
Submitted:
08 March 2023
Posted:
09 March 2023
You are already at the latest version
Abstract
Plesiomonas shigelloides is the only species in the genus and has zoonotic importance due to its serious implications resulting from the consumption of contaminated seafood. This is the first report on the genomic features of the whole-genome sequence (WGS) of P. shigelloides strain V-78 recovered from diseased rainbow trout, Oncorhynchus mykiss. The genome of P. shigelloides V-78 consists of 4,478,098 base pairs (bp), which encode 3730 proteins and has a G+C content of 51.1%. The bioinformatics analysis of WGS of V-78 confirmed the presence of 121 tRNA genes and 42 rRNA genes (15 genes for 5S rRNA, 13 genes for 16S rRNA and 14 genes for 23S rRNA). Comprehensive genome analyses revealed that the strain encodes for secondary metabolites, antimicrobial resistance, and virulence genes. The strain V-78 has 31 known antibiotic resistance models, which encode many antimicrobial resistances. Also, strain V-78 has 42 different virulence genes such as adhesion, secretion system, and motility. The digital DNA-DNA hybridization value against P. shigelloides NCTC 10360 was 74.2%, while the average nucleotide identity value was 97.1%. Based on the scrutinized analysis of genomic data, strain V-78 should be considered a novel subspecies of P. shigelloides, for which Plesiomonas shigelloides subsp. oncorhynchi is proposed.
Keywords:
Whole genome sequence
; Plesiomonas genus
; Plesiomoniasis
; Aquaculture
1. Introduction
Plesiomonas shigelloides, formerly known as Pseudomonas shigelloides, has a debatable taxonomic history and is currently classified in the family Enterobacteriaceae as the only member of the genus Plesiomonas [1]. This bacterium naturally inhabits freshwater and marine environments as well as animals dwelling those ecosystems including fish, amphibia and insects [2]. P. shigelloides occasionally causes systemic infections, such as acute diarrhea and extraintestinal diseases, mainly in immunocompromised individuals. The natural habitat of P. shigelloides is associated with its transmission to human body since this agent enters the body either through contact with contaminated water or seafood. Asymptomatic carriage of the agent is common among freshwater and marine species such as oysters, shrimp, trout, sea bass or sea bream.
A large number of freshwater plesiomonads strains have been commonly reported from rainbow trout [3,4], carp [5], and tilapia [6,7,8,9]. Studies of the intestinal tracts of several freshwater fish suggest that the genus Plesiomonas is one of the most common species composing the bacterial microbiota of these vertebrates (58, 59). Plesiomonas has not yet been associated with post-natural disaster infections (45). However, indirect evidence suggests that plesiomonads could be involved in illnesses after major natural aquatic disasters [9]. High mortality rate in trout due to P. shigelloides alone [10] and along with other bacteria like Flavobacterium spp. and Aeromonas hydrophila were also reported [11]. This pathogen was also identified as one of the main pathogens in cultured sturgeons [12]; it was also isolated from fishes with clinical symptoms during mass mortality of Ctenopharyngodo nidellus [5] and Orechromis nilotica [13] providing more evidence that P. shigelloides might be highly pathogenic for these cultured fishes. Besides cultured species, 100% mortality rate was recorded in cichlid ornamental fish due to infection caused by P. shigelloides [14].
While the agent is assumed to be a natural member of microbiota or responsible for disease, especially found along with other fish pathogens, the information about the genomic characteristics of P. shigelloides is very limited. Almost all present data strongly suggest that the genus and species designation P. shigelloides is composed of a collection of homogeneous bacteria at the phenotypic and molecular levels [9]. A population study of a diverse collection of 77 P. shigelloides strains from different geographic as well as environmental settings indicated a monophyletic clade nested within the Enterobacteriaceae [15,16]. Complete genome information has noted P. shigelloides strains isolated from humans or terrestrial animals but not in fish [9,17,18]. The lack of information causes a gap in the relation of the agent isolated from fish to humans in zoonotic potency, transmission routes and virulence. The Plesiomonas genus strains, has commonly been reported from aquaculture and fish by 16S rRNA or 23S rRNA gene sequencing or housekeeping genes such as hugA [19,20]. Successful bacterial identification reports show P. shigelloides prevalence in fish, but the paucity of genomic information limits its exact role of it by means of virulence characteristics and antimicrobial resistance gene transfer.
To the best of our knowledge, there was only one report on the genome analysis and deep molecular characterization of P. shigelloides isolates recovered from rainbow trout [21]. We aimed to characterize P. shigelloides strains isolated from fish by genome analysis and annotate the first genome of strain P. shigelloides V-78 as a novel subspecies. In addition, virulence and antimicrobial resistance genes detected in the genome of strains will help understand the pathogenicity of this agent for fish, notably found in other fish pathogens.
2. Material and Method
2.1. Bacteria Isolation and Biochemical Characterization
Isolates were collected from rainbow trout (Oncorhynchus mykiss) farms using conventional flow-through systems with non-disinfected surface water and spring water in the Central Anatolia Aegean region of Turkey. Besides rainbow trout isolates, another group of strains were also collected from ornamental freshwater fish which were transferred to the fish disease laboratory between 2018-2021. The bacterial isolates were collected during a fish health surveillance program from internal organs of diseased fish (showing darkening in color, exophthalmia and ulceration on skin and fin) of different weights, such as 0,5-1 g and 200 g rainbow trout, and from species of freshwater ornamental fish. Two strains, designed as 63 and V-78 isolated from the samples collected from Aegean region in 2018 and Central Anatolia in 2013, respectively, were selected for comprehensive genome analyses.
Isolates were biochemically characterized by Gram-staining, motility, oxidase and catalase reaction and glucose fermentation of O/F medium (Merck 103865). Hemolytic characteristic was tested on 5% sheep blood-added agar, and sensitivity to Vibriostatic agent were also tested (Oxoid DD0014 and DD0015). Growth in the presence of NaCl was tested in tryptic soy broth (TSB, Merck 105459) supplemented with 0-3% NaCl (w/v). Optimum growth temperature range was tested in the range of 4-50°C on TSB medium. In addition, the ability to grow on Thiosulfate Citrate Bile Sucrose (TCBS) agar (Merck, 103854) and to hydrolyse gelatin were determined by following published methods [22].
2.2. DNA Extraction, PCR and Sequence Analysis
The genomic DNA of the isolates was extracted using a QIAamp DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The amount and purity of DNA in each sample were measured at 260 nm and 260/280 nm wavelengths with a spectrophotometer (Multiskan Go, Thermo).
The isolates were identified by MLSA analysis using 16S rRNA, atpA (ATP synthase α subunit), gyrB (DNA gyrase β subunit), pryH (Uridylate kinase) and recA (Recombinase A) gene sequences [22,23]. Polymerase chain reaction (PCR) was performed as described in previous works [22]. All PCR products were sequenced by Macrogen Korea (Republic of Korea). The DNA fragments used for the analysis are 1400 bp for 16S rRNA, 570 bp for gyrB, 501 bp for pyrH, 462 for recA and 489 for atpA.
2.3. Genome Sequencing, Assembly and Identification
For comprehensive genome-based analyses, the whole-genome sequence of V-78 was obtained as described previously [17]. The genome of strain V-78 was sequenced with the Oxford Nanopore MinION system (Oxford Nanopore Technologies, Oxford, UK). The high-quality reads of the genome were assembled into contigs by de novo assembly using the Canu v. 1.7.1 for strain V-78. The draft genome sequence data was submitted to the GenBank. The annotation of the contigs longer than 1000 bp was achieved by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [24]. In addition, the genome of strain V-78 was annotated on the Rapid Annotations Using Subsystems Technology (RAST) server (https://rast.nmpdr.org/) [25] using RASTtk pipeline [26]. The type strain genomes server pipeline (TYGS, https://tygs.dsmz. de/) was employed to perform phylogenomic analysis and determine the digital DNA-DNA hybridization (dDDH) values between the genome of strain V-78 and those of the type strains deposited in the DSMZ database.
2.4. Comparative Genome Analysis
The publicly available genomes of P. shigelloides in the NCBI GenBank database were retrieved for comparative genome analysis. From 37 genomes, a total of 19 P. shigelloides genomes whose isolation source is known were selected for further analyses. The Codon Tree method selects single-copy Bacterial and Viral Bioinformatic resource center (BV-BRC) programs and analyzes aligned proteins and coding DNA from single-copy genes using the program RAxML (https://www.bv-brc.org/app/PhylogeneticTree [27]. For comprehensive genomic characterization, secondary metabolite biosynthetic gene clusters and antibiotic resistance models encoded by the genome of strain V-78 were determined by using antiSMASH version 6.1.1 (https://antismash.secondarymetabolites.org/#!/start) [28] and Antibiotic Resistant Target Seeker version 2 (ARTS) (https://arts.ziemertlab.com/) [29], respectively. To determine the virulence genes of the strains, VFDB (virulence factor of pathogenic bacteria) online database [http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi] was used. The GenBank files of P. shigelloides genomes were subjected to BLASTn against the VFDB with default settings. In addition, the genomic islands, the clusters of genes of probable horizontal transfer origin, were predicted by IslandCompare software available at https://islandcompare.ca/ [30]. The compilation of taxonomic, functional and ecological features of strain V-78 was acquired by Protologger web server available at http://protologger.de/ [31].
2.5. Antimicrobial Susceptibility and Resistance Gene Characteristics
The isolates were tested for their susceptibility to the antimicrobials by an agar disk diffusion method as was essentially described in the Clinical and Laboratory Standards Institute (CLSI) guideline M42-A [32], using Mueller-Hinton Agar (MHA, BBL-Becton Dickinson). The antibacterial susceptibility patterns of isolates were performed using disks containing the antibacterial agents: Trimethoprim/Sulfamethoxazole (SXT, 25 µg), Amoxicillin (AML, 25 µg), Doxycycline (DO, 30 µg), Tetracycline (TE, 30 µg), Oxytetracycline (OT, 30 µg), Oxolinic Acid (OA, 2 µg), Enrofloxacin (ENR, 5 µg), Ciprofloxacin (CIP, 5 µg), Flumequine (UB, 30 µg), Cefalexin (CN, 10 µg), Ampicillin (AMP, 10 µg), Erythromycin (E, 15 µg), Lincomycin (MY, 15 µg) and Florfenicol (FFC, 30 µg). All disks were obtained from Oxoid Ltd. (Basingstoke, Hampshire, England). The bacterial isolates were suspended in sterile 0.85% saline to obtain a 0.5 McFarland (bioMerieux S.A.) turbidity and inoculated on the test media. The plates were incubated for 24–28 h at 28°C. The control strain E. coli ATCC 25922 was incubated at 28°C for 24-28h [32].
3. Results
3.1. Biochemical Characterization
The strains were Gram-negative, short bacilli, motile, oxidase and catalase positive, glucose fermentative, non-hemolytic on sheep blood and sensitive to Vibriostatic agent (O/129 150µg). The tolerance tests for growth in the presence of NaCl and in a range of temperatures showed that the strains could grow in the presence of up to 1.5% NaCl and at the temperature range of 4-45°C. Unlike Vibrios, the isolates V-63 and V-78 did not grow on TCBS medium and could not hydrolyze gelatin.
3.2. PCR and Sequence Analysis
The strains were identified as Plesiomonas spp. by the 16S rRNA gene sequence analysis. For a better identification the following genes were sequenced: gyrB, pryH, recA and atpA, suggested in PubMLST database for Vibrio sp. These genes were analyzed by Blast in the NCBI database and the closest match were P. shigelloides. The sequences were accessed to the PubMLST database and sequence types were attained 133, 103, 127, 93, for V-63 and 136, 104, 128, 95, for V-78, by gyrB, pryH, recA and atpA, respectively. The isolates were identified, ST: 177 and 178 for V-63 and V-78, respectively, in the PubMLST database due to the similarity of the Vibrioneaceae genus. The strains are closely related to P. shigelloides reference strain, sharing at least 99% similarity for the mentioned genes.
3.3. Phylogenomics
Genome-based identification of V-78 strain was done with the TYGS web server and identified as P. shigelloides (Figure 1). The dDDH value for strain V-78 compared with its most closely related type strain P. shigelloides NCTC 10360T was calculated as 74.2%.
At the time of writing the manuscript (February 2023), a total of 37 Plesiomonas shigelloides genomes were available in the GenBank database. However, six of these genomes were assembled from metagenomics data, 2 genomes lacked information for the isolate source, and three genomes did not have information for the country of origin. Therefore, it was determined that eight genomes were obtained from the isolates from various fish species (Table 1). The codon tree was built by including 19 genomes. The codon tree reveals phylogenomic heterogeneity within the genus Plesiomonas inferred from the alignment of coding DNA from single-copy genes and amino acid sequences The strain V-78 were located in a distinct branch of the tree (Figure 2).
The genome of strain V-78 harbors 13 gene sequences coding for 16S rRNA whose pairwise identity level with that of P. shigelloides NCTC 10360T ranged from 99.38% to 99.93%. These rRNA gene sequences differed also by length ranging from 1534 to 1541 and intra-strain similarity levels of 99.28-100%.
The genomes of closely related species were retrieved form the Genome Taxonomy Database (GTDB) by the Protologger to perform pairwise analyses for the average nucleotide identity (ANI) and percentage of conserved proteins (POCP). The ANI values were calculated by FastANI (v1.2) algorithm [33] while the POCP values were acquired by custom Python scripts available at the GitHub repository by the Protologger (Table S1). The genome of strain V-78 shared 97.1% ANI value with the type strain of P. shigelloides while that value with the other closely related taxa were found to be lower than 80% implying that the genus exhibit considerable distance to the other genera (Figure S1). The highest POCP value of 68.1% was calculated between the genome of strain V-78 and that of the type strain of P. shigelloides as expected. The other POCP values were below the threshold of 50% for genus demarcation [34] proving the distinctness of the genus Plesiomonas (Table 2).
3.4. Functional and Ecological Analyses
Functional and ecological features of strain V-78 was predicted by the Protologger software. Of 5454 coding sequences identified, 235 genes encode for transporter proteins, while 50 genes are responsible for secretion. The number of unique enzymes encoded by the genome of V-78 was determined as 920. Arbutin, glucose, salicin, sucrose and trehalose were predicted to be utilized by the strain as carbon sources. In addition, strain V-78 could produce acetate from acetyl-CoA (EC:2.3.1.8, 2.7.2.1) and propionate from propanoyl-CoA (EC:2.3.1.8, 2.7.2.1) as predicted by the Protologger. The flagellar proteins identified in the genome are FlhA, FlhB, FlgB, FlgC, FlgD, FlgE, FlgF, FlgG, FlgJ, FlgK, FlgL, FliD, FliF, FliG, FliM, MotA and MotB. The strain could produce L-cysteine and acetate by utilizing sulfide and L-serine (EC:2.3.1.30, 2.5.1.47). The genome also has pathways for L-glutamate production from ammonia via L-glutamine (EC:6.3.1.2, 1.4.1.-) and biosynthetic genes for biotin, folate and riboflavin synthesis. A total of 157 carbohydrate-active enzymes (CAZymes), mainly in glycoside hydrolase (GH) families, were identified within the genome of strain V-78. Glycoside transferase (GT), carbohydrate esterase (CE), polysaccharide lyase (PL) and carbohydrate-binding module (CBM) families were also predicted in the genome.
For an ecological analysis, both the 16S rRNA gene and whole genome sequences were submitted to the Protologger [31]. The genome of strain V-78 was compared with a database of 49,094 metagenome-assembled genomes (MAGs) collected from various habitats, as described by Hitch et al. [31]. Briefly, the comparison was performed using MASH [35], and the results were filtered by using a distance threshold of <0.05. In addition, the 16S rRNA gene was employed to perform a comparative analysis involving operational taxonomic units (OTUs) generated from 19,000 amplicon datasets from 19 different habitats obtained from the IMNGS database [36]. Only one MAG (MAG-ID: HeQ_2017__SZAXPI029476-50__bin_9.fa) collected from an adult human stool [37] clustered with the genome of strain V-78. Habitat distribution and preference analysis based on comparing 16S rRNA gene amplicons revealed that strain V-78 was detected in all 19 environments in the IMNGS database with a total of 38,163,501 OTUs (Figure 3). The highest detection ratio was observed within the rhizosphere amplicons, while the mean relative abundance in human gut amplicons was the highest compared to the other environments (Table 3).
3.5. Secondary Metabolites
Secondary metabolite biosynthesis gene clusters encoded by the genome of strain V-78 were identified by antiSMASH server. The strain has two gene clusters coding for a betalactone and a thiopeptide. The betalactone gene cluster shared 2% similarity with the gene cluster of gausemycin A/B, a lipoglycopeptide antibiotic, while the thiopeptide gene cluster showed 33% similarity to menaquinone biosynthetic gene cluster. In addition, the RAST annotation revealed that the genome of strain V-78 encodes genes for anthranilate phosphoribosyltransferase, phosphoribosylanthranilate isomerase, tryptophan synthase alpha and beta chains, which are responsible for synthesis of auxin-type plant hormones.
3.6. Antimicrobial Susceptibility and Resistance Gene Characteristics
Strains were fully resistant to Lincomycin, while full susceptibility were determined against Sulfamethoxazole+trimetprim and Ciprofloxacin. The quality control strains, A. salmonicida subps. salmonicida and E. coli fell the suggested values in CLSI (Table 4). However, cut-off values are not presented in EUCAST [38] for Amoxicillin, Doxycycline, Tetracycline, Oxytetracycline, Oxolinic Acid, Enrofloxacin, Flumequine, Cefalexin, Ampicillin, Erythromycin, Lincomycin, and Florfenicol so the strains could not be evaluated to be susceptible or resistant (Table 4).
3.7. Antimicrobial Resistance Genes and Virulence
A total of 24 antimicrobial resistance models and targets were determined by the ARTS server across 19 genomes. The resistance models detected include ATP-binding cassette (ABC) antibiotic efflux pump, CARB-PSE beta-lactamases (class a), chloramphenicol acetyltransferase (CAT), ABC transporter, membrane fusion proteins of the MexEF-OprN and MexGHI-OpmD multidrug efflux complexes, major facilitator superfamily (MFS) antibiotic efflux pump and resistance-nodulation-cell division (RND) antibiotic efflux pump. Of 24 resistance models and targets, class A beta-lactamase (RF0053) and tetracycline resistance MFS efflux pump were encoded only in the genome of strain V-78. The former is responsible for the resistance through hydrolysis of beta-lactam antibiotics such as penicillins, cephalosporins and carbapenems, while the latter enables the bacteria to pump out tetracycline or tetracycline derivatives selectively (Table S2).
Identifying the genomic islands encoding mainly virulence factors, antimicrobial resistance genes and other adaptation elements is a useful strategy for adopting a population-based approach to genomic epidemiology and characterization [30]. The IslandCompare tool was used to identify and compare the genomic islands residing in the genomes of P. shigelloides strains. All strains differed in terms of size, number, location and identity of the genomic islands encoded in their genomes (Figure 4).
When the virulence gene of the P. shigelloides strains was examined, a total of 83 virulence genes were detected in the various P. shigelloides genomes obtained from the GenBank database and our isolates. Into the 83 virulence genes, only 20 virulence genes were commonly found in all strains encoding motility, immune modulation, T2SS (Type II secretion system), adherence, immunogenic lipoprotein A, stress survival, biofilm formation, stress protein, early escape from the phagosome of macrophages, secreted toxins and enzymes into the extracellular fluid. The virulence genes according to the each analyzed isolate are presented in supplementary Table S3. Different from other P. shigelloides strains, tviB gene encoding to prevent antibody-mediated opsonization, increase resistance to host peroxide and resistance to complement activation by the alternate pathway and complement-mediated lysis was detected only the strain V-78 and MS-17-188 (catfish). The lgtF and flgG genes encoding for immune modulation and motility, respectively, were detected only in the genomes of strain V-78 and strain CAPA003 isolated from Oreochromis niloticus. Functioning to a tripartite multidrug efflux pump essential for resistance to β-lactams (penicillin G and nafcillin), macrolides (erythromycin) and host-derived compounds (peptide LL-37) and progesterone, and essential for growth of gonococci in the lower genital tract of experimentally infected female mice, the mtrD gene was found in V-78, Zfcc051 (isolated from zebra fish) and CAPA003. In addition to fbpC gene, encodes a periplasmic-binding protein-dependent iron transport system necessary for the utilization of iron bound to transferrin or iron chelates was found only in the genome of strain V-78.
4. Discussion
Plesiomonas shigelloides is a unique Gram-negative, polar-flagellated pathogenic bacterium naturally found in freshwater ecosystems, such as rivers, lakes, and surface waters, and marine estuaries in tropical and temperate climates [39]. Unlike other aquatic pathogens, the isolation of P. shigelloides might be enhanced by an incubation temperature of 42–44°C since most aeromonads or other psychrophilic microorganisms could not tolerate this temperature range. The temperature tolerance of bacteria, especially in high conditions (42–44°C), causes high mortality in freshwater ornamental fish species living in climate habitats; besides cold-water fishes such as rainbow trout [40]. In our study, the isolation of P. shigelloides from rainbow trout and freshwater ornamental fish species supports a broad host range and temperature-tolerant characteristics. Although P. shigelloides is grouped in Enterobacteriaceae, the agent is oxidase and catalase positive. It also shares high similarity with the genera Vibrio and Pseudomonas, especially regarding sensitivity for Vibriostatic agent, which we detected in our study. Besides susceptibility to Vibriostatic agent, we identified our isolates by gyrB, pryH, recA and atpA, which were specifically characterized for Vibrio species. Previously, the species were placed chronologically into the genera Pseudomonas, Aeromonas, and Vibrio [41]. Habs and Schubert [42] initially established the genus Plesiomonas in the family Vibrionaceae based on a number of its unique features and its similarity to the genus Aeromonas (“plesio,” neighbor, “monas,” Aeromonas). The genus Plesiomonas has several common characteristics with the members of the family Vibrionaceae, such as being polarly flagellated, facultatively anaerobic, and cytochrome oxidase positive. The similarity between our study and the taxonomical history of P. shigelloides enable the misidentification of this species as Vibrio sp. As commonly reported that P. shigelloides is the only member of Plesiomonas genus, the strains have a genetic homology and close relationship between each other. Unlike the close phylogenetic relationship among P. shigelloides strains, Gu et al. [43] reported that the strains had a significant variability based on isolation source revealed by Random Amplified Polymorphic DNA (RAPD) analysis. In our study, the strains obtained from the GenBank database clustered in a phylogenetic group; however, our strain was separated from all genomes uploaded in the GenBank database. Although the genome of strain V-78 encodes 5454 genes, higher than the genomes of the other strains analysed, it has the lowest number of the core genes among the others, which implies its flexibility for horizontal gene transfer events. The genomic island comparison also approved the versatility of the P. shigelloides genomes since each genome has various genomic islands scattered across the genome.
The members of P. shigelloides naturally occur in freshwater and marine habitats. However, the comprehensive analysis of taxonomic, functional and ecological features revealed that strain V-78 mostly matched with the 16S amplicons obtained from rhizosphere metagenomes followed by freshwater and insect gut metagenomes. Both CAZymes and secondary metabolites, such as plant hormones produced by P. shigelloides strains, may contribute to their adaptation to various environments, from plant rhizosphere to insect gut.
From a taxonomical point of view, strain V-78 is certainly a member of the genus Plesiomonas. However, the overall genome relatedness indices, i.e., dDDH, ANI and POCP, revealed that strain V-78 might be differentiated from the type strain and other strains of P. shigelloides. The dDDH, ANI and POCP values between strain V-78 and the type strain of P. shigelloides were calculated as 74.2%, 97.1% and 68.1%, respectively. These relatively low values imply that strain V-78 must be considered a subspecies of P. shigelloides.
In humans, most enteric Plesiomonas infections are self-limiting and do not need a specific antimicrobial treatment. Some severe dysenteric and chronic intestinal infections, however, benefit from antimicrobial therapy. In these cases, fluoroquinolones and trimethoprim are generally considered to be the best oral agents because they have been shown to shorten the clinical course of Plesiomonas diarrhoea [44,45]. For fish, because P. shigelloides is an agent that seems to be an opportunistic pathogen or host of microbiota, veterinarians do not need an antimicrobial treatment when the mortality rate is low. However, empiric treatments have been done in high mortality cases due to lacking identification or ignoring this species. Unfortunately, CLSI or EUCAST do not publish a breakpoint for antimicrobials commonly used in fish health rather than ciprofloxacin and Trimethoprim/Sulfamethoxazole. We have tested our strains on 14 different antimicrobial agents belonging to nine groups; unfortunately, we only determined our isolates as susceptible or resistant to only two agents. The isolates commonly tested and reported for antimicrobials used in human health as susceptible or not, but few reports suggested antimicrobials for fish isolates [46]. Due to the ubiquitous characteristics of P. shigelloides, we do not suggest an empirical treatment or antimicrobial usage for fish strains, because the strains could easily gain antimicrobial resistance, as revealed by the genomic island analysis. We also suggest that epidemiological cut-off values (ECVs) should be established to determine the antimicrobial susceptibility of P. shigelloides strains. While there is a significant gap in the antimicrobial susceptibility values of P. shigelloides, we detected 19 to 34 antimicrobial resistance genes in the genome of P. shigelloides strains we employed. Unlike other strains obtained from the GenBank database, the genome of strain V-78 harbours a relatively low number of antimicrobial resistance genes.
As an important opportunistic pathogen, the pathogenesis of P. shigelloides can be correlated to multiple virulence genes, which encode secreted proteins and toxins. A variety of virulence factors have been associated with the pathogenesis of this microorganism, including β-hemolysins [47,48,49], enterotoxins [50], cholera-like toxins [51] and possible endotoxins [52]. Among them, ast or act encode enterotoxins in many bacteria, which damage intestinal epithelial cells, lyse red blood cells, and cause gastroenteritis, water-like diarrhea and death [53,54]. In our studies, the analyzed P. shigelloides strains harbored 20 common virulence genes including cheD, flhA, fliI, fliN, flip, gmhA/lpcA, gspD, gspE, gspG, htpB, IlpA, katB, kdsA, lpxC, lpxD, luxS, mgtB, clpC, rfaD, xcpR, which may be related to the pathogenicity in humans, animals and fish. Although various virulence gene determinants were determined in P. shigelloides strains that originated from different sources, this is the first comprehensive report presenting virulence genes in detail. We also present all virulence genes, totalling 83, found in genomes of all employed strains. Our strain, V-78, has a difference for antimicrobial resistance genes from other employed strains, but all strains have a similar count of virulence genes between 39 to 43. The findings for virulence genes suggested that P. shigelloides has an important threat to all living organisms by means of similar counts but different virulence genes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1. Average nucleotide identity values between the genome of strain V-78 and closely related type strains inferred by the Protologger using FastANI (v1.2) algorithm; Table S2. Antibiotic resistance models detected in the genomes of Plesiomonas shigelloides strains by the ARTS server; Table S3. Common and strain based virulence genes detected in the analyzed genomes; Figure S1. The genome-based tree was created in the Protologger software after the genome was checked for completeness and contamination using CheckM (v1.0.12) [55]. Then, a concatenated protein sequence tree approach employing multiple marker genes was applied by the Protologger using GTDB-Tk (r89) (v1.2.0) for placement within the GTDB taxonomy system via recognition of marker genes to assign a domain [56].
Author Contributions
Conceptualization, M.D, I.B.S., E.G.V. and H.A.; Methodology, M.D, I.B.S, S.A. and H.A.; Software, I.B.S., and H.A.; Validation, M.D, I.B.S., E.G.V. and H.A.; Formal analysis, M.D, I.B.S., and H.A.; Investigation, M.D, I.B.S., S.A.; Resources, M.D, I.B.S., S.A.; Data curation, M.D, I.B.S, and H.A.; Writing—original draft preparation, M.D, I.B.S., E.G.V. and H.A.; Writing—review & editing, M.D, I.B.S., S.A., E.G.V. and H.A.; Visualization, M.D, I.B.S., and H.A.; Supervision, M.D, I.B.S., E.G.V.; Project administration, M.D, I.B.S., S.A.; Funding acquisition, M.D, I.B.S., S.A. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This study includes isolates recovered from rainbow trout which were collected by previous projects and from ornamental fish species founded by Bursa Uludag University, Coordinatorship of scientific research projects with project number TGA-2022-1052. Analysis and materials were analyzed supporting by Bursa Uludag University, Coordinatorship of scientific research projects with project number TGA-2022-1052.
Conflicts of Interest
The authors declare no conflict of interest.
- Protologue: Emended description of Plesiomonas shigelloides (Bader 1954) Habs and Schubert 1962
- N.L. fem. dim. n. Shigella, a generic name; L. adj. suff. -oides, ressembling, similar; from Gr. neut. adj. suff. -eides, resembling, similar; from Gr. neut. n. eîdos, that which is seen, form, shape, figure; N.L. adj. shigelloides, Shigella-like
- The description is the same as reported by Janda (2015) with the following modifications:
- The genome size of the type strain is 3.4 Mb and genomic G+C content is 52.0%. Type strain: ATCC 14029; CCUG 410; CIP 63.5; DSM 8224; LMG 4242; NCCB 80007; NCTC 10360.
- Description of Plesiomonas shigelloides subsp. shigelloides subsp. nov.
- N.L. fem. dim. n. Shigella, a generic name; L. adj. suff. -oides, ressembling, similar; from Gr. neut. adj. suff. -eides, resembling, similar; from Gr. neut. n. eîdos, that which is seen, form, shape, figure; N.L. adj. shigelloides, Shigella-like
- The description is as given by Janda (2015) with the following modifications: The genome size of the type strain is 3.4 Mb and genomic G+C content is 52.0%. Genome size of strains in the subspecies ranges from 3.0 to 4.08. Genomic GC content ranges from 50.9 to 52.4%. Type strain: ATCC 14029; CCUG 410; CIP 63.5; DSM 8224; LMG 4242; NCCB 80007; NCTC 10360.
- Description of Plesiomonas shigelloides subsp. oncorhynchi subsp. nov.
- on.co.rhyn’chi. N.L. gen. masc. n. oncorhynchi, of Oncorhynchus, named after the rainbow trout, Oncorhynchus mykiss, from which the type strain was isolated
- Gram-negative, short bacilli, motile, oxidase and catalase positive, glucose fermentative, non-hemolytic on sheep blood. Able to tolerate up to 1.5% NaCl and grow at a temperature range of 4-45°C. Negative for gelatin hydrolysis.
- The genome size is 4.4 Mb and genomic G+C content is 51.1%. The GenBank accession number for the whole genome is JAQFHV000000000.
References
- Steinberg, J.P.; Lutgring, J.D.; Burd, E.M. Other Gram-Negative and Gram-Variable Bacilli - ClinicalKey. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier, 2020; Vol. 9, pp. 2847–2864.
- Willems, A.; Mergaert, J.; Swings, J. Variovorax. In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., DeVos, P., Dedysh, S., Hedlund, B., Kämpfer, P., Rainey, F., Trujillo, M.E., Bowman, J.P., Brown, D.R., Glöckner, F.O., et al., Eds.; American Cancer Society: Hoboken, NJ, USA, 2015; Vol. 324, pp. 1–11; ISBN 978-1-118-96060-8.
- Huber, I.; Spanggaard, B.; Appel, K.F.; Rossen, L.; Nielsen, T.; Gram, L. Phylogenetic analysis and in situ identification of the intestinal microbial community of rainbow trout (Oncorhynchus mykiss, Walbaum). J. Appl. Microbiol. 2004, 96, 117–132. [CrossRef]
- Salgado-Miranda, C.; Palomares, E.; Jurado, M.; Marín, A.; Vega, F.; Soriano-Vargas, E. Isolation and Distribution of Bacterial Flora in Farmed Rainbow Trout from Mexico. J. Aquat. Anim. Health 2010, 22, 244–247. [CrossRef]
- Hu, Q.; Lin, Q.; Shi, C.; Fu, X.; Li, N.; Liu, L.; Wu, S. Isolation and identification of a pathogenic Plesiomonas shigelloides from diseased grass carp. Wei sheng wu xue bao= Acta Microbiol. Sin. 2014, 54, 229–35.
- Pakingking, R.; Palma, P.; Usero, R. Quantitative and qualitative analyses of the bacterial microbiota of tilapia (Oreochromis niloticus) cultured in earthen ponds in the Philippines. World J. Microbiol. Biotechnol. 2015, 31, 265–275. [CrossRef]
- Nadirah, M.; Ruhil, H.H.; Jalal, K.C.A.; Najiah, M. Occurrence of Plesiomonas shigelloides in Cultured Red Hybrid Tilapia (Oreochromis niloticus) from Tropical Rivers, East Coast Malaysia. Pak. J. Biol. Sci. 2012, 15, 600–603. [CrossRef]
- Liu, Z.; Ke, X.; Lu, M.; Gao, F.; Cao, J.; Zhu, H.; Wang, M. Identification and pathological observation of a pathogenic Plesiomonas shigelloides strain isolated from cultured tilapia (Oreochromis niloticus). Wei sheng wu xue bao= Acta Microbiol. Sin. 2015, 55, 96–106.
- Michael Janda, J.; Abbott, S.L.; McIver, C.J. Plesiomonas Shigelloides Revisited. Clin. Microbiol. Rev. 2016, 29, 349–374. [CrossRef]
- Cruz, J.M.; Saraiva, A.; Eiras, J.C.; Branco, R.; Sousa, J.C. An Outbreak of Plesiomonas Shigelloides in Farmed Rainbow Trout, Salmo Gairdneri Richardson, in Portugal. Bull. Eur. Assoc. Fish Pathol. 1986.
- Vladík, P.; Vítovec, J. Plesiomonas shigelloides in rainbow troup septicemia. Vet. Med. (Praha).1974, 19, 297–301.
- Wang, X.; Xu, L.; Cao, H.; Wang, J.; Wang, S. Identification and drug sensitivity of a Plesiomonas shigelloides isolated from diseased sturgeons. Wei sheng wu xue bao= Acta Microbiol. Sin. 2013, 53, 723–729.
- Liu, Y.; Rzeszutek, E.; van der Voort, M.; Wu, C.-H.; Thoen, E.; Skaar, I.; Bulone, V.; Dorrestein, P.C.; Raaijmakers, J.M.; de Bruijn, I. Diversity of Aquatic Pseudomonas Species and Their Activity against the Fish Pathogenic Oomycete Saprolegnia. PLOS ONE 2015, 10, e0136241. [CrossRef]
- Nisha, R.G.; Rajathi, V.; Manikandan, R.; Prabhu, N.M. Isolation of Plesiomonas Shigelloides from Infected Cichlid Fishes Using 16S RRNA Characterization and Its Control with Probiotic Pseudomonas Sp. Acta Sci. Vet. 2014, 42, 1–7.
- Ruimy, R.; Breittmayer, V.; Elbase, P.; Lafay, B.; Boussemart, O.; Gauthier, M.; Christen, R. Phylogenetic Analysis and Assessment of the Genera Vibrio, Photobacterium, Aeromonas, and Plesiomonas Deduced from Small-Subunit rRNA Sequences. Int. J. Syst. Evol. Microbiol. 1994, 44, 416–426. [CrossRef]
- Salerno, A.; Delétoile, A.; Lefevre, M.; Ciznar, I.; Krovacek, K.; Grimont, P.; Brisse, S. Recombining Population Structure of Plesiomonas shigelloides ( Enterobacteriaceae ) Revealed by Multilocus Sequence Typing. J. Bacteriol. 2007, 189, 7808–7818. [CrossRef]
- Abdelhamed, H.; Ozdemir, O.; Tekedar, H.C.; Arick, M.A.; Hsu, C.-Y.; Karsi, A.; Lawrence, M.L. Complete Genome Sequence of Multidrug-Resistant Plesiomonas shigelloides Strain MS-17-188. Genome Announc. 2018, 6. [CrossRef]
- Piqué, N.; Aquilini, E.; Alioto, T.; Miñana-Galbis, D.; Tomás, J.M. Genome Sequence of Plesiomonas shigelloides Strain 302-73 (Serotype O1). Genome Announc. 2013, 1. [CrossRef]
- Behera, B.K.; Bera, A.K.; Paria, P.; Das, A.; Parida, P.K.; Kumari, S.; Bhowmick, S.; Das, B.K. Identification and pathogenicity of Plesiomonas shigelloides in Silver Carp. Aquaculture 2018, 493, 314–318. [CrossRef]
- Herrera, F.C.; Santos, J.A.; Otero, A.; García-López, M.-L. Occurrence of Plesiomonas shigelloides in displayed portions of saltwater fish determined by a PCR assay based on the hugA gene. Int. J. Food Microbiol. 2006, 108, 233–238. [CrossRef]
- Martins, A.F.M.; Fontana, H.; Moreira, B.M.; Bonelli, R.R. Draft Genome Sequence of a Tetracycline-Resistant Plesiomonas shigelloides Strain Isolated from Aquaculture-Reared Tilapia. Genome Announc. 2018, 7, e00832-18. [CrossRef]
- Duman, M.; Buján, N.; Altun, S.; Romalde, J.L.; Saticioglu, I.B. Population genetic and evolution analysis of Vibrio isolated from Turkish fish farms. Aquaculture 2023, 562, 738728. [CrossRef]
- PubMLST, P. databases for molecular typing and microbial genome diversity Vibrio Spp. | PubMLST Available online: https://pubmlst.org/organisms/vibrio-spp (accessed on 6 March 2023).
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [CrossRef]
- Mungan, M.D.; Alanjary, M.; Blin, K.; Weber, T.; Medema, M.H.; Ziemert, N. ARTS 2.0: feature updates and expansion of the Antibiotic Resistant Target Seeker for comparative genome mining. Nucleic Acids Res. 2020, 48, W546–W552. [CrossRef]
- Bertelli, C.; Gray, K.L.; Woods, N.; Lim, A.C.; Tilley, K.E.; Winsor, G.L.; Hoad, G.R.; Roudgar, A.; Spencer, A.; Peltier, J.; et al. Enabling genomic island prediction and comparison in multiple genomes to investigate bacterial evolution and outbreaks. Microb. Genom. 2022, 8, 000818. [CrossRef]
- A Hitch, T.C.; Riedel, T.; Oren, A.; Overmann, J.; Lawley, T.D.; Clavel, T. Automated analysis of genomic sequences facilitates high-throughput and comprehensive description of bacteria. ISME Commun. 2021, 1, 1–16. [CrossRef]
- 2006; 32. CLSI - Clinical and Laboratory Standards Institute VET03-A Methods for Antimicrobial Disk Susceptibility Testing of Bacteria Isolated From Aquatic Animals; Approved Guideline A Guideline for Global Application Developed through the Clinical and Laboratory Standards Institute Consensus Process; 2006;
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [CrossRef]
- Qin, Q.-L.; Xie, B.-B.; Zhang, X.-Y.; Chen, X.-L.; Zhou, B.-C.; Zhou, J.; Oren, A.; Zhang, Y.-Z. A Proposed Genus Boundary for the Prokaryotes Based on Genomic Insights. J. Bacteriol. 2014, 196, 2210–2215. [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132.
- Lagkouvardos, I.; Joseph, D.; Kapfhammer, M.; Giritli, S.; Horn, M.; Haller, D.; Clavel, T. IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 2016, 6, srep33721. [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e20. [CrossRef]
- The European Committee on Antimicrobial Susceptibility Testing—EUCAST. Available online: http://www.eucast.org/clinical_breakpoints/ (accessed on 15 November 2018).
- Monteil, H.; Harf-Monteil, C. Plesiomonas Shigelloides: Une Bactérie Exotique. La Lett. l’infectiologue 1997, 12.
- Levin, R.E. Plesiomonas shigelloides- An Aquatic Food Borne Pathogen: A Review of its Characteristics, Pathogenicity, Ecology, and Molecular Detection. Food Biotechnol. 2008, 22, 189–202. [CrossRef]
- Miller, M.L.; Koburger, J.A.; MARY L. MILLER and JOHN A. KOBURGER*Food Science and Human Nutrition, University of Florida, Gainesville, Florida 32611; Nedoluha, P.C.; Owens, S.; Russek-Cohen, E.; Westhoff, D.C. Plesiomonas shigelloides: An Opportunistic Food and Waterborne Pathogen. J. Food Prot. 1985, 48, 449–457. [CrossRef]
- Habs, H.; Schubert, R.H. Uber Die Biochemischen Merkmale Und Die Taxonomische Stellung von Pseudomonas Shigelloides (Bader). Zentralblatt Fur Bakteriol. Parasitenkd. Infekt. und Hyg. Abteılung 1 1962, 186, 316.
- Gu, W.; Gonzalez-Rey, C.; Krovacek, K.; Levin, R.E. Genetic Variability among Isolates of Plesiomonas Shigelloides from Fish, Human Clinical Sources and Fresh Water, Determined by RAPD Typing. Food Biotechnol. 2006, 20, 1–12. [CrossRef]
- Holmberg, S.D.; Wachsmuth, I.K.; Hickman-Brenner, F.W.; Blake, P.A.; Farmer, J.J. Plesiomonas Enteric Infections in the United States. Ann. Intern. Med. 1986, 105, 690–694. [CrossRef]
- Kain, K.C.; Kelly, M.T. Clinical features, epidemiology, and treatment of Plesiomonas shigelloides diarrhea. J. Clin. Microbiol. 1989, 27, 998–1001. [CrossRef]
- Jiang, J.; Liu, Y.; Yan, L.; Yan, Q.; Wen, X.; Cao, S.; Huang, Y.; Huang, X.; Ma, X.; Han, X.; et al. Identification and pathogenicity of Plesiomonas shigelloides from Acipenser dabryanus in China. Aquac. Res. 2021, 52, 2286–2293. [CrossRef]
- Janda, J.M.; Abbott, S.L. Expression of hemolytic activity by Plesiomonas shigelloides. J. Clin. Microbiol. 1993, 31, 1206–1208. [CrossRef]
- Baratéla, K.C.; Saridakis, H.O.; Gaziri, L.C.J.; Pelayo, J.S. Effects of medium composition, calcium, iron and oxygen on haemolysin production by Plesiomonas shigelloides isolated from water. J. Appl. Microbiol. 2001, 90, 482–487. [CrossRef]
- Falcón, R.; Carbonell, G. V; Figueredo, P.M.S.; Butiao, F.; Saridakis, H.O.; Pelayo, J.S.; Yano, T. Intracellular vacuolation induced by culture filtrates of Plesiomonas shigelloides isolated from environmental sources. J. Appl. Microbiol. 2003, 95, 273–278. [CrossRef]
- Abbott, S.L.; Kokka, R.P.; Janda, J.M. Laboratory investigations on the low pathogenic potential of Plesiomonas shigelloides. J. Clin. Microbiol. 1991, 29, 148–153. [CrossRef]
- Gardner, S.E.; Fowlston, S.E.; George, W.L. In Vitro Production of Cholera Toxin-Like Activity by Plesiomonas shigelloides. J. Infect. Dis. 1987, 156, 720–722. [CrossRef]
- Kaszowska, M.; Stojkovic, K.; Niedziela, T.; Lugowski, C. The O-antigen of Plesiomonas shigelloides serotype O36 containing pseudaminic acid. Carbohydr. Res. 2016, 434, 1–5. [CrossRef]
- Hoel, S.; Vadstein, O.; Jakobsen, A.N. Species Distribution and Prevalence of Putative Virulence Factors in Mesophilic Aeromonas spp. Isolated from Fresh Retail Sushi. Front. Microbiol. 2017, 8, 931. [CrossRef]
- Qian, Q.; Chen, Z.; Xu, J.; Zhu, Y.; Xu, W.; Gao, X.; Jiang, Q.; Zhang, X. Pathogenicity of Plesiomonas shigelloides causing mass mortalities of largemouth bass (Micropterus salmoides) and its induced host immune response. Fish Shellfish. Immunol. 2023, 132, 108487. [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [CrossRef]
Figure 1.
Tree inferred with FastME 2.1.6.1 from GBDP distances calculated from genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values > 60 % from 100 replications, with an average branch support of 65.1 %. The tree was rooted at the midpoint.
Figure 1.
Tree inferred with FastME 2.1.6.1 from GBDP distances calculated from genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values > 60 % from 100 replications, with an average branch support of 65.1 %. The tree was rooted at the midpoint.
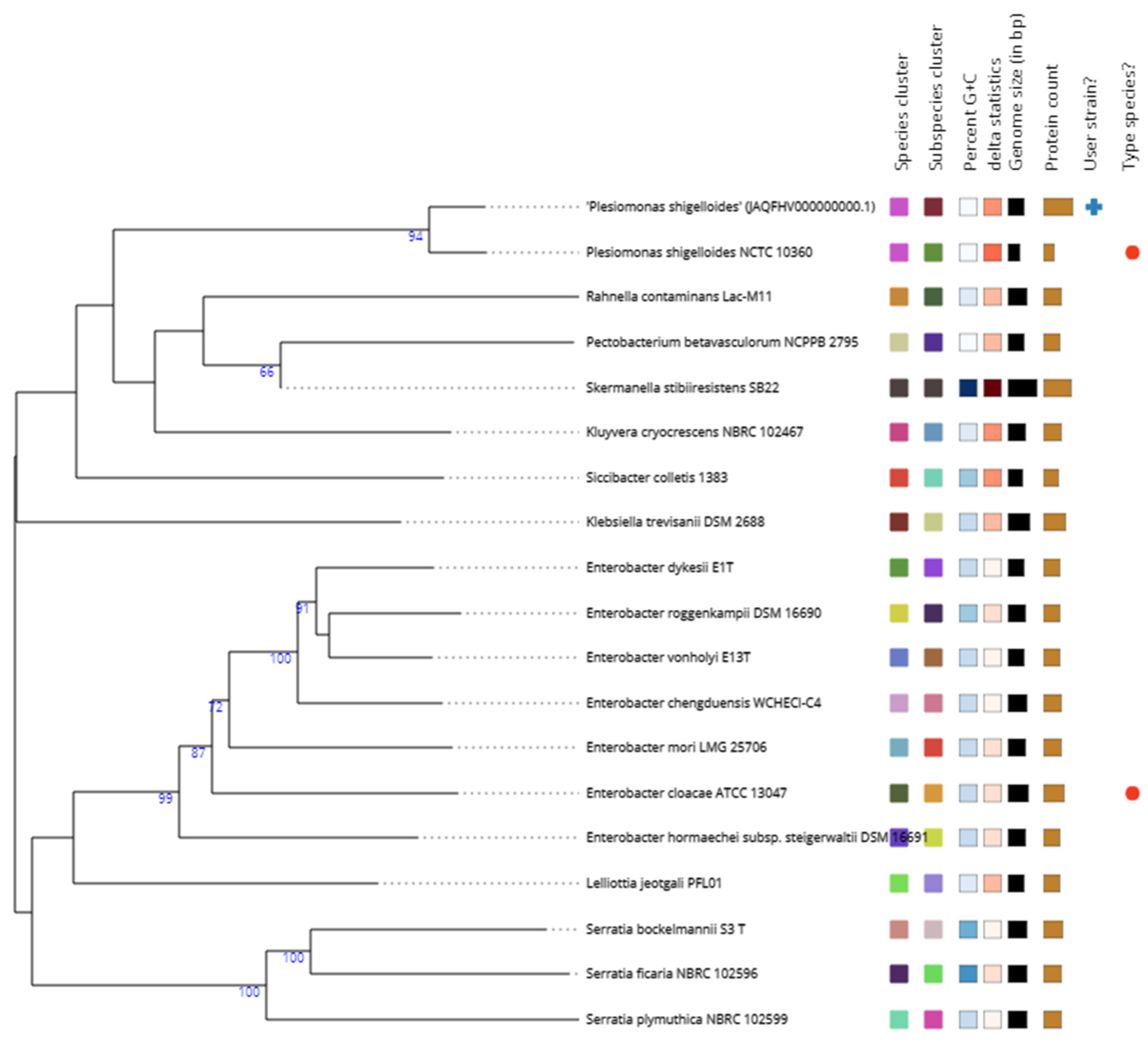
Figure 2.
The codon tree based on single-copy genes found in the genomes of strain V-78 and its close relatives within the species P. shigelloides built using the BV-BRC server (https:// www.bv-brc.org/app/PhylogeneticTree).
Figure 2.
The codon tree based on single-copy genes found in the genomes of strain V-78 and its close relatives within the species P. shigelloides built using the BV-BRC server (https:// www.bv-brc.org/app/PhylogeneticTree).
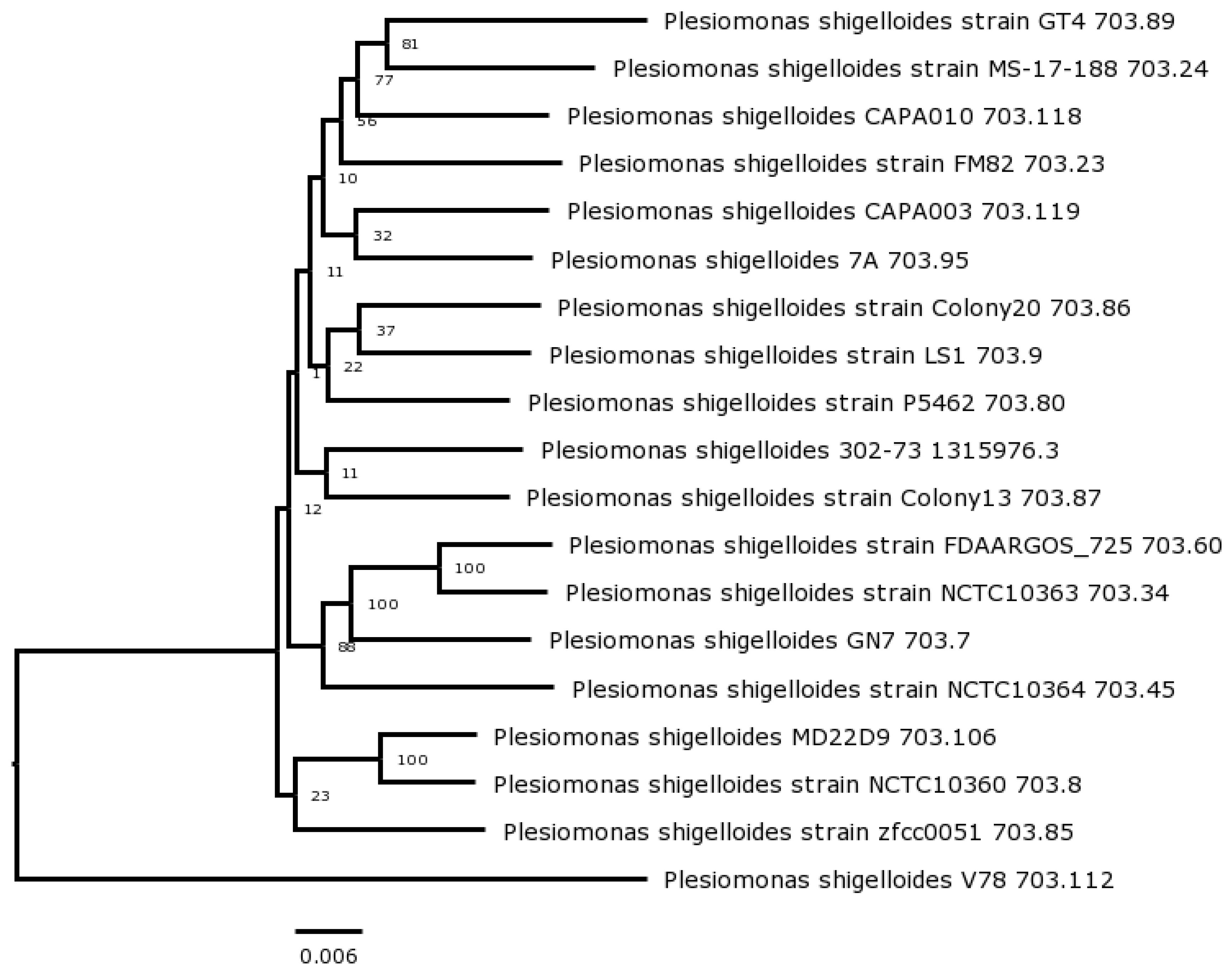
Figure 3.
Habitat distribution and preference analysis based on comparing 16S rRNA gene amplicons.
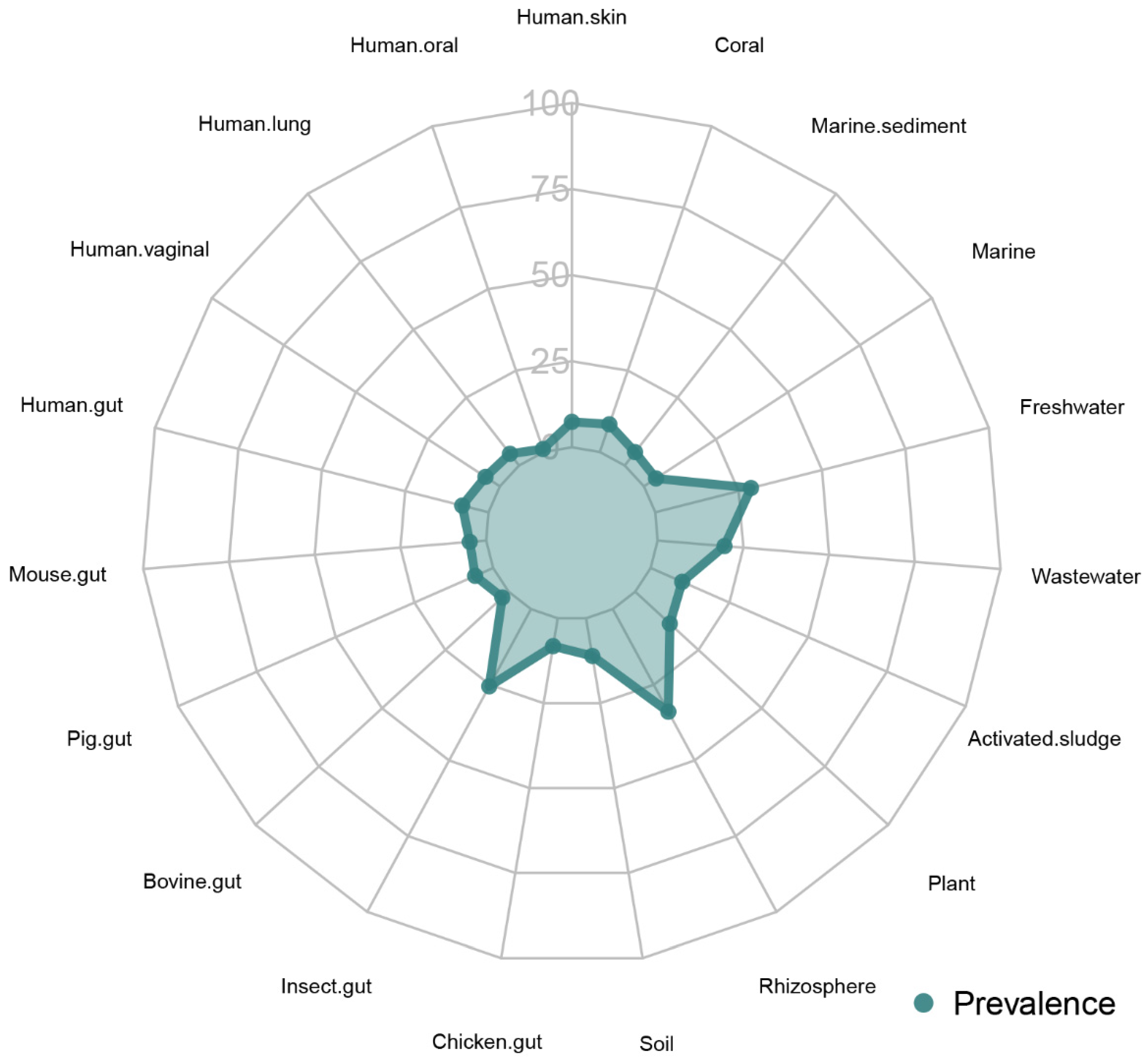
Figure 4.
Genomic islands encoded in 19 Plesiomonas genomes.
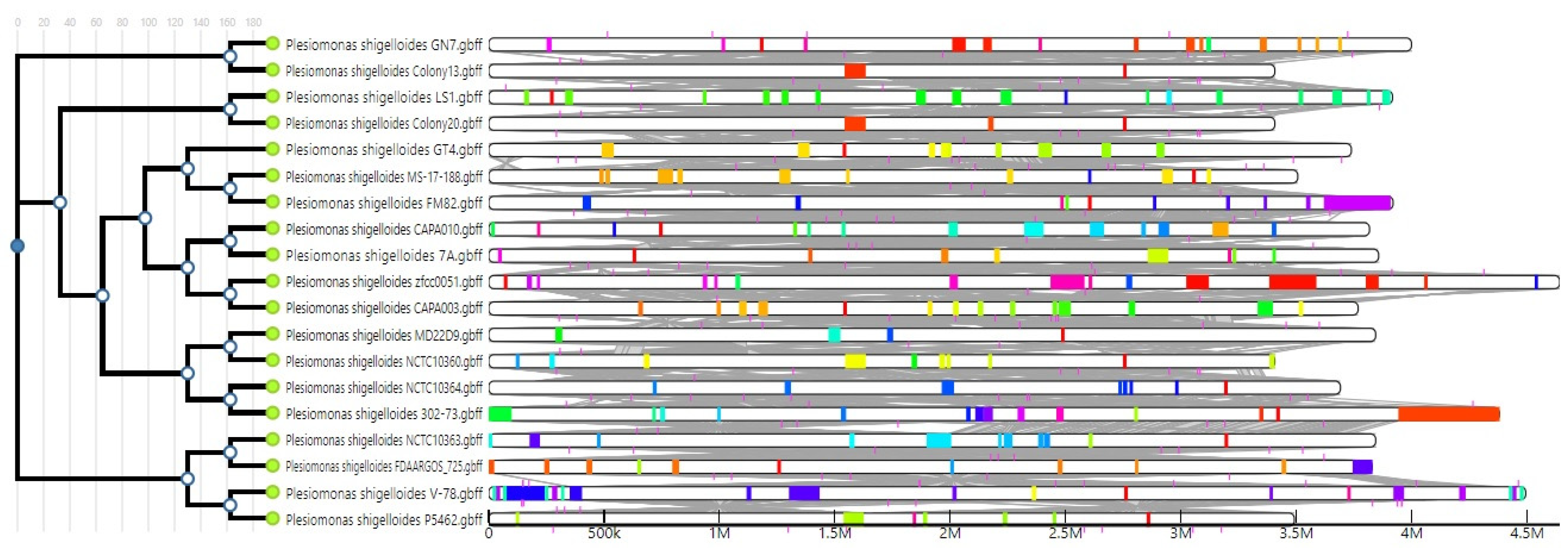

Table 1.
P. shigelloides strains compared in this study.
| Strain name | Host | Country | Total Genes | Housekeeping Genes | Known Resistance | Virulence Genes |
|---|---|---|---|---|---|---|
| zfcc0051 | Zebra Fish | USA | 3818 | 596 | 31 | 41 |
| V-78 | Rainbow trout | Turkey | 5454 | 297 | 19 | 41 |
| P5462 | Gentoo penguins | Hong Kong | 3287 | 596 | 33 | 40 |
| NCTC10364 | Human | Unknown | 3098 | 584 | 32 | 42 |
| NCTC10363 | Human | Unknown | 3264 | 599 | 30 | 40 |
| NCTC10360 | Dog | Unknown | 2886 | 583 | 28 | 40 |
| MS-17-188 | Catfish | USA | 3426 | 595 | 32 | 42 |
| MD22D9 | Macrobdella decora | USA | 3190 | 595 | 32 | 40 |
| LS1 | Percocypris pingi | China | 3484 | 590 | 33 | 43 |
| GT4 | Oreochromis niloticus | Brazil | 3134 | 600 | 34 | 43 |
| GN7 | Water | Malaysia | 3434 | 596 | 33 | 42 |
| FM82 | Fish | Brazil | 3233 | 593 | 32 | 41 |
| FDAARGOS_725 | Human | USA | 3250 | 597 | 32 | 40 |
| Colony20 | Food | Thailand | 2598 | 576 | 28 | 43 |
| Colony13 | Human | Thailand | 2648 | 580 | 28 | 39 |
| CAPA010 | Oreochromis niloticus | Peru | 3176 | 599 | 33 | 42 |
| 7A | River | Sweden | 3298 | 597 | 32 | 41 |
| 302-73 | Human | Japan | 3398 | 592 | 32 | 40 |
| CAPA003 | Oreochromis niloticus | Peru | 3185 | 596 | 33 | 41 |
The Plesiomonas isolates from fish were highlighted in bold.
Table 2.
Percentage of conserved proteins (POCP) values from related type strains as calculated by the Protologger.
Table 2.
Percentage of conserved proteins (POCP) values from related type strains as calculated by the Protologger.
| Strain number | Type strain | POCP (%) |
|---|---|---|
| Strain V-78 | Plesiomonas shigelloides | 68.1 |
| Yersinia entomophaga | 43.2 | |
| Obesumbacterium proteus | 42.4 | |
| Hafnia paralvei | 42.1 | |
| Buttiauxella agrestis | 41.8 | |
| Citrobacter werkmanii | 41.8 | |
| Buttiauxella noackiae | 41.8 | |
| Citrobacter pasteurii | 41.7 | |
| Salmonella enterica | 41.7 | |
| Buttiauxella brennerae | 41.6 | |
| Citrobacter freundii | 41.5 | |
| Enterobacter soli | 41.2 | |
| Serratia marcescens | 40.9 | |
| Serratia grimesii | 40.8 | |
| Pectobacterium carotovorum | 40.8 | |
| Serratia proteamaculans | 40.6 | |
| Buttiauxella ferragutiae | 40.6 | |
| Serratia nematodiphila | 40.5 | |
| Citrobacter braakii | 40.5 | |
| Serratia odorifera | 40.3 | |
| Chania multitudinisentens | 40.3 | |
| Serratia quinivorans | 40.2 | |
| Yersinia pestis | 40.2 | |
| Serratia fonticola | 40.2 | |
| Buttiauxella gaviniae | 40.1 | |
| Enterobacter hormaechei | 40.0 | |
| Proteus hauseri | 39.9 | |
| Serratia ficaria | 39.9 | |
| Serratia plymuthica | 39.9 | |
| Cronobacter sakazakii | 39.8 | |
| Citrobacter amalonaticus | 39.7 | |
| Pectobacterium parmentieri | 39.5 | |
| Enterobacter cloacae | 39.4 | |
| Dickeya chrysanthemi | 39.4 | |
| Raoultella planticola | 39.0 | |
| Klebsiella pneumoniae | 38.9 | |
| Raoultella ornithinolytica | 38.8 | |
| Lonsdalea quercina | 38.3 | |
| Pantoea agglomerans | 38.1 | |
| Enterobacter ludwigii | 38.0 | |
| Rouxiella chamberiensis | 38.0 | |
| Pantoea allii | 37.2 | |
| Rouxiella silvae | 37.2 | |
| Rouxiella badensis | 37.0 | |
| Sodalis praecaptivus | 36.3 | |
| Pantoea cypripedii | 35.3 |
Table 3.
Habitat preference and distribution analysis of strain V-78 based on the 16S rRNA gene amplicons obtained from the IMNGS database by the Protologger software.
Table 3.
Habitat preference and distribution analysis of strain V-78 based on the 16S rRNA gene amplicons obtained from the IMNGS database by the Protologger software.
| Environment | Detection ratio (%) | Mean relative abundance (%) | Standard deviation (%) |
|---|---|---|---|
| Rhizosphere | 33.9 | 0.71 | 3.47 |
| Freshwater | 28.7 | 3.29 | 12.49 |
| Insect gut | 25.5 | 4.42 | 16.15 |
| Wastewater | 19.5 | 0.20 | 0.55 |
| Plant | 13.7 | 3.26 | 11.07 |
| Soil | 11.1 | 0.20 | 1.37 |
| Activated sludge | 10.0 | 0.02 | 0.05 |
| Coral | 8.5 | 0.38 | 0.66 |
| Chicken gut | 8.2 | 0.23 | 1.17 |
| Human gut | 7.9 | 6.17 | 16.59 |
| Human skin | 7.4 | 0.22 | 0.67 |
| Pig gut | 5.7 | 0.04 | 0.08 |
| Human vaginal | 5.0 | 0.02 | 0.04 |
| Marine sediment | 4.9 | 0.10 | 0.21 |
| Mouse gut | 4.8 | 0.15 | 0.33 |
| Human lung | 4.3 | 0.38 | 1.61 |
| Marine | 4.2 | 0.05 | 0.22 |
| Bovine gut | 2.5 | 0.03 | 0.05 |
| Human oral | 0.9 | 0.20 | 0.36 |
Table 4.
The inhibition zones of antimicrobial agents for quality control and P. shigelloides strains.
Table 4.
The inhibition zones of antimicrobial agents for quality control and P. shigelloides strains.
| Family | Antibiotics |
Disk content (µg) |
A. salmonicida subps. salmonicida ATCC 33658 |
E. coli ATCC 25922 |
P. shigelloides | ||
| V63 | V78 | AF160 | |||||
| Sulfonamides | Trimethoprim/Sulfamethoxazole (SXT) | 25 | 23 | 27 | 25 (S) | 25 (S) | 21 (S) |
| Aminopenicillins | Amoxicillin (AML)* | 25 | 32 | 23 | 12 | 13 | 25 |
| Tetracyclines | Doxycycline (DO)* | 30 | 31 | 25 | 22 | 20 | 15 |
| Tetracycline (TE)* | 30 | 35 | 30 | 15 | 13 | 12 | |
| Oxytetracycline (OT)* | 30 | 30 | 29 | 10 | 0 (R) | 0 (R) | |
| Quinolone | Oxolinic Acid (OA)* | 2 | 35 | 28 | 24 | 25 | 10 |
| Fluoroquinolone | Enrofloxacin (ENR)* | 5 | 40 | 37 | 31 | 35 | 25 |
| Ciprofloxacin (CIP) | 5 | 50 | 40 | 33 (S) | 33 (S) | 25 (S) | |
| Flumequine (UB)* | 30 | 40 | 35 | 26 | 30 | 28 | |
| Cephalosporins | Cefalexin (CN)* | 10 | 25 | 25 | 20 | 20 | 21 |
| Penicillins | Ampicillin (AMP)* | 10 | 32 | 20 | 11 | 12 | 10 |
| Macrolides | Erythromycin (E)* | 15 | 22 | 8 | 16 | 11 | 10 |
| Clindamycin | Lincomycin (MY)* | 15 | 0 (R) | 0 (R) | 0 (R) | 0 (R) | 0 (R) |
| Chloramphenicol | Florfenicol (FFC)* | 30 | 40 | 24 | 11 | 12 | 12 |
| *Cut-off values are not available on EUCAST | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



