I.M. Functions of HIV envelope glycans. Trends Biochem. Sci. 1994, 19, 65–70. [Google Scholar] [CrossRef]
- I Fernandes, A.; Gregoriadis, G. The effect of polysialylation on the immunogenicity and antigenicity of asparaginase: implication in its pharmacokinetics. Int. J. Pharm. 2001, 217, 215–224. [Google Scholar] [CrossRef]
- FIEDLER, W. , CRESTA, S., SCHULZE-BERGKAMEN, H., DE DOSSO, S., WEIDMANN, J., TESSARI, A., BAUMEISTER, H., DANIELCZYK, A., DIETRICH, B., GOLETZ, S., ZURLO, A., SALZBERG, M., SESSA, C. & GIANNI, L. 2018. Phase I study of tomuzotuximab, a glycoengineered therapeutic antibody against the epidermal growth factor receptor, in patients with advanced carcinomas. ESMO Open, 3, e000303.
- Filipe, V.; Que, I.; Carpenter, J.F.; Löwik, C.; Jiskoot, W. In Vivo Fluorescence Imaging of IgG1 Aggregates After Subcutaneous and Intravenous Injection in Mice. Pharm. Res. 2013, 31, 216–227. [Google Scholar] [CrossRef]
- Fineberg, S.E.; Galloway, J.A.; Fineberg, N.S.; Rathbun, M.J.; Hufferd, S. Immunogenicity of recombinant DNA human insulin. Diabetologia 1983, 25, 465–469. [Google Scholar] [CrossRef]
- Flintegaard, T.V.; Thygesen, P.; Rahbek-Nielsen, H.; Levery, S.B.; Kristensen, C.; Clausen, H.; Bolt, G. N-Glycosylation Increases the Circulatory Half-Life of Human Growth Hormone. Endocrinology 2010, 151, 5326–5336. [Google Scholar] [CrossRef]
- Flynn, G.C.; Chen, X.; Liu, Y.D.; Shah, B.; Zhang, Z. Naturally occurring glycan forms of human immunoglobulins G1 and G2. Mol. Immunol. 2010, 47, 2074–2082. [Google Scholar] [CrossRef]
- FOSSA, S. D. , LEHNE, G., GUNDERSON, R., HJELMAAS, U. & HOLDENER, E. E. 1992. Recombinant interferon alpha-2A combined with prednisone in metastatic renal-cell carcinoma: treatment results, serum interferon levels and the development of antibodies. Int J Cancer, 50, 868-70.
- Fox, J.E.; Volpe, L.; Bullaro, J.; Kakkis, E.D.; Sly, W.S. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol. Genet. Metab. 2015, 114, 203–208. [Google Scholar] [CrossRef]
- Friedman, B.; Vaddi, K.; Preston, C.; Mahon, E.; Cataldo, J.R.; McPherson, J.M. A comparison of the pharmacological properties of carbohydrate remodeled recombinant and placental-derived beta-glucocerebrosidase: implications for clinical efficacy in treatment of Gaucher disease. . 1999, 93, 2807–16. [Google Scholar]
- FUKUDA, M. N. , SASAKI, H. & FUKUDA, M. 1989. Survival of recombinant erythropoietin in the circulation: the role of carbohydrates. Blood, 73, 84-89.
- Furbish, F.; Steer, C.J.; Krett, N.L.; Barranger, J.A. Uptake and distribution of placental glucocerebrosidase in rat hepatic cells and effects of sequential deglycosylation. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 1981, 673, 425–434. [Google Scholar] [CrossRef]
- FUTERMAN, A. H. & VAN MEER, G. 2004. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol, 5, 554-65.
- Galili, U.; E Mandrell, R.; Hamadeh, R.M.; Shohet, S.B.; Griffiss, J.M. Interaction between human natural anti-alpha-galactosyl immunoglobulin G and bacteria of the human flora. Infect. Immun. 1988, 56, 1730–1737. [Google Scholar] [CrossRef]
- Garcia, A.R.; DaCosta, J.M.; Pan, J.; Muenzer, J.; Lamsa, J.C. Preclinical dose ranging studies for enzyme replacement therapy with idursulfase in a knock-out mouse model of MPS II. Mol. Genet. Metab. 2007, 91, 183–190. [Google Scholar] [CrossRef] [PubMed]
- García-García, A.; Serna, S.; Yang, Z.; Delso, I.; Taleb, V.; Hicks, T.; Artschwager, R.; Vakhrushev, S.Y.; Clausen, H.; Angulo, J.; et al. FUT8-Directed Core Fucosylation of N-glycans Is Regulated by the Glycan Structure and Protein Environment. ACS Catal. 2021, 11, 9052–9065. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Jain, N.K. Reduced hematopoietic toxicity, enhanced cellular uptake and altered pharmacokinetics of azidothymidine loaded galactosylated liposomes. J. Drug Target. 2006, 14, 1–11. [Google Scholar] [CrossRef]
- GERDES, C. A. , NICOLINI, V. G., HERTER, S., VAN PUIJENBROEK, E., LANG, S., ROEMMELE, M., MOESSNER, E., FREYTAG, O., FRIESS, T., RIES, C. H., BOSSENMAIER, B., MUELLER, H. J. & UMANA, P. 2013. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res, 19, 1126-38.
- Ghaderi, D.; E Taylor, R.; Padler-Karavani, V.; Diaz, S.; Varki, A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat. Biotechnol. 2010, 28, 863–867. [Google Scholar] [CrossRef]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef]
- Goh, J.B.; Ng, S.K. Impact of host cell line choice on glycan profile. Crit. Rev. Biotechnol. 2018, 38, 851–867. [Google Scholar] [CrossRef]
- Golay, J.; Andrea, A.E.; Cattaneo, I. Role of Fc Core Fucosylation in the Effector Function of IgG1 Antibodies. Front. Immunol. 2022, 13, 929895. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Chatzikleanthous, D.; Lou, G.; Giusti, F.; Bonci, A.; Taccone, M.; Brazzoli, M.; Gallorini, S.; Ferlenghi, I.; Berti, F.; et al. Mannosylation of LNP Results in Improved Potency for Self-Amplifying RNA (SAM) Vaccines. ACS Infect. Dis. 2019, 5, 1546–1558. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Barton, N.W.; Pastores, G.; Dambrosia, J.M.; Banerjee, T.K.; McKee, M.A.; Parker, C.; Schiffmann, R.; Hill, S.C.; Brady, R.O. Enzyme Therapy in Type 1 Gaucher Disease: Comparative Efficacy of Mannose-Terminated Glucocerebrosidase from Natural and Recombinant Sources. Ann. Intern. Med. 1995, 122, 33–39. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Golembo, M.; Shaaltiel, Y. Taliglucerase alfa: An enzyme replacement therapy using plant cell expression technology. Mol. Genet. Metab. 2014, 112, 1–8. [Google Scholar] [CrossRef]
- Grigorian, A.; Lee, S.-U.; Tian, W.; Chen, I.-J.; Gao, G.; Mendelsohn, R.; Dennis, J.W.; Demetriou, M. Control of T Cell-mediated Autoimmunity by Metabolite Flux to N-Glycan Biosynthesis. J. Biol. Chem. 2007, 282, 20027–20035. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Chaffey, P.K.; Wei, X.; Gulbranson, D.R.; Ruan, Y.; Wang, X.; Li, Y.; Ouyang, Y.; Chen, L.; Zeng, C.; et al. Chemically Precise Glycoengineering Improves Human Insulin. ACS Chem. Biol. 2018, 13, 73–81. [Google Scholar] [CrossRef] [PubMed]
- GUDELJ, I. , LAUC, G. & PEZER, M. 2018. Immunoglobulin G glycosylation in aging and diseases. Cell Immunol, 333, 65-79.
- Halbert, C.L.; Allen, J.M.; Miller, A.D. Adeno-Associated Virus Type 6 (AAV6) Vectors Mediate Efficient Transduction of Airway Epithelial Cells in Mouse Lungs Compared to That of AAV2 Vectors. J. Virol. 2001, 75, 6615–6624. [Google Scholar] [CrossRef] [PubMed]
- Handa, A.; Muramatsu, S.-I.; Qiu, J.; Mizukami, H.; Brown, K.E. Adeno-associated virus (AAV)-3-based vectors transduce haematopoietic cells not susceptible to transduction with AAV-2-based vectors. J. Gen. Virol. 2000, 81, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Harmatz, P.; Giugliani, R.; Schwartz, I.; Guffon, N.; Teles, E.L.; Miranda, M.C.S.; Wraith, J.E.; Beck, M.; Arash, L.; Scarpa, M.; et al. Enzyme replacement therapy for mucopolysaccharidosis VI: A phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J. Pediatr. 2006, 148, 533–539. [Google Scholar] [CrossRef] [PubMed]
- HARMATZ, P. , KRAMER, W. G., HOPWOOD, J. J., SIMON, J., BUTENSKY, E., SWIEDLER, S. J. & MUCOPOLYSACCHARIDOSIS, V. I. S. G. 2005. Pharmacokinetic profile of recombinant human N-acetylgalactosamine 4-sulphatase enzyme replacement therapy in patients with mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): a phase I/II study. Acta Paediatr Suppl, 94, 61-8; discussion 57.
- Harmatz, P.; Whitley, C.B.; Waber, L.; Pais, R.; Steiner, R.; Plecko, B.; Kaplan, P.; Simon, J.; Butensky, E.; Hopwood, J.J. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J. Pediatr. 2004, 144, 574–580. [Google Scholar] [CrossRef]
- Harmatz, P.; Whitley, C.B.; Wang, R.Y.; Bauer, M.; Song, W.; Haller, C.; Kakkis, E. A novel Blind Start study design to investigate vestronidase alfa for mucopolysaccharidosis VII, an ultra-rare genetic disease. Mol. Genet. Metab. 2018, 123, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Hart, F.; Danielczyk, A.; Goletz, S. Human Cell Line-Derived Monoclonal IgA Antibodies for Cancer Immunotherapy. Bioengineering 2017, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- HATFIELD, G. , TEPLIAKOVA, L., GINGRAS, G., STALKER, A., LI, X., AUBIN, Y. & TAM, R. Y. 2022. Specific location of galactosylation in an afucosylated antiviral monoclonal antibody affects its FcgammaRIIIA binding affinity. Front Immunol, 13, 972168.
- Hébert, E. Mannose-6-phosphate/Insulin-like Growth Factor II Receptor Expression and Tumor Development. Biosci. Rep. 2006, 26, 7–17. [Google Scholar] [CrossRef] [PubMed]
- HENDRIKSZ, C. , SANTRA, S., JONES, S. A., GEBERHIWOT, T., JESAITIS, L., LONG, B., QI, Y., HAWLEY, S. M. & DECKER, C. 2018. Safety, immunogenicity, and clinical outcomes in patients with Morquio A syndrome participating in 2 sequential open-label studies of elosulfase alfa enzyme replacement therapy (MOR-002/MOR-100), representing 5 years of treatment. Mol Genet Metab, 123, 479-487.
- Hendriksz, C.J.; Burton, B.; Fleming, T.R.; Harmatz, P.; Hughes, D.; Jones, S.A.; Lin, S.-P.; Mengel, E.; Scarpa, M.; Valayannopoulos, V.; et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 2014, 37, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, A.; Bohorova, N.; Bohorov, O.; Goodman, C.; Kim, D.; Pauly, M.H.; Velasco, J.; Whaley, K.J.; Piedra, P.A.; Gilbert, B.E.; et al. Glycan variants of a respiratory syncytial virus antibody with enhanced effector function and in vivo efficacy. Proc. Natl. Acad. Sci. 2014, 111, 5992–5997. [Google Scholar] [CrossRef]
- Hirobe, S.; Imaeda, K.; Tachibana, M.; Okada, N. The Effects of Chimeric Antigen Receptor (CAR) Hinge Domain Post-Translational Modifications on CAR-T Cell Activity. Int. J. Mol. Sci. 2022, 23, 4056. [Google Scholar] [CrossRef]
- Hodoniczky, J.; Zheng, Y.Z.; James, D.C. Control of Recombinant Monoclonal Antibody Effector Functions by Fc N-Glycan Remodeling in Vitro. Biotechnol. Prog. 2005, 21, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Houde, D.; Peng, Y.; Berkowitz, S.A.; Engen, J.R. Post-translational Modifications Differentially Affect IgG1 Conformation and Receptor Binding. Mol. Cell. Proteom. 2010, 9, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- HUANG, H. Y. , LIAO, H. Y., CHEN, X., WANG, S. W., CHENG, C. W., SHAHED-AL-MAHMUD, M., LIU, Y. M., MOHAPATRA, A., CHEN, T. H., LO, J. M., WU, Y. M., MA, H. H., CHANG, Y. H., TSAI, H. Y., CHOU, Y. C., HSUEH, Y. P., TSAI, C. Y., HUANG, P. Y., CHANG, S. Y., CHAO, T. L., KAO, H. C., TSAI, Y. M., CHEN, Y. H., WU, C. Y., JAN, J. T., CHENG, T. R., LIN, K. I., MA, C. & WONG, C. H. 2022. Vaccination with SARS-CoV-2 spike protein lacking glycan shields elicits enhanced protective responses in animal models. Sci Transl Med, 14, eabm0899.
- Huang, Y.; Zhang, H.-L.; Li, Z.-L.; Du, T.; Chen, Y.-H.; Wang, Y.; Ni, H.-H.; Zhang, K.-M.; Mai, J.; Hu, B.-X.; et al. FUT8-mediated aberrant N-glycosylation of B7H3 suppresses the immune response in triple-negative breast cancer. Nat. Commun. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- Hubbard, A.L.; Stukenbrok, H. An electron microscope autoradiographic study of the carbohydrate recognition systems in rat liver. II. Intracellular fates of the 125I-ligands. J. Cell Biol. 1979, 83, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Bergmann, U.; Kornmann, M.; Lopez, M.; Beger, H.G.; Korc, M. Altered Expression of Insulin-like Growth Factor II Receptor in Human Pancreatic Cancer. Pancreas 1997, 15, 367–373. [Google Scholar] [CrossRef]
- Jacobs, P.P.; Geysens, S.; Vervecken, W.; Contreras, R.; Callewaert, N. Engineering complex-type N-glycosylation in Pichia pastoris using GlycoSwitch technology. Nat. Protoc. 2008, 4, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kesharwani, P.; Garg, N.K.; Jain, A.; Jain, S.A.; Jain, A.K.; Nirbhavane, P.; Ghanghoria, R.; Tyagi, R.K.; Katare, O.P. Galactose engineered solid lipid nanoparticles for targeted delivery of doxorubicin. Colloids Surfaces B: Biointerfaces 2015, 134, 47–58. [Google Scholar] [CrossRef]
- Jefferis, R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Discov. 2009, 8, 226–234. [Google Scholar] [CrossRef]
- Jefferis, R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 2009, 30, 356–362. [Google Scholar] [CrossRef]
- Jefferis, R. Isotype and glycoform selection for antibody therapeutics. Arch. Biochem. Biophys. 2012, 526, 159–166. [Google Scholar] [CrossRef]
- JEPPSSON, J. O. , LARSSON, C. & ERIKSSON, S. 1975. Characterization of alpha1-antitrypsin in the inclusion bodies from the liver in alpha 1-antitrypsin deficiency. N Engl J Med, 293, 576-9.
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Kakkis, E.; Matynia, A.; Jonas, A.; Neufeld, E. Overexpression of the Human Lysosomal Enzyme α-L-Iduronidase in Chinese Hamster Ovary Cells. Protein Expr. Purif. 1994, 5, 225–232. [Google Scholar] [CrossRef] [PubMed]
- KAKKIS, E. D. , MUENZER, J., TILLER, G. E., WABER, L., BELMONT, J., PASSAGE, M., IZYKOWSKI, B., PHILLIPS, J., DOROSHOW, R., WALOT, I., HOFT, R. & NEUFELD, E. F. 2001. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med, 344, 182-8.
- Kaludov, N.; Brown, K.E.; Walters, R.W.; Zabner, J.; Chiorini, J.A. Adeno-Associated Virus Serotype 4 (AAV4) and AAV5 Both Require Sialic Acid Binding for Hemagglutination and Efficient Transduction but Differ in Sialic Acid Linkage Specificity. J. Virol. 2001, 75, 6884–6893. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Fukui, R.; Suzuki, Y.; Kajihara, Y.; Kinoshita, M.; Kakehi, K.; Hojo, H.; Tezuka, K.; Tsuji, T. Definitive evidence that a single N-glycan among three glycans on inducible costimulator is required for proper protein trafficking and ligand binding. Biochem. Biophys. Res. Commun. 2010, 391, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-Inflammatory Activity of Immunoglobulin G Resulting from Fc Sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-Y.; Shin, K.K.; Kim, H.H.; Min, J.-K.; Ji, E.S.; Kim, J.Y.; Kwon, O.; Oh, D.-B. Lysosomal Targeting Enhancement by Conjugation of Glycopeptides Containing Mannose-6-phosphate Glycans Derived from Glyco-engineered Yeast. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.; Achord, D.T.; Sly, W.S. Phosphohexosyl components of a lysosomal enzyme are recognized by pinocytosis receptors on human fibroblasts. Proc. Natl. Acad. Sci. 1977, 74, 2026–2030. [Google Scholar] [CrossRef]
- Karottki, K.J.l.C.; Hefzi, H.; Xiong, K.; Shamie, I.; Hansen, A.H.; Li, S.; Pedersen, L.E.; Li, S.; Lee, J.S.; Lee, G.M.; et al. Awakening dormant glycosyltransferases in CHO cells with CRISPRa. Biotechnol. Bioeng. 2019, 117, 593–598. [Google Scholar] [CrossRef] [PubMed]
- KARPUSAS, M. , WHITTY, A., RUNKEL, L. & HOCHMAN, P. 1998. The structure of human interferon-beta: implications for activity. Cell Mol Life Sci, 54, 1203-16.
- Kim, B.; Sun, R.; Oh, W.; Kim, A.M.J.; Schwarz, J.R.; Lim, S.O. Saccharide analog, 2-deoxy-d-glucose enhances 4-1BB-mediated antitumor immunity via PD-L1 deglycosylation. Mol. Carcinog. 2020, 59, 691–700. [Google Scholar] [CrossRef]
- Kim, C.; Seo, J.; Chung, Y.; Ji, H.-J.; Lee, J.; Sohn, J.; Lee, B.; Jo, E.-C. Comparative study of idursulfase beta and idursulfase in vitro and in vivo. J. Hum. Genet. 2016, 62, 167–174. [Google Scholar] [CrossRef]
- Kim, T.H.; Jin, H.; Kim, H.W.; Cho, M.-H.; Cho, C.S. Mannosylated chitosan nanoparticle–based cytokine gene therapy suppressed cancer growth in BALB/c mice bearing CT-26 carcinoma cells. Mol. Cancer Ther. 2006, 5, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Nicolino, M.; Voit, T.; Rogers, R.C.; Tsai, A.C.-H.; Waterson, J.; Herman, G.E.; Amalfitano, A.; Thurberg, B.L.; Richards, S.; et al. Chinese hamster ovary cell-derived recombinant human acid α-glucosidase in infantile-onset Pompe disease. J. Pediatr. 2006, 149, 89–97. [Google Scholar] [CrossRef] [PubMed]
- KIVISAKK, P. , ALM, G. V., FREDRIKSON, S. & LINK, H. 2000. Neutralizing and binding anti-interferon-beta (IFN-beta) antibodies. A comparison between IFN-beta-1a and IFN-beta-1b treatment in multiple sclerosis. Eur J Neurol, 7, 27-34.
- Kolbeck, R.; Kozhich, A.; Koike, M.; Peng, L.; Andersson, C.K.; Damschroder, M.M.; Reed, J.L.; Woods, R.; Dall'Acqua, W.W.; Stephens, G.L.; et al. MEDI-563, a humanized anti–IL-5 receptor α mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J. Allergy Clin. Immunol. 2010, 125, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Konrad, M.W.; Childs, A.L.; Merigan, T.C.; Borden, E.C. Assessment of the antigenic response in humans to a recombinant mutant interferon beta. J. Clin. Immunol. 1987, 7, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Krambeck, F.J.; Bennun, S.V.; Andersen, M.R.; Betenbaugh, M.J. Model-based analysis of N-glycosylation in Chinese hamster ovary cells. PLOS ONE 2017, 12, e0175376. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Kumpel, B.M.; Rademacher, T.W.; A Rook, G.; Williams, P.J.; Wilson, I.B. Galactosylation of human IgG monoclonal anti-D produced by EBV-transformed B-lymphoblastoid cell lines is dependent on culture method and affects Fc receptor-mediated functional activity. . 1994, 5, 143–51. [Google Scholar] [PubMed]
- Kwon, K.-S.; Yu, M.-H. Effect of glycosylation on the stability of α1-antitrypsin toward urea denaturation and thermal deactivation. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 1997, 1335, 265–272. [Google Scholar] [CrossRef]
- Lachmann, R.H. Enzyme replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 2011, 23, 588–593. [Google Scholar] [CrossRef]
- Largent, B.L.; Walton, K.M.; A Hoppe, C.; Lee, Y.C.; Schnaar, R.L. Carbohydrate-specific adhesion of alveolar macrophages to mannose-derivatized surfaces. J. Biol. Chem. 1984, 259, 1764–1769. [Google Scholar] [CrossRef]
- Larocca, A.P.; Leung, S.C.; Marcus, S.G.; Colby, C.B.; Borden, E.C. Evaluation of neutralizing antibodies in patients treated with recombinant interferon-beta ser. . 1989, S51–60. [Google Scholar]
- Lassiter, G.; Bergeron, C.; Guedry, R.; Cucarola, J.; Kaye, A.M.; Cornett, E.M.; Kaye, A.D.; Varrassi, G.; Viswanath, O.; Urits, I. Belantamab Mafodotin to Treat Multiple Myeloma: A Comprehensive Review of Disease, Drug Efficacy and Side Effects. Curr. Oncol. 2021, 28, 640–660. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-Glycan Number and Degree of Branching Cooperate to Regulate Cell Proliferation and Differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Laube, F. Mannose-6-phosphate/insulin-like growth factor-II receptor in human melanoma cells: effect of ligands and antibodies on the receptor expression. . 2009, 29, 1383–8. [Google Scholar] [PubMed]
- Lee, H.S.; Qi, Y.; Im, W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep. 2015, 5, srep08926–8926. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jin, X.; Zhang, K.; Copertino, L.; Andrews, L.; Baker-Malcolm, J.; Geagan, L.; Qiu, H.; Seiger, K.; Barngrover, D.; et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology 2003, 13, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, C.; Xia, Y.; Bertino, A.; Glaspy, J.; Roberts, M.; Kuter, D.J. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2001, 98, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; DiLillo, D.J.; Bournazos, S.; Giddens, J.P.; Ravetch, J.V.; Wang, L.-X. Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. 2017, 114, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Chiang, A.W.T.; Hansen, A.H.; Arnsdorf, J.; Schoffelen, S.; Sorrentino, J.T.; Kellman, B.P.; Bao, B.; Voldborg, B.G.; Lewis, N.E. A Markov model of glycosylation elucidates isozyme specificity and glycosyltransferase interactions for glycoengineering. Curr. Res. Biotechnol. 2020, 2, 22–36. [Google Scholar] [CrossRef]
- Linehan, S.A.; Martínez-Pomares, L.; Stahl, P.D.; Gordon, S. Mannose Receptor and Its Putative Ligands in Normal Murine Lymphoid and Nonlymphoid Organs: In Situ Expression of Mannose Receptor by Selected Macrophages, Endothelial Cells, Perivascular Microglia, and Mesangial Cells, but not Dendritic Cells. J. Exp. Med. 1999, 189, 1961–1972. [Google Scholar] [CrossRef]
- LIS, H. & SHARON, N. 1993. Protein glycosylation. Structural and functional aspects. Eur J Biochem, 218, 1-27.
- LIU, H. , NOWAK, C., ANDRIEN, B., SHAO, M., PONNIAH, G. & NEILL, A. 2017. Impact of IgG Fc-Oligosaccharides on Recombinant Monoclonal Antibody Structure, Stability, Safety, and Efficacy. Biotechnol Prog, 33, 1173-1181.
- Liu, J.; Wang, G.; Liu, L.; Wu, R.; Wu, Y.; Fang, C.; Zhou, X.; Jiao, J.; Gu, Y.; Zhou, H.; et al. Study of the interactions of a novel monoclonal antibody, mAb059c, with the hPD-1 receptor. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- LIU, K. , TAN, S., JIN, W., GUAN, J., WANG, Q., SUN, H., QI, J., YAN, J., CHAI, Y., WANG, Z., DENG, C. & GAO, G. F. 2020. N-glycosylation of PD-1 promotes binding of camrelizumab. EMBO Rep, 21, e51444.
- Liu, L. Antibody Glycosylation and Its Impact on the Pharmacokinetics and Pharmacodynamics of Monoclonal Antibodies and Fc-Fusion Proteins. J. Pharm. Sci. 2015, 104, 1866–1884. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sambrooks, C.; Shrimal, S.; Khodier, C.; Flaherty, D.P.; Rinis, N.; Charest, J.C.; Gao, N.; Zhao, P.; Wells, L.; A Lewis, T.; et al. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat. Chem. Biol. 2016, 12, 1023–1030. [Google Scholar] [CrossRef]
- Lu, D.; Xu, Z.; Zhang, D.; Jiang, M.; Liu, K.; He, J.; Ma, D.; Ma, X.; Tan, S.; Gao, G.F.; et al. PD-1 N58-Glycosylation-Dependent Binding of Monoclonal Antibody Cemiplimab for Immune Checkpoint Therapy. Front. Immunol. 2022, 13, 826045. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.L.; Pereira, D.S.; Zhu, Z.; Hicklin, D.J.; Bohlen, P. Monoclonal antibody therapeutics and apoptosis. Oncogene 2003, 22, 9097–9106. [Google Scholar] [CrossRef]
- Lundahl, M.L.E.; Fogli, S.; Colavita, P.E.; Scanlan, E.M. Aggregation of protein therapeutics enhances their immunogenicity: causes and mitigation strategies. RSC Chem. Biol. 2021, 2, 1004–1020. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Guan, X.; Li, Y.; Shang, S.; Li, J.; Tan, Z. Protein Glycoengineering: An Approach for Improving Protein Properties. Front. Chem. 2020, 8, 622. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.Y.; A Mikolajczak, S.; Yoshida, T.; Yoshida, R.; Kelvin, D.J.; Ochi, A. CD28 T cell costimulatory receptor function is negatively regulated by N-linked carbohydrates. Biochem. Biophys. Res. Commun. 2004, 317, 60–67. [Google Scholar] [CrossRef]
- Maack, T.; Johnson, V.; Kau, S.T.; Figueiredo, J.; Sigulem, D. Renal filtration, transport, and metabolism of low-molecular-weight proteins: A review. Kidney Int. 1979, 16, 251–270. [Google Scholar] [CrossRef]
- MACHER, B. A. & GALILI, U. 2008. The Galalpha1,3Galbeta1,4GlcNAc-R (alpha-Gal) epitope: a carbohydrate of unique evolution and clinical relevance. Biochim Biophys Acta, 1780, 75-88.
- Madigan, V.J.; Asokan, A. Engineering AAV receptor footprints for gene therapy. Curr. Opin. Virol. 2016, 18, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Madireddi, S.; Eun, S.-Y.; Lee, S.-W.; Nemčovičová, I.; Mehta, A.K.; Zajonc, D.M.; Nishi, N.; Niki, T.; Hirashima, M.; Croft, M. Galectin-9 controls the therapeutic activity of 4-1BB–targeting antibodies. J. Exp. Med. 2014, 211, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I.; Green, M.D. Pharmacokinetic and Pharmacodynamic Considerations in the Development of Therapeutic Proteins. Clin. Pharmacokinet. 2005, 44, 331–347. [Google Scholar] [CrossRef] [PubMed]
- MAJEWSKA, N. I. , TEJADA, M. L., BETENBAUGH, M. J. & AGARWAL, N. 2020. N-Glycosylation of IgG and IgG-Like Recombinant Therapeutic Proteins: Why Is It Important and How Can We Control It? Annu Rev Chem Biomol Eng, 11, 311-338.
- Markov, O.V.; Mironova, N.L.; Shmendel, E.V.; Serikov, R.N.; Morozova, N.G.; Maslov, M.A.; Vlassov, V.V.; Zenkova, M.A. Multicomponent mannose-containing liposomes efficiently deliver RNA in murine immature dendritic cells and provide productive anti-tumour response in murine melanoma model. J. Control. Release 2015, 213, 45–56. [Google Scholar] [CrossRef] [PubMed]
- MARTINEZ-POMARES, L. 2012. The mannose receptor. J Leukoc Biol, 92, 1177-86.
- Mary, B.; Maurya, S.; Arumugam, S.; Kumar, V.; Jayandharan, G.R. Post-translational modifications in capsid proteins of recombinant adeno-associated virus (AAV) 1-rh10 serotypes. FEBS J. 2019, 286, 4964–4981. [Google Scholar] [CrossRef] [PubMed]
- Mary, B.; Maurya, S.; Kumar, M.; Bammidi, S.; Kumar, V.; Jayandharan, G.R. Molecular Engineering of Adeno-Associated Virus Capsid Improves Its Therapeutic Gene Transfer in Murine Models of Hemophilia and Retinal Degeneration. Mol. Pharm. 2019, 16, 4738–4750. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Keiser, K.; Nair, J.K.; Charisse, K.; Manoharan, R.M.; Kretschmer, P.; Peng, C.G.; Kel’in, A.V.; Kandasamy, P.; Willoughby, J.L.; et al. siRNA Conjugates Carrying Sequentially Assembled Trivalent N-Acetylgalactosamine Linked Through Nucleosides Elicit Robust Gene Silencing In Vivo in Hepatocytes. ACS Chem. Biol. 2015, 10, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- MAYEUX, P. & CASADEVALL, N. 2003. Antibodies to endogenous and recombinant erythropoietin.. In: MOLINEUX, G., FOOTE, M. A. & ELLIOTT, S. G. (eds.) Erythropoietins and Erythropoiesis. Milestones in Drug Therapy. Basel: Birkhäuser Basel.
- McCafferty, E.H.; Scott, L.J. Vestronidase Alfa: A Review in Mucopolysaccharidosis VII. BioDrugs 2019, 33, 233–240. [Google Scholar] [CrossRef] [PubMed]
- MEREITER, S. , BALMANA, M., CAMPOS, D., GOMES, J. & REIS, C. A. 2019. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell, 36, 6-16.
- MEURIS, L. , SANTENS, F., ELSON, G., FESTJENS, N., BOONE, M., DOS SANTOS, A., DEVOS, S., ROUSSEAU, F., PLETS, E., HOUTHUYS, E., MALINGE, P., MAGISTRELLI, G., CONS, L., CHATEL, L., DEVREESE, B. & CALLEWAERT, N. 2014. GlycoDelete engineering of mammalian cells simplifies N-glycosylation of recombinant proteins. Nat Biotechnol, 32, 485-9.
- Mevel, M.; Bouzelha, M.; Leray, A.; Pacouret, S.; Guilbaud, M.; Penaud-Budloo, M.; Alvarez-Dorta, D.; Dubreil, L.; Gouin, S.G.; Combal, J.P.; et al. Chemical modification of the adeno-associated virus capsid to improve gene delivery. Chem. Sci. 2020, 11, 1122–1131. [Google Scholar] [CrossRef]
- Meyer, N.L.; Chapman, M.S. Adeno-associated virus (AAV) cell entry: structural insights. Trends Microbiol. 2021, 30, 432–451. [Google Scholar] [CrossRef]
- Mietzsch, M.; Broecker, F.; Reinhardt, A.; Seeberger, P.H.; Heilbronn, R. Differential Adeno-Associated Virus Serotype-Specific Interaction Patterns with Synthetic Heparins and Other Glycans. J. Virol. 2014, 88, 2991–3003. [Google Scholar] [CrossRef] [PubMed]
- Mimura, Y.; Katoh, T.; Saldova, R.; O'Flaherty, R.; Izumi, T.; Mimura-Kimura, Y.; Utsunomiya, T.; Mizukami, Y.; Yamamoto, K.; Matsumoto, T.; et al. Glycosylation engineering of therapeutic IgG antibodies: challenges for the safety, functionality and efficacy. Protein Cell 2018, 9, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Minnelli, C.; Cianfruglia, L.; Laudadio, E.; Galeazzi, R.; Pisani, M.; Crucianelli, E.; Bizzaro, D.; Armeni, T.; Mobbili, G. Selective induction of apoptosis in MCF7 cancer-cell by targeted liposomes functionalised with mannose-6-phosphate. J. Drug Target. 2017, 26, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Mitra, N.; Sinha, S.; Ramya, T.N.; Surolia, A. N-linked oligosaccharides as outfitters for glycoprotein folding, form and function. Trends Biochem. Sci. 2006, 31, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Mkhikian, H.; Grigorian, A.; Li, C.F.; Chen, H.-L.; Newton, B.; Zhou, R.W.; Beeton, C.; Torossian, S.; Tatarian, G.G.; Lee, S.-U.; et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat. Commun. 2011, 2, 334. [Google Scholar] [CrossRef] [PubMed]
- Mollé, L.M.; Smyth, C.H.; Yuen, D.; Johnston, A.P.R. Nanoparticles for vaccine and gene therapy: Overcoming the barriers to nucleic acid delivery. WIREs Nanomed. Nanobiotechnology 2022, 14, e1809. [Google Scholar] [CrossRef]
- Mondal, N.; Silva, M.; Castano, A.P.; Maus, M.V.; Sackstein, R. Glycoengineering of chimeric antigen receptor (CAR) T-cells to enforce E-selectin binding. J. Biol. Chem. 2019, 294, 18465–18474. [Google Scholar] [CrossRef]
- Moorkens, E.; Meuwissen, N.; Huys, I.; Declerck, P.; Vulto, A.G.; Simoens, S. The Market of Biopharmaceutical Medicines: A Snapshot of a Diverse Industrial Landscape. Front. Pharmacol. 2017, 8, 314. [Google Scholar] [CrossRef]
- Morell, A.G.; Gregoriadis, G.; Scheinberg, I.H.; Hickman, J.; Ashwell, G. The Role of Sialic Acid in Determining the Survival of Glycoproteins in the Circulation. J. Biol. Chem. 1971, 246, 1461–1467. [Google Scholar] [CrossRef]
- Morell, A.G.; A Irvine, R.; Sternlieb, I.; Scheinberg, I.H.; Ashwell, G. Physical and chemical studies on ceruloplasmin. V. Metabolic studies on sialic acid-free ceruloplasmin in vivo.. 1968, 243, 155–9. [Google Scholar] [CrossRef]
- MOSSNER, E. , BRUNKER, P., MOSER, S., PUNTENER, U., SCHMIDT, C., HERTER, S., GRAU, R., GERDES, C., NOPORA, A., VAN PUIJENBROEK, E., FERRARA, C., SONDERMANN, P., JAGER, C., STREIN, P., FERTIG, G., FRIESS, T., SCHULL, C., BAUER, S., DAL PORTO, J., DEL NAGRO, C., DABBAGH, K., DYER, M. J., POPPEMA, S., KLEIN, C. & UMANA, P. 2010. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood, 115, 4393-402.
- Muenzer, J.; Gucsavas-Calikoglu, M.; McCandless, S.E.; Schuetz, T.J.; Kimura, A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Abstr. 2007 Meet. Soc. Inherit. Metab. Disord. 2007, 90, 329–337. [Google Scholar] [CrossRef] [PubMed]
- MUENZER, J. , LAMSA, J. C., GARCIA, A., DACOSTA, J., GARCIA, J. & TRECO, D. A. 2002. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr Suppl, 91, 98-9.
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.; Arakawa, T.; Aoki, K.; Elmore, R.; Rohde, M.; Boone, T.; Strickland, T. The effect of carbohydrate on the structure and stability of erythropoietin. J. Biol. Chem. 1991, 266, 23022–23026. [Google Scholar] [CrossRef] [PubMed]
- NEUFELD, E. F. & MUENZER, J. 1989. The metabolic basis of inherited disease, New York, McGraw-Hill.
- Ng, R.; Govindasamy, L.; Gurda, B.L.; McKenna, R.; Kozyreva, O.G.; Samulski, R.J.; Parent, K.N.; Baker, T.S.; Agbandje-McKenna, M. Structural Characterization of the Dual Glycan Binding Adeno-Associated Virus Serotype 6. J. Virol. 2010, 84, 12945–12957. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Tangvoranuntakul, P.; Varki, A. Effects of Natural Human Antibodies against a Nonhuman Sialic Acid That Metabolically Incorporates into Activated and Malignant Immune Cells. J. Immunol. 2005, 175, 228–236. [Google Scholar] [CrossRef]
- NIMMERJAHN, F. , ANTHONY, R. M. & RAVETCH, J. V. 2007. Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc Natl Acad Sci U S A, 104, 8433-7.
- Nimmerjahn, F.; Ravetch, J.V. Anti-Inflammatory Actions of Intravenous Immunoglobulin. Annu. Rev. Immunol. 2008, 26, 513–533. [Google Scholar] [CrossRef]
- Nishida-Aoki, N.; Tominaga, N.; Kosaka, N.; Ochiya, T. Altered biodistribution of deglycosylated extracellular vesicles through enhanced cellular uptake. J. Extracell. Vesicles 2020, 9, 1713527. [Google Scholar] [CrossRef]
- Nishiyama, T.; Kimura, N.; Jitsuhara, Y.; Uchida, M.; Ochi, F.; Yamaguchi, H. N-Glycans protect proteins from protease digestion through their binding affinities for aromatic amino acid residues. J. Biochem. 2000, 127, 427–433. [Google Scholar] [CrossRef]
- Nose, M.; Wigzell, H. Biological significance of carbohydrate chains on monoclonal antibodies. Proc. Natl. Acad. Sci. 1983, 80, 6632–6636. [Google Scholar] [CrossRef]
- O'Neil, B.H.; Allen, R.; Spigel, D.R.; Stinchcombe, T.E.; Moore, D.T.; Berlin, J.D.; Goldberg, R.M. High Incidence of Cetuximab-Related Infusion Reactions in Tennessee and North Carolina and the Association With Atopic History. J. Clin. Oncol. 2007, 25, 3644–3648. [Google Scholar] [CrossRef] [PubMed]
- Ogata, S.; Shimizu, C.; Franco, A.; Touma, R.; Kanegaye, J.T.; Choudhury, B.P.; Naidu, N.N.; Kanda, Y.; Hoang, L.T.; Hibberd, M.L.; et al. Treatment Response in Kawasaki Disease Is Associated with Sialylation Levels of Endogenous but Not Therapeutic Intravenous Immunoglobulin G. PLOS ONE 2013, 8, e81448. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-B. Glyco-engineering strategies for the development of therapeutic enzymes with improved efficacy for the treatment of lysosomal storage diseases. BMB Rep. 2015, 48, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Oh-Eda, M.; Hasegawa, M.; Hattori, K.; Kuboniwa, H.; Kojima, T.; Orita, T.; Tomonou, K.; Yamazaki, T.; Ochi, N. O-linked sugar chain of human granulocyte colony-stimulating factor protects it against polymerization and denaturation allowing it to retain its biological activity. J. Biol. Chem. 1990, 265, 11432–11435. [Google Scholar] [CrossRef]
- Okada, M.; Chikuma, S.; Kondo, T.; Hibino, S.; Machiyama, H.; Yokosuka, T.; Nakano, M.; Yoshimura, A. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-tumor Immune Responses of T Cells. Cell Rep. 2017, 20, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Nakai, M.; Nakayama, C.; Yanagi, H.; Matsui, H.; Noguchi, H.; Namiki, M.; Sakai, J.; Kadota, K.; Fukui, M.; et al. Purification and characterization of three forms of differently glycosylated recombinant human granulocyte-macrophage colony-stimulating factor. Arch. Biochem. Biophys. 1991, 286, 562–568. [Google Scholar] [CrossRef] [PubMed]
- OKERBLOM, J. & VARKI, A. 2017. Biochemical, Cellular, Physiological, and Pathological Consequences of Human Loss of N-Glycolylneuraminic Acid. Chembiochem, 18, 1155-1171.
- Orange, J.S.; Hossny, E.M.; Weiler, C.R.; Ballow, M.; Berger, M.; Bonilla, F.A.; Buckley, R.; Chinen, J.; Elgamal, Y.; Mazer, B.D.; et al. Use of intravenous immunoglobulin in human disease: A review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J. Allergy Clin. Immunol. 2006, 117, S525–S553. [Google Scholar] [CrossRef] [PubMed]
- Padler-Karavani, V.; Yu, H.; Cao, H.; Chokhawala, H.; Karp, F.; Varki, N.; Chen, X.; Varki, A. Diversity in specificity, abundance, and composition of anti-Neu5Gc antibodies in normal humans: Potential implications for disease. Glycobiology 2008, 18, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Pignata, C.; Vajro, P.; Salerno, M. New strategies for the treatment of lysosomal storage diseases (Review). Int. J. Mol. Med. 2012, 31, 11–20. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F. Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations. Int. J. Mol. Sci. 2020, 21, 2975. [Google Scholar] [CrossRef]
- Park, E.I.; Manzella, S.M.; Baenziger, J.U. Rapid Clearance of Sialylated Glycoproteins by the Asialoglycoprotein Receptor. J. Biol. Chem. 2003, 278, 4597–4602. [Google Scholar] [CrossRef] [PubMed]
- PARK, E. I. , MI, Y., UNVERZAGT, C., GABIUS, H. J. & BAENZIGER, J. U. 2005. The asialoglycoprotein receptor clears glycoconjugates terminating with sialic acid alpha 2,6GalNAc. Proc Natl Acad Sci U S A, 102, 17125-9.
- Park, H.; Kim, J.; Lee, Y.K.; Kim, W.; You, S.K.; Do, J.; Jang, Y.; Oh, D.-B.; Kim, J.I.; Kim, H.H. Four unreported types of glycans containing mannose-6-phosphate are heterogeneously attached at three sites (including newly found Asn 233) to recombinant human acid alpha-glucosidase that is the only approved treatment for Pompe disease. Biochem. Biophys. Res. Commun. 2018, 495, 2418–2424. [Google Scholar] [CrossRef]
- Zacco, E.; Paul, A.; Segal, D. Glycans to improve efficacy and solubility of protein aggregation inhibitors. Neural Regen. Res. 2021, 16, 2215–2216. [Google Scholar] [CrossRef] [PubMed]
- Pavelić, K.; Kolak, T.; Kapitanović, S.; Radošević, S.; Spaventi. ; Krušlin, B.; Pavelić, J. Gastric cancer: the role of insulin-like growth factor 2 (IGF 2) and its receptors (IGF 1R and M6-P/IGF 2R). J. Pathol. 2003, 201, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Calabrese, C.; Vatrella, A.; Busceti, M.T.; Garofalo, E.; Lombardo, N.; Terracciano, R.; Pelaia, G. Benralizumab: From the Basic Mechanism of Action to the Potential Use in the Biological Therapy of Severe Eosinophilic Asthma. BioMed Res. Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.A.; Chan, K.F.; Lin, P.C.; Song, Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. mAbs 2018, 10, 693–711. [Google Scholar] [CrossRef] [PubMed]
- PERLMAN, S. , VAN DEN HAZEL, B., CHRISTIANSEN, J., GRAM-NIELSEN, S., JEPPESEN, C. B., ANDERSEN, K. V., HALKIER, T., OKKELS, S. & SCHAMBYE, H. T. 2003. Glycosylation of an N-terminal extension prolongs the half-life and increases the in vivo activity of follicle stimulating hormone. J Clin Endocrinol Metab, 88, 3227-35.
- Peschke, B.; Keller, C.W.; Weber, P.; Quast, I.; Lünemann, J.D. Fc-Galactosylation of Human Immunoglobulin Gamma Isotypes Improves C1q Binding and Enhances Complement-Dependent Cytotoxicity. Front. Immunol. 2017, 8, 646–646. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.B.; Meng, W.S. Protein aggregation and immunogenicity of biotherapeutics. Int. J. Pharm. 2020, 585, 119523–119523. [Google Scholar] [CrossRef] [PubMed]
- PORTER, S. 2001. Human immune response to recombinant human proteins. J Pharm Sci, 90, 1-11.
- Pound, J.D.; Lund, J.; Jefferis, R. Aglycosylated chimaeric human IgG3 can trigger the human phagocyte respiratory burst. Mol. Immunol. 1993, 30, 233–241. [Google Scholar] [CrossRef]
- Pratt, K.P.; Thompson, A.R. B-Cell and T-Cell Epitopes in Anti-factor VIII Immune Responses. Clin. Rev. Allergy Immunol. 2009, 37, 80–95. [Google Scholar] [CrossRef]
- Qian, J.; Liu, T.; Yang, L.; Daus, A.; Crowley, R.; Zhou, Q. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole–quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal. Biochem. 2007, 364, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.E.; Rolling, F.; Li, C.; Conrath, H.; Xiao, W.; Xiao, X.; Samulski, R.J. Cross-Packaging of a Single Adeno-Associated Virus (AAV) Type 2 Vector Genome into Multiple AAV Serotypes Enables Transduction with Broad Specificity. J. Virol. 2002, 76, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Rajeev, K.G.; Nair, J.K.; Jayaraman, M.; Charisse, K.; Taneja, N.; O'Shea, J.; Willoughby, J.L.S.; Yucius, K.; Nguyen, T.; Shulga-Morskaya, S.; et al. Hepatocyte-Specific Delivery of siRNAs Conjugated to Novel Non-nucleosidic Trivalent N-Acetylgalactosamine Elicits Robust Gene Silencing in Vivo. ChemBioChem 2015, 16, 903–908. [Google Scholar] [CrossRef]
- Raju, T.S. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr. Opin. Immunol. 2008, 20, 471–478. [Google Scholar] [CrossRef]
- Raju, T.; Briggs, J.B.; Borge, S.M.; Jones, A.J.S. Species-specific variation in glycosylation of IgG: evidence for the species-specific sialylation and branch-specific galactosylation and importance for engineering recombinant glycoprotein therapeutics. Glycobiology 2000, 10, 477–486. [Google Scholar] [CrossRef]
- Raman, R.; Tharakaraman, K.; Sasisekharan, V.; Sasisekharan, R. Glycan–protein interactions in viral pathogenesis. Curr. Opin. Struct. Biol. 2016, 40, 153–162. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Rennke, H.G.; Cotran, R.S.; A Venkatachalam, M. Role of molecular charge in glomerular permeability. Tracer studies with cationized ferritins. J. Cell Biol. 1975, 67, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Ringe, R.P.; Pugach, P.; Cottrell, C.A.; LaBranche, C.C.; Seabright, G.E.; Ketas, T.J.; Ozorowski, G.; Kumar, S.; Schorcht, A.; van Gils, M.J.; et al. Closing and Opening Holes in the Glycan Shield of HIV-1 Envelope Glycoprotein SOSIP Trimers Can Redirect the Neutralizing Antibody Response to the Newly Unmasked Epitopes. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Rinis, N.; Golden, J.E.; Marceau, C.D.; Carette, J.E.; Van Zandt, M.C.; Gilmore, R.; Contessa, J.N. Editing N-Glycan Site Occupancy with Small-Molecule Oligosaccharyltransferase Inhibitors. Cell Chem. Biol. 2018, 25, 1231–1241. [Google Scholar] [CrossRef]
- Ross, D.; Brown, T.; Harper, R.; Pamarthi, M.; Nixon, J.; Bromirski, J.; Li, C.-M.; Ghali, R.; Xie, H.; Medvedeff, G.; et al. Production and characterization of a novel human recombinant alpha-1-antitrypsin in PER.C6 cells. J. Biotechnol. 2012, 162, 262–273. [Google Scholar] [CrossRef]
- ROSSI, M. , PARENTI, G., DELLA CASA, R., ROMANO, A., MANSI, G., AGOVINO, T., ROSAPEPE, F., VOSA, C., DEL GIUDICE, E. & ANDRIA, G. 2007. Long-term enzyme replacement therapy for pompe disease with recombinant human alpha-glucosidase derived from chinese hamster ovary cells. J Child Neurol, 22, 565-73.
- Royo, F.; Cossio, U.; de Angulo, A.R.; Llop, J.; Falcon-Perez, J.M. Modification of the glycosylation of extracellular vesicles alters their biodistribution in mice. Nanoscale 2019, 11, 1531–1537. [Google Scholar] [CrossRef]
- Rujas, E.; Cui, H.; Sicard, T.; Semesi, A.; Julien, J.-P. Structural characterization of the ICOS/ICOS-L immune complex reveals high molecular mimicry by therapeutic antibodies. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- RUNKEL, L. , MEIER, W., PEPINSKY, R. B., KARPUSAS, M., WHITTY, A., KIMBALL, K., BRICKELMAIER, M., MULDOWNEY, C., JONES, W. & GOELZ, S. E. 1998. Structural and functional differences between glycosylated and non-glycosylated forms of human interferon-beta (IFN-beta). Pharm Res, 15, 641-9.
- Rushworth, J.L.; Montgomery, K.S.; Cao, B.; Brown, R.; Dibb, N.J.; Nilsson, S.K.; Chiefari, J.; Fuchter, M.J. Glycosylated Nanoparticles Derived from RAFT Polymerization for Effective Drug Delivery to Macrophages. ACS Appl. Bio Mater. 2020, 3, 5775–5786. [Google Scholar] [CrossRef]
- Sakuraba, H.; Murata-Ohsawa, M.; Kawashima, I.; Tajima, Y.; Kotani, M.; Ohshima, T.; Chiba, Y.; Takashiba, M.; Jigami, Y.; Fukushige, T.; et al. Comparison of the effects of agalsidase alfa and agalsidase beta on cultured human Fabry fibroblasts and Fabry mice. J. Hum. Genet. 2005, 51, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Evanno, G.; Lim, N.; Rousse, J.; Le Berre, L.; Nicot, A.; Bach, J.-M.; Brouard, S.; Harris, K.M.; Ehlers, M.R.; et al. Anti-Gal and Anti-Neu5Gc Responses in Nonimmunosuppressed Patients After Treatment With Rabbit Antithymocyte Polyclonal IgGs. Transplantation 2017, 101, 2501–2507. [Google Scholar] [CrossRef]
- Sallustio, S.; Stanley, P. Novel genetic instability associated with a developmental regulated glycosyltransferase locus in Chinese hamster ovary cells. Somat. Cell Mol. Genet. 1989, 15, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Efficient Presentation of Soluble Antigen by Cultured Human Dendritic Cells Is Maintained by Granulocyte/Macrophage Colony-stimulating Factor Plus Interleukin 4 and Downregulated by Tumor Necrosis Factor alpha. J. Immunol. 2018, 200, 887–896. [Google Scholar] [CrossRef]
- SARENEVA, T. , PIRHONEN, J., CANTELL, K. & JULKUNEN, I. 1995. N-glycosylation of human interferon-gamma: glycans at Asn-25 are critical for protease resistance. Biochem J, 308 ( Pt 1), 9-14.
- SARMAY, G. , LUND, J., ROZSNYAY, Z., GERGELY, J. & JEFFERIS, R. 1992. Mapping and comparison of the interaction sites on the Fc region of IgG responsible for triggering antibody dependent cellular cytotoxicity (ADCC) through different types of human Fc gamma receptor. Mol Immunol, 29, 633-9.
- SASAWATARI, S. , OKAMOTO, Y., KUMANOGOH, A. & TOYOFUKU, T. 2020. Blockade of N-Glycosylation Promotes Antitumor Immune Response of T Cells. J Immunol, 204, 1373-1385.
- Sato, Y.; Beutler, E. Binding, internalization, and degradation of mannose-terminated glucocerebrosidase by macrophages. J. Clin. Investig. 1993, 91, 1909–1917. [Google Scholar] [CrossRef]
- Sazinsky, S.L.; Ott, R.G.; Silver, N.W.; Tidor, B.; Ravetch, J.V.; Wittrup, K.D. Aglycosylated immunoglobulin G1variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. 2008, 105, 20167–20172. [Google Scholar] [CrossRef]
- SCHACHTER, H. 2000. The joys of HexNAc. The synthesis and function of N- and O-glycan branches. Glycoconj J, 17, 465-83.
- SCHIFFMANN, R. , KOPP, J. B., AUSTIN, H. A., 3RD, SABNIS, S., MOORE, D. F., WEIBEL, T., BALOW, J. E. & BRADY, R. O. 2001. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA, 285, 2743-9.
- Schiffmann, R.; Murray, G.J.; Treco, D.; Daniel, P.; Sellos-Moura, M.; Myers, M.; Quirk, J.M.; Zirzow, G.C.; Borowski, M.; Loveday, K.; et al. Infusion of α-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc. Natl. Acad. Sci. 2000, 97, 365–370. [Google Scholar] [CrossRef] [PubMed]
- SCHLESINGER, P. H. , DOEBBER, T. W., MANDELL, B. F., WHITE, R., DESCHRYVER, C., RODMAN, J. S., MILLER, M. J. & STAHL, P. 1978. Plasma clearance of glycoproteins with terminal mannose and N-acetylglucosamine by liver non-parenchymal cells. Studies with beta-glucuronidase, N-acetyl-beta-D-glucosaminidase, ribonuclease B and agalacto-orosomucoid. Biochem J, 176, 103-9.
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125 (Suppl. 2), S41–S52. [Google Scholar] [CrossRef] [PubMed]
- SCHWAB, I. & NIMMERJAHN, F. 2013. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol, 13, 176-89.
- SEITE, J. F. , SHOENFELD, Y., YOUINOU, P. & HILLION, S. 2008. What is the contents of the magic draft IVIg? Autoimmun Rev, 7, 435-9.
- Seo, J.; Oh, D.-B. Mannose-6-phosphate glycan for lysosomal targeting: various applications from enzyme replacement therapy to lysosome-targeting chimeras. Anim. Cells Syst. 2022, 26, 84–91. [Google Scholar] [CrossRef] [PubMed]
- SHAALTIEL, Y. , BARTFELD, D., HASHMUELI, S., BAUM, G., BRILL-ALMON, E., GALILI, G., DYM, O., BOLDIN-ADAMSKY, S. A., SILMAN, I., SUSSMAN, J. L., FUTERMAN, A. H. & AVIEZER, D. 2007. Production of glucocerebrosidase with terminal mannose glycans for enzyme replacement therapy of Gaucher's disease using a plant cell system. Plant Biotechnol J, 5, 579-90.
- Shamie, I.; Duttke, S.H.; Karottki, K.J.l.C.; Han, C.Z.; Hansen, A.H.; Hefzi, H.; Xiong, K.; Li, S.; Roth, S.J.; Tao, J.; et al. A Chinese hamster transcription start site atlas that enables targeted editing of CHO cells. NAR Genom. Bioinform. 2021, 3, lqab061. [Google Scholar] [CrossRef]
- Shao, B.; Li, C.-W.; Lim, S.-O.; Sun, L.; Lai, Y.-J.; Hou, J.; Liu, C.; Chang, C.-W.; Qiu, Y.; Hsu, J.-M.; et al. Deglycosylation of PD-L1 by 2-deoxyglucose reverses PARP inhibitor-induced immunosuppression in triple-negative breast cancer. 2018, 8, 1837–+.
- Sharma, V.K.; Osborn, M.F.; Hassler, M.R.; Echeverria, D.; Ly, S.; Ulashchik, E.A.; Martynenko-Makaev, Y.V.; Shmanai, V.V.; Zatsepin, T.S.; Khvorova, A.; et al. Novel Cluster and Monomer-Based GalNAc Structures Induce Effective Uptake of siRNAs in Vitro and in Vivo. Bioconjugate Chem. 2018, 29, 2478–2488. [Google Scholar] [CrossRef]
- Sharon, D.; Kamen, A. Advancements in the design and scalable production of viral gene transfer vectors. Biotechnol. Bioeng. 2017, 115, 25–40. [Google Scholar] [CrossRef]
- Sheeley, D.M.; Merrill, B.M.; Taylor, L.C. Characterization of Monoclonal Antibody Glycosylation: Comparison of Expression Systems and Identification of Terminal α-Linked Galactose. Anal. Biochem. 1997, 247, 102–110. [Google Scholar] [CrossRef]
- Shen, S.; Bryant, K.D.; Brown, S.M.; Randell, S.H.; Asokan, A. Terminal N-Linked Galactose Is the Primary Receptor for Adeno-associated Virus 9. J. Biol. Chem. 2011, 286, 13532–13540. [Google Scholar] [CrossRef]
- Shental-Bechor, D.; Levy, Y. Effect of glycosylation on protein folding: A close look at thermodynamic stabilization. Proc. Natl. Acad. Sci. 2008, 105, 8256–8261. [Google Scholar] [CrossRef]
- Shepherd, V.L.; Campbell, E.J.; Senior, R.M.; Stahl, P.D. Characterization of the mannose/fucose receptor on human mononuclear phagocytes. . 1982, 32, 423–31. [Google Scholar]
- Shi, H.H.; Goudar, C.T. Recent advances in the understanding of biological implications and modulation methodologies of monoclonal antibody N-linked high mannose glycans. Biotechnol. Bioeng. 2014, 111, 1907–1919. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, D.; Li, F.; Zhang, Z.; Wang, S.; Xuan, Y.; Ping, Y.; Zhang, Y. Targeting glycosylation of PD-1 to enhance CAR-T cell cytotoxicity. J. Hematol. Oncol. 2019, 12, 1–4. [Google Scholar] [CrossRef]
- SHIELDS, R. L. , LAI, J., KECK, R., O'CONNELL, L. Y., HONG, K., MENG, Y. G., WEIKERT, S. H. & PRESTA, L. G. 2002. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem, 277, 26733-40.
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The Absence of Fucose but Not the Presence of Galactose or Bisecting N-Acetylglucosamine of Human IgG1 Complex-type Oligosaccharides Shows the Critical Role of Enhancing Antibody-dependent Cellular Cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef]
- Shitara, K. Potelligent Antibodies as Next Generation Therapeutic Antibodies. YAKUGAKU ZASSHI 2009, 129, 3–9. [Google Scholar] [CrossRef]
- Sinclair, A.M.; Elliott, S. Glycoengineering: The effect of glycosylation on the properties of therapeutic proteins. J. Pharm. Sci. 2005, 94, 1626–1635. [Google Scholar] [CrossRef]
- Sly, W.S. Receptor-mediated transport of acid hydrolases to lysosomes. Curr. Top. Cell. Regul. 1985, 26, 27–38. [Google Scholar]
- Sohn, Y.; Lee, J.M.; Park, H.-R.; Jung, S.-C.; Park, T.H.; Oh, D.-B. Enhanced sialylation and in vivo efficacy of recombinant human α-galactosidase through in vitro glycosylation. BMB Rep. 2013, 46, 157–162. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Cho, S.Y.; Park, S.W.; Kim, S.J.; Ko, A.-R.; Kwon, E.-K.; Han, S.J.; Jin, D.-K. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter Syndrome). Orphanet J. Rare Dis. 2013, 8, 42–42. [Google Scholar] [CrossRef]
- Solá, R.J.; Griebenow, K. Influence of modulated structural dynamics on the kinetics of α-chymotrypsin catalysis. FEBS J. 2006, 273, 5303–5319. [Google Scholar] [CrossRef]
- Solá, R.J.; Griebenow, K. Effects of glycosylation on the stability of protein pharmaceuticals. J. Pharm. Sci. 2009, 98, 1223–1245. [Google Scholar] [CrossRef]
- Solá, R.J.; Rodriguez-Martinez, J.A.; Griebenow, K. Modulation of protein biophysical properties by chemical glycosylation: biochemical insights and biomedical implications. Cell. Mol. Life Sci. 2007, 64, 2133–2152. [Google Scholar] [CrossRef]
- Sondermann, P.; Pincetic, A.; Maamary, J.; Lammens, K.; Ravetch, J.V. General mechanism for modulating immunoglobulin effector function. Proc. Natl. Acad. Sci. 2013, 110, 9868–9872. [Google Scholar] [CrossRef]
- Song, X.; Zhou, Z.; Li, H.; Xue, Y.; Lu, X.; Bahar, I.; Kepp, O.; Hung, M.-C.; Kroemer, G.; Wan, Y. Pharmacologic Suppression of B7-H4 Glycosylation Restores Antitumor Immunity in Immune-Cold Breast Cancers. Cancer Discov. 2020, 10, 1872–1893. [Google Scholar] [CrossRef]
- Spahn, P.N.; Hansen, A.H.; Hansen, H.G.; Arnsdorf, J.; Kildegaard, H.F.; Lewis, N.E. A Markov chain model for N-linked protein glycosylation – towards a low-parameter tool for model-driven glycoengineering. Metab. Eng. 2015, 33, 52–66. [Google Scholar] [CrossRef]
- Spahn, P.N.; Hansen, A.H.; Kol, S.; Voldborg, B.G.; Lewis, N.E. Predictive glycoengineering of biosimilars using a Markov chain glycosylation model. Biotechnol. J. 2016, 12. [Google Scholar] [CrossRef]
- Stahl, P.; Gordon, S. Expression of a mannosyl-fucosyl receptor for endocytosis on cultured primary macrophages and their hybrids. J. Cell Biol. 1982, 93, 49–56. [Google Scholar] [CrossRef]
- Stahl, P.D. The Macrophage Mannose Receptor: Current Status. Am. J. Respir. Cell Mol. Biol. 1990, 2, 317–318. [Google Scholar] [CrossRef]
- Stahl, P.D. The mannose receptor and other macrophage lectins. Curr. Opin. Immunol. 1992, 4, 49–52. [Google Scholar] [CrossRef]
- Stahl, P.D.; Rodman, J.S.; Miller, M.J.; Schlesinger, P.H. Evidence for receptor-mediated binding of glycoproteins, glycoconjugates, and lysosomal glycosidases by alveolar macrophages. Proc. Natl. Acad. Sci. 1978, 75, 1399–1403. [Google Scholar] [CrossRef]
- Stockert, R.J. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol. Rev. 1995, 75, 591–609. [Google Scholar] [CrossRef]
- Strasser, L.; Boi, S.; Guapo, F.; Donohue, N.; Barron, N.; Rainbow-Fletcher, A.; Bones, J. Proteomic Landscape of Adeno-Associated Virus (AAV)-Producing HEK293 Cells. Int. J. Mol. Sci. 2021, 22, 11499. [Google Scholar] [CrossRef]
- Ströh, L.J.; Stehle, T. Glycan Engagement by Viruses: Receptor Switches and Specificity. Annu. Rev. Virol. 2014, 1, 285–306. [Google Scholar] [CrossRef]
- Su, D.; Zhao, H.; Xia, H. Glycosylation-modified erythropoietin with improved half-life and biological activity. Int. J. Hematol. 2010, 91, 238–244. [Google Scholar] [CrossRef]
- Summerford, C.; Samulski, R.J. Membrane-Associated Heparan Sulfate Proteoglycan Is a Receptor for Adeno-Associated Virus Type 2 Virions. J. Virol. 1998, 72, 1438–1445. [Google Scholar] [CrossRef]
- Sun, L.; Li, C.-W.; Chung, E.M.; Yang, R.; Kim, Y.-S.; Park, A.H.; Lai, Y.-J.; Yang, Y.; Wang, Y.-H.; Liu, J.; et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res 2020, 80, 2298–2310. [Google Scholar] [CrossRef]
- Sun, R.; Kim, A.M.J.; Lim, S.-O. Glycosylation of Immune Receptors in Cancer. Cells 2021, 10, 1100. [Google Scholar] [CrossRef]
- Sun, R.; Kim, A.M.J.; Murray, A.A.; Lim, S.-O. N-Glycosylation Facilitates 4-1BB Membrane Localization by Avoiding Its Multimerization. Cells 2022, 11, 162. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Takahashi, K.; Donovan, M.J.; Rogers, R.A.; Ezekowitz, R.A.B. Distribution of murine mannose receptor expression from early embryogenesis through to adulthood. Cell Tissue Res. 1998, 292, 311–323. [Google Scholar] [CrossRef]
- Tams, J.W.; Vind, J.; Welinder, K.G. Adapting protein solubility by glycosylation.: N-Glycosylation mutants of Coprinus cinereus peroxidase in salt and organic solutions. Biochim. et Biophys. Acta (BBA) - Protein Struct. Mol. Enzym. 1999, 1432, 214–221. [Google Scholar] [CrossRef]
- Tangvoranuntakul, P.; Gagneux, P.; Diaz, S.; Bardor, M.; Varki, N.; Varki, A.; Muchmore, E. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc. Natl. Acad. Sci. 2003, 100, 12045–12050. [Google Scholar] [CrossRef]
- Tao, M.H.; Morrison, S.L. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 1989, 143, 2595–2601. [Google Scholar] [CrossRef]
- Tate, M.D.; Job, E.R.; Deng, Y.-M.; Gunalan, V.; Maurer-Stroh, S.; Reading, P.C. Playing Hide and Seek: How Glycosylation of the Influenza Virus Hemagglutinin Can Modulate the Immune Response to Infection. Viruses 2014, 6, 1294–1316. [Google Scholar] [CrossRef]
- Taylor, M.; Drickamer, K. Structural requirements for high affinity binding of complex ligands by the macrophage mannose receptor. J. Biol. Chem. 1993, 268, 399–404. [Google Scholar] [CrossRef]
- TEKOAH, Y. , TZABAN, S., KIZHNER, T., HAINRICHSON, M., GANTMAN, A., GOLEMBO, M., AVIEZER, D. & SHAALTIEL, Y. 2013. Glycosylation and functionality of recombinant beta-glucocerebrosidase from various production systems. Biosci Rep, 33.
- TEMAM, S. , SPICER, J., FARZANEH, F., SORIA, J. C., OPPENHEIM, D., MCGURK, M., HOLLEBECQUE, A., SARINI, J., HUSSAIN, K., SOEHRMAN BROSSARD, S., MANENTI, L., EVERS, S., DELMAR, P., DI SCALA, L., MANCAO, C., FEUERHAKE, F., ANDRIES, L., OTT, M. G., PASSIOUKOV, A. & DELORD, J. P. 2017. An exploratory, open-label, randomized, multicenter study to investigate the pharmacodynamics of a glycoengineered antibody (imgatuzumab) and cetuximab in patients with operable head and neck squamous cell carcinoma. Ann Oncol, 28, 2827-2835.
- Tian, W.; Ye, Z.; Wang, S.; Schulz, M.A.; Van Coillie, J.; Sun, L.; Chen, Y.-H.; Narimatsu, Y.; Hansen, L.; Kristensen, C.; et al. The glycosylation design space for recombinant lysosomal replacement enzymes produced in CHO cells. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- TIELS, P. , BARANOVA, E., PIENS, K., DE VISSCHER, C., PYNAERT, G., NERINCKX, W., STOUT, J., FUDALEJ, F., HULPIAU, P., TANNLER, S., GEYSENS, S., VAN HECKE, A., VALEVSKA, A., VERVECKEN, W., REMAUT, H. & CALLEWAERT, N. 2012. A bacterial glycosidase enables mannose-6-phosphate modification and improved cellular uptake of yeast-produced recombinant human lysosomal enzymes. Nat Biotechnol, 30, 1225-31.
- Tobinai, K.; Klein, C.; Oya, N.; Fingerle-Rowson, G. A Review of Obinutuzumab (GA101), a Novel Type II Anti-CD20 Monoclonal Antibody, for the Treatment of Patients with B-Cell Malignancies. Adv. Ther. 2016, 34, 324–356. [Google Scholar] [CrossRef]
- Togawa, T.; Takada, M.; Aizawa, Y.; Tsukimura, T.; Chiba, Y.; Sakuraba, H. Comparative study on mannose 6-phosphate residue contents of recombinant lysosomal enzymes. Mol. Genet. Metab. 2014, 111, 369–373. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Gutierrez, M.; Grubb, J.H.; Oikawa, H.; Dung, V.C.; Ohashi, A.; Nishioka, T.; Yamada, M.; Yamada, M.; et al. Characterization and pharmacokinetic study of recombinant human N-acetylgalactosamine-6-sulfate sulfatase. Mol. Genet. Metab. 2007, 91, 69–78. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Travis, J.; Owen, M.; George, P.; Carrell, R.; Rosenberg, S.; A Hallewell, R.; Barr, P.J. Isolation and properties of recombinant DNA produced variants of human alpha 1-proteinase inhibitor. J. Biol. Chem. 1985, 260, 4384–4389. [Google Scholar] [CrossRef]
- TRYGGVASON, K. & WARTIOVAARA, J. 2005. How does the kidney filter plasma? Physiology (Bethesda), 20, 96-101.
- Uchio, T.; Baudyš, M.; Liu, F.; Song, S.C.; Kim, S.W. Site-specific insulin conjugates with enhanced stability and extended action profile. Adv. Drug Deliv. Rev. 1999, 35, 289–306. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Timossi, C.; Damián-Matsumura, P.; Dias, J.A. Role of Glycosylation in Function of Follicle-Stimulating Hormone. Endocrine 1999, 11, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Umaña, P.; Jean–Mairet, J.; Moudry, R.; Amstutz, H.; Bailey, J.E. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 1999, 17, 176–180. [Google Scholar] [CrossRef]
- UNVERZAGT, C. , ANDRE, S., SEIFERT, J., KOJIMA, S., FINK, C., SRIKRISHNA, G., FREEZE, H., KAYSER, K. & GABIUS, H. J. 2002. Structure-activity profiles of complex biantennary glycans with core fucosylation and with/without additional alpha 2,3/alpha 2,6 sialylation: synthesis of neoglycoproteins and their properties in lectin assays, cell binding, and organ uptake. J Med Chem, 45, 478-91.
- van de Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196, 1435–1441. [Google Scholar] [CrossRef]
- Hamer, C.J.V.D.; Morell, A.G.; Scheinberg, I.H.; Hickman, J.; Ashwell, G. Physical and chemical studies on ceruloplasmin. IX. The role of galactosyl residues in the clearance of ceruloplasmin from the circulation.. 1970, 245, 4397–402. [Google Scholar]
- VAN DEN HOUT, J. M. , KAMPHOVEN, J. H., WINKEL, L. P., ARTS, W. F., DE KLERK, J. B., LOONEN, M. C., VULTO, A. G., CROMME-DIJKHUIS, A., WEISGLAS-KUPERUS, N., HOP, W., VAN HIRTUM, H., VAN DIGGELEN, O. P., BOER, M., KROOS, M. A., VAN DOORN, P. A., VAN DER VOORT, E., SIBBLES, B., VAN CORVEN, E. J., BRAKENHOFF, J. P., VAN HOVE, J., SMEITINK, J. A., DE JONG, G., REUSER, A. J. & VAN DER PLOEG, A. T. 2004. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics, 113, e448-57.
- Varki, A. Are humans prone to autoimmunity? Implications from evolutionary changes in hominin sialic acid biology. J. Autoimmun. 2017, 83, 134–142. [Google Scholar] [CrossRef]
- VARKI, A. , CUMMINGS, R. D., ESKO, J. D., STANLEY, P., HART, G. W., AEBI, M., MOHNEN, D., KINOSHITA, T., PACKER, N. H., PRESTEGARD, J. H., SCHNAAR, R. L. & SEEBERGER, P. H. 2022. Cold Spring Harbor (NY).
- Varki, A.; Kornfeld, S. Structural studies of phosphorylated high mannose-type oligosaccharides. J. Biol. Chem. 1980, 255, 10847–10858. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520–520. [Google Scholar] [CrossRef] [PubMed]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef]
- Wadhwa, M.; Knezevic, I.; Kang, H.-N.; Thorpe, R. Immunogenicity assessment of biotherapeutic products: An overview of assays and their utility. Biologicals 2015, 43, 298–306. [Google Scholar] [CrossRef]
- Walsh, G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. [Google Scholar] [CrossRef]
- Walters, R.W.; Pilewski, J.M.; Chiorini, J.A.; Zabner, J. Secreted and Transmembrane Mucins Inhibit Gene Transfer with AAV4 More Efficiently than AAV5. J. Biol. Chem. 2002, 277, 23709–23713. [Google Scholar] [CrossRef]
- Walters, R.W.; Yi, S.M.P.; Keshavjee, S.; Brown, K.E.; Welsh, M.J.; Chiorini, J.A.; Zabner, J. Binding of Adeno-associated Virus Type 5 to 2,3-Linked Sialic Acid Is Required for Gene Transfer. J. Biol. Chem. 2001, 276, 20610–20616. [Google Scholar] [CrossRef]
- Wang, C.; Eufemi, M.; Turano, C.; Giartosio, A. Influence of the Carbohydrate Moiety on the Stability of Glycoproteins. Biochemistry 1996, 35, 7299–7307. [Google Scholar] [CrossRef]
- Wang, C.-C.; Chen, J.-R.; Tseng, Y.-C.; Hsu, C.-H.; Hung, Y.-F.; Chen, S.-W.; Chen, C.-M.; Khoo, K.-H.; Cheng, T.-J.; Cheng, Y.-S.E.; et al. Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc. Natl. Acad. Sci. 2009, 106, 18137–18142. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Wang, J.; Lozier, J.; Johnson, G.; Kirshner, S.; Verthelyi, D.; Pariser, A.; Shores, E.; Rosenberg, A. Neutralizing antibodies to therapeutic enzymes: considerations for testing, prevention and treatment. Nat. Biotechnol. 2008, 26, 901–908. [Google Scholar] [CrossRef]
- Wang, M.; Wang, J.; Wang, R.; Jiao, S.; Wang, S.; Zhang, J.; Zhang, M. Identification of a monoclonal antibody that targets PD-1 in a manner requiring PD-1 Asn58 glycosylation. Commun. Biol. 2019, 2, 1–10. [Google Scholar] [CrossRef]
- Wang, R.Y.; Franco, J.F.d.S.; Lopez-Valdez, J.; Martins, E.; Sutton, V.R.; Whitley, C.B.; Zhang, L.; Cimms, T.; Marsden, D.; Jurecka, A.; et al. The long-term safety and efficacy of vestronidase alfa, rhGUS enzyme replacement therapy, in subjects with mucopolysaccharidosis VII. Mol. Genet. Metab. 2020, 129, 219–227. [Google Scholar] [CrossRef]
- Wang, S.; Xu, H.; Xu, J.; Zhang, Y.; Liu, Y.; Deng, Y.-H.; Chen, D. Sustained Liver Targeting and Improved Antiproliferative Effect of Doxorubicin Liposomes Modified with Galactosylated Lipid and PEG-Lipid. Aaps Pharmscitech 2010, 11, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tang, X.; Han, J.; Ding, Y.; Guo, W.; Pei, M. Biodegradable Self-Assembled Nanoparticles of Galactose-Containing Amphiphilic Triblock Copolymers for Targeted Delivery of Paclitaxel to HepG2 Cells. Macromol. Biosci. 2016, 16, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Protein aggregation and its inhibition in biopharmaceutics. Int. J. Pharm. 2005, 289, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Wanzeck, K.; Boyd, K.L.; McCullers, J.A. Glycan Shielding of the Influenza Virus Hemagglutinin Contributes to Immunopathology in Mice. Am. J. Respir. Crit. Care Med. 2011, 183, 767–773. [Google Scholar] [CrossRef]
- WASHBURN, N. , SCHWAB, I., ORTIZ, D., BHATNAGAR, N., LANSING, J. C., MEDEIROS, A., TYLER, S., MEKALA, D., COCHRAN, E., SARVAIYA, H., GAROFALO, K., MECCARIELLO, R., MEADOR, J. W., 3RD, RUTITZKY, L., SCHULTES, B. C., LING, L., AVERY, W., NIMMERJAHN, F., MANNING, A. M., KAUNDINYA, G. V. & BOSQUES, C. J. 2015. Controlled tetra-Fc sialylation of IVIg results in a drug candidate with consistent enhanced anti-inflammatory activity. Proc Natl Acad Sci U S A, 112, E1297-306.
- Wasley, L.C.; Timony, G.; Murtha, P.; Stoudemire, J.; Dorner, A.J.; Caro, J.; Krieger, M.; Kaufman, R.J. The importance of N- and O-linked oligosaccharides for the biosynthesis and in vitro and in vivo biologic activities of erythropoietin. . 1991, 77, 2624–32. [Google Scholar]
- WATANABE, Y. , ALLEN, J. D., WRAPP, D., MCLELLAN, J. S. & CRISPIN, M. 2020. Site-specific glycan analysis of the SARS-CoV-2 spike. Science, 369, 330-333.
- Watson, S.; Marx, J.B. Mogamulizumab-kpkc: A Novel Therapy for the Treatment of Cutaneous T-Cell Lymphoma. J. Adv. Pr. Oncol. 2019, 10, 883–888. [Google Scholar] [CrossRef]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef]
- Weber, W.; Steube, K.; Gross, V.; Tran-Thi, T.-A.; Decker, K.; Gerok, W.; Heinrich, P.C. Unglycosylated rat α1-proteinase inhibitor has a six-fold shorter plasma half-life than the mature glycoprotein. Biochem. Biophys. Res. Commun. 1985, 126, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Weenen, C.; Peña, J.E.; Pollak, S.V.; Klein, J.; Lobel, L.; Trousdale, R.K.; Palmer, S.; Lustbader, E.G.; Ogden, R.T.; Lustbader, J.W. Long-Acting Follicle-Stimulating Hormone Analogs Containing N-Linked Glycosylation Exhibited Increased Bioactivity Compared with O-Linked Analogs in Female Rats. J. Clin. Endocrinol. Metab. 2004, 89, 5204–5212. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef]
- Wide, L.; Wikström, B.; Eriksson, K. A New Principle Suggested for Detection of Darbepoetin-α (NESP) Doping. Upsala J. Med Sci. 2003, 108, 229–238. [Google Scholar] [CrossRef]
- E Wileman, T.; Lennartz, M.R.; Stahl, P.D. Identification of the macrophage mannose receptor as a 175-kDa membrane protein. Proc. Natl. Acad. Sci. 1986, 83, 2501–2505. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Pazos, R.; Royo, F.; Gonzalez, E.; Roura-Ferrer, M.; Martinez, A.; Gamiz, J.; Reichardt, N.-C.; Falcon-Perez, J.M. Assessing the role of surface glycans of extracellular vesicles on cellular uptake. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Woof, J.M.; Burton, D.R. Human antibody–Fc receptor interactions illuminated by crystal structures. Nat. Rev. Immunol. 2004, 4, 89–99. [Google Scholar] [CrossRef]
- Wraith, J.E. Enzyme replacement therapy with idursulfase in patients with mucopolysaccharidosis type II. Acta Paediatr. 2008, 97, 76–78. [Google Scholar] [CrossRef] [PubMed]
- WRAITH, J. E. , CLARKE, L. A., BECK, M., KOLODNY, E. H., PASTORES, G. M., MUENZER, J., RAPOPORT, D. M., BERGER, K. I., SWIEDLER, S. J., KAKKIS, E. D., BRAAKMAN, T., CHADBOURNE, E., WALTON-BOWEN, K. & COX, G. F. 2004. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr, 144, 581-8.
- WU, C. Y. , CHENG, C. W., KUNG, C. C., LIAO, K. S., JAN, J. T., MA, C. & WONG, C. H. 2022. Glycosite-deleted mRNA of SARS-CoV-2 spike protein as a broad-spectrum vaccine. Proc Natl Acad Sci U S A, 119.
- Wu, Z.; Miller, E.; Agbandje-McKenna, M.; Samulski, R.J. α2,3 and α2,6 N-Linked Sialic Acids Facilitate Efficient Binding and Transduction by Adeno-Associated Virus Types 1 and 6. J. Virol. 2006, 80, 9093–9103. [Google Scholar] [CrossRef]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. 2016, 113, 10304–10309. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Akai, K.; Kawanishi, G.; Ueda, M.; Masuda, S.; Sasaki, R. Effects of site-directed removal of N-glycosylation sites in human erythropoietin on its production and biological properties. J. Biol. Chem. 1991, 266, 20434–20439. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sawamura, M.; Wada, F.; Harada-Shiba, M.; Obika, S. Serial incorporation of a monovalent GalNAc phosphoramidite unit into hepatocyte-targeting antisense oligonucleotides. Bioorganic Med. Chem. 2016, 24, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Yang, Z.; Duan, J.; Wang, J.; Liu, Q.; Shang, R.; Yang, X.; Lu, P.; Xia, C.; Wang, L.; Dou, K. Superparamagnetic iron oxide nanoparticles modified with polyethylenimine and galactose for siRNA targeted delivery in hepatocellular carcinoma therapy. Int. J. Nanomed. 2018, 13, 1851–1865. [Google Scholar] [CrossRef]
- YANO, H. , ISHIDA, T., INAGAKI, A., ISHII, T., DING, J., KUSUMOTO, S., KOMATSU, H., IIDA, S., INAGAKI, H. & UEDA, R. 2007. Defucosylated anti CC chemokine receptor 4 monoclonal antibody combined with immunomodulatory cytokines: a novel immunotherapy for aggressive/refractory Mycosis fungoides and Sezary syndrome. Clin Cancer Res, 13, 6494-500.
- Yehuda, S.; Padler-Karavani, V. Glycosylated Biotherapeutics: Immunological Effects of N-Glycolylneuraminic Acid. Front. Immunol. 2020, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Yoo, E.M.; Chintalacharuvu, K.R.; Penichet, M.L.; Morrison, S.L. Myeloma expression systems. J. Immunol. Methods 2002, 261, 1–20. [Google Scholar] [CrossRef]
- Yoshida, M.; Takimoto, R.; Murase, K.; Sato, Y.; Hirakawa, M.; Tamura, F.; Sato, T.; Iyama, S.; Osuga, T.; Miyanishi, K.; et al. Targeting Anticancer Drug Delivery to Pancreatic Cancer Cells Using a Fucose-Bound Nanoparticle Approach. PLOS ONE 2012, 7, e39545. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Brown, D.; Reed, C.; Chung, S.; Lutman, J.; Stefanich, E.; Wong, A.; Stephan, J.-P.; Bayer, R. Production, characterization and pharmacokinetic properties of antibodies with N-linked Mannose-5 glycans. mAbs 2012, 4, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-D.; Gan, J.C. Effects of progressive desialylation on the survival of human plasma α1-anti-trypsin in rat circulation. Int. J. Biochem. 1978, 9, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.Q.; Yang, D.; Hong, J.; Tejada-Simon, M.V.; Rivera, V.M.; Zhang, J.Z. Immunoregulation and blocking antibodies induced by interferon beta treatment in MS. Neurology 2000, 55, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, M.; Xu, X.; Zhang, Y.; Liu, Y.; Zhao, M.; Li, P.; Chen, J.; Fukuda, T.; Gu, J.; et al. Loss of core fucosylation enhances the anticancer activity of cytotoxic T lymphocytes by increasing PD-1 degradation. Eur. J. Immunol. 2020, 50, 1820–1833. [Google Scholar] [CrossRef]
- ZHANG, Q. , JOUBERT, M. K., POLOZOVA, A., DE GUZMAN, R., LAKAMSANI, K., KINDERMAN, F., XIANG, D., SHAMI, A., MISCALICHI, N., FLYNN, G. C. & KUHNS, S. 2020b. Glycan engineering reveals interrelated effects of terminal galactose and core fucose on antibody-dependent cell-mediated cytotoxicity. Biotechnol Prog, 36, e3045.
- Zheng, K.; Bantog, C.; Bayer, R. The impact of glycosylation on monoclonal antibody conformation and stability. mAbs 2011, 3, 568–576. [Google Scholar] [CrossRef]
- Zheng, L.; Yang, Q.; Li, F.; Zhu, M.; Yang, H.; Tan, T.; Wu, B.; Liu, M.; Xu, C.; Yin, J.; et al. The Glycosylation of Immune Checkpoints and Their Applications in Oncology. Pharmaceuticals 2022, 15, 1451. [Google Scholar] [CrossRef]
- Zhou, Q.; Qiu, H. The Mechanistic Impact of N-Glycosylation on Stability, Pharmacokinetics, and Immunogenicity of Therapeutic Proteins. J. Pharm. Sci. 2019, 108, 1366–1377. [Google Scholar] [CrossRef]
- Zhou, Q.; Shankara, S.; Roy, A.; Qiu, H.; Estes, S.; McVie-Wylie, A.; Culm-Merdek, K.; Park, A.; Pan, C.; Edmunds, T. Development of a simple and rapid method for producing non-fucosylated oligomannose containing antibodies with increased effector function. Biotechnol. Bioeng. 2007, 99, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Doria-Rose, N.A.; Cheng, C.; Stewart-Jones, G.B.; Chuang, G.-Y.; Chambers, M.; Druz, A.; Geng, H.; McKee, K.; Kwon, Y.D.; et al. Quantification of the Impact of the HIV-1-Glycan Shield on Antibody Elicitation. Cell Rep. 2017, 19, 719–732. [Google Scholar] [CrossRef]
- Zhou, X.; Hu, W.; Qin, X. The Role of Complement in the Mechanism of Action of Rituximab for B-Cell Lymphoma: Implications for Therapy. Oncol. 2008, 13, 954–966. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Mahato, R.I. Targeted Delivery of siRNA to Hepatocytes and Hepatic Stellate Cells by Bioconjugation. Bioconjugate Chem. 2010, 21, 2119–2127. [Google Scholar] [CrossRef]
- Zhu, Y.; Jiang, J.-L.; Gumlaw, N.K.; Zhang, J.; Bercury, S.D.; Ziegler, R.J.; Lee, K.; Kudo, M.; Canfield, W.M.; Edmunds, T.; et al. Glycoengineered Acid α-Glucosidase With Improved Efficacy at Correcting the Metabolic Aberrations and Motor Function Deficits in a Mouse Model of Pompe Disease. Mol. Ther. 2009, 17, 954–963. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; Kyazike, J.; Zhou, Q.; Thurberg, B.L.; Raben, N.; Mattaliano, R.J.; Cheng, S.H. Conjugation of Mannose 6-Phosphate-containing Oligosaccharides to Acid α-Glucosidase Improves the Clearance of Glycogen in Pompe Mice. J. Biol. Chem. 2004, 279, 50336–50341. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; Mcvie-Wylie, A.; Jiang, C.; Thurberg, B.L.; Raben, N.; Mattaliano, R.J.; Cheng, S.H. Carbohydrate-remodelled acid α-glucosidase with higher affinity for the cation-independent mannose 6-phosphate receptor demonstrates improved delivery to muscles of Pompe mice. Biochem. J. 2005, 389, 619–628. [Google Scholar] [CrossRef]
- Zimran, A.; Loveday, K.; Fratazzi, C.; Elstein, D. A pharmacokinetic analysis of a novel enzyme replacement therapy with Gene-Activated® human glucocerebrosidase (GA-GCB) in patients with type 1 Gaucher disease. Blood Cells, Mol. Dis. 2007, 39, 115–118. [Google Scholar] [CrossRef]
Figure 1.
Most protein-based drugs undergo N-linked or O-linked glycosylation.
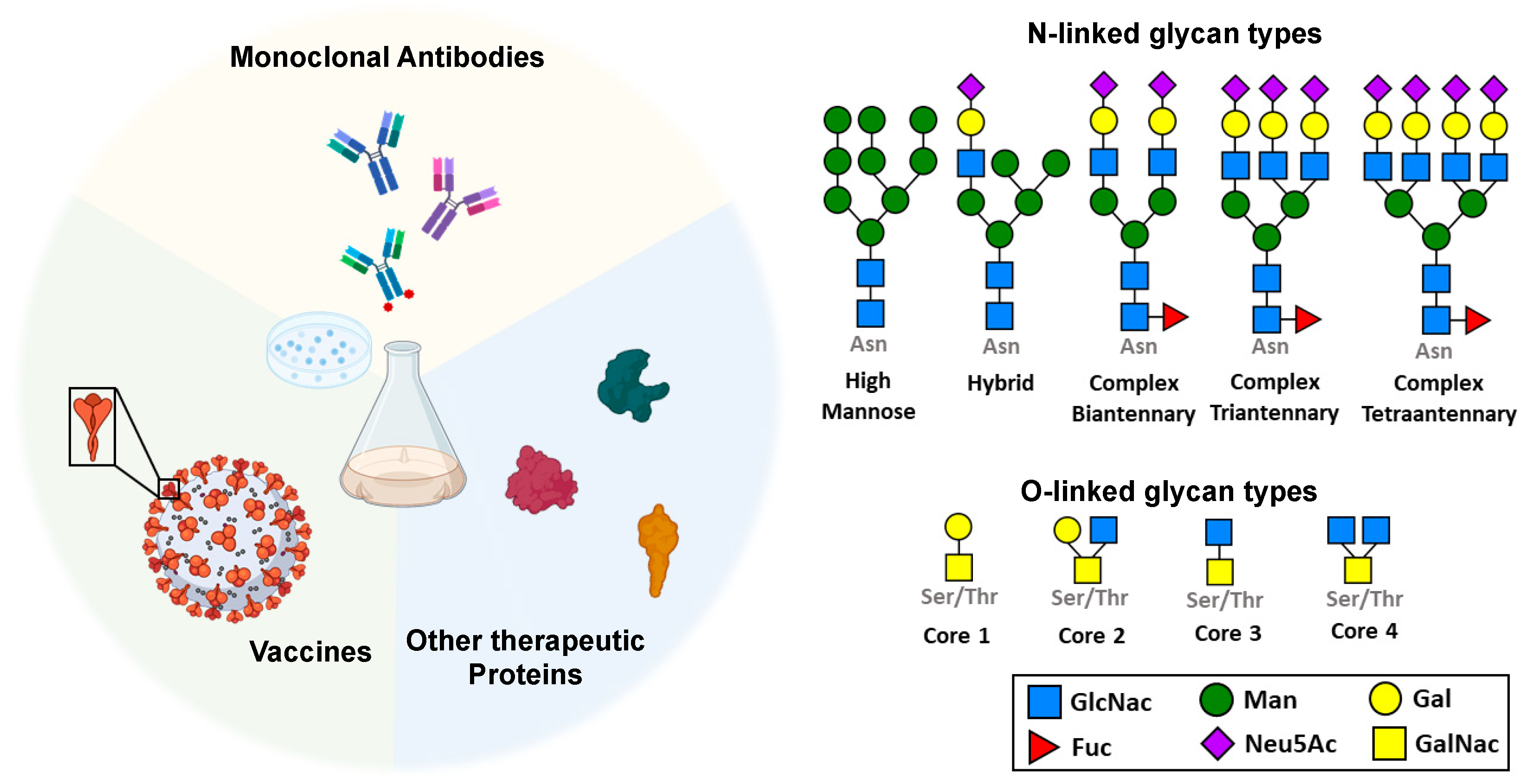
Figure 2.
Protein glycosylation is a critical quality attribute that has a significant impact on the stability, efficacy, and safety of the drug. (A) Sialylation of therapeutic proteins prolongs circulatory half-life by helping them evade capture by lectin receptors that facilitate clearance from the bloodstream. The presence of the terminal sialic acid masks the sugars that would otherwise be bound by asialoglycoprotein receptors (terminal Gal/GalNac) and mannose receptors (terminal Man/Fuc/GlcNac). (B) The presence of Mannose-6-Phosphate residues on therapeutic proteins facilitate better targeting and uptake by cells—such as macrophages—that express Mannose-6-Phosphate receptors. (C) Some glycan structures are associated with adverse immune responses among human subjects due to their structural dissimilarity to human-derived glycans. Such immunogenic glycans include the 𝛼-Gal Epitope and the Neu5Gc residue—both of which are derived from non-human, mammalian protein expression systems.
Figure 2.
Protein glycosylation is a critical quality attribute that has a significant impact on the stability, efficacy, and safety of the drug. (A) Sialylation of therapeutic proteins prolongs circulatory half-life by helping them evade capture by lectin receptors that facilitate clearance from the bloodstream. The presence of the terminal sialic acid masks the sugars that would otherwise be bound by asialoglycoprotein receptors (terminal Gal/GalNac) and mannose receptors (terminal Man/Fuc/GlcNac). (B) The presence of Mannose-6-Phosphate residues on therapeutic proteins facilitate better targeting and uptake by cells—such as macrophages—that express Mannose-6-Phosphate receptors. (C) Some glycan structures are associated with adverse immune responses among human subjects due to their structural dissimilarity to human-derived glycans. Such immunogenic glycans include the 𝛼-Gal Epitope and the Neu5Gc residue—both of which are derived from non-human, mammalian protein expression systems.
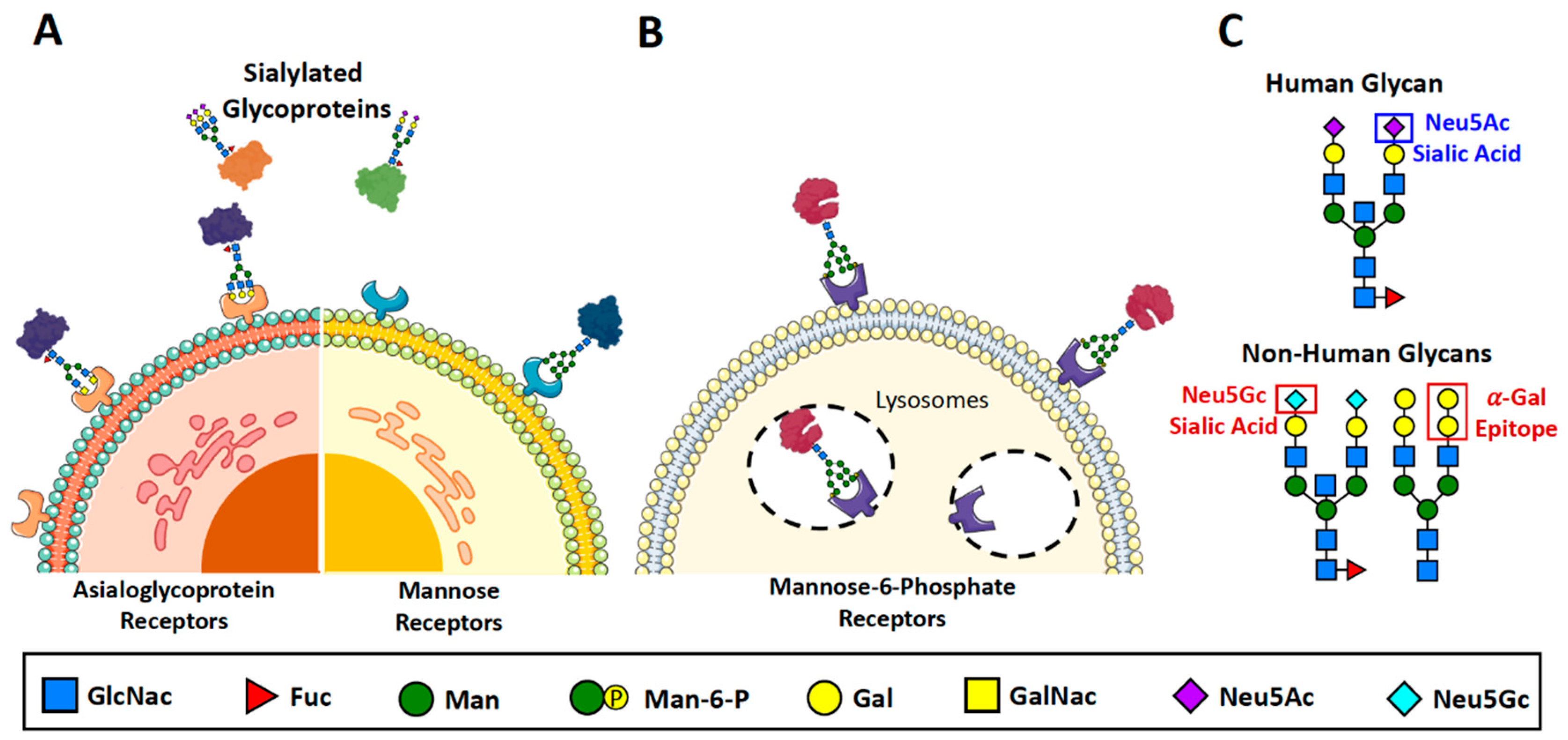
Figure 3.
Fc Glycosylation impacts monoclonal antibody effector function. All IgG antibodies are N-glycosylated at Asn-297 on their Fc region. The absence or presence of core fucose, bisecting GlcNac, terminal mannose/galactose/sialic acid residues on the Fc Asn-297 glycosite are crucial to regulating antibody function.
Figure 3.
Fc Glycosylation impacts monoclonal antibody effector function. All IgG antibodies are N-glycosylated at Asn-297 on their Fc region. The absence or presence of core fucose, bisecting GlcNac, terminal mannose/galactose/sialic acid residues on the Fc Asn-297 glycosite are crucial to regulating antibody function.
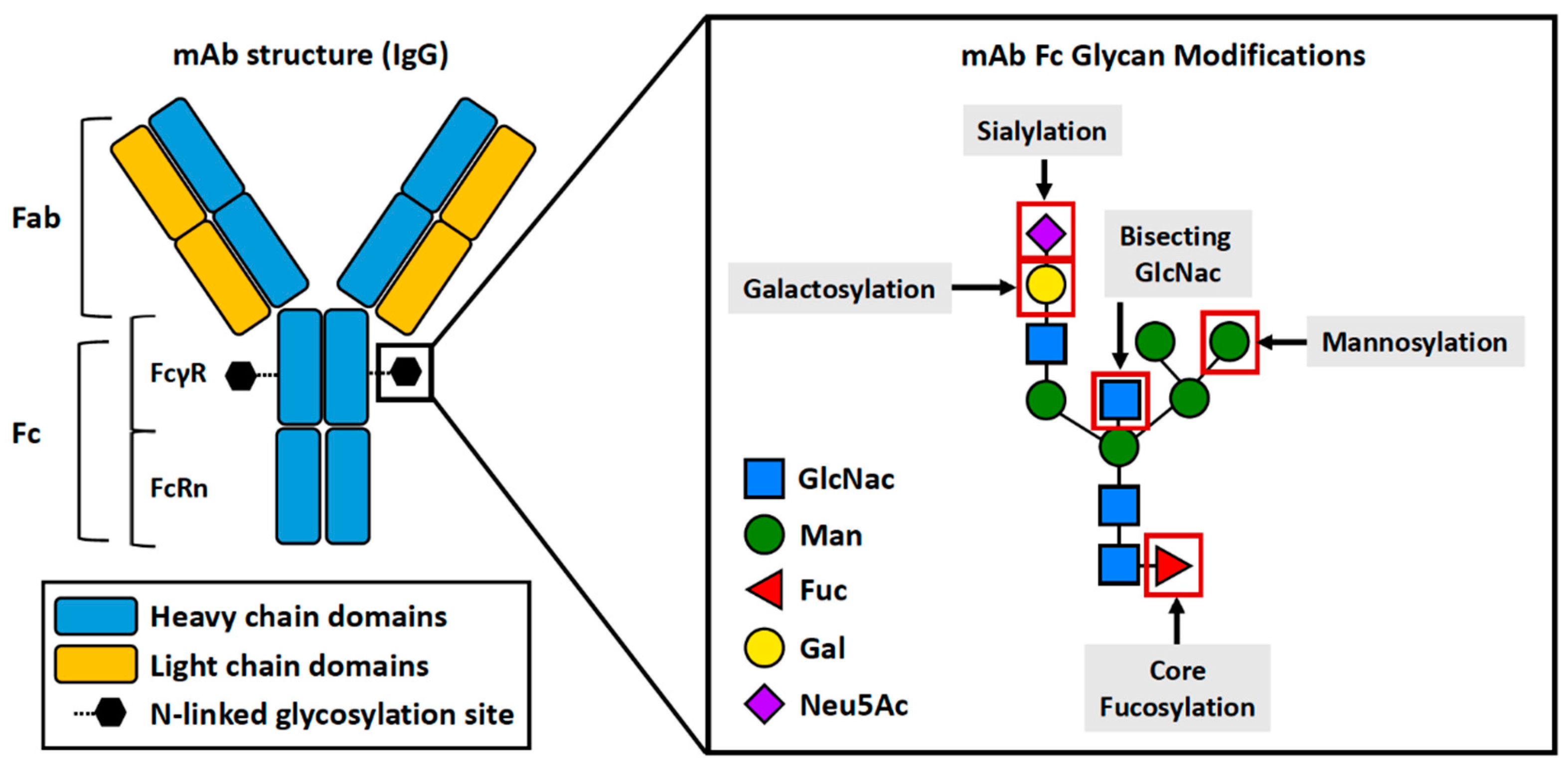
Figure 4.
Glycosylation impacts therapeutic efficacy of biologics beyond protein therapeutics. (A) Anticancer T-cell therapies rely on interactions between the T-cell and the tumor cell or antigen presenting cell. Some of these important interactions include CD28⇔CD80/CD86 which positively regulates T-cell activation and promote downstream anti-tumor activity, while PD1⇔PDL1 and CTLA-4⇔CD80/CD86 negatively regulate T-cell activation and reduce anti-tumor activity. N-linked glycan heterogeneity on these interacting proteins significantly impacts their binding affinity, expression level and cellular localization and therefore play a role in ensuring the therapeutic efficacy of such cell-based treatment modalities. (B) The presence/absence and identity of N-linked glycans on AAV capsid proteins can affect host cell infectivity, vector yield and immune response. Furthermore, the primary receptors of all AAV serotypes are O-linked and N-linked glycans such as terminal sialic acid and terminal galactose residues found on the host cell surface. N- and O-linked glycosylation, therefore, are key factors that modulate AAV transduction efficiency and tissue tropism. (C) Because different cell types usually express a distinct array of glycan receptors, conjugation of glycan structures to naked oligonucleotide-based drugs and delivery platforms such as nanoparticles and extracellular vesicles can improve targeting efficiency as well as modulate biodistribution in vivo. .
Figure 4.
Glycosylation impacts therapeutic efficacy of biologics beyond protein therapeutics. (A) Anticancer T-cell therapies rely on interactions between the T-cell and the tumor cell or antigen presenting cell. Some of these important interactions include CD28⇔CD80/CD86 which positively regulates T-cell activation and promote downstream anti-tumor activity, while PD1⇔PDL1 and CTLA-4⇔CD80/CD86 negatively regulate T-cell activation and reduce anti-tumor activity. N-linked glycan heterogeneity on these interacting proteins significantly impacts their binding affinity, expression level and cellular localization and therefore play a role in ensuring the therapeutic efficacy of such cell-based treatment modalities. (B) The presence/absence and identity of N-linked glycans on AAV capsid proteins can affect host cell infectivity, vector yield and immune response. Furthermore, the primary receptors of all AAV serotypes are O-linked and N-linked glycans such as terminal sialic acid and terminal galactose residues found on the host cell surface. N- and O-linked glycosylation, therefore, are key factors that modulate AAV transduction efficiency and tissue tropism. (C) Because different cell types usually express a distinct array of glycan receptors, conjugation of glycan structures to naked oligonucleotide-based drugs and delivery platforms such as nanoparticles and extracellular vesicles can improve targeting efficiency as well as modulate biodistribution in vivo. .
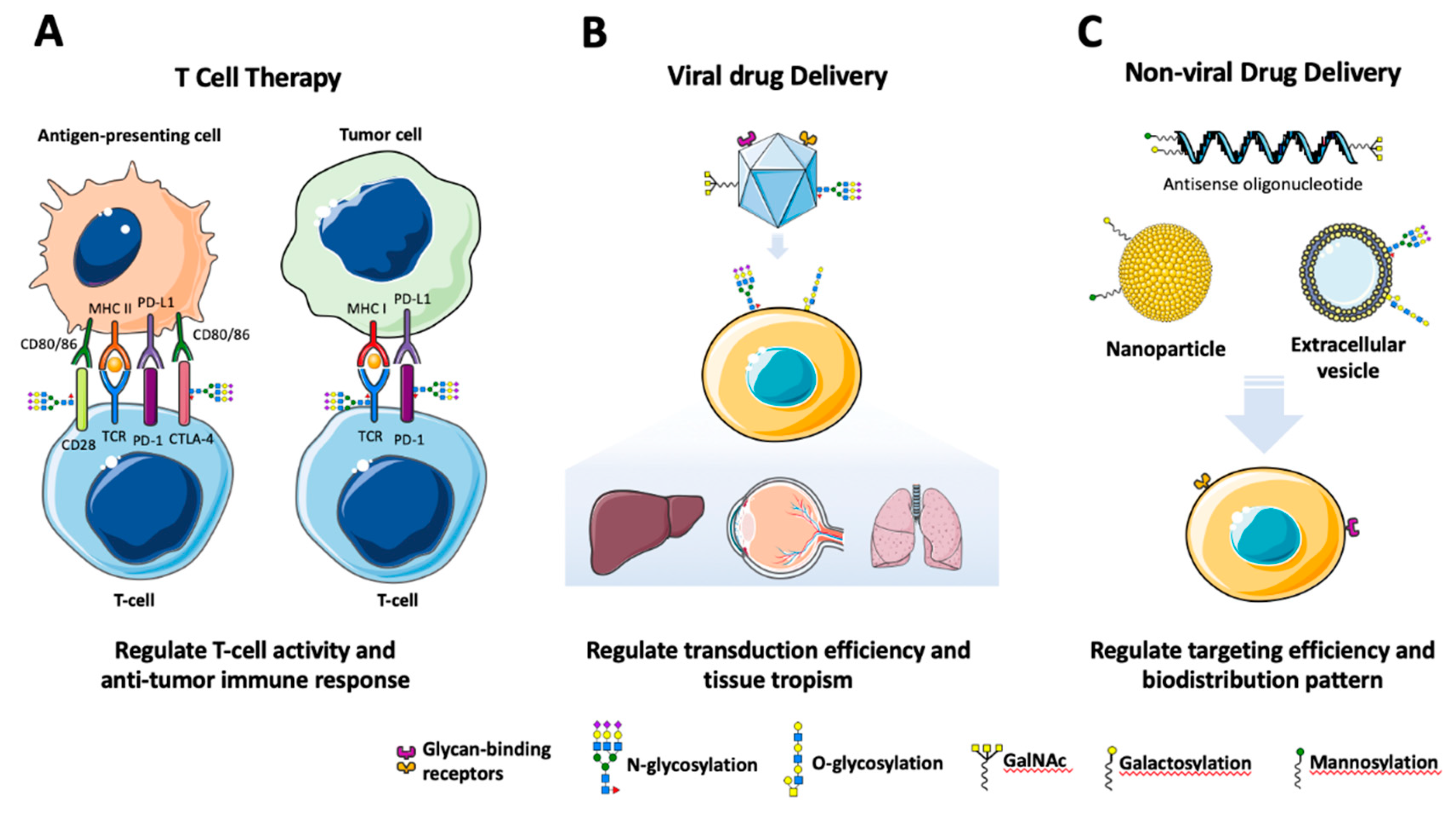
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



