
Submitted:
14 March 2023
Posted:
15 March 2023
You are already at the latest version
Abstract
Infectious bursal disease (IBD) is a viral poultry disease known worldwide for impacting the economy and food security. The disease is endemic in Nigeria, with reported outbreaks in vaccinated poultry flocks. To gain insight into the dynamics of infectious bursal disease virus (IBDV) evolution in Nigeria, near-complete genomes of four IBDVs were evaluated. Amino acid sequences in the hypervariable region of the VP2 revealed conserved markers (222A, 242I, 256I, 294I and 299S), associated with very virulent (vv) IBDV, including the serine-rich heptapeptide motif (SWSASGS). Based on the newly proposed classification for segments A and B, the IBDVs clustered in the A3B5 group (where A3 are IBDVs with vvIBDV-like segment A and B5 are from non-vvIBDV-like segment B) forming a monophyletic subcluster. Unique amino acid mutations with yet-to-be-determined biological functions have been observed in both segments. Amino acid sequences of the Nigerian IBDVs showed that they are reassortant viruses. Circulation of reassortant IBDVs may be responsible for the vaccination failures observed in the Nigerian poultry population. Close monitoring of changes in the IBDV genome is recommended to nip deleterious changes in the bud, through the identification and introduction of the most appropriate vaccine candidates and advocacy/extension programs for properly implementing disease control.
Keywords:
genome sequence
; infectious bursal disease virus
; Nigeria
; phylogenetic analysis
; poultry
; segments A and B
1. Introduction
Infectious bursal disease (IBD) is a viral poultry disease that is known worldwide not only for its devastating effects on poultry [1,2] but, also, for its impact on other aspects of human existence vis-a-vis economy and food security. In terms of susceptibility, young chicks between 3 and 6 weeks of age are most susceptible to IBD infections, typically characterized by the destruction of B lymphocytes in the lymphoid organ, resulting in death. Hence, surviving chicks become immunosuppressed, making them susceptible to other diseases [2]. Although, infections have been reported in older chickens of up to 20 weeks [3,4].
The aetiologic agent, Infectious Bursal Disease Virus (IBDV), is a non-enveloped RNA virus assigned to the family Birnaviridae, genus Avibirnavirus [5]. The IBDV genome is bi-segmented [6], comprising the larger segment A (3.2 kbp) encoding VP2, VP4, VP3 [7,8] and the smaller overlapping open reading frame encoding a non-structural protein, VP5 [9,10]. Segment B (2.8 kbp) encodes the RNA-dependent RNA polymerase VP1 [11].
Based on pathogenicity, two serotypes of IBDV are known; serotype 1, which is pathogenic to chickens, and serotype 2, a nonpathogenic strain [12]. Serotype 1 consists of four groups: classical, antigenic variant, very virulent and attenuated [13,14]. As an RNA virus, IBDV is prone to mutation and recombination, culminating in reassortant(s) and recombinant(s) [15,16]. The classification of IBDV was initially based on the different strains that existed, namely: classical, variant, and very virulent. However, Michel and Jackwood [17] proposed the classification of IBDV serotype 1 into seven genogroups (G1-G7) based on the VP2 hypervariable region sequences. Recently, Islam et al. [18] proposed a further classification based on the phylogenetic analysis of both segments A and B. In details, two genetic fragments were considered to build the dataset: a 366 bp portion of segment A that include the VP2 hypervariable region (nt 785-1150) and a 508 bp fragment of segment B focused on VP1 N-terminal domain and finger subdomain of the central polymerase (nt 328-835). This classification produced eight genogroups for segment A (A1-A8) with serotype 2 designated as A0, and five genogroups (B1-B5) for segment B, with serotype 2 designated as B1. Another classification by Wang et al. [19] identified nine genogroups for segment A (A1-A9) and five (B1-B5) for segment B.
In Nigeria, IBD is endemic, with reports of outbreaks in vaccinated poultry flocks [4,20,21]. In most IBD outbreaks in Nigeria, the vvIBDV strain has been implicated [20,21,22,23,24,25,26]. In 2016, the presence of reassortant IBDV was first confirmed in Nigeria [21]. It has been speculated that the reassortant viruses arose from the exchange of genetic materials between the very virulent field and attenuated (vaccine) strains of IBDV. The presence of reassortant viruses is one of the factors that can give rise to vaccination failure, therefore their detection could be one of the reasons for IBDV vaccination failure in Nigeria. The presence of the vvIBDV and the reassortant IBDV have been associated with the failure of IBDV vaccination even under strict management practices [27].
Here, we sequenced near complete genomes of four IBDV collected between 2017-2019 with the aim of gaining insight into the evolutionary dynamics of the virus. This information is critical to develop an effective control strategy to reduce the threat of the disease.
2. Materials and Methods
2.1. Sources of samples
Suspected outbreaks of IBD from four different geographical locations in Nigeria were investigated from 2017 to 2019. Epidemiological information on the outbreaks were documented. At postmortem, bursae of Fabricius (bF) were collected from the poultry for processing.
2.2. IBD virus antigen detection
The bF was processed into 20% (w/v) homogenate as previously described [25]. The supernatant was analyzed using the agar gel precipitin test (AGPT) alongside standard IBDV antigen and antiserum (Charles Rivers, USA). The agar plates were incubated in a humidified incubator at 37oC for 48 hours. Aliquots of the supernatant were stored at -80oC until shipped to Istituto Zooprofilattico Sperimentale delle Venezie, Padua, Italy for molecular analysis.
2.3. Sequencing
According to the manufacturer’s instructions, total RNA was extracted from the bursal homogenate samples using the QIAsymphony RNA kit (Qiagen, USA). RNA from one of the four samples was reverse-transcribed into double-stranded cDNA (cDNAds) using Maxima H Minus Double-stranded cDNA synthesis kit (ThermoFisher, Waltham, MA) to be sequenced using a metagenomic approach. RNA from the other three samples was reverse-transcribed and amplified using SuperScript™ III One-Step RT-PCR System with Platinum™ Taq High Fidelity DNA Polymerase (ThermoFisher, Waltham, MA) and specific primers to amplify segment A (IBDV-VV_A-83-F 5’- AACTCCTCCTTCTACAATGC-3’; IBDV-VV_A-3123-R 5’- CTGTGTTGGAGCATTGGG-3’) and segment B (IBDV-VV_B-91-F 5’- TCTTCTTGATGATTCTRCCAC -3’; IBDV-B-2782-R 5’- TCTTCTTGAGTGGTTCCC -3’). Double-stranded cDNA and amplicons were purified using Agencourt AMPure XP (Beckman Coulter™) and quantified with Qubit™ DNA HS Assay (Thermo Fisher Scientific, USA). Sequencing libraries were generated using the Illumina Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) following the manufacturer’s instructions and sequenced using the Illumina MiSeq platform (2×300 bp PE mode).
2.4. Bioinformatic Analysis
Illumina read quality was assessed using FastQC v0.11.2. Raw data were filtered by removing a) reads with more than 10% of undetermined (“N”) bases; b) reads with more than 100 bases with Q score below 7; c) duplicated paired end reads. The remaining reads were clipped from Illumina adaptors with Scythe v0.991(https://github.com/vsbuffalo/scythe) and, only for samples processed with the target amplification approach, from PCR primers with Trimmomatic v0.32 [28] before being trimmed with Sickle v1.33 (https://github.com/najoshi/sickle). Reads shorter than 80 bases or unpaired after previous filters are discarded.
Reads associated with the sample sequenced using the metagenomic approach were further processed by taxonomic assignment using BLAST v2.10.0+ [29] alignment against the integrated NT database (version 23 February 2020) and Diamond v0.9.17 [30] alignment against the integrated NR database (version 23 February 2020). Alignment hits with e-values greater than 1 x 10-3 were discarded. The taxonomical level of each read was determined by the lowest common ancestor-based algorithm implemented in MEGAN v6.18.50 [31]. Reads not classified taxonomically as belonging to IBDV were discarded.
High quality reads that passed previous filters were aligned against the IBDV reference genome (for the samples sequences with target amplification approach: AF051837.1 for segment A, AB368969.1 for segment B; for the sample sequenced with the metagenomic approach: AF092943.1 for segment A, KF569804.1 for segment B) using BWA v0.7.12 [32] and standard parameters. Alignments were processed using SAMtools v0.1.19 [33] to convert them into BAM format and sort them by position.
SNPs were called using LoFreq v2.1.2 [34]. According to LoFreq usage recommendations, the alignment was first processed with Picard-tools v2.1.0 (http://picard.sourceforge.net) and GATK v3.5 [35] in order to correct potential errors, realign reads around indels and recalibrate base quality. LoFreq was then run-on fixed alignment with an option “--call-indels” to produce a vcf file containing both SNPs and indels. From the final set of variants, indels with a frequency lower than 50% and SNPs with a frequency lower than 25% were discarded. To produce the consensus sequence, we changed the reference genome in agreement with the following rules: a) for a position j, if coverage was not high enough to reliable call variants, we added an "N" base; b) for a position j, if coverage was high enough to reliable call variants, but no SNP was called, we added reference genome base at position j; c) for a position j, if coverage was enough to reliable call variants and at least one SNP was called, we added the nucleotide using the IUPAC nucleotide code (http://www.bioinformatics.org/sms/iupac.html) in accordance with bases present.
The sequences have been submitted to the GenBank database under accession numbers OP311682 – OP311689.
2.5. Phylogenetic Analysis
The phylogenetic analysis was carried out following the criteria suggested by Islam et al. [18]. More specifically, for the VP2 hypervariable region (366 bp) of segment A, the dataset included i) 151 sequences representatives of the eight genogroups of serotype 1, ii) sequences obtained from BLAST results and iii) one genogroup of serotype 2. Serotype 1 sequences included: A1a (classical attenuated) and A1b (classical virulent), A2 (US antigenic variant), A3 (very virulent), A4 (early European and recent South American distinct IBDV, dIBDV), A5 (atypical or recombinant Mexican), A6 (atypical Italian), A7 (early Australian) and A8 (Australian variant). The dataset for segment B of the VP1 N-terminal domain and the finger subdomain of the central polymerase fragment (508 bp) includes 134 sequences representatives of the 5 distinct genogroups, namely B1 (classic-like), B2 (very virulent-like), B3 (early Australian-like), B4 (Polish & Tanzanian), B5 (Nigerian) and sequences obtained from the BLAST search. Alignment for both datasets was performed using FFT-NS-i (Standard) implemented in MAFFT V.7.0 (https://mafft.cbrc.jp/alignment/server/; [36]. The phylogenetic trees were obtained using IQ-TREE 1.6.10 [37], with 1000 ultra-fast bootstrap replicates and the best fit model automatically identified by the software; in details, for segment A phylogenetic tree, the best-fit model chosen according to BIC was SYM+I+G4, whereas GTR+F+G4 model was automatically selected for segment B. FigTree v.1.4.3 (https://github.com/rambaut/figtree/releases) program was used to view phylogenetic trees graphically.
3. Results
3.1. Flock History
3.2. Detection of IBD viruses
In all the samples (n = 15) tested by AGPT, precipitin bands were observed between 24 to 48 hours post-incubation. Only 4 samples identified as being from different outbreaks were considered for sequencing.
3.3. Phylogenetic analysis
The phylogenetic tree of the VP2 fragment (366 bp) from segment A shows that all four samples clustered into A3 genogroup (Figure 2), which contains very virulent pathotypes, and are closely related to viruses collected between 2009 and 2016 in Nigeria. The Nigerian viruses form two separate well-supported monophyletic groups within the A3 genogroup. The viruses analyzed in this study belong to both Nigerian groups, suggesting a persistent circulation of two variants in the country. In particular, sample VRD-18-71_19VIR8426-6 clustered with strains collected in Nigeria from 2011 to 2014, showing the highest nucleotide sequence identity with the Nigerian virus ANAMBRA54/NG/2012 (99.2%); while samples VRD-17-460_19VIR8426-5, VRD-18-119_19VIR8426-8 and VRD-19-048_19VIR8426-10 fall into a separate group comprising Nigerian IBDV samples collected from 2009 to 2016; in detail, VRD-17-460_19VIR8426-5 shows the highest nucleotide sequence identity (98.9%) with ABUJA93/NG/2013, while VRD-18-119_19VIR8426-8 and VRD-19-048_19VIR8426-10 are closely related to NG592/2016 (99.4% and 99.2% respectively).
The phylogenetic tree based on partial segment B sequence (508 nt) (Figure 3) shows that all four samples are grouped in the B5 genogroup, with Nigerian samples circulating in 2009-2019. Sample VRD-17-460_19VIR8426-5 and VRD-19-048_19VIR8426-10 show the highest identity (99.1%) with samples isolated in Nigeria in 2010-2014, VRD-18-119_19VIR8426-8 with PLATEAU53/NG/2010 (99.4%) and sample VRD-18-71_19VIR8426-6 with ABUJA93/NG/2013 (99.4%).
3.4. Nucleotide sequence analysis of segments A and B
The consensus sequences obtained were compared to the GenBank database using the BLAST algorithm (https://blast.ncbi.nlm.nih.gov/Blast.cgi); the results are summarized in Table 2.
For the four Nigerian IBDV isolates examined in this study, nucleotide analysis of the complete segment A (VP2-4-3 polyprotein) and segment B (VP1), to each other range from 94.9% - 99.1% and 97.1% - 98.8% respectively.
3.5. Amino acid sequence analysis of segments A and B
Amino acid sequences of both segments of the four Nigerian isolates were compared with other IBDV sequences from GenBank comprising of very virulent, variant, attenuated and reassortant strains. In all, thirty-one amino acid substitutions were observed in the VP5 of the four Nigerian isolates, with nine of these substitutions being unique only to Nigerian IBDVs (Table S1). The polyprotein (VP2-VP4-VP3) had thirty-five substitutions (Table S2), ten of which were unique to the Nigerian IBDVs. The VP1 had a total of twenty substitutions, with eleven of which were unique to Nigerian IBDVs.
The VP2 amino acid sequences of the four Nigerian IBDVs under investigation had the markers typical of vvIBDV (222A, 242I, 256I, 294I and 299S). Also, the serine-rich heptapeptide region (SWSASGS) spanning from amino acid position 326 to 332 adjacent to the second hydrophilic region of the VP2 gene were conserved as established in other vvIBDVs. At amino acid position 300, the substitution EA was observed in three of the four Nigerian IBDVs examined, while the fourth had substitution, E→Q. Amino acid substitution at position 300 has been implicated for vaccination failure.
Analysis of the VP4 amino acid sequences of the four Nigerian IBDV and others from GenBank revealed H471Q and Q486L mutations in three of the four Nigeria IBDV previously reported [23]. Interestingly, the Nigerian IBDV, VRD-18-71_19VIR8426-6, had unique amino acid mutations F599Y, D614E and K642Q (Table S2) that were not observed in the other three IBD viruses under investigation or hitherto reported in IBDV from Nigeria. Another substitution, N745S, was observed in three of the four Nigerian IBD viruses and two previously reported Nigerian IBDVs used for comparison and in a reassortant IBDV from China (HLJ-0504) (data not shown).
Sequence analysis of VP3 revealed amino acid substitutions E761G (Table S2) in three of the four Nigerian IBD viruses. These substitutions were also present in some previously reported Nigerian IBD viruses [23]. Position 767 of one of the four Nigerian IBD viruses had this mutation, S767G, also present in the previously reported Nigerian IBDV, T09 [22]. At amino acid positions 777 and 778, the following substitutions, N→S and V→A, were only seen in VRD-19-048_19VIR8426-10. Mutation at position V990A (Table S2) was observed in all four Nigerian IBDVs.
Sequence analysis of the VP1 of the four Nigerian IBDV used in this study revealed the following mutations: V4I in one of four, as in other IBDVs, notably attenuated and reassortant strains used for comparison, T23S in three of the four, E119D and V141I in all four. The triplet amino acid at positions 145, 146 and 147 (Table S3) of the four Nigerian IBDVs showed similar QEG mutations as previously reported [21]. Positions 150 and 158 had D→E and N→S mutations, with the first being unique to Nigerian IBDV and some IBDV from China, whilst the second is also unique to Nigerian viruses. At position 163, the substitution A→V was observed in all the Nigerian IBDV examined and a reassortant IBDV strain GX-NNZ-11, while the following mutations at positions D219E, E242D, M390L, A391T, D393E and P562S were observed in all four (Table S3). The substitution, R695K, was observed in three of the four Nigerian IBD viruses. At position 697, V→T occurred in three out of four while Nasarawa 8426-8/2018 Nigeria had V→I and Cross River/19VIR8426-6/2018 only had K761R mutation (Table S3).
4. Discussion
This study provided insight into the sequences from four complete genomes of IBDV from different outbreaks and locations in Nigeria. Unique amino acid mutations with as yet unknown biological functions have been identified. Most of the previous molecular studies on IBDVs in Nigeria are based on partial sequence analyses of either the hvVP2 or the VP2 and VP1 genes [20,21,22,23,24,26]. As of February 2023, only two complete genome sequence of segment A [22,23] and segment B [22] of Nigerian IBDV exist in the GenBank. Sequence analysis of both VP2 and VP1 has been advocated to determine the virulence of IBDV and to identify genetic reassortments that may occur within the genome segment B, as well as mutations in segment A that occur in vvIBDV [15,38,39]. The complete genome sequence analysis of IBDV has helped to advance our understanding of the dynamics, epidemiology, and evolution of IBDV to aid in its prevention and control [40,41,42]. This study has, therefore, further elucidated the evolution of IBDV in Nigeria.
Phylogenetic analysis using the nucleotide sequences of the four Nigerian IBDVs studied, showed that they are reassortant viruses. Previous studies in the country have identified reassortant IBDV strain, with segment A derived from vvIBDV and segment B derived from non-vvIBDV [21,26]. The reassorted viruses produced a novel lineage unique to Nigeria [21] and recently, Islam et al. [18] re-assigned the Nigerian reassortant IBD viruses into a new group B5. Based on the hypervariable region of the VP2, all four IBDV belong to the very virulent strains. Previous molecular studies on IBDV in Nigeria reported the presence of the vvIBDV strains circulating in poultry [20,21,22,23,24,25,26]. Using the criteria for classifying IBD viruses proposed by Michel and Jackwood [17], Nigerian viruses clustered with the G3 group of IBDV. However, based on the recent IBDV classification by Islam et al. [18], Nigerian IBDVs exclusively formed a monophyletic cluster designated as A3B5 (where A3: IBDV isolate with vvIBDV-like segment A and B5: IBDV isolate with non-vvIBDV-like segment B).
Segment A (VP2-4-3 polyprotein) of the four isolates showed a high percentage similarity (94.9 – 99.5%) with each other, and to previously reported IBDV from Nigeria (95.1 – 98.7%). The VP5 of the four Nigerian viruses showed a higher percentage nucleotide homology of 99.0 – 99.5% with each other with each other and 98.0 – 98.9% with other previously reported IBDV VP5 protein sequences from Nigeria. For now, this might indicate a level of stability within this region of the virus in Nigeria, albeit not at 100% compared to previously reported IBDVs from Nigeria, as IBDV is well known for its amino acid mutation rate and its ability to evolve [25]. The same can be attributed to the numbers obtained for the polyprotein sequences. The percentage similarity obtained when the four Nigerian IBDV isolates were compared to other IBDV strains retrieved from the GenBank shows that they more closely match the vvIBDV strains than others. The results of the nucleotide sequence analysis of the segment B of the four Nigerian IBDV showed a low percentage nucleotide identity with a previously reported Nigerian IBDV (T09), UK661 and even the classic D78. The first two viruses are vvIBDV with segments A and B derived from vvIBDV-like sequences, while the second, a classic IBDV, has segments A and B derived from non-vvIBDV-like sequences.
In the amino acid sequences of segment A of the four Nigerian IBDV isolates used in this study, the hvVP2 region, extending from amino acid 206-350, which is responsible for tissue culture adaptation, antigenic variation, and antigenicity, exhibited the markers: A222, I242, I256, I294, S299, Q253, D279 and A284 are typical of very virulent IBDV [43,44,45]. Also, the serine-rich heptapeptide region from aa326-332 was conserved in all four, indicating that they belong to the vvIBDV [46]. Within this region, few previously reported amino acid substitutions Q219T [21,26], G254S, T269S and E300A/Q [21,22,23,26] were observed in a single, some or all the four Nigerian viruses. The occurrence of three of these mutations (Q219T, G254S and E300A/Q) within the hydrophilic loops of the virus can affect their antigenicity, which in turn can lead to a drift [47]. Also, the four Nigerian IBDVs have these amino acid residues I272, M290, Q324 and S330, which are intrinsic to vvIBDV [48,49]. Apart from the delineated amino acid substitutions within the hvVP2 region of the four Nigerian IBDVs, the amino acid VP2 sequences from aa1-452 were conserved as observed in the vvIBDVs.
Amino acid residues conserved for vvIBDVs in the VP4 region are 541I, 680Y, 685N, 715S and 751D [39]. The VP4 amino acid sequences of the four Nigerian viruses had amino acids that were conserved for vvIBDV at these positions, except at position 685 where three out of the four Nigerian IBDVs had N (asparagine) replaced with S (serine). Within the Nigerian IBDV VP4, other amino acid substitutions occurred at the following positions: H471Q, Q486L, F599Y, D614E, K642Q, N745S, in either a single, some, or all four viruses. These substitutions were also observed in some previously reported IBDV from Nigeria (Table S2). Apart from being the viral protease responsible for the cleavage of the polyprotein, VP4 plays a vital role in IBDV replication, growth, and maturation [50]. Therefore, any variation observed within this region may affect the virulence of the virus [49,51]. These mutations observed in the VP4 sequence of the Nigerian viruses need further investigation to determine their effect on viral pathogenicity.
The VP3, one of the major structural proteins of IBDV, acts as a scaffolding protein that binds the viral double-stranded RNA and, in collaboration with VP1, mediates recovery from infectious IBDV [52]. Also, the VP3 may play a role in receptor-mediated virus-cell attachment and virulence of IBDV [53]. The four Nigerian IBDVs examined had five amino acid substitutions (Table S2) compared to the vvIBDVs. Some of the mutations were observed in previously reported IBDV from Nigeria except for these substitutions; N777S and V778A were only seen in Akwa Ibom/19VIR8426-10/2019. Q783, E919, P981, V990 and A1005 are typical amino acids found in vvIBDVs and are conserved in the four Nigerian IBDVs, with the exception of the A990 substitution found in non-vvIBDVs. This mutation (A990) has been reported to decrease IBDV replication and efficacy during challenge [54].
The VP5 amino acid sequences of three Nigerian IBDV isolates examined had thirteen substitutions. Other amino acids within the VP5 were conserved, as seen in vvIBDVs. Two Nigerian isolates, Nasarawa/19VIR8426-8/2018 and Plateau/19VIR8426-5/2017 VP5 amino acid sequences, started at position 16 while that of Akwa Ibom/19VIR8426-10/2019 was at position 2. Amino acid sequences of vvIBDV VP5 are 149 long, while those of non-vvIBDV are 145 long [55]. Conserved amino acid residues for vvIBDVs include; E18, R49, F78, P129 and W137 [56]. Others are S18, S49, N78, L129, R137 [49], R45, F74, P125 and W133 [48]. A few mutations were observed in the three Nigerian viruses examined, present in either only one (R21G, G109S, S119P, H122D) out of the three Nigerian viruses or in all the three (T108A) and previously reported Nigerian viruses (G45R, S53T) (Table S1). Some share it with viruses from other countries used for comparison, in particular, the three Nigerian IBDVs share W80G with IBDVs from USA, Algeria and Tunisia. Some of the substitutions observed in the three Nigerian viruses are novel compared to other previously reported Nigerian IBDVs. Some of the mutations do not correspond with the positions mapped on the VP5 to differentiate the vvIBDV strains from other strains. IBDV VP5, a non-structural protein, although not essential for viral replication, plays an essential role in its pathogenesis, prevents apoptosis, and plays a role in the adaptive evolution of IBDV [10,57,58]. The role of the mutations observed in the three Nigerian IBDVs in terms of pathogenicity is still unknown.
The complete genome sequence of the VP1 of the four Nigerian IBDVs has advanced our understanding of the evolution of reassortant IBDVs in Nigeria. Currently, there is only one full-length genome sequence of the Nigerian IBDV VP1 in the GenBank [22]; others are partial sequences. Reassortment events have been associated with vaccine failure in IBDV [27]. In addition, the VP1 of IBDV plays a role in the pathogenicity of the virus, an event previously attributed only to VP2 [59]. Though, reassortant IBDVs have been reported in Nigeria [21,26], those studies used partial sequences of the VP1 to arrive at their conclusions. However, the only full-length sequence of the Nigerian IBDV VP1 sequence in the GenBank [22] is not a reassortant IBDV. Hence, this present study provided insight into a complete VP1 reassortant IBDV from Nigeria. The recent Nigerian IBDVs VP1 showed some of the previously reported mutations that are unique to Nigerian IBDVs, including others that were not reported previously because the partial sequences of the VP1 were used in the analysis.
As reported by Cui et al. [60], the amino acids conserved for vvIBDVs are 4V, 13K, 61I, 145T, 146D, 147N, 242E, 287A, 390M, 508K, 511S, 546P, 562P, 646S, 687P, 695R. Analysis of the VP1 amino acid sequences of the four Nigerian viruses revealed twenty-one (Table S3) amino acid substitutions compared to vvIBDV UK661. Eight of the amino acid substitutions were unique to the four Nigerian IBDV isolates. The implication of these mutations on the pathogenicity of the virus deserved further investigations.
To better understand the evolutionary dynamics of the VP1 of Nigerian viruses, the VP1 amino acid sequences of the four Nigerian IBDVs used in this study were aligned and compared to the partial VP1 sequences of previous Nigerian reassortant IBDVs (aas 26-299). A total of eleven amino acid substitutions were observed between the recent VP1 amino acids and the previously reported VP1 amino acids. The substitutions observed in the previously reported Nigerian VP1 amino acids were not seen in the recent ones used for this study. Of these eleven substitutions, seven were from the same states (Plateau and Akwa Ibom) used in this most recent study, while the remaining four were from other states not sampled in this study. A substitution N158S observed in Plateau/19VIR8426-5/2017 was completely absent from the previously reported Nigerian VP1 IBDV from the same state. Results from previously reported Nigerian IBDV VP1 amino acid sequences aligned (aa 100-248) and compared showing a total of eleven substitutions showed that six out of the eleven substitutions in the VP1 sequences reported by Nwagbo et al. [21] from Plateau, Bauchi and Uyo (capital of Akwa Ibom) were absent from sequences from the same state from Arowolo et al., [26]. It will be recalled that IBDV samples by Nwagbo et al. were collected between 2009 to 2014, while those by Arowolo et al. were from 2019. A substitution T174A that was observed in a sample from Plateau in 2013 was absent from samples from the same state in 2019, rather T174S seen in a sample from Kaduna in 2012 appeared in samples from Plateau and Bauchi in 2019. Bauchi, Plateau and Kaduna states in Nigeria share common boundaries. Likewise, A144T appeared in samples from Bauchi and Plateau in 2019 but was completely absent from samples from the same state between 2009 to 2014..
5. Conclusions
The complete genome sequences of four Nigerian Infectious bursal disease viruses were determined and molecular markers typical of very virulent IBDV strains were identified. Likewise, segment B of the four Nigerian viruses further confirmed the presence of reassortant strains of IBDV with a combination of this unique amino acid combination QEG at the triplet positions (145 – 147), which appears to be stable since its first report in Nigeria. These recent IBDVs identified five new amino acid mutations compared to previously reported IBDVs from Nigeria, confirming the continuous evolution of IBDV in Nigeria. This study also highlights the need for constant epidemiological investigation of IBDV to keep track of any new or sudden changes over time as these may impact vaccine efficiency.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Table S1: Amino acid substitutions at the VP5; Table S2: Amino acid substitutions at the VP2, VP4 and VP3; Table S3: Amino acid substitutions at the VP1.
Author Contributions
Conceptualization, I.S. and I.N..; methodology, I.N., L.S., A.S.; software, A.F, G.Z., A.M.; formal analysis, A.M., A.F., G.Z; investigation, N.M., C.M..; writing—original draft preparation, I.N., A.M., I.S.; writing—review and editing, A.F., C.M., L.S., G.Z, N.M., A.S.; supervision, I.S, A.F., A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
All data that supports the findings of this study are available in the main manuscript and the Supplementary Materials (Table S1: Amino acid substitutions at the VP5; Table S2: Amino acid substitutions at the VP2, VP4 and VP3; Table S3: Amino acid substitutions at the VP1).
Acknowledgments
We thank the management of NVRI for the provision of reagents for the analysis. We are grateful to Daspan Amos and Jesse Jonathan for excellent technical assistance and Mayowa Olabode for the map. We appreciate Isabella Monne for critically reading the manuscript and for her constructive comments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rautenschlein, S.; Alkie, T.N. Infectious bursal disease virus in poultry: current status and future prospects. Vet. Med. Res. Reports 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Eterradossi, N.; Saif, Y.M. Infectious bursal disease. In Diseases of poultry; Swayne, D.E., Ed.; John Wiley & Sons: New Jersey, US, 2019; pp. 257–283. ISBN 978-1-119-37117-5. [Google Scholar]
- Okoye, J.O.; Uzoukwu, M. An outbreak of infectious bursal disease among chickens between 16 and 20 weeks old. Avian Dis. 1981, 25, 1034–8. [Google Scholar] [CrossRef] [PubMed]
- Shekaro, A.; Josiah, I.E. Infectious bursal disease outbreak in fifteen weeks old pullets in Kaduna, Nigeria. J. Anim. Prod. Adv. 2015, 5, 636–644. [Google Scholar] [CrossRef]
- Delmas, B.; Attoui, H.; Ghosh, S.; Malik, Y.S.; Mundt, E.; Vakharia, V.N. ICTV virus taxonomy profile: Birnaviridae. J. Gen. Virol. 2019, 100, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Scholtissek, C.; Becht, H. The genome of infectious bursal disease virus consists of two segments of double-stranded RNA. J. Virol. 1979, 31, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Barrett, S.A.; Fahey, K.J. The characterization and molecular cloning of the double-stranded RNA genome of an Australian strain of infectious bursal disease virus. Virology 1985, 143, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Birghan, C. A non-canonical Lon proteinase lacking the ATPase domain employs the Ser-Lys catalytic dyad to exercise broad control over the life cycle of a double-stranded RNA virus. EMBO J. 2000, 19, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, E.; Maraver, A.; Espinosa, I.; Fernández-Arias, A.; Rodriguez, J.F. VP5, the Nonstructural Polypeptide of Infectious Bursal Disease Virus, Accumulates within the Host Plasma Membrane and Induces Cell Lysis. Virology 2000, 277, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Méndez, F.; de Garay, T.; Rodríguez, D.; Rodríguez, J.F. Infectious Bursal Disease Virus VP5 Polypeptide: A Phosphoinositide-Binding Protein Required for Efficient Cell-to-Cell Virus Dissemination. PLoS One 2015, 10, e0123470. [Google Scholar] [CrossRef] [PubMed]
- Shwed, P.S.; Dobos, P.; Cameron, L.A.; Vakharia, V.N.; Duncan, R. Birnavirus VP1 Proteins Form a Distinct Subgroup of RNA-Dependent RNA Polymerases Lacking a GDD Motif. Virology 2002, 296, 241–250. [Google Scholar] [CrossRef]
- McFerran, J.B.; McNulty, M.S.; McKillop, E.R.; Connor, T.J.; McCracken, R.M.; Collins, D.S.; Allan, G.M. Isolation and serological studies with infectious bursal disease viruses from fowl, turkeys and ducks: Demonstration of a second serotype. Avian Pathol. 1980, 9, 395–404. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, T.P. Acute infectious bursal disease in poultry: A review. Avian Pathol. 2000, 29, 175–194. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Islam, M.R.; Raue, R. Research on infectious bursal disease—the past, the present and the future. Vet. Microbiol. 2003, 97, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Nouën, C.L.; Rivallan, G.; Toquin, D.; Darlu, P.; Morin, Y.; Beven, V.; de Boisseson, C.; Cazaban, C.; Comte, S.; Gardin, Y.; et al. Very virulent infectious bursal disease virus: reduced pathogenicity in a rare natural segment-B-reassorted isolate. J. Gen. Virol. 2006, 87, 209–216. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xiong, Z.; Yang, L.; Guan, D.; Yang, X.; Wei, P. Molecular epidemiology studies on partial sequences of both genome segments reveal that reassortant infectious bursal disease viruses were dominantly prevalent in southern China during 2000-2012. Arch. Virol. 2014, 159, 3279–3292. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.O.; Jackwood, D.J. Classification of infectious bursal disease virus into genogroups. Arch. Virol. 2017, 162, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Nooruzzaman, M.; Rahman, T.; Mumu, T.T.; Rahman, M.M.; Chowdhury, E.H.; Eterradossi, N.; Müller, H. A unified genotypic classification of infectious bursal disease virus based on both genome segments. Avian Pathol. 2021, 50, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, L.; Jiang, N.; Gao, L.; Li, K.; Gao, Y.; Liu, C.; Cui, H.; Pan, Q.; Zhang, Y.; et al. An improved scheme for infectious bursal disease virus genotype classification based on both genome-segments A and B. J. Integr. Agric. 2021, 20, 1372–1381. [Google Scholar] [CrossRef]
- Owolodun, O.A.; Yakubu, B.; Jambol, A.R.; Audu, B.J.; Dogonyaro, B.B.; Luka, P.D. Further evidence for very virulent infectious bursal disease virus in vaccinated chickens in Nigeria. Trop. Anim. Health Prod. 2015, 47, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Nwagbo, I.O.; Shittu, I.; Nwosuh, C.I.; Ezeifeka, G.O.; Odibo, F.J.C.; Michel, L.O.; Jackwood, D.J. Molecular characterization of field infectious bursal disease virus isolates from Nigeria. Vet. World 2016, 9, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Owoade, A.A.; Mulders, M.N.; Kohnen, J.; Ammerlaan, W.; Muller, C.P. High sequence diversity in infectious bursal disease virus serotype 1 in poultry and turkey suggests West-African origin of very virulent strains. Arch. Virol. 2004, 149, 653–672. [Google Scholar] [CrossRef] [PubMed]
- Adamu, J.; Owoade, A.A.; Abdu, P.A.; Kazeem, H.M.; Fatihu, M.Y. Characterization of field and vaccine infectious bursal disease viruses from Nigeria revealing possible virulence and regional markers in the VP2 minor hydrophilic peaks. Avian Pathol. 2013, 42, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Luka, P.D.; Yakubu, B.; Jambol, A.R.; Audu, B.J.; Dogonyaro, B.B.; Owolodun, O.A. Detection and differentiation of infectious bursal disease virus from the outbreaks in two layer farms by PCR-RFLP in Jos, Nigeria. Vet. World 2014, 7, 30–33. [Google Scholar] [CrossRef]
- Nwagbo, I.O.; Shittu, I.; Qasim, A.M.M.; Joannis, T.M. Continuous circulation and mutation at the hypervariable region of the VP2 gene of very virulent infectious bursal disease virus in Nigeria. Niger. Vet. J. 2018, 39, 143. [Google Scholar] [CrossRef]
- Arowolo, O.A.; George, U.E.; Luka, P.D.; Maurice, N.A.; Atuman, Y.J.; Shallmizhili, J.J.; Shittu, I.; Oluwayelu, D.O. Infectious bursal disease in Nigeria: continuous circulation of reassortant viruses. Trop. Anim. Health Prod. 2021, 53, 271. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Pathak, D.; Ramamurthy, N.; Maity, H.K.; Chellappa, M.M. Infectious bursal disease virus in chickens: prevalence, impact, and management strategies. Vet. Med. Res. Reports 2019, 10, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–10. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLOS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, H.; Chen, G.; Liu, H.; Wang, S.; Xia, D.; Yu, Y.; Zhang, Y.; Jiang, J.; Ma, J.; et al. Identification and assessment of pathogenicity of a naturally reassorted infectious bursal disease virus from Henan, China. Poult. Sci. 2019, 98, 6433–6444. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Wu, T.; Li, H.; Fan, L.; Li, K.; Gao, L.; Wang, Y.; Gao, Y.; Liu, C.; Cui, H.; et al. Pathogenic Characterization and Full Length Genome Sequence of a Reassortant Infectious Bursal Disease Virus Newly Isolated in Pakistan. Virol. Sin. 2019, 34, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, J.; Yan, Z.; Liu, D.; Ji, J.; Qin, J.; Li, H.; Ma, J.; Bi, Y.; Xie, Q. Complete Genome Sequence Analysis of a Natural Reassortant Infectious Bursal Disease Virus in China. J. Virol. 2012, 86, 11942–11943. [Google Scholar] [CrossRef] [PubMed]
- Raja, P.; Senthilkumar, T.M.A.; Parthiban, M.; Thangavelu, A.; Gowri, A.M.; Palanisammi, A.; Kumanan, K. Complete Genome Sequence Analysis of a Naturally Reassorted Infectious Bursal Disease Virus from India. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Drissi Touzani, C.; Fellahi, S.; Fassi Fihri, O.; Gaboun, F.; Khayi, S.; Mentag, R.; Lico, C.; Baschieri, S.; El Houadfi, M.; Ducatez, M. Complete genome analysis and time scale evolution of very virulent infectious bursal disease viruses isolated from recent outbreaks in Morocco. Infect. Genet. Evol. 2020, 77, 104097. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J.; Sreedevi, B.; LeFever, L.J.; Sommer-Wagner, S.E. Studies on naturally occurring infectious bursal disease viruses suggest that a single amino acid substitution at position 253 in VP2 increases pathogenicity. Virology 2008, 377, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Escaffre, O.; Le Nouën, C.; Amelot, M.; Ambroggio, X.; Ogden, K.M.; Guionie, O.; Toquin, D.; Müller, H.; Islam, M.R.; Eterradossi, N. Both Genome Segments Contribute to the Pathogenicity of Very Virulent Infectious Bursal Disease Virus. J. Virol. 2013, 87, 2767–80. [Google Scholar] [CrossRef] [PubMed]
- Shehata, A.A.; Sultan, H.; Halami, M.Y.; Talaat, S.; Vahlenkamp, T.W. Molecular characterization of very virulent infectious bursal disease virus strains circulating in Egypt from 2003 to 2014. Arch. Virol. 2017, 162, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Eterradossi, N.; Arnauld, C.; Toquin, D.; Rivallan, G. Critical amino acid changes in VP2 variable domain are associated with typical and atypical antigenicity in very virulent infectious bursal disease viruses. Arch. Virol. 1998, 143, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J.; Sommer-Wagner, S.E. Amino acids contributing to antigenic drift in the infectious bursal disease Birnavirus (IBDV). Virology 2011, 409, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Soubies, S.; Courtillon, C.; Briand, F.-X.; Allée, C.; Amelot, M.; De Boisseson, C.; Lucas, P.; Blanchard, Y.; Belahouel, A.; et al. Infectious bursal disease virus in Algeria: Detection of highly pathogenic reassortant viruses. Infect. Genet. Evol. 2018, 60, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Lachheb, J.; Jbenyeni, A.; Nsiri, J.; Larbi, I.; Ammouna, F.; El behi, I.; Ghram, A. Full-length genome sequencing of a very virulent infectious bursal disease virus isolated in Tunisia. Poult. Sci. 2021, 100, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, L.; Chen, Y.; Lu, Z.; Gao, L.; Wang, Y.; Gao, Y.; Gao, H.; Cui, H.; Li, K.; et al. Cyclophilin A Interacts with Viral VP4 and Inhibits the Replication of Infectious Bursal Disease Virus. Biomed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.F.; Heine, H.G.; Sapats, S.I.; Parede, L.; Ignjatovic, J. Characterisation of an Indonesian very virulent strain of infectious bursal disease virus. Arch. Virol. 2002, 147, 1303–1322. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Wang, Y.; Zhang, E.; Han, X.; Yu, Z.; Liu, H. VP1 and VP3 Are Required and Sufficient for Translation Initiation of Uncapped Infectious Bursal Disease Virus Genomic Double-Stranded RNA. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Li, R.; Wang, H.; Huang, G.; Zhang, M. Residues of 862, 921 of VP3 are associated with virulence in infectious bursal disease virus. Nat. Sci. 2010, 02, 718–725. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, X.; Kang, Z.; Yu, F.; Qin, L.; Gao, H.; Gao, Y.; Wang, X. A single amino acid in the C-terminus of VP3 protein influences the replication of attenuated infectious bursal disease virus in vitro and in vivo. Antiviral Res. 2010, 87, 223–229. [Google Scholar] [CrossRef]
- Hernández, M.; Villegas, P.; Hernández, D.; Banda, A.; Maya, L.; Romero, V.; Tomás, G.; Pérez, R. Sequence variability and evolution of the terminal overlapping VP5 gene of the infectious bursal disease virus. Virus Genes 2010, 41, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Mundt, E. Tissue culture infectivity of different strains of infectious bursal disease virus is determined by distinct amino acids in VP2. J. Gen. Virol. 1999, 80, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.L.; Omar, A.R.; Hair-Bejo, M.; Aini, I.; Seow, H.F. Comparative analysis of viral RNA and apoptotic cells in bursae following infection with infectious bursal disease virus. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Goodwin, M.A.; Vakharia, V.N. Generation of a Mutant Infectious Bursal Disease Virus That Does Not Cause Bursal Lesions. J. Virol. 1998, 72, 2647–2654. [Google Scholar] [CrossRef] [PubMed]
- Le Nouën, C.; Toquin, D.; Müller, H.; Raue, R.; Kean, K.M.; Langlois, P.; Cherbonnel, M.; Eterradossi, N. Different Domains of the RNA Polymerase of Infectious Bursal Disease Virus Contribute to Virulence. PLoS One 2012, 7, e28064. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Ma, S.-J.; Zhang, Y.-G.; Li, X.-S.; Gao, X.-Y.; Cui, B.-A.; Chen, H.-Y. Genomic sequence analysis of a new reassortant infectious bursal disease virus from commercial broiler flocks in central China. Arch. Virol. 2013, 158, 1973–1978. [Google Scholar] [CrossRef]
Figure 1.
Map of Nigeria showing the locations where the samples were obtained for the study.
Figure 2.
Phylogenetic tree of nucleotide sequences of partial segment A of IBDV based on VP2 hypervariable region. Maximum likelihood method with the best-fit model (SYM+I+G4) automatically identified by IQ-TREE v.1.6.12. All genogroups except A3, and A0 have collapsed. The Ultra-fast bootstrap values higher than 70% are indicated near the nodes.
Figure 2.
Phylogenetic tree of nucleotide sequences of partial segment A of IBDV based on VP2 hypervariable region. Maximum likelihood method with the best-fit model (SYM+I+G4) automatically identified by IQ-TREE v.1.6.12. All genogroups except A3, and A0 have collapsed. The Ultra-fast bootstrap values higher than 70% are indicated near the nodes.
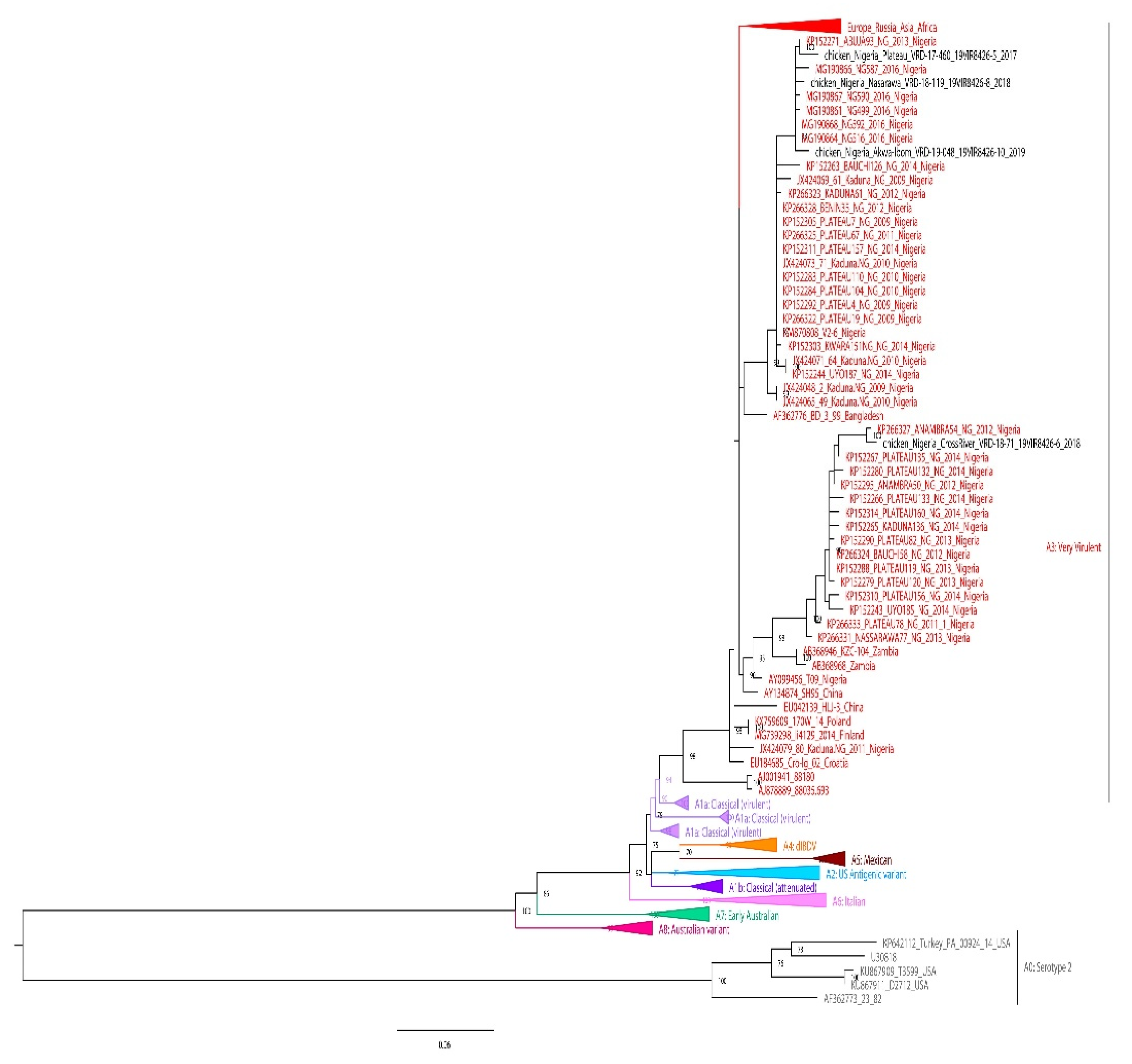
Figure 3.
Phylogenetic tree of nucleotide sequences of partial segment B of IBDV based on VP1 N-terminal domain and finger subdomain of central polymerase. Maximum likelihood method with the best-fit model (GTR+F+R4) automatically identified by IQ-TREE v.1.6.12. All genogroups except B5 (brown color) have collapsed. The Ultra-fast bootstrap values higher than 70% are indicated near the nodes.
Figure 3.
Phylogenetic tree of nucleotide sequences of partial segment B of IBDV based on VP1 N-terminal domain and finger subdomain of central polymerase. Maximum likelihood method with the best-fit model (GTR+F+R4) automatically identified by IQ-TREE v.1.6.12. All genogroups except B5 (brown color) have collapsed. The Ultra-fast bootstrap values higher than 70% are indicated near the nodes.
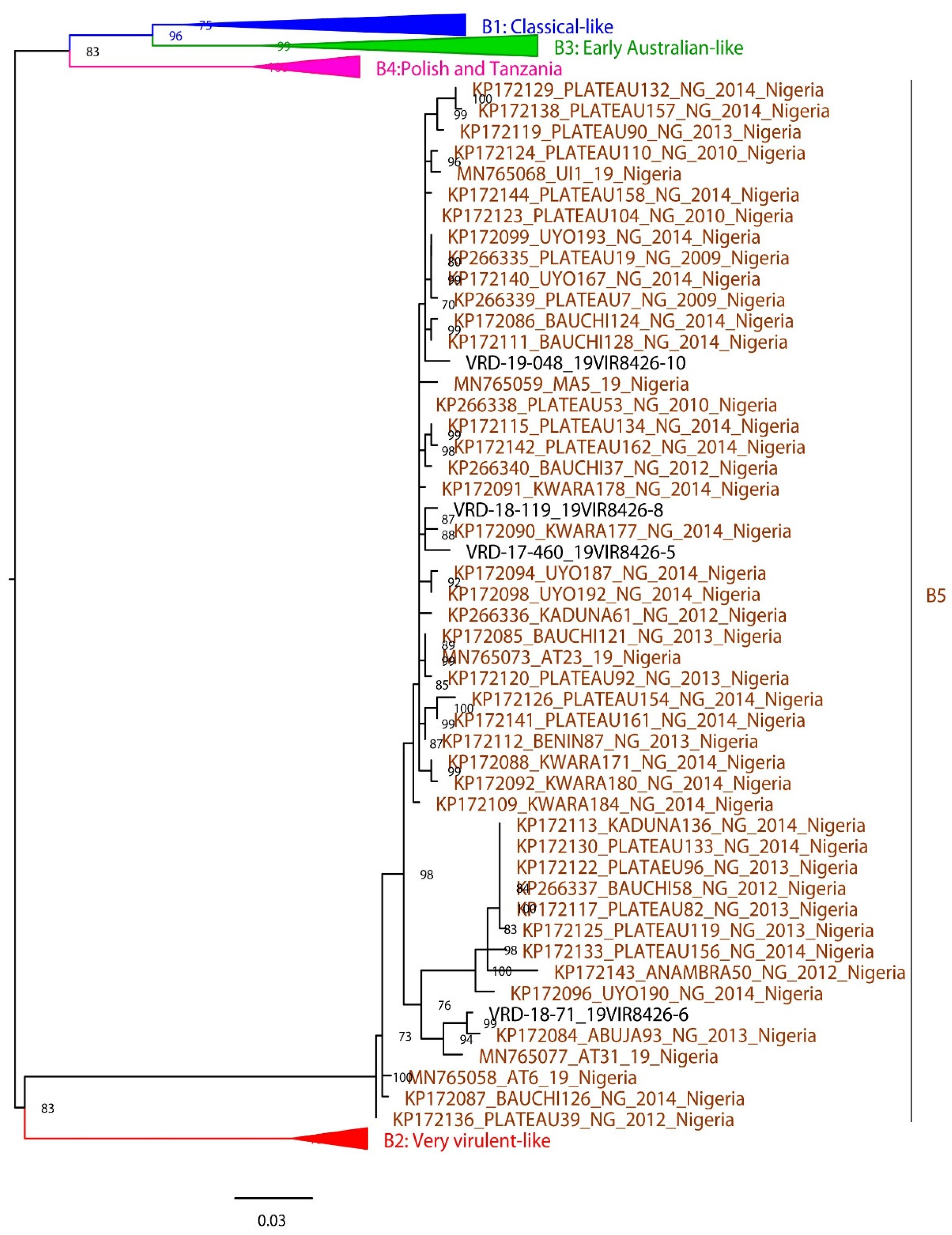

Table 1.
Epidemiological information on the IBD outbreaks from the investigated locations in Nigeria.
Table 1.
Epidemiological information on the IBD outbreaks from the investigated locations in Nigeria.
| Virus id | Year | Outbreak location | Specie | Vaccination status |
| VRD/17/460_19VIR8426-5 | 2017 | Plateau | Chicken | Not vacinated |
| VRD/18/71_19VIR8426-6 | 2018 | Cross-River | Chicken | IBDV twice |
| VRD/18/119_19VIR8426-8 | 2018 | Nasarawa | Chicken | IBDV twice |
| VRD/19/048_19VIR8426-10 | 2019 | Akwa-Ibom | Chicken | IBDV twice |
| Virus ID | Segment | Accession Number | % Similarity | Description | Accession Number | Country | |||
|---|---|---|---|---|---|---|---|---|---|
| VRD/17/460_19VIR8426-5 | A | OP311683 | 98.08 | IBDV 80/Kaduna.NG/2011 | JX424079.1 | Nigeria | |||
| B | OP311687 | 90.09 | IBDV strain 150133/3.2 | MF969112.1 | Algeria | ||||
| VRD/18/71_19VIR8426-6 | A | OP311684 | 96.61 | IBDV strain T09 | AY099456.1 | Nigeria | |||
| B | OP311688 | 90.43 | IBDV HuB-1 | GQ449693.1 | China | ||||
| VRD/18/119_19VIR8426-8 | A | OP311685 | 98.08 | IBDV 80/Kaduna.NG/2011 | JX424079.1 | Nigeria | |||
| B | OP311689 | 90.20 | IBDV strain 150133/3.2 | MF969112.1 | Algeria | ||||
| VRD/19/048_19VIR8426-10 | A | OP311682 | 98.16 | IBDV 80/Kaduna.NG/2011 | JX424079.1 | Nigeria | |||
| B | OP311686 | 92.91 | IBDV strain 150133/3.2 | MF969112.1 | Algeria |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



