
Submitted:
26 March 2023
Posted:
27 March 2023
You are already at the latest version
Abstract
Biocontainment techniques for genetically modified yeasts (GMYs) are pivotal due to the importance of these organisms for biotechnological processes and also due to the design of new yeast strains by using synthetic biology tools and technologies. Due to the large genetic modifications that many yeast strains display, it is highly desirable to avoid the leakage of GMY cells into natural environments and, consequently, the spread of synthetic genes and circuits by horizontal or vertical gene transfer mechanisms within the microorganisms. Moreover, it is also desirable to avoid that patented yeast gene technologies spread outside the production facility. In this review, it was evaluated the different biocontainment technologies currently available for GMYs. Interestingly, uniplex-type biocontainment approaches (UTBAs), which relies on nutrient auxotrophies induced by gene mutation or deletion, or the expression of simple kill switches apparatus, are still the major biocontainment approaches still in use with GMY. While bacteria like Escherichia coli account for advanced biocontainment technologies based on synthetic biology and multiplex-type biocontainment approaches (MTBAs), GMYs are distant from this scenario due to many reasons. Thus, a comparison of different UTBAs and MTBAs applied for GMY and genetically engineered microorganisms (GEMs) was made, indicating the major advances of biocontainment techniques for GMYs.
Keywords:
Biocontainment
; Genetically Engineered Microorganisms
; Genetically Modified Yeasts
; Synthetic Biology
; Gene Circuits
; Auxotrophies
; Kill Switches
1. Introduction
Genetically engineered or modified microorganisms (GEMs/GMMs) are the workhorse for basic and applied research as well as in industry. GEMs are employed in the food industry to improve protein synthesis or to generate small molecules that impact the nutritional value of a food, like flavor enhancers, oligosaccharides, vitamins, and amino acids [1]. GEMs are also employed for food enzyme production, including enzymes like lactase, amylase, proteases, and phospholipases [1,2]. Another biotechnological field that benefits from the application of GEMS is bioremediation, which allows the removal of pollutants (e.g., heavy metals) from water and soil [3], and finally different GEMs species are employed for the production of clinically important peptides/proteins (e.g., insulin) [4], vaccines [5], biofertilizers [6,7], biocontrol [8], and biofuels [9].
Considering eukaryotic GEMs, the yeasts constitute an important group of microorganisms with many industrial applications. Yeasts are used as a cell factory for the production of chemicals and biologicals due to its ability to grow on inexpensive culture media and have a well established fermentative technology [10]. One of the most popular yeast employed as GEM is Saccharomyces cerevisiae. S. cerevisiae has many different biotechnology applications, from the production of traditional fermented foods [11,12] to cell factories for the synthesis of chemicals and pharmaceuticals, such as bioethanol, propanol, butanol, artemisinic acid, and insulin precursor [10,13,14]. In addition to its biotechnological importance, S. cerevisiae is considered an eukaryotic model organism for basic and applied research [15,16]. Other yeast species that have biotechnological importance include the halophilic/halotolerant yeast species like Debaryomyces hansenii, which can metabolize different types of carbon sources and synthesize high levels of lipids, xylitol, and flavonoids, making it an attractive model for metabolic engineering [17,18]. By its turn, methylotrophic yeasts (e.g., Pichia pastoris and Ogataea polymorpha) are very efficient in producing heterologous protein in industrial scale, with many strains and molecular tools available for genetic/synthetic biology engineering [19,20]. Other examples of non-Saccharomyces with industrial importance include Arxula adeninivorans and Yarrowia lipolytica for the production of therapeutic heterologous proteins [21]. In addition, yeast hybrid strains/chassis from the Saccharomyces sensu stricto complex for industrial purposes are gaining industrial importance [22]. Finally, advances in synthetic biology are leading to the generation of “synthetic microorganisms” by using the so-called “bottom-up approach”, where isolated and well characterized biochemical components are modularly assembled in order to design artificial cells with specific phenotypes [23]. Another synthetic biology approach is the “top-down method”, where a microbial genome is reduced to its essential genes, allowing to factory any desired DNA sequence to “tailor-made” a specific phenotype [24]. The top-down approach is currently being applied by the international Synthetic Yeast Genome project (known as the “Yeast 2.0” or “Sc2.0 project”), whose major objectives are to redesign all chromosomes of S. cerevisiae and generate the first synthetic eukaryote [24,25].
Despite the importance of genetically modified yeasts (GMYs) there are concerns that the widespread use of GMYs could lead to a potential exchange of modified DNA molecules with other microorganisms in an ecosystem [26,27]. These concerns can be potentialized by considering that yeasts can exchange parts of their genomes with other microorganisms by interkingdom horizontal gene transfer [28], resulting in the widespread use of a transgene and/or a synthetic DNA molecule (Figure 1). Thus, highly effective biocontainment/safeguard strategies are needed to keep GMYs restricted to a laboratory and/or industrial environments (here defined as “production facilities”) [27,29] and restricting the “escape frequency” of GMYs. The escape frequency is the consequence of a combination of different molecular mechanisms (e.g., mutagenesis, gene loss, recombination) as well as environmental/evolutionary processes that result in the widespread of GMY/GEM into the environment (Figure 1). The National Institutes of Health (NIH) guidelines for research involving recombinant or synthetic nucleic acid molecules recommends an escape frequency of one cell per 108 cells as standard [30].
Another major issue regarding the massive use of GMY/GEM is that the simple physico-chemical inactivation of cell biomass (e.g., by heat and/or pH treatment) do not avoid the DNA stability/persistence in the environment after its release from the cell (Figure 1). Data gathered from the PCR analysis of soil fertilized with the waste product of industrial GMY/GEM fermentations showed the presence of genes related to antibiotic resistance commonly found in synthetic plasmids [31]. Additionally, engineered DNA molecules can be detected in soil after several days or months after its introduction due to the intrinsic high stability of DNA molecules [32,33]. Interestingly, none of these studies observed the horizontal transfer of released engineered DNA molecules to the native soil microbiomes; however, the chance of horizontal gene transfer from released synthetic DNA molecules in the environment can not be excluded especially if those molecules confer phenotype advantage to the native microbiome.
Different molecular approaches can be employed for GMY biocontainment, including auxotrophic mutations, synthetic molecular mechanisms (xenobiology), and kill switches based on the expression of toxin-antitoxin [27,34] or exo-/endonucleases [29,35] (Figure 1). However, all of these approaches are not fail proof, since molecular and evolutive mechanisms lead to inactivation of biocontainment devices [36] (Figure 1). This is especially true when a biocontainment approach makes use of a single component/device (in this work defined as “uniplex-type biocontainment approach” or UTBA; Table 1 and Figure 1), like natural auxotrophic markers. On the other hand, the combination of different biocontainment approaches (“multiplex-type biocontainment approach” or MTBA; Table 1) reduces its potential escape frequency at the expense of low cell fitness (Table 1) [27,34]. Paradoxically, the reduction in cell fitness increases the frequency of inactivated biocontainment devices [37]. Thus, an effective biocontainment strategy should consider different escape mechanisms, including mutagenic drift, environmental supplementation of nutrients and/or essential molecules, and horizontal gene transfer (HGT) [38].
The purpose of this review manuscript is to focus on different UTBA and MTBA strategies for GMY biocontainment, and what approaches have been or not applied for industrially important GMYs.
2. Uniplex-type biocontainment approaches
2.1. Natural auxotrophic markers
Natural auxotrophy (Figure 1) is a simple and widespread UTBA technique commonly applied to prevent the releasing and proliferation of GMYs outside the production facility. In general terms, natural auxotrophy can be defined as a nutritional deficiency induced by mutated genes resulting in an GMY strain that depends on the addition of the nutrient on its growth media [39]. The yeast S. cerevisiae has been used as a model for auxotrophic markers due to the facility of inducing deletion and/or point mutations in different genes associated with nutrient metabolism [39]. In this sense, genes linked to the metabolism of amino acids like L-histidine, L-leucine, L-tryptophan, and L-methionine [39], and nitrogen bases (e.g., adenine and uracil) [40] have been used as auxotrophic markers for decades.
Considering nitrogen base auxotrophies, the allele ade2-1, which contains a nonsense mutation (Glu64STOP) [41], is widely found in different laboratorial S. cerevisiae strains and confer adenine dependence. A major phenotypic characteristic of ade2-1 is the development of a red ochre color due to the intracellular accumulation of oxidized adenine-associated metabolic intermediates [40,42], a phenotype that can be useful for red-white colony screening [43], redox biology [44] and drug discovery [40].
The depletion of adenine reserves in yeast cells increase trehalose synthesis and lead to cell cycle arrest, recapitulating the protective effects observed for desiccation stress tolerance. As a consequence, the adenine-deficient cells become viable for a longer period of time and evade biocontainment [42]. Complementally, it was observed that adenine auxotrophy increases mutagenesis rate in yeast cells [45], a condition that is observed for other auxotrophic markers like leu2 and/or lys2 alleles. Yeast cells carrying leu2 and/or lys2 alleles, when subjected to L-lysine or L-leucine starvation, display an increase in the number of respiratory deficient cells (rho- cells) due to the accumulation of mutations in mitochondrial genome, a condition termed “adaptive mutation” [46]. Additionally, adaptive mutations are linked to auxotrophy marker reversion to the wild-type state, as observed for his4 marker in yeast under selective pressure [47].
In order to avoid the reversion of auxotrophic markers, a series of deletion gene markers have been generated for S. cerevisiae [48], as well as for other non-Saccharomyces species, like Kluyveromyces lactis [49]. However, it has been observed in some non-Saccharomyces species that auxotrophic gene deletion leads to a bradytroph/leak auxotroph phenotype, like those observed for PHA2 in Pichia pastoris, which is linked to L-phenylalanine biosynthesis [50]. In this case, the auxotrophic phe- cells are able to survive under L-phenylalanine starvation due to alternative L-phenylalanine biosynthesis mechanisms [50].
Considering the impact of auxotrophy in yeast metabolism, the effect of adaptive mutation in the selection of prototrophic cells, and bradytrophy due to the presence of poorly described biochemical pathways in non-Saccharomyces species, natural auxotrophy per se has strong limitations regarding its use as a biocontainment strategy. In addition, the presence of other microorganisms in the environment and/or production facility (Figure 1) can provide the auxotrophy-dependent nutrients for GMY mutants, bypassing the auxotrophy requirement [51].
A solution to overcome the natural auxotrophies limitations is the use of synthetic amino acids and nitrogen bases not available outside the production facility and can not be biologically supplied. This approach, termed “xenobiology” or “synthetic auxotrophies” (Figure 1), make use of top-down synthetic biology techniques to engineering GMYs and create new biocontainment strategies [35]. Another approach to circumvent the limitations of natural auxotrophies is the use of a so-called “conditional gene essentiality” (Figure 1), which employs promoter engineering to modify genes linked to nutrient metabolism and strictly regulate their expression by using synthetic molecules and/or orthogonal RNA polymerases [52].
2.2. Xenobiology and synthetic auxotrophies
Xenobiology focuses on the development of synthetic biological devices and systems that utilize non-canonical amino acids (ncAAs), nucleic acids with non-standard sugar backbone (xeno-nucleic acids or XNA) and non-natural nitrogen base pairs for different purposes, including the design of synthetic metabolic processes (neo-metabolism) [53], and biocontainment [52]. Many of xenobiology devices are designed by principle using orthogonality where synthetic components (e.g., proteins, RNAs, DNAs, and small molecules) are engineered for a purposed function and will not interfere with the natural biochemistry of a host cell [53,54]. The orthogonality also ensures that these components will not be used by natural biological systems, making it useful as a biocontainment strategy [55,56].
Biocontainment-based xenobiology is mostly centered on the use of different ncAAs for protein synthesis for both GEM and GMY. For example, the application of genetic code expansion (GCE) or orthogonal translation systems (OTSs) techniques [57,58] has been used with success in Escherichia coli biocontainment [56].
In bacteria, GCE/OTS relies on a orthogonal pyrrolysyl-tRNA synthetase/tRNAPylCUA (PylRS/tRNAPylCUA) pair derived from Methanosarcina barkeri, M. mazei or Methanocaldococcus jannaschii to incorporate ncAAs in proteins [59,60], in the reassignment of the UAG amber codon, a rare stop codon in both E. coli and S. cerevisiae [61], and the deletion of the release factor 1 (RF1). Considering GCE for E. coli biocontainment, the genome of this bacterium was refactored by the introduction of a reassigned UAG codon into 22 essential genes together with a Methanocaldococcus jannaschii PylRS that is able to incorporate L-phenylalanine derivatives into tRNAPylCUA [56], generating a synthetic auxotrophy. Data from this work indicated that the synthetic E. coli auxotroph cells were dependent on the addition of L-phenylalanine derivatives; moreover, these synthetic auxotrophs have undetectable escape frequencies in both solid and liquid culture media [56]. Additional work related to the creation of E. coli synthetic auxotroph strains have been made [38], pointing to the feasibility of this biocontainment technique for bacteria.
The generation of yeast ncAAS-dependent synthetic auxotrophs was achieved with very limited success [58,62]. Yeast are naturally able to incorporate ncAAs into proteins [61,63] and a GCE/OTS technique for S. cerevisiae was developed by Chin et al. [62]. In this work, the authors engineering orthogonal codons, anticodons and tRNA synthetase, including an E. coli tyrosine-tRNA synthetase (TyrRS) and a amber suppressor tRNATyrCUA, to generate a library of TyrRS mutants that pairs only with ncAAs [62]. Once this TyrRS library was transformed into a S. cerevisiae strain, five different ncAAs were incorporated into human superoxide dismutase 1 protein (hSOD1) [62].
Other relevant works related to the development of GCE/OTS for S. cerevisiae and Pichia pastoris have been made by using the E. coli TyrRS/tRNATyrCUA or leucyl- (Leu)RS/tRNALeu5CUA pairs for ncAA incorporation into proteins [60,64,65,66,67,68,69,70,71,72]. However, the use of GCE/OTS in yeast have some major challenges, including the low expression of tRNAPylCUA in yeast due to the absence of intragenic promoter sequences A- and B-boxes [58,60]; moreover, the eukaryotic release factor 1 (eRF1), codified in yeast by the SUP45 essential gene [73], compose the Sup45p-Sup35p complex that is necessary to end translation by binding into all three stop codons in yeast cells [74]. Comparatively, the E. coli RF1, which recognizes the UAG/UAA codons, can be deleted without major physiological impacts into the cell due to the functional superimposition with the release factor 2 (RF2) [75].
In order to circumvent the limitations inherently associated with the implementation of a GCE/OST-based biocontainment in GMY, different approaches were applied, like (i) prospecting new archeal PylRS with higher ncAA incorporation efficiency in S. cerevisiae [76], (ii) selecting yeast strains with specific mutations (e.g., yil014c-aΔ and alo1Δ) for increasing ncAAs incorporation into tRNAPylCUA [77], (iii) improving E. coli TyrRS and LeuRS with enhanced ncAAs polyspecificity and efficiency by using random mutagenesis and directed selection [78], (iv) the use of an OTS based on recognition of quadruplet codons by an engineered orthogonal ribosome that allows to expand the genetic code to 256 codons; this quadruplet codon-base OTS have been implemented with more or less success in E. coli [58,79,80] and mammalian cells [81], and (v), the use of synthetic/unnatural nitrogen base pairs (UBPs) to expand the genetic code and increase the repertoire of ncAAs that can be used for protein synthesis in E. coli [82].
There are different approaches when considering the use of UPBs and XNAs for xenobiology and GMY/GEM biocontainment [83]. In fact, it is expected that XNA technology could be efficient for GMY/GEM biocontainment since the building blocks of XNAs (e.g., nucleobases, sugar moieties and phosphate-modified groups) can not be find in natural environments and the GMY/GEM cells should be able to incorporate these building blocks into new XNA polymers by the usage of specialized polymerization enzymes and transmembrane proteins able to uptake this precursors [84,85]. Thus, UBPs and XNAs can be like a “genetic firewall” [84], where HGT events could be avoided to the restrained aspects of XNA technology.
The developments on XNA technologies follows two major mainstreams: (i) the use of unnatural nucleobases, sugar moieties and phosphate-modified groups (XNA substrates) to incorporate into XNAs or hybrid XNA/DNA/RNA polymers by canonical DNA and/or RNA polymerases [55,86] and (ii) design new DNA and/or RNA enzymes (XNAzymes) able to metabolize XNA polymers and/or substrates with an “alien” chemistry, like threose nucleic acid (TNA), cyclohexenyl nucleic acid (CeNA), arabino nucleic acid (ANA), 2′-fluoro-arabino nucleic acid (FANA), glycol nucleic acid (GNA), and locked nucleic acid (LNA) [87,88,89,90,91,92,93,94]. In all cases, the XNAs should display orthogonality in vivo with little or, preferentially, none interaction with the canonical components of DNA/RNA metabolism [95]. Unfortunately, many different XNA technologies were not implemented in vivo, which renders its usage for GMY/GEM biocontainment still far from technical and practical viewpoint [96]. Finally, both GCE/OTS and XNA technologies were not tested in an open and uncontrolled environment (Figure 1), making its behavior unpredictable for real biocontainment applications [26].
Another approach related to the induction of synthetic auxotrophies in E. coli is based on the selection of essential proteins whose structure and activity is dependent on the presence of a small molecule ligands, like the so called “synthetic auxotrophs based on ligand-dependent essential genes” (SLiDE) technique [97]. In this work, the authors were able to select five essential genes in E. coli by applying protein engineering and saturation mutagenesis and generate proteins dependent on benzothiazole. The authors related a very low escape frequency (< 3 x 10-11) under laboratory assays [97]. Similar works on bacterial synthetic auxotrophs have been made, including phosphite-dependent Synechococcus elongatus [98] and Pseudomonas putida [99]. In S. cerevisiae it has been identified a series of mutations in CDC10 gene, which codify an essential septin protein that can be rescued in the presence of small molecules, like guanidinium ion [100,101]. Although the authors of this work have not applied the CDC10 conditional mutants for biocontainment, the data may indicate new techniques based on chemical rescue for GMY biocontainment.
2.3. Conditional gene essentiality
The conditional essentiality is, similarly to auxotrophy or xenobiology, a plethora of molecular techniques where essential genes that codify for proteins and enzymes needed for cell growth and maintenance are modified to have its expression regulated by external agents [52]. In this regard, the conditional gene essentiality has been applied for biocontainment in S. cerevisiae, E. coli and other bacteria with a low escape frequency (Figure 1) [27,52]. In S. cerevisiae, conditional gene essentiality was employed to regulate the expression of histone genes [27], fatty acid synthetase, mitochondrial and cytoplasmic histidine tRNA synthetase (HisRS), RNA polymerase II subunit B, and interorganellar chaperones and GTPases related to vesicular transport and fusion [102]. These genes have their original regulatory sequences replaced by a combination of promoters whose activities are modulated by galactose and estradiol [27]. Moreover, the orthogonality is ensured in this biocontainment system by providing a “fail-safe” recombination-induced lethality mechanism based on small molecule dependent Cre recombinase [27,103].
2.4. Orthogonal DNA replication and RNA transcription
Another potential strategy for GMY biocontainment is to implement orthogonal DNA and RNA polymerases [51]. In yeast, an orthogonal DNA replication system derived from the Kluyveromyces lactis cytoplasmic plasmid pGKL1/2 named “OrthoRep” have been applied for studies of in vivo continuous evolution of target genes due to the high error-prone activity of DNA polymerase 1 (DNAP1) and DNA polymerase 2 (DNAP2), both codified by pGLK1/2 [104,105,106]. Similar in vitro works were also described by using the orthogonal DNA polymerase derived from bacteriophage phi29 to generate a self-contained synthetic transcription and translation-coupled DNA replication system [107,108,109]. Additionally, orthogonal DNA replication systems, including XNA polymerases and/or engineered DNA polymerases [110], are being developed for xenobiology applications. However, the use of different orthogonal DNA replication mechanisms as a biocontainment system for GMY/GEM was not reported until now, pointing to an unexplored research field (Figure 1).
Like orthogonal DNA replication systems, orthogonal RNA transcription systems are also underexplored in the context of GMY/GEM biocontainment. Some examples include orthogonal genic circuits based on the use of bacteriophage T7 RNA polymerase in bacteria and yeast for different biotechnological purposes [111,112]. However, no biocontainment applications using T7 RNA polymerase-based techniques for GMY/GEM were described until now (Figure 1).
2.5. Nuclease-based kill switches
Kill switches are defined as gene circuits activated by specific environmental inputs, resulting in the expression of lethal genes that lead to cell death [113]. Different kill switch designs have been used for GEM biocontainment (Figure 1), including gene circuits that respond to environment changes by activating/deactivating a type II toxin-antitoxin system CcdB/CcdA [113]. Besides it, specific and unspecific nucleases, auxotrophies, as well as type I toxin-antitoxin pairs have been employed to engineer kill switches circuits for biocontainment purposes, many of them in a MTBA format (Table 1) [56,114,115,116,117,118] in order to reduce the GEM escape frequency.
Considering nucleases for kill switch design, it has been shown its usefulness for bacteria biocontainment. For example, the expression of EcoRI endonuclease-EcoRI methylase pair combined with conditional gene essentiality in E. coli considerably lowers the escape frequency [117]. In this sense, combining EcoRI and mf-Lon protease in a strictly regulated gene circuit allowed the development of the “Deadman” kill switch, which considerably lowered the escape frequency associated with a high genetic stability [114]. Other non-specific nucleases, like nucA from Serratia marcescens [119] or nuclease A from Staphylococcus aureus have been employed in different bacterial biocontainment projects [118].
In S. cerevisiae, a kill switch based on the conditional expression of S. marcescens nucA under the control of a glucose-repressed ADH2/GAPDH hybrid promoter has been proposed [120]. This work showed that under non repressible conditions, the nucA was efficiently expressed leading to yeast cell death in both laboratorial and soil microcosm assays [120]. Interestingly, type II restriction enzymes have been expressed in yeast to study DNA damage and repair mechanisms [121,122,123]. Data from the expression of type II restriction enzymes in yeast cells point to a low survival and DNA damage tolerance, specially when blunt ends are induced by type II restriction enzymes (e.g., PvuII) [123]; however, the use of type II restriction enzymes as part of a kill switch mechanism in yeast cells for biocontainment purposes was not described until now.
In addition to conventional exo- and endonucleases, the CRISPR-associated nucleases (Cas) that have been used in E. coli to design kill switches. These kill switches gene circuits make use of Cas3 [124] or Cas9 [125] and were successfully used to biocontain E. coli cells, both displaying an escape frequency ≤ 10-8 cells. A major advantage of the Cas-based kill switches is the use of guide RNAS (gRNAs) to select specific DNA sequence targets or microorganism strains, allowing to selectively eliminate the target strain from microbiome [125,126]. Until now, no Cas-based kill switches were reported for GMY.
2.6. Kill switches based on type I and II toxin-antitoxin systems
Another kill switch extensively applied in GEM biocontainment is based on type I and II toxin-antitoxin (TA) systems. These kill switch-based TA systems efficiently lead to controlled cell death outside the production facility and can be easily engineered in GEM/GYM cells (Figure 1). What makes the TA systems so attractive for biocontainment applications is its pleiotropy, which targets different molecular mechanisms, like DNA and mRNA synthesis, cell cycle, nucleotide synthesis, and protein translation [127,128].
The prokaryotic TA system is classified into seven types numbered from I to VII [130], which act in different cell mechanisms. For example, type I toxin-antitoxin system prevents toxin RNA translation through the binding of sRNA [129], while type II toxin-antitoxin system is based on the interaction of an endoribonuclease and an inhibitor, forming a stable complex [131]. By its turn, type III toxin-antitoxin system consists of the inhibition of the toxin protein by an antitoxin RNA [132]. Type IV toxin-antitoxin system is composed of two proteins that do not form a complex; instead, the antitoxin acts as an antagonist of the toxin at its cellular targets [133]. The type V toxin-antitoxin system differs from the others by having its antitoxin cleave the toxin mRNA [134]. Type VI is composed of an antitoxin-based proteolytic adapter that degrades the toxin-based protein [135], while type VII is composed of an antitoxin that enzymatically neutralizes the toxin by post-translational modification [130].
Type I and II are historically employed for biocontainment gene circuit design, especially for E. coli [136]. For example, the type I hok-sok pair have been applied for the design of conditional suicide of plasmid-containing E. coli cells in phosphate- [137] or tryptophan-limited [136] environments. Additionally, type I TA systems have been used in synthetic biology projects related to GEM biocontainment [118]. Similarly, type II TA systems have been applied into kill switch circuits based on the use of ccdB-ccdA pair regulated by a bi-stable cI/Cro memory switch (‘essentializer’ circuit) and by a cold-inducible promoter (‘cryodeath’ circuit) [113]. Finally, type II TA systems have been applied for the design of ‘plasmid addiction’ systems to prevent the lost of plasmids under production facility [138]
In addition to GEM, the use of type I and I TA systems was proven to be effective for GMY biocontainment, like the expression of the relE-relB system in S. cerevisiae [139]. When not being neutralized by the RelB antitoxin, the RelE toxin inhibits protein synthesis by cleaving mRNA that are being translated on the ribosome [140]. When RelB is neutralizing RelE it displaces the toxin’’s C-terminal and forms the RelBE complex [141]. To express this TA system in S. cerevisiae two recombinant plasmids were constructed, one containing the relE toxin gene under the control of galactose-induced GAL1 promoter (pKP727) and another containing relE regulated by GAL1 promoter and the relB antitoxin gene under the control of methionine-repressible MET25 promoter (pKP1006). Yeast cells transformed with the plasmid pKP727 showed visible growth inhibitory effects induced by the relE toxin; on the other hand, yeast transformed with the plasmid pKP1006 showed a higher growth rate. This data indicates that the toxic effects of relE can be minimized by relB antitoxin in yeast [139]. Similar to the relE-relB system, the ribonuclease-associated toxin Kid (killing determinant) and its antitoxin Kis (killing suppressor) [142] have been used in S. cerevisiae as a potential biocontainment system. For this purpose, an expression system containing the antitoxin Kis controlled by the MET25 promoter and the Kid toxin controlled by the Cu2+-induced CUP1 promoter was cloned into the recombinant integrative plasmid pRS303. In the presence of both methionine and Cu2+, the expression of Kis antitoxin is inhibited while the expression of Kid toxin is induced, leading to cell death [143].
Another TA system already tested in yeast is based on the ε-ζ (epsilon-zeta) genes from the gram-positive bacteria Streptococcus pyogenes [144]. The ε-ζ genes are organized in an operon together with the ω (omega) gene, which codify a repressor that modulates the transcription of the ζ toxin, ε antitoxin and its own [144]. This TA system was expressed in a yeast two-hybrid system without including the ω sequence, through which the toxicity of the ζ protein was shown as well as the efficiency of the ε protein in antagonizing its toxin [145]. Despite the efficiency of ε-ζ pair in to induce yeast cell death, its use as a biocontainment system for GMY was not attempted until now.
Finally, it was observed that the expression of the chpK-chpI TA pair from Lepstospira interrogans in E. coli and S. cerevisiae modulates their cell growth [146,147]. However, the application of chpK-chpI TA pair as a biocontainment system was not reported until now. In addition to S. cerevisiae, the expression of endoribonuclease-associated toxin mazF from the E. coli mazEF module [148] in Pichia pastoris result in cell death and can be useful as a counter-selectable marker for genetic modification [149].
3. Multiplex-type biocontainment approaches
Multiplex-type biocontainment approaches (MTBA) (Table 1) have several advantages over UTBAs. First, the combined use of different molecular mechanisms can reduce the selective evolutionary pressure observed for the majority of UTBAs that results in mutagenic drift, HGT events and small molecule supplementation by an ecologic niche that inactivate the biocontainment-associated gene or gene circuits [38]. Interestingly, an early work on E. coli showed that duplicating the biocontainment-associaciated gene could lower the mutational events that lead to biocontainment inactivation [150]. The same authors suggest that the use of multiple copies of biocontainment-associated genes, with different regulatory mechanisms and independent molecular targets could also diminish the populational evolutionary pressure to negatively select mutated, non-functional biocontainment circuits [150]. In this sense, the use of synthetic biology approaches, like redesign the organism genome by incorporating expanded genetic code alphabets, the application of orthogonal DNA/RNA devices and/or systems, or XNA technologies could be a solution to reduce the evolutionary pressure to inactivate the biocontainment in GMY/GEM populations [27,52,53,54,83,84]. Unfortunately, such approaches are far away for direct biocontainment applications in eukaryotic cells, especially yeasts. Even for prokaryotes, these technologies are mainly restricted to E. coli and many of them were never tested in non-laboratorial conditions. Even considering the production facility environment, it has been reported that synthetic transcriptional circuits based in the boolean gates ‘AND’ and ‘NOR’ have altered responses in laboratory-scale fermentations, negatively impacting both biomass production and gene circuit expression [151]. However, some successful applications of multiplex technologies in biocontainment were already described, which includes the ‘Deadman’ and ‘Passcode’ kill switches in E. coli [114] and the transcriptional and recombinational control of essential genes in S. cerevisiae [27]. Another approach is the use of overlapping ‘safeguards’ composed by engineered riboregulators that control the expression of essential genes and engineered nucleases that cleaves E. coli genome when in the absence of exogenously supplied synthetic small molecules [117]. These examples reinforce the idea that redundancy combined with the use of different molecular approaches are crucial factors for the design of a resilient biocontainment technology.
4. Conclusions and perspectives
More than ever, the development of new biocontainment technologies for GMY is imperative facing the availability of synthetic biology tools and/or fermentation technologies. Data gathered so far showed that the technologies available for GMY biocontainment are far away from what is available for GEMs (e.g., E. coli). While technologies like xenobiology and MTBAs are available for E. coli, the same is not true for yeasts that mainly rely on natural auxotrophies or other non-redundant UTBAs for biocontainment. The major factors that contributes to the lack of new GMY-linked biocontainment technologies are the complexity of eukaryotic genome, the intracellular compartmentalization and biochemical specialization of organelles, as well as natural mechanisms that increase the genetic diversity of the yeast population, like meiosis or even non-homologous recombination mechanisms, which leads to the inactivation of biocontainment circuits. However, with the advances in top-down synthetic biology approaches for eukaryote genomes (e.g., ‘Yeast 2.0′), it is expected that technologies like xenobiology and genetic code refactoring become available for the development of different UTBAs and MTBAs for GMY.
Funding
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq [grant number 314558/2020-9], by “Programa Pesquisador Gaúcho - PqG” from Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul - FAPERGS [Edital FAPERGS 07/2021 and grant number 21/2551-0001958-1], and by “Programa de Redes Inovadoras de Tecnologias Estratégicas do Rio Grande Do Sul – RITEs-RS” from Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul - FAPERGS [Edital FAPERGS 06/2021 and grant number 22/2551-0000397-4]. The sponsors have no role in the study design; collection, analysis, and interpretation of data; writing of the report; or decision to submit the article for publication.
References
- Hanlon, P.; Sewalt, V. GEMs: Genetically Engineered Microorganisms and the Regulatory Oversight of Their Uses in Modern Food Production. Crit. Rev. Food Sci. Nutr. 2021, 61, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Olempska-Beer, Z.S.; Merker, R.I.; Ditto, M.D.; DiNovi, M.J. Food-Processing Enzymes from Recombinant Microorganisms—a Review. Regul. Toxicol. Pharmacol. 2006, 45, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Tu, H.; Chen, T. Potential Application of Living Microorganisms in the Detoxification of Heavy Metals. Foods. 2022, 11, 1905. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Rai, S.; Singh, D.; Upadhyay, R.S. Application of Soil Microorganisms for Agricultural and Environmental Sustainability: A Review. Plant, Soil and Microbes in Tropical Ecosystems; Dubey, S.K., Verma, S.K., Eds.; Rhizosphere Biology; Springer: Singapore, 2021; pp. 151–175. ISBN 9789811633645. [Google Scholar]
- Shin, M.-K.; Yoo, H.S. Animal Vaccines Based on Orally Presented Yeast Recombinants. Vaccine. 2013, 31, 4287–4292. [Google Scholar] [CrossRef] [PubMed]
- Adetunji, C.O.; Anani, O.A.; Olaniyan, O.T.; Bodunrinde, R.E.; Osemwegie, O.O.; Ubi, B.E. Chapter 14 - Sustainability of Biofertilizers and Other Allied Products from Genetically Modified Microorganisms. Biomass, Biofuels, Biochemicals; Varjani, S., Pandey, A., Bhaskar, T., Mohan, S.V., Tsang, D.C.W., Eds.; Elsevier: Amsterdam UK, 2022; pp. 363–393. ISBN 978-0-323-89855-3. [Google Scholar]
- Ali, R.; Zulaykha, K.D.; Sajjad, N. Genetically Modified Microbes as Biofertilizers. Bioremediation and Biotechnology, Vol 4: Techniques for Noxious Substances Remediation; Bhat, R.A., Hakeem, K.R., Eds.; Springer Nature: Cham, 2020; pp. 275–293. ISBN 978-3-030-48690-7. [Google Scholar]
- Kebede, G.; Abera, S.; Haregu, S.; Yeshitila, A.; Palanivel, H. Genetically Modified Bacteria for Alleviating Agrochemical Impact on the Environment. Agrochemicals in Soil and Environment: Impacts and Remediation; Naeem, M., Bremont, J.F.J., Ansari, A.A., Gill, S.S., Eds.; Springer Nature: Singapore, 2022; pp. 565–583. ISBN 9789811693106. [Google Scholar]
- Ruchala, J.; Kurylenko, O.O.; Dmytruk, K.V.; Sibirny, A.A. Construction of Advanced Producers of First- and Second-Generation Ethanol in Saccharomyces cerevisiae and Selected Species of Non-Conventional Yeasts (Scheffersomyces stipitis, Ogataea polymorpha). J. Ind. Microbiol. Biotechnol. 2020, 47, 109–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Huang, M.; Nielsen, J. Exploring the Potential of Saccharomyces cerevisiae for Biopharmaceutical Protein Production. Curr. Opin. Biotechnol. 2017, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kavšček, M.; Stražar, M.; Curk, T.; Natter, K.; Petrovič, U. Yeast as a Cell Factory: Current State and Perspectives. Microb. Cell Factories. 2015, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Budroni, M.; Zara, G.; Ciani, M.; Comitini, F. Saccharomyces and Non-Saccharomyces Starter Yeasts. Brew. Technol. 2017. [Google Scholar] [CrossRef]
- Parapouli, M.; Vasileiadis, A.; Afendra, A.-S.; Hatziloukas, E. Saccharomyces cerevisiae and Its Industrial Applications. AIMS Microbiol. 2020, 6, 1–31. [Google Scholar] [CrossRef]
- Nielsen, J. Production of Biopharmaceutical Proteins by Yeast. Bioengineered. 2013, 4, 207–211. [Google Scholar] [CrossRef]
- Petranovic, D.; Nielsen, J. Can Yeast Systems Biology Contribute to the Understanding of Human Disease? Trends Biotechnol. 2008, 26, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Mustacchi, R.; Hohmann, S.; Nielsen, J. Yeast Systems Biology to Unravel the Network of Life. Yeast. 2006, 23, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Andreu, C.; Zarnowski, R.; Del Olmo, M. Recent Developments in the Biology and Biotechnological Applications of Halotolerant Yeasts. World J. Microbiol. Biotechnol. 2022, 38, 27. [Google Scholar] [CrossRef] [PubMed]
- Strucko, T.; Andersen, N.L.; Mahler, M.R.; Martínez, J.L.; Mortensen, U.H. A CRISPR/Cas9 Method Facilitates Efficient Oligo-Mediated Gene Editing in Debaryomyces hansenii. Synth. Biol. 2021, 6, ysab031. [Google Scholar] [CrossRef]
- Potvin, G.; Ahmad, A.; Zhang, Z. Bioprocess Engineering Aspects of Heterologous Protein Production in Pichia pastoris: A Review. Biochem. Eng. J. 2012, 64, 91–105. [Google Scholar] [CrossRef]
- Yan, C.; Yu, W.; Zhai, X.; Yao, L.; Guo, X.; Gao, J.; Zhou, Y.J. Characterizing and Engineering Promoters for Metabolic Engineering of Ogataea polymorpha. Synth. Syst. Biotechnol. 2022, 7, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Gellissen, G.; Kunze, G.; Gaillardin, C.; Cregg, J.; Berardi, E.; Veenhuis, M.; Vanderklei, I. New Yeast Expression Platforms Based on Methylotrophic Hansenula polymorpha and Pichia pastoris and on Dimorphic Arxula adeninivorans and Yarrowia lipolytica – A Comparison. FEMS Yeast Res. 2005, 5, 1079–1096. [Google Scholar] [CrossRef] [PubMed]
- Borneman, A.R.; Pretorius, I.S. Genomic Insights into the Saccharomyces Sensu Stricto Complex. Genetics. 2015, 199, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Hirschi, S.; Ward, T.R.; Meier, W.P.; Müller, D.J.; Fotiadis, D. Synthetic Biology: Bottom-Up Assembly of Molecular Systems. Chem. Rev. 2022, acs.chemrev.2c00339. [CrossRef]
- Venter, J.C.; Glass, J.I.; Hutchison, C.A.; Vashee, S. Synthetic Chromosomes, Genomes, Viruses, and Cells. Cell. 2022, 185, 2708–2724. [Google Scholar] [CrossRef]
- Pretorius, I.S.; Boeke, J.D. Yeast 2.0—Connecting the Dots in the Construction of the World’s First Functional Synthetic Eukaryotic Genome. FEMS Yeast Res. 2018, 18, foy032. [Google Scholar] [CrossRef]
- Arnolds, K.L.; Dahlin, L.R.; Ding, L.; Wu, C.; Yu, J.; Xiong, W.; Zuniga, C.; Suzuki, Y.; Zengler, K.; Linger, J.G.; et al. Biotechnology for Secure Biocontainment Designs in an Emerging Bioeconomy. Curr. Opin. Biotechnol. 2021, 71, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Agmon, N.; Choi, W.J.; Ubide, A.; Stracquadanio, G.; Caravelli, K.; Hao, H.; Bader, J.S.; Boeke, J.D. Intrinsic Biocontainment: Multiplex Genome Safeguards Combine Transcriptional and Recombinational Control of Essential Yeast Genes. Proc. Natl. Acad. Sci. 2015, 112, 1803–1808. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, P.; Gonçalves, C. Horizontal Gene Transfer in Yeasts. Curr. Opin. Genet. Dev. 2022, 76, 101950. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; de Lorenzo, V. Synthetic Bugs on the Loose: Containment Options for Deeply Engineered (Micro)organisms. Curr. Opin. Biotechnol. 2016, 38, 90–96. [Google Scholar] [CrossRef]
- NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines) - April 2019. 2019.
- Andersen, J.T.; Schäfer, T.; Jørgensen, P.L.; Møller, S. Using Inactivated Microbial Biomass as Fertilizer: The Fate of Antibiotic Resistance Genes in the Environment. Res. Microbiol. 2001, 152, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.M.; Johnsen, P.J.; Bensasson, D.; Daffonchio, D. Release and Persistence of Extracellular DNA in the Environment. Environ. Biosafety Res. 2007, 6, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Recorbet, G.; Picard, C.; Normand, P.; Simonet, P. Kinetics of the Persistence of Chromosomal DNA from Genetically Engineered Escherichia coli Introduced into Soil. Appl. Environ. Microbiol. 1993, 59, 4289–4294. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R.R.; Patel, J.R.; Interiano, A.L.; Rovner, A.J.; Isaacs, F.J. Multilayered Genetic Safeguards Limit Growth of Microorganisms to Defined Environments. Nucleic Acids Res. 2015, 43, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Chan, C.T.Y.; Slomovic, S.; Collins, J.J. Next-Generation Biocontainment Systems for Engineered Organisms. Nat. Chem. Biol. 2018, 14, 530–537. [Google Scholar] [CrossRef]
- Knudsen, S.M.; Karlström, O.H. Development of Efficient Suicide Mechanisms for Biological Containment of Bacteria. Appl. Environ. Microbiol. 1991, 57, 85–92. [Google Scholar] [CrossRef]
- Kong, W.; Wanda, S.-Y.; Zhang, X.; Bollen, W.; Tinge, S.A.; Roland, K.L.; Curtiss, R. Regulated Programmed Lysis of Recombinant Salmonella in Host Tissues to Release Protective Antigens and Confer Biological Containment. Proc. Natl. Acad. Sci. 2008, 105, 9361–9366. [Google Scholar] [CrossRef] [PubMed]
- Mandell, D.J.; Lajoie, M.J.; Mee, M.T.; Takeuchi, R.; Kuznetsov, G.; Norville, J.E.; Gregg, C.J.; Stoddard, B.L.; Church, G.M. Biocontainment of Genetically Modified Organisms by Synthetic Protein Design. Nature. 2015, 518, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Pronk, J.T. Auxotrophic Yeast Strains in Fundamental and Applied Research. Appl. Environ. Microbiol. 2002, 68, 2095. [Google Scholar] [CrossRef] [PubMed]
- Kokina, A.; Ozolina, Z.; Liepins, J. Purine Auxotrophy: Possible Applications beyond Genetic Marker. Yeast. 2019, 36, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Ralser, M.; Kuhl, H.; Ralser, M.; Werber, M.; Lehrach, H.; Breitenbach, M.; Timmermann, B. The Saccharomyces cerevisiae W303-K6001 Cross-Platform Genome Sequence: Insights into Ancestry and Physiology of a Laboratory Mutt. Open Biol. 2, 120093. [CrossRef]
- Kokina, A.; Kibilds, J.; Liepins, J. Adenine Auxotrophy - Be Aware: Some Effects of Adenine Auxotrophy in Saccharomyces cerevisiae Strain W303-1A. FEMS Yeast Res. 2014, 14, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Nickoloff, J.A. Nonselective URA3 Colony-Color Assay in Yeast Ade1 or Ade2 Mutants. BioTechniques. 1997, 23, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Bharathi, V.; Girdhar, A.; Prasad, A.; Verma, M.; Taneja, V.; Patel, B.K. Use of Ade1 and Ade2 Mutations for Development of a Versatile Red/White Colour Assay of Amyloid-Induced Oxidative Stress in Saccharomyces cerevisiae: Use of Ade1 and Ade2 Mutations for Development of Colour Assay. Yeast. 2016, 33, 607–620. [Google Scholar] [CrossRef]
- Achilli, A.; Matmati, N.; Casalone, E.; Morpurgo, G.; Lucaccioni, A.; Pavlov, Y.I.; Babudri, N. The Exceptionally High Rate of Spontaneous Mutations in the Polymerase Delta Proofreading Exonuclease-Deficient Saccharomyces cerevisiae Strain Starved for Adenine. BMC Genet. 2004, 5, 34. [Google Scholar] [CrossRef]
- Heidenreich, E.; Wintersberger, U. Starvation for a Specific Amino Acid Induces High Frequencies of Rho− Mutants in Saccharomyces cerevisiae. Curr. Genet. 1997, 31, 408–413. [Google Scholar] [CrossRef]
- Hall, B.G. Selection-Induced Mutations Occur in Yeast. Proc. Natl. Acad. Sci. 1992, 89, 4300–4303. [Google Scholar] [CrossRef]
- Brachmann, C.B.; Davies, A.; Cost, G.J.; Caputo, E.; Li, J.; Hieter, P.; Boeke, J.D. Designer Deletion Strains Derived from Saccharomyces cerevisiae S288C: A Useful Set of Strains and Plasmids for PCR-Mediated Gene Disruption and Other Applications. Yeast Chichester Engl. 1998, 14, 115–132. [Google Scholar] [CrossRef]
- Heinisch, J.J.; Buchwald, U.; Gottschlich, A.; Heppeler, N.; Rodicio, R. A Tool Kit for Molecular Genetics of Kluyveromyces lactis Comprising a Congenic Strain Series and a Set of Versatile Vectors. FEMS Yeast Res. 2010, 10, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Winkler, C.M.; Kolmbauer, M.; Pichler, H.; Schwab, H.; Emmerstorfer-Augustin, A. Pichia pastoris Protease-Deficient and Auxotrophic Strains Generated by a Novel, User-Friendly Vector Toolbox for Gene Deletion. Yeast. 2019, 36, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhaoyang Zhang; Bin Jia; Yuan, Y. Current Advances of Biocontainment Strategy in Synthetic Biology. Chin. J. Chem. Eng. 2022. [CrossRef]
- Kim, D.; Lee, J.W. Genetic Biocontainment Systems for the Safe Use of Engineered Microorganisms. Biotechnol. Bioprocess Eng. 2020, 25, 974–984. [Google Scholar] [CrossRef]
- Nieto-Domínguez, M.; Nikel, P.I. Intersecting Xenobiology and Neometabolism To Bring Novel Chemistries to Life. ChemBioChem. 2020, 21, 2551–2571. [Google Scholar] [CrossRef] [PubMed]
- Odar, C.; Winkler, M.; Wiltschi, B. Fluoro Amino Acids: A Rarity in Nature, yet a Prospect for Protein Engineering. Biotechnol. J. 2015, 10, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Handal-Marquez, P.; Anupama, A.; Pezo, V.; Marlière, P.; Herdewijn, P.; Pinheiro, V.B. Beneath the XNA World: Tools and Targets to Build Novel Biology. Curr. Opin. Syst. Biol. 2020, 24, 142–152. [Google Scholar] [CrossRef]
- Rovner, A.J.; Haimovich, A.D.; Katz, S.R.; Li, Z.; Grome, M.W.; Gassaway, B.M.; Amiram, M.; Patel, J.R.; Gallagher, R.R.; Rinehart, J.; et al. Recoded Organisms Engineered to Depend on Synthetic Amino Acids. Nature. 2015, 518, 89–93. [Google Scholar] [CrossRef]
- de la Torre, D.; Chin, J.W. Reprogramming the Genetic Code. Nat. Rev. Genet. 2021, 22, 169–184. [Google Scholar] [CrossRef]
- Sanders, J.; Hoffmann, S.A.; Green, A.P.; Cai, Y. New Opportunities for Genetic Code Expansion in Synthetic Yeast. Curr. Opin. Biotechnol. 2022, 75, 102691. [Google Scholar] [CrossRef]
- Wan, W.; Tharp, J.M.; Liu, W.R. Pyrrolysyl-TRNA Synthetase: An Ordinary Enzyme but an Outstanding Genetic Code Expansion Tool. Biochim. Biophys. Acta 2014, 1844, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Hancock, S.M.; Uprety, R.; Deiters, A.; Chin, J.W. Expanding the Genetic Code of Yeast for Incorporation of Diverse Unnatural Amino Acids via a Pyrrolysyl-TRNA Synthetase/TRNA Pair. J. Am. Chem. Soc. 2010, 132, 14819–14824. [Google Scholar] [CrossRef] [PubMed]
- Wiltschi, B. Incorporation of Non-Canonical Amino Acids into Proteins in Yeast. Fungal Genet. Biol. 2016, 89, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Cropp, T.A.; Anderson, J.C.; Mukherji, M.; Zhang, Z.; Schultz, P.G. An Expanded Eukaryotic Genetic Code. Science. 2003, 301, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Budisa, N.; Wenger, W.; Wiltschi, B. Residue-Specific Global Fluorination of Candida antarctica Lipase B in Pichia pastoris. Mol. Biosyst. 2010, 6, 1630–1639. [Google Scholar] [CrossRef]
- Ai, H.-W.; Shen, W.; Brustad, E.; Schultz, P.G. Genetically Encoded Alkenes in Yeast. Angew. Chem. Int. Ed. 2010, 49, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, L. New Methods Enabling Efficient Incorporation of Unnatural Amino Acids in Yeast. J. Am. Chem. Soc. 2008, 130, 6066–6067. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Schultz, P.G.; Brock, A. An Improved System for the Generation and Analysis of Mutant Proteins Containing Unnatural Amino Acids in Saccharomyces cerevisiae. J. Mol. Biol. 2007, 371, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Brustad, E.; Bushey, M.L.; Brock, A.; Chittuluru, J.; Schultz, P.G. A Promiscuous Aminoacyl-TRNA Synthetase That Incorporates Cysteine, Methionine, and Alanine Homologs into Proteins. Bioorg. Med. Chem. Lett. 2008, 18, 6004–6006. [Google Scholar] [CrossRef]
- Tippmann, E.M.; Schultz, P.G. A Genetically Encoded Metallocene Containing Amino Acid. Tetrahedron. 2007, 63, 6182–6184. [Google Scholar] [CrossRef]
- Lemke, E.A.; Summerer, D.; Geierstanger, B.H.; Brittain, S.M.; Schultz, P.G. Control of Protein Phosphorylation with a Genetically Encoded Photocaged Amino Acid. Nat. Chem. Biol. 2007, 3, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Summerer, D.; Chen, S.; Wu, N.; Deiters, A.; Chin, J.W.; Schultz, P.G. A Genetically Encoded Fluorescent Amino Acid. Proc. Natl. Acad. Sci. 2006, 103, 9785–9789. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Guo, J.; Lemke, E.A.; Dimla, R.D.; Schultz, P.G. Genetic Incorporation of a Small, Environmentally Sensitive, Fluorescent Probe into Proteins in Saccharomyces cerevisiae. J. Am. Chem. Soc. 2009, 131, 12921–12923. [Google Scholar] [CrossRef] [PubMed]
- Young, T.S.; Ahmad, I.; Brock, A.; Schultz, P.G. Expanding the Genetic Repertoire of the Methylotrophic Yeast Pichia pastoris. Biochemistry. 2009, 48, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, H.J.; Maicas, E.; Friesen, J.D. Isolation of the SUP45 Omnipotent Suppressor Gene of Saccharomyces cerevisiae and Characterization of Its Gene Product. Mol. Cell. Biol. 1985, 5, 816–822. [Google Scholar] [PubMed]
- Stansfield, I.; Jones, K.M.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Poznyakovski, A.I.; Paushkin, S.V.; Nierras, C.R.; Cox, B.S.; Ter-Avanesyan, M.D.; Tuite, M.F. The Products of the SUP45 (ERF1) and SUP35 Genes Interact to Mediate Translation Termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.-R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically Recoded Organisms Expand Biological Functions. Science. 2013, 342, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, J.T.; Lahiri, P.; Stout, M.I.; Van Deventer, J.A. Exploration of Methanomethylophilus Alvus Pyrrolysyl-TRNA Synthetase Activity in Yeast. ACS Synth. Biol. 2022, 11, 1824–1834. [Google Scholar] [CrossRef]
- Zackin, M.T.; Stieglitz, J.T.; Van Deventer, J.A. Genome-Wide Screen for Enhanced Noncanonical Amino Acid Incorporation in Yeast. ACS Synth. Biol. 2022, 11, 3669–3680. [Google Scholar] [CrossRef]
- Stieglitz, J.T.; Van Deventer, J.A. High-Throughput Aminoacyl-TRNA Synthetase Engineering for Genetic Code Expansion in Yeast. ACS Synth. Biol. 2022, 11, 2284–2299. [Google Scholar] [CrossRef]
- Dunkelmann, D.L.; Oehm, S.B.; Beattie, A.T.; Chin, J.W. A 68-Codon Genetic Code to Incorporate Four Distinct Non-Canonical Amino Acids Enabled by Automated Orthogonal MRNA Design. Nat. Chem. 2021, 13, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Hankore, E.D.; Zhang, L.; Chen, Y.; Liu, K.; Niu, W.; Guo, J. Genetic Incorporation of Noncanonical Amino Acids Using Two Mutually Orthogonal Quadruplet Codons. ACS Synth. Biol. 2019, 8, 1168–1174. [Google Scholar] [CrossRef]
- Niu, W.; Schultz, P.G.; Guo, J. An Expanded Genetic Code in Mammalian Cells with a Functional Quadruplet Codon. ACS Chem. Biol. 2013, 8, 1640–1645. [Google Scholar] [CrossRef]
- Fischer, E.C.; Hashimoto, K.; Zhang, Y.; Feldman, A.W.; Dien, V.T.; Karadeema, R.J.; Adhikary, R.; Ledbetter, M.P.; Krishnamurthy, R.; Romesberg, F.E. New Codons for Efficient Production of Unnatural Proteins in a Semi-Synthetic Organism. Nat. Chem. Biol. 2020, 16, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Nie, P.; Bai, Y.; Mei, H. Synthetic Life with Alternative Nucleic Acids as Genetic Materials. Molecules. 2020, 25, 3483. [Google Scholar] [CrossRef]
- Torres, L.; Krüger, A.; Csibra, E.; Gianni, E.; Pinheiro, V.B. Synthetic Biology Approaches to Biological Containment: Pre-Emptively Tackling Potential Risks. Essays Biochem. 2016, 60, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Herdewijn, P.; Marlière, P. Toward Safe Genetically Modified Organisms through the Chemical Diversification of Nucleic Acids. Chem. Biodivers. 2009, 6, 791–808. [Google Scholar] [CrossRef] [PubMed]
- Dhami, K.; Malyshev, D.A.; Ordoukhanian, P.; Kubelka, T.; Hocek, M.; Romesberg, F.E. Systematic Exploration of a Class of Hydrophobic Unnatural Base Pairs Yields Multiple New Candidates for the Expansion of the Genetic Alphabet. Nucleic Acids Res. 2014, 42, 10235–10244. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V.B.; Holliger, P. The XNA World: Progress towards Replication and Evolution of Synthetic Genetic Polymers. Curr. Opin. Chem. Biol. 2012, 16, 245–252. [Google Scholar] [CrossRef]
- Hendrix, C.; Rosemeyer, H.; Verheggen, I.; Van Aerschot, A.; Seela, F.; Herdewijn, P. 1′, 5′ -Anhydrohexitol Oligonucleotides: Synthesis, Base Pairing and Recognition by Regular Oligodeoxyribonucleotides and Oligoribonucleotides. Chem. - Eur. J. 1997, 3, 110–120. [Google Scholar] [CrossRef]
- Schöning, K.U.; Scholz, P.; Guntha, S.; Wu, X.; Krishnamurthy, R.; Eschenmoser, A. Chemical Etiology of Nucleic Acid Structure: The α-Threofuranosyl-(3′→2′) Oligonucleotide System. Science. 2000, 290, 1347–1351. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Verbeure, B.; Luyten, I.; Lescrinier, E.; Froeyen, M.; Hendrix, C.; Rosemeyer, H.; Seela, F.; Van Aerschot, A.; Herdewijn, P. Cyclohexene Nucleic Acids (CeNA): Serum Stable Oligonucleotides That Activate RNase H and Increase Duplex Stability with Complementary RNA. J. Am. Chem. Soc. 2000, 122, 8595–8602. [Google Scholar] [CrossRef]
- Noronha, A.M.; Wilds, C.J.; Lok, C.N.; Viazovkina, K.; Arion, D.; Parniak, M.A.; Damha, M.J. Synthesis and Biophysical Properties of Arabinonucleic Acids (ANA): Circular Dichroic Spectra, Melting Temperatures, and Ribonuclease H Susceptibility of ANA·RNA Hybrid Duplexes. Biochemistry 2000, 39, 7050–7062. [Google Scholar] [CrossRef] [PubMed]
- Kalota, A.; Karabon, L.; Swider, C.R.; Viazovkina, E.; Elzagheid, M.; Damha, M.J.; Gewirtz, A.M. 2′-Deoxy-2′-Fluoro-β-d-Arabinonucleic Acid (2′F-ANA) Modified Oligonucleotides (ON) Effect Highly Efficient, and Persistent, Gene Silencing. Nucleic Acids Res. 2006, 34, 451–461. [Google Scholar] [CrossRef]
- Zhang, L.; Peritz, A.; Meggers, E. A Simple Glycol Nucleic Acid. J. Am. Chem. Soc. 2005, 127, 4174–4175. [Google Scholar] [CrossRef]
- Singh, S.K.; Koshkin, A.A.; Wengel, J.; Nielsen, P. LNA (Locked Nucleic Acids): Synthesis and High-Affinity Nucleic Acid Recognition. Chem. Commun. 1998, 455–456. [Google Scholar] [CrossRef]
- Liu, C.; Cozens, C.; Jaziri, F.; Rozenski, J.; Maréchal, A.; Dumbre, S.; Pezo, V.; Marlière, P.; Pinheiro, V.B.; Groaz, E.; et al. Phosphonomethyl Oligonucleotides as Backbone-Modified Artificial Genetic Polymers. J. Am. Chem. Soc. 2018, 140, 6690–6699. [Google Scholar] [CrossRef]
- Diwo, C.; Budisa, N. Alternative Biochemistries for Alien Life: Basic Concepts and Requirements for the Design of a Robust Biocontainment System in Genetic Isolation. Genes. 2019, 10, 17. [Google Scholar] [CrossRef]
- Lopez, G.; Anderson, J.C. Synthetic Auxotrophs with Ligand-Dependent Essential Genes for a BL21(DE3) Biosafety Strain. ACS Synth. Biol. 2015, 4, 1279–1286. [Google Scholar] [CrossRef]
- Motomura, K.; Sano, K.; Watanabe, S.; Kanbara, A.; Gamal Nasser, A.-H.; Ikeda, T.; Ishida, T.; Funabashi, H.; Kuroda, A.; Hirota, R. Synthetic Phosphorus Metabolic Pathway for Biosafety and Contamination Management of Cyanobacterial Cultivation. ACS Synth. Biol. 2018, 7, 2189–2198. [Google Scholar] [CrossRef]
- Asin-Garcia, E.; Batianis, C.; Li, Y.; Fawcett, J.D.; de Jong, I.; dos Santos, V.A.P.M. Phosphite Synthetic Auxotrophy as an Effective Biocontainment Strategy for the Industrial Chassis Pseudomonas putida. Microb. Cell Factories. 2022, 21, 156. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.R.; Steingesser, M.G.; Weems, A.D.; Khan, A.; Gladfelter, A.; Bertin, A.; McMurray, M.A. Guanidine Hydrochloride Reactivates an Ancient Septin Hetero-Oligomer Assembly Pathway in Budding Yeast. eLife. 2020, 9, e54355. [Google Scholar] [CrossRef] [PubMed]
- Hassell, D.S.; Steingesser, M.G.; Denney, A.S.; Johnson, C.R.; McMurray, M.A. Chemical Rescue of Mutant Proteins in Living Saccharomyces cerevisiae Cells by Naturally Occurring Small Molecules. G3 GenesGenomesGenetics. 2021, 11, jkab252. [Google Scholar] [CrossRef] [PubMed]
- Agmon, N.; Tang, Z.; Yang, K.; Sutter, B.; Ikushima, S.; Cai, Y.; Caravelli, K.; Martin, J.A.; Sun, X.; Choi, W.J.; et al. Low Escape-Rate Genome Safeguards with Minimal Molecular Perturbation of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. 2017, 114, E1470–E1479. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, D.L.; Gottschling, D.E. The Mother Enrichment Program: A Genetic System for Facile Replicative Life Span Analysis in Saccharomyces cerevisiae. Genetics. 2009, 183, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, G.A.; Gabriel, K.N.; Ravikumar, A.; Javanpour, A.A.; Liu, C.C. Mutually Orthogonal DNA Replication Systems In Vivo. ACS Synth. Biol. 2018, 7, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, A.; Arzumanyan, G.A.; Obadi, M.K.A.; Javanpour, A.A.; Liu, C.C. Scalable, Continuous Evolution of Genes at Mutation Rates above Genomic Error Thresholds. Cell. 2018, 175, 1946–1957.e13. [Google Scholar] [CrossRef] [PubMed]
- Strak, M.J.R.; Boyd, A.; Mileham, A.J.; Ramonos, M.A. The Plasmid-Encoded Killer System of Kluyveromyces lactis: A Review. Yeast. 1990, 6, 1–29. [Google Scholar] [CrossRef] [PubMed]
- van Nies, P.; Westerlaken, I.; Blanken, D.; Salas, M.; Mencía, M.; Danelon, C. Self-Replication of DNA by Its Encoded Proteins in Liposome-Based Synthetic Cells. Nat. Commun. 2018, 9, 1583. [Google Scholar] [CrossRef]
- Sakatani, Y.; Ichihashi, N.; Kazuta, Y.; Yomo, T. A Transcription and Translation-Coupled DNA Replication System Using Rolling-Circle Replication. Sci. Rep. 2015, 5, 10404. [Google Scholar] [CrossRef]
- Libicher, K.; Hornberger, R.; Heymann, M.; Mutschler, H. In Vitro Self-Replication and Multicistronic Expression of Large Synthetic Genomes. Nat. Commun. 2020, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Medina, E.; Yik, E.J.; Herdewijn, P.; Chaput, J.C. Functional Comparison of Laboratory-Evolved XNA Polymerases for Synthetic Biology. ACS Synth. Biol. 2021, 10, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Ellington, A.D. Construction of Synthetic T7 RNA Polymerase Expression Systems. Methods. 2018, 143, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Morse, N.J.; Wagner, J.M.; Reed, K.B.; Gopal, M.R.; Lauffer, L.H.; Alper, H.S. T7 Polymerase Expression of Guide RNAs in Vivo Allows Exportable CRISPR-Cas9 Editing in Multiple Yeast Hosts. ACS Synth. Biol. 2018, 7, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Stirling, F.; Bitzan, L.; O’Keefe, S.; Redfield, E.; Oliver, J.W.K.; Way, J.; Silver, P.A. Rational Design of Evolutionarily Stable Microbial Kill Switches. Mol. Cell. 2017, 68, 686–697.e3. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.T.Y.; Lee, J.W.; Cameron, D.E.; Bashor, C.J.; Collins, J.J. “Deadman” and “Passcode” Microbial Kill Switches for Bacterial Containment. Nat. Chem. Biol. 2016, 12, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, T.M.; Ricci, D.P.; Park, D.M.; Jiao, Y.; Yung, M.C. Comparison of Kill Switch Toxins in Plant-Beneficial Pseudomonas fluorescens Reveals Drivers of Lethality, Stability, and Escape. ACS Synth. Biol. 2022, 11, 3785–3796. [Google Scholar] [CrossRef] [PubMed]
- Wright, O.; Delmans, M.; Stan, G.-B.; Ellis, T. GeneGuard: A Modular Plasmid System Designed for Biosafety. ACS Synth. Biol. 2015, 4, 307–316. [Google Scholar] [CrossRef]
- Gallagher, R.R.; Patel, J.R.; Interiano, A.L.; Rovner, A.J.; Isaacs, F.J. Multilayered Genetic Safeguards Limit Growth of Microorganisms to Defined Environments. Nucleic Acids Res. 2015, 43, 1945–1954. [Google Scholar] [CrossRef]
- Whitford, C.M.; Dymek, S.; Kerkhoff, D.; März, C.; Schmidt, O.; Edich, M.; Droste, J.; Pucker, B.; Rückert, C.; Kalinowski, J. Auxotrophy to Xeno-DNA: An Exploration of Combinatorial Mechanisms for a High-Fidelity Biosafety System for Synthetic Biology Applications. J. Biol. Eng. 2018, 12, 13. [Google Scholar] [CrossRef]
- Ahrenholtz, I.; Lorenz, M.G.; Wackernagel, W. A Conditional Suicide System in Escherichia coli Based on the Intracellular Degradation of DNA. Appl. Environ. Microbiol. 1994, 60, 3746–3751. [Google Scholar] [CrossRef] [PubMed]
- Balan, A.; Schenberg, A.C.G. A Conditional Suicide System for Saccharomyces cerevisiae Relying on the Intracellular Production of the Serratia marcescens Nuclease. Yeast. 2005, 22, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.K.; Kirchner, J.M.; Resnick, M.A. Requirement for End-Joining and Checkpoint Functions, but Not RAD52-Mediated Recombination, after EcoRI Endonuclease Cleavage of Saccharomyces cerevisiae DNA. Mol. Cell. Biol. 1998, 18, 1891–1902. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.; Rine, J. Regulated Expression of Endonuclease EcoRI in Saccharomyces cerevisiae: Nuclear Entry and Biological Consequences. Proc. Natl. Acad. Sci. 1985, 82, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Westmoreland, J.W.; Summers, J.A.; Holland, C.L.; Resnick, M.A.; Lewis, L.K. Blunt-Ended DNA Double-Strand Breaks Induced by Endonucleases PvuII and EcoRV Are Poor Substrates for Repair in Saccharomyces cerevisiae. DNA Repair. 2010, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Caliando, B.J.; Voigt, C.A. Targeted DNA Degradation Using a CRISPR Device Stably Carried in the Host Genome. Nat. Commun. 2015, 6, 6989. [Google Scholar] [CrossRef]
- Rottinghaus, A.G.; Ferreiro, A.; Fishbein, S.R.S.; Dantas, G.; Moon, T.S. Genetically Stable CRISPR-Based Kill Switches for Engineered Microbes. Nat. Commun. 2022, 13, 672. [Google Scholar] [CrossRef] [PubMed]
- Rottinghaus, A.G.; Vo, S.; Moon, T.S. Computational Design of CRISPR Guide RNAs to Enable Strain-Specific Control of Microbial Consortia. Proc. Natl. Acad. Sci. 2023, 120, e2213154120. [Google Scholar] [CrossRef]
- Pantoja Angles, A.; Valle-Pérez, A.U.; Hauser, C.; Mahfouz, M.M. Microbial Biocontainment Systems for Clinical, Agricultural, and Industrial Applications. Front. Bioeng. Biotechnol. 2022, 10, 830200. [Google Scholar] [CrossRef]
- Yeo, C.C.; Abu Bakar, F.; Chan, W.T.; Espinosa, M.; Harikrishna, J.A. Heterologous Expression of Toxins from Bacterial Toxin-Antitoxin Systems in Eukaryotic Cells: Strategies and Applications. Toxins. 2016, 8, 49. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Park, J.-H.; Inouye, M. Toxin-Antitoxin Systems in Bacteria and Archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yao, J.; Sun, Y.-C.; Wood, T.K. Type VII Toxin/Antitoxin Classification System for Antitoxins That Enzymatically Neutralize Toxins. Trends Microbiol. 2021, 29, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Kamruzzaman, M.; Wu, A.Y.; Iredell, J.R. Biological Functions of Type II Toxin-Antitoxin Systems in Bacteria. Microorganisms. 2021, 9, 1276. [Google Scholar] [CrossRef] [PubMed]
- Fineran, P.C.; Blower, T.R.; Foulds, I.J.; Humphreys, D.P.; Lilley, K.S.; Salmond, G.P.C. The Phage Abortive Infection System, ToxIN, Functions as a Protein-RNA Toxin-Antitoxin Pair. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Tan, Q.; Awano, N.; Wu, K.-P.; Inouye, M. YeeU Enhances the Bundling of Cytoskeletal Polymers of MreB and FtsZ, Antagonizing the CbtA (YeeV) Toxicity in Escherichia coli. Mol. Microbiol. 2012, 84, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lord, D.M.; Cheng, H.-Y.; Osbourne, D.O.; Hong, S.H.; Sanchez-Torres, V.; Quiroga, C.; Zheng, K.; Herrmann, T.; Peti, W.; et al. A New Type V Toxin-Antitoxin System Where MRNA for Toxin GhoT Is Cleaved by Antitoxin GhoS. Nat. Chem. Biol. 2012, 8, 855–861. [Google Scholar] [CrossRef]
- Aakre, C.D.; Phung, T.N.; Huang, D.; Laub, M.T. A Bacterial Toxin Inhibits DNA Replication Elongation through a Direct Interaction with the β Sliding Clamp. Mol. Cell. 2013, 52, 617–628. [Google Scholar] [CrossRef]
- Bej, A.K.; Perlin, M.H.; Atlas, R.M. Model Suicide Vector for Containment of Genetically Engineered Microorganisms. Appl. Environ. Microbiol. 1988, 54, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Schweder, T.; Schmidt, I.; Herrmann, H.; Neubauer, P.; Hecker, M.; Hofmann, K. An Expression Vector System Providing Plasmid Stability and Conditional Suicide of Plasmid-Containing Cells. Appl. Microbiol. Biotechnol. 1992, 38, 91–93. [Google Scholar] [CrossRef]
- Kroll, J.; Klinter, S.; Schneider, C.; Voss, I.; Steinbüchel, A. Plasmid Addiction Systems: Perspectives and Applications in Biotechnology. Microb. Biotechnol. 2010, 3, 634–657. [Google Scholar] [CrossRef]
- Kristoffersen, P.; Jensen, G.B.; Gerdes, K.; Piškur, J. Bacterial Toxin-Antitoxin Gene System as Containment Control in Yeast Cells. Appl. Environ. Microbiol. 2000, 66, 5524–5526. [Google Scholar] [CrossRef]
- Pedersen, K.; Zavialov, A.V.; Pavlov, M.Y.; Elf, J.; Gerdes, K.; Ehrenberg, M. The Bacterial Toxin RelE Displays Codon-Specific Cleavage of MRNAs in the Ribosomal A Site. Cell. 2003, 112, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-Y.; Zhang, Y.; Inouye, M.; Ikura, M. Inhibitory Mechanism of Escherichia coli RelE-RelB Toxin-Antitoxin Module Involves a Helix Displacement Near an MRNA Interferase Active Site. J. Biol. Chem. 2009, 284, 14628–14636. [Google Scholar] [CrossRef]
- Monti, M.C.; Hernández-Arriaga, A.M.; Kamphuis, M.B.; López-Villarejo, J.; Heck, A.J.R.; Boelens, R.; Díaz-Orejas, R.; van den Heuvel, R.H.H. Interactions of Kid–Kis Toxin–Antitoxin Complexes with the ParD Operator-Promoter Region of Plasmid R1 Are Piloted by the Kis Antitoxin and Tuned by the Stoichiometry of Kid–Kis Oligomers. Nucleic Acids Res. 2007, 35, 1737–1749. [Google Scholar] [CrossRef]
- de la Cueva-Méndez, G.; Mills, A.D.; Clay-Farrace, L.; Díaz-Orejas, R.; Laskey, R.A. Regulatable Killing of Eukaryotic Cells by the Prokaryotic Proteins Kid and Kis. EMBO J. 2003, 22, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Zielenkiewicz, U.; Cegłowski, P. The Toxin-Antitoxin System of the Streptococcal Plasmid PSM19035. J. Bacteriol. 2005, 187, 6094–6105. [Google Scholar] [CrossRef]
- Zielenkiewicz, U.; Kowalewska, M.; Kaczor, C.; Cegłowski, P. In Vivo Interactions between Toxin-Antitoxin Proteins Epsilon and Zeta of Streptococcal Plasmid PSM19035 in Saccharomyces cerevisiae. J. Bacteriol. 2009, 191, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Picardeau, M.; Le Dantec, C.; Richard, G.-F.; Saint Girons, I. The Spirochetal ChpK-Chromosomal Toxin–Antitoxin Locus Induces Growth Inhibition of Yeast and Mycobacteria. FEMS Microbiol. Lett. 2003, 229, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Picardeau, M.; Ren, S.; Saint Girons, I. Killing Effect and Antitoxic Activity of The Leptospira Interrogans Toxin-Antitoxin System In Escherichia coli. J. Bacteriol. 2001, 183, 6494–6497. [Google Scholar] [CrossRef]
- Engelberg-Kulka, H.; Hazan, R.; Amitai, S. MazEF: A Chromosomal Toxin-Antitoxin Module That Triggers Programmed Cell Death in Bacteria. J. Cell Sci. 2005, 118, 4327–4332. [Google Scholar] [CrossRef]
- Yang, J.; Jiang, W.; Yang, S. MazF as a Counter-Selectable Marker for Unmarked Genetic Modification of Pichia pastoris. FEMS Yeast Res. 2009, 9, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, S.; Saadbye, P.; Hansen, L.H.; Collier, A.; Jacobsen, B.L.; Schlundt, J.; Karlström, O.H. Development and Testing of Improved Suicide Functions for Biological Containment of Bacteria. Appl. Environ. Microbiol. 1995, 61, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Moser, F.; Broers, N.J.; Hartmans, S.; Tamsir, A.; Kerkman, R.; Roubos, J.A.; Bovenberg, R.; Voigt, C.A. Genetic Circuit Performance under Conditions Relevant for Industrial Bioreactors. ACS Synth. Biol. 2012, 1, 555–564. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram of major uniplex-type biocontainment approaches (UTBAs) applied for genetically modified yeast (GMY). Under different selective pressures induced by UTBAs, GMY cells are kept active/live in a production facility, where the absence of these selective pressures leads to cell death in an environment. However, a small fraction of GMY cells have the potential to escape the UTBA mechanisms, where different molecular mechanisms, like mutagenesis, horizontal gene transfer (HGT), DNA recombination, and gene loss, results in GMY adaptability to the environment. In some cases, the environment-associated microbiome could promote GMY adaptability by releasing nutrients that bypass UTBA (e.g., natural auxotrophies). On the other hand, GMY cell death can release synthetic/transgenic DNA molecules that remain stable in the environment and can be incorporated by HGT into different environment-associated microorganisms and thus changing the microbiome. The colors of UTBAs (inset) indicate the experimental status of the technology for GMYs: black, technology extensively tested inside and outside production facility; gray, experimental technology not tested outside production facility; red, technology not applied for GMYs and other microorganisms.
Figure 1.
Diagram of major uniplex-type biocontainment approaches (UTBAs) applied for genetically modified yeast (GMY). Under different selective pressures induced by UTBAs, GMY cells are kept active/live in a production facility, where the absence of these selective pressures leads to cell death in an environment. However, a small fraction of GMY cells have the potential to escape the UTBA mechanisms, where different molecular mechanisms, like mutagenesis, horizontal gene transfer (HGT), DNA recombination, and gene loss, results in GMY adaptability to the environment. In some cases, the environment-associated microbiome could promote GMY adaptability by releasing nutrients that bypass UTBA (e.g., natural auxotrophies). On the other hand, GMY cell death can release synthetic/transgenic DNA molecules that remain stable in the environment and can be incorporated by HGT into different environment-associated microorganisms and thus changing the microbiome. The colors of UTBAs (inset) indicate the experimental status of the technology for GMYs: black, technology extensively tested inside and outside production facility; gray, experimental technology not tested outside production facility; red, technology not applied for GMYs and other microorganisms.
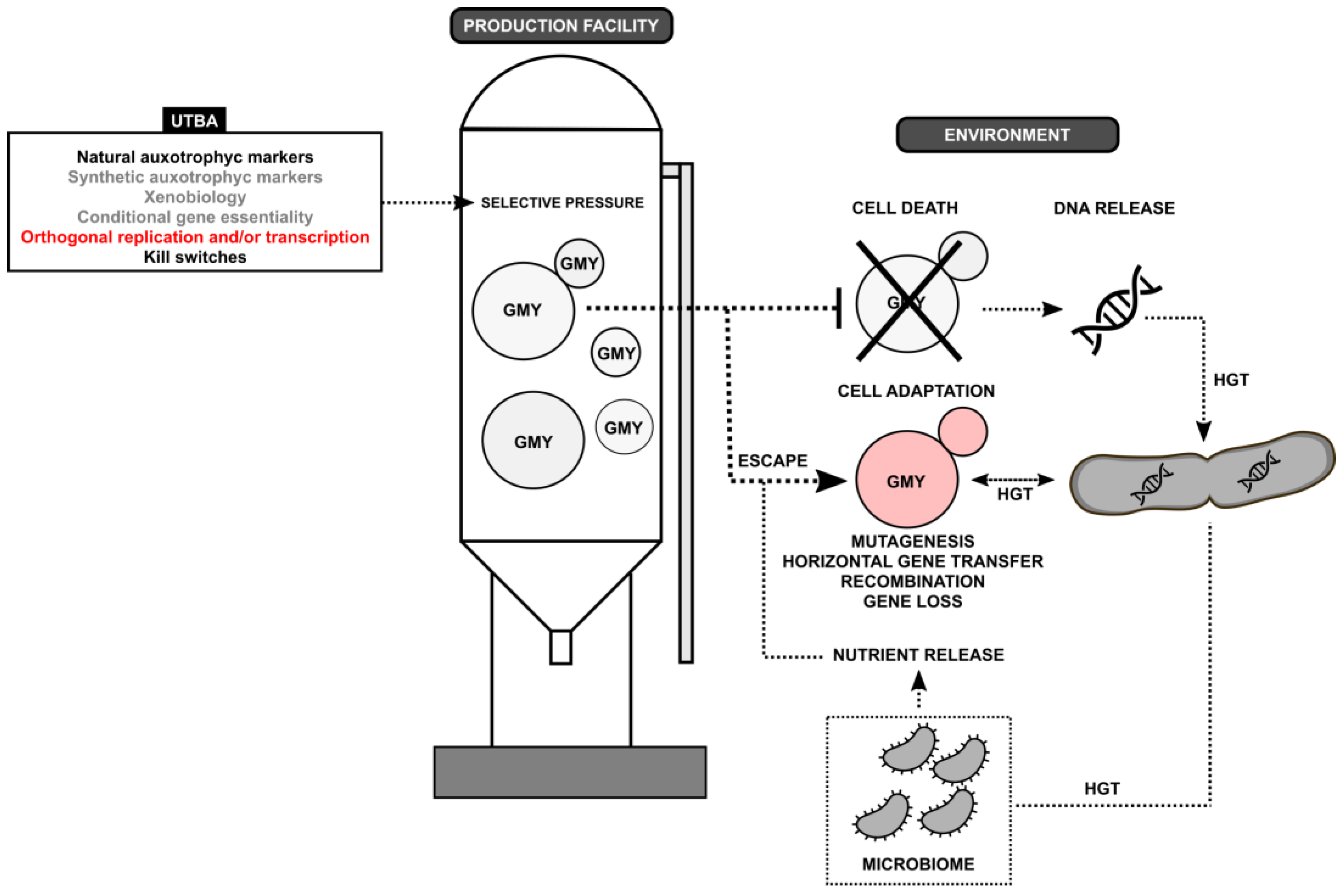

Table 1.
Advantages and disadvantages of different biocontainment approaches for genetically modified yeasts.
Table 1.
Advantages and disadvantages of different biocontainment approaches for genetically modified yeasts.
| Biocontainment | Advantages | Disadvantages |
|---|---|---|
| Uniplex-type approach (UTBA) |
|
|
| Multiplex-type approach (MTBA) |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



