
Submitted:
18 March 2023
Posted:
20 March 2023
You are already at the latest version
Abstract
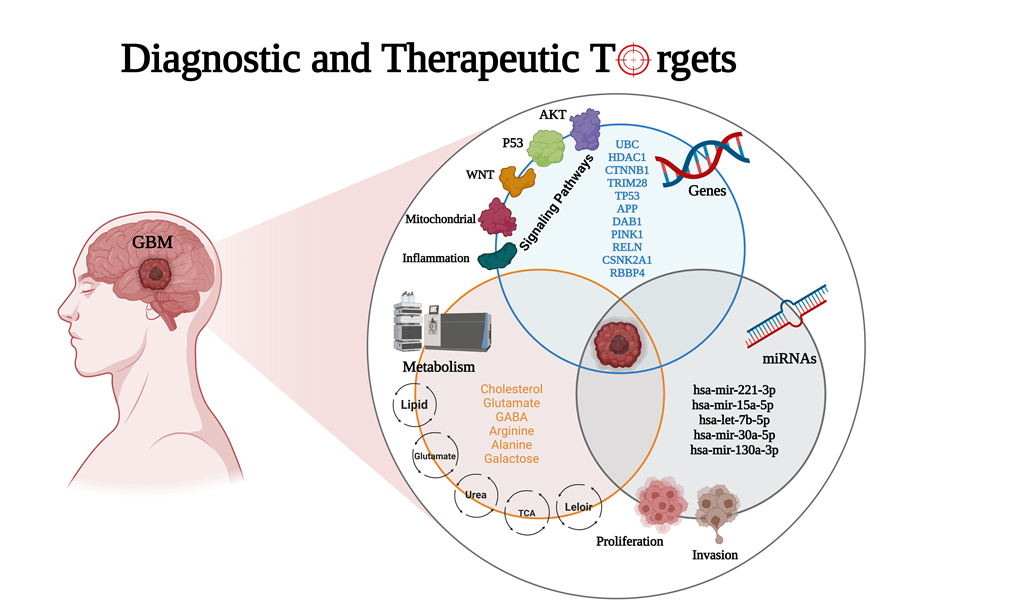
The most aggressive primary malignant brain tumor in adults is glioblastoma (GBM), which has poor overall survival (OS). There is a high relapse rate among patients with GBM despite maxi-mally safe surgery, radiation therapy, temozolomide (TMZ), and aggressive treatment. Hence, there is an urgent and unmet clinical need for new approaches to managing GBM. The current study identified modules (MYC, EGFR, PIK3CA, SUZ12, and SPRK2) involved in GBM disease through the NeDRex plugin. Furthermore, hub genes were identified in a comprehensive interaction network containing 7,560 proteins related to GBM disease and 3,860 proteins associated with signaling pathways involved in GBM. By integrating the results of the aforementioned analyses and performing centrality analysis again, eleven key genes involved in GBM disease were identi-fied. ProteomicsDB or Gliovis databases were used for determining the gene expression in normal or tumor brain tissue. The NetworkAnalyst and the mGWAS-Explorer tools identified miRNAs, SNPs, and metabolites associated with these 11 genes. Moreover, a literature review of recent studies revealed other lists of metabolites related to GBM disease. The enrichment analysis of iden-tified genes, miRNAs, and metabolites associated with GBM disease was done using ExpressAna-lyst, miEAA, and MetaboAnalyst tools. Further investigation of metabolite roles in GBM was done through the pathway, joint pathway, and network analyses. The results of this study identified 11 genes (UBC, HDAC1, CTNNB1, TRIM28, CSNK2A1, RBBP4, TP53, APP, DAB1, PINK1, and RELN), five miRNAs (hsa-mir-221-3p, hsa-mir-30a-5p, hsa-mir-15a-5p, hsa-mir-130a-3p, hsa-let-7b-5p), six metabolites (HDL, N6-acetyl-L-lysine, cholesterol, formate, N, N-dimethylglycine/xylose and X2. piperidinone) and 15 distinct signaling pathways that play an indispensable role in the GBM disease development. To establish early diagnostic methods and plan personalized GBM treatment strategies, the identified top genes-miRNAs and metabolite signatures can be targeted.
Keywords:
glioblastoma
; biomarker selection
; metabolomics
; pathway analysis
; personalized therapy
; network analysis
; inflammationomics
1. Introduction
Glioblastoma (GBM) is the most common high-grade primary malignant brain tumor with a poor prognosis [1,2]. It is urgently necessary to develop new therapeutic strategies for GBM with approved treatments due to its poor survival rates [3,4]. Innovative clinical trials of GBM are being conducted utilizing improved genetic and epigenetic profiling, in response to decades of basic science investment in the disease [5,6]. Proneural, neural, classical, and mesenchymal are the four molecular subtypes of GBM [7]. The complex genetic profile of GBM is revealed by multi-omics studies of the Cancer Genome Atlas Research Network (TCGA), the Chinese Glioma Genome Atlas (CGGA), and other databases. Genetic alterations, gene transcription, and DNA methylation are molecular markers to determine prognosis and therapy selection. Molecular subtype signatures with higher resolution are essential for more effective personalized therapy [8].
There is a possibility of developing a treatment for GBM if a gene signature can be identified [9]. This can be useful for diagnosis, treatment, prognosis prediction, and drug development. By analyzing the differential gene expression of astrocytomas or non-GBM gliomas, the researchers could identify a 33-gene signature of GBM, using the GSE108474 database by Gene Set Enrichment Analysis (GSEA) and machine learning analysis. The 33 discovered signature genes included the downregulated genes CHST9, CSDC2, ENHO, FERMT1, IGFN1, LINC00836, MGAT4C, SHANK2, and VIPR2, as well as the overexpressed genes COL6A2, ABCC3, COL8A1, FAM20A, ADM, CTHRC1, PDPN, IBSP, MIR210HG, GPX8, MYL9, and PDLIM4. According to CELLO2GO's protein functional analysis, these signature genes may be involved in controlling several biological processes, such as the formation of anatomical structures, cell proliferation and adhesion, and signal transduction, and many of the genes were annotated as being receptive to stress [10]. Historically, GBMs were considered among the most heterogeneous tumors due to their diverse cellular organization and histological appearance. In addition to Telomerase Reverse Transcriptase (TERT) promoter mutations, they commonly carry copy number changes in chromosomes 7 and 10 (+7/−10). Genetic changes such as amplifying Epidermal Growth Factor Receptor (EGFR), Platelet-Derived Growth Factor Receptor Alpha (PDGFRA), and Cyclin-Dependent Kinases 4 and 6 (CDK4/6), deletions or inactivating mutations of TP53, Phosphatase and Tensin Homolog (PTEN), Neurofibromin 1 (NF1), and CDKN2A/B could induce variability of tumors among GBM patients [11]. Besides the crucial role that gene signatures play in GBM pathogenesis, their miRNAs can also play a pivotal role in the disease. To distinguish GBM from other CNS malignancies, a comprehensive, integrated analysis of microarray data was conducted. One hundred seventy-six samples from 118 individuals who had been diagnosed with GBM were included in the study for identifying dysregulated miRNAs. The only associations with GBM were found for the miRNAs hsa-miR-21-3p, hsa-miR-338-5p, hsa-miR-485-5p, hsa-miR-491-5p, and hsa-miR-1290. This characteristic was thoroughly described, focusing on tumor invasion, progression, and patient survival. Therefore, these five naturally occurring molecules, which exhibit differential expression in GBM, are proposed as prospective therapeutic targets. They affect various genes implicated in important signaling pathways, such as MAPK/ERK, calcium, PI3K/AKT, mTOR, and Wnt [12]. Additionally, the integrating bioinformatics and clinical analyses demonstrated the potential use of miR-1224-5p, as a prognostic and therapeutic biomarker in GBM [13]. Based on the results of another study, PLK1, CCNA2, CCNB2, and AURKA were selected as potential diagnostic marker genes based on crosstalk genes in the KEGG, PPI network, and WGCNA studies. According to the survival study, a low overall survival (OS) rate was substantially correlated with increased mRNA expression of PLK1, CCNA2, and AURKA. In particular, it was discovered that hsa-let-7b-5p functions as a key miRNA, controlling potential glioma-related genes. It was verified that hsa-let-7b-5p could obstruct glioma cell motility, invasion, and cell cycle [14].
In this study, the mechanistic investigation of GBM disease is improved and the results on the transcriptomics, metabolomics, and proteogenomics value as multi-omics approach contribution in biomarker discovery for GBM diagnosis/therapeutics/prognostic are addressed based on the phenotypic insight. Our findings provide a new point of reference for the prognostic prediction of GBM and contribute to an in-depth understanding of the molecular mechanisms in GBM development. Furthermore, these novel signature genes might be exploited as therapeutic targets for GBM.
2. Materials and Methods
2.1. Disease Module Identification
GBM-related disease modules were found using the NeDRex plugin version 1.0.0 (https://nedrex.net/), implemented in the Cytoscape platform (version 3.7.2). Two different NeDRex algorithms, MuST (Multi-Steiner trees) and DIAMOnD (DIseAse MOdule Detection), were used for this investigation. To extract a connected subnetwork engaged in the disease pathways, MuST combines a variety of approximate Steiner tree calculations that are not all unique [15,16]. Based on the idea that the connectivity importance for known disease proteins is highly distinctive, the DIAMOnD algorithm determines the disease module surrounding a set of known disease genes or proteins (seeds) [17]. Under the headings of " Set of genes obtained from the two algorithms of MuST and DIAMOnD are listed and used in the study” (122 and 305 genes, respectively) (Supplementary Tables S1 and S2), the genes collected from various algorithms were each given special consideration. A schematic figure of the bioinformatics approaches designed in this study is shown in Figure 1.
2.2. GBM-Related Protein-Protein Interaction Network
Several databases, including DisGeNET (https://www.disgenet.org) [18,19], STRING (https://string-db.org) [20], KEGG (https://www.genome.jp/kegg) [21], and the Human microRNA Disease Database (HMDD) (https://www.cuilab.cn/hmdd) [22], were used to identify significant proteins, associated in GBM disease. Four keywords and disease ID were used to extract data from DisGeNET, including GBM multiforme (C1621958), brain GBM (C0349543), brain stem GBM (C1332610), and GBM multiforme, somatic (C4016231). The STRING database was checked, using GBM multiforme and brain GBM multiforme keywords. The KEGG database was used to extract information about the pathways involved in the development of GBM disease, including glioma (hsa05214), mTOR signaling pathway (hsa04150), p53 signaling pathway (hsa04115), cell cycle (hsa04110), cytokine-cytokine receptor interaction (hsa04060), and signaling pathways of calcium (hsa04020), ErbB (hsa04012), and MAPK (hsa04010). The acquired results were combined for network reconstruction to provide a list of 7,560 proteins (Supplementary Table S3). In the next step, the protein-protein interaction network between these 7,560 cases was reconstructed using the GeneMANIA plugin [23] and Cytoscape software (version 3.7.2). The following step involved a centrality analysis with various characteristics, including degree, closeness, betweenness, centroid, eigenvector, bridge, and eccentricity [24]. The first 20 proteins from each centrality with the highest scores were chosen (Supplementary Table S4) and then combined to determine the most important proteins. Consequently, 48 fundamental proteins were found and chosen for additional examination after processing 7,560 initial entries (Supplementary Table S5).
2.3. Network Reconstruction of GBM-Related Signaling Pathways
A third analysis level was also established to learn about GBM disease and determine the essential proteins. Two techniques were established to accomplish this: i) A literature review on genes associated with epithelial-mesenchymal transition (EMT), cytoskeleton remodeling, autophagy, secretory autophagy, and metabolism and ii) Compiling the genes from 78 distinct signaling pathways. Table 1 lists the names of these pathways. The data from steps: i and ii were combined in the following step to creating a different list of 3,860 genes (Supplementary Table S6). In the next step, centrality analysis was performed similarly to the previous step (Supplementary Table S7), and 20 proteins with the highest scores from each of the centralities were selected and combined. After processing 3,860 initial entries, 35 essential proteins were discovered and selected for further analysis (Supplementary Table S8).
2.4. Combining the Findings from the Aforementioned Four Stages of Research and Integrated Database
The final gene list and network related to the GBM disease were created by combining the results of four earlier study methodologies: MuST algorithm (122 genes), DIAMOnD algorithm (305 genes), glioblastoma-related protein-protein interaction network (48 genes), and network analysis of signaling pathways associated with GBM (35 genes). In the end, 351 genes (Supplementary Table S9) were integrated, and a network was reconstructed using the GeneMANIA plugin and Cytoscape software (version 3.7.2). Similar to the previous steps, we performed a centrality analysis and selected five proteins with the highest scores from each centrality (Supplementary Table S10). Using the results of this stage, 11 essential genes (Table 2) related to GBM disease were identified, and a miRNA-gene regulatory network was drawn via NetworkAnalyst (https://www.networkanalyst.ca/) [25,26] and miRTarBase v8.0 [27]. Then, using NetworkAnalyst, miRNA centrality analysis was carried out based on degree and betweenness. Using integration results, we identified five essential miRNAs based on network characteristics (Table 3).
2.5. Study of Eleven Critical Proteins in Normal Brain and Brain Tumor Expression Datasets
To ensure the expression of eleven critical proteins in the normal brain tissue and also investigate the mRNA expression (Log2) of these genes in eleven different situations (Non-tumor, GBM, wild-type, mutant, primary, secondary, recurrent, classical, mesenchymal, neural, and proneural), we used ProteomicsDB (https://www.proteomicsdb.org/) [28,29] and Gliovis (http://gliovis.bioinfo.cnio.es) [30], as a unique web-based tool to expeditiously access data related to brain research, respectively. Gliovis database can also be used to investigate gene expression correlations. In ProteomicsDB, transcriptomic data from the Human Protein Atlas (https://www.proteinatlas.org/) [31] and BGEE (https://bgee.org/) [32] can be integrated. At the same time, raw expression data in the Gliovis database came from various sources: ArrayExpress (https://www.ebi.ac.uk/arrayexpress/, accessed October 31, 2016) [33], Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, accessed October 31, 2016) [34], and Firebrowse (http://firebrowse.org, accessed October 31, 2016) [35].
2.6. Identification of Significant Metabolites and Snps That Interact with Eleven Essential Genes
We identified significant SNPs and metabolites, interacting with eleven essential genes using the mGWAS-Explorer database (https://www.mgwas.ca/) [36], a user-friendly web-based tool that connects SNPs, metabolites, genes, and their known disease relationships using sophisticated network visual analytics. To more deeply identify the role of metabolites in glioma and GBM, we extracted the list of essential metabolites (182 cases) (Supplementary Table S11) from recent studies [37,38]. In the next step, pathway, joint-pathway, and network analyses were performed using MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/) database [39,40].
2.7. Enrichment Analysis
Gene ontology and pathway enrichment analyses were carried out, using the ExpressAnalyst (https://www.expressanalyst.ca/) [25], microRNA enrichment analysis and annotation (miEAA) (https://www.ccb.uni-saarland.de/mieaa tool/) [41,42], and MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/) [39] databases, respectively. To better understand the results, FDR < 0.05 was used to interpret the outcomes of the analysis.
3. Results
3.1. The Network Obtained from the NeDRex Plugin to Identify Disease Modules
Proteins in GBM were identified by delineating the disease network through the NeDRex plugin (Figure 2). In the following, step disease modules were determined by two distinct algorithms (MuST and DIAMOnD). The MuST algorithm identified five disease modules around the essential genes MYC, EGFR, PIK3CA, SUZ12, and SPRK2. IRAK1, PTK2, and BMI1 also represent bridging roles between MYC-EGFR, EGFR-PIK3R1, and SUZ12-SPRK2, respectively (Figure 3). The DIAMOnD algorithm identified only one module with a high number of genes (305 genes), compared to the MuST algorithm (122 genes).
3.2. miRNA-Gene Regulatory Network Analysis
The interaction network of eleven essential proteins identified with corresponding miRNAs is shown in Figure 4. After the centrality analysis, it was inevitable to investigate the interaction of the five prominent miRNAs (hsa-mir-221-3p, hsa-mir-30a-5p, hsa-mir-15a-5p, hsa-mir-130a-3p, hsa-let-7b-5p) with eleven identified genes (Figure 5). The two miRNAs (hsa-mir-221-3p, hsa-mir-30a-5p) showed higher degree and betweenness centrality levels, and their targets (TP53, CTNNB1, UBC, TRIM28, HDAC1) seem to play a more critical role in GBM than others.
3.3. Analyzing the Status of Identified Gene Expression in Healthy and Malignant Brain Tissue
Using microarray and RNA-Seq data, transcriptomics results from proteomicsDB revealed that all eleven identified genes were expressed in healthy brain tissue (Figure 6). In addition, Gliovis' findings revealed that except for the CSNK2A1, the remaining ten genes displayed a significant variation between the GBM and the non-tumor condition. Six genes represent an increased expression in the GBM state, whereas four genes were downregulated. Furthermore, all eleven specific genes were altered during the primary stage of the tumor (Table 4). As a consequence of the correlation analysis, ten positive correlations were found between UBC (APP), HDAC1 (TP53, RBBP4, TRIM28, CTNNB1), RBBP4 (CTNNB1, TP53), TRIM28 (TP53, RBBP4, CSNK2A1), and eight negative correlations were found between RBBP4 (APP), PINK1 (TRIM28, RBBP4, TP53, HDAC1), and DAB1 (UBC, HDAC1, TP53) (Figure 7).
3.4. Enrichment Analysis
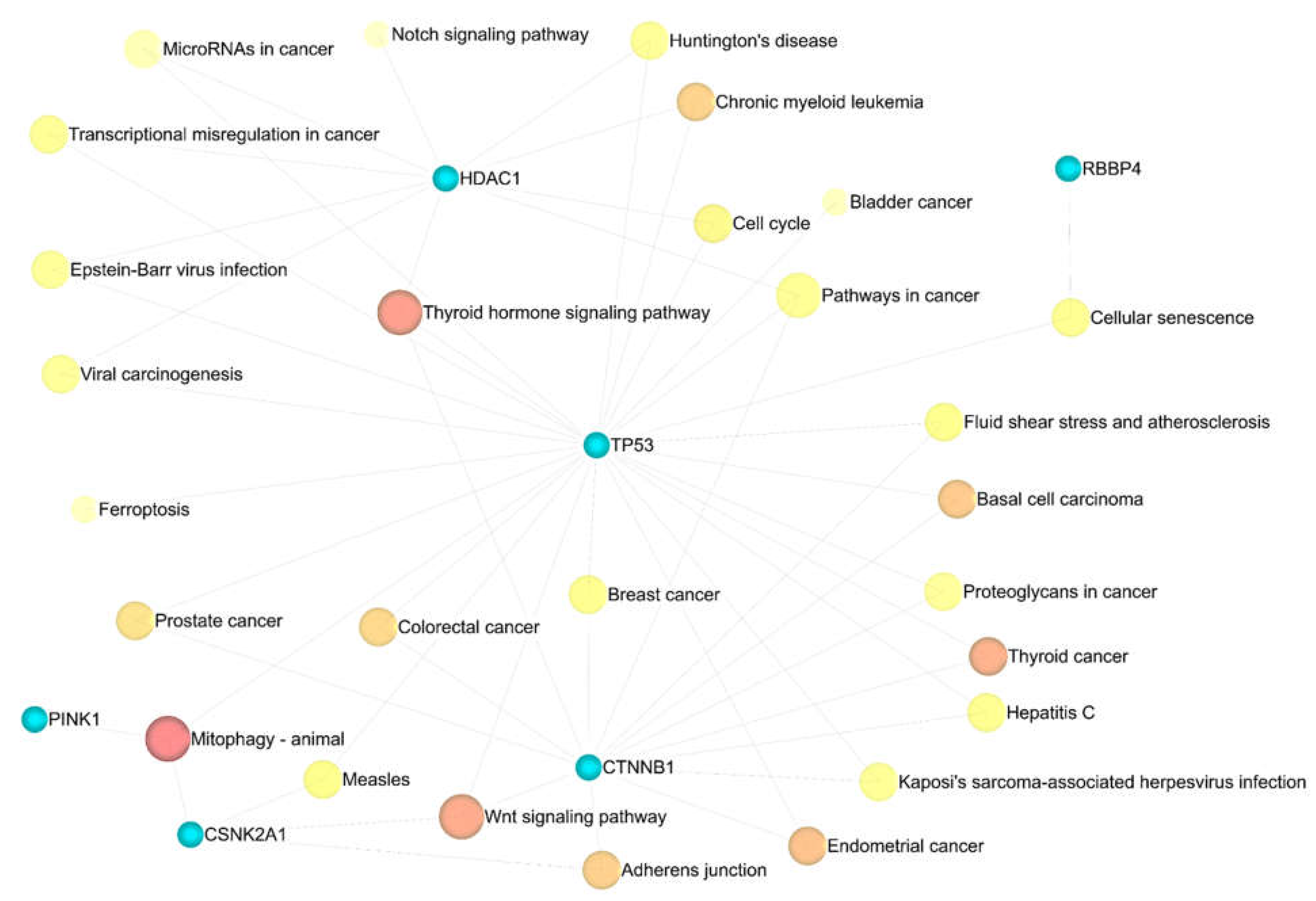
Eleven significant GSEA results were categorized into four axes. Negative control of the cellular process (FDR = 0.00165), negative control of the cell cycle (FDR = 0.00165), protein phosphorylation (FDR = 0.00165), negative control of the biological process (FDR = 0.00165), and negative control of the apoptotic process (FDR = 0.00165) were significant in the biological process (Supplementary Table S12). Enzyme binding (FDR = 0.000316), positive transcription regulation (FDR = 0.0055), DNA-dependent negative transcription regulation (FDR = 0.0188), and DNA-dependent transcription from RNA polymerase II promoter (FDR = 0.0315), all played a crucial role in molecular function (Supplementary Table S13). Nuclear chromatin (FDR = 4.04E-06), nuclear chromosome part (FDR = 3.01E-05), nuclear chromosome (FDR = 3.63E-05), chromatin (FDR = 3.63E-05), and histone deacetylase complex (FDR = 0.000161) in cellular components (Supplementary Table S14) were indispensable, as well as mitophagy (FDR = 0.00978) and Wnt signaling pathway (FDR = 0.046) in KEGG pathway enrichment analysis (Supplementary Table S15). Additionally, miEAA-related results showed that fatty acid biosynthesis (0.0078415), galactose metabolism (0.0078415), mucin-type O-glycan biosynthesis (0.0233921), autophagy (0.0264018), might also be crucial in GBM disease (Supplementary Table S16).
Figure 8.
The results of gene ontology analysis between eleven identified genes. (A) Molecular function, (B) Cellular components. Darker shade has a greater significance.
Figure 8.
The results of gene ontology analysis between eleven identified genes. (A) Molecular function, (B) Cellular components. Darker shade has a greater significance.
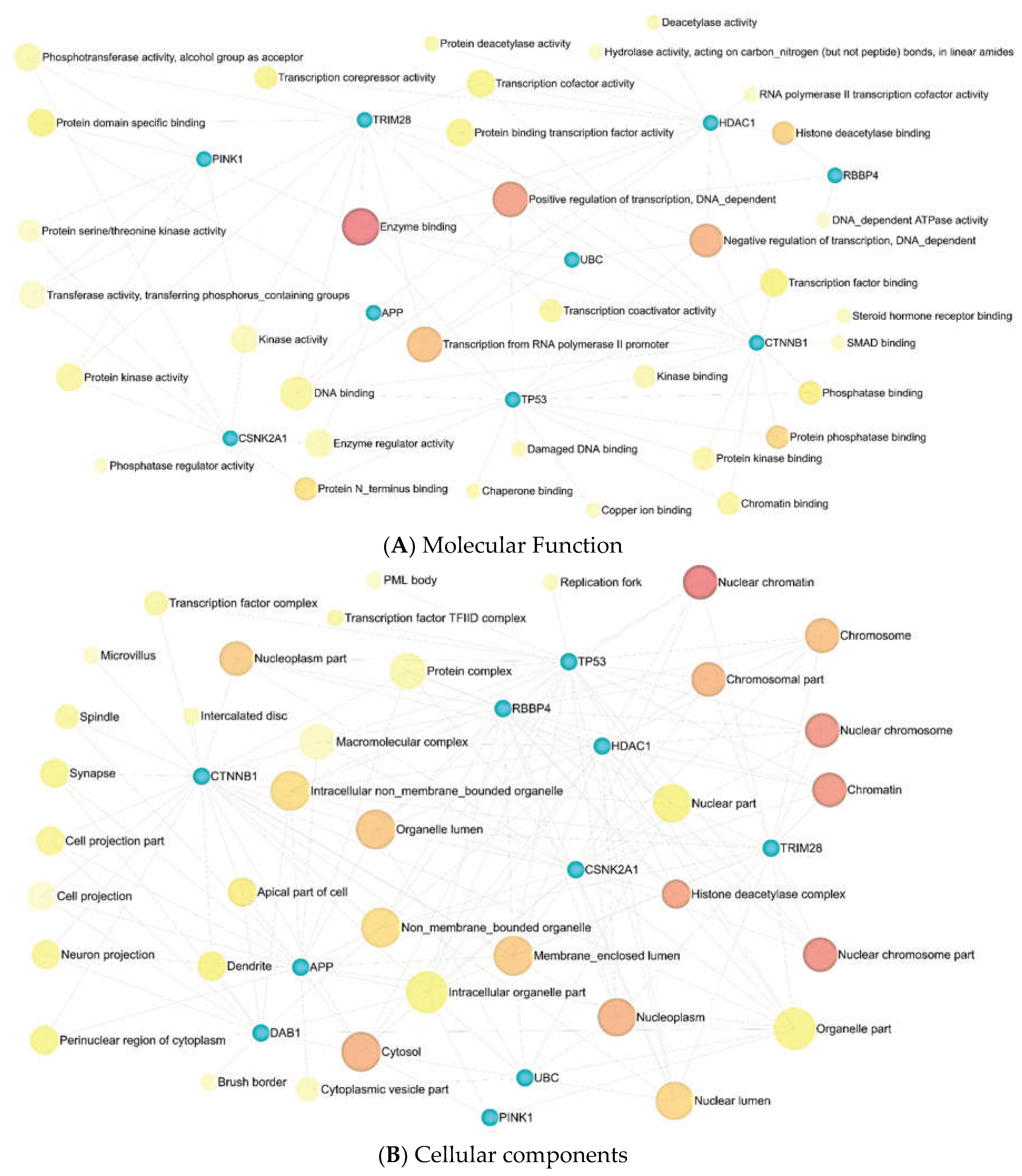
Figure 9.
The association of the KEGG pathway enrichment analysis results with eleven identified genes. Darker shade has a greater significance.
Figure 9.
The association of the KEGG pathway enrichment analysis results with eleven identified genes. Darker shade has a greater significance.
The third level of analysis was done using metabolites, KEGG, and SMPDB databases [43]. The FDR-based three main pathways obtained from KEGG (Supplementary Table S17) were aminoacyl-tRNA biosynthesis (1.39E-08), arginine biosynthesis (3.94E-07), alanine, aspartate, and glutamate metabolism (2.03E-06). In comparison, three other main pathways were identified through the SMPDB databases (Supplementary Table S18), including glutamate metabolism (9.34E-09), urea cycle (9.34E-09), and arginine and proline metabolism (3.24E-08) (Figure 10).
3.5. Metabolic Pathway Analysis
The outcomes of metabolic pathway enrichment analysis and pathway topology analysis were combined for this investigation. Four different sorts of results were produced per factors taken into account, and Table 5 displays the various criteria taken into consideration.
The results were analyzed and summarized based on three criteria: 1. Selection of the first 5 cases in each of the centralities - Relative-betweenness Centrality (R-b C) and Out-degree Centrality (O-d C)- based on the highest score, obtained in the impact parameter, 2. In the next step, the results were classified based on FDR, and 5 cases (phenylalanine, tyrosine, and tryptophan biosynthesis, synthesis, and degradation of ketone bodies, one-carbon (1C) pool by folate, trehalose degradation, glycerol phosphate shuttle) were excluded because they were not significant in the metabolic pathway analysis (red color), 3. The common items in both cases (R-b C, O-d C) or databases (KEGG, SMPDB) were considered vital and further discussed. Two items were obtained from the KEGG database (Nitrogen metabolism, alanine, aspartate, and glutamate metabolism) and three from the SMPDB database (alanine metabolism, aspartate metabolism, and malate-aspartate shuttle) (Table 6, Supplementary Tables S19–S22).
3.6. Joint Pathway Analysis
We used joint pathway analysis to analyze eleven identified essential genes and 182 distinctive metabolites within metabolic pathways, simultaneously. Based on the topology measure used, three types of results were obtained (Table 7). The results were analyzed and summarized based on four criteria: 1. Selection of the first 10 cases in each of the centralities based on the highest score obtained in the impact parameter, 2. In the next step, the results were classified based on FDR, and 5 cases (1C pool by folate, p53 signaling pathway, phosphatidylinositol signaling system, longevity regulating pathway, and mitophagy-animal) were excluded because they were not significant in the joint pathway analysis, 3. Considering the results obtained from all three centralities, the two items (citrate cycle and arginine biosynthesis) observed in all three, were selected and discussed further (Table 8, Supplementary Tables S23–S26). The results were combined in one table to get a comprehensive overview of the pathway enrichment analysis at different levels (eleven essential proteins, five miRNAs, and 182 metabolites), pathway, and joint pathway analyses (Table 9).
3.7. Gene-Metabolite Interaction Network
Drawing the interaction network between genes and metabolites showed that APP and TP53 genes are related to each other through 5 factors (adenosine triphosphate, ADP, glycerol, L-glutamic acid, and L-lysine). Moreover, the connection between RELN, CTNNB1, and CSNK2A1 with APP has been shown through Gamma-aminobutyric acid (GABA), palmitic Acid, and glycerol, respectively (Figure 11).
3.8. Identification of SNPs-Related Metabolites and Genes
Using eleven critical genes as input data in the mGWAS-Explorer database, six metabolites and 23 SNPs were identified (Table 10, Supplementary Table S27). Additionally, we determined the top 25 SNPs via 182 metabolites, as input data in MetaboAnalyst 5.0 database (Figure 12).
4. Discussion
GBM is incurable cancer characterized by poor survival rates and a high recurrence rate caused by glial cells that surround and support neurons. In terms of treatment resistance, complexity, and lethality, it is one of the deadliest types of cancer. Even though surgery followed by radiotherapy and chemotherapy are the standard treatments for GBM, its prognosis is dismal, with a median survival time of only 15 months [44]. In spite of extensive research into the pathogenesis of GBM, the disease continues to have a poor outcome due to a lack of understanding of genetic risk factors. Gene sequencing studies have made it possible to learn much more about GBM genetics and epigenetics in recent years [45]. Identifying practical molecular biomarkers is crucial to help clinicians treat glioma patients as effectively as possible. Due to changes in miRNA target binding sites and the miRNA processing machinery in tumor cells, miRNAs have been discovered to be intimately associated with malignancies. The most significant benefit of biomarkers is their assistance in clinical decision-making [46]. A significant approach for diagnosing and treating GBM is to use molecular biomarkers. In the current study, genes, miRNAs, and metabolites were used to develop a panel of predictive biomarkers for GBM.
Based on the biological pathways, the identified genes in the GBM biomarker panel were classified into different signaling pathways such as the AKT (MYC, BMI1, EGFR, PIK3CA, and PTK2, UBC), the inflammation (IRAK1 and APP), the P53 (HDAC1, P53, and TRIM28), the WNT (CTNNB1), and the mitochondrial signaling pathways (DAB1, PINK1, and RELN). The Akt pathway, or PI3K-Akt pathway, plays an imperative role in fundamental cellular processes such as protein synthesis, proliferation, and survival. Additionally, AKT regulates angiogenesis and metabolism [47]. Most human cancers are associated with a transcription factor called MYC, a member of the bHLHZip family. Some cellular functions are controlled by MYC, including cell growth and proliferation, differentiation, and programmed cell death. There are also numerous human tumors with elevated levels of MYC, including GBM [48]. There is a correlation between Myc expression and glioma grade, and 60 to 80% of GBM exhibit elevated Myc [49,50]. Moreover, it was shown that the inhibition of Myc in gliomas reduces proliferation and increases apoptosis [51]. The BMI-1 is another important gene related to the AKT signaling pathway. The BMI1 gene belongs to the polycomb group (PcG) gene family and is a transcriptional repressor of several genes that govern cell proliferation and differentiation throughout life [52,53,54,55,56]. It was first discovered that BMI1 cooperated with the oncogene c-MYC during murine lymphomagenesis [57,58]. BMI1 has been shown to regulate glioblastoma stem cells (GSCs) via differential gene networks in CD133POS brain tumor-initiating cells [59]. According to the results of another study, GSCs were targeted via the combination of BMI1 and EZH2. It was found that proneural GSCs are preferentially sensitive to EZH2 disruption, while mesenchymal GSCs are more sensitive to BMI1 inhibition. EZH2 and BMI1 targeting proved more effective than either agent alone in GBM due to the presence of both proneural and mesenchymal GSCs in GBM [60].
GBM pathogenesis is also affected by EGFR. Oncogenes such as EGFR are frequently amplified in GBMs. There is evidence that EGFR overexpression is associated with more aggressive GBM phenotypes in most primary GBMs, as well as some secondary GBMs [61]. An analysis of the TCGA GBM database uncovered a subgroup with EGFR amplification and TP53 mutations that are almost mutually exclusive, suggesting EGFR is involved in regulating the function of wild-type p53 (wt-p53). EGFR signaling inhibits the function of wt-p53 in GBM by facilitating the interaction between p53 and DNA-dependent protein kinase catalytic subunit (DNA-PKcs) [62]. The overexpression of YT521-B homology domain family 2 (YTHDF2) clinically was correlated with poor prognosis in a glioma patient. As a result of EGFR activation in most GBMs, YTHDF2 is overexpressed through the EGFR/SRC/ERK pathway. Signaling through EGFR/SRC/ERK stabilizes YTHDF2 by phosphorylating serine 39 and threonine 381 [63].
For AKT to be activated, phosphatidylinositol 3-kinase (PI3K) must first be activated, which is activated by several upstream signaling pathways, including insulin receptors, receptor tyrosine kinases, G protein-coupled receptors, and cytokine receptors. Infiltrative gliomas frequently activate the PI3K signaling pathway, which promotes the growth and survival of cells. It has been reported that 6–15% of glioblastomas contain activating mutations in the PIK3CA. There is evidence that PIK3CA activating mutations are associated with an earlier recurrence of GBM in adults and a shorter survival time [64]. PTK2, also known as focal adhesion kinase 1 (Fak1), is associated with focal adhesions and mediates signal transduction from integrin receptors to the MAPK and PI3k/Akt pathways [65,66]. PTEN may dephosphorylate PTK2, thereby regulating cell spreading, migration, and invasion [67]. Furthermore, a strong expression of the PTK2 protein was demonstrated by immunohistochemical analysis in most anaplastic astrocytomas and glioblastomas [68]. Also, elevated PTK2 protein levels were detected in astrocytic gliomas [69]. Ubiquitin-dependent mechanisms could be exploited therapeutically for GBM. It was indicated that the ubiquitin system is involved in core signaling pathways, including EGFR, TGF-β, p53, and stemness-related pathways in GBM [70]. In addition, it was shown that inhibition of ubiquitin signaling could reverse metabolic reprogramming and suppresses GBM growth. The regulation of protein stability by the ubiquitin-proteasome system (UPS) represents an important control mechanism of cell growth. UPS deregulation is mechanistically linked to the development and progression of various human cancers, including GBM. Praja2, a RING E3 ubiquitin ligase, is preferentially expressed in primary GBM lesions, expressing the wild-type isocitrate dehydrogenase 1 gene (IDH1). The researchers found that praja2 ubiquitylates and degrades the kinase suppressor of Ras 2 (KSR2). As a consequence, praja2 restrains the activity of downstream AMP-dependent protein kinase in GBM cells and attenuates oxidative metabolism [71].
Inflammation plays a crucial role in tumor development, which is why interleukin-1 receptor-associated kinases (IRAK), indispensable mediators of interleukin-1 receptor (IL1R) and Toll-like receptor (TLR)-inflammatory signaling, may contribute to the biological function of human cancers. According to the study, IRAK expression was extensively altered and related to patient survival in pan-cancer. Furthermore, in vitro and in vivo studies have demonstrated that the highest expressed form of IRAK1 in low-grade gliomas (LGG) inhibits cell apoptosis and increases malignancy [72]. In mammals, amyloid precursor protein (APP) and amyloid precursor-like protein 1 (APLP1) and amyloid precursor-like protein 2 (APLP2) are highly conserved [73]. Neuronal homeostasis, development, and neural transmission are critical functions of APP and APLP in Alzheimer's disease (AD). Cyclooxygenase-2 (COX-2), cytosolic phospholipase, interleukin-1β (IL-1β), and the β-amyloid precursor protein of proinflammatory and neurodegenerative genes were found to be up-regulated in the American Tissue Culture Collection of glioma and GBM. It has been shown that these genes are associated with inflammatory signaling cascades and gliosis in AD. Molecular genetic studies of the pathogenic signaling axis of βAPP–COX-2–CPL–IL-1β could reveal additional new therapeutic targets for the treatment of this devastating and lethal neurological condition [74]. In addition, GBM is positively associated with mortality in AD [75]. In glioma mouse models, immunostaining revealed that amyloid-β (1–42) deposition is observed in glioma tumors and nearby blood vessels. APLP2 is also closely related to gliomas and can be detected using thioflavin [76].
There is an up-regulation of the HDAC class I isoforms HDAC1 and HDAC2 in GBM cell lines, compared with non-neoplastic brain tissues [77,78]. GBM cells are inhibited from proliferating, migrating, and invading when HDAC1 and HDAC2 expressions are silenced. Similarly, HDAC3 is overexpressed in aggressive glioma cell lines and is associated with poor prognosis and OS of GBM patients [79]. A selective histone deacetylase inhibitor was shown to induce autophagy and cell death in GBM cells by downregulating SCNN1A [80]. The HDAC family was clinically significant for gliomas. A significant correlation was found between most members of the HDAC family and glioma grade, IDH1 mutation, and 1p/19q co-deletion. Among the HDAC1-related signatures for precise prognosis prediction in glioma, HDAC1 indicates prognosis and immune infiltration [81]. GBM is commonly associated with TP53 mutations, which are associated with poorer prognoses and a poorer response to conventional therapies (chemoradiotherapy). Approximately 84% of GBM patients exhibit dysregulation of the p53-ARF-MDM2 pathway, a finding that is confirmed in 94% of GBM cell lines adopted for in vitro assays [82,83]. There is a transcriptional co-repressor known as TRIM28 that is involved in the regulation of cancer. The expression of TRIM28 in gliomas was significantly higher than in non-glioma controls [84]. Further, expression of TRIM28 was positively correlated with tumor malignancy and associated with poor OS and progression-free survival (PF). The results of another study indicated that TRIM28 promotes the proliferation of GBM cells and activates autophagy [85]. Diverse cellular functions are mediated by PI3K/Akt-WNT signaling interactions in GBM, including cell proliferation, EMT, metabolism, and angiogenesis [47]. Canonical WNT signaling plays an important role in cell proliferation and development, and its aberrant activation has been linked to tumorigenesis [47]. The main effector of the WNT canonical pathway is CTNNB1/β-catenin, which has cooperative properties with transcription factors from the TCF/LEF (transcription factor/lymphoid enhancer binding factor) families to control gene expression via WNT signaling [86]. A study has shown that inhibiting WNT-CTNNB1 signaling enhances the SQSTM1 expression and sensitizes GBM cells to autophagy blockers [87].
According to the study, both RELN and its main downstream effector, DAB1, are silenced in GBM compared to non-neoplastic tissue, and their mRNA expression is inversely correlated with the grade of the disease [88]. According to the results of another study, reelin, and DAB1 transcripts were more abundant in the peritumoral area and in peritumoral-derived CSCs that originated from the GBM tumor core. It is possible that reelin signaling plays a role in the pathology of GBM and in the recurrence of tumors that typically originate from the peritumoral region [89]. There is evidence that PINK1 is an important regulator of the Warburg effect and a negative regulator of the growth of GBM. Loss of PINK1 contributes to the Warburg effect by stabilizing hypoxia-inducible factor-1A (HIF-1A) and reducing pyruvate kinase muscle isozyme 2 (PKM2) activity, both critical regulators of aerobic glycolysis. Through FOXO3a, a master regulator of oxidative stress and superoxide dismutase 2, PINK1 suppresses ROS and tumor growth. A loss of PINK1 has been observed in many human brain tumors, including GBM, and has been correlated with poor patient survival. In orthotopic mouse xenograft models and a transgenic drosophila GBM model, PINK1 overexpression attenuates GBM growth in vivo. In this regard, PINK1 is a negative regulator of growth and the Warburg effect in GBM [90]. A loss-of-function mutation in PINK1 causes mitochondrial defects and the degeneration of dopaminergic neurons, a hallmark of Parkinson's disease (PD). As opposed to this, increased expression of PINK1 was observed in different cancers, suggesting that PINK1 plays a role in neurodegeneration and tumorigenesis [91]. Mitophagy, a selective autophagy of mitochondria, is crucial for quality control since it can efficiently degrade, remove and recycle malfunctioning or damaged mitochondria [92]. It has been demonstrated that rapamycin, an inhibitor of mTOR, can rescue mitochondrial alterations. Specifically, rapamycin induces the expression of genes that promote mitochondrial autophagy (PINK1, PARKIN, ULK1, and AMBRA1) and mitochondrial fission (FIS1, and DRP1) [93]. In addition, it has been demonstrated that platelet-derived growth factor (PDGF) signaling induces N6-methyladenosine (m6A) accumulation in GSCs to regulate mitophagy. A PDGF ligand stimulates the transcription of early growth response 1 (EGR1), which promotes the proliferation and self-renewal of GSCs by inducing methyltransferase-like 3 (METTL3). Through the regulation of m6A modification of optineurin (OPTN), the PDGF-METTL3 axis inhibits mitophagy. In GBM patients, forced expression of OPTN mimics inhibition of PDGF, and higher OPTN levels predict a longer survival time [94].
There were three categories of miRNAs identified in the GBM biomarker panel: Proliferation (hsa-mir-221-3p), invasion (hsa-mir-15a-5p and hsa-let-7b-5p), and proliferation and invasion (hsa-mir-30a-5p and hsa-mir-130a-3p). In various types of human cancer, including GBM, miR-221, and miR-222 (miR-221/222) are frequently up-regulated. MiR-221/222 regulates cell growth and cell cycle progression through targeting of p27 and p57, according to recent studies. In GBM, miR-221/222, which targets the p53 upregulated modulator of apoptosis (PUMA), was reported to induce cell survival [95]. There is evidence that chronic miR-221/222-mediated downregulation of MGMT may result in cells being unable to repair genetic damage. The presence of miR-221/222 oncogenic potential may improve the prognosis of GBM [96]. Furthermore, decreased EGFR and increased miR-221 were associated with increased resistance to temozolomide (TMZ) and radiotherapy in GBM [97], compared to normal brain tissues (NBTs). MiR-30a-5p is overexpressed in glioma cell lines and glioma samples, with its expression level positively correlated with tumor grade [98]. WWP1 (WW domain containing E3 ubiquitin-protein ligase 1) has also been shown to suppress NF-κB activation, which is strongly associated with the development of glioma. Moreover, the study's results have revealed the positive feedback loop of miR-30a-5p-WWP1-NF-κB in regulating glioma development [99]. According to researchers, Wnt/β-catenin–miR-30a-5p–NCAM regulatory axis plays an essential role in controlling glioma cell invasion and tumorigenesis. It was shown that the Wnt/β-catenin pathway activates miR-30a-5p through the direct binding of β-catenin/TCF4 to two sites in the promoter region of miR-30a-5p. As well, miR-30a-5p can inhibit the expression of neural cell adhesion molecule (NCAM) by directly targeting two sites in the 3′-untranslated regions (3′-UTR) of NCAM mRNA [100].
The proliferation and invasion of GBM cells are mediated by several critical molecules, such as cell adhesion molecule 1 (CADM1). CADM1 expression is decreased in GBM patients and GBM cell lines, and CADM1 overexpression inhibits the proliferation of GBM cells [101,102]. According to these findings, CADM1 effectively suppresses the proliferation of GBM. MiR15a5p was shown to promote the proliferation and invasion of T98G GBM cells by targeting CADM1 [103]. The specificity protein 1 (Sp1) is aberrantly expressed in GBM and involved in the development and metastasis of the disease. In GBM cell lines, researchers found that the Sp1 expression was upregulated while miR-130a-3p expression was downregulated. Further, increased levels of miR-130a-3p inhibited GBM cell proliferation, migration, and TMZ resistance [104]. The study also demonstrated that hsa-let-7b-5p could inhibit glioma cell migration, invasion, and cell cycle [105].
In the GBM biomarker panel, five categories of metabolites were identified: Lipid metabolism (cholesterol), glutamate metabolism (glutamate and GABA), tricarboxylic acid (TCA) cycle (alanine), urea cycle (arginine), and Leloir cycle (galactose). There is a link between metabolic syndrome and several types of cancer, including GBM. An analysis of a New Zealand cohort of GBM patients showed that metabolic syndrome is associated with reduced OS. In light of this finding, there is a greater likelihood that GBM results from metabolic pathogenesis [106]. According to studies, lipid metabolism plays a critical role in the pathogenesis of GBM. Among the members of the apolipoprotein family, apolipoprotein C1 (ApoC1) is crucial for the metabolism of both very-low-density lipoprotein (VLDL) and high-density lipoprotein (HDL) cholesterols. Recent research has indicated that ApoC1 may be a viable therapeutic target for solid malignancies. Based on the study's findings, Apoc1 could enhance glioma metastasis by increasing EMT and activating STAT3 [107]. In another study, the level of serum LDL cholesterol pre-surgery was a prognostic factor for the outcome of patients with GBM [108]. Cholesterol is another important molecule that would have the potential role of repurposed drugs. Due to the discovery that many cancers, including GBM, reprogrammed cholesterol metabolism, cholesterol metabolism has become a promising potential target for therapy. As GBM cells require external cholesterol for survival, as well as lipid droplets for rapid growth, different strategies have been proposed to inhibit cholesterol metabolism, including inhibition of cholesterol uptake and promotion of cholesterol efflux by activating liver X receptors (LXRs), disruption of cellular cholesterol trafficking, inhibition of SREBP signaling, inhibition of cholesterol esterification, and may potentially counteract with glial tumor growth [109,110]. There is an association between obesity-related pathologies of the central nervous system (CNS) like neuroinflammation and reactive gliosis and a high-fat diet (HFD)-related elevation of saturated fatty acids like palmitic acid (PA) in neurons and astrocytes of the brain. In neurons and astrocytes, PA causes apoptosis by increasing oxidative stress. As a result of these findings, it appears that HFD may cause neuronal and astrocytic damage, suggesting that CNS pathologies involving neuroinflammation and reactive gliosis may be associated with HFD [111]. It has also been demonstrated that lipid accumulation and oxidation play a role in GBM. Monounsaturated fatty acids have been found to promote GBM proliferation by modulating triglyceride metabolism [112]. A knockdown of carnitine palmitoyltransferase 1A (CPT1A), a critical enzyme in fatty acid oxidation (FAO), also reduced tumor growth and increased survival, according to in vivo studies [113].
The excitatory neurotransmitter glutamate plays a significant role in the proliferation, growth, and movement of brain tumor cells. Glutaminase produces a large amount of glutamate in glioma cells, which converts glutamate from glutamine and increases intracellular Ca2+ through P2 × 7Rs [114]. Moreover, high levels of glutamate have been found to cause brain edema and seizures in glioma patients. In GBM cells, GLAST, a glutamate-aspartate transporter expressed by astrocytes and involved in glutamate uptake, is highly expressed on the plasma membrane. There is a significant correlation between its expression and shortened patient survival. GBM xenografts were shown to be restricted in their progression and invasion when GLAST expression was inhibited [115]. NADH shuttles, such as the malate-aspartate shuttle (MAS) and the glycerol-3-phosphate shuttle, can shuttle the reducing equivalents of cytosolic NADH into mitochondria. NADH shuttles are crucial in increasing mitochondrial energy production, as is widely accepted. An interesting finding revealed that NADH shuttles have a primary function in cancer cells, to maintain glycolysis by reducing cytosolic NADH/NAD+ ratios. A widely used MAS inhibitor, AOAA (aminooxy acetic acid), decreased intracellular ATP levels, altered the cell cycle, and increased the apoptosis and necrosis of C6 glioma cells, without affecting the survival of primary astrocyte cultures [116]. Glutamate and glutamine are linked to the proline pathway. L-proline is a multifunctional amino acid that plays an essential role in primary metabolism and physiological functions. Proline is oxidized to glutamate in the mitochondria, and the FAD-containing enzyme proline oxidase (PO) catalyzes the first step in the L-proline degradation pathway. It was shown that PO might play a regulatory role in glutamatergic neurotransmission by affecting the cellular concentration of glutamate [117]. Study results indicate that serine and glycine levels are higher in low-nutrient regions of GBM tumors than in other regions. A study of the metabolic and functional properties of GBM cells revealed that serine availability and 1C metabolism support the survival of glioma cells, following glutamine deprivation. The synthesis of serine was mediated by autophagy rather than glycolysis [118].
ATP by glycolysis and the TCA cycle are associated with oxidative phosphorylation (OXPHOS) through the breakdown of pyruvate or fatty acids to meet the growing energy demand of cancer cells. Recently, it was shown that SMI EPIC-0412 could effectively perturb the TCA cycle, which participated in the combination therapy of cytosolic phospholipase A2 (cPLA2)-inhibitor AACOCF3, and hexokinase II (HK2)-inhibitor 2-DG to disrupt the GBM energy metabolism for targeted metabolic treatments. ATP production was significantly declined in glioma cells when treated with monotherapy (EPIC-0412 or AACOCF3), dual therapy (EPIC-0412 + AACOCF3), or triple therapy (EPIC-0412 + AACOCF3 +2-DG) regimen [119]. In the TCA cycle, glutamate is recycled to synthesize glutamate, which in turn is converted to GABA. Neurotransmitters, such as GABA, are thought to play a crucial role in GBM behavior. LGGs may be inhibited by GABAAR activation through depolarization of the membrane caused by Na+–K+–2Cl− co-transporter NKCC1. It is possible, however, that the decreased number of mRNA encoding GABAAR subunits and loss of GABAAR in GBM may indicate a relationship between the number of functional GABAAR and the severity of the disease [120,121,122]. In addition, GBM may counteract the attenuation of GABA on cell proliferation through a decrease in the expression of GABAAR [123,124].
It has also been demonstrated that succinic semialdehyde dehydrogenase (SSADH) expression may contribute to the oxidation and/or consumption of GABA in gliomas; GABA oxidation capacity may also contribute to tumor proliferation and worse outcomes. Further, the IDH mutation, as well as the production of D-2-hydroxyglutarate (2-HG), inhibits the oxidation of GABA in glioma cells [125]. Additionally, it was shown that GBM patients with high expression of glycolysis-related genes such as HK2 and PKM2, and low expression of mitochondrial metabolism-related genes, such as SDHB and COX5A, which are associated with TCA cycle and oxidative phosphorylation (OXPHOS), respectively, had poor patient survival. In contrast to LGG, expression levels of genes involved in mitochondrial oxidative metabolism in GBM are markedly increased; however, they are lower than those in normal brains [126]. It has been shown that dysregulated alanine could serve as a potential predictive marker for glioma [127]. Alanine, a glucogenic amino acid, enters the metabolic stream through enzymatic conversion to pyruvate to provide energy and replenish the nutrient reservoir for rapidly proliferating tumor cells [128]. It is used to make proteins, and transfer RNA (tRNA) plays an important role in the epigenetic regulation of gene expression in tumors. A number of cell processes are mediated by tRNAs, including cell proliferation, signaling pathways, and stress responses, suggesting that tRNAs may also play a role in tumorigenesis and cancer progression [129]. As tRNA methyltransferase 1 (METTL1) levels increase with increasing glioma grade, the expression levels of METTL1 may be used to predict the prognosis of gliomas [130]. There has also been a link between the synthesis of tRNA in GBM and de novo GTP biosynthesis due to the increased expression of Impdh2 [131]. It has also been shown that the upregulation of Impdh2 is positively correlated with an increase in glioma malignancy and negatively correlated with the survival of patients [131].
Arginine is another amino acid substrate actively metabolized by tumor cells to promote tumor growth and immunosuppression. L-arginine plays an important role in the urea cycle and modulates immune function and tumor metabolism. Arginase 1 (ARG1) and cytokine-inducible nitric oxide synthase (iNOS) are substrates for L-arginine. In the urea cycle, ARG1 converts L-arginine into urea and ornithine. The iNOS enzyme converts L-arginine to citrulline and nitric oxide (NO), which is necessary for the immune system to direct anti-tumor functions [132,133]. Arginine transporters appear to be in abundance in GBM, as evidenced by the accumulation of byproducts of arginine metabolism [134,135]. The results indicate that arginine metabolism is functional and may be sensitive to targeted depletion. Recent research demonstrated that pegylated human recombinant ARG1 depleted arginine in glioma cells and induced cytotoxicity [136]. Likewise, these results demonstrate that radiotherapy's efficacy in treating GBM is potentiated by concomitant treatment with ADI-PEG20 in a non-arginine-auxotrophic cellular background (argininosuccinate synthase 1 positive). As a result of elevated NO, ADI-PEG20 enhanced the sensitivity of argininosuccinate synthetase 1-positive GBM to ionizing radiation, which generated cytotoxic peroxynitrites and promoted glioma-associated macrophage/microglial infiltration into tumors, changing their classical anti-inflammatory (protumor) phenotype into one of pro-inflammatory (anti-tumor) [137].
The proliferation of GBM is dependent on the availability of extracellular nutrients. As a result of inadequate tumor perfusion, glucose, and glutamine are in short supply. Due to this metabolic remodeling, GBMs scavenge alternative nutrients from the tumor microenvironment to sustain their growth and proliferation. Glut3 and Glut14 are sugar transporters expressed in GBM. GBM cells are capable of scavenging galactose (Gal) from the circulation and extracellular space, as a suitable substrate for Glut3/Glut14. The Leloir pathway provides GBM cells with an alternative energy source by transporting and metabolizing Gal at physiological Glc concentrations [138]. Additionally, D-galactose (D-gal), a reducing sugar, has been shown to induce senescence in GBM cells by inactivating the Yes-associated protein (YAP) and Cyclin-dependent kinase 6 (YAP-CDK6) signaling pathways [139]. Another study examined the effects of 4-deoxy-4-fluoro galactose (4DFG), a galactose-based antimetabolite, on glioma metabolism in vitro and in vivo. In this study, they found that low concentrations of 4DFG (5 µM) could inhibit glycolytic and mitochondrial flux by approximately 12% [140].
5. Conclusions and Future Direction
In summary, we identified eleven important genes and five miRNAs that have a critical role in the pathogenesis of GBM. In addition, we found essential metabolites that drive GBM development. These findings highlight the importance of genes, miRNAs, and metabolites in GBM progression. Metabolic analysis of various metabolic profiles provides a powerful and feasible method to monitor dynamic changes in tumor metabolism and response to therapy during disease progression. Metabolism will continue to be incorporated extensively into future cancer diagnostics and therapy because it provides unique insight into how malignant tumors reprogram their metabolism. To understand metabolic changes, one needs to know the mechanisms involved. To find out which genes or proteins lead to or are associated with metabolic changes, it is possible to combine transcriptome analysis with proteomic analysis to find potential targets for tumor treatment. At one point, tumor cells express high levels of IL -1R and TLR inflammatory proteins, which can lead to the infiltration of tumor cells into the blood. This increase in cytokine expression may allow tumor cells to survive and spread to other parts of the body. In addition, tumor cells may secrete factors that promote blood metastasis. To track the progression of GBM, it is important to monitor the level of inflammation in the blood. To determine how inflammation affects GBM progression, transcriptome, and proteomic analyzes should be performed to identify key genes and proteins that contribute to inflammation, as well as metabolites that support inflammation, such as proinflammatory cytokines, which are associated with GBM progression. Therefore, understanding how inflammation influences GBM progression is critical for the development of new therapeutic strategies. Targeting miRNAs or metabolites may exploit a vulnerability in GBM progression, which could be considered a viable therapeutic strategy. Further in vitro and in vivo studies are needed to validate our findings. We hope that our results will contribute to the development of diagnostic and therapeutic biomarkers for GBM in the near future and could open new avenues to personalized treatment via the tailoring of formulation and repurposed drug approach.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
A.B.B., and H.L.-N. prepared the draft of the manuscript and equally contributed to data curation, investigation, methodology, resource application, software application, data validation and visualization, writing, review, and editing. S.C.d.S.R. (Simone C da Silva Rosa), C.V. (Carla Vitorino), M.S. (Maciej Swiat), and Rui Vitorino (R.V.) critically evaluated the data analysis and linking to GBM; E.W. (Emilia Wiechec) participated in the discussion and genetic evaluation of the data, S.G. (Saeid Ghavami) and Z.J. (Zahra Jamalpoor) prevised and lead the team, finalized initial and revision of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fekrirad Z.;Barzegar Behrooz A.;Ghaemi S.;Khosrojerdi A.;Zarepour A.;Zarrabi A.;Arefian E., Ghavami S. Immunology Meets Bioengineering: Improving the Effectiveness of Glioblastoma Immunotherapy. Cancers 2022, 14. [CrossRef]
- Samiei E.;Seyfoori A.;Toyota B.;Ghavami S., Akbari M. Investigating Programmed Cell Death and Tumor Invasion in a Three-Dimensional (3D) Microfluidic Model of Glioblastoma. Int J Mol Sci. 2020, 21. [CrossRef]
- Shojaei S.;Koleini N.;Samiei E.;Aghaei M.;Cole L.K.;Alizadeh J.;Islam M.I.;Vosoughi A.R.;Albokashy M.;Butterfield Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [CrossRef]
- Shojaei S.;Alizadeh J.;Thliveris J.;Koleini N.;Kardami E.;Hatch G.M.;Xu F.;Hombach-Klonisch S.;Klonisch T., Ghavami S. Statins: A new approach to combat temozolomide chemoresistance in glioblastoma. J Investig Med. 2018, 66, 1083–1087. [CrossRef]
- Rong L.;Li N., Zhang Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 1–18.
- Hajiahmadi S.;Lorzadeh S.;Iranpour R.;Karima S.;Rajabibazl M.;Shahsavari Z., Ghavami S. Temozolomide, Simvastatin and Acetylshikonin Combination Induces Mitochondrial-Dependent Apoptosis in GBM Cells, Which Is Regulated by Autophagy. Biology 2023, 12. [CrossRef]
- Sharifzad F.;Ghavami S.;Verdi J.;Mardpour S.;Mollapour Sisakht M.;Azizi Z.;Taghikhani A.;Los M.J.;Fakharian E.;Ebrahimi M.; et al. Glioblastoma cancer stem cell biology: Potential theranostic targets. Drug Resist Update 2019, 42, 35–45. [CrossRef]
- Zhang P.;Xia Q.;Liu L.;Li S., Dong L. Current opinion on molecular characterization for GBM classification in guiding clinical diagnosis, prognosis, and therapy. Front. Mol. Biosci. . 2020, 7, 562798.
- Sharifzad F.;Yasavoli-Sharahi H.;Mardpour S.;Fakharian E.;Nikuinejad H.;Heydari Y.;Mardpour S.;Taghikhani A.;Khellat R.;Vafaei S.; et al. Neuropathological and genomic characterization of glioblastoma-induced rat model: How similar is it to humans for targeted therapy? J Cell Physiol. 2019, 234, 22493–22504.
- Lu C.-H.;Wei S.-T.;Liu J.-J.;Chang Y.-J.;Lin Y.-F.;Yu C.-S., Chang S.L.-Y. Recognition of a Novel Gene Signature for Human Glioblastoma. Int. J. Mol. Sci. 2022, 23, 4157.
- Yabo Y.A.;Niclou S.P., Golebiewska A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro-Oncol. 2022, 24, 669–682. [CrossRef]
- Basso J.;Paggi M.G.;Fortuna A.;Vitorino C., Vitorino R. Deciphering specific miRNAs in brain tumors: A 5-miRNA signature in glioblastoma. Mol. Genet. Genom. 2022, 297, 507–521. [CrossRef]
- Wei X.;Zhang Q.-m.;Liu C.;Wu S.;Nong W.-x.;Ge Y.-y.;Lin L.-n.;Li F.;Xie X.-x., Luo B. Microrna-1224-5p Is a Potential Prognostic and Therapeutic Biomarker in Glioblastoma: Integrating Bioinformatics and Clinical Analyses. Curr. Med. Sci. 2022, 42, 584–596.
- Xi X.;Chu Y.;Liu N.;Wang Q.;Yin Z.;Lu Y., Chen Y. Joint bioinformatics analysis of underlying potential functions of hsa-let-7b-5p and core genes in human glioma. J. Transl. Med. 2019, 17, 1–16.
- Sadegh S.;Skelton J.;Anastasi E.;Bernett J.;Blumenthal D.B.;Galindez G.;Salgado-Albarrán M.;Lazareva O.;Flanagan K., Cockell S. Network medicine for disease module identification and drug repurposing with the NeDRex platform. Nat. Commun. 2021, 12, 1–12. [CrossRef]
- Shannon P.;Markiel A.;Ozier O.;Baliga N.S.;Wang J.T.;Ramage D.;Amin N.;Schwikowski B., Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [CrossRef]
- Ghiassian S.D.;Menche J., Barabási A.-L. A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Comput. Biol. 2015, 11, e1004120. [CrossRef]
- Piñero J.;Bravo À.;Queralt-Rosinach N.;Gutiérrez-Sacristán A.;Deu-Pons J.;Centeno E.;García-García J.;Sanz F., Furlong L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, gkw943. [CrossRef]
- Piñero J.;Ramírez-Anguita J.M.;Saüch-Pitarch J.;Ronzano F.;Centeno E.;Sanz F., Furlong L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [CrossRef]
- Szklarczyk D.;Gable A.L.;Nastou K.C.;Lyon D.;Kirsch R.;Pyysalo S.;Doncheva N.T.;Legeay M.;Fang T., Bork P. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612.
- Kanehisa M.;Furumichi M.;Tanabe M.;Sato Y., Morishima K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [CrossRef]
- Huang Z.;Shi J.;Gao Y.;Cui C.;Zhang S.;Li J.;Zhou Y., Cui Q. HMDD v3. 0: A database for experimentally supported human microRNA–disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017.
- Warde-Farley D.;Donaldson S.L.;Comes O.;Zuberi K.;Badrawi R.;Chao P.;Franz M.;Grouios C.;Kazi F., Lopes C.T. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [CrossRef]
- Latifi-Navid H.;Soheili Z.S.;Samiei S.;Sadeghi M.;Taghizadeh S.;Pirmardan E.R., Ahmadieh H. Network analysis and the impact of Aflibercept on specific mediators of angiogenesis in HUVEC cells. J. Cell. Mol. Med. 2021, 25, 8285–8299. [CrossRef]
- Zhou G.;Soufan O.;Ewald J.;Hancock R.E.;Basu N., Xia J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic acids research 2019, 47, W234–W241.
- Xia J.;Gill E.E., Hancock R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844.
- Huang H.-Y.;Lin Y.-C.-D.;Li J.;Huang K.-Y.;Shrestha S.;Hong H.-C.;Tang Y.;Chen Y.-G.;Jin C.-N., Yu Y. miRTarBase 2020: Updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 2020, 48, D148–D154.
- Samaras P.;Schmidt T.;Frejno M.;Gessulat S.;Reinecke M.;Jarzab A.;Zecha J.;Mergner J.;Giansanti P., Ehrlich H.-C. ProteomicsDB: A multi-omics and multi-organism resource for life science research. Nucleic Acids Res. 2020, 48, D1153–D1163. [CrossRef]
- Schmidt T.;Samaras P.;Frejno M.;Gessulat S.;Barnert M.;Kienegger H.;Krcmar H.;Schlegl J.;Ehrlich H.-C., Aiche S. ProteomicsDB. Nucleic Acids Res. 2018, 46, D1271–D1281.
- Bowman R.L.;Wang Q.;Carro A.;Verhaak R.G., Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncol. 2017, 19, 139–141.
- Thul P.J., Lindskog C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244.
- Bastian F.B.;Roux J.;Niknejad A.;Comte A.;Fonseca Costa S.S.;De Farias T.M.;Moretti S.;Parmentier G.;De Laval V.R., Rosikiewicz M. The Bgee suite: Integrated curated expression atlas and comparative transcriptomics in animals. Nucleic Acids Res. 2021, 49, D831–D847. [CrossRef]
- Brazma A.;Parkinson H.;Sarkans U.;Shojatalab M.;Vilo J.;Abeygunawardena N.;Holloway E.;Kapushesky M.;Kemmeren P., Lara G.G. ArrayExpress—A public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003, 31, 68–71. [CrossRef]
- Edgar R.;Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [CrossRef]
- Deng M.;Brägelmann J.;Kryukov I.;Saraiva-Agostinho N., Perner S. FirebrowseR: An R client to the Broad Institute’s Firehose Pipeline. Database 2017, 2017. [CrossRef]
- Chang L.;Zhou G.;Ou H., Xia J. mGWAS-Explorer: Linking SNPs, Genes, Metabolites, and Diseases for Functional Insights. Metabolites 2022 12, 526.
- Deshmukh R.;Allega M.F., Tardito S. A map of the altered glioma metabolism. Trends Mol. Med. 2021, 27, 1045–1059. [CrossRef]
- Jaroch K.;Modrakowska P., Bojko B. Glioblastoma Metabolomics—In Vitro Studies. Metabolites 2021, 11, 315. [CrossRef]
- Pang Z.;Chong J.;Zhou G.;de Lima Morais D.A.;Chang L.;Barrette M.;Gauthier C.;Jacques P.-É.;Li S., Xia J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396.
- Pang Z.;Zhou G.;Ewald J.;Chang L.;Hacariz O.;Basu N., Xia J. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protocols 2022, 17, 1735–1761.
- Backes C.;Khaleeq Q.T.;Meese E., Keller A. miEAA: microRNA enrichment analysis and annotation. Nucleic Acids Res. 2016, 44, W110–W116. [CrossRef]
- Kern F.;Fehlmann T.;Solomon J.;Schwed L.;Grammes N.;Backes C.;Van Keuren-Jensen K.;Craig D.W.;Meese E., Keller A. miEAA 2.0: Integrating multi-species microRNA enrichment analysis and workflow management systems. Nucleic Acids Res. 2020, 48, W521–W528. [CrossRef]
- Jewison T.;Su Y.;Disfany F.M.;Liang Y.;Knox C.;Maciejewski A.;Poelzer J.;Huynh J.;Zhou Y., Arndt D. SMPDB 2.0: Big improvements to the Small Molecule Pathway Database. Nucleic Acids Res. 2014, 42, D478-D484. [CrossRef]
- Ostrom Q.T.;Bauchet L.;Davis F.G.;Deltour I.;Fisher J.L.;Langer C.E.;Pekmezci M.;Schwartzbaum J.A.;Turner M.C., Walsh K.M. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncol. 2014, 16, 896–913. [CrossRef]
- Deweerdt S. Below the surface. 2018.
- Zhou Q.;Liu J.;Quan J.;Liu W.;Tan H., Li W. MicroRNAs as potential biomarkers for the diagnosis of glioma: A systematic review and meta-analysis. Cancer Sci. 2018, 109, 2651–2659.
- Barzegar Behrooz A.;Talaie Z.;Jusheghani F.;Łos M.J.;Klonisch T., Ghavami S. Wnt and PI3K/Akt/mTOR survival pathways as therapeutic targets in glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [CrossRef]
- Ruiz-Pérez M.V.;Henley A.B., Arsenian-Henriksson M. The MYCN protein in health and disease. Genes 2017, 8, 113. [CrossRef]
- Orian J.;Vasilopoulos K.;Yoshida S.;Kaye A.;Chow C., Gonzales M. Overexpression of multiple oncogenes related to histological grade of astrocytic glioma. Br. J. Cancer 1992, 66, 106–112. [CrossRef]
- Herms J.W.;von Loewenich F.D.;Behnke J.;Markakis E., Kretzschmar H.A. c-Myc oncogene family expression in glioblastoma and survival. Surg. Neurol. 1999, 51, 536–542. [CrossRef]
- Annibali D.;Whitfield J.R.;Favuzzi E.;Jauset T.;Serrano E.;Cuartas I.;Redondo-Campos S.;Folch G.;Gonzàlez-Juncà A., Sodir N.M. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 2014, 5, 1–11. [CrossRef]
- Alkema M.;Wiegant J.;Raap A.K.;Bems A., van Lohuizen M. Characterization and chromosomal localization of the human proto-oncogene BMI-1. Hum. Mol. Genet. 1993, 2, 1597–1603.
- Bracken A.P.;Dietrich N.;Pasini D.;Hansen K.H., Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [CrossRef]
- Sauvageau M., Sauvageau G. Polycomb group proteins: Multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 2010, 7, 299–313. [CrossRef]
- Schuringa J.J., Vellenga E. Role of the polycomb group gene BMI1 in normal and leukemic hematopoietic stem and progenitor cells. Curr. Opin. Hematol. 2010, 17, 294–299. [CrossRef]
- Valk-Lingbeek M.E.;Bruggeman S.W., van Lohuizen M. Stem cells and cancer: The polycomb connection. Cell 2004, 118, 409–418. [CrossRef]
- Haupt Y.;Alexander W.S.;Barri G.;Klinken S.P., Adams J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in Eμ-myc transgenic mice. Cell 1991, 65, 753–763. [CrossRef]
- Van Lohuizen M.;Verbeek S.;Scheljen B.;Wientjens E.;van der Guidon H., Berns A. Identification of cooperating oncogenes in Eμ-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [CrossRef]
- Vora P.;Seyfrid M.;Venugopal C.;Qazi M.A.;Salim S.;Isserlin R.;Subapanditha M.;O’Farrell E.;Mahendram S., Singh M. Bmi1 regulates human glioblastoma stem cells through activation of differential gene networks in CD133+ brain tumor initiating cells. J. Neuro-Oncol. 2019, 143, 417–428. [CrossRef]
- Jin X.;Kim L.J.;Wu Q.;Wallace L.C.;Prager B.C.;Sanvoranart T.;Gimple R.C.;Wang X.;Mack S.C., Miller T.E. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat. Med. 2017, 23, 1352–1361.
- Xu H.;Zong H.;Ma C.;Ming X.;Shang M.;Li K.;He X.;Du H., Cao L. Epidermal growth factor receptor in glioblastoma. Oncol. Lett. 2017, 14, 512–516.
- Ding J.;Li X.;Khan S.;Zhang C.;Gao F.;Sen S.;Wasylishen A.R.;Zhao Y.;Lozano G., Koul D. EGFR suppresses p53 function by promoting p53 binding to DNA-PKcs: A noncanonical regulatory axis between EGFR and wild-type p53 in glioblastoma. Neuro-Oncol. 2022, 24, 1712–1725.
- Fang R.;Chen X.;Zhang S.;Shi H.;Ye Y.;Shi H.;Zou Z.;Li P.;Guo Q., Ma L. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat. Commun. 2021, 12, 1–17.
- Tanaka S.;Batchelor T.T.;Iafrate A.J.;Dias-Santagata D.;Borger D.R.;Ellisen L.W.;Yang D.;Louis D.N.;Cahill D.P., Chi A.S. PIK3CA activating mutations are associated with more disseminated disease at presentation and earlier recurrence in glioblastoma. Acta Neuropathol. Commun. 2019, 7, 1–8. [CrossRef]
- Abbi S., Guan J. Focal adhesion kinase: Protein interactions and cellular functions. Histol. Histopathol. 2002.
- Parsons J.T.;Martin K.H.;Slack J.K.;Taylor J.M., Weed S.A. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene 2000, 19, 5606–5613. [CrossRef]
- Tamura M.;Gu J.;Matsumoto K.;Aota S.-i.;Parsons R., Yamada K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [CrossRef]
- Jones G.;Machado Jr J.;Tolnay M., Merlo A. PTEN-independent induction of caspase-mediated cell death and reduced invasion by the focal adhesion targeting domain (FAT) in human astrocytic brain tumors which highly express focal adhesion kinase (FAK). Cancer Res. 2001, 61, 5688–5691.
- Knobbe C.B., Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-lnositol-3′-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003, 13, 507–518.
- Scholz N.;Kurian K.M.;Siebzehnrubl F.A., Licchesi J.D. Targeting the ubiquitin system in glioblastoma. Front. Oncol. 2020, 10, 574011. [CrossRef]
- Delle Donne R.;Iannucci R.;Rinaldi L.;Roberto L.;Oliva M.A.;Senatore E.;Borzacchiello D.;Lignitto L.;Giurato G., Rizzo F. Targeted inhibition of ubiquitin signaling reverses metabolic reprogramming and suppresses glioblastoma growth. Commun. Biol. 2022, 5, 1–14. [CrossRef]
- Li J.;Sun Y.;Ma Y.;Zhao X.;Sun X.;Wang Y., Zhang X. Comprehensive Pan-Cancer Analysis of IRAK Family Genes Identifies IRAK1 as a Novel Oncogene in Low-Grade Glioma. J. Oncol. 2022, 2022.
- Lee H.N.;Jeong M.S., Jang S.B. Molecular Characteristics of Amyloid Precursor Protein (APP) and Its Effects in Cancer. Int. J. Mol. Sci. 2021, 22, 4999.
- Culicchia F.;Cui J.-G.;Li Y.Y., Lukiw W.J. Upregulation of β-amyloid precursor protein expression in glioblastoma multiforme. Neuroreport 2008, 19, 981–985.
- Lehrer S. Glioblastoma and dementia may share a common cause. Med. Hypotheses 2010, 75, 67–68. [CrossRef]
- Kucheryavykh L.Y.;Ortiz-Rivera J.;Kucheryavykh Y.V.;Zayas-Santiago A.;Diaz-Garcia A., Inyushin M.Y. Accumulation of innate amyloid beta peptide in glioblastoma tumors. Int. J. Mol. Sci. 2019, 20, 2482. [CrossRef]
- Li S.;Chen X.;Mao L.;Zahid K.R.;Wen J.;Zhang L.;Zhang M.;Duan J.;Duan J., Yin X. Histone deacetylase 1 promotes glioblastoma cell proliferation and invasion via activation of PI3K/AKT and MEK/ERK signaling pathways. Brain Res. 2018, 1692, 154–162.
- Zhang Z.;Wang Y.;Chen J.;Tan Q.;Xie C.;Li C.;Zhan W., Wang M. Silencing of histone deacetylase 2 suppresses malignancy for proliferation, migration, and invasion of glioblastoma cells and enhances temozolomide sensitivity. Cancer Chemother. Pharmacol. 2016, 78, 1289–1296.
- Zhong S.;Fan Y.;Wu B.;Wang Y.;Jiang S.;Ge J.;Hua C.;Zhao G.;Chen Y., Xu H. HDAC3 expression correlates with the prognosis and grade of patients with glioma: A diversification analysis based on transcriptome and clinical evidence. World Neurosurg. 2018, 119, e145–e158.
- Chang H.H.;Chang Y.-Y.;Tsai B.-C.;Chen L.-J.;Chang A.-C.;Chuang J.-Y.;Gean P.-W., Hsueh Y.-S. A Selective Histone Deacetylase Inhibitor Induces Autophagy and Cell Death via SCNN1A Downregulation in Glioblastoma Cells. Cancers 2022, 14, 4537. [CrossRef]
- Fan Y.;Peng X.;Wang Y.;Li B., Zhao G. Comprehensive analysis of HDAC family Identifies HDAC1 as A Prognostic and Immune Infiltration Indicator and HDAC1-related Signature for Prognosis Estimation in Glioma. Front. Mol. Biosci. 2021, 737.
- Zhang Y.;Dube C.;Gibert Jr M.;Cruickshanks N.;Wang B.;Coughlan M.;Yang Y.;Setiady I.;Deveau C., Saoud K. The p53 pathway in glioblastoma. Cancers 2018, 10, 297.
- Sukumar U.K.;Massoud T.F., Paulmurugan R. p53 supplementation as a targeted cancer gene therapy for glioblastoma. Glioblastoma Resistance to Chemotherapy: Molecular Mechanisms and Innovative Reversal Strategies: Elsevier; 2021. p. 773-86.
- Qi Z.-X.;Cai J.-J.;Chen L.-C.;Yue Q.;Gong Y.;Yao Y., Mao Y. TRIM28 as an independent prognostic marker plays critical roles in glioma progression. J. Neuro-Oncol. 2016, 126, 19–26.
- Peng Y.;Zhang M.;Jiang Z., Jiang Y. TRIM28 activates autophagy and promotes cell proliferation in glioblastoma. OncoTargets Ther. 2019, 12, 397. [CrossRef]
- Clevers H., Nusse R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205.
- Nàger M.;Sallán M.C.;Visa A.;Pushparaj C.;Santacana M.;Macià A.;Yeramian A.;Cantí C., Herreros J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [CrossRef]
- Schulze M.;Violonchi C.;Swoboda S.;Welz T.;Kerkhoff E.;Hoja S.;Brüggemann S.;Simbürger J.;Reinders J., Riemenschneider M.J. RELN signaling modulates glioblastoma growth and substrate-dependent migration. Brain Pathol. 2018, 28, 695–709. [CrossRef]
- Biamonte F.;Sica G.;Filippini A., D’Alessio A. Evidence of Reelin Signaling in GBM and Its Derived Cancer Stem Cells. Brain Sci. 2021, 11, 745. [CrossRef]
- Agnihotri S.;Golbourn B.;Huang X.;Remke M.;Younger S.;Cairns R.A.;Chalil A.;Smith C.A.;Krumholtz S.-L., Mackenzie D. PINK1 Is a Negative Regulator of Growth and the Warburg Effect in GlioblastomaPINK1 Inhibits Glioblastoma Growth. Cancer Res. 2016, 76, 4708–4719.
- Lee K.-S., Lu B. Targeting PINK1 and MQC in brain tumors. Oncotarget 2014, 5, 2864.
- Wang Y.;Liu H.-H.;Cao Y.-T.;Zhang L.-L.;Huang F., Yi C. The role of mitochondrial dynamics and mitophagy in carcinogenesis, metastasis and therapy. Front. Cell Dev. Biol. 2020, 8, 413.
- Lenzi P.;Ferese R.;Biagioni F.;Fulceri F.;Busceti C.L.;Falleni A.;Gambardella S.;Frati A., Fornai F. Rapamycin ameliorates defects in mitochondrial fission and mitophagy in glioblastoma cells. Int. J. Mol. Sci. 2021, 22, 5379. [CrossRef]
- Lv D.;Gimple R.C.;Zhong C.;Wu Q.;Yang K.;Prager B.C.;Godugu B.;Qiu Z.;Zhao L., Zhang G. PDGF signaling inhibits mitophagy in glioblastoma stem cells through N6-methyladenosine. Dev. Cell 2022.
- Zhang C.-Z.;Zhang J.-X.;Zhang A.-L.;Shi Z.-D.;Han L.;Jia Z.-F.;Yang W.-D.;Wang G.-X.;Jiang T., You Y.-P. MiR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Mol. Cancer 2010, 9, 1–9.
- Quintavalle C.;Mangani D.;Roscigno G.;Romano G.;Diaz-Lagares A.;Iaboni M.;Donnarumma E.;Fiore D.;De Marinis P., Soini Y. MiR-221/222 target the DNA methyltransferase MGMT in glioma cells. PLoS ONE 2013, 8, e74466.
- Areeb Z.;Stuart S.F.;West A.J.;Gomez J.;Nguyen H.;Paradiso L.;Zulkifli A.;Jones J.;Kaye A.H., Morokoff A.P. Reduced EGFR and increased miR-221 is associated with increased resistance to temozolomide and radiotherapy in glioblastoma. Sci. Rep. 2020, 10, 1–12. [CrossRef]
- Wang K.;Jia Z.;Zou J.;Zhang A.;Wang G.;Hao J.;Wang Y.;Yang S., Pu P. Analysis of hsa-miR-30a-5p expression in human gliomas. Pathol. Oncol. Res. 2013, 19, 405–411.
- Zhao P.;Wang M.;An J.;Sun H.;Li T., Li D. A positive feedback loop of miR-30a-5p-WWP1-NF-κB in the regulation of glioma development. Int. J. Biochem. Cell Biol. 2019, 112, 39–49.
- Wang Z.;Dai X.;Chen Y.;Sun C.;Zhu Q.;Zhao H.;Liu G.;Huang Q., Lan Q. MiR-30a-5p is induced by Wnt/β-catenin pathway and promotes glioma cell invasion by repressing NCAM. Biochem. Biophys. Res. Commun. 2015, 465, 374–380.
- Isobe T.;Hisamori S.;Hogan D.J.;Zabala M.;Hendrickson D.G.;Dalerba P.;Cai S.;Scheeren F.;Kuo A.H., Sikandar S.S. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. Elife 2014, 3, e01977.
- Zhang X.;Li W.;Kang Y.;Zhang J., Yuan H. SynCAM, a novel putative tumor suppressor, suppresses growth and invasiveness of glioblastoma. Mol. Biol. Rep. 2013, 40, 5469–5475.
- Kong F.;Li X.;Li S.;Sheng D.;Li W., Song M. MicroRNA-15a-5p promotes the proliferation and invasion of T98G glioblastoma cells via targeting cell adhesion molecule 1. Oncol. Lett. 2021, 21, 1.
- Wang Z.;Li Z.;Fu Y.;Han L., Tian Y. MiRNA-130a-3p inhibits cell proliferation, migration, and TMZ resistance in glioblastoma by targeting Sp1. Am. J. Transl. Res. 2019, 11, 7272.
- Xi X.;Chu Y.;Liu N.;Wang Q.;Yin Z.;Lu Y., Chen Y. Joint bioinformatics analysis of underlying potential functions of hsa-let-7b-5p and core genes in human glioma. J. Transl. Med. 2019, 17, 1–16.
- McManus E.J.;Frampton C.;Tan A., Phillips M.C. Metabolics risk factors in a New Zealand glioblastoma cohort. Neuro-Oncol. Pract. 2022, 9, 43–49. [CrossRef]
- Liang R.;Zhang G.;Xu W.;Liu W., Tang Y. ApoC1 promotes glioma metastasis by enhancing epithelial-mesenchymal transition and activating the STAT3 pathway. Neurol. Res. 2022, 1-8.
- Liang R.;Li J.;Li M.;Yang Y.;Wang X.;Mao Q., Liu Y. Clinical significance of pre-surgical serum lipid levels in patients with glioblastoma. Oncotarget 2017, 8, 85940.
- Guo X.;Zhou S.;Yang Z.;Li Z.-A.;Hu W.;Dai L.;Liang W., Wang X. Cholesterol metabolism and its implication in glioblastoma therapy. J. Cancer 2022, 13, 1745.
- Villa G.R.;Hulce J.J.;Zanca C.;Bi J.;Ikegami S.;Cahill G.L.;Gu Y.;Lum K.M.;Masui K., Yang H. An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell 2016, 30, 683–693. [CrossRef]
- Ng Y.-W., Say Y.-H. Palmitic acid induces neurotoxicity and gliatoxicity in SH-SY5Y human neuroblastoma and T98G human glioblastoma cells. PeerJ. 2018, 6, e4696.
- Taïb B.;Aboussalah A.M.;Moniruzzaman M.;Chen S.;Haughey N.J.;Kim S.F., Ahima R.S. Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 2019, 9, 1–14.
- Sperry J.;Condro M.C.;Guo L.;Braas D.;Vanderveer-Harris N.;Kim K.K.;Pope W.B.;Divakaruni A.S.;Lai A., Christofk H. Glioblastoma utilizes fatty acids and ketone bodies for growth allowing progression during ketogenic diet therapy. Iscience 2020, 23, 101453. [CrossRef]
- So J.-S.;Kim H., Han K.-S. Mechanisms of Invasion in Glioblastoma: Extracellular Matrix, Ca2+ Signaling, and Glutamate. Front. Cell. Neurosci. 2021, 190.
- Corbetta C.;Di Ianni N.;Bruzzone M.G.;Patanè M.;Pollo B.;Cantini G.;Cominelli M.;Zucca I.;Pisati F., Poliani P.L. Altered function of the glutamate–aspartate transporter GLAST, a potential therapeutic target in glioblastoma. Int. J. Cancer 2019, 144, 2539–2554.
- Wang C.;Chen H.;Zhang M.;Zhang J.;Wei X., Ying W. Malate-aspartate shuttle inhibitor aminooxyacetic acid leads to decreased intracellular ATP levels and altered cell cycle of C6 glioma cells by inhibiting glycolysis. Cancer Lett. 2016, 378, 1–7.
- Cappelletti P.;Tallarita E.;Rabattoni V.;Campomenosi P.;Sacchi S., Pollegioni L. Proline oxidase controls proline, glutamate, and glutamine cellular concentrations in a U87 glioblastoma cell line. PLoS ONE 2018, 13, e0196283. [CrossRef]
- Tanaka K.;Sasayama T.;Nagashima H.;Irino Y.;Takahashi M.;Izumi Y.;Uno T.;Satoh N.;Kitta A., Kyotani K. Glioma cells require one-carbon metabolism to survive glutamine starvation. Acta Neuropathol. Commun. 2021, 9, 1–14. [CrossRef]
- Yang S.;Zhao J.;Cui X.;Zhan Q.;Yi K.;Wang Q.;Xiao M.;Tan Y.;Hong B., Fang C. TCA-phospholipid-glycolysis targeted triple therapy effectively suppresses ATP production and tumor growth in glioblastoma. Theranostics 2022, 12, 7032.
- Jung E.;Alfonso J.;Osswald M.;Monyer H.;Wick W., Winkler F. Emerging intersections between neuroscience and glioma biology. Nat. Neurosci. 2019, 22, 1951–1960. [CrossRef]
- Smits A.;Jin Z.;Elsir T.;Pedder H.;Nistér M.;Alafuzoff I.;Dimberg A.;Edqvist P.-H.;Pontén F., Aronica E. GABA-A channel subunit expression in human glioma correlates with tumor histology and clinical outcome. PLoS ONE 2012, 7, e37041. [CrossRef]
- Babateen O.;Jin Z.;Bhandage A.;Korol S.V.;Westermark B.;Nilsson K.F.;Uhrbom L.;Smits A., Birnir B. Etomidate, propofol and diazepam potentiate GABA-evoked GABAA currents in a cell line derived from human glioblastoma. Eur. J. Pharmacol. 2015, 748, 101–107. [CrossRef]
- Huang Q.;Chen L.;Liang J.;Huang Q., Sun H. Neurotransmitters: Potential Targets in Glioblastoma. Cancers 2022, 14, 3970.
- D'Urso P.I.;D'Urso O.F.;Storelli C.;Mallardo M.;Gianfreda C.D.;Montinaro A.;Cimmino A.;Pietro C., Marsigliante S. miR-155 is up-regulated in primary and secondary glioblastoma and promotes tumour growth by inhibiting GABA receptors. Int. J. Oncol. 2012, 41, 228–234. [CrossRef]
- Hujber Z.;Horváth G.;Petővári G.;Krencz I.;Dankó T.;Mészáros K.;Rajnai H.;Szoboszlai N.;Leenders W.P., Jeney A. GABA, glutamine, glutamate oxidation and succinic semialdehyde dehydrogenase expression in human gliomas. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [CrossRef]
- Stanke K.M.;Wilson C., Kidambi S. High Expression of Glycolytic Genes in Clinical Glioblastoma Patients Correlates with Lower Survival. Front. Mol. Biosci. 2021, 8. [CrossRef]
- Firdous S.;Abid R.;Nawaz Z.;Bukhari F.;Anwer A.;Cheng L.L., Sadaf S. Dysregulated alanine as a potential predictive marker of glioma—An insight from untargeted HRMAS-NMR and machine learning data. Metabolites 2021, 11, 507. [CrossRef]
- Ijare O.;Baskin D., Pichumani K. Cbmt-01. Alanine Fuels Energy Metabolism of Glioblastoma Cells. Neuro-Oncology 2019, 21, vi32. [CrossRef]
- Gupta T.;Malkin M., Huang S. tRNA function and dysregulation in cancer. Front. Cell Dev. Biol. 2022, 1128. [CrossRef]
- Li L.;Yang Y.;Wang Z.;Xu C.;Huang J., Li G. Prognostic role of METTL1 in glioma. Cancer Cell Int. 2021, 21, 1–20.
- Kofuji S.;Hirayama A.;Eberhardt A.O.;Kawaguchi R.;Sugiura Y.;Sampetrean O.;Ikeda Y.;Warren M.;Sakamoto N., Kitahara S. IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat. Cell Biol. 2019, 21, 1003–1014. [CrossRef]
- Mohan A.A.;Tomaszewski W.H.;Haskell-Mendoza A.P.;Hotchkiss K.M.;Singh K.;Reedy J.L.;Fecci P.E.;Sampson J.H., Khasraw M. Targeting Immunometabolism in Glioblastoma. Front. Oncol. 2021, 11, 2260. [CrossRef]
- Rath M.;Müller I.;Kropf P.;Closs E.I., Munder M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front. Immunol. 2014, 5, 532. [CrossRef]
- Kobayashi K.;Ohnishi A.;Promsuk J.;Shimizu S.;Kanai Y.;Shiokawa Y., Nagane M. Enhanced tumor growth elicited by L-type amino acid transporter 1 in human malignant glioma cells. Neurosurgery 2008, 62, 493–504. [CrossRef]
- Chinnaiyan P.;Kensicki E.;Bloom G.;Prabhu A.;Sarcar B.;Kahali S.;Eschrich S.;Qu X.;Forsyth P., Gillies R. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012, 72, 5878–5888. [CrossRef]
- Khoury O.;Ghazale N.;Stone E.;El-Sibai M.;Frankel A.E., Abi-Habib R.J. Human recombinant arginase I (Co)-PEG5000 [HuArgI (Co)-PEG5000]-induced arginine depletion is selectively cytotoxic to human glioblastoma cells. J. Neuro-Oncol. 2015, 122, 75–85. [CrossRef]
- Hajji N.;Garcia-Revilla J.;Soto M.S.;Perryman R.;Symington J.;Quarles C.C.;Healey D.R.;Guo Y.;Orta-Vázquez M.L., Mateos-Cordero S. Arginine deprivation alters microglial polarity and synergizes with radiation to eradicate non-arginine-auxotrophic glioblastoma tumors. J. Clin. Investig. 2022, 132. [CrossRef]
- Sharpe M.A.;Ijare O.B.;Baskin D.S.;Baskin A.M.;Baskin B.N., Pichumani K. The leloir cycle in glioblastoma: Galactose scavenging and metabolic remodeling. Cancers 2021, 13, 1815. [CrossRef]
- Xu X.;Shen X.;Feng W.;Yang D.;Jin L.;Wang J.;Wang M.;Ting Z.;Xue F., Zhang J. D-galactose induces senescence of glioblastoma cells through YAP-CDK6 pathway. Aging 2020, 12, 18501.
- Sharpe M.;Baskin A.;Baskin B.;Baskin D., Raghavan S., editors. Targeting Glioblastoma's Galactose Scavenging Pathway. Neuro-Oncology; 2021: Oxford Univ Press Inc Journals Dept, 2001 Evans Rd, Cary, NC 27513 USA.
Figure 1.
An overview of the bioinformatics approaches used in this study. We created four gene lists from several different study levels. By merging these gene lists and performing various analyses, 11 genes and five key miRNAs were identified.
Figure 1.
An overview of the bioinformatics approaches used in this study. We created four gene lists from several different study levels. By merging these gene lists and performing various analyses, 11 genes and five key miRNAs were identified.
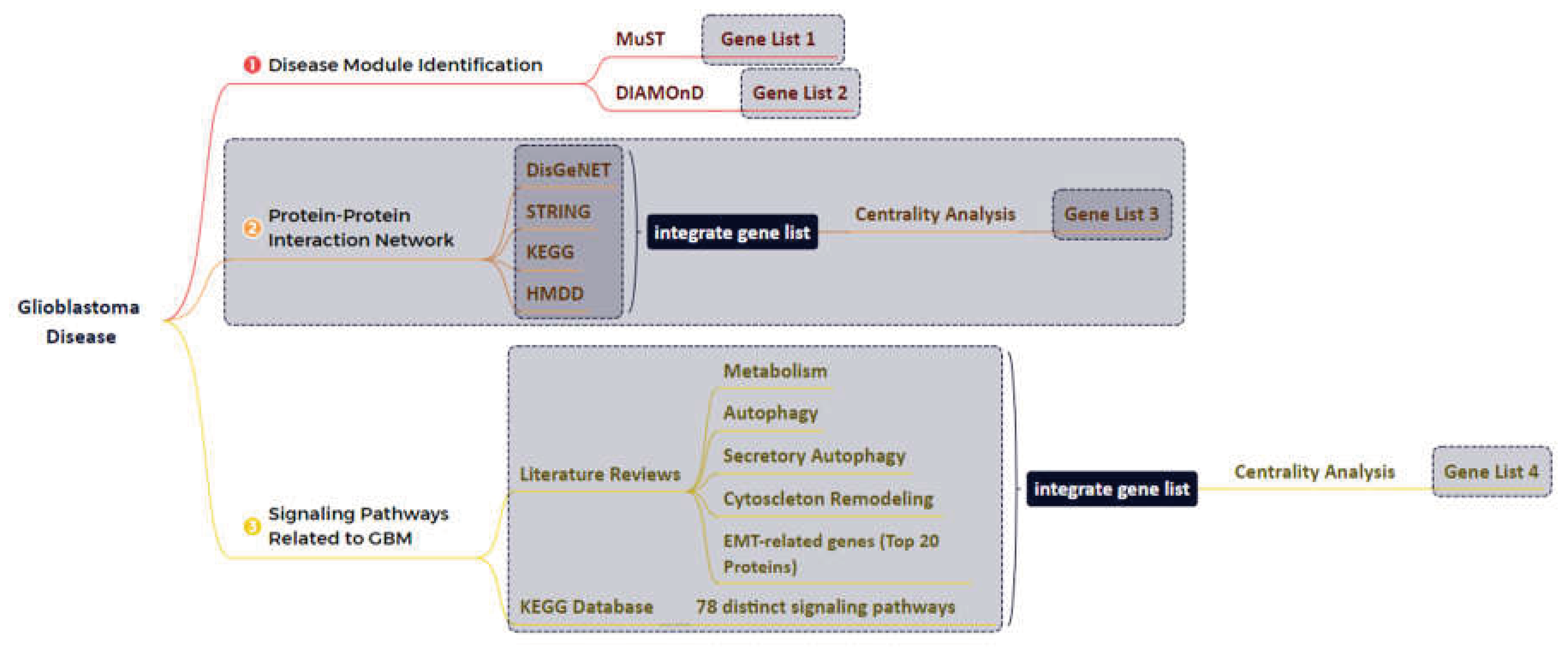
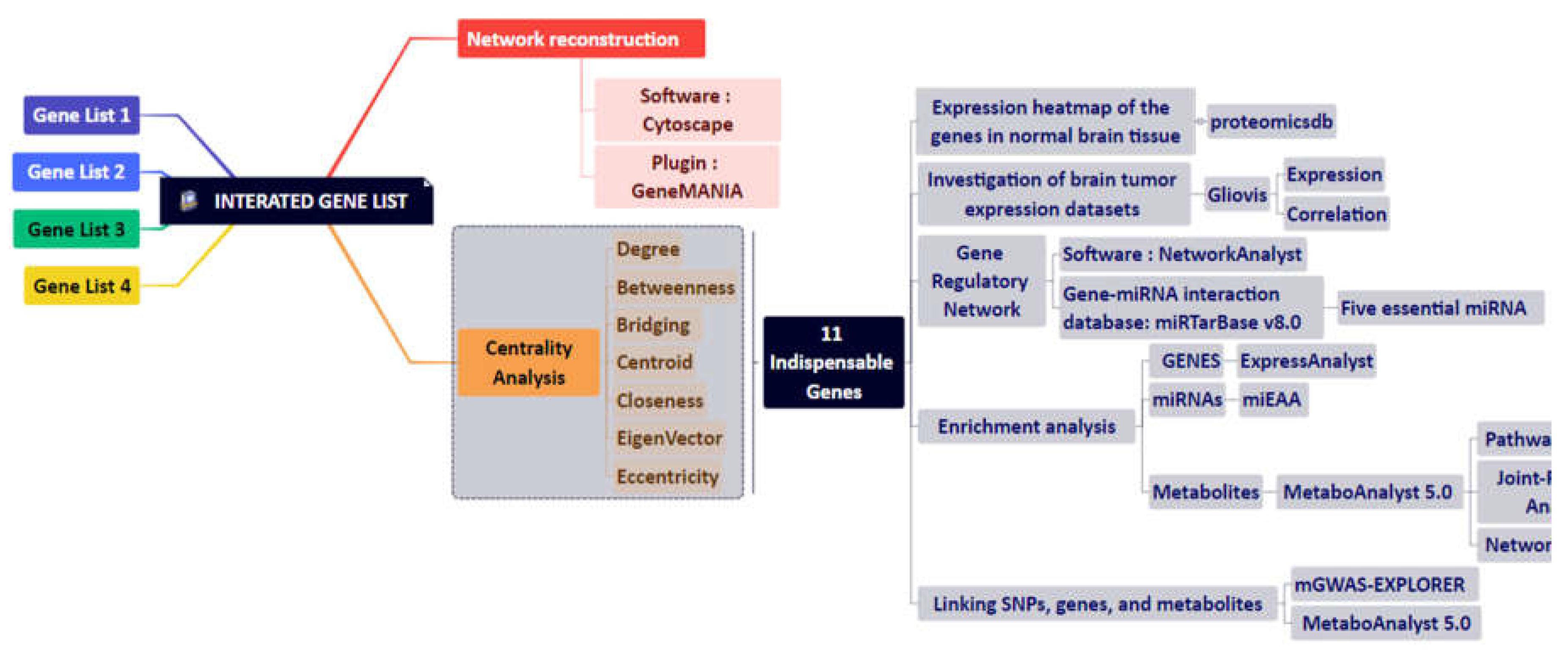
Figure 2.
Glioblastoma-related proteins identified by the NeDRex plugin.
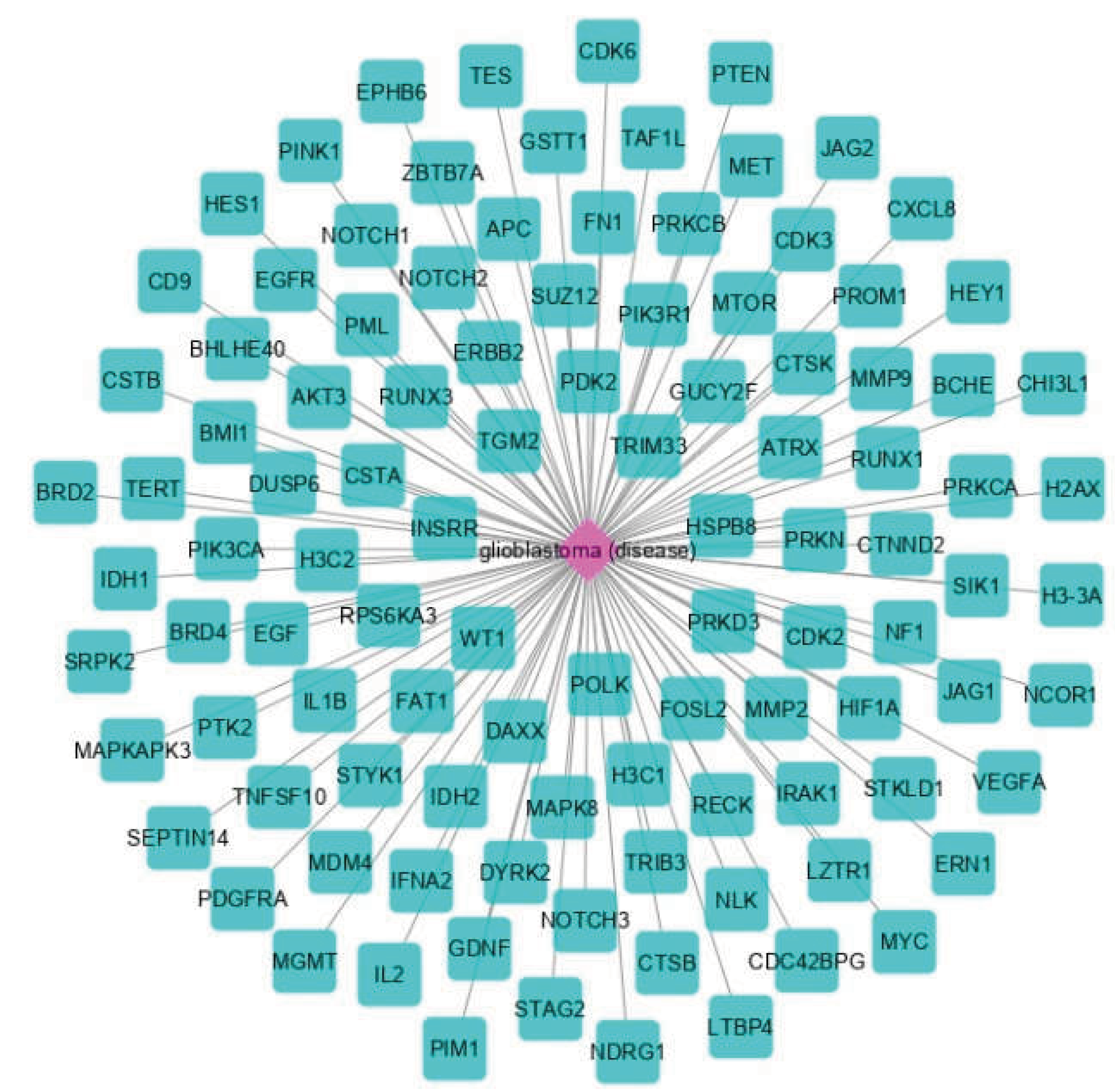
Figure 3.
Modules of disease identified by the MuST algorithm (A) and genes (B) that interconnect them. Five of the essential genes were identified: MYC, EGFR, PIK3CA, SUZ12, and SPRK2. Also, IRAK1, PTK2, and BMI1 represent bridging roles between disease modules.
Figure 3.
Modules of disease identified by the MuST algorithm (A) and genes (B) that interconnect them. Five of the essential genes were identified: MYC, EGFR, PIK3CA, SUZ12, and SPRK2. Also, IRAK1, PTK2, and BMI1 represent bridging roles between disease modules.
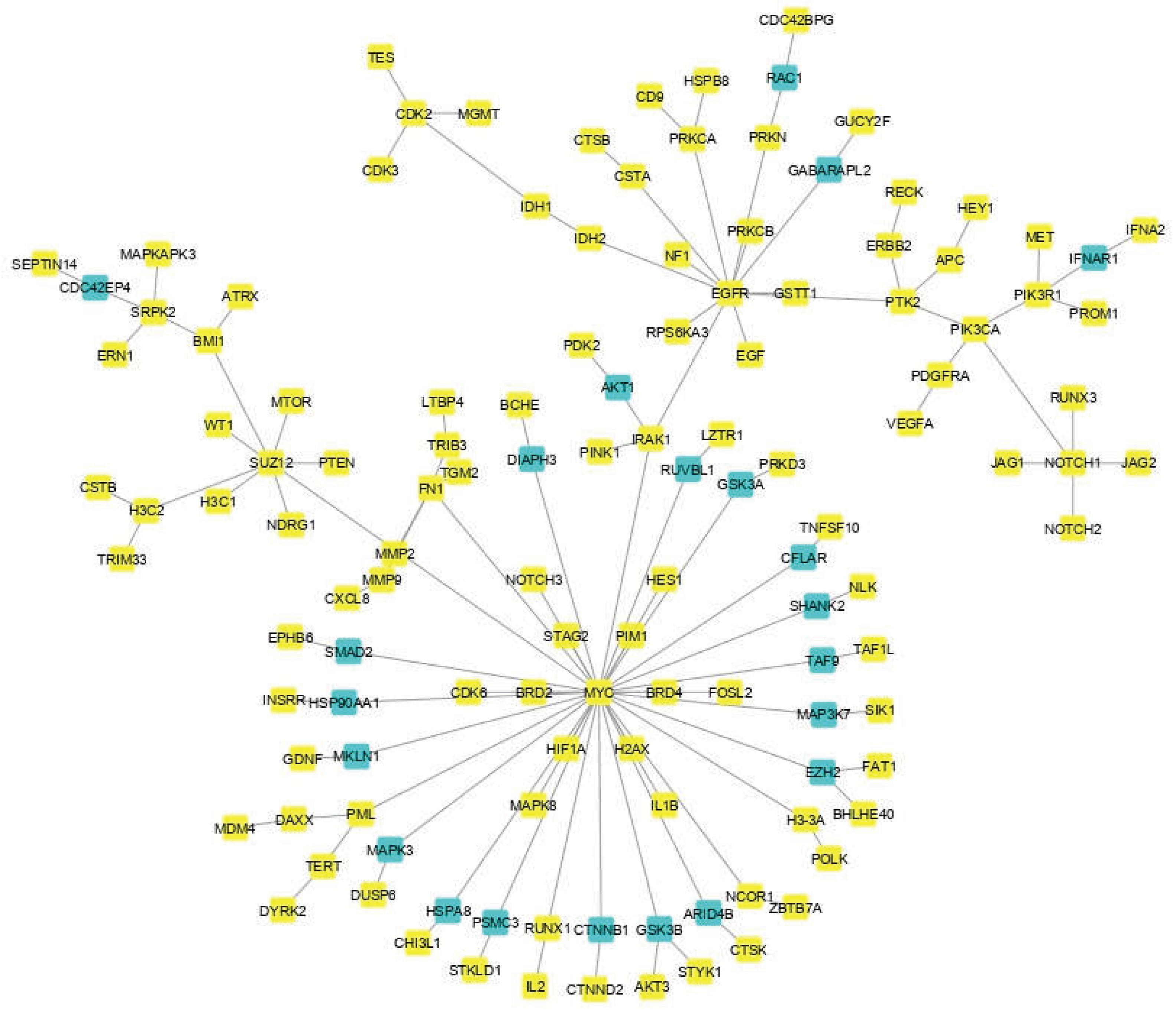
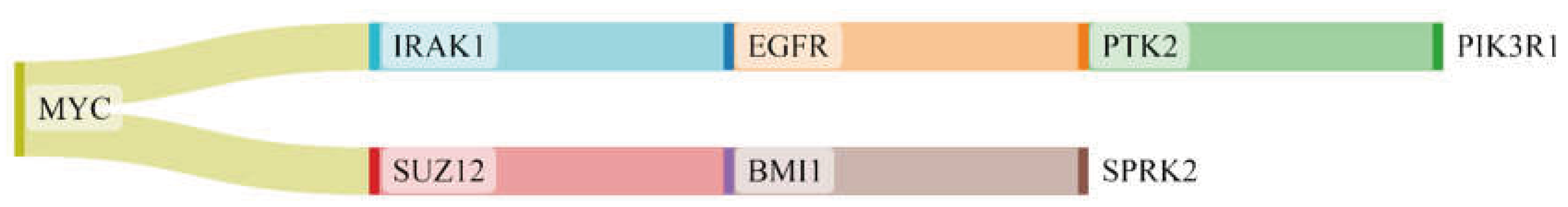
Figure 4.
Gene regulatory network obtained from eleven identified proteins. A wide range of interacting genes-miRNAs has been determined.
Figure 4.
Gene regulatory network obtained from eleven identified proteins. A wide range of interacting genes-miRNAs has been determined.
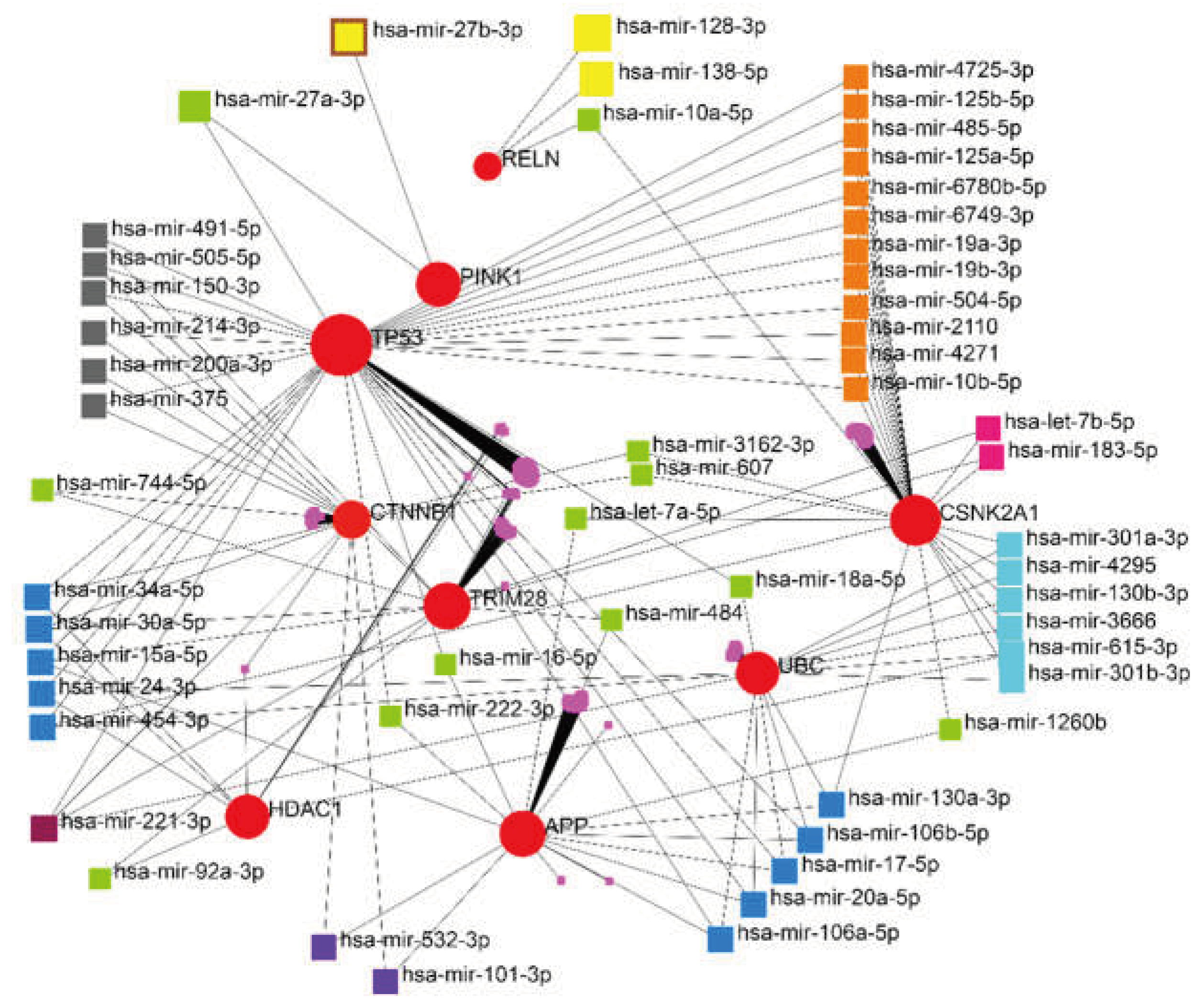
Figure 5.
The scheme of the relationship between the five identified miRNAs and their targets. hsa-mir-221-3p, and hsa-mir-30a-5p showed higher degree and betweenness centrality levels.
Figure 5.
The scheme of the relationship between the five identified miRNAs and their targets. hsa-mir-221-3p, and hsa-mir-30a-5p showed higher degree and betweenness centrality levels.
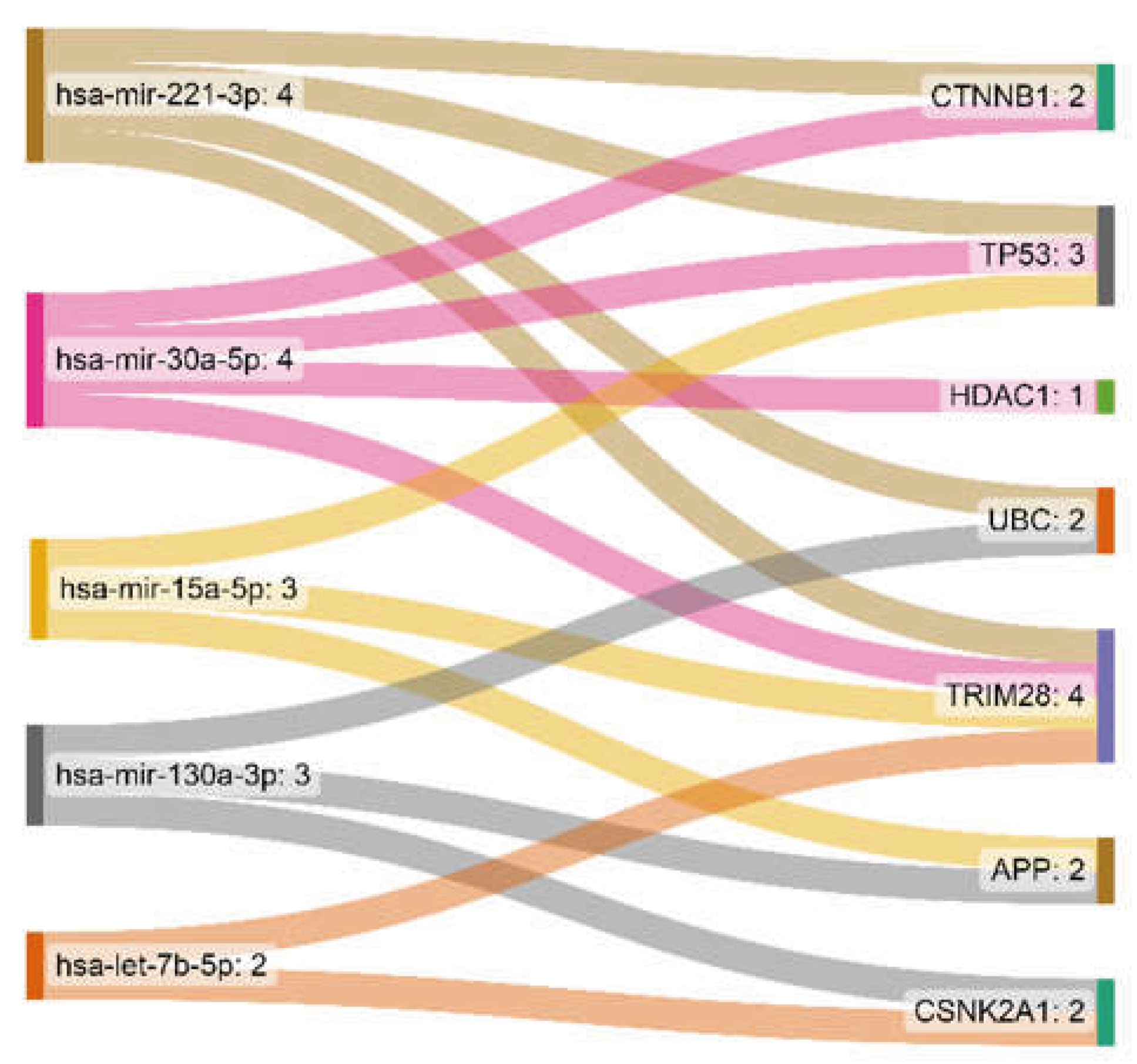
Figure 6.
The heatmap of the expression of eleven genes in normal brain tissue. A. Based on a microarray; B. Based on RNA-Seq.
Figure 6.
The heatmap of the expression of eleven genes in normal brain tissue. A. Based on a microarray; B. Based on RNA-Seq.
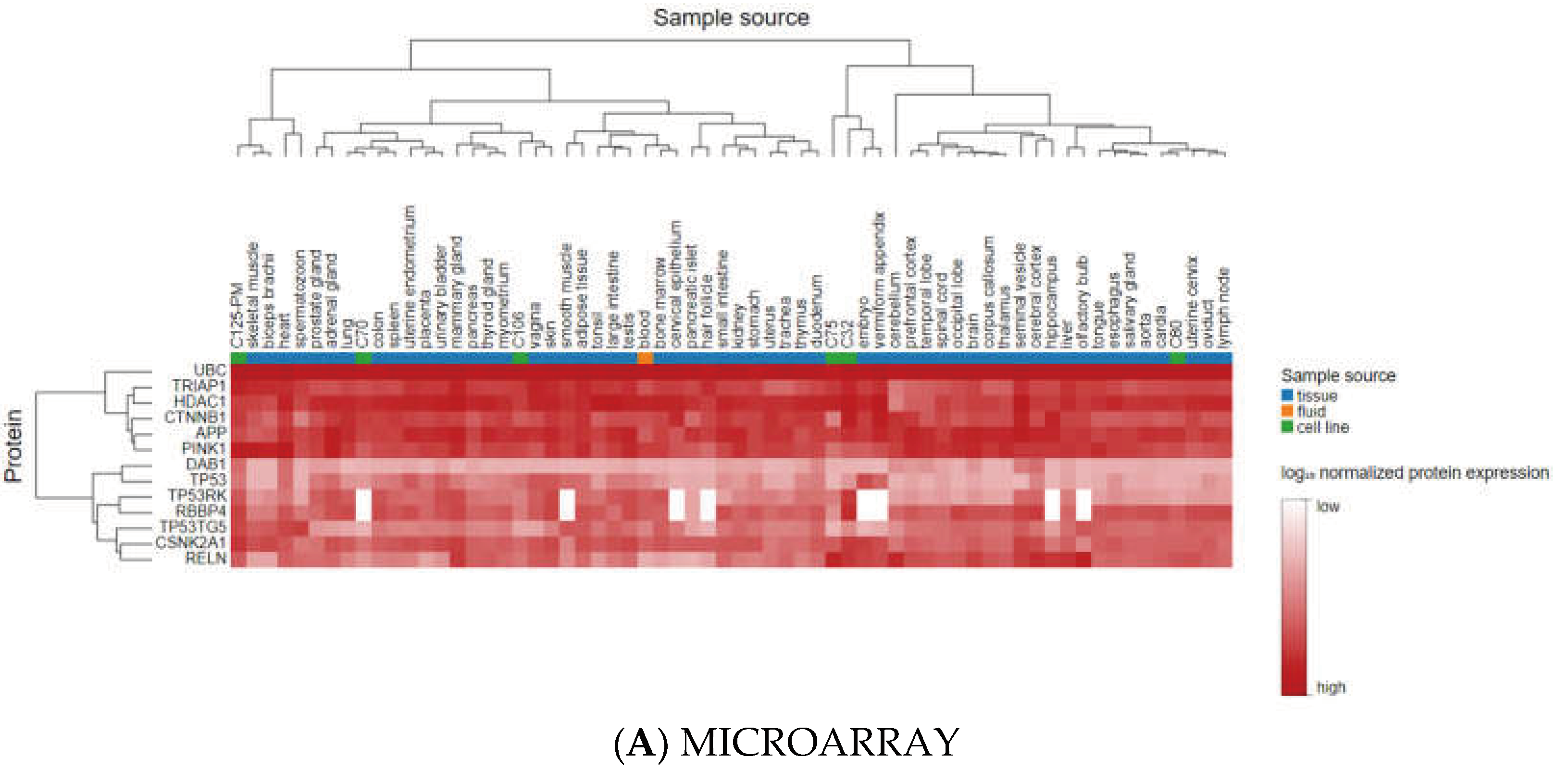
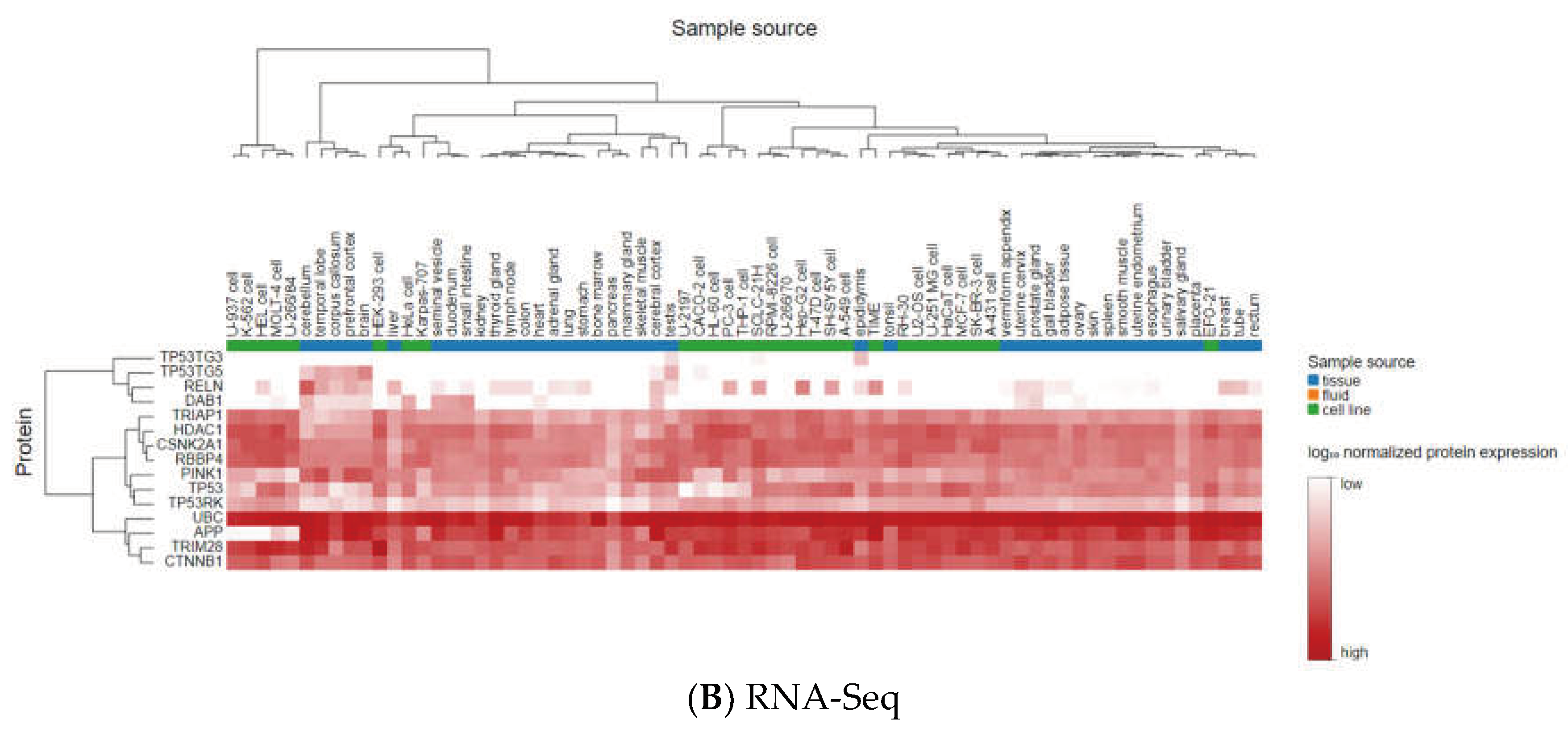
Figure 7.
The results of correlation analysis between eleven identified genes. Ten positive correlations were found between UBC (APP), HDAC1 (TP53, RBBP4, TRIM28, CTNNB1), RBBP4 (CTNNB1, TP53), TRIM28 (TP53, RBBP4, CSNK2A1), and eight negative correlations were found between RBBP4 (APP), PINK1 (TRIM28, RBBP4, TP53, HDAC1), and DAB1 (UBC, HDAC1, TP53).
Figure 7.
The results of correlation analysis between eleven identified genes. Ten positive correlations were found between UBC (APP), HDAC1 (TP53, RBBP4, TRIM28, CTNNB1), RBBP4 (CTNNB1, TP53), TRIM28 (TP53, RBBP4, CSNK2A1), and eight negative correlations were found between RBBP4 (APP), PINK1 (TRIM28, RBBP4, TP53, HDAC1), and DAB1 (UBC, HDAC1, TP53).
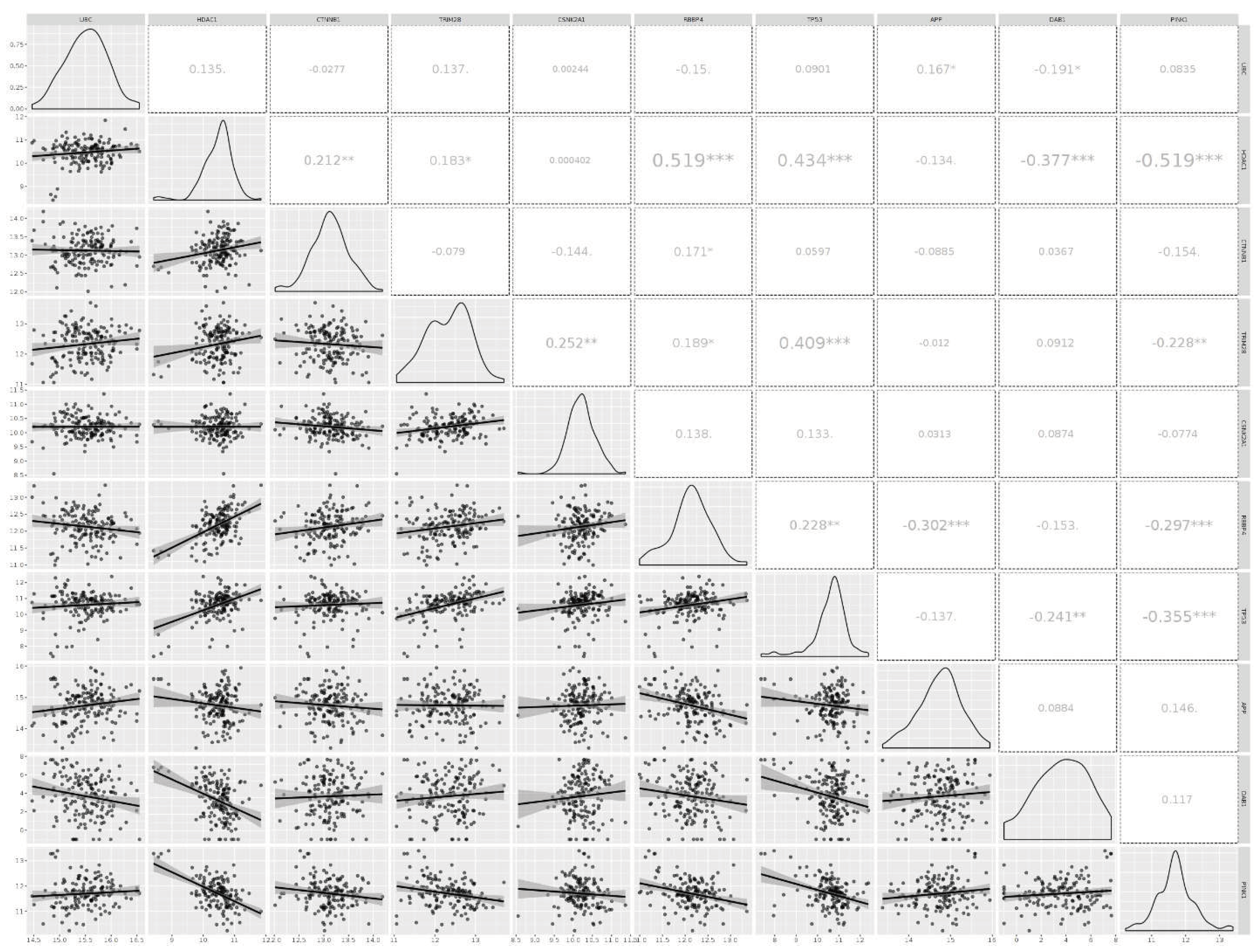
Figure 10.
The association of the results of metabolic pathway enrichment analysis with 182 metabolites: (A) Based on the KEGG database, (B) Based on the SMPDB database.
Figure 10.
The association of the results of metabolic pathway enrichment analysis with 182 metabolites: (A) Based on the KEGG database, (B) Based on the SMPDB database.
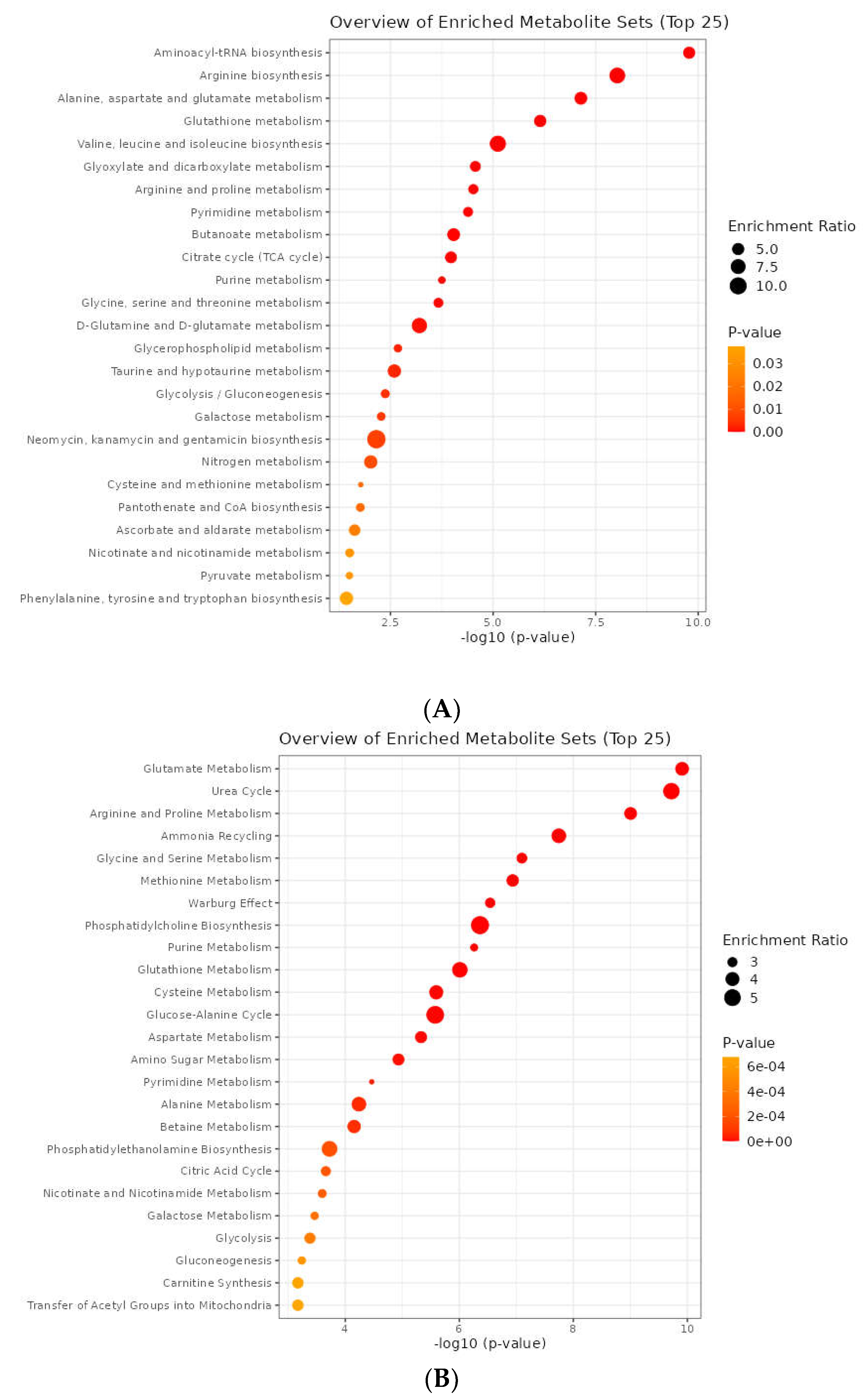
Figure 11.
Gene-Metabolite Interaction Network. The factors involved in the relationship between TP53, CTNNB1, CSNK2A1, and RELN with APP were further investigated.
Figure 11.
Gene-Metabolite Interaction Network. The factors involved in the relationship between TP53, CTNNB1, CSNK2A1, and RELN with APP were further investigated.
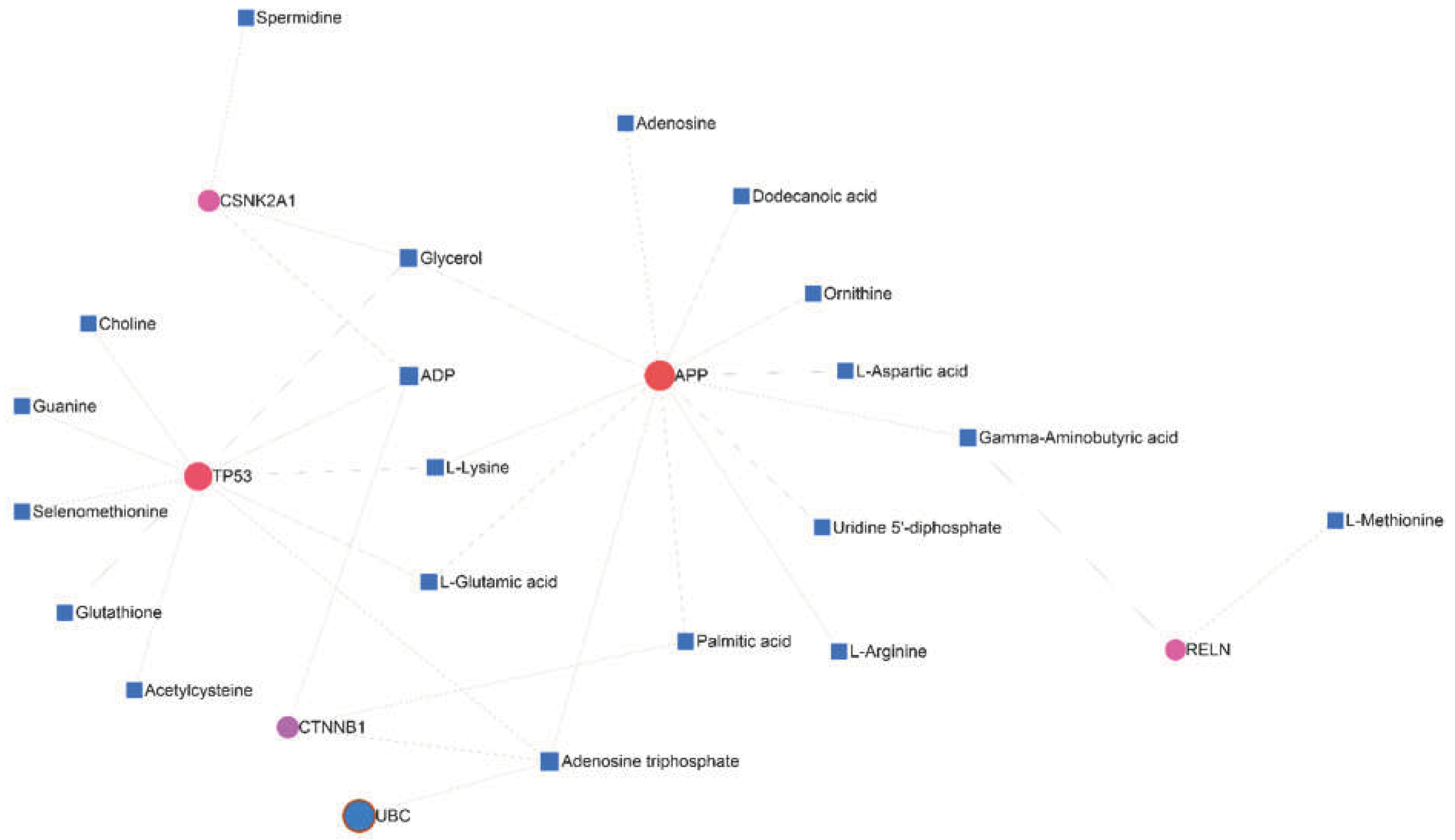
Figure 12.
The Determination of the top 25 SNPs via the MetaboAnalyst 5.0 database.
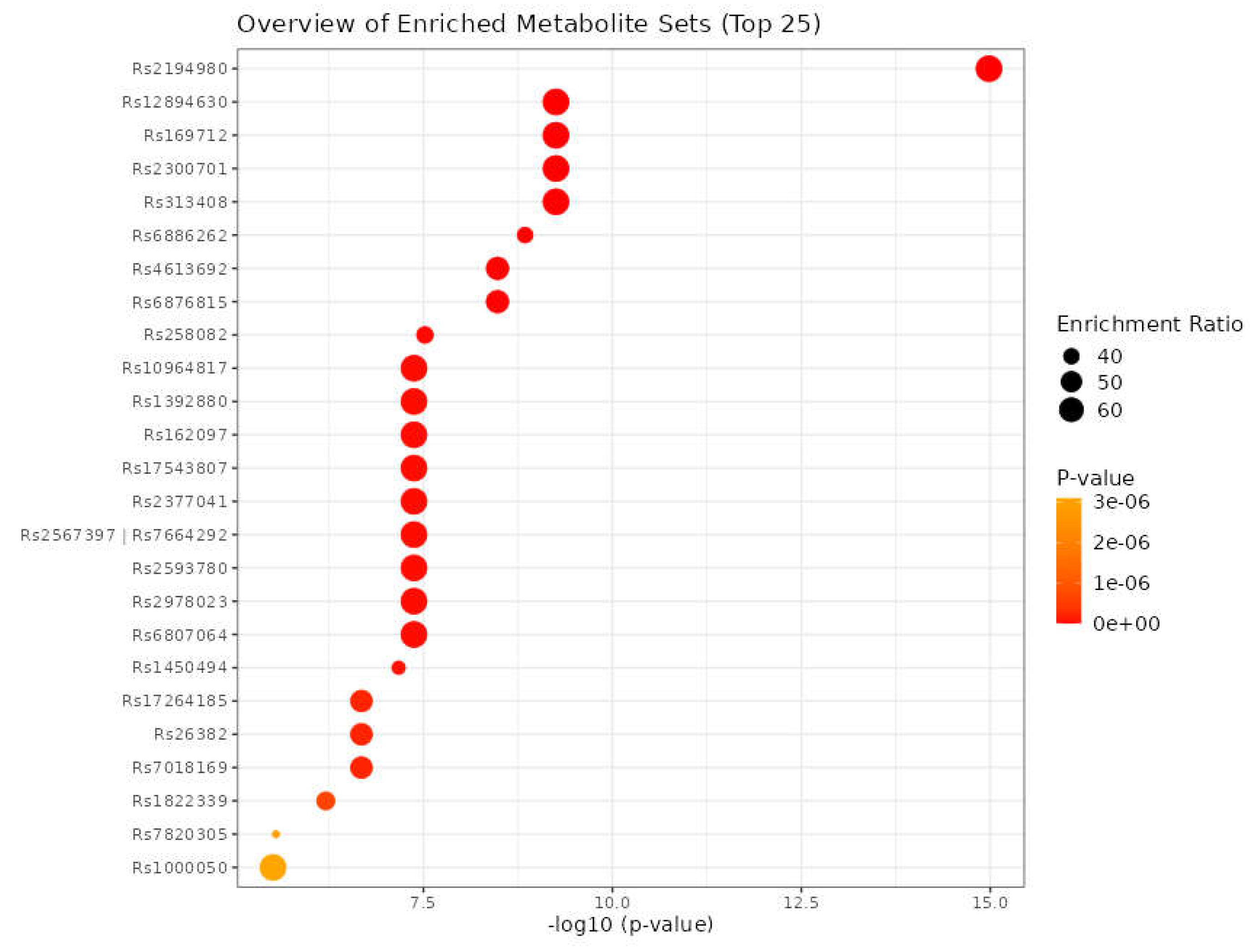

Table 1.
Data collection using 78 signaling pathways.
| 1 | VEGF signaling pathway- hsa04370 | 40 | TNF signaling pathway-hsa04668 |
|---|---|---|---|
| 2 | PI3K-Akt signaling pathway-hsa04151 | 41 | Citrate cycle (T.C.A. cycle)-hsa00020 |
| 3 | Ras signaling pathway- hsa04014 | 42 | Glycolysis / Gluconeogenesis-hsa00010 |
| 4 | TGF-beta signaling pathway- hsa04350 | 43 | Oxidative phosphorylation-hsa00190 |
| 5 | HIF-1 signaling pathway- hsa04066 | 44 | Starch and sucrose metabolism-hsa00500 |
| 6 | AMPK signaling pathway- hsa04152 | 45 | Pentose phosphate pathway- hsa00030 |
| 7 | MAPK signaling pathway - hsa04010 | 46 | Pyruvate metabolism- hsa00620 |
| 8 | Rap1 signaling pathway - hsa04015 | 47 | Insulin signaling pathway- hsa04910 |
| 9 | Wnt signaling pathway - hsa04310 | 48 | Lysosome-hsa04142 |
| 10 | Notch signaling pathway-hsa04330 | 49 | Phospholipase D signaling pathway- hsa04072 |
| 11 | Hedgehog signaling pathway -hsa04340 | 50 | Mitophagy- hsa04137 |
| 12 | Hippo signaling pathway -hsa04390 | 51 | Signaling pathways regulating pluripotency of stem cells- hsa04550 |
| 13 | JAK-STAT signaling pathway -hsa04630 | 52 | Cell adhesion molecules- hsa04514 |
| 14 | Apelin signaling pathway - hsa04371 | 53 | Cell cycle -hsa04110 |
| 15 | NF-kappa B signaling pathway-hsa04064 | 54 | ECM-receptor interaction-hsa04512 |
| 16 | TNF signaling pathway - hsa04668 | 55 | PD-L1 expression and PD-1 checkpoint pathway in cancer- hsa05235 |
| 17 | FoxO signaling pathway - hsa04068 | 56 | Pathways in cancer-hsa05200 |
| 18 | Phosphatidylinositol signaling system - hsa04070 | 57 | Transcriptional misregulation in cancer-hsa05202 |
| 19 | mTOR signaling pathway - hsa04150 | 58 | Central carbon metabolism in cancer- hsa05230 |
| 20 | p53 signaling pathway-hsa04115 | 59 | IL-17 signaling pathway-hsa04657 |
| 21 | Apoptosis-hsa04210 | 60 | Necroptosis-hsa04217 |
| 22 | Ubiquitin-mediated proteolysis- hsa04120 | 61 | Cellular senescence - hsa04218 |
| 23 | Cell cycle- hsa04110 | 62 | Chemokine signaling pathway-hsa04062 |
| 24 | Regulation of actin cytoskeleton - hsa04810 | 63 | Transcriptional misregulation in cancer-hsa05202 |
| 25 | Calcium signaling pathway- hsa04020 | 64 | ECM-receptor interaction- hsa04512 |
| 26 | T cell receptor signaling pathway- hsa04660 | 65 | Proteoglycans in cancer-hsa05205 |
| 27 | Focal adhesion- hsa04510 | 66 | Choline metabolism in cancer-hsa05231 |
| 28 | Adherens junction- hsa04520 | 67 | PD-L1 expression and PD-1 checkpoint pathway in cancer-hsa05235 |
| 29 | Gap junction-hsa04540 | 68 | Ferroptosis-hsa04216 |
| 30 | Tight junction- hsa04530 | 69 | Cholesterol metabolism- map04979 |
| 31 | Arachidonic acid metabolism- hsa00590 | 70 | Lipid and atherosclerosis-map05417 |
| 32 | Autophagy-hsa04140 | 71 | Fat digestion and absorption - map04975 |
| 33 | Regulation of lipolysis in adipocytes- hsa04923 | 72 | Vitamin digestion and absorption - map04977 |
| 34 | Cytokine-cytokine receptor interaction-hsa04060 | 73 | Aldosterone synthesis and secretion - map04925 |
| 35 | Proteasome- hsa03050 | 74 | Primary bile acid biosynthesis - map00120 |
| 36 | B cell receptor signaling pathway- hsa04662 | 75 | Cortisol synthesis and secretion - map04927 |
| 37 | Complement and coagulation cascades- hsa04610 | 76 | Bile secretion - map04976 |
| 38 | Toll-like receptor signaling pathway-hsa04620 | 77 | Ovarian steroidogenesis - map04913 |
| 39 | RIG-I-like receptor signaling pathway- hsa04622 | 78 | Steroid biosynthesis - map00100 |
Table 2.
The identification of eleven critical genes through the integration of results and network analysis. The list of important genes is based on their centrality. Deg: Degree, Bet: Betweenness, Bri: Bridge, Cent: Centroid, Close: Closeness, and EiVe: EigenVector.
Table 2.
The identification of eleven critical genes through the integration of results and network analysis. The list of important genes is based on their centrality. Deg: Degree, Bet: Betweenness, Bri: Bridge, Cent: Centroid, Close: Closeness, and EiVe: EigenVector.
| Gene Name | description | Deg | Bet | Bridg | Cent | Close | EiVe |
|---|---|---|---|---|---|---|---|
| UBC | Ubiquitin C [Source: HGNC Symbol; Acc: HGNC:12468 | + | + | -- | -- | + | + |
| HDAC1 | Histone deacetylase 1 [Source: HGNC Symbol; Acc: HGNC:4852 | + | -- | -- | -- | + | + |
| CTNNB1 | Catenin beta 1 [Source: HGNC Symbol; Acc: HGNC:2514 | + | -- | -- | -- | + | + |
| TRIM28 | Tripartite motif-containing 28 [Source: HGNC Symbol; Acc: HGNC:16384 | -- | + | -- | -- | + | + |
| CSNK2A1 | casein kinase two alpha 1 [Source: HGNC Symbol; Acc: HGNC:2457 | -- | -- | -- | -- | + | + |
| RBBP4 | RB binding protein 4, chromatin remodeling factor [Source: HGNC Symbol; Acc: HGNC:9887 | + | -- | -- | -- | -- | -- |
| TP53 | Tumor protein p53 [Source:HGNC Symbol;Acc:HGNC:11998 | + | -- | -- | -- | -- | -- |
| APP | Amyloid beta precursor protein [Source: HGNC Symbol; Acc: HGNC:620 | -- | + | -- | -- | -- | -- |
| DAB1 | DAB1, reelin adaptor protein [Source: HGNC Symbol; Acc: HGNC:2661 | -- | + | -- | -- | -- | -- |
| PINK1 | PTEN-induced putative kinase 1 [Source: HGNC Symbol; Acc: HGNC:14581 | -- | + | -- | -- | -- | -- |
| RELN | Reelin | literature review + miRNA-gene regulatory network | |||||
Table 3.
The identification of five Key miRNAs by consideration of two parameters (degree and betweenness centralities).
Table 3.
The identification of five Key miRNAs by consideration of two parameters (degree and betweenness centralities).
| Label | Degree | Betweenness |
|---|---|---|
| hsa-mir-221-3p | 4 | 5682.13 |
| hsa-mir-30a-5p | 4 | 2373.43 |
| hsa-mir-15a-5p | 3 | 3710.08 |
| hsa-mir-130a-3p | 3 | 3589.18 |
| hsa-let-7b-5p | 2 | 2523.74 |
Table 4.
The expression status of eleven genes identified in GBM disease. Six genes represent a significantly increased expression in the GBM state, whereas four genes were downregulated. All eleven specific genes were altered during the primary stage of the tumor.
Table 4.
The expression status of eleven genes identified in GBM disease. Six genes represent a significantly increased expression in the GBM state, whereas four genes were downregulated. All eleven specific genes were altered during the primary stage of the tumor.
| Gene Name | Non- tumor | GBM | Pairwise t-test (GBM-Non-tumor) p.adj (p-value with Bonferroni correction) |
Primary | Secondary | Recurrent |
|---|---|---|---|---|---|---|
| UBC | -- | + | 1.8E-03 | + | -- | -- |
| HDAC 1 | -- | + | 7.8E-18 | + | -- | -- |
| CTNNB1 | -- | + | 6.0E-03 | + | -- | -- |
| TRIM28 | -- | + | 1.1E-03 | + | -- | -- |
| CSNK2A1 | -- | + | 6.9E-01 (ns) | + | -- | -- |
| RBBP4 | -- | + | 3.2E-05 | + | -- | -- |
| TP53 | -- | + | 1.6E-13 | + | -- | -- |
| APP | + | -- | 1.2E-03 | + | -- | -- |
| DAB1 | + | -- | 4.0E-04 | + | -- | -- |
| PINK1 | + | -- | 2.9E-10 | + | -- | -- |
| RELN | + | -- | 5.7E-08 | + | -- | -- |
Table 5.
The application of multiple criteria in the metabolic pathway analysis.
| Result | Visualization methods | Enrichment method | Topology analysis | Reference metabolome | Pathway library |
|---|---|---|---|---|---|
| 1 | Scatter plot | Hypergeometric test | Relative-betweenness centrality R-b C |
All compounds in the selected pathway library | Homo sapiens (KEGG) |
| 2 | Scatter plot | Hypergeometric test | Out-degree Centrality O-d C |
All combinations in the selected pathway library | Homo sapiens (KEGG) |
| 3 | Scatter plot | Hypergeometric test | Relative-betweenness centrality | All compounds in the selected pathway library | Homo sapiens (SMPDB) |
| 4 | Scatter plot | Hypergeometric test | Out-degree Centrality | All combinations in the selected pathway library | Homo sapiens (SMPDB) |
Table 6.
The final results from the metabolic pathway analysis. Further discussion of two items from the KEGG database (nitrogen metabolism, alanine, aspartate, and glutamate metabolism), and three from the SMPDB database (alanine metabolism, aspartate metabolism, and malate-aspartate shuttle) due to vitality.
Table 6.
The final results from the metabolic pathway analysis. Further discussion of two items from the KEGG database (nitrogen metabolism, alanine, aspartate, and glutamate metabolism), and three from the SMPDB database (alanine metabolism, aspartate metabolism, and malate-aspartate shuttle) due to vitality.
| KEGG Database | SMPDB Database | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Result 1 | R-b C impact |
FDR | Result 2 | O-d C impact |
FDR | Result 3 | R-b C impact |
FDR | Result 4 | O-d C impact |
FDR |
| Final Decision (FD) | Final Decision (FD) | Final Decision (FD) | Final Decision (FD) | ||||||||
| Nitrogen metabolism | 1 | 0.043213 | Arginine biosynthesis | 0.8125 | 1.90E-07 | Alanine metabolism | 1 | 0.010641 | Malate-aspartate shuttle | 0.63333 | 0.013128 |
| FD: + | FD: -- | FD: + | FD: + | ||||||||
| Phenylalanine, tyrosine, and tryptophan biosynthesis | 1 | 0.12885 | Alanine, aspartate and glutamate metabolism | 0.75 | 1.73E-07 | Trehalose degradation | 0.84211 | 0.18355 | Phosphatidylcholine biosynthesis | 0.56707 | 0.00011577 |
| FD: -- | FD: + | FD: -- | FD: -- | ||||||||
| Synthesis and degradation of ketone bodies | 0.86667 | 0.18716 | Valine, leucine, and isoleucine biosynthesis | 0.75 | 8.82E-05 | Aspartate metabolism | 0.8 | 0.0044894 | Transfer of acetyl groups into mitochondria | 0.54167 | 0.010641 |
| FD: -- | FD: -- | FD: + | FD: -- | ||||||||
| Alanine, aspartate and glutamate metabolism | 0.81732 | 1.73E-07 | Nitrogen metabolism | 0.75 | 0.043213 | Glycerol phosphate shuttle | 0.7619 | 0.3023 | Ammonia recycling | 0.49306 | 0.00011577 |
| FD: + | FD: + | FD: -- | FD: -- | ||||||||
| One-carbon pool by folate | 0.80793 | 0.46957 | Phenylalanine, tyrosine, and tryptophan biosynthesis | 0.75 | 0.12885 | Malate-Aspartate Shuttle | 0.71429 | 0.013128 | Cardiolipin biosynthesis | 0.49057 | 0.013128 |
| FD:-- | FD:-- | FD: + | FD: -- | ||||||||
Table 7.
The application of multiple criteria in the joint pathway analysis.
| Result | Enrichment method | Topology measure | Integration method |
|---|---|---|---|
| 1 | Hypergeometric test | Degree centrality | Combined score |
| 2 | Betweenness centrality | ||
| 3 | Closeness centrality |
Table 8.
The final results from joint pathway analysis. The two items (citrate cycle and arginine biosynthesis) observed in all three centralities were selected and discussed further.
Table 8.
The final results from joint pathway analysis. The two items (citrate cycle and arginine biosynthesis) observed in all three centralities were selected and discussed further.
| Title | Degree | Betweenness | Closeness |
|---|---|---|---|
| Alanine, aspartate and glutamate metabolism | + | + | -- |
| Citrate cycle (TCA cycle) | + | + | + |
| Arginine biosynthesis | + | + | + |
| Synthesis and degradation of ketone bodies | + | -- | + |
| Pyruvate metabolism | + | -- | + |
| Purine metabolism | + | + | -- |
| Glutathione metabolism | + | + | -- |
| Pyrimidine metabolism | + | + | -- |
| Glycolysis or gluconeogenesis | -- | + | + |
Table 9.
The summary of all results obtained from the pathway enrichment analysis at different levels of pathway, and joint pathway analyses.
Table 9.
The summary of all results obtained from the pathway enrichment analysis at different levels of pathway, and joint pathway analyses.
| Enrichment Analysis | Pathway analysis | Joint pathway analysis | ||
|---|---|---|---|---|
| Eleven Genes | Five miRNAs | 182 Metabolites | ||
| Mitophagy | Fatty acid biosynthesis | Aminoacyl-tRNA biosynthesis | Nitrogen metabolism | Citrate cycle (TCA cycle) |
| Wnt signaling pathway | Galactose metabolism | Arginine biosynthesis | Alanine, aspartate and glutamate metabolism | Arginine biosynthesis |
| Mucin-type O-glycan biosynthesis | Alanine, aspartate and glutamate metabolism | Malate-aspartate shuttle | ||
| Autophagy | Glutamate metabolism | |||
| Urea cycle | ||||
| Arginine and proline metabolism | ||||
Table 10.
The metabolites and SNPs from the mGWAS-Explorer database.
| Metabolite | SNP |
|---|---|
| HDL | rs111929233, rs7298751 |
| N6-acetyllysine | rs12602273, rs12603869, rs12945970, rs12947788, rs12949655, rs12951053, rs1642782, rs17881556, rs1794284, rs2078486, rs5819163 |
| Cholesterol | rs35608584, rs111929233 |
| Formate | rs17520463 |
| N, N-Dimethylglycine/Xylose | rs41450451 |
| X2.piperidinone | rs75787097, rs75524270, rs79232054, rs145435197, rs74901488, rs117235978 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



