
Submitted:
12 March 2023
Posted:
13 March 2023
You are already at the latest version
Abstract
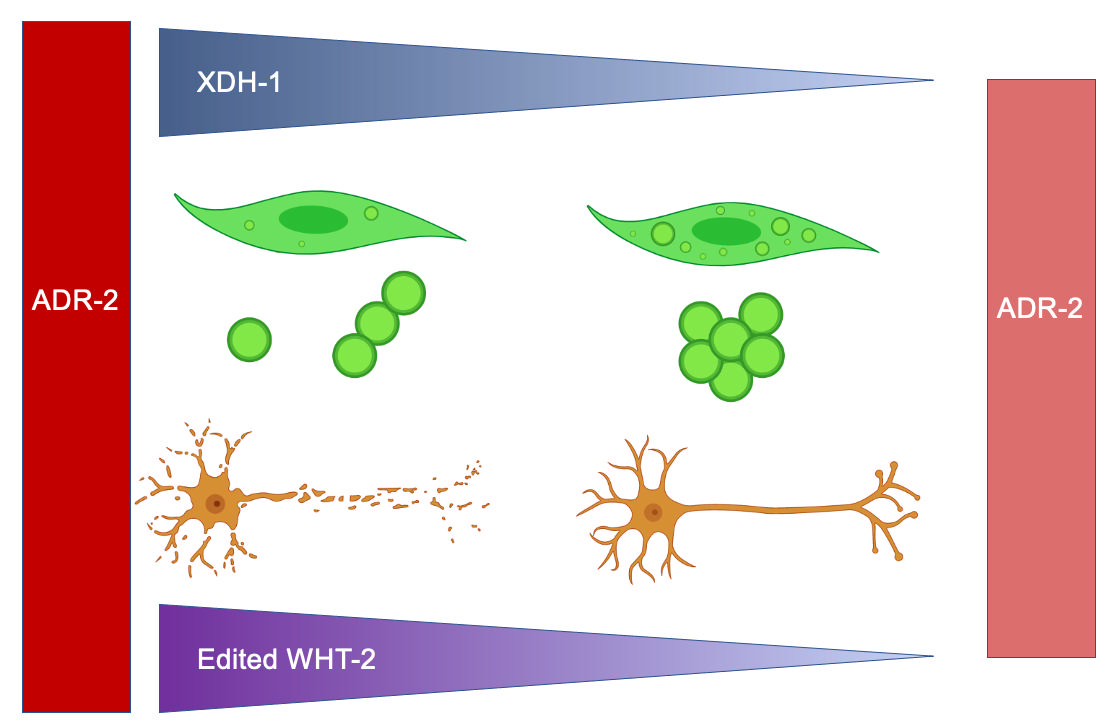
Differential RNA editing by adenosine deaminases that act on RNA (ADARs) has been implicat-ed in several neurological disorders, including Parkinson’s disease (PD). Here, we report results of an RNAi screen of genes differentially regulated in adr-2 mutants, normally encoding the only catalytically active ADAR in Caenorhabditis elegans, ADR-2. Subsequent analysis of candidate genes that alter the misfolding of human α-synuclein (α-syn) and dopaminergic neurodegenera-tion, two PD pathologies, reveal that reduced expression of xdh-1, the ortholog of human xanthine dehydrogenase (XDH), is protective against α-synuclein-induced dopaminergic neurodegenera-tion. Further RNAi experiments show that WHT-2, the worm ortholog of the human ABCG2 transporter and a predicted interactor of XDH-1, is the rate-limiting factor in the ADR-2, XDH-1, WHT-2 system for dopaminergic neuroprotection. Proteomic analysis indicates that the editing of one nucleotide in wht-2 RNA leads to the substitution of threonine with alanine at residue 124 in the WHT-2 protein, changing its structure. Thus, we propose a model where wht-2, is edited by ADR-2 which promotes optimal export of uric acid, a known substrate of WHT-2 and a product of XDH-1 activity. In the absence of editing, uric acid export is limited provoking a reduction in xdh-1 transcription to limit uric acid production and maintain cellular homeostasis. In turn, ele-vation of uric acid is protective against dopaminergic neuronal cell death. These data indicate that modifying specific targets of RNA editing may represent a promising therapeutic strategy for PD.
Keywords:
alpha-synuclein
; ADAR
; C. elegans
; dopamine
; neurodegeneration
; Parkinson's disease
; RNA editing
; xanthine dehydrogenase
; ABC transporter
1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, afflicting 1-2% of adults over age 65 worldwide [1]. PD is pathologically characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) accompanied by the misfolding of α-syn into beta sheets, forming Lewy bodies in the midbrain. Mitochondrial dysfunction is an important factor contributing to PD, occurring when there is an imbalance among reactive oxygen species (ROS) and antioxidants, resulting in cellular stress. Inhibition of complex I in the mitochondrial electron transport chain induces enhanced ROS generation [2], ultimately inhibiting activity of the proteasome, lysosomes, and mitochondria [3]. Further, enhanced cellular ROS promotes the misfolding of α-syn and perturbs protein clearance mechanisms, exacerbating disease [4]. Such pathologies have been found in the SNpc of PD patients [7,8].
Unfortunately, many hallmark symptoms of PD such as bradykinesia, tremor, and abnormal gait remain undetectable until a substantial amount of dopaminergic cell death has occurred [7]. Despite robust efforts to develop therapeutics for PD, treatment remains focused on the alleviation of symptoms rather than ceasing disease progression. Moreover, the exact pathology underlying the foundation of this disease remains unclear. To date, twenty genes have been deemed causative of PD, while genome wide association studies have identified 90 variants associated with PD risk [8]. However, the majority of PD cases are idiopathic with disease spurring from interactions between genetic networks and environmental factors [9].
Adenosine to inosine (A-to-I) editing is the mechanism in which adenosine is converted to inosine in regions of double-stranded RNA; this base change is catalyzed by a family of enzymes known as adenosine deaminases that act on RNA (ADARs) [10]. The chemical properties of inosine are similar to those of guanosine, so it is treated as guanosine by translational machinery [11]. RNA editing by ADARs profoundly increases transcriptomic and proteomic diversity. ADARs are imperative for the healthy development and function of the nervous system [12], and ADAR protein dysfunction has been implicated in a variety of neurological diseases including amyotrophic lateral sclerosis, epilepsy, developmental epileptic encephalopathy, and Aicardi-Goutières syndrome, among others [13]. Furthermore, knockouts of ADAR genes in model organisms have exhibited profound neurological dysfunction. For example, ADAR2 knockout mice are prone to seizures and have a shortened lifespan [14], and Adar mutant Drosophila display synaptic and neurotransmission defects [15]. Notably, a recent study in post-mortem brain tissue revealed differential editing in mRNA of PD patients that succumbed to disease [16]. Thus, it is plausible that ADARs have a salient role in PD.
The nematode roundworm, C. elegans, is an exceptional model organism to study progressive neurodegenerative diseases such as PD due to its hermaphroditic reproduction, large brood size, short generation time (~3 days) and ease of maintenance [17,18]. Additionally, the entire connectome of C. elegans has been delineated [19]. Thus, C. elegans provides researchers with the ability to rapidly analyze isogenic populations and precisely quantify neurodegeneration in individuals disadvantaged by α-syn, the central protein associated with dopaminergic neurodegeneration in PD. In C. elegans, two ADAR genes with human orthologs have been identified, adr-1 and adr-2. Analyses of these genes indicated that both have distinct roles in RNA editing and are essential for normal nervous system function in C. elegans [20]. However, ADR-2 is the only active adenosine deaminase, as knockout of adr-2 abolishes all A-to-I RNA editing [21]. In contrast, ADR-1 acts as a regulator of ADR-2 by facilitating the binding of ADR-2 to its targets [21,22]. Previous work on ADARs in C. elegans has resulted in the identification of genes that are both regulated and edited by ADR-2, including xdh-1 and wht-2, respectively [23].
Xanthine dehydrogenase (XDH) is an enzyme that is responsible for the catabolism of hypoxanthine to xanthine and xanthine to uric acid, the final two steps in the purine catabolic pathway [24,25]. Located intracellularly in peroxisomes and the cytosol, XDH has two subunits, each consisting of four redox center domains: a molybdopterin center, two iron sulfur centers, a flavin adenine dinucleotide (FAD) center, and two iron-sulfur subunits [26]. NADH and uric acid are produced as byproducts of XDH activity. Additionally, XDH can be converted to xanthine oxidase (XO) both irreversibly and reversibly via proteolysis or the oxidation of two cysteine residues, respectively. This interconversion occurs in the presence of urea or when concentrations of guanidine hydrochloride are low. Oxygen is required for XO activity in lieu of NAD, yielding superoxide anion, hydrogen peroxide, and uric acid [26]. This difference in cofactors is attributed to the capacity for XDH to rapidly react with NAD and slowly with oxygen, while XO does the opposite [27]. This stems from the conformational change that results upon XDH to XO conversion—in simplest terms, the XDH has a higher binding affinity than XO [26]. As a result, XO is a major generator of ROS (superoxide anion and hydrogen peroxide), which significantly contributes to oxidative stress.
ATP binding cassette subfamily G member 2 (ABCG2), the human ortholog of worm WHT-2, is an efflux transporter containing a hydrophobic transmembrane domain and a nucleotide binding domain [28]. It can be found in various tissues such as the intestine, kidney and brain. Specifically, it is expressed on the apical side of endothelial cells in the blood brain barrier (BBB). It has also been shown to be located in mitochondrial cristae [29]. ABCG2 is a transporter of many endogenous substrates such as urate, steroids, and heme, as well as xenobiotic compounds. Importantly, it has been shown to be upregulated in response to oxidative stress as it is able to efflux substances that generate ROS [30].
Here, we show experimental evidence that reduction of functional XDH in dopamine neurons is protective against neurodegeneration attributed to oxidative stress induced by α-syn. Further, we propose a network in which ADR-2 edits WHT-2/ABCG2, altering its structure and, thus, its ability to export uric acid from the cell. In turn, XDH is downregulated to maintain homeostasis and prevent potential deleterious effects of excess uric acid.
2. Materials and Methods
2.1. C. elegans Strains
All strains were maintained according to standard procedures [31]. For RNAi experiments, UA49 [Punc-54::α-syn::GFP, rol-6 (baIn2)] and UA196 {sid-1(pk3321); Pdat-1::sid-1, Pmyo-2::mCherry; Pdat-1 ::GFP, Pdat-1 ::α-syn} were used to knock down genes in aggregate or neurons, respectively. Other strains used in this study include UA44 (Pdat-1 ::α-syn, Pdat-1 ::GFP), RB2379 (xdh-1((ok3134)), RB886 (adr-2(ok735)). BY250 (vtIs7[Pdat-1::GFP], kindly provided by Randy Blakely, and UA457 adr-2(ok735); Pdat-1 ::α-syn, Pdat-1 ::GFP, also referred to as UA44; adr-2(ok735), gifted by Heather Hundley, were also utilized in this investigation. Crosses to generate worm strains used in this study are summarized in Table 1.
2.2. RNAi and Analysis of Protein Misfolding
RNAi by feeding was performed as described [32] and UA49 worms were scored semi-quantitatively for aggregate size and number two days post-hatch. For each gene, two replicates of forty worms each were immobilized with 2mM levamisole, transferred onto an agarose pad, and analyzed. The size of misfolded proteins was categorized as small, medium or large, and the number of aggregates was denoted as few, multiple or many. These categories were then quantified by multiplying by 1 for small or few, 2 for medium or multiple, and 3 for large or many to get a scaled score for size and number. These scores were added together to get an overall aggregate score, and the percent difference of each experiment compared to the empty vector control was determined.
2.3. GO Term Analysis of Candidate Genes
Gene enrichment analysis were performed to identify tissues, phenotypes, and gene ontology (GO) terms associated with the annotations of candidate genes. All analyses were performed using the WormBase enrichment suite. [32]
2.4. RNAi and Analysis of Neurodegeneration
RNAi by feeding was performed [33] and UA196 worms were scored for neurodegeneration on day 7 post-hatch. Three biological replicates of 30 worms each were immobilized with 2mM levamisole, transferred onto an agarose pad, and analyzed. The four CEP and two ADE neurons of each worm were analyzed for the presence of a cell body and intact axon process. Worms were grouped by the number of degenerated neurons. [34]
2.5. Analysis of xdh-1(ok3134) Mutants for Neurodegeneration
UA44 and UA455 [(Pdat-1::α-syn and Pdat-1::α-syn + xdh-1(ok3134), respectively] worms were grown on OP50-1 bacteria and were scored on day 7 post-hatch. Worms were transferred to fresh plates on days 4, 5 and 6 to ensure that only target worms were analyzed and not confused with progeny. Three replicates of 30 worms each were immobilized with 2mM levamisole, transferred onto an agarose pad, and analyzed. The four CEP and two ADE neurons of each worm were analyzed for the presence of a cell body and intact axon process. Worms with all six neurons intact were considered wildtype, and those with one or more degenerated neurons were noted as degenerative.
2.6. Transgenic line construction and analysis of neurodegeneration
Worms overexpressing xdh-1driven by the dat-1 promoter were generated by injecting both the Pdat-1:xdh-1 construct (kindly gifted by Atsushi Kuhara) and rol-6 co-injection marker at a concentration of 50 ng/μL each (100 ng/ μL total) into N2 worms. Three stable lines were created and then crossed to UA44. Three replicates consisting of 30 worms of each of three stable lines were analyzed as described above (section 2.5), and the number of worms with wildtype and degenerated dopaminergic neurons was determined. Data from analysis of three replicates from each of the three stable lines (Figure A3) were averaged and considered as one biological replicate for statistical purposes.
2.7. Body Wall Muscle and Dopaminergic Neuron Image Acquisition
Images of worm body wall muscles and dopaminergic neurons were obtained by placing worms in a 7 μL drop of 10 mM levamisole (dissolved in 0.5x S basal buffer) on a glass coverslip. The coverslip with the worms in levamisole was inverted and placed on a 2% agarose pad attached to a microscope slide, paralyzing worms to allow for visualization. Fluorescence microscopy was performed with a Nikon Eclipse E800 epifluorescence microscope equipped with an Endow GFP HYQ filter cube. Images were captured using a Cool Snap CCD camera (Photometrics) with Metamorph software (Molecular Devices).
2.8. RT-qPCR of α-syn expression
Total RNA from UA44 and UA455 worms was isolated seven days post-hatching. RNA from 100 worms from three replicates of each group was isolated using TRI reagent (Molecular Research Center). Samples were rid of genomic DNA contamination with 1 μl of DNaseI (Promega) treatment for 60 minutes at 37 ̊C, then with DNase Stop solution for 10 minutes at 65 ̊C. 1μg of RNA was used for cDNA synthesis using the iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad) following the manufacturer’s protocol. RT-qPCR was performed using IQ-SYBR Green Supermix (Bio-Rad) with the Bio-Rad CFX96 Real-Time System. Reactions consisted of 7.5 μl of IQ SYBR Green Supermix, 200 nM of forward and reverse primers, and 5 ng of cDNA, to a final volume of 15 μl. Following thermocycling, a melting curve analysis was performed using the default setting of CFX96 Real-Time System. Single melt peaks were observed for each targeted gene. The PCR efficiency for each primer pair was calculated from standard curves generated using serial dilutions: Eα-syn = 99.0%, Esnb-1 = 99.8%, Etba-1 = 99.5%, Eama-1 = 103.1%. The expression levels of α-syn were normalized to three reference genes: snb-1, tba-1, and ama-1. No template control (NTC) and no reverse transcriptase control (NRT) exhibited no amplification. All reference genes used were analyzed by geNorm (genorm.cmgg.be) and passed for target stability. Three independent biological replicates, with three technical replicates each, were tested for both worm strains. Data analysis was executed using the “do my qPCR calculation” web tool [35].
2.8. Quantification of ROS
A DCF-DA assay was completed with N2 (wildtype), xdh-1(ok3134), UA44 [Pdat-1::α-syn]and UA455 [Pdat-1::α-syn + xdh-1(ok3134)], animals seven days post-hatch. An acetylated form of fluorescein that is able to diffuse into the cell was used; once in the cell, the acetate group of this fluorescein are removed by cellular ROS, producing a fluorescent end-product [36]. Worms were washed three times prior to the start of the experiment. The assay was completed using a microplate reader, shaking at 37 degrees. 50uL of M9 and 50uL of 100uM DCF-DA were added to each sample, for a final concentration of 50uM DCF-DA. Plate spectroscopy readouts were recorded at an excitation of 485nm and an emission of 525 nm every 15 minutes for a total of 2.5 hours. Normalization of DCF-DA signal to cell population was conducted prior to the start of the assay. Three biological replicates and two technical replicates were completed for each worm strain.
2.9. Proteomic analysis of unedited and edited wht-2
Previously, two editing events in the wht-2 gene (IV: 1147035 and IV: 11473419), corresponding to nucleotides that are edited by ADR-2, were identified in mRNA from C. elegans neural cells [37]. To determine the impact of editing at these sites, adenosines were changed to guanosines since translational machinery treated inosines as guanosines. Using EMBOSS [38], the modified RNA sequence that reflects the impact of editing was translated in silico, and the edited and unedited amino acid sequences were aligned to determined differences between the two sequences. C. elegans wht-2 and human ABCG2 were also aligned using EMBOSS to determine whether the identified editing site is conserved in humans. AlphaFold [39] and ChimeraX [40] were used to visualize the impact of this amino acid change on the WHT-2 protein.
2.10. Statistical Analysis
For aggregate analyses, each worm was categorized as having either small, medium, or large, and either few, multiple or many aggregates. These categories were then quantified by multiplying by 1 for small or few, 2 for medium or multiple, and 3 for large or many to get a scaled score for size and number. These scores were added together to get an overall aggregate score, and the percent difference of each experiment compared to the empty vector control was determined. The top 14 hits were deemed candidates. For all neuronal analysis, the mean and standard error of the mean (SEM) were determined, and the data were analyzed via the Chi-Square test, unpaired T test, one-way ANOVA with Tukey’s post hoc test or two-way ANOVA with Sidak’s post hoc test (GraphPad Prism, version 8.0.1). Values below 0.05 were considered significant.
3. Results
3.1. Genes Regulated by ADR-2 Alter α-syn Misfolding.
169 genes were previously reported to be up- or downregulated in C. elegans mutant for adr-2; thus, expression of these genes is regulated by ADR-2 [25]. To identify presumptive effectors of the accumulation and misfolding of α-syn, genes regulated by ADR-2 with human orthologs were analyzed in an isogenic C. elegans strain in which α-syn and GFP are fused and co-expressed in the body wall muscle, the largest cell type in this species [41]. In such worms, misfolded α-syn is evident in the body wall and increases or decreases in aggregated α-syn can be easily evaluated. All genes with human orthologs were screened via RNAi in two-day old worms. 40 worms were subjected to RNAi in duplicate for each evaluated gene, and the body wall of individual worms were analyzed for aggregate size and number (Figure 1). The fourteen genes that resulted in the greatest change in aggregation were considered strong candidates for further examination for their role in PD. Among these genes, only F52E1.2 and fmo-5 are upregulated in the adr-2 mutant—all other genes are downregulated.
3.2. Modifiers of protein misfolding are associated with FAD and iron binding.
To functionally analyze the fourteen identified candidates, a gene enrichment analyses was performed to identify highly associated biological pathways, molecular mechanisms, and phenotypes (Figure 2A). Of these, the two most significant terms were flavin adenine dinucleotide (FAD) binding and iron ion binding (Figure 2B)—four genes were affiliated with FAD binding, and three were connected to iron binding. Notably, FAD and iron are essential for the function of complex II of the electron transport chain. In this step, the citric acid cycle intermediate succinate is oxidized to form fumarate; FAD accepts two electrons from this reaction, which are then passed to iron-sulfur clusters and subsequently to coenzyme Q. It has been shown that reactive oxygen species (ROS) can be produced at this site if electrons are delayed in the iron-sulfur clusters, or if electrons move in the reverse direction, toward complex I [42,43]. ROS have been tied to oxidative stress as they prompt dysfunction of the ubiquitin-proteasome system as well as mito- and autophagic-mechanisms, known contributors to PD [44]. Further, PD has been shown to be associated with increased expression of transferrin receptor 1 [45] and reduced expression of ferroportin-1 [46], leading to enhanced iron deposition. Moreover, α-syn has been shown to interact with genes associated with the import and export of iron, which detrimentally alters iron homeostasis in dopaminergic neurons [47].
3.3. Select modifiers of protein misfolding impact neurodegeneration when knocked down in dopamine neurons.
Other significant processes connected to these candidates include function of the NADH dehydrogenase complex, ATP synthesis coupled electron transport, and respirasome activity—all affiliated with the electron transport chain (Figure 2A). Since these genes are so strongly correlated with PD pathologies and altered protein-misfolding of α-syn, it was hypothesized that they likely represent functional effectors dopaminergic neurodegeneration. To determine the impact of the candidate genes on dopaminergic neuronal death, RNAi of each candidate gene was performed in an RNAi-sensitive worm strain in which knockdown is delimited to only the dopamine neurons—these worms also overexpress WT human α-syn and GFP, independently, under the control of the dopamine transporter (Pdat-1) promoter, and allow for age- and dose-dependent analysis of neurodegeneration {34] Following treatment, the four cephalic (CEP) and two anterior deirid (ADE) dopaminergic neurons of all worms in each population were analyzed via fluorescent microscopy, and individual worms were subsequently categorized as having 0 degenerated neurons, 1 degenerated neuron, 2 degenerated neurons, 3 degenerated neurons or 4-6 degenerated neurons. This experimental paradigm facilitated the examination of the neuronal health of the entire population of worms in each group (Figure 2C). Of the candidates, one gene, papl-1, enhanced neurodegeneration, while four, xdh-1, acdh-1, pho-1 and F52E1.2 were protective when knocked down (Table 1). Of the four protective genes, xdh-1, the ortholog of human xanthine dehydrogenase (XDH), was the most significant. Further, all the genes in which a decrease in expression led to a neuronal phenotype have a role in enzymatic activity. Of the candidates which were neuroprotective on knockdown, xdh-1 had the strongest protective effect. Additionally, RNAi of candidate genes was done in worms where knockdown occurs in all tissue types excluding neurons (Figure A1). xdh-1 was not protective in this strain, indicating that the protective phenotype is tissue specific, and a decrease in gene expression must occur in dopamine neurons for protection to result.
3.4. Loss of XDH leads to neuroprotection through the reduction of ROS.
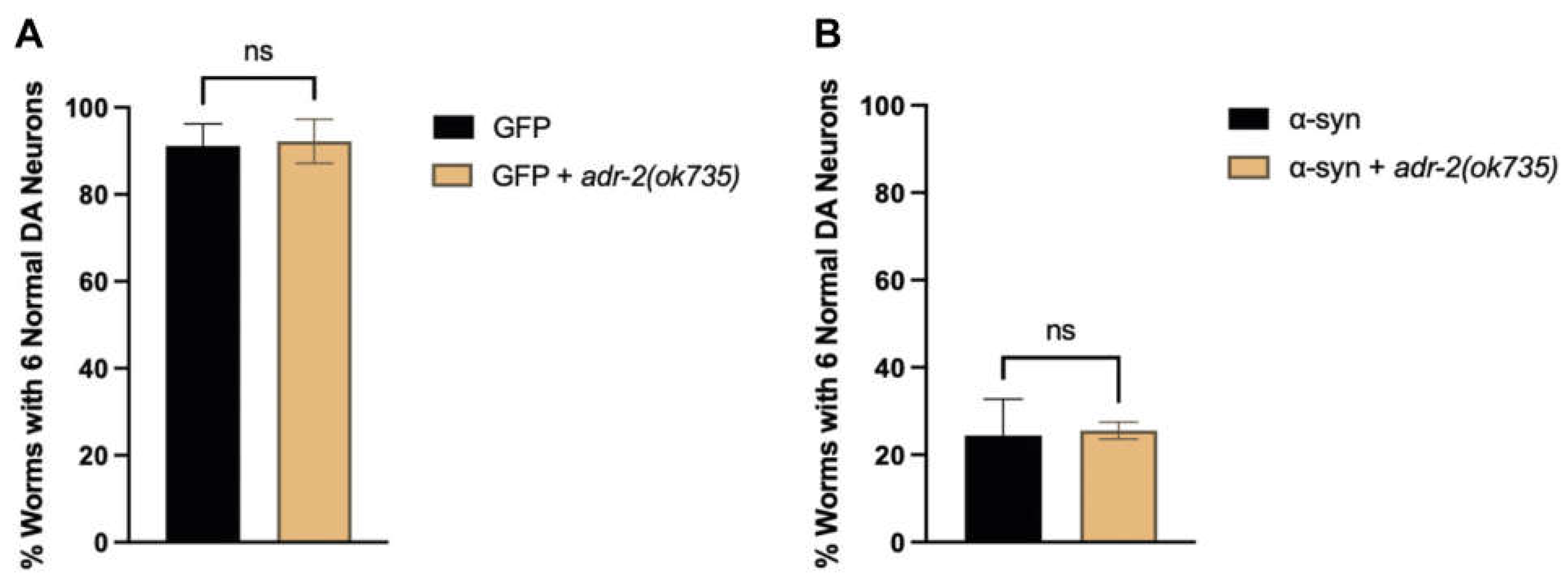
To further explore the relationship between xdh-1 and neuroprotection, worms overexpressing GFP and α-syn under the control of the DA neuron-specific dat-1 promoter were crossed to xdh-1 mutants. xdh-1 mutants displayed substantial protection against α-syn-induced neurodegeneration seven days post-hatch (Figure 3A-C). In fact, the number of xdh-1 mutants with six wildtype dopamine neurons was nearly double that of worms lacking the mutation. qPCR of α-syn was done to ensure that the observed neuroprotection was not due to silencing of the α-syn transgene (Figure A2). Additionally, xdh-1 mutants were crossed to worms expressing GFP alone under control of the dat-1 promoter; these worms did not display enhanced dopamine neurodegeneration (Figure A3). Thus, knockout of xdh-1 is robustly neuroprotective against α-syn toxicity but is not detrimental to dopamine neuronal health in the absence of α-syn. To determine the impact of increased xdh-1 expression on dopaminergic neuronal health, a transgenic worm strain in which dopaminergic overexpression of α-syn, GFP and xdh-1 was generated and subsequently analyzed for neurodegeneration. These worms exhibited significantly more neurodegeneration than worms only expressing α-syn and GFP in dopaminergic neurons four days post-hatch (Figure 3D-E).
Although xdh-1 is translated to form XDH, it regularly interconverts post-translationally to an alternate enzymatic product, xanthine oxidase (XO). This conversion may occur reversibly through the oxidation of cysteine to form disulfide bridges, or reversibly via proteolysis [26,48]. While the final product of purine catabolism by both XDH and XO is uric acid, XDH activity also generates NADH whereas XO function yields ROS (Figure 3F). Thus, it is plausible that worms mutant for xdh-1 exhibit neuroprotective phenotypes due to a reduction of ROS production. To confirm this possibility, an assay to measure ROS in worms expressing and not expressing α-syn in dopamine neurons with wildtype and mutant xdh-1 was completed seven days post-hatch. Indeed, worms harboring a deletion knockout in the xdh-1 locus exhibited a substantial decrease in ROS, conceivably leading to neuroprotection (Figure 3G). The opposite was found in animals not expressing α-syn: xdh-1(ok3134) mutants displayed enhanced ROS in comparison to wildtype worms, implying that ROS reduction in the xdh-1 mutants is α-syn specific.

Table 2.
Summary of hits of RNAi screen. Genes that produced a phenotype upon RNAi in dopaminergic neurons include xdh-1, pho-1, F52E1.2, and papl-1. The gene name, human orthologs [49], differential expression in adr-2 mutant worms [25], change in protein misfolding upon RNAi (Figure 1A), change in neurodegeneration upon RNAi (Figure 2C), and a general description of each gene are provided [50,51].
Table 2.
Summary of hits of RNAi screen. Genes that produced a phenotype upon RNAi in dopaminergic neurons include xdh-1, pho-1, F52E1.2, and papl-1. The gene name, human orthologs [49], differential expression in adr-2 mutant worms [25], change in protein misfolding upon RNAi (Figure 1A), change in neurodegeneration upon RNAi (Figure 2C), and a general description of each gene are provided [50,51].
| Gene Name | Human Ortholog(s) | Differential Expression in adr-2 Mutant |
Change in Protein Misfolding |
Change in Dopamine Neuron Degeneration |
Description |
|---|---|---|---|---|---|
| xdh-1 | XDH | Downregulated | +8.55% | Protective (p = 0.0002***) |
Catalyzes the final two steps of purine catabolism; low oxidase activity toward aldehydes; produces ROS. |
| pho-1 | ACP2, ACPT | Downregulated | +10.92% | Protective (p = 0.0198*) |
Converts orthophosphoric monoesters to alcohol and phosphate via hydrolysis; role in dephosphorylation. |
| F52E1.2 | CLEC4A, CLEC4C, CLEC4D, CLEC4E, CLEC6A, ASGR1 | Upregulated | +11.93% | Protective (p = 0.0452*) |
Promotes carbohydrate binding activity; involved in cell signaling, adhesion, glycoprotein degradation and production, inflammation, immune response. |
| papl-1 | ACP7 | Downregulated | +9.195% | Enhanced (p = 0.0361*) |
Promotes acid phosphatase activity; enables metal ion binding. |
3.5. A target of ADR-2 editing, wht-2, interacts in a network with xdh-1 to mediate PD-associated pathologies.
Although XDH-1 is downregulated in the absence of ADR-2 function, it is not edited [52]; thus, it is likely that the decrease in xdh-1 expression in adr-2 mutants is due to the interaction of XDH-1 with another protein that is a substrate of ADR-2. To determine which gene is edited by ADR-2 that interacts with XDH-1, a genetic interaction network that includes 18,183 predicted interactions in C. elegans [53] was cross-referenced with genes edited by ADR-2 [52]. Specifically, genes that interact with XDH-1 and are also targets of ADR-2 were identified. Only one gene, wht-2, encoding the worm ortholog of the human ABCG2 transporter, met these criteria. Consequently, wht-2 has previously been associated with gout [32], a condition that results from high levels of serum urate [54], an end-product of hypoxanthine or xanthine catabolism by XDH-1. Thus, wht-2 was deemed a promising link between ADR-2 and XDH-1.
To further elucidate the potential relationship between ADR-2, XDH-1 and WHT-2, RNAi of all three genes was performed in dopamine neurons and evaluated for neurodegeneration seven days post-hatch. xdh-1 knockdown was completed as proof of principle—as expected, roughly double the number of worms with decreased xdh-1 expression in dopaminergic neurons had six intact dopamine neurons seven days post-hatch than those in the control group (Figure 4A). However, knockdown of adr-2 in both models did not lead to a significant phenotype; this is surprising, since 169 genes are differentially expressed upon the loss of adr-2, including xdh-1. Further, it has been shown that loss of ADAR function in neurons is detrimental in other species; for example, ADAR2 null mice exhibit seizures and die prematurely [14]. To further explore this unexpected result, worms with GFP (Figure A4A) and those with α-syn and GFP (Figure A4B) expressed under the dat-1 promoter were crossed to adr-2 knockout worms. Upon analysis, it was confirmed that loss of ADR-2 is neither protective nor detrimental to dopamine neuron health in both the presence and absence of α-syn in C. elegans. Further, xdh-1 and adr-2 were simultaneously knocked down in dopamine neurons, and worms showed the same level of neuroprotection as xdh-1 knockdown alone (Figure 4A). Additionally, simultaneous “double-knockdown” of xdh-1 and wht-2 in dopamine neurons mirrored that of wht-2 alone. These data suggest that adr-2, wht-2 and xdh-1 act in the same pathway to confer neuroprotection.
This experimental paradigm was repeated in the protein misfolding assay with worms expressing α-syn and GFP in the body wall muscles. Knockdown of adr-2 did not alter protein misfolding (Figure 4B), while RNAi of xdh-1 and adr-2 + xdh-1 lead to comparable changes in protein misfolding. Similarly, knockdown of wht-2 and xdh-1 + wht-2 had the same results. These data indicate that these genes comprise a regulatory network that impacts PD pathologies in a tissue-specific manner. Upon editing by ADR-2, the structure of WHT-2 is best suited for the export of uric acid, a product of purine catabolism by XDH. In contrast, in the absence of editing, the structure of WHT-2 limits uric acid export. As a result, there is a downregulation of XDH transcription to limit the amount of uric acid produced and maintain cellular homeostasis. However, the elevation of uric acid is protective against dopaminergic neuron cell death by increasing aggregated α-syn, the less cytotoxic form of this protein.
3.6. A-to-I RNA editing of wht-2 is predicted to alter WHT-2 protein structure.
To further analyze the impact of RNA editing on the interaction between WHT-2 and XDH-1, a proteomic analysis of unedited and edited WHT-2 was performed. Previously, two sites in the wht-2 gene were identified as targets of RNA edited by ADARs, located at IV: 1147035 and IV: 11473419 in the C. elegans genome [37]. Because inosine is treated as guanosine by translational machinery, the RNA sequence of the wht-2 gene at the identified sites was changed to guanosine to reflect editing. The resulting sequence was translated, and the non-edited and edited amino acid sequences were compared. While the impact of editing at the second site is synonymous, editing at the first site results in a non-synonymous change from threonine to alanine at amino acid residue 124, located in the ABC-transporter domain of the protein (Figure 5A and Figure 5B). This analysis agrees with the proposed model in which it is hypothesized that WHT-2 protein structure changes upon editing, since threonine is a polar and alanine is non-polar. To determine whether the region of WHT-2 that is edited in C. elegans is conserved in humans, the DNA sequences of the two genes were aligned using EMBOSS Needle [38,55]. This analysis revealed that the genes are 41.7% homologous, and the region containing the editing site is highly conserved (Figure 5C). AlphaFold [39] and ChimeraX [40] were used to visualize this amino acid change and determine the potential functional impact of RNA editing at this site. The switch from threonine to alanine at WHT-2 residue 124 modifies hydrogen bonds in the region of the protein, thus affecting the overall structure of the protein (Figure 5D-E). Intriguingly, the substitution of threonine with alanine has been implicated in protein misfolding in the aggregation of proteins in PD and AD [56], in agreement with the results of wht-2 RNAi wht-2 in the worm body wall. These data further indicate that RNA editing impacts export of uric acid by WHT-2, and that this mechanism is likely present in human PD patients. Thus, we propose a model in which editing of wht-2 RNA leads to optimal uric acid export, the end-product of XDH-1 enzymatic activity. In contrast, in the absence of editing, uric acid cannot be efficiently exported, resulting in downregulation of XDH-1 to maintain cellular homeostasis (Figure 6).
4. Discussion
Despite the lack of complete understanding of the causative factors of PD, it is accepted that sporadic PD cases arise because of genetic and environmental factors. Indeed, rigorous work in both the clinical and laboratory settings have convincingly connected PD with processes such as oxidative stress and mitochondrial dysfunction. Moreover, numerous studies in various cellular and organismal models have implicated ADARs in neurological diseases, but few have directly correlated RNA editing with disease. Our present study builds upon this body of evidence by connecting ADARs to disease pathologies in multiple C. elegans models of PD, and by predicting the impact of editing at the protein level on α-syn protein misfolding and α-syn-induced neurodegeneration.
We have demonstrated that several genes regulated by ADARs are involved in α-syn misfolding and dopaminergic neuronal cell death, two pathological hallmarks of PD. We have shown that knockdown of these genes altered the misfolding of α-syn, and such modifiers of protein misfolding were most highly associated with FAD and iron ion binding. When knocked down in the dopamine neurons of worms co-expressing α-syn and GFP under the control of the Pdat-1 promoter, four presumptive modifiers of protein misfolding altered neurodegeneration: xdh-1, acdh-1, pho-1, F52E1.2 and papl-1. All these genes were protective when knocked down except papl-1, which enhanced dopaminergic neuronal cell death. Consistent with recent reports suggesting that monomeric and oligomeric α-syn is more cytotoxic than the aggregated form [57,58], the genes that were neuroprotective enhanced protein misfolding when knocked down. Further, we have shown that knockout of xdh-1, which conferred the strongest observed effect upon knockdown, was indeed protective in our neuronal PD model. Finally, we illustrate that WHT-2, which interacts with XDH-1 and is edited by ADR-2, appears to act in the same pathway as XDH-1 to protect dopaminergic neurons from α-syn-induced neurotoxicity. Concurrent knockdown of xdh-1 and wht-2 by RNAi indicates that the neuroprotection associated with loss or absence of xdh-1 is not contingent on adr-2 expression. Thus, we propose a model in which the absence of editing leads to ineffective WHT-2 export of uric acid, which is produced by XDH-1, in turn reducing the toxicity of α-syn and protecting dopaminergic neurons from degeneration (Figure 6).
XDH, the human ortholog of XDH-1, is responsible for the breakdown of hypoxanthine to xanthine and xanthine to uric acid, the final two steps of purine catabolism. Uric acid has been associated with neuroprotection, as it serves as a scavenger of free radicals, in turn protecting against ROS. Notably, higher serum urate has been associated with lower risk of PD and slower progression of PD [59]. Additionally, it has been demonstrated that uric acid inhibits ROS accumulation and improves mitochondrial function in rat hippocampal neurons [60]. Further, uric acid is a powerful iron chelator, and dysregulation with iron metabolism has been correlated with oxidative stress in PD [61]. Drugs that promote uric acid and iron accumulation have shown moderate success in clinical trials, but both strategies for treating PD have resulted in iatrogenic effects. For instance, uric acid accretion has resulted in kidney stones containing uric acid crystals [25,62] and iron chelation has propagated anemia [63]. Given that both iron and uric acid are associated with XDH activity, it is plausible that this enzyme is a promising therapeutic target for PD if its activity is regulated specifically in the SNpc.
ABCG2, the human ortholog of C. elegans WHT-2, exports uric acid, among many other endogenous and xenobiotic substrates. Our study illustrates that RNAi of wht-2 alone and wht-2 and xdh-1 simultaneously in dopamine neurons of worm expressing α-syn and GFP leads to comparable levels of neuroprotection. Thus, it is possible that XDH-1 activity is reduced to limit uric acid production when WHT-2 is dysfunctional, to avoid the deleterious effects of hyperuricemia. Both ABCG2 and XDH inhibition have been associated with hyperuricemia and gout, providing further evidence of our hypothesis that these proteins act in the same pathway. Moreover, simultaneous RNAi of both genes did not have an additive phenotypic effect in either of our PD models—additional affirmation of this conjecture.
Reports have indicated that the 3’ UTRs of many genes are edited by ADARs, as these regions contain Alu elements [64]. ABCG2 is known to be regulated by various miRNAs, including at the 3’ UTR. One study in the S1 colon cancer cell line revealed that a miRNA binds to the 3’ UTR of ABCG2, thereby decreasing expression [65]. miRNAs are known to repress translation, through various modes of regulation including preventing translational initiation, which does not necessarily impact transcription of the target gene [66]. It is possible that editing by ADARs inhibits the binding of this miRNA to the 3’ UTR of ABCG2, leading to increased ABCG2 translation. Such events could lead to an increase of XDH activity, as more uric acid produced by XDH could be excreted out of the cell by ABCG2. However, in the absence of ADAR activity, the putative miRNA can bind to the gene encoding ABCG2, leading to reduced translation of ABCG2 and the dampening of uric acid transport out of the cell. This would potentially lead to a decrease in XDH expression to maintain cellular uric acid homeostasis (Figure 6). This prospective mechanism would explain why xdh-1 is downregulated upon loss of adr-2 in C. elegans, while wht-2 mRNA levels are not altered [25]. Of course, this hypothesis must be validated experimentally.
It is interesting that knockdown of xdh-1 and wht-2 lead to enhanced neuroprotection and increased protein misfolding. It is possible that the intracellular elevation in uric acid is protective against dopaminergic neuronal cell death by increasing the aggregation of α-syn, the less cytotoxic form of this protein. However, whether increased misfolding is associated with enhanced or diminished disease progression remains controversial. It has been shown that soluble α-syn monomers and oligomers, which precede fibril formation, are more cytotoxic than larger inclusions due to their propensity to disrupt cell membrane permeability and promote neuroinflammation [57]. Further, analysis of post-mortem brain tissue has indicated that more monomers and oligomers are found in PD patients in comparison to age-matched controls [67]. Contrastingly, other researchers have associated increased protein misfolding with more severe disease [68,69]. While our results suggest that enhanced misfolding of α-syn is commensurate to increased dopaminergic neuronal health, these data are due to gene expression in the body wall muscle. RNAi of xdh-1 was cell autonomous, and it is possible that RNAi exclusively in the body wall may yield different results. The generation of a model that expresses α-syn and GFP in the body wall muscles and is RNAi sensitive solely in the body wall muscles, in addition to more studies on the role of α-syn misfolding in PD, have the potential to resolve this issue.
Notably, loss of ADR-2 activity did not impact α-syn-induced neurodegeneration, especially due to supporting evidence that aberrant ADAR activity has detrimental neurological effects across species, including C. elegans. For example, it has been shown that C. elegans that lack editing by ADARs exhibit reduced chemotaxis [23]. However, chemotaxis in C. elegans is regulated by ASE sensory neurons, not dopaminergic neurons [70]. Since dopamine neurons were the only neuronal cell type evaluated in our study, it is possible that aberrant ADR-2 activity does indeed impact the health of other neuronal cell types. Further, it is surprising that loss of ADR-2 did not impact aggregation of α-syn. This may be due to a lack of expression of adr-2 in the body wall—high levels of ADR-2 activity have been reported in the C. elegans nervous system specifically [22,25], but expression patterns in other tissues have not been extensively studied. It is also possible that the absence of observable phenotypes attributable to adr-2 deletion in neurons and body wall muscles expressing α-syn may be an additive effect of changes in expression of all targets of ADARs—simply put, some genes may lead to enhanced neurodegeneration or misfolding, or vice versa, cumulatively leading to no net change.
Here, we describe a relationship between ADR-2, WHT-2, and XDH-1 that impacts PD pathologies. Further, we identify precisely how RNA editing by ADARs affects the folding of the WHT-2 protein, presumably changing xdh-1 expression. While additional work is needed to further elucidate this mechanism, the targeting of ABCG2, the human ortholog of WHT-2, by ADARs may serve as a promising therapeutic strategy for the treatment of PD. Because ADARs target many genes in different tissues, the targeted altering of editing or edited substrate proteins, opposed to a complete abolishment of editing, is presumably a better approach. Nonetheless, RNA editing represents an exciting area of enquiry with substantial implications for biomedical research. The foundational studies on ADAR function in C. elegans have established this system as an exceptional model for investigation of these transformative enzymes as putative modifying factors in neurological diseases such as PD [20,71]. Moreover, the cost-effective application of C. elegans towards translational goals is expanding as an outcome of the increasingly proven preclinical utility of worm disease models [72,73,74].
Supplementary Materials
All study data are included in the article or in Appendix A.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, L.A.S., L.A.B., K.A.C. and G.A.C.; methodology, L.A.S., L.A.B., K.A.C. and G.A.C..; software, L.A.S.; validation, L.A.S., K.A.C., and G.A.C..; formal analysis, L.A.S., K.A.C. and G.A.C.; investigation, L.A.S., L.E.M., K.P., and L.M.S. ; resources, K.A.C. and G.A.C..; data curation; writing—original draft preparation, L.A.S..; writing—review and editing, L.A.S., K.A.C., and G.A.C.; visualization, L.A.S. and K.A.C..; supervision, K.A.C. and G.A.C..; project administration, L.A.S., K.A.C., and G.A.C..; funding acquisition, L.A.S., L.E.M., K.A.C., G.A.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by a travel award from The Company of Biologists and by a summer teaching release award from The University of Alabama Department of Biological Sciences to L.A.S; a summer research fellowship from the Parkinson’s Foundation supported L.E.M.; K.P. and L.M.S. were supported by a gift from the Hill Crest Foundation.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
We wish to thank Heather A. Hundley and the Hundley Lab for sharing their expertise on ADARs, and Roger L. Chang and the Chang Lab for their insight on proteomic analysis. Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). We also thank Wormbase. We would like to acknowledge all members of the Caldwell Lab, especially Jennifer L. Thies and Anthony L. Gaeta for their scientific insights. We are also grateful to Bradley A. Deem for editorial assistance.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Figure A1.
Analysis of neuronal health in worm populations upon RNAi of candidate genes in all tissue types excluding neurons. Candidate genes were knocked down in all cell types excluding neurons of C. elegans expressing α-syn and GFP under control of the Pdat-1 promoter. Subsequent scoring of dopamine neuron health and worm population analysis indicate that RNAi of F52E1.2 was robustly protective. Knockdown of ugt-16 was substantially protective, while RNAi of folt-2, fmo-5 and acdh-1 were significantly protective against α-syn induced dopamine neurodegeneration. No genes enhanced dopamine neuron death when knocked down. Synchronized worms were analyzed as 7-day old adults (three independent experiments, n = 30 per independent groups). Chi-Square analysis was performed where each candidate gene knockdown value was compared to EV and are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30).
Figure A1.
Analysis of neuronal health in worm populations upon RNAi of candidate genes in all tissue types excluding neurons. Candidate genes were knocked down in all cell types excluding neurons of C. elegans expressing α-syn and GFP under control of the Pdat-1 promoter. Subsequent scoring of dopamine neuron health and worm population analysis indicate that RNAi of F52E1.2 was robustly protective. Knockdown of ugt-16 was substantially protective, while RNAi of folt-2, fmo-5 and acdh-1 were significantly protective against α-syn induced dopamine neurodegeneration. No genes enhanced dopamine neuron death when knocked down. Synchronized worms were analyzed as 7-day old adults (three independent experiments, n = 30 per independent groups). Chi-Square analysis was performed where each candidate gene knockdown value was compared to EV and are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30).
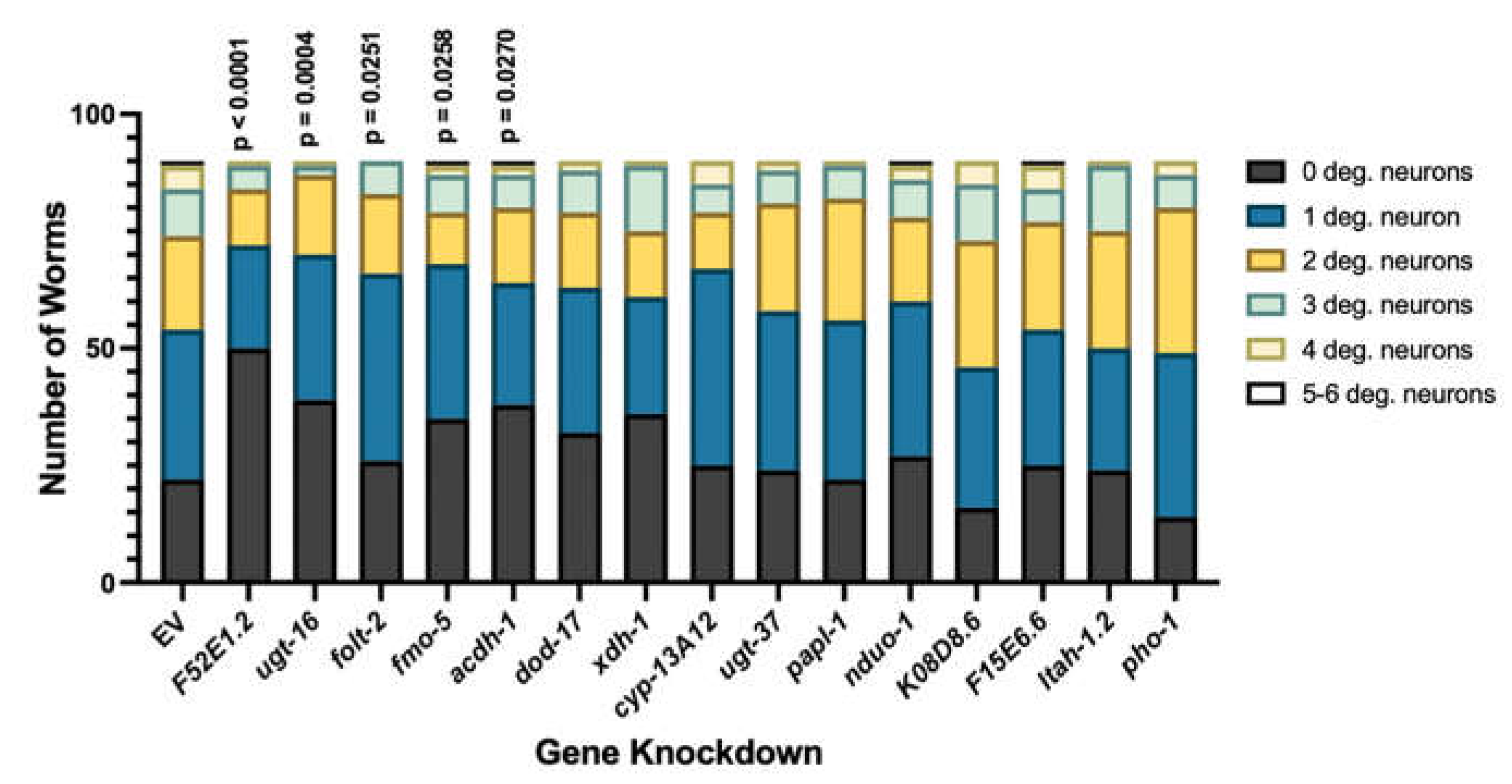
Figure A2.
qPCR of α-syn in worms independently expressing α-syn and GFP under control of the Pdat-1 promoter with and without functional xdh-1 seven days post-hatch. α-syn expression was normalized to three housekeeping genes: snb-1, tba-1, and ama-1. (Three independent experiments, n = 30 per independent groups, three technical replicates). Worms with the xdh-1(ok3134) mutation did not show a difference in α-syn expression compared to those with wildtype xdh-1 in the α-syn background. mRNA data analyzed by Student’s t-test and presented as mean +/- SEM of three biological replicates, with three technical replicates each; at least 100 animals were used for each replicate. These data were normalized to the α-syn control.
Figure A2.
qPCR of α-syn in worms independently expressing α-syn and GFP under control of the Pdat-1 promoter with and without functional xdh-1 seven days post-hatch. α-syn expression was normalized to three housekeeping genes: snb-1, tba-1, and ama-1. (Three independent experiments, n = 30 per independent groups, three technical replicates). Worms with the xdh-1(ok3134) mutation did not show a difference in α-syn expression compared to those with wildtype xdh-1 in the α-syn background. mRNA data analyzed by Student’s t-test and presented as mean +/- SEM of three biological replicates, with three technical replicates each; at least 100 animals were used for each replicate. These data were normalized to the α-syn control.
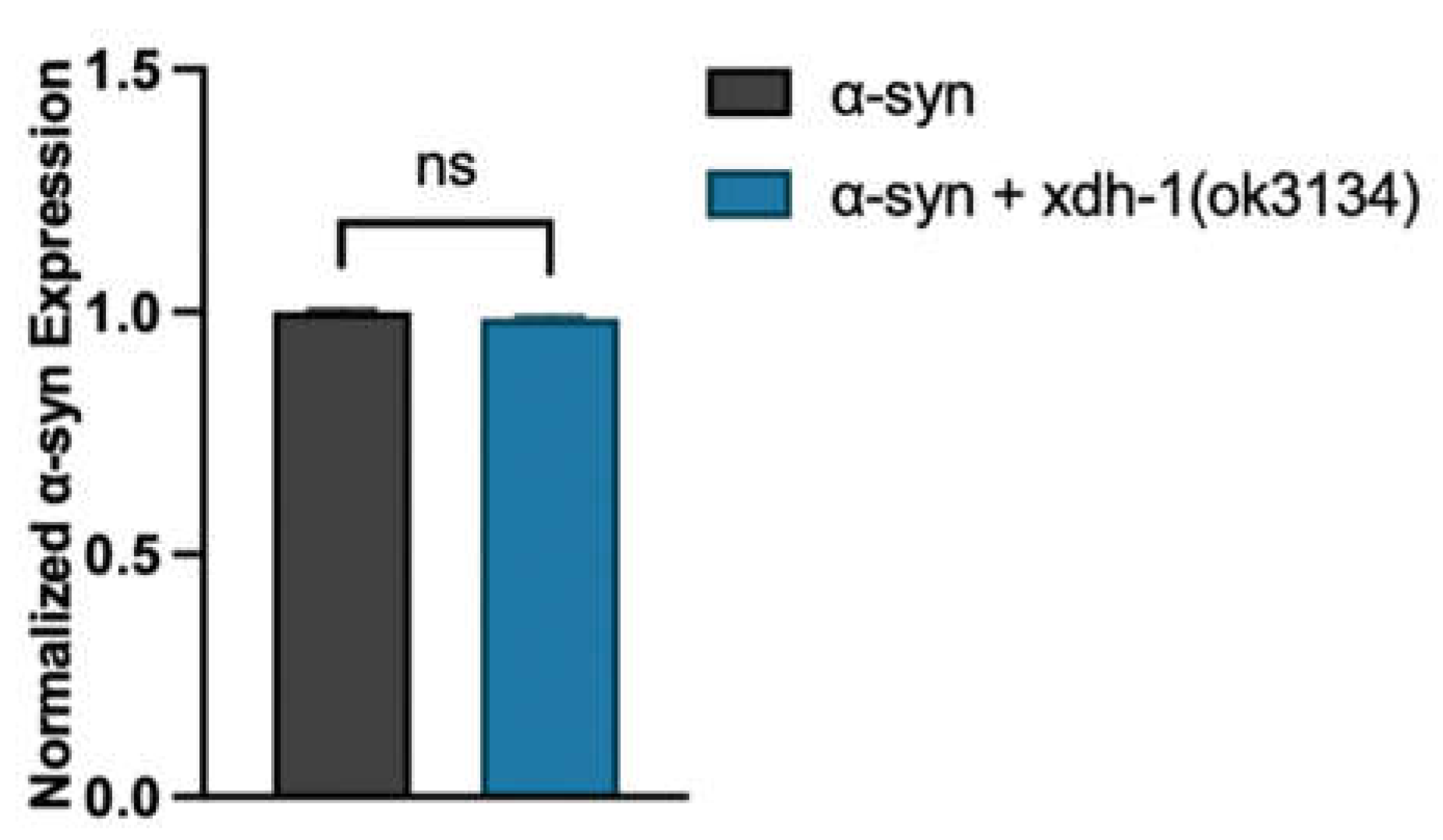
Figure A3.
Knockout of xdh-1 in Pdat-1 ::GFP worms does not impact dopaminergic neuron health. Animals with wildtype or mutant xdh-1 were crossed to isogenic worms expressing only GFP in dopamine neurons under control of Pdat-1. The four CEP and two ADE dopaminergic neurons were analyzed for the presence or absence of intact cell bodies and axonal processes in synchronized worms seven days post-hatch (three independent experiments, n = 30 per independent groups). Worms with the xdh-1(ok3134) mutation do not show a change in dopaminergic neuronal health. Student’s t-test where GFP + xdh-1(ok3134) was compared to GFP alone (three independent experiments, n = 30 per independent groups; N=3; n=30); ns >0.05.
Figure A3.
Knockout of xdh-1 in Pdat-1 ::GFP worms does not impact dopaminergic neuron health. Animals with wildtype or mutant xdh-1 were crossed to isogenic worms expressing only GFP in dopamine neurons under control of Pdat-1. The four CEP and two ADE dopaminergic neurons were analyzed for the presence or absence of intact cell bodies and axonal processes in synchronized worms seven days post-hatch (three independent experiments, n = 30 per independent groups). Worms with the xdh-1(ok3134) mutation do not show a change in dopaminergic neuronal health. Student’s t-test where GFP + xdh-1(ok3134) was compared to GFP alone (three independent experiments, n = 30 per independent groups; N=3; n=30); ns >0.05.
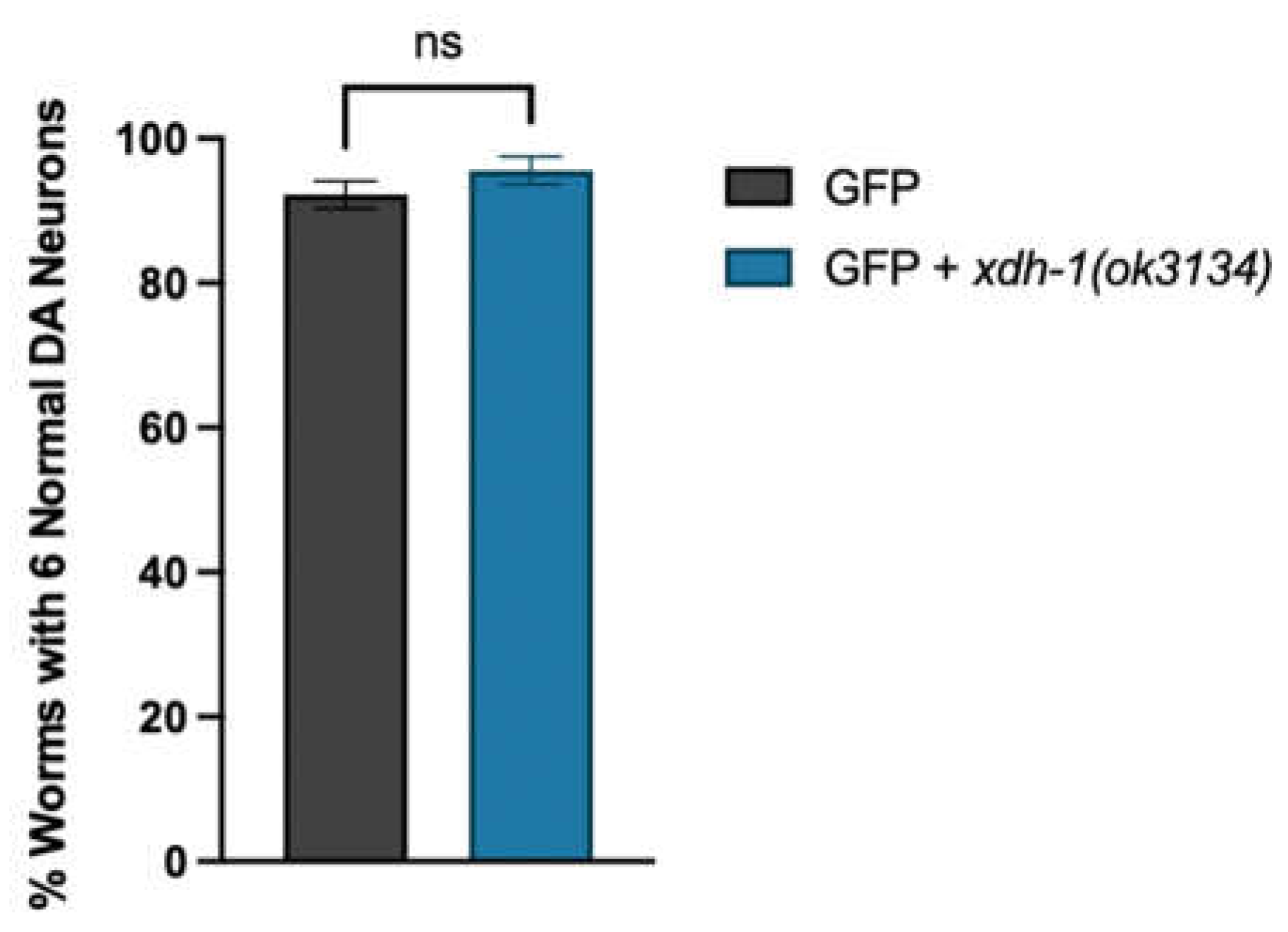
Figure A4.
Knockout of adr-2 does not impact dopaminergic neuronal health. (A) Animals with wildtype or mutant adr-2 were crossed to isogenic worms expressing GFP in dopamine neurons, and GFP + α-syn, independently, under control of the Pdat-1 promoter. The four CEP and two ADE dopaminergic neurons were analyzed for the presence or absence of intact cell bodies and axonal processes in synchronized worms seven days post-hatch (three independent experiments, n = 30 per independent groups). Worms with the adr-2(ok735) mutation do not show a change in dopaminergic neuronal health in both the absence (A) or presence (B) of α-syn. Student’s t-test where GFP + adr-2(ok735) was compared to GFP alone (three independent experiments, n = 30 per independent groups; N=3; n=30) in the absence (A) or presence (B) of α-syn; ns >0.05.
Figure A4.
Knockout of adr-2 does not impact dopaminergic neuronal health. (A) Animals with wildtype or mutant adr-2 were crossed to isogenic worms expressing GFP in dopamine neurons, and GFP + α-syn, independently, under control of the Pdat-1 promoter. The four CEP and two ADE dopaminergic neurons were analyzed for the presence or absence of intact cell bodies and axonal processes in synchronized worms seven days post-hatch (three independent experiments, n = 30 per independent groups). Worms with the adr-2(ok735) mutation do not show a change in dopaminergic neuronal health in both the absence (A) or presence (B) of α-syn. Student’s t-test where GFP + adr-2(ok735) was compared to GFP alone (three independent experiments, n = 30 per independent groups; N=3; n=30) in the absence (A) or presence (B) of α-syn; ns >0.05.
References
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, Genotype, and Worldwide Genetic Penetrance of LRRK2-Associated Parkinson’s Disease: A Case-Control Study. The Lancelet Neurology 2008. [CrossRef] [PubMed]
- Mythri, R.B.; Jagatha, B.; Pradhan, N.; Andersen, J.; Bharath, M.M.S. Mitochondrial Complex I Inhibition in Parkinson’s Disease: How Can Curcumin Protect Mitochondria? Antioxidants and Redox Signaling 2007, 9. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V. Oxidative Stress and Parkinson’s Disease. Frontiers in Neuroanatomy 2015. [CrossRef]
- Cook, C.; Stetler, C.; Petrucelli, L. Disruption of Protein Quality Control in Parkinson’s Disease. Cold Spring Harbor Perspective Medicine 2012. [CrossRef]
- Hattori, N.; Tanaka, M.; Ozawa, T.; Mizuno, Y. Immunohistochemical Studies on Complexes I, II, III, and IV of Mitochondria in Parkinson’s Disease. Annals of Neurology 1991. [CrossRef]
- Hattingen, E.; Magerkurth, J.; Pilatus, U.; Mozer, A.; Seifried, C.; Steinmetz, H.; Zanella, F.; Hilker, R. Phosphorus and Proton Magnetic Resonance Spectroscopy Demonstrates Mitochondrial Dysfunction in Early and Advanced Parkinson’s Disease. Brain 2009. [CrossRef]
- Sveinbjornsdottir, S. The Clinical Symptoms of Parkinson’s Disease. Journal of Neurochemistry 2016. [CrossRef] [PubMed]
- Li, S.; Jia, C.; Li, T.; Le, W. Hot Topics in Recent Parkinson’s Disease Research: Where We Are and Where We Should Go. Neuroscience Bulletin 2021. [CrossRef]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; KA, M.-Z. Parkinson’s Disease; Subcellular Biochemistry, 2012;
- Wang, I.X.; So, E.; Devlin, J.L.; Zhao, Y.; Wu, M.; Cheung, V.G. ADAR Regulates RNA Editing, Transcript Stability, and Gene Expression. Cell Reports 2013, 5. [Google Scholar] [CrossRef]
- Reich, D.P.; Tyc, K.M.; Bass, B.L. C. C. elegans ADARs Antagonize Silencing of Cellular DsRNAs by the Antiviral RNAi Pathway. Genes and Development 2018. [CrossRef]
- Morse, D.P.; Aruscavage, P.J.; Bass, B.L. RNA Hairpins in Noncoding Regions of Human Brain and Caenorhabditis elegans mRNA Are Edited by Adenosine Deaminases That Act on RNA. PNAS 2002. [CrossRef]
- Yang, Y.; Okada, S.; Sakurai, M. Adenosine-to-Inosine RNA Editing in Neurological Development and Disease. RNA Biology 2021. [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point Mutation in an AMPA Receptor Gene Rescues Lethality in Mice Deficient in the RNA-Editing Enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Khan, A.; Paro, S.; McGurk, L.; Sambrani, N.; Hogg, M.C.; Brindle, J.; Pennetta, G.; Keegan, L.P.; O’Connell, M.A. Membrane and Synaptic Defects Leading to Neurodegeneration in Adar Mutant Drosophila Are Rescued by Increased Autophagy. BMC Biology 2020, 18, 15. [Google Scholar] [CrossRef]
- Pozdyshev, D.V.; Zharikova, A.A.; Medvedeva, M.V.; Muronetz, V.I. Differential Analysis of A-to-I mRNA Edited Sites in Parkinson’s Disease. Genes 2021, 13, 14. [Google Scholar] [CrossRef]
- Calahorro, F.; Ruiz-Rubio, M. Caenorhabditis elegans as an Experimental Tool for the Study of Complex Neurological Diseases: Parkinson’s Disease, Alzheimer’s Disease and Autism Spectrum Disorder. Invertebrate Neuroscience 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling Neurodegeneration in Caenorhabditis elegans. Disease Models & Mechanisms 2020, 13, dmm046110. [Google Scholar] [CrossRef]
- Towlson, E.K.; Vértes, P.E.; Ahnert, S.E.; Schafer, W.R.; Bullmore, E.T. The Rich Club of the C. elegans Neuronal Connectome. Journal of Neuroscience 2013, 33. [Google Scholar] [CrossRef]
- Tonkin, L.A.; Saccomanno, L.; Morse, D.P.; Brodigan, T.; Krause, M.; BL, B. RNA Editing by ADARs Is Important for Normal Behavior in Caenorhabditis elegans. EMBO Journal 2002. [CrossRef] [PubMed]
- Washburn, M.C.; Kakaradov, B.; Sundararaman, B.; Wheeler, E.; Hoon, S.; Yeo, G.W.; HA, H. The DsRBP and Inactive Editor ADR-1 Utilizes DsRNA Binding to Regulate A-to-I RNA Editing across the C. elegans Transcriptome. elegans Transcriptome. Cell Reports 2014. [CrossRef]
- Rajendren, S.; Manning, A.C.; Al-Awadi, H.; Yamada, K.; Takagi, Y.; HA, H. A Protein-Protein Interaction Underlies the Molecular Basis for Substrate Recognition by an Adenosine-to-Inosine RNA-Editing Enzyme. Nucleic Acids Research 2018. [CrossRef]
- Deffit, S.N.; Yee, B.A.; Manning, A.C.; Rajendren, S.; Vadlamani, P.; Wheeler, E.C.; Domissy, A.; Washburn, M.C.; Yeo, G.W.; HA, H. The C. elegans neural editome reveals an ADAR target mRNA required for proper chemotaxis. eLife 2017. [CrossRef]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine dehydrogenase/xanthine oxidase and oxidative stress 1997, 20, 127-140. [CrossRef]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine Oxidoreductase: One Enzyme for Multiple Physiological Tasks. Redox Biology 2021, 41, 101882. [Google Scholar] [CrossRef]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian Xanthine Oxidoreductase - Mechanism of Transition from Xanthine Dehydrogenase to Xanthine Oxidase. FEBS Journal 2008, 275, 3278–3289. [Google Scholar] [CrossRef]
- Furuhashi, M. New Insights into Purine Metabolism in Metabolic Diseases: Role of Xanthine Oxidoreductase Activity. American Journal of Physiology - Endocrinology and Metabolism 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Kim, W.S.; Weickert, C.S.; Garner, B. Role of ATP-Binding Cassette Transporters in Brain Lipid Transport and Neurological Disease; Journal of Neurochemistry, 2008, 104,1145-1166. [CrossRef]
- Kobuchi, H.; Moriya, K.; Ogino, T.; Fujita, H.; Inoue, K.; Shuin, T.; Yasuda, T.; Utsumi, K.; Utsumi, T. Mitochondrial Localization of ABC Transporter ABCG2 and Its Function in 5-Aminolevulinic Acid-Mediated Protoporphyrin IX Accumulation. PLOS One 2012. [CrossRef]
- Kukal, S.; Guin, D.; Rawat, C.; Bora, S.; Mishra, M.K.; Sharma, P.; Paul, P.R.; Kanojia, N.; Grewal, G.K.; Kukreti, S.; et al. Multidrug Efflux Transporter ABCG2: Expression and Regulation. Cell and Molecular Life Sciences 2021. [CrossRef]
- Stiernagle, T. Maintenance of C. elegans. WormBook : the online review of C. elegans biology 2006. [CrossRef]
- Angeles-Albores, D.; Ryn, L.; Chan, J.; PW, S. Tissue Enrichment Analysis for C. elegans Genomics. elegans Genomics. BMC Bioinformatics 2016. [CrossRef] [PubMed]
- Timmons, L.; Court, D.L.; Fire, A. Ingestion of Bacterially Expressed dsRNAs Can Produce Specific and Potent Genetic Interference in Caenorhabditis elegans. Gene 2001, 263. [Google Scholar] [CrossRef] [PubMed]
- Harrington, AJ; Knight, AL; Caldwell, GA; Caldwell, KA. Caenorhabditis elegans as a Model System for Identifying Effectors of α-Synuclein Misfolding and Dopaminergic Cell Death Associated with Parkinson’s Disease. Methods 53, 220–225. [CrossRef]
- Tournayre, J.; Reichstadt, M.; Parry, L.; Fafournoux, P.; Jousse, C. “Do My QPCR Calculation”, a Web Tool. Bioinformation 2019, 15, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, D.; Asaduzzaman, M.; Young, F. Real Time Monitoring and Quantification of Reactive Oxygen Species in Breast Cancer Cell Line MCF-7 by 2′,7′–Dichlorofluorescin Diacetate (DCFDA) Assay. Journal of Pharmacological and Toxicological Methods 2018, 94. [Google Scholar] [CrossRef]
- Goldstein, B.; Agranat-Tamir, L.; Light, D.; Ben-Naim Zgayer, O.; Fishman, A.; Lamm, A.T. A-to-I RNA Editing Promotes Developmental Stage–Specific Gene and LncRNA Expression. Genome Research 2017, 27, 462–470. [Google Scholar] [CrossRef]
- Rice EMBOSS: The European Molecular Biology Open Software Suite (2000) Rice, P. Longden, I. and Bleasby, A. Trends in Genetics. 16, pp. 276--277.
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Research 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Science 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-Based RNAi Screen Identifies Neuroprotective Genes in a Parkinson’s Disease Model. PNAS 2008, 105, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Larosa, V.; Remacle, C. Insights into the Respiratory Chain and Oxidative Stress. Bioscience Reports 2018, 38, BSR20171492. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Sruti Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biology 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M. The Role of Oxidative Stress in Parkinson’s Disease. 2013, 3, 461-491. [CrossRef]
- Mash, D.C.; Pablo, J.; Buck, B.E.; Sanchez-Ramos, J., Weiner, W.J. Distribution and Number of Transferrin Receptors in Parkinson’s Disease and in MPTP-Treated Mice. Experimental Neurology 1991. 114, 73-81. [CrossRef]
- Song, N.; Wang, J.; Jiang, H.; Xie, J. Ferroportin 1 but Not Hephaestin Contributes to Iron Accumulation in a Cell Model of Parkinson’s Disease. Free Radical Biology & Medicine 2010, 48, 332–241. [Google Scholar] [CrossRef]
- Patel, D.; Xu, C.; Nagarajan, S.; Liu, Z.; Hemphill, W.O.; Shi, R.; Uversky, V.N.; Caldwell, G.A.; Caldwell, K.A.; Witt, S.N. Alpha-Synuclein Inhibits Snx3–Retromer-Mediated Retrograde Recycling of Iron Transporters in S. cerevisiae and C. elegans Models of Parkinson’s Disease. Human Molecular Genetics 2018, 27, 1514–1532. [Google Scholar] [CrossRef]
- Della Corte, E.; Stirpe, F. The Regulation of Rat Liver Xanthine Oxidase. Involvement of thiol groups in the conversion of the enzyme activity from dehydrogenase (type D) into oxidase (type O) and purification of the enzyme 1972, 2, 83-84. [CrossRef]
- Shaye, D.D.; Greenwald, I. Ortholist: A Compendium of C. elegans Genes with Human Orthologs. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Research 2022, 50. [Google Scholar] [CrossRef]
- Howe, K.; Davis, P.; Paulini, M.; Tuli, M.A.; Williams, G.; Yook, K.; Durbin, R.; Kersey, P.; Sternberg, P.W. WormBase. Worm 2012, 1. [Google Scholar] [CrossRef]
- Deffit, S.N.; Yee, B.A.; Manning, A.C.; Rajendren, S.; Vadlamani, P.; Wheeler, E.C.; Domissy, A.; Washburn, M.C.; Yeo, G.W.; Hundley, H.A. The C. elegans Neural Editome Reveals an ADAR Target MRNA Required for Proper Chemotaxis. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Sternberg, P.W. Genome-Wide Prediction of C. elegans Genetic Interactions. Science 2006, 311, 1481–1484. [Google Scholar] [CrossRef]
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. The Lancet 2021, 397. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI Bioinformatics Web and Programmatic Tools Framework. Nucleic Acids Research 2015, 43, W580–W584. [Google Scholar] [CrossRef]
- Podoly, E.; Hanin, G.; Soreq, H. Alanine-to-Threonine Substitutions and Amyloid Diseases: Butyrylcholinesterase as a Case Study. Chemico-Biological Interactions 2010, 187, 64–71. [Google Scholar] [CrossRef]
- Kim, D.H.; Jongchan, L.; Mok, K.H.; Lee, J.H.; Han, K.H. Salient Features of Monomeric Alpha-Syn Revealed by NMR Spectroscopy. Biomolecules 2020, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, K. Monomeric α-Synuclein Induces Blood-Brain Barrier Dysfunction through Activated Brain Pericytes Releasing Inflammatory Mediators in Vitro. Microvascular Research, 2019, 124, 61–66. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The Epidemiology of Parkinson’s Disease: Risk Factors and Prevention. Lancet Neurology 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Yu, Z.F.; Bruce-Keller, A.J.; Goodman, Y.; Mattson, M.P. Uric Acid Protects Neurons against Excitotoxic and Metabolic Insults in Cell Culture, and against Focal Ischemic Brain Injury in Vivo. Journal of Neuroscience Research 1998, 53, 613–625. [Google Scholar] [CrossRef]
- Ghio, A.J.; Kennedy, T.P.; Stonehuerner, J.; Carter, J.D.; Skinner, K.A.; Parks, D.A.; Hoidal, J.R. Iron Regulates Xanthine Oxidase Activity in the Lung. American Journal of Physiology - Lung Cellular and Molecular Physiology 2002, 283. [Google Scholar] [CrossRef] [PubMed]
- Investigators, S.-P.-. 3 Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 2021, 326, 926–939. [Google Scholar] [CrossRef]
- Devos, D.; Cabantchik, Z.I.; Moreau, C.; Daniel, V.; Mahoney-Sanchez, L.; Bouchaoui, H.; Gouel, F.; Rolland, A.S.; Duce, J.A.; Devedijan, J.C.; et al. Conservative Iron Chelation for Neurodegenerative Diseases Such as Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Journal of Neural Transmission 2020, 27, 189–203. [Google Scholar] [CrossRef]
- Gu, T.; Buaas, F.W.; Simons, A.K.; Ackert-Bicknell, C.L.; Braun, R.E.; Hibbs, M.A. Canonical A-to-I and C-to-U RNA Editing Is Enriched at 3’UTRs and MicroRNA Target Sites in Multiple Mouse Tissues. PLOS ONE 2012, 7, e33720. [Google Scholar] [CrossRef]
- To, K.; Zhan, Z.; Litman, T.; Bates, S.E. Regulation of ABCG2 Expression at the 3’ Untranslated Region of Its MRNA through Modulation of Transcript Stability and Protein Translation by a Putative MicroRNA in the S1 Colon Cancer Cell Line. Molecular and Cellular Biology, 28, 5147-5161. [CrossRef]
- Gu, S.; Kay, M. How Do miRNAs Mediate Translational Repression? Silence 2010, 1, 11. [Google Scholar] [CrossRef]
- Emin, D.; Zhang, Y.P.; Lobanova, E.; Miller, A.; Li, X.; Xia, Z.; Dakin, H.; Sideris, D.I.; Lam, J.Y.; Ranasinghe, R.T.; et al. Small Soluble α-Synuclein Aggregates Are the Toxic Species in Parkinson’s Disease. Nature Communications 2022, 13, 5512. [Google Scholar] [CrossRef] [PubMed]
- Mougenot, A.-L.; Nicot, S.; Bencsik, A.; Morignat, E.; Verchère, J.; Lakhdar, L.; Legastelois, S.; Baron, T. Prion-like Acceleration of a Synucleinopathy in a Transgenic Mouse Model. Neurobiology of Aging 2012, 33, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-Like Mechanisms in Parkinson’s Disease. Frontiers in Neuroscience 2019, 13. [Google Scholar] [CrossRef]
- Luo, L.; Wen, Q.; Ren, J.; Hendricks, M.; Gershow, M.; Qin, Y.; Greenwood, J.; Soucy, E.R.; Klein, M.; Smith-Parker, H.K.; et al. Dynamic Encoding of Perception, Memory, and Movement in a C. elegans Chemotaxis Circuit. Neuron 2014, 82, 1115–1128. [Google Scholar] [CrossRef]
- Erdmann, E.A.; Mahapatra, A.; Mukherjee, P.; Yang, B.; Hundley, H.A. To Protect and Modify Double-Stranded RNA – the Critical Roles of ADARs in Development, Immunity and Oncogenesis. Critical Reviews in Biochemistry and Molecular Biology 2021, 56, 54–87. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, A.L.; Caldwell, K.A.; Caldwell, G.A. Found in Translation: The Utility of C. elegans Alpha-Synuclein Models of Parkinson’s Disease. Brain Sciences 2019, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- Mew, M.; Caldwell, K.A.; Caldwell, G.A. From Bugs to Bedside: Functional Annotation of Human Genetic Variation for Neurological Disorders Using Invertebrate Models. Human Molecular Genetics 2022, 31, R37–R46. [Google Scholar] [CrossRef] [PubMed]
- P.A. Kropp; R. Bauer; I. Zafra; Golden, A. Caenorhabditis elegans for Rare Disease Modeling and Drug Discovery: Strategies and Strengths. Disease Models & Mechanisms 2021, 14, dmm049010. [CrossRef]
Figure 1.
RNAi of genes differentially regulated in adr-2 mutants alter protein misfolding. (A) Isogenic worm strain expressing α-syn::GFP in the body wall muscle cells of C. elegans were fed E. coli expressing candidate gene RNAi constructs and scored for aggregate size and number, which were combined to generate an overall aggregate score. Synchronized worms were analyzed two days post-hatch, and a percent change for each gene was calculated in comparison to empty vector control. The top 14 genes that resulted in the greatest cumulative percent change in aggregation are shown in red. (B) RNAi of xdh-1 (bottom) in worms expressing α-syn::GFP in the body wall muscles under control of the Punc-54 promoter showed increased aggregate size and number in comparison to empty vector control (top). Examples of scoring criteria: A = small aggregate, B = medium aggregate, C = large aggregate, D = nuclei of body wall muscle cell.
Figure 1.
RNAi of genes differentially regulated in adr-2 mutants alter protein misfolding. (A) Isogenic worm strain expressing α-syn::GFP in the body wall muscle cells of C. elegans were fed E. coli expressing candidate gene RNAi constructs and scored for aggregate size and number, which were combined to generate an overall aggregate score. Synchronized worms were analyzed two days post-hatch, and a percent change for each gene was calculated in comparison to empty vector control. The top 14 genes that resulted in the greatest cumulative percent change in aggregation are shown in red. (B) RNAi of xdh-1 (bottom) in worms expressing α-syn::GFP in the body wall muscles under control of the Punc-54 promoter showed increased aggregate size and number in comparison to empty vector control (top). Examples of scoring criteria: A = small aggregate, B = medium aggregate, C = large aggregate, D = nuclei of body wall muscle cell.
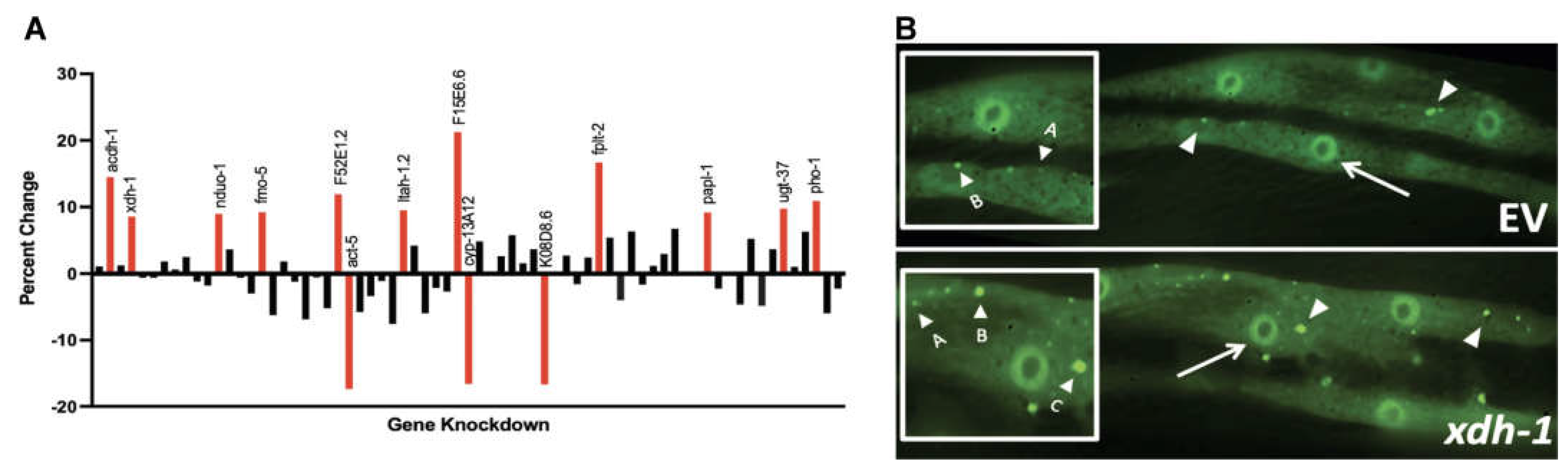
Figure 2.
ADR-2-regulated candidate modifiers of α-syn misfolding are most highly associated with FAD and iron ion binding and impact dopamine neuronal health. (A) Graphical representation of biological processes (www.wormbase.org), molecular functions and cellular components associated with genes differentially regulated in adr-2 mutants and alter α-syn misfolding. (B) GO terms enriched in candidate genes (C) Candidate genes were knocked down in dopaminergic neurons of C. elegans expressing α-syn and GFP under control of the Pdat-1 promoter. Subsequent scoring of dopaminergic neuronal health and worm population analysis indicate that RNAi of xdh-1, acdh-1, pho-1 and F52E1.2 were protective against α-syn induced dopaminergic neurodegeneration, while papl-1 knockdown enhanced dopaminergic neuronal death. Worms were analyzed as 7-day old adults. Chi-Square analysis was performed where each candidate gene knockdown value was compared to EV and are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30).
Figure 2.
ADR-2-regulated candidate modifiers of α-syn misfolding are most highly associated with FAD and iron ion binding and impact dopamine neuronal health. (A) Graphical representation of biological processes (www.wormbase.org), molecular functions and cellular components associated with genes differentially regulated in adr-2 mutants and alter α-syn misfolding. (B) GO terms enriched in candidate genes (C) Candidate genes were knocked down in dopaminergic neurons of C. elegans expressing α-syn and GFP under control of the Pdat-1 promoter. Subsequent scoring of dopaminergic neuronal health and worm population analysis indicate that RNAi of xdh-1, acdh-1, pho-1 and F52E1.2 were protective against α-syn induced dopaminergic neurodegeneration, while papl-1 knockdown enhanced dopaminergic neuronal death. Worms were analyzed as 7-day old adults. Chi-Square analysis was performed where each candidate gene knockdown value was compared to EV and are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30).
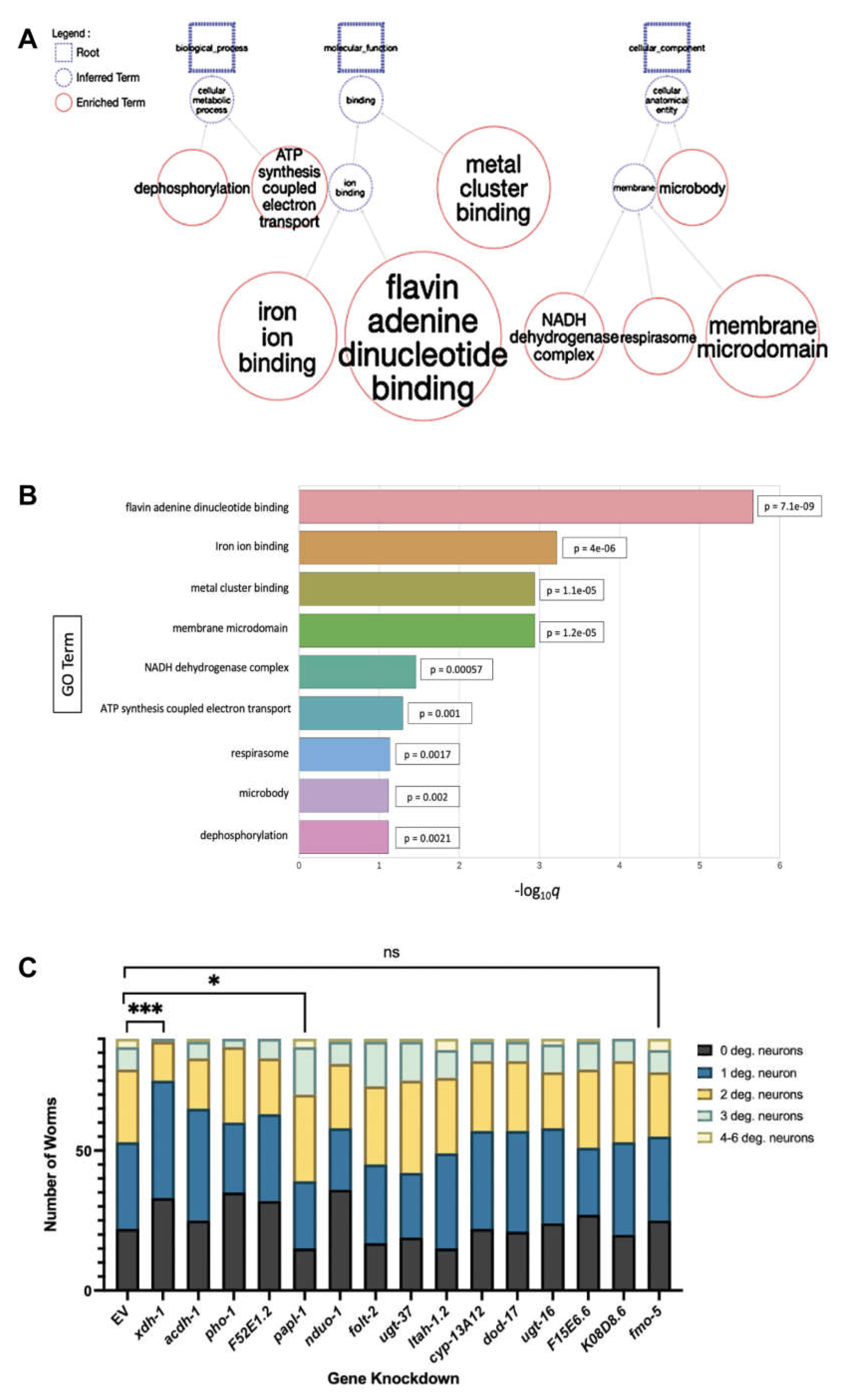
Figure 3.
XDH-1 modulates neuroprotection by impacting ROS production. (A) C. elegans expressing α-syn + GFP in dopaminergic neurons, wildtype, and mutant for xdh-1. Arrows represent wildtype dopamine neurons with a cell body and intact axonal process, and arrowheads represent degenerated dopamine neurons. The xdh-1 mutation confers protection against α-syn induced neurodegeneration. (B-C) Animals with wildtype or mutant xdh-1 were crossed to isogenic worms expressing α-syn + GFP in dopamine neurons. The four CEP and two ADE dopaminergic neurons were analyzed for the presence or absence of intact cell bodies and axonal processes in synchronized worms seven days post-hatch (three independent experiments, n = 30 per independent groups). Worms with the xdh-1(ok3134) mutation exhibit neuroprotection against α-syn. (B) The number of degenerated neurons in each individual worm of the total population. Chi-Square analysis was performed where α-syn + xdh-1(ok3134) was compared to α-syn alone and values are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30). (C) The percent of worms with 6 normal dopaminergic neurons. Student’s t-test where α-syn + xdh-1(ok3134) was compared to α-syn alone and values are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30). (D-E) Worms overexpressing xdh-1 under control of the Pdat-1 promoter were crossed to worms expressing α-syn + GFP in dopaminergic neurons. Subsequently, animals were synchronized and scored for neurodegeneration. Worms with increased xdh-1 expression in dopaminergic neurons showed decreased neuroprotection four days post-hatch. (D) The number of degenerated neurons in each individual worm of the total population. Chi square analysis was performed where α-syn + Pdat-1::xdh-1 was compared to α-syn alone and values are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30). (E) The percent of worms with 6 normal dopaminergic neurons. Student’s t-test where α-syn + Pdat-1::xdh-1 was compared to α-syn alone and values are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30). (F) In the purine catabolic pathway, xanthine dehydrogenase (XDH) and xanthine oxidase (XO) catalyze hypoxanthine to xanthine and xanthine to uric acid. XDH and XO are interconvertible and are encoded by a single gene. For XDH activity, NAD is required as a cofactor, and NADH and uric acid are produced. In contrast, molecular oxygen is needed for XO to function, and uric acid and superoxide anions result. Reactive oxygen species (ROS) contribute to mitochondrial dysfunction, oxidative stress, and ultimately cell death. Created with Biorender.com. (G) Wildtype N2 worms, xdh-1(ok3134) worms, and worms expressing α-syn in dopaminergic neurons with and without the xdh-1(ok3134) mutant, were treated with a specialized fluorescein that moves into the cell and promotes the removal of acetate groups by cellular esterase. The product of this can be oxidized by ROS to form a fluorescent molecule to allow for quantification of ROS. Plate spectroscopy readouts were recorded at an excitation of 485nm and an emission of 525 nm every 15 minutes for a total of 2.5 hours. Normalization of DCF-DA signal to cell population was conducted prior to the start of the assay. Three biological replicates and two technical replicates were completed for all worm strains. After 2.5 hours, worms expressing α-syn and GFP in dopaminergic neurons that are mutant for xdh-1 produce lower ROS than worms without the mutation. Since ROS contribute to oxidative stress, mitochondrial dysfunction, and ultimately neuronal death, this decrease in ROS in the xdh-1(ok3134) mutant could contribute to neuroprotection. xdh-1 worms lacking α-syn exhibited enhanced ROS production at 2.5 hours, indicating that changes in ROS production due to XDH/XO activity is α-syn specific. Significance was obtained using a One-way ANOVA with a Tukey’s post hoc test and are represented by exact p values (N= 3; n=30).
Figure 3.
XDH-1 modulates neuroprotection by impacting ROS production. (A) C. elegans expressing α-syn + GFP in dopaminergic neurons, wildtype, and mutant for xdh-1. Arrows represent wildtype dopamine neurons with a cell body and intact axonal process, and arrowheads represent degenerated dopamine neurons. The xdh-1 mutation confers protection against α-syn induced neurodegeneration. (B-C) Animals with wildtype or mutant xdh-1 were crossed to isogenic worms expressing α-syn + GFP in dopamine neurons. The four CEP and two ADE dopaminergic neurons were analyzed for the presence or absence of intact cell bodies and axonal processes in synchronized worms seven days post-hatch (three independent experiments, n = 30 per independent groups). Worms with the xdh-1(ok3134) mutation exhibit neuroprotection against α-syn. (B) The number of degenerated neurons in each individual worm of the total population. Chi-Square analysis was performed where α-syn + xdh-1(ok3134) was compared to α-syn alone and values are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30). (C) The percent of worms with 6 normal dopaminergic neurons. Student’s t-test where α-syn + xdh-1(ok3134) was compared to α-syn alone and values are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30). (D-E) Worms overexpressing xdh-1 under control of the Pdat-1 promoter were crossed to worms expressing α-syn + GFP in dopaminergic neurons. Subsequently, animals were synchronized and scored for neurodegeneration. Worms with increased xdh-1 expression in dopaminergic neurons showed decreased neuroprotection four days post-hatch. (D) The number of degenerated neurons in each individual worm of the total population. Chi square analysis was performed where α-syn + Pdat-1::xdh-1 was compared to α-syn alone and values are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30). (E) The percent of worms with 6 normal dopaminergic neurons. Student’s t-test where α-syn + Pdat-1::xdh-1 was compared to α-syn alone and values are represented by exact p values (three independent experiments, n = 30 per independent groups; N=3; n=30). (F) In the purine catabolic pathway, xanthine dehydrogenase (XDH) and xanthine oxidase (XO) catalyze hypoxanthine to xanthine and xanthine to uric acid. XDH and XO are interconvertible and are encoded by a single gene. For XDH activity, NAD is required as a cofactor, and NADH and uric acid are produced. In contrast, molecular oxygen is needed for XO to function, and uric acid and superoxide anions result. Reactive oxygen species (ROS) contribute to mitochondrial dysfunction, oxidative stress, and ultimately cell death. Created with Biorender.com. (G) Wildtype N2 worms, xdh-1(ok3134) worms, and worms expressing α-syn in dopaminergic neurons with and without the xdh-1(ok3134) mutant, were treated with a specialized fluorescein that moves into the cell and promotes the removal of acetate groups by cellular esterase. The product of this can be oxidized by ROS to form a fluorescent molecule to allow for quantification of ROS. Plate spectroscopy readouts were recorded at an excitation of 485nm and an emission of 525 nm every 15 minutes for a total of 2.5 hours. Normalization of DCF-DA signal to cell population was conducted prior to the start of the assay. Three biological replicates and two technical replicates were completed for all worm strains. After 2.5 hours, worms expressing α-syn and GFP in dopaminergic neurons that are mutant for xdh-1 produce lower ROS than worms without the mutation. Since ROS contribute to oxidative stress, mitochondrial dysfunction, and ultimately neuronal death, this decrease in ROS in the xdh-1(ok3134) mutant could contribute to neuroprotection. xdh-1 worms lacking α-syn exhibited enhanced ROS production at 2.5 hours, indicating that changes in ROS production due to XDH/XO activity is α-syn specific. Significance was obtained using a One-way ANOVA with a Tukey’s post hoc test and are represented by exact p values (N= 3; n=30).
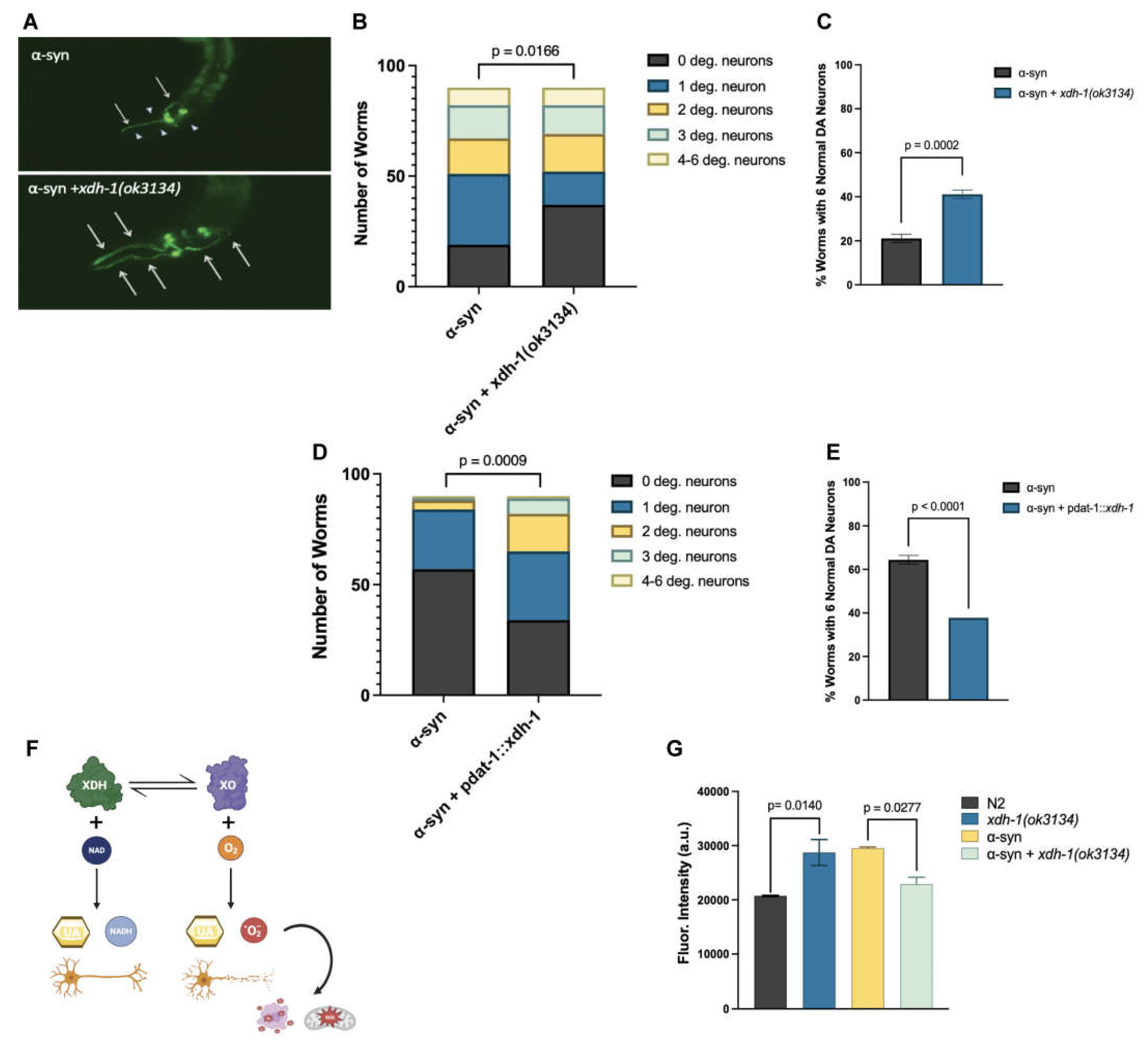
Figure 4.
xdh-1, wht-2 and adr-2 comprise a genetic regulatory network that affects PD pathologies in a tissue-specific manner. (A) Synchronized worms were scored for dopamine neurodegeneration seven days post-hatch. Knockdown of xdh-1 and xdh-1 + adr-2 indicate that adr-2 does not impact xdh-1 activity, while knockdown of wht-2 + xdh-1 indicate that xdh-1 does not regulate wht-2 activity. These experiments were completed via RNAi knockdown in dopaminergic neurons of C. elegans expressing α-syn + GFP under control of the Pdat-1 promoter (three independent experiments, n = 30 per independent groups; N=3; n=30). Significance was obtained using a One-way ANOVA with a Tukey post hoc test; ns >0.05. (B) The same experimental paradigm was performed in worms expressing α-syn and GFP in the body wall muscles. Synchronized two-day old worms were scored for aggregate size and number, which were combined to generate an overall aggregate score. The relation between adr-2, xdh-1 and wht-2 in the body wall match that shown in dopaminergic neurons, implying that wht-2 is the rate-limiting member of this system (three independent experiments, n = 30 per independent groups; N=3; n=30). A One-way ANOVA with a Tukey post hoc analysis was employed, ns >0.05.
Figure 4.
xdh-1, wht-2 and adr-2 comprise a genetic regulatory network that affects PD pathologies in a tissue-specific manner. (A) Synchronized worms were scored for dopamine neurodegeneration seven days post-hatch. Knockdown of xdh-1 and xdh-1 + adr-2 indicate that adr-2 does not impact xdh-1 activity, while knockdown of wht-2 + xdh-1 indicate that xdh-1 does not regulate wht-2 activity. These experiments were completed via RNAi knockdown in dopaminergic neurons of C. elegans expressing α-syn + GFP under control of the Pdat-1 promoter (three independent experiments, n = 30 per independent groups; N=3; n=30). Significance was obtained using a One-way ANOVA with a Tukey post hoc test; ns >0.05. (B) The same experimental paradigm was performed in worms expressing α-syn and GFP in the body wall muscles. Synchronized two-day old worms were scored for aggregate size and number, which were combined to generate an overall aggregate score. The relation between adr-2, xdh-1 and wht-2 in the body wall match that shown in dopaminergic neurons, implying that wht-2 is the rate-limiting member of this system (three independent experiments, n = 30 per independent groups; N=3; n=30). A One-way ANOVA with a Tukey post hoc analysis was employed, ns >0.05.
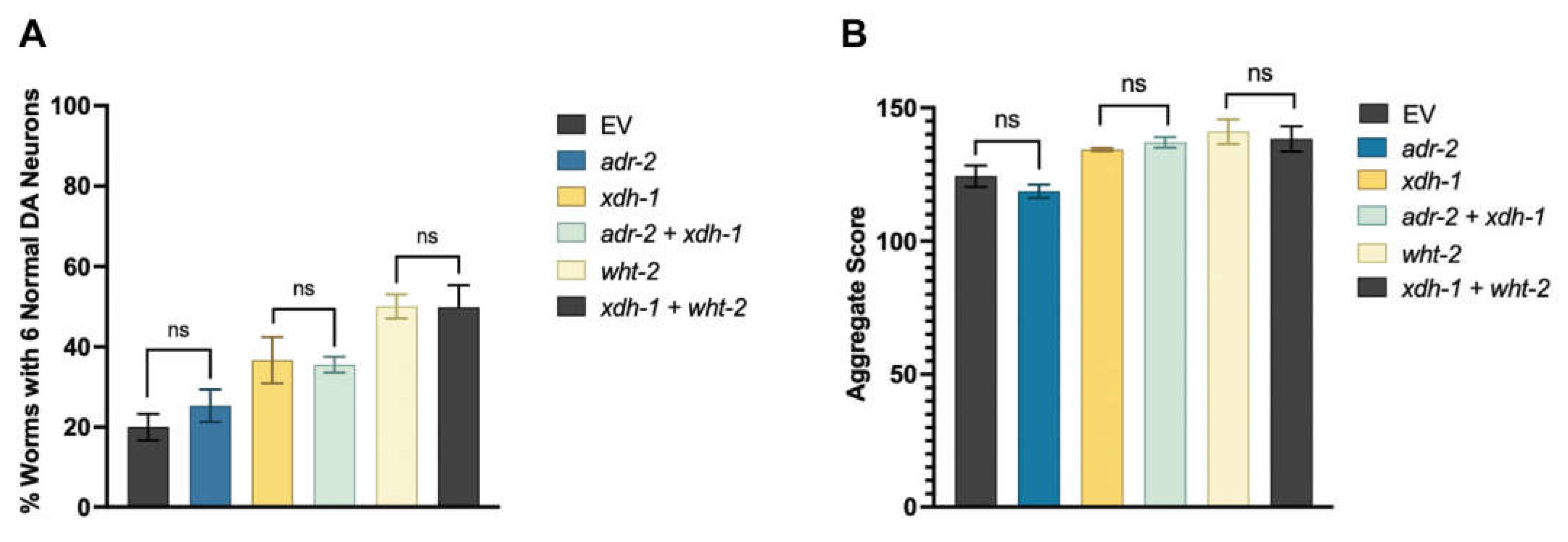
Figure 5.
RNA editing of wht-2 leads to a non-synonymous amino acid substitution that is predicted to alter protein folding. (A) Editing sites in the wht-2 gene. wht-2 consists of 12 exons located at genomic position IV: 11470436 to IV: 11473419 in the C. elegans genome. Two bases in the coding mRNA are edited, with one resulting in an amino acid substitution upon translation. (B) Comparison of the wht-2 RNA and amino acid sequences in the presence and absence of editing at IV: 11470944. Key indicates AlphaFold model prediction confidence. (C) Pairwise alignment between C. elegans wht-2 (top) and human ABCG2 (bottom) DNA sequences. The identified editing site, indicated in red, is located in a conserved region. (D) The complete structure of the unedited WHT-2 protein. Arrow indicates residue 124 where threonine is switched to alanine when RNA is edited, outlined in green. (E-F) Changes in protein structure at residue 124 when WHT-2 is not edited (D) versus edited (E). Hydrogen bonding (shown in green) and protein folding are altered because of editing.
Figure 5.
RNA editing of wht-2 leads to a non-synonymous amino acid substitution that is predicted to alter protein folding. (A) Editing sites in the wht-2 gene. wht-2 consists of 12 exons located at genomic position IV: 11470436 to IV: 11473419 in the C. elegans genome. Two bases in the coding mRNA are edited, with one resulting in an amino acid substitution upon translation. (B) Comparison of the wht-2 RNA and amino acid sequences in the presence and absence of editing at IV: 11470944. Key indicates AlphaFold model prediction confidence. (C) Pairwise alignment between C. elegans wht-2 (top) and human ABCG2 (bottom) DNA sequences. The identified editing site, indicated in red, is located in a conserved region. (D) The complete structure of the unedited WHT-2 protein. Arrow indicates residue 124 where threonine is switched to alanine when RNA is edited, outlined in green. (E-F) Changes in protein structure at residue 124 when WHT-2 is not edited (D) versus edited (E). Hydrogen bonding (shown in green) and protein folding are altered because of editing.
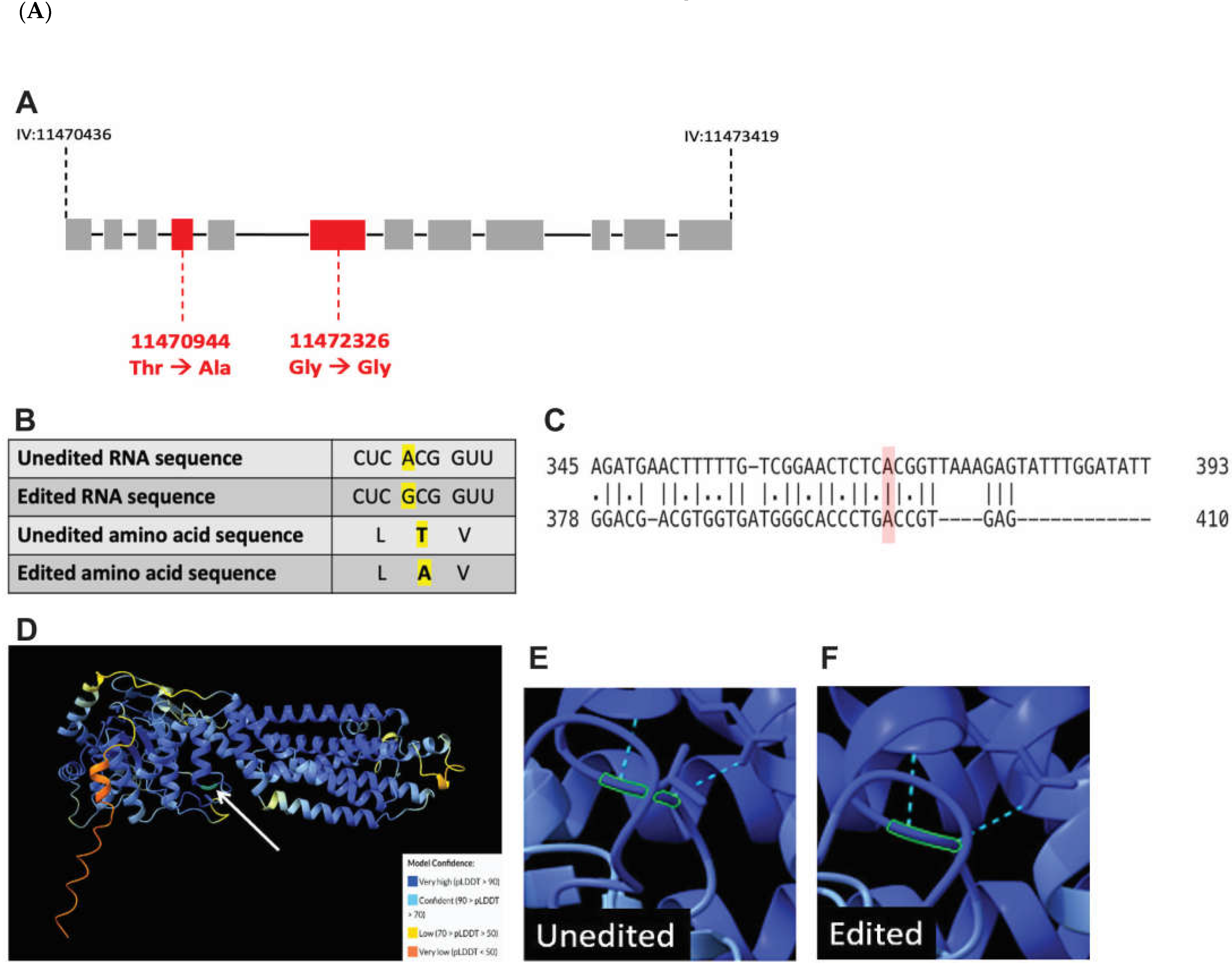
Figure 6.
A proposed model of ADR-2, WHT-2 and XDH-1 activity. In the presence of ADR-2, WHT-2 is edited which allows for optimal export of uric acid, a product of purine catabolism by XDH-1. In the absence of editing by ADR-2, the structure of WHT-2 is altered such that uric acid cannot efficiently be exported out of the cell. To maintain cellular homeostasis, there is a downregulation of xdh-1 transcription. Created with Biorender.com.
Figure 6.
A proposed model of ADR-2, WHT-2 and XDH-1 activity. In the presence of ADR-2, WHT-2 is edited which allows for optimal export of uric acid, a product of purine catabolism by XDH-1. In the absence of editing by ADR-2, the structure of WHT-2 is altered such that uric acid cannot efficiently be exported out of the cell. To maintain cellular homeostasis, there is a downregulation of xdh-1 transcription. Created with Biorender.com.
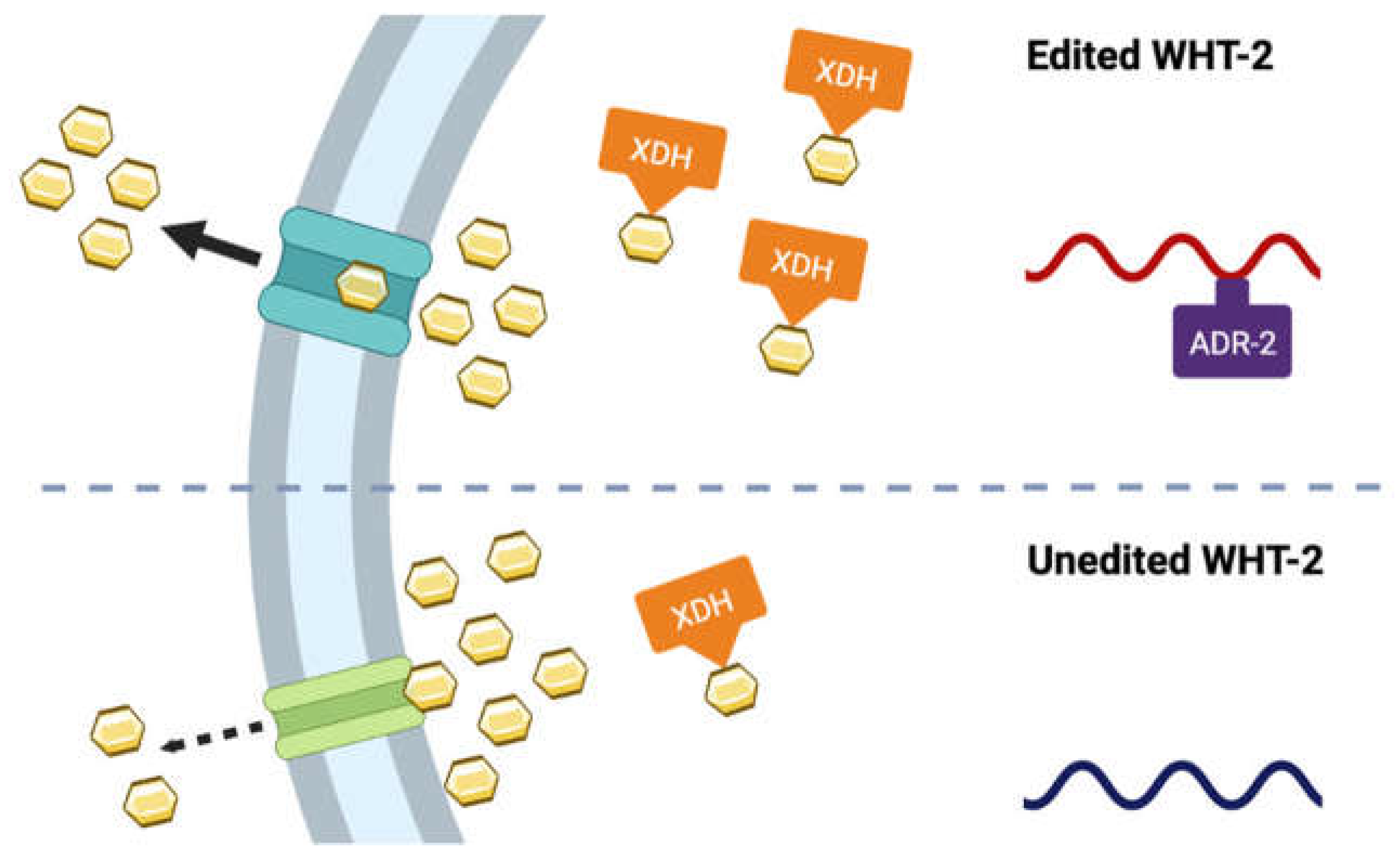
Table 1.
Summary of crosses completed to generate new strains used in this study.
| Strain | Genotype | Strains Crossed |
| UA455 | xdh-1(ok3134); baIn11[Pdat-1::α-syn, Pdat-1 ::GFP] | RB2379 x UA44 |
| UA456 | xdh-1(ok3134); vtIs7 [Pdat-1::GFP] | RB2379 x BY250 |
| UA457 | adr-2(ok735); vtIs7[Pdat-1::α-syn, Pdat-1 ::GFP] | RB886 x BY250 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



